
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 23 August 2021
Sec. Cardiovascular Genetics and Systems Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.696362
This article is part of the Research Topic Cardiovascular Involvement in Autoimmune Diseases, Volume II View all 5 articles
 Yujing Wu1,2,3
Yujing Wu1,2,3 Zhenzhong Zheng1,2*Xiantong Cao1Qing Yang1,2Vikram Norton4Avner Adini4
Zhenzhong Zheng1,2*Xiantong Cao1Qing Yang1,2Vikram Norton4Avner Adini4 Amit K. Maiti5
Amit K. Maiti5 Irit Adini6
Irit Adini6 Hao Wu4*
Hao Wu4*Cardiomyopathy often leads to dilated cardiomyopathy (DCM) when caused by viral myocarditis. Apoptosis is long considered as the principal process of cell death in cardiomyocytes, but programmed necrosis or necroptosis is recently believed to play an important role in cardiomyocyte cell death. We investigated the role of necroptosis and its interdependency with other processes of cell death, autophagy, and apoptosis in a rat system of experimental autoimmune myocarditis (EAM). We successfully created a rat model system of EAM by injecting porcine cardiac myosin (PCM) and showed that in EAM, all three forms of cell death increase considerably, resulting in the deterioration of cardiac conditions with an increase in inflammatory infiltration in cardiomyocytes. To explore whether necroptosis occurs in EAM rats independent of autophagy, we treated EAM rats with a RIP1/RIP3/MLKL kinase-mediated necroptosis inhibitor, Necrostatin-1 (Nec-1). In Nec-1 treated rats, cell death proceeds through apoptosis but has no significant effect on autophagy. In contrast, autophagy inhibitor 3-Methyl Adenine (3-MA) increases necroptosis, implying that blockage of autophagy must be compensated through necroptosis. Caspase 8 inhibitor zVAD-fmk blocks apoptosis but increases both necroptosis and autophagy. However, all necroptosis, apoptosis, and autophagy inhibitors independently reduce inflammatory infiltration in cardiomyocytes and improve cardiac conditions. Since apoptosis or autophagy is involved in many important cellular aspects, instead of suppressing these two major cell death processes, Nec1 can be developed as a potential therapeutic target for inflammatory myocarditis.
Dilated cardiomyopathy (DCM) is a myocardial disease that is caused by viral infection or autoimmune or genetic factors. Some viral myocarditis (VMC) can develop into DCM, which leads to intractable congestive heart failure (CHF) and even sudden death (1). The development of VMC to DCM is mainly caused by progressive autoimmune injury. When cardiomyocytes are infected by pathogenic microorganisms, they generate an antigen epitope and trigger an autoimmune response (2).
Cell death is a fundamental process that has been classified as necroptosis, apoptosis, and autophagy (3). Over the years, necroptosis has been widely considered to be an unregulated, passive type of cell death that has been closely associated with many diseases. Its role in myocardial ischemia–reperfusion injury, cerebral ischemia, neuronal degeneration, immune diseases, tumors, and DCM has been established (4–7). It has long been known that apoptosis is vital to the maintenance of proper adaptive immune function. More recently, the contributions of additional cell death processes including cellular autophagy and necroptosis have been implicated in controlling both innate and adaptive immune functions (8).
Most studies involving cardiomyocyte cell death mainly focused on apoptosis. Necroptosis is a regulated, caspase-independent form of cell death, mediated by TNFR (tumor necrosis factor receptor), RIP1 (receptor interacting protein kinase 1), RIP3 (receptor interacting protein kinase 3), and MLKL (mixed lineage kinase domain-like). Accumulating evidence demonstrate that RIP1 and RIP3 form a RIP1–RIP3 complex (necrosome) that phosphorylates each other (9). MLKL oligomerizes and translocates to plasma membrane to form the RIP1/RIP3/MLKL complex to induce necroptosis (10). MLKL comprises an N-terminal helical bundle domain (HBD) and a C-terminal kinase-like domain of which HBD is crucial for the oligomerization of MLKL (11). The MLKL oligomerization results in ion channel dysfunction at the plasma membrane that causes an ion imbalance in both interior and exterior compartments of the membrane, which finally triggers necroptosis (12). When caspase-8 is inhibited with the apoptosis inhibitor zVAD-fmk, or suppressed with other agents, the RIP1 kinase combines with RIP3 to form necrosome. The kinase activity of RIP3, oligomerization of MLKL, and the necrosome formation are now considered to be crucial in the necroptosis signaling pathway (10, 13, 14).
Experimental autoimmune myocarditis (EAM) is an effective animal model to study the mechanism of myocardial injury. In the present study, we established an animal model of EAM in Lewis rats by immunizing a susceptible rat strain with porcine cardiac myosin (PCM) together with a strong adjuvant (15). The principle aim of this present study is to evaluate whether necroptosis is activated in EAM. Overall, our data show that the necroptotic pathway is triggered in EAM through the increased expression of RIP1, RIP3, and MLKL. As a result, the altered expression of these proteins deteriorates cardiac conditions and increases infiltration of inflammatory cells. Furthermore, we explored the possible effect of a small-molecule inhibitor of the necroptosis pathway, necrostatin-1 (Nec-1, a RIP1 kinase inhibitor), in myocardial cell injury and cardiac conditions in EAM (16). As expected, Nec-1 blocks RIP1/RIP3/MLKL-mediated necroptosis, reduces myocardial inflammatory infiltration, and improves cardiac conditions in EAM in vivo. Nec-1 does not show a significant effect on autophagy but shifts the response toward apoptosis. In contrast, suppression of autophagy by 3-MA increases necroptosis modulating proteins. Furthermore, apoptosis blockage by zVAD-fmk increases both necroptosis and autophagy. These results highlight the interrelation of necroptosis, apoptosis, and autophagy pathway genes in EAM and shed light on the pathogenic mechanism of myocarditis as well as indicate a novel therapeutic strategy for treatment of patients with myocarditis.
Thirty-eight male Lewis rats, aged 8–9 weeks, weighing 200–220 g, specific-pathogen-free, were obtained from Vital River Laboratories (VRL), China, and all works were performed at Jiangxi Hypertension Research Institute, China. The study protocol was approved by the Institutional Review Board of the First Affiliated Hospital of Nanchang University. All experiments were conducted in accordance with the institutional guidelines for animal research and comply with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publ. no. 85-23, revised 1996).
All rats are euthanized and sacrificed with the compliance of animal research ethical guidelines, and we strictly followed the instructions. Briefly, all rats in each group were administered, through subcutaneous injection, a single dose of 20% urethane (ethyl carbamate) solution (3 ml/kg of rat). Rats were then dissected, and their hearts were removed with incisions to the heart along with the coronal slitting. Half of the heart (longitudinal section keeping all four chambers) was fixed in 4% paraformaldehyde for histological examination; the other half of the heart was divided into two parts (again, longitudinally keeping all four chambers), one half is stored at −80°C for later Western Blotting tests, and the other half is saved in tissue preservation solution for RNA isolation.
