
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 23 June 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.701229
This article is part of the Research Topic Lipids and Inflammation in Health and Disease View all 26 articles
 Takuro Miyazaki*
Takuro Miyazaki* Akira Miyazaki
Akira MiyazakiLymphatic vessels are necessary for maintaining tissue fluid balance, trafficking of immune cells, and transport of dietary lipids. Growing evidence suggest that lymphatic functions are limited under hypercholesterolemic conditions, which is closely related to atherosclerotic development involving the coronary and other large arteries. Indeed, ablation of lymphatic systems by Chy-mutation as well as depletion of lymphangiogenic factors, including vascular endothelial growth factor-C and -D, in mice perturbs lipoprotein composition to augment hypercholesterolemia. Several investigations have reported that periarterial microlymphatics were attracted by atheroma-derived lymphangiogenic factors, which facilitated lymphatic invasion into the intima of atherosclerotic lesions, thereby modifying immune cell trafficking. In contrast to the lipomodulatory and immunomodulatory roles of the lymphatic systems, the critical drivers of lymphangiogenesis and the details of lymphatic insults under hypercholesterolemic conditions have not been fully elucidated. Interestingly, cholesterol-lowering trials enable hypercholesterolemic prevention of lymphatic drainage in mice; however, a causal relationship between hypercholesterolemia and lymphatic defects remains elusive. In this review, the contribution of aberrant lymphangiogenesis and lymphatic cholesterol transport to hypercholesterolemic atherosclerosis was highlighted. The causal relationship between hypercholesterolemia and lymphatic insults as well as the current achievements in the field were discussed.
It is widely known that plasma dyslipidemia, which refers to elevation of plasma cholesterol and/or triglyceride levels, impacts chronic inflammatory diseases, such as type 2 diabetes mellitus and obesity. In particular, hypercholesterolemia can be responsible for lethal ischemic diseases, including acute coronary syndrome and stroke, which is the leading cause of death globally (1), as it is responsible for atherosclerotic vascular disease. Accordingly, controlling lipoprotein cholesterol is necessary for the primary and secondary prevention of ischemic diseases (2). The typical dyslipidemia pattern in type 2 diabetes comprises decreased high-density lipoprotein (HDL) cholesterol and, occasionally, elevated low-density lipoprotein (LDL) cholesterol levels (3). As a result, these metabolic diseases influence each other and worsen overall disease status. Oxidatively- and enzymatically-modified LDL can directly induce cytotoxic responses in blood vessels as well as metabolic organs. Much research has focused on such insults under the dyslipidemic conditions.
The lymphatic vessel network, which is spread throughout the body, is necessary for sustaining systemic fluid balance, intestinal absorption of fats, and draining waste products from the peripheral tissues. Lymphatic vessels, which are comprised of lymphatic endothelial cells (LECs), enable the production of chemokines to attract immune cells to the interstitial regions within the tissues (4), and interact with innate and adaptive immune cells. Among the LEC-derived chemokines, CCL21 has a pivotal role in the recruitment of dendritic cells (DCs) (5, 6). LECs can impact the adaptive immune system to regulate peripheral tolerance and immunomodulation (7). Microlymphatics around large blood vessels were discovered more than a 100 years ago (8). Notably, lymphatic dysfunction caused by Chy mutation or the deficiency of lymphangiogenesis factors reportedly potentiates atherosclerotic lesion progression (9, 10). This appears to be due to the limitation of reverse cholesterol transport. Recently, it was documented that hypercholesterolemia potentiates lymphangiogenesis around the atherosclerotic arteries, thereby modifying immune cell trafficking in lymphatic systems and atherosclerotic lesions (11). In contrast, the exact mechanisms underlying lymphatic insults as well as the causal relationship between hypercholesterolemia-driven lymphangiogenesis and atherosclerotic diseases are largely obscure. In this review, we focused on the relationship between hypercholesterolemia-driven lymphatic defects and atherosclerosis. The dysfunctional regulation of LECs under hypercholesterolemic conditions is also discussed.
In the late 18th century, Hoggan et al. discovered microlymphatics adjacent to the arterial wall (8). About a century later, it was reported that dysfunctional lymphatic drainage in allogenic transplanted hearts could be responsible for the progression of coronary atherosclerosis (12). More recently, it was reported that microlymphatics are enriched in the adventitial regions of human atherosclerotic plaques (13, 14). Notably, the density of the lymphatics is positively correlated with the severity of atherosclerosis; thus, it is thought that inflammatory elements, such as cytokines, chemokines, and growth factors, may induce lymphangiogenesis within atherosclerotic plaques (13). In addition to the adventitial regions, lymphatics appear to be detectable within the intraplaque regions of human carotid arteries (15). While some studies documented the enrichment of the lymphatics in atherosclerotic lesions, several other studies challenged this. Eliska et al. could detect microlymphatics in the periadventitial regions in human coronary arteries, whereas the vessels did not penetrate into the vascular walls even in normal and atheroprone arteries (16). Nakano et al. noted that microlymphatics in human coronary atheromas, which are mainly detectable in adventitial regions, were not correlated with the severity of atherosclerosis as well as the expression of lymphatic driver vascular endothelial growth factor-C (VEGF-C) and VEGF-D (17). Apoe-deficient hypercholesterolemic mice showed abundant adventitial lymphatics in comparison with age-matched wild-type mice, while these were reduced during the progression of the lesions (18). They interpreted that soluble VEGFR-2, which is upregulated in advanced atherosclerotic lesions, interrupts VEGF-C-induced lymphangiogenesis. Collectively, the drivers as well as mechanisms underlying the hypercholesterolemic regulation of periarterial microlymphatics, are currently unclear. Whereas the lymphatic regulation within other organs in hypercholesterolemic mice is mostly unclear, sinus lymphatic vessels within lymph nodes are shown to have hyperplastic appearance under hypercholesterolemic conditions (19).