Rats were kept under standard conditions with a mean temperature of 21 ± 2°C with a mean relative humidity of 50 ± 20% and a defined day-and-night rhythm of 12 h light and 12 h dark. Animals were randomized to control group 1 [21 days, 3 weeks (3W), n = 4], control group 2 (56 days, 8W, n = 4), and experimental group (n = 30). Experimental group rats were assigned (Figure 1) to five groups: the EAM group (21 days, 3W, n = 5; 56 days, 8W, n = 5), the necrostatin-1 group (Nec-1, n = 5), the zVAD-fmk group (zVAD-fmk, n = 5), the 3-Methyladenine group (3-MA, n = 5), and the DMSO group (n = 5). Rats of the control group (3W, n = 4), EAM group (3W, n = 5), Nec-1 group (n = 5), 3-MA group (n = 5), and DMSO group (n = 5) were sacrificed on 21 days (3W). Rats of the control group (8W, n = 4) and EAM group (8W, n = 5) were sacrificed on 56 days (8W) after the first injection.
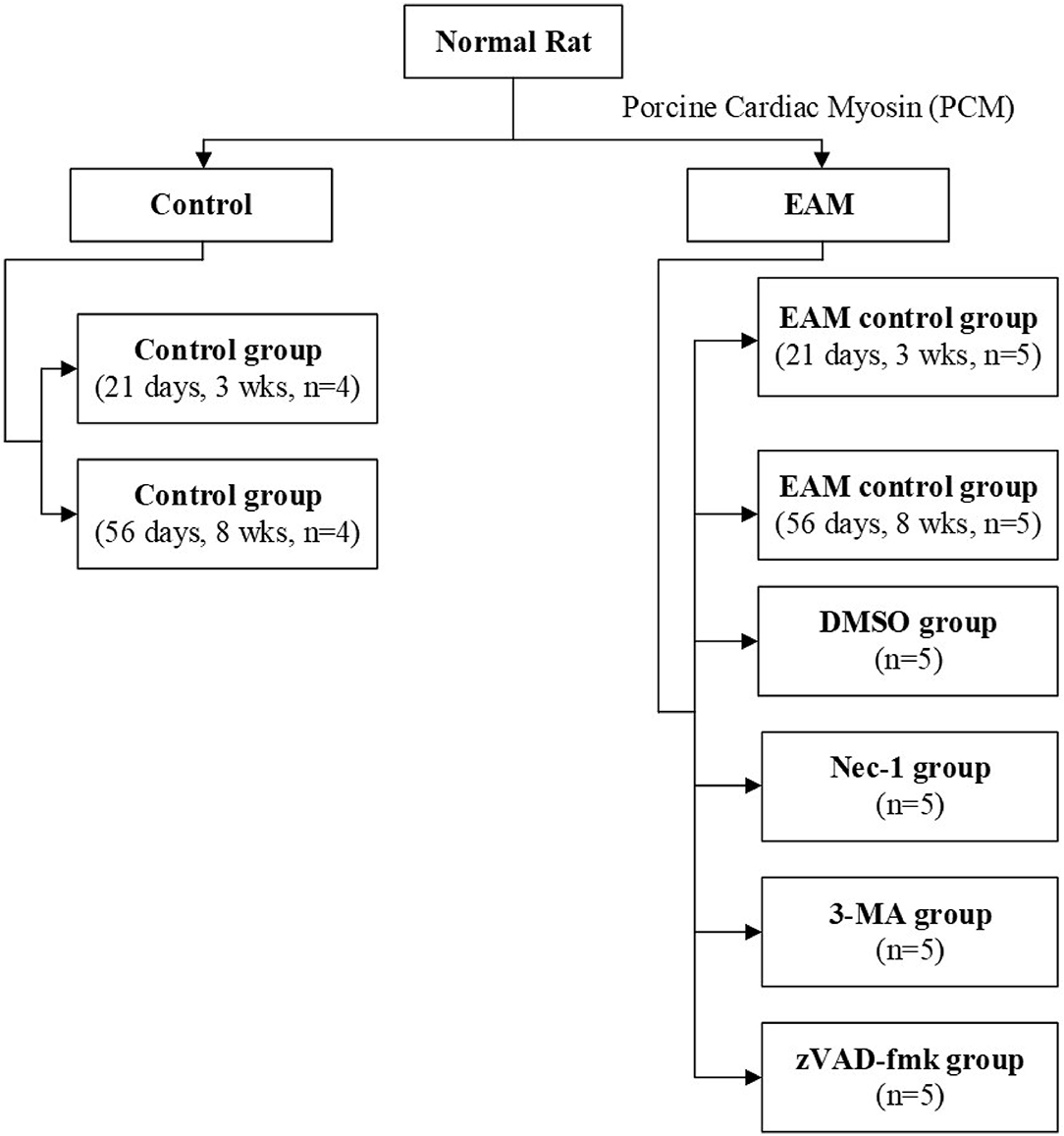
Figure 1. Flowchart of rats used in experiments.
PCM (Sigma Aldrich, M0531) was diluted with 0.1 M PBS and finally emulsified with Complete Freund's Adjuvant (CFA, Sigma Aldrich, F5881) with a ratio of 1:1. Experimental group rats were subcutaneously injected with 0.2 ml of the PCM-CFA emulsion containing 1 mg of PCM in the footpad with a 7-day interval between injections (on days 0, 7, 14, and so on). Rats of the control group were subcutaneously injected with 0.1 ml of CFA alone on days 0 and 7. Control rats were sacrificed on 21 days (3W) and 56 days (8W) after the first injection.
Nec-1 (MedChem Express, HY-15760), zVAD-fmk (MedChem Express, HY-16658), and 3-MA (MedChem Express, HY-19312) were dissolved in 0.1% dimethylsulfoxide (DMSO, Sigma Aldrich, D2650) with 0.1 M phosphate-buffered saline (PBS). The drug dosages are: Nec-1 group, 0.6 mg/kg/day; zVAD-fmk group, 1.0 mg/kg/day; 3-MA group, 15 mg/kg/day; and DMSO group, 0.1% DMSO. Rats were injected intraperitoneally on day 0 and day 8, respectively. On day 21, rats were sacrificed after the first injection.
Transthoracic echocardiography was performed using a Sonos 5500 ultrasound machine (Phillips, USA.) with a 12-MHz phased array transducer with real-time digital acquisition, storage, and review capabilities. On day 21 and day 56, rats were anesthetized intraperitoneally with 3% chloral hydrate (0.01 ml/g) and the chests were shaved. Rats were placed on a shallow left lateral position. Two-dimensional mode, M-mode, and Doppler flow images were obtained in parasternal long/short-axis view. The left ventricular end-systolic and end-diastolic internal diameters (LVESd, LVEDd) were measured over the course of at least three consecutive cardiac cycles. The left ventricular ejection fraction (LVEF) and fractional shortening (FS) were calculated.
The hearts were fixed in 4% paraformaldehyde in PBS and embedded in paraffin wax. Tissue sections (5 μm thick) were cut and stained with hematoxylin and eosin (H&E) to determine the level of inflammation. To evaluate the tissue-specific expression of RIP1, RIP3, MLKL, and LC3 I and II, we performed immunohistochemistry. In brief, control and the respective experimental group of tissue sections were incubated with mouse monoclonal anti-RIP1 antibodies (abcam, ab72139), rabbit polyclonal anti-RIP3 antibodies (abcam, ab62344), rabbit polyclonal anti-MLKL (Affinity Bioscience, DF7412), or rabbit polyclonal anti-LC3 I and II (Cell Signaling Technology, 12741S). The specificity of these antibodies was confirmed by Western blots (data not shown). The degree of myocardial infiltration and fibrosis was determined by two independent investigators in a double-blinded manner, and histology was scored as follows: 0, no infiltration; 1, <25%; 2, 25 to 50%; 3, 51 to 75%; 4, >75%.
All hearts were snap frozen and stored at −80°C. For the preparation of total RNA, the tissue was homogenized in Trizol reagent, extracted thrice with phenol-chloroform, and the total RNA was dissolved in 10 μl of RNase-free water. cDNA was prepared with PrimeScript™ RT reagent Kit with gDNA Eraser according to the kit specific protocol. Briefly, 10.0 μl of RNA, 2.0 μl of 5× gDNA Eraser Buffer, and 1.0 μl of gDNA Eraser were incubated at 42°C for 2 min. Reverse transcription was done with 1.0 μl of PrimeScript RT Enzyme Mix and 1.0 μl of RT Primer (random) Mix with 5 μl of 5× PrimeScript Buffer and 4.0 μl of 10 mM dNTP and made up the volume with RNase-free dH2O, incubated at 37°C for 1 h and then denatured at 85°C for 1 min. Real-time PCR was done with SYBR green method with SYBR Premix Taq. Briefly, 2× qPCR mix with SYBR Premix Taq 5 μl, primer (2.5 μm) 1 μl, cDNA 1 μl, ROX 0.2 μl, and dH2O (2.8 μl) with PCR cycle: 95°C, 1 min; 95°C, 40 s, 15 s → 58°C, and 20 s → 72°C. Dissolution curve was at 60°C → 95°C, with an increase of 1°C every 20 s. Forward and reverse primer sequences for each gene are given in Supplementary Table 1.
For Western analysis, total proteins were extracted using radio-immunoprecipitation assay (RIPA) lysis buffer (Solarbio, R0020). Protein was quantified by Varioskan (Thermo Scientific, USA) using a BCA protein assay kit (Beyotime, P0012S). Total protein was denatured by boiling for 10 min. Protein (40 μg) was separated by 10% SDS-PAGE and transferred to nitrocellulose filter membrane (Millipore, USA). After blocking with 5% non-fat milk/PBST for 1 h at room temperature, the membranes were incubated with the following primary antibodies in the respective experimental groups: mouse monoclonal anti-RIP1 antibodies (abcam, ab72139, 1:1,000 in 2% milk PBS), rabbit polyclonal anti-RIP3 antibodies (abcam, ab62344, 1:1,000 in 2% milk PBS), rabbit polyclonal anti-MLKL (Affinity Bioscience, DF7412, 1:1,000 in 2% milk PBS), and rabbit polyclonal anti-LC3 I and II (Cell Signaling Technology, 12741S, 1:1,000 in 2% milk PBS) for 12–16 h. Then, Peroxidase-Conjugated AffiniPure Goat anti-Rabbit IgG (H + L) (ZSGE-BIO, ZB-2301, 1:2,000 in 2% PBS) or Peroxidase-Conjugated AffiniPure Goat anti-Mouse IgG (H + L) (ZSGE-BIO, ZB-2305, 1:2,000 in 2% PBS) was added depending on the origin of primary antibodies for 2 h at room temperature and subsequently developed using an enhanced chemiluminescence reagent (Beyotime, P0018A). The signals emitted for chemiluminescence were detected using the ImageQuant LAS 4000 (Bio-Rad, 00746947) and analyzed with ImageJ software (NIH). The results were normalized to the corresponding densitometry signal of GAPDH (Abcam, 9484).
Data are presented as mean ± SEM. When comparing between two groups, data were statistically analyzed with a Student's two-tailed t-test. Differences among groups were determined by ANOVA followed by the Bonferroni's post-hoc test using SPSS version 20 (IBM SPSS, Armonk, NY) and GraphPad Prism 5 (GraphPad Software, Inc., CA, USA). p-values < 0.05 were considered statistically significant.
Necroptosis is emerging as an important process of cell death in various cardiovascular pathological conditions. Although necroptosis influences the pathophysiological process during myocardial ischemia–reperfusion injury, acute myocardial infarction, acute heart failure, and atherosclerosis, no compelling evidence is reported for the association of necroptosis with EAM (17–19). To investigate whether necroptosis takes place in EAM, we designed experiments to understand the mechanisms of RIP1/RIP3/MLKL-mediated necroptosis and tested its effects in EAM in vivo. At 8–10 weeks of age of rats, there were no significant differences between the control group and experimental group in body weight and cardiac conditions (data not shown). PCM-treated rats during the phase of immunization on 3 and 8W had significant decreases in cardiac conditions and increases in myocardial inflammation compared to the control groups as revealed by echocardiographic examination of EF and FS (Figure 2) and immunohistological score of the myocarditis rats (Supplementary Figure 1). However, the EF values observed for control rats are higher than the value (64 ± 6.9) of Sprague–Dawley rats (20) but the Lewis rats considerably differ from Sprague–Dawley rats in heart size. EF measurements in Lewis rats are not reported elsewhere (21). Our measurement of EF value (85.152 ± 1.154%) may be relevant to our earlier report of EF value (81 ± 3.5%) in Lewis rats (22). However, deviation from 85.152 ± 1.154% to 63.9 ± 4.0% in EAM is significant, although it is within the normal range of the EF value of Sprague–Dawley rats.