Atherosclerosis is a vascular disease in which cholesterol-enriched atherosclerotic lesions develop within the arterial intima, resulting in narrowing of the luminal vascular walls (20). Structural changes involving the arterial lumen lead to thrombus formation due to perturbation of laminar blood flow (21), resulting in arterial occlusion. In addition to blood clotting, arterial occlusion is induced via rupture of unstable atherosclerotic plaques (22). Vascular endothelial cell dysfunction is associated with the initiation of atherosclerosis (23). In the early stage of atherosclerosis, endothelial barrier functions are disrupted by environmental stressors, such as oxidative stress and inflammatory substances (24). This reduced barrier function is responsible for the infiltration of leukocytes and extravasation of plasma ingredients, including oxidative LDL. In particular, monocyte-derived macrophages tend to accumulate within the vascular walls. Monocytes normally patrol the circulatory system and are attracted by endothelial cell-derived chemoattractants, such as monocyte chemoattractant protein-1. After recruitment into the intimal space, monocytes differentiate into macrophages, which are further converted to cholesterol-enriched foamy macrophages (25). Since excessive accumulation of intracellular cholesterol induces cytotoxicity, deposition of LDL cholesterol in the vascular wall accelerates the necrosis of macrophages to form cholesterol-enriched unstable plaques, which contain large necrotic cores. During atherogenesis, lymphangiogenic drivers, including VEGF-C, appear to be enriched within atherosclerotic lesions (17, 18).
Recently, several studies have highlighted lymphatic defects under hypercholesterolemic conditions as well as the contribution of lymphatic systems to hypercholesterolemia (Table 1). Martel et al. documented the role of aortic microlymphatics in cholesterol drainage from atheromas (9). They transplanted atheroprone aortae derived from D6-cholesterol-loaded Apoe-deficient mice into Apoe-deficient recipient mice. The leakage of D6-cholesterol from the transplanted tissue was then monitored. As a result, lymphangiogenesis was detected within the connecting region between the recipient and implanted vessels, and D6-cholesterol was transferred to recipient mice. Both lymphangiogenesis and cholesterol drainage from the transplanted tissues were prevented via the administration of anti-VEGFR3 blocking antibody, suggesting that cholesterol drainage under hypercholesterolemic conditions is mediated through angiogenic microlymphatics. Vuorio et al. documented that insufficiency of lymphatic vessels by transgenic induction of sVEGFR3 or Chy mutant augments hypercholesterolemia in atherogenic mice (10). They crossed low-density lipoprotein receptor/apolipoprotein B48-double knockout mice with sVEGFR3- transgenic mice or Chy-mutant mice to induce lymphatic insufficiency in hypercholesterolemic mice. As a result, sVEGFR3-induced lymphatic insufficiency facilitated the progression of atherosclerotic lesions. Rademakers et al. reported that dissection of microlymphatics, which connect the carotid artery and regional lymph nodes, resulted in the expansion of carotid atherosclerotic lesions (11). Since surgical lymphatic dissection appears to induce accumulation of CD3+ T cells, but not macrophages, adventitial microlymphatics may be a route for emigrating CD3+ T cells from the lesions. Collectively, lymphatic insufficiency exacerbates atherosclerotic lesion progression within arteries.
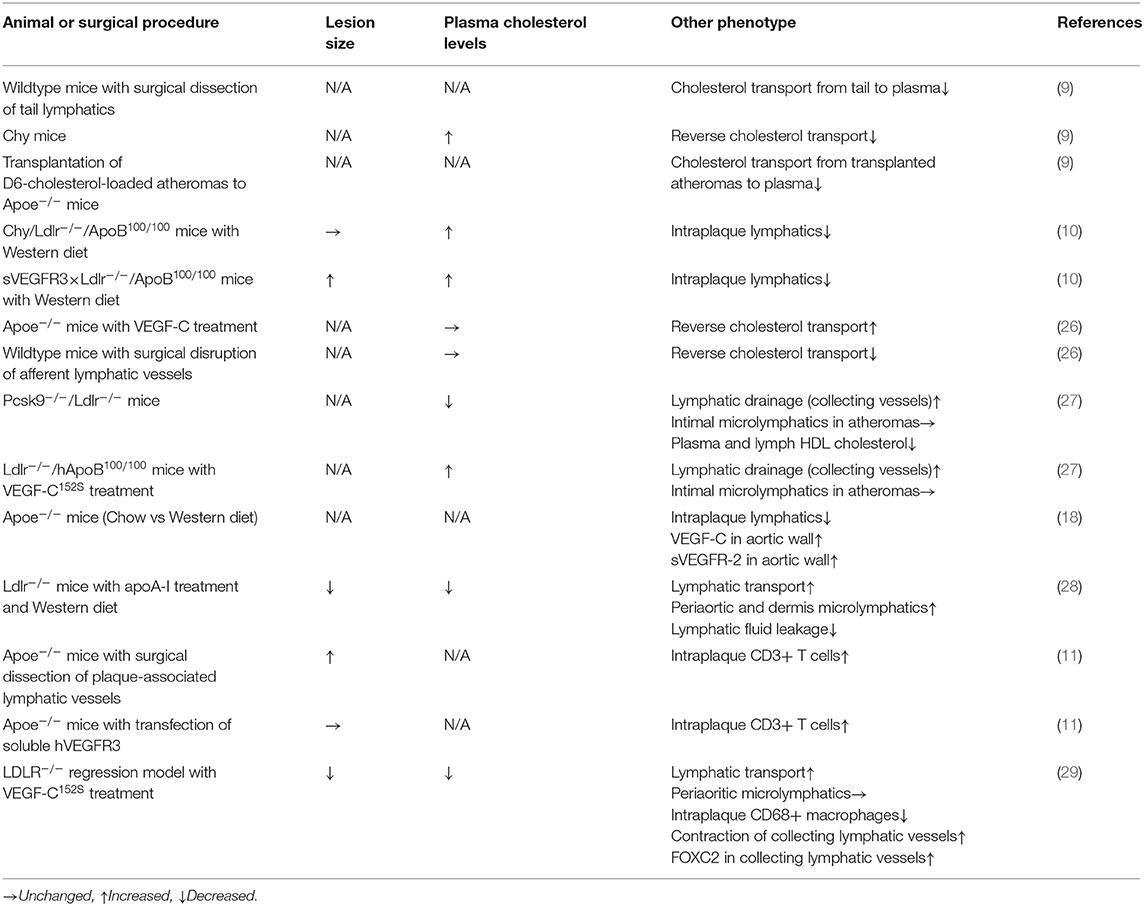
Table 1. Lymphatic function-related assessment and atherosclerosis-related phenotypes in normocholesterolemic and hypercholesterolemic animal models.