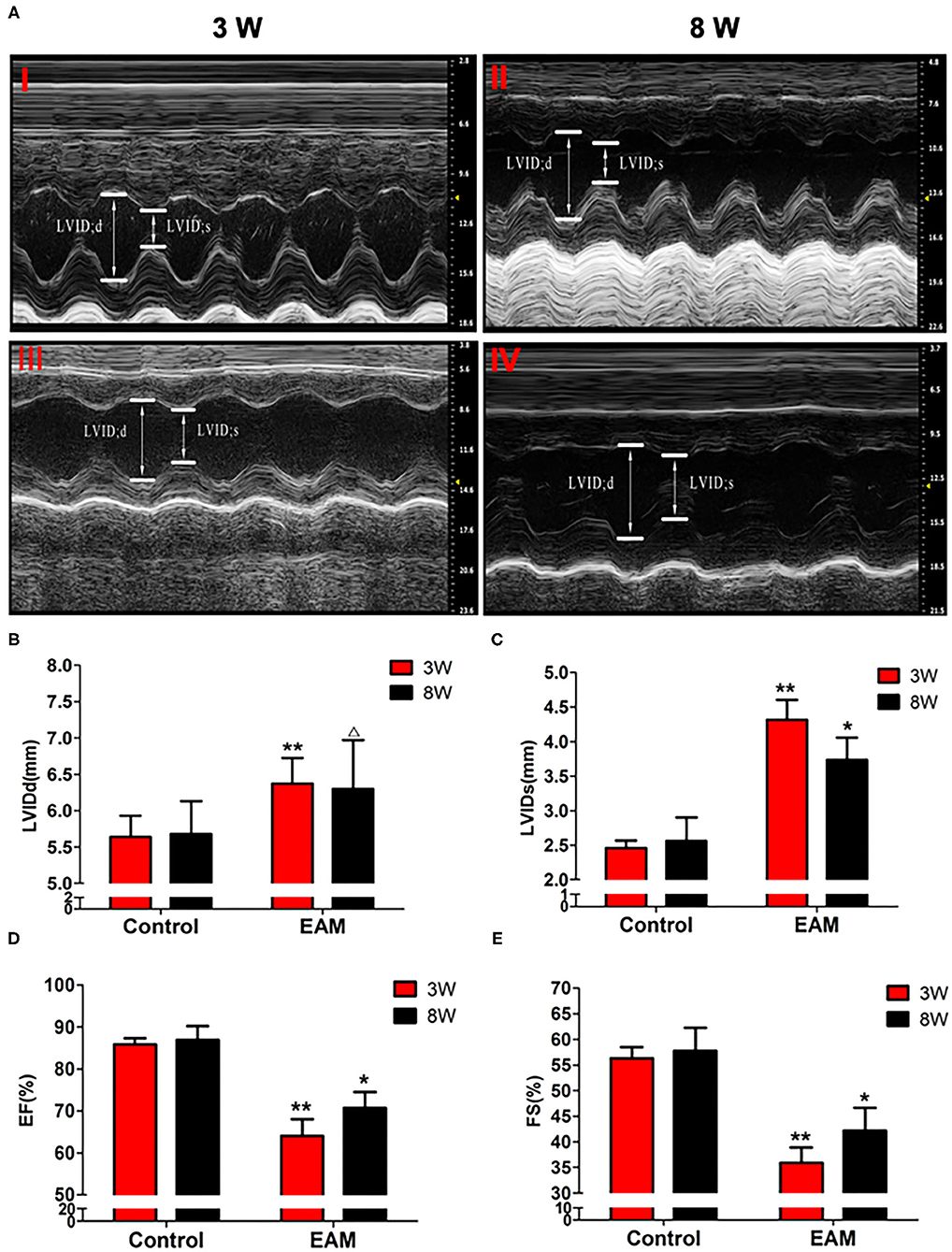
Figure 2. The level of LVIDd/LVIDs/EF/FS in “early” and “late” stages of myocarditis in control and EAM mice by echocardiography. (A) Control groups (top panel, 3W, I; 8W; II) and EAM mice (bottom panel, 3W,I II; 8W, IV). (B–E) Quantification of LVIDd (B), LVIDs (C), EF (D), and FS (E) in “early” and “late” stages of myocarditis from control groups and EAM mice. n = 4 rat in control group, n = 5 rat per group (B–E). All data are presented as mean ± SEM; a Student's t-test was performed to control group and EAM group. *p < 0.05 vs. control group; **p < 0.01 vs. EAM + DMSO group; Δp > 0.05 vs. EAM group.
Hearts from the EAM group of rats consistently showed dense inflammatory infiltrates on 3W and obvious myocardial fibrosis on 8W, whereas hearts from control group animals had only sparse lymphocytic infiltrations on both 3 and 8W. The deterioration of both cardiac conditions and myocardial inflammation in the experimental model over control group indicated that the EAM model was successfully established. Liu et al. (23) created a similar experimental model of Lewis rats with PCM that also showed similar inflammatory molecules in cardiac tissue with the increase of systemic serum inflammation with the increase of IFNΓ, IL2, IL4, and IL10 (23).
Next, we evaluated the potential effects of RIP1/RIP3/MLKL-induced necroptosis in myocardial injury in the EAM rat model. We investigated the expression of RIP1, RIP3, and MLKL in the early “stages” (day 21, 3W) and “late” stages (day 56, 8W) in EAM-induced myocardial injury. Notably, the mRNA and protein expression level of RIP1, RIP3, and MLKL were dramatically elevated in the EAM group compared to the control group on both 3 and 8W, as they were observed with Western blotting (Figures 3A–D) and RT-QPCR results for 3W (Figures 5A–C). Immunohistochemical examination also showed similar increases of RIP1, RIP3, and MLKL in the EAM group (Figures 7A–D). These results suggest that in both “early” and, “late” EAM rats, RIP1/RIP3-mediated necroptosis plays a critical role in developing myocardial injury and inflammatory infiltrates, although it is unclear whether necroptosis is the primary or principal pathway of cell death in EAM rats.
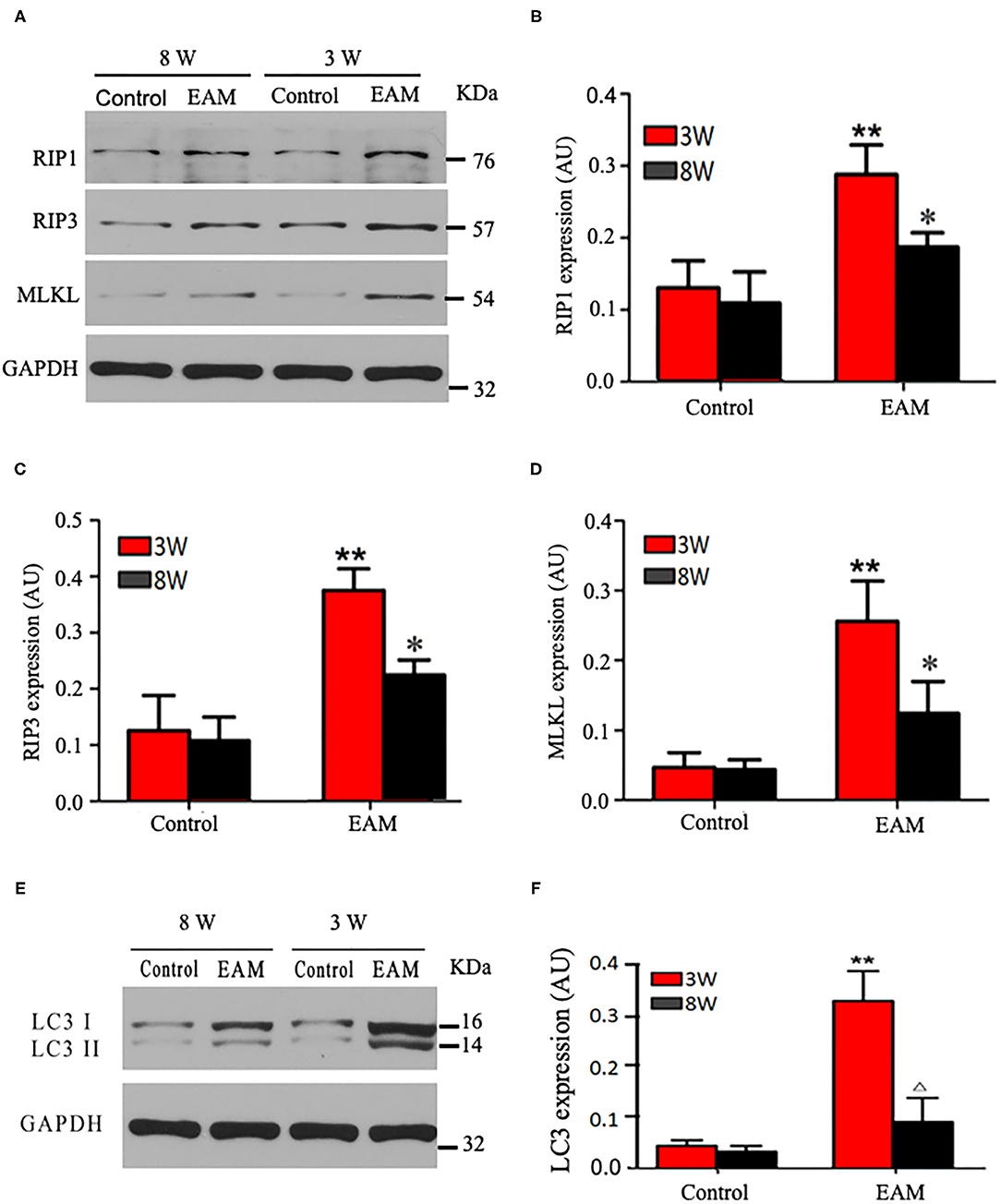
Figure 3. Protein expression levels of RIP1, RIP3, and MLKL in myocardial tissues of acute and chronic stages detected by Western blot. (A) Western blot analysis of protein expression of RIP1/RIP3/MLKL in rat myocardium during “early” and “late” stages. (B–D) Quantification of RIP1 (B), RIP3 (C), MLKL (D) in (A). (E) Western blot analysis of marker protein expression of autophagosome LC-3 I and LC-3 II in rat myocardium during “early” and “late” stages. (F) Quantification of LC-3 I and LC-3 II in e. All data are presented as mean ± SEM. A Student's t-test was performed. **p < 0.05 means compared with control group (3W); *p < 0.05 means vs. control Group (8 weeks) comparison.
Next, we further tested the expression of CASPASE 3, BAX, BCL2, and LC3 I and II, for assessing apoptosis and autophagy, respectively, in the myocardium. CASPASE 3 is the primary executioner of the apoptosis pathway with increased BAX expression while increased BCL2 expression serves to protect cells from apoptosis. LC3 I and II, microtubule-associated proteins, are the accepted biological markers of autophagy pathway, and their expression in myocardium should indicate autophagosome formation, reflecting the extent of autophagy. The increase of CASPASE 3 and BAX and the decrease of BCL2 [Supplementary Figure 3; Figures 5D–F (EAM group, 3W)] in myocardial cells indicate an increase in apoptosis in EAM rats. Also, the increased protein expression of LC3 I and II (Figures 3E,F; Supplementary Figure 2) in both “early” and “late” stages of EAM rat myocardial tissue are observed in comparison to control groups on 3 and 8W. Taken together, these data confirm that in EAM rats, all three types of programmed cell death, necroptosis, apoptosis, and autophagy occur in vivo. In addition, the EAM group in “early” stages (3W) developed severe cardiac dysfunction (Figure 2), including decrease of EF and FS, ventricular dilation, and hypertrophy with reduced RIP1, RIP3, and MLKL expression. Thus, this group of rats (3W) could serve as an excellent EAM model for future use.
Nec-1 is a small molecule specific inhibitor of necroptosis that uniquely inhibits the activation of RIP1 and the interaction of RIP1 with RIP3 to form the RIP1–RIP3 complex to subsequently block necroptosis (24, 25). To understand whether Nec-1 inhibits RIP1/RIP3/MLKL-dependent necroptosis in EAM rats in vivo and whether this inhibition is independent of autophagy, we treated “early” stages of EAM group (3W) rats with necrostatin injection (Nec-1 group). Strikingly, we observed that in the Nec-1 group, cardiac conditions were improved, inflammatory infiltration was ameliorated, myocardial cell injury was attenuated, and, thus, most of the pathological lesions of DCM in Nec-1 group were alleviated compared to the EAM-DMSO group (Figures 4, 8A–F). We assessed the expression of RIP1, RIP3, MLKL, CASPASE 3, BAX, BCL2, and LC3 I and II in Nec-1-treated rats. The results of RT-QPCR, Western blotting, and immunohistochemical studies revealed that the expression levels of RIP1, RIP3, and MLKL were significantly decreased (Figures 5A–C, 6A–D, 7A–D) in the Nec-1 group compared to the EAM + DMSO group and there were no differences in the expression levels of these genes and proteins (RIP1, RIP3, and MLKL) between the non-treated EAM and DMSO groups. Meanwhile, we also observed that treatment with Nec-1 increased the mRNA expression of CASPASE 3 and BAX and decreased BCL2 expression (Figures 5D–F). Notably, it did not obviously affect the expression level of LC3 I and II in the Nec-1 group compared to the EAM-DMSO group (Figures 6E,F). However, the expression levels of these two genes and proteins were also unchanged in the non-treated EAM and DMSO groups. These results demonstrate that administration of Nec-1 in the “early” stages of EAM inhibits RIP1/RIP3/MLKL-dependent necroptosis, increases apoptosis, but has no effect on the autophagy pathway in the EAM Nec-1 group in vivo. Nevertheless, Nec-1 significantly preserved cardiac function and reduced inflammatory infiltrates in the EAM model, highlighting the importance of its cardioprotective effects, suggesting that it could be developed as future therapeutic and diagnostic targets for inflammatory myocarditis.