Reverse cholesterol transport is indispensable for the clearance of excessive cholesterol from peripheral tissues (30). Mechanistically, ATP-binding cassette transporter A1 and ATP-binding cassette transporter G1 in peripheral tissues integrate intracellular cholesterol into apolipoprotein A-I (ApoA-I), a constitutive apolipoprotein of HDL, thereby generating HDL cholesterol. Subsequently, plasma and lymph HDL cholesterol is incorporated into hepatocytes through scavenger receptor B1 (SR-B1), and HDL receptors (31). Accordingly, defective reverse cholesterol transport induces elevation of plasma cholesterol levels, leading to severe hypercholesterolemia. Some studies have documented that lymphatic dysfunction leads to defective reverse cholesterol transport. Hypercholesterolemia in Apoe-deficient mice reportedly abrogates lymphatic drainage of intravenously injected HDL as well as its transport to the liver (26). SR-B1 in LECs plays a central role in lymphatic reverse cholesterol transport, since this molecule can mediate transcytosis of HDL in the cells (26). Furthermore, administration of ApoA-I in hypercholesterolemic mice suppressed atherosclerosis development, accompanied by resolution of lymphatic hyperpermeability involving collecting lymphatic vessels (28). While ApoA-I upregulates VEGFR3 in cultured LECs, the exact mechanisms by which ApoA-I retains lymphatic function remain elusive. Since the VEGF-C/VEGFR3 signaling pathway reportedly increase permeability in LECs (32), causal relationship between VEGFR3 and the ApoA-I-induced lymphatic protection is unknown. In addition to VEGFR3 induction, ApoA-I appears to potentiate platelet adhesion to LECs. Platelets are necessary for blood/lymphatic vessel separation during embryogenesis (33) and maintenance of the lymphvenous junction throughout life (34). Since platelets reportedly confer stability to LECs through physical interaction between C-type lectin-like receptor 2 in platelets and podoplanin in LECs, it is speculated that ApoA-I-induces platelet adhesion to LECs, stabilizing lymphatic vessels. Similar to ApoA-I, deficiency of proprotein convertase subtilisin/kexin type 9 (PCSK9), a negative regulator of LDL receptor, recovered lymphatic drainage in hypercholesterolemic mice concomitantly with the suppression of atherosclerotic lesions (27). Indeed, the LDL receptor, which is expressed in LECs in mice, is downregulated in hypercholesterolemic mice concomitantly with elevation of plasma PCSK9 levels. Consistently, downregulated LDL receptors in LECs are recovered via the targeted deficiency of PCSK9 (27). Retaining LDL receptors in LECs was not associated with the density of lymphatic vessels. Thus, LDL receptors may contribute to lymphatic integrity rather than lymphangiogenic functions. Furthermore, administration of ezetimibe, a cholesterol-lowering drug, in hypercholesterolemic mice retained lymphatic drainage (26). These observations suggest that hypercholesterolemia, accompanying with declining plasma HDL-associated ApoA-I, may disrupt the integrity of LECs and their barrier functions, particularly in large lymphatic vessels, concomitantly with dysfunction of SR-B1 in microlymphatics.
Since lymphatic insufficiency induced by transgenic overexpression of sVEGFR3 reportedly elevated plasma cholesterol levels in hypercholesterolemic mice, particularly regarding VLDL and LDL fractions (10), it is likely that endogenous VEGF-C and/or VEGF-D signaling pathways may contribute to the clearance of these lipoproteins even under hypercholesterolemic conditions. Lymphatic vessels are reportedly associated with endogenous lipoprotein metabolism through VLDL and LDL, as well as intestinal absorption of chylomicrons (35). Therefore, VEGFR3-mediated lymphatic modulation may be attributed to cholesterol transport toward peripheral tissues through VLDL and LDL. Since VLDL and LDL modification by lymphatic insufficiency appear to be independently of HDL (10), these actions are probably independent of HDL-associated reverse cholesterol transport. In addition to overexpressing soluble VEGFR3, targeted deficiency of VEGF-D exacerbates hypercholesterolemia (36). Indeed, VEGF-D deficiency substantially elevates plasma cholesterol, particularly in chylomicron and chylomicron remnant fractions. In this case, VEGF-D appears to contribute to the hepatic transcriptional regulation of lipid handling elements as well as the incorporation of chylomicron remnants into the liver, independent of its lymphangiogenic effects. Accordingly, VEGF-D deficiency did not accelerate atherosclerosis progression in hypercholesterolemic mice. Hence, lymphangiogenic factors, at least VEGF-D, may have pleiotropic lipomodulatory functions, in addition to their lymph-modulatory functions. Collectively, hypercholesterolemia appears to abrogate transcytosis of HDL within microlymphatics as well as integrity in large lymphatic vessels as note above, whereas the contribution of periarterial microlymphatics to atherogenesis and the exact pathophysiologic mechanisms underlying the hypercholesterolemic lymphatic insults are largely unknown.
VEGF-C is a robust lymphangiogenic factor, which is indispensable for lymphatic development (37). Several lines of evidence suggest that VEGF-C is associated with lymphatic patterning under hypercholesterolemic conditions. In addition to VEGF-C, several modulatory elements that can be involved in lymphangiogenesis under hypercholesterolemia have been reported to date.
VEGF-C is abundantly expressed in foamy macrophages and smooth muscle cells in human coronary atherosclerotic lesions (17). Furthermore, VEGF-C is reportedly elevated in moderate to advanced atheromas involving hypercholesterolemic mice (18). However, it is noteworthy that adventitial microlymphatics regress during lesion progression. Targeted delivery of VEGF-C to atherosclerotic lesions failed to induce adventitial lymphangiogenesis (38). Accordingly, the association between VEGF-C accumulation during atherogenesis and periarterial lymphangiogenesis remains unclear. Pretreatment of hypercholesterolemic mice with VEGF-C reportedly improved the contraction rate of collecting lymphatic vessels and lymphatic transfer of inflammatory cells, thereby suppressing atherosclerotic lesions (29). This suggests that VEGF-C may impact the trafficking of immune cells via the collecting vessels under hypercholesterolemic conditions rather than through prolymphangiogenic actions in periarterial regions.
VEGF-D has a close structural and functional similarity to VEGF-C (39) and possesses robust angiogenic and lymphangiogenic activity. Similar to VEGF-C, VEGF-D action is mediated through VEGFR3 (40), though it does not play a major role in lymphatic development in mice (41). As noted above, targeted deficiency of VEGF-D in hypercholesterolemic mice elevated plasma cholesterol and triglyceride levels without altering lymphangiogenesis and atherosclerosis (36). Transgenic induction of soluble VEGFR3, which can interrupt VEGF-C/D-induced signaling, was unable to inhibit adventitial lymphangiogenesis (11). Hence, the lymphangiogenic effects of VEGFR3 signaling under hypercholesterolemic conditions remain elusive.