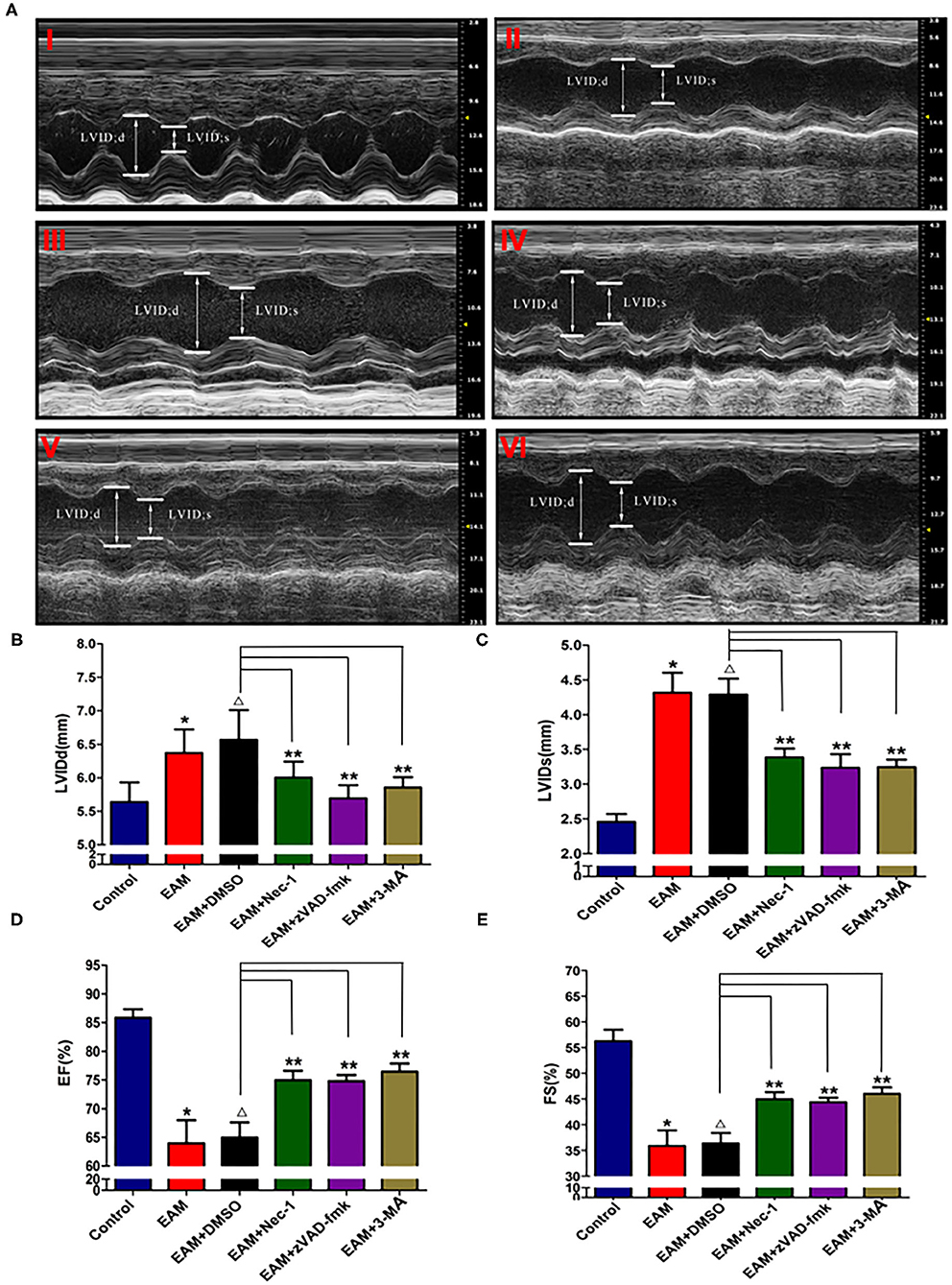
Figure 4. The level of LVIDd/LVIDs/EF/FS in “early” stage of myocarditis in EAM mice with inhibitors by echocardiography. (A) Echocardiographic images of control and EAM mice (3W) (I—control, II—EAM, III—EAM + DMSO, IV—EAM + Nec-1, V—EAM + zVAD-fmk, VI—EAM+3Ma). (B–E) Quantification of LVIDd, LVIDs, EF, and FS in the “early” stage of myocarditis from control groups. A horizontal bar is shown to indicate compared groups. All data are presented as mean ± SEM. A Student's t-test was performed for the control group and the EAM group, *p < 0.05 vs. control group **p < 0.01 vs. EAM + DMSO group, Δp > 0.05 vs. EAM group.
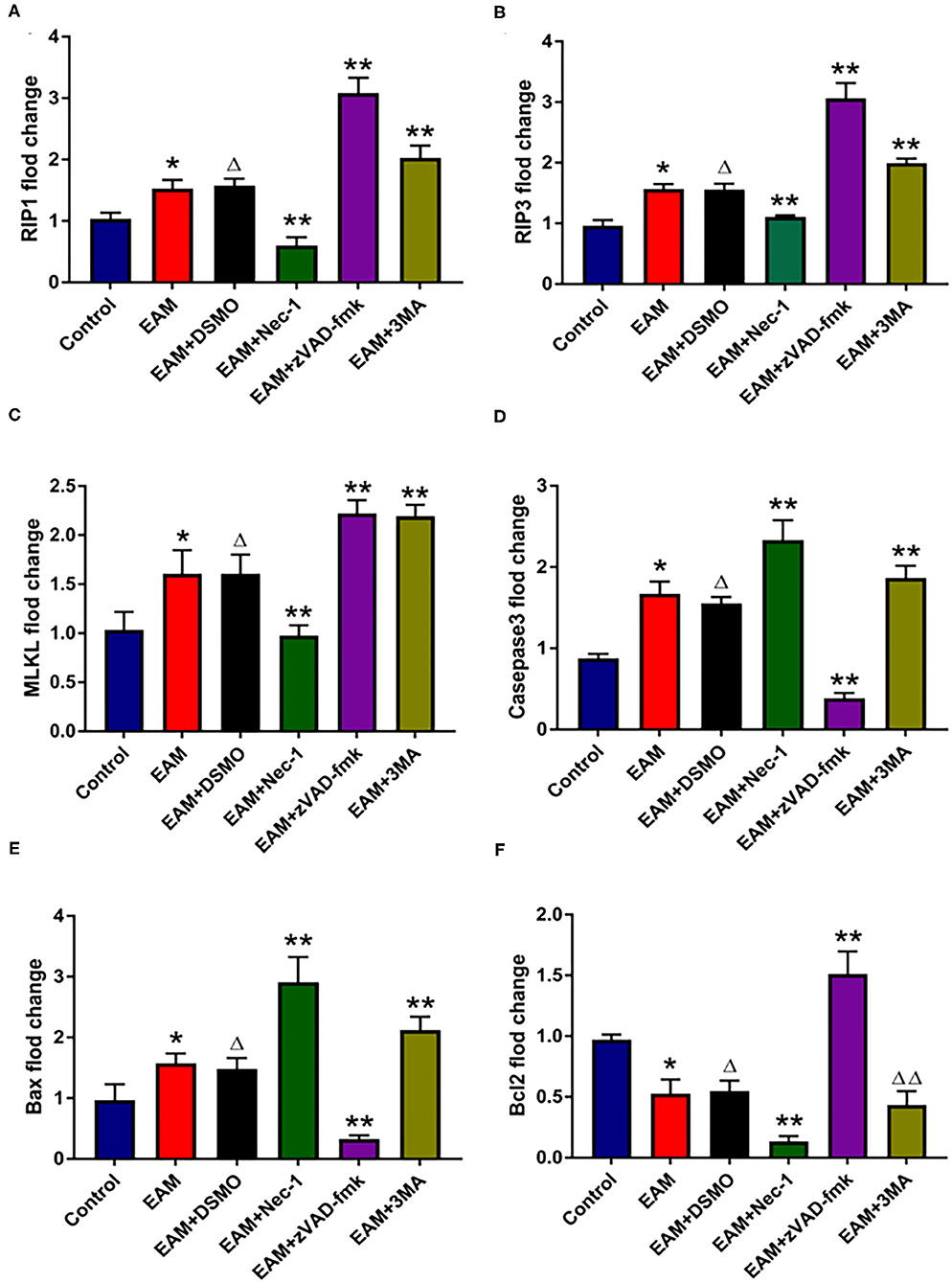
Figure 5. The relative expression of mRNA in myocardial tissue of each group detected by quantitative real-time PCR. (A–C) Real-time PCR analysis of RIP1, RIP3, and MLKL in each group, and inhibited by necrosis inhibitor Nec-1, and 3-MA but increased by zVAD-fmk. For EAM, 3W rats are used. (D–F) Real-time PCR analysis of Caspase 3, Bax, and Bcl2 in each group. The necrosis inhibitor Nec-1 and 3-MA increase the mRNA expression levels of Caspase 3/Bax mRNA in myocardial tissues. *p < 0.05 vs. control group; **p < 0.01 vs. EAM + DMSO group, Δp > 0.05 vs. EAM group, ΔΔp > 0.05 vs. EAM + DMSO group. A Student's t-test was performed for the control group and the EAM group, one-way analysis of variance (ANOVA) followed by the LSD test; data are presented as mean ± SEM.
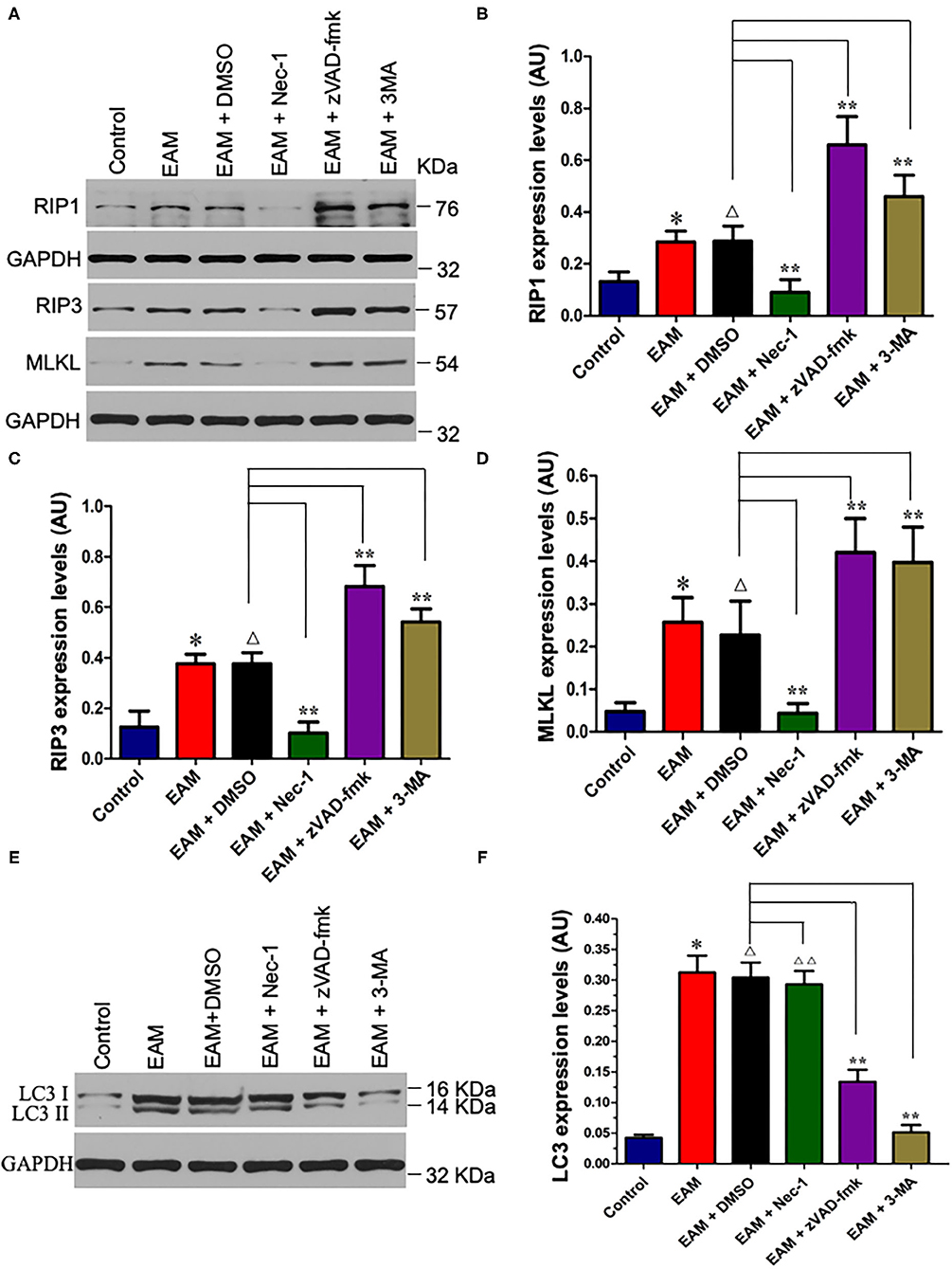
Figure 6. Nec-I inhibits but 3-MA increases the expression levels of RIP1, RIP3, and MLKL in EAM. (A) Western analysis of protein expression levels of RIP1, RIP3, and MLKL in myocardial tissues from the control group; EAM (3W) group; EAM + DMSO group; and EAM + Nec-I, EAM + zVAD-fmk, and EAM + 3MA drug group. (B–D) Quantification of protein levels of RIP1 in b, RIP3 in c, and MLKL in d. Horizontal lines above bars indicate groups compared. All data are presented as mean ± SEM. A Student's t-test was performed for the control group and EAM group, one-way analysis of variance (ANOVA) followed by the LSD test. *p < 0.05 vs. control group; **p < 0.01 vs. EAM + DMSO group, Δp > 0.05 vs. EAM group. Δp > 0.05 vs. EAM + Nec-1 group, Δp > 0.05 vs. EAM + 3-MA group. (E,F) Quantification of LC-3 I and II. Western analysis of LC-3 I and II expression levels treated by inhibitors of Nec-1 and 3-MA. (D) Quantification of LC-3 I and II of EAM (3W); ΔΔp < 0.05 means compared with the control group (3 weeks); **p > 0.05 means compared with the control group (8 weeks).