Sphingosine-1-phosphate (S1P) is a bioactive lipid synthesized from ceramide (42). In the initial step of its synthesis, ceramidase converts ceramide into sphingosine. Subsequently, sphingosine kinase (Sphk)1 and Sphk2 phosphorylate sphingosine to form S1P (43). Five subtypes of S1P receptors, including S1PR1- S1PR5, have been reported to be involved in various physiological and pathophysiological events, such as cardiovascular regulation, immune regulation, neurodevelopment, neuroprotection, and fibrogenic responses (44). Among them, S1PR1 was originally cloned from vascular endothelial cells (45). Notably, S1P is abundantly carried on HDL (46) and is believed to contribute to the antiatherogenic action of HDL (47). Lyve1-driven ablation of Sphk1 and lacking Sphk2 interrupt the production of S1P in LECs, thereby depleting S1P within the lymph, but not in the plasma (48). Such lymphatic S1P depletion induces aberrant lymphatic morphology involving the trachea and diaphragm, and interrupts lymphocyte egress from peripheral tissues, indicating that LEC-derived S1P plays a crucial role in lymphatic patterning and lymphocyte trafficking. Consistently, S1P is reportedly involved in the transmigration of lymphocytes in LECs through S1PR2/ERK-mediated regulation of junctional proteins, including VE-cadherin, occludin, zonulin-1, and VCAM1 expression (49). Importantly, S1P has been shown to decline in LNs of hypercholesterolemic mice (19). Moreover, hypercholesterolemia appears to accelerate lymphangiogenesis within LNs and impair lymphocyte egress from these LNs. Although intervention of S1P signaling in hypercholesterolemic mice has not been performed so far, S1P depletion in hypercholesterolemic mice can impact lymphatic pattering and lymphocyte trafficking.
Lysophosphatidic acid (LPA), a multifunctional bioactive lysophospholipid, is involved in the pathogenesis of atherosclerosis (50–53). LPA can be generated via several enzymatic pathways (54). For instance, LPA is synthesized from lysophosphatidylcholine by the action of autotaxin (lysophospholipase D). Alternatively, phospholipase A2 mediates the conversion of phosphatidic acid to LPA through hydrolysis of its sn-2 acyl chain. In addition to the synthetic pathways noted above, LPA can be derived from glycerol-3-phosphate by glycerol-3-phosphate acyltransferase-induced addition of fatty acids toward the sn-1 position. Currently, six species of G-protein-coupled LPA receptors, LPA1-LPA6, have been reported (51). Among these receptors, LPA4 is involved in lymphatic vessel formation during embryogenesis (55). Moreover, LPA contributes to NF-κB-mediated induction of IL-8 in human dermal LECs, thereby potentiating lymphangiogenesis (56). Furthermore, it has been documented that LPA1 is coupled with S1PR1 to induce S1PR1/β-arrestin coupling and inhibit Gαi signaling (57). This potentiates the disorganization of intercellular junctions in sinus-lining LECs within LNs, which facilitates the lymphatic transfer of lymphocytes toward the LNs. Of note, LPA reportedly accumulates within atherosclerotic lesions in human and hypercholesterolemic mice (58, 59). While the prolymphangiogenic roles of LPA in hypercholesterolemic conditions have not been proved in human and animal experimental models, it is speculated that LPA in atheromas attracts adjacent LECs to induce lymphangiogenesis.
Nitric oxide (NO) is primarily derived from vascular endothelial cells through endothelial NO synthase (eNOS) and its substrate L-arginine (60), and is associated with vasodilation when adjusting regional pressure-flow balance (61–63). In addition to its vasomotor functions, NO possesses robust angiogenic effects on vascular endothelial cells (64, 65). Mechanistically, treatment of vascular endothelial cells with NO leads to cGMP-dependent activation of protein kinase G through guanylate cyclase to upregulate target molecules (66). Furthermore, NO stabilizes hypoxia-inducible factor-1α and the subsequent production of VEGF-A (67). Importantly, VEGF-C activates eNOS to generate NO in LECs. Indeed, treatment of human dermal LECs with VEGF-C potentiates lymphangiogenesis through the activation of eNOS (68). Targeted deficiency of eNOS or pharmacological inhibition of eNOS suppresses peritumoral lymphatic hyperplasia (68). Moreover, treatment of LECs with NO donors potentiates lymphangiogenic tube formation concomitantly with elevation of cGMP levels in affected cells (69). Singla et al. identified matrix protein R-spondin 2 (RSPO2) as a lymphangiogenesis inhibitor (70). RSPO2 expression was noted in vascular endothelial cells and LECs, which counteracted the VEGF-C-induced activation of AKT, resulting in the depletion of NO and subsequent lymphangiogenesis. Since a variety of vasoactive elements can potentiate NO production, it is possible that perturbation of these elements under hypercholesterolemic conditions disturbs lymphangiogenesis.
There is another possibility of NO-mediated pathogenic lymphatic modifications. Liao et al. documented that NO derived from iNOS-overexpressing CD11b+ myeloidal DCs, which accumulated around the subcutaneous lymphatic vessels during oxazolone-induced contact sensitization, could weaken rhythmic lymphatic contraction (71). Intriguingly, lymphatic contraction appears to be associated with T cell activation as well as the pathogenesis of oligodendrocyte glycoprotein peptide 35–55-induced autoimmune encephalomyelitis (71), suggesting that lymphatic contraction has an immunomodulatory role during inflammation. While VEGF-C reportedly improves contractile functions in collecting lymphatic vessels, as noted above (29), there is no evidence of perilymphatic accumulation of DCs and aberrant contractile regulation of collecting lymphatic vessels under hypercholesterolemic conditions. Hence, future studies should investigate NO homeostasis and immune cell composition around large lymphatic vessels.
Calpain, a superfamily of intracellular Ca2+-dependent proteases, can be defined as molecules possessing a calpain-like cysteine protease sequence (CysPc) motif. Currently, 15 species of calpain isozymes have been identified in mammals (72, 73). Among the calpain subtypes, calpain-1 and calpain-2 are classified into conventional calpains and comprise a heterodimer of the small regulatory subunit CAPNS1 and their unique catalytic subunits CAPN1 and CAPN2, respectively (74). Conventional calpains can be intracellularly activated in response to various physiological and pathogenic stressors, such as lysophospholipids, hypoxia, cytokines, and growth factors, thereby proteolyzing functional cellular proteins through limited proteolytic cleavage (74, 75). As a result of proteolysis, calpain enables the modification of cellular functions and phenotypes. Our previous study showed that conventional calpains in vascular endothelial cells are involved in pathological angiogenesis in oxygen-induced retinopathy and cancer allograft models in mice (76). Mechanistically, activation of the conventional calpains in vascular endothelial cells elicits shedding of suppressor of cytokine signaling protein 3, resulting in the sensitization of Janus kinase/signal transducer and activators of transcription systems. Importantly, these inflammatory signaling pathways are associated with VEGF-C production. Since VEGF-C accumulatively accelerates VEGF-A-induced angiogenic responses in vascular endothelial cells (77), the calpain-activated cytokine signaling can synergize with VEGF-A-driven signaling cascades. As a result, calpain overactivation accelerates the aforementioned proliferative insults in mice. Of note, calpain-2 potentiates lymphangiogenesis in human dermal LECs through NO production (78). Importantly, conventional calpains can be upregulated by lysophosphatidylcholine under hypercholesterolemic conditions (79). Thus, it is speculated that these molecules are activated even in LECs under hypercholesterolemic conditions. In contrast to the conventional calpains, the contribution of unconventional calpains to lymphangiogenesis has not been reported so far, although they are reportedly involved in a variety of physiological and pathophysiological events (80–82).
Growing evidence suggest that lymphatic defects under hypercholesterolemic conditions are likely to interrupt reverse cholesterol transport (Figure 1). This may be due to dysfunctional HDL transcytosis in microlymphatics and impaired lymphatic drainage involving collecting lymphatic vessels. To the best of our knowledge, impaired cholesterol transport is likely to be the primary cause of atherosclerosis modification by lymphatic insults. Concomitantly with the prevention of reverse cholesterol transport, lymphangiogenesis is disrupted under hypercholesterolemic conditions. Such lymphangiogenic insufficiency is unlikely to be dependent on the depletion of VEGF-C and VEGF-D. Although impaired lymphangiogenesis can be responsible for the limitation of lymphocyte trafficking, further research is needed to investigate the relationship between limited lymphocyte trafficking and immune responses, such as regulatory T cell-driven immunosuppression. It is noteworthy that LECs reportedly exert antigen presentation to modulate DCs and T cells, thereby modifying adaptive immunity (7), which presumably causes lymphocyte dysfunction to accelerate atherosclerosis progression. In addition, several dyslipidemic lipid mediators, such as lysophospholipids, modify the lymphatic structure and function. Several lipid-lowering drugs and apoA-I are reportedly effective in improving lymphatic functions, including lymphatic drainage and reverse cholesterol transport; accordingly, it is possible that hypercholesterolemia can precede lymphatic insults, which is presumably mediated through dyslipidemic lipid mediators. Hence, future investigations are necessary to explore the causal relationship between pathophysiologic lymphatic regulation and hypercholesterolemia-driven atherogenesis.
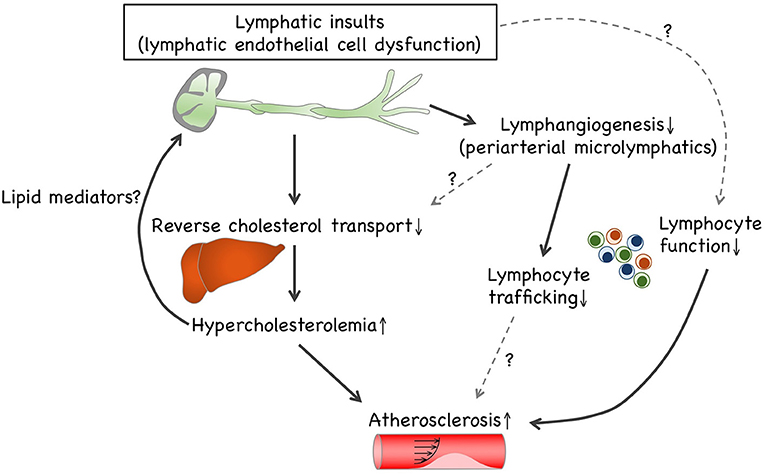
Figure 1. Causal relationship between lymphatic insults and hypercholesterolemia. Lymphatic insults under hypercholesterolemic conditions are likely to interrupt reverse cholesterol transport, which can be owed to the turbulence of lymphatic drainage within collecting lymphatic vessels and to impaired lymphangiogenesis involving periaortic microlymphatics. Additionally, lymphangiogenesis is disrupted under hypercholesterolemic conditions independently of VEGF-C and VEGF-D depletion. Such lymphangiogenic insufficiency can be responsible for limited lymphocyte trafficking. LECs enable exerting antigen presentation to modulate dendritic cells and T cells, thereby modifying adaptive immunity. Therefore, hypercholesterolemic lymphatic insults presumably cause lymphocyte dysfunction that accelerates atherosclerosis progression. In addition, several dyslipidemic lipid mediators, such as lysophospholipids, enable modification of lymphatic patterning and functions. Since cholesterol-lowering trials reportedly recover hypercholesterolemic lymphatic insults, it is possible that hypercholesterolemia can precede lymphatic insults, presumably owing to lipid mediators.
A growing body of evidence suggests that lymphatic defects are responsible for hypercholesterolemia. At the same time, hypercholesterolemia itself appears to elicit lymphatic insults. In addition, several investigations support the notion that lymphangiogenesis around the atheroprone artery exerts lymphocyte migration from atherosclerotic lesions, which can be sustained by VEGF-C. Since the lymphangiogenic role of VEGF-C in periarterial regions has been challenged by some researchers, exploring alternative lymphatic drivers under hypercholesterolemic conditions is necessary. While several bioactive lipids enable the induction of lymphangiogenesis, lipid composition in the lymphatic environment under hypercholesterolemic conditions has not been fully elucidated. Thus, defining lipid composition and its temporal changes in the lymphatic environment during hypercholesterolemia is a promising approach for understanding the causal relationship. In addition to the research exploring environmental factors that induce hypercholesterolemic lymphatic insults, pathogenic intracellular mechanisms in LECs should be investigated in future studies. Employing Cre-loxP systems enables the elucidation of the effects of lymphatic defects on atherogenesis in mice. Defining lipid compositions and intracellular mechanistic insight will be helpful for developing a strategy for the prevention of atherosclerotic diseases and for estimating the personal susceptibility to atherosclerotic diseases.
TM wrote the manuscript. AM revised the manuscript. Both authors contributed to the article and approved the submitted version.
This study was supported in part by the Japan Society for the Promotion of Science KAKENHI (grant numbers 21K08585 to AM and 19K08590 to TM), a research grant from Bristol-Myers Squibb (to TM), and a research grant from the Naito Memorial Foundation (to TM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
2. Packard C, Chapman MJ, Sibartie M, Laufs U, Masana L. Intensive low-density lipoprotein cholesterol lowering in cardiovascular disease prevention: opportunities and challenges. Heart. (2021). doi: 10.1136/heartjnl-2020-318760. [Epub ahead of print].
3. Bahiru E, Hsiao R, Phillipson D, Watson KE. Mechanisms and treatment of dyslipidemia in diabetes. Curr Cardiol Rep. (2021) 23:26. doi: 10.1007/s11886-021-01455-w
4. Telinius N, Hjortdal VE. Role of the lymphatic vasculature in cardiovascular medicine. Heart. (2019) 105:1777–84. doi: 10.1136/heartjnl-2018-314461
5. Teijeira A, Rouzaut A, Melero I. Initial afferent lymphatic vessels controlling outbound leukocyte traffic from skin to lymph nodes. Front Immunol. (2013) 4:433. doi: 10.3389/fimmu.2013.00433
6. Johnson LA, Jackson DG. Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis. (2014) 17:335–45. doi: 10.1007/s10456-013-9407-0
7. Card CM, Yu SS, Swartz MA. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Invest. (2014) 124:943–52. doi: 10.1172/JCI73316
8. Hoggan G, Hoggan FE. The Lymphatics of the walls of the larger blood-vessels and lymphatics. J Anat Physiol. (1882) 17:1–23.
9. Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. (2013) 123:1571–9. doi: 10.1172/JCI63685
10. Vuorio T, Nurmi H, Moulton K, Kurkipuro J, Robciuc MR, Ohman M, et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol. (2014) 34:1162–70. doi: 10.1161/ATVBAHA.114.302528
11. Rademakers T, van der Vorst EP, Daissormont IT, Otten JJ, Theodorou K, Theelen TL, et al. Adventitial lymphatic capillary expansion impacts on plaque T cell accumulation in atherosclerosis. Sci Rep. (2017) 7:45263. doi: 10.1038/srep45263
12. Miller AJ, DeBoer A, Palmer A. The role of the lymphatic system in coronary atherosclerosis. Med Hypotheses. (1992) 37:31–6. doi: 10.1016/0306-9877(92)90009-2
13. Drozdz K, Janczak D, Dziegiel P, Podhorska M, Patrzałek D, Ziółkowski P, et al. Adventitial lymphatics of internal carotid artery in healthy and atherosclerotic vessels. Folia Histochem Cytobiol. (2008) 46:433–6. doi: 10.2478/v10042-008-0083-7
14. Kholová I, Dragneva G, Cermáková P, Laidinen S, Kaskenpää N, Hazes T, et al. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest. (2011) 41:487–97. doi: 10.1111/j.1365-2362.2010.02431.x
15. Kutkut I, Meens MJ, McKee TA, Bochaton-Piallat ML, Kwak BR. Lymphatic vessels: an emerging actor in atherosclerotic plaque development. Eur J Clin Invest. (2015) 45:100–8. doi: 10.1111/eci.12372
16. Eliska O, Eliskova M, Miller AJ. The absence of lymphatics in normal and atherosclerotic coronary arteries in man: a morphologic study. Lymphology. (2006) 39:76–83.
17. Nakano T, Nakashima Y, Yonemitsu Y, Sumiyoshi S, Chen YX, Akishima Y, et al. Angiogenesis and lymphangiogenesis and expression of lymphangiogenic factors in the atherosclerotic intima of human coronary arteries. Hum Pathol. (2005) 36:330–40. doi: 10.1016/j.humpath.2005.01.001
18. Taher M, Nakao S, Zandi S, Melhorn MI, Hayes KC, Hafezi-Moghadam A. Phenotypic transformation of intimal and adventitial lymphatics in atherosclerosis: a regulatory role for soluble VEGF receptor 2. FASEB J. (2016) 30:2490–9. doi: 10.1096/fj.201500112
19. Tay MHD, Lim SYJ, Leong YFI, Thiam CH, Tan KW, Torta FT, et al. Halted lymphocyte egress via efferent lymph contributes to lymph node hypertrophy during hypercholesterolemia. Front Immunol. (2019) 10:575. doi: 10.3389/fimmu.2019.00575
20. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
21. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. (2011) 91:327–87. doi: 10.1152/physrev.00047.2009
22. Oikonomou EK, West HW, Antoniades C. Cardiac computed tomography: assessment of coronary inflammation and other plaque features. Arterioscler Thromb Vasc Biol. (2019) 39:2207–19. doi: 10.1161/ATVBAHA.119.312899
23. Duan H, Zhang Q, Liu J, Li R, Wang D, Peng W, et al. Suppression of apoptosis in vascular endothelial cell, the promising way for natural medicines to treat atherosclerosis. Pharmacol Res. (2021) 168:105599. doi: 10.1016/j.phrs.2021.105599
24. Sun HJ, Wu ZY, Nie XW, Bian JS. Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Front Pharmacol. (2019) 10:1568. doi: 10.3389/fphar.2019.01568
25. Miyazaki T, Miyazaki A. Impact of dysfunctional protein catabolism on macrophage cholesterol handling. Curr Med Chem. (2019) 26:1631–43. doi: 10.2174/0929867325666180326165234
26. Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. (2013) 17:671–84. doi: 10.1016/j.cmet.2013.04.002
27. Milasan A, Dallaire F, Mayer G, Martel C. Effects of LDL Receptor modulation on lymphatic function. Sci Rep. (2016) 6:27862. doi: 10.1038/srep27862
28. Milasan A, Jean G, Dallaire F, Tardif JC, Merhi Y, Sorci-Thomas M, et al. Apolipoprotein A-I modulates atherosclerosis through lymphatic vessel-dependent mechanisms in mice. J Am Heart Assoc. (2017) 6:9. doi: 10.1161/JAHA.117.006892
29. Milasan A, Smaani A, Martel C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr(-/-) mice. Atherosclerosis. (2019) 283:106–19. doi: 10.1016/j.atherosclerosis.2019.01.031
30. Pownall HJ, Rosales C, Gillard BK, Gotto AM, Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat Rev Cardiol. (2021). doi: 10.1038/s41569-021-00538-z
31. Rosenson RS, Brewer HB, Jr., Ansell BJ, Barter P, Chapman MJ, Heinecke JW, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. (2016) 13:48–60. doi: 10.1038/nrcardio.2015.124
32. Tacconi C, Correale C, Gandelli A, Spinelli A, Dejana E, D'Alessio S, et al. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology. (2015) 148:1438–51.e8. doi: 10.1053/j.gastro.2015.03.005
33. Uhrin P, Zaujec J, Breuss JM, Olcaydu D, Chrenek P, Stockinger H, et al. Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood. (2010) 115:3997–4005. doi: 10.1182/blood-2009-04-216069
34. Hess PR, Rawnsley DR, Jakus Z, Yang Y, Sweet DT, Fu J, et al. Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J Clin Invest. (2014) 124:273–84. doi: 10.1172/JCI70422
35. Hokkanen K, Tirronen A, Ylä-Herttuala S. Intestinal lymphatic vessels and their role in chylomicron absorption and lipid homeostasis. Curr Opin Lipidol. (2019) 30:370–6. doi: 10.1097/MOL.0000000000000626
36. Tirronen A, Vuorio T, Kettunen S, Hokkanen K, Ramms B, Niskanen H, et al. Deletion of lymphangiogenic and angiogenic growth factor VEGF-D leads to severe hyperlipidemia and delayed clearance of chylomicron remnants. Arterioscler Thromb Vasc Biol. (2018) 38:2327–37. doi: 10.1161/ATVBAHA.118.311549
37. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. (2004) 5:74–80. doi: 10.1038/ni1013
38. Silvestre-Roig C, Lemnitzer P, Gall J, Schwager S, Toska A, Yvan-Charvet L, et al. Arterial delivery of VEGF-C stabilizes atherosclerotic lesions. Circ Res. (2021) 128:284–6. doi: 10.1161/CIRCRESAHA.120.317186
39. Achen MG, Jeltsch M, Kukk E, Mäkinen T, Vitali A, Wilks AF, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA. (1998) 95:548–53. doi: 10.1073/pnas.95.2.548
40. Rissanen TT, Markkanen JE, Gruchala M, Heikura T, Puranen A, Kettunen MI, et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res. (2003) 92:1098–106. doi: 10.1161/01.RES.0000073584.46059.E3
41. Baldwin ME, Halford MM, Roufail S, Williams RA, Hibbs ML, Grail D, et al. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol Cell Biol. (2005) 25:2441–9. doi: 10.1128/MCB.25.6.2441-2449.2005
42. Mendelson K, Evans T, Hla T. Sphingosine 1-phosphate signaling. Development. (2014) 141:5–9. doi: 10.1242/dev.094805
43. Ouyang J, Shu Z, Chen S, Xiang H, Lu H. The role of sphingosine 1-phosphate and its receptors in cardiovascular diseases. J Cell Mol Med. (2020) 24:10290–301. doi: 10.1111/jcmm.15744
44. Cartier A, Hla T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science. (2019) 366:6463. doi: 10.1126/science.aar5551
45. Hla T, Maciag T. An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J Biol Chem. (1990) 265:9308–13. doi: 10.1016/S0021-9258(19)38849-0
46. Okajima F. Plasma lipoproteins behave as carriers of extracellular sphingosine 1-phosphate: is this an atherogenic mediator or an anti-atherogenic mediator? Biochim Biophys Acta. (2002) 1582:132–7. doi: 10.1016/S1388-1981(02)00147-6
47. Levkau B. HDL-S1P: cardiovascular functions, disease-associated alterations, and therapeutic applications. Front Pharmacol. (2015) 6:243. doi: 10.3389/fphar.2015.00243
48. Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. (2010) 207:17–27. doi: 10.1084/jem.20091619
49. Xiong Y, Piao W, Brinkman CC, Li L, Kulinski JM, Olivera A, et al. CD4 T cell sphingosine 1-phosphate receptor (S1PR)1 and S1PR4 and endothelial S1PR2 regulate afferent lymphatic migration. Sci Immunol. (2019) 4:33. doi: 10.1126/sciimmunol.aav1263
50. Schober A, Siess W. Lysophosphatidic acid in atherosclerotic diseases. Br J Pharmacol. (2012) 167:465–82. doi: 10.1111/j.1476-5381.2012.02021.x
51. Zhou Y, Little PJ, Ta HT, Xu S, Kamato D. Lysophosphatidic acid and its receptors: pharmacology and therapeutic potential in atherosclerosis and vascular disease. Pharmacol Ther. (2019) 204:107404. doi: 10.1016/j.pharmthera.2019.107404
52. Siess W, Tigyi G. Thrombogenic and atherogenic activities of lysophosphatidic acid. J Cell Biochem. (2004) 92:1086–94. doi: 10.1002/jcb.20108
53. Cui MZ. Lysophosphatidic acid effects on atherosclerosis and thrombosis. Clin Lipidol. (2011) 6:413–26. doi: 10.2217/clp.11.38
54. Geraldo LHM, Spohr T, Amaral RFD, Fonseca A, Garcia C, Mendes FA, et al. Role of lysophosphatidic acid and its receptors in health and disease: novel therapeutic strategies. Signal Transduct Targeted Ther. (2021) 6:45. doi: 10.1038/s41392-020-00367-5
55. Sumida H, Noguchi K, Kihara Y, Abe M, Yanagida K, Hamano F, et al. LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood. (2010) 116:5060–70. doi: 10.1182/blood-2010-03-272443
56. Mu H, Calderone TL, Davies MA, Prieto VG, Wang H, Mills GB, et al. Lysophosphatidic acid induces lymphangiogenesis and IL-8 production in vitro in human lymphatic endothelial cells. Am J Pathol. (2012) 180:2170–81. doi: 10.1016/j.ajpath.2012.03.003
57. Hisano Y, Kono M, Cartier A, Engelbrecht E, Kano K, Kawakami K, et al. Lysolipid receptor cross-talk regulates lymphatic endothelial junctions in lymph nodes. J Exp Med. (2019) 216:1582–98. doi: 10.1084/jem.20181895
58. Tanaka H, Zaima N, Sasaki T, Yamamoto N, Inuzuka K, Yata T, et al. Lysophosphatidylcholine acyltransferase-3 expression is associated with atherosclerosis progression. J Vasc Res. (2017) 54:200–8. doi: 10.1159/000473879
59. Bot M, de Jager SC, MacAleese L, Lagraauw HM, van Berkel TJ, Quax PH, et al. Lysophosphatidic acid triggers mast cell-driven atherosclerotic plaque destabilization by increasing vascular inflammation. J Lipid Res. (2013) 54:1265–74. doi: 10.1194/jlr.M032862
60. Cyr AR, Huckaby LV, Shiva SS, Zuckerbraun BS. Nitric oxide and endothelial dysfunction. Crit Care Clin. (2020) 36:307–21. doi: 10.1016/j.ccc.2019.12.009
61. Smeda JS, VanVliet BN, King SR. Stroke-prone spontaneously hypertensive rats lose their ability to auto-regulate cerebral blood flow prior to stroke. J Hypertens. (1999) 17:1697–705. doi: 10.1097/00004872-199917120-00006
62. Wang X, Loutzenhiser RD, Cupples WA. Frequency modulation of renal myogenic autoregulation by perfusion pressure. Am J Physiol Regul Integr Comp Physiol. (2007) 293:R1199–204. doi: 10.1152/ajpregu.00281.2007
63. Abu-Amarah I, Ajikobi DO, Bachelard H, Cupples WA, Salevsky FC. Responses of mesenteric and renal blood flow dynamics to acute denervation in anesthetized rats. Am J Physiol. (1998) 275:R1543–52. doi: 10.1152/ajpregu.1998.275.5.R1543
64. Pipili-Synetos E, Sakkoula E, Maragoudakis ME. Nitric oxide is involved in the regulation of angiogenesis. Br J Pharmacol. (1993) 108:855–7. doi: 10.1111/j.1476-5381.1993.tb13476.x
65. Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. (1994) 94:2036–44. doi: 10.1172/JCI117557
66. Sessa WC. Molecular control of blood flow and angiogenesis: role of nitric oxide. J Thromb Haemost. (2009) 7:35–7. doi: 10.1111/j.1538-7836.2009.03424.x
67. Dulak J, Józkowicz A. Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxid Redox Signal. (2003) 5:123–32. doi: 10.1089/152308603321223612
68. Lahdenranta J, Hagendoorn J, Padera TP, Hoshida T, Nelson G, Kashiwagi S, et al. Endothelial nitric oxide synthase mediates lymphangiogenesis and lymphatic metastasis. Cancer Res. (2009) 69:2801–8. doi: 10.1158/0008-5472.CAN-08-4051
69. Kajiya K, Huggenberger R, Drinnenberg I, Ma B, Detmar M. Nitric oxide mediates lymphatic vessel activation via soluble guanylate cyclase alpha1beta1-impact on inflammation. FASEB J. (2008) 22:530–7. doi: 10.1096/fj.07-8873com
70. Singla B, Lin HP, Chen A, Ahn W, Ghoshal P, Cherian-Shaw M, et al. Role of R-spondin 2 in arterial lymphangiogenesis and atherosclerosis. Cardiovasc Res. (2021) 117:1489–509. doi: 10.1093/cvr/cvaa244
71. Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, et al. Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci USA. (2011) 108:18784–9. doi: 10.1073/pnas.1116152108
72. Ono Y, Saido TC, Sorimachi H. Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov. (2016) 15:854–76. doi: 10.1038/nrd.2016.212
73. Ono Y, Sorimachi H. Calpains: an elaborate proteolytic system. Biochim Biophys Acta. (2012) 1824:224–36. doi: 10.1016/j.bbapap.2011.08.005
74. Miyazaki T, Miyazaki A. Dysregulation of calpain proteolytic systems underlies degenerative vascular disorders. J Atheroscler Thromb. (2018) 25:1–15. doi: 10.5551/jat.RV17008
75. Miyazaki T, Akasu R, Miyazaki A. Calpain proteolytic systems counteract endothelial cell adaptation to inflammatory environments. Inflamm Regen. (2020) 40:5. doi: 10.1186/s41232-020-00114-x
76. Miyazaki T, Taketomi Y, Saito Y, Hosono T, Lei XF, Kim-Kaneyama JR, et al. Calpastatin counteracts pathological angiogenesis by inhibiting suppressor of cytokine signaling 3 degradation in vascular endothelial cells. Circ Res. (2015) 116:1170–81. doi: 10.1161/CIRCRESAHA.116.305363
77. Pepper MS, Mandriota SJ, Jeltsch M, Kumar V, Alitalo K. Vascular endothelial growth factor (VEGF)-C synergizes with basic fibroblast growth factor and VEGF in the induction of angiogenesis in vitro and alters endothelial cell extracellular proteolytic activity. J Cell Physiol. (1998) 177:439–52.
78. Prangsaengtong O, Senda K, Doki Y, Park JY, Jo M, Sakurai H, et al. Calpain 1 and−2 play opposite roles in cord formation of lymphatic endothelial cells via eNOS regulation. Hum Cell. (2012) 25:36–44. doi: 10.1007/s13577-012-0042-7
79. Miyazaki T, Taketomi Y, Takimoto M, Lei XF, Arita S, Kim-Kaneyama JR, et al. m-Calpain induction in vascular endothelial cells on human and mouse atheromas and its roles in VE-cadherin disorganization and atherosclerosis. Circulation. (2011) 124:2522−2. doi: 10.1161/CIRCULATIONAHA.111.021675
80. Miyazaki T, Tonami K, Hata S, Aiuchi T, Ohnishi K, Lei XF, et al. Calpain-6 confers atherogenicity to macrophages by dysregulating pre-mRNA splicing. J Clin Invest. (2016) 126:3417–32. doi: 10.1172/JCI85880
81. Hata S, Abe M, Suzuki H, Kitamura F, Toyama-Sorimachi N, Abe K, et al. Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet. (2010) 6:e1001040. doi: 10.1371/journal.pgen.1001040
Keywords: plasma dyslipidemia, LPA, S1P, calpain, nitric oxide synthase
Citation: Miyazaki T and Miyazaki A (2021) Hypercholesterolemia and Lymphatic Defects: The Chicken or the Egg? Front. Cardiovasc. Med. 8:701229. doi: 10.3389/fcvm.2021.701229
Received: 27 April 2021; Accepted: 28 May 2021;
Published: 23 June 2021.
Edited by:
Vasily Sukhorukov, Research Institute of Human Morphology, RussiaReviewed by:
Helge Wiig, University of Bergen, NorwayCopyright © 2021 Miyazaki and Miyazaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takuro Miyazaki, dGFrdUBwaGFybS5zaG93YS11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.