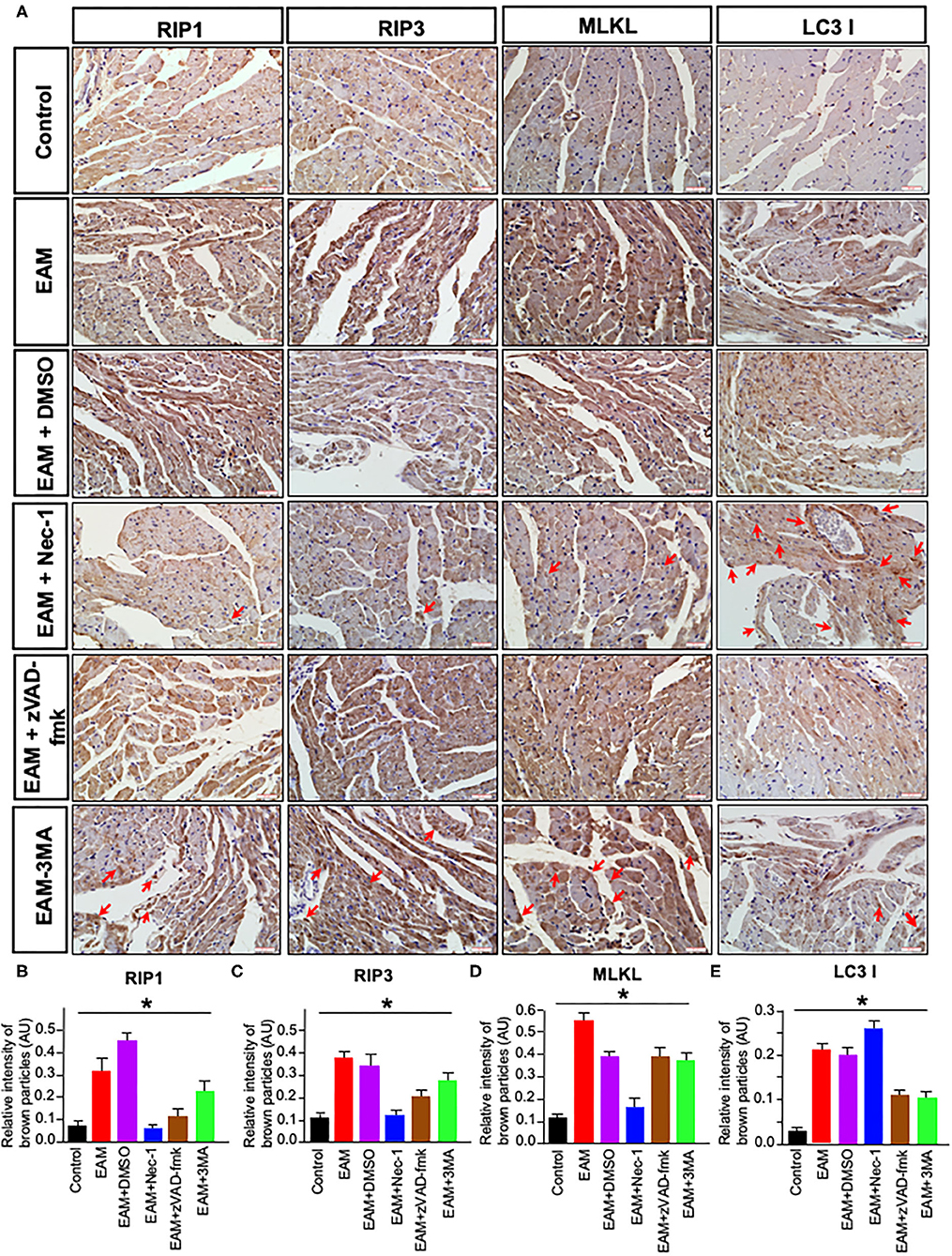
Figure 7. Immunohistochemical staining of RIP1/RIP3/MLKL and LC3B in rat myocardium. (A) Imunohistochemistry staining images of RIP1/RIP3/MLKL and LC3B. Red arrows point the brown particle deposition. Control: control group; EAM: EAM (3W); IMM + DMSO; EAM + Nec-1: necroptosis inhibitor group; EAM-zVAD-fmk, apoptosis inhibitor group, EAM + 3-MA: autophagy inhibitor group. RIP1, RIP3, MLKL, and LC3B. (B–E) Quantity of brown–red particles' intensity in the cytoplasm of RIP1, RIP3, MLKL, and LC3 I, respectively. The necrosis inhibitor Nec-1 decreased the intensities in RIP1, RIP3, and MLKL, but not in LC3 I and II. However, the autophagy inhibitor 3-MA inhibited the intensity of LC3B. A Student's t-test was performed in each group. *p < 0.05 are obtained for RIP1, when comparing EAM with EAM + Nec-1, EAM + zVAD-fmk, 3-MA; for RIP3, EAM with EAM + Nec-1, EAM + zVAD-fmk, and EAM + 3-MA; for MLKL, EAM with EAM-Nec-1, EAM + zVAD-fmk, and EAM-3-MA; for LC3B, EAM and EAM + Nec-1 and EAM and EAM +3-MA.
Autophagy, a caspase-independent pathway, is physiologically a cellular mechanism for cell growth regulation with internal homeostasis. Recent studies show that autophagy also occurs during necrosis (26). Nevertheless, it remains a conjecture whether 3-MA can shift cell death from autophagy toward RIP1/RIP3/MLKL-mediated necroptosis. To test this, we treated the “early” stages of EAM rats with the autophagy inhibitor 3-MA (3-MA group) and then assessed the expression of RIP1, RIP3, MLKL, and LC3 I and II. Our results demonstrated that LC3 I and II levels were decreased (Figures 6E,F), whereas RIP1, RIP3, and MLKL levels were elevated in the 3-MA rat groups compared to the EAM-DMSO group (Figures 6A–D). Immunohistochemical studies for these proteins also showed similar patterns as expression differences (Figure 7). These results indicate that the blockade of autophagy by 3-MA can induce the RIP1/RIP3/MLKL-mediated necroptosis pathway in EAM rats in “early” stages. Autophagy blockade by 3-MA also induces apoptosis as observed by the increased expression of Caspase3 and BAX.
Next, we also observed that cardiac conditions (measured by EF and FS) in the 3-MA group were significantly increased compared to the EAM-DMSO group as shown in the echocardiographic examination on 3W (Figure 4). In addition, HE staining showed that in the 3-MA group, inflammatory infiltration was reduced, which alleviated myocardial cells compared to the DMSO group (Figure 8). These results demonstrate that treatment with 3-MA also significantly preserves cardiac function and reduces inflammatory response in EAM.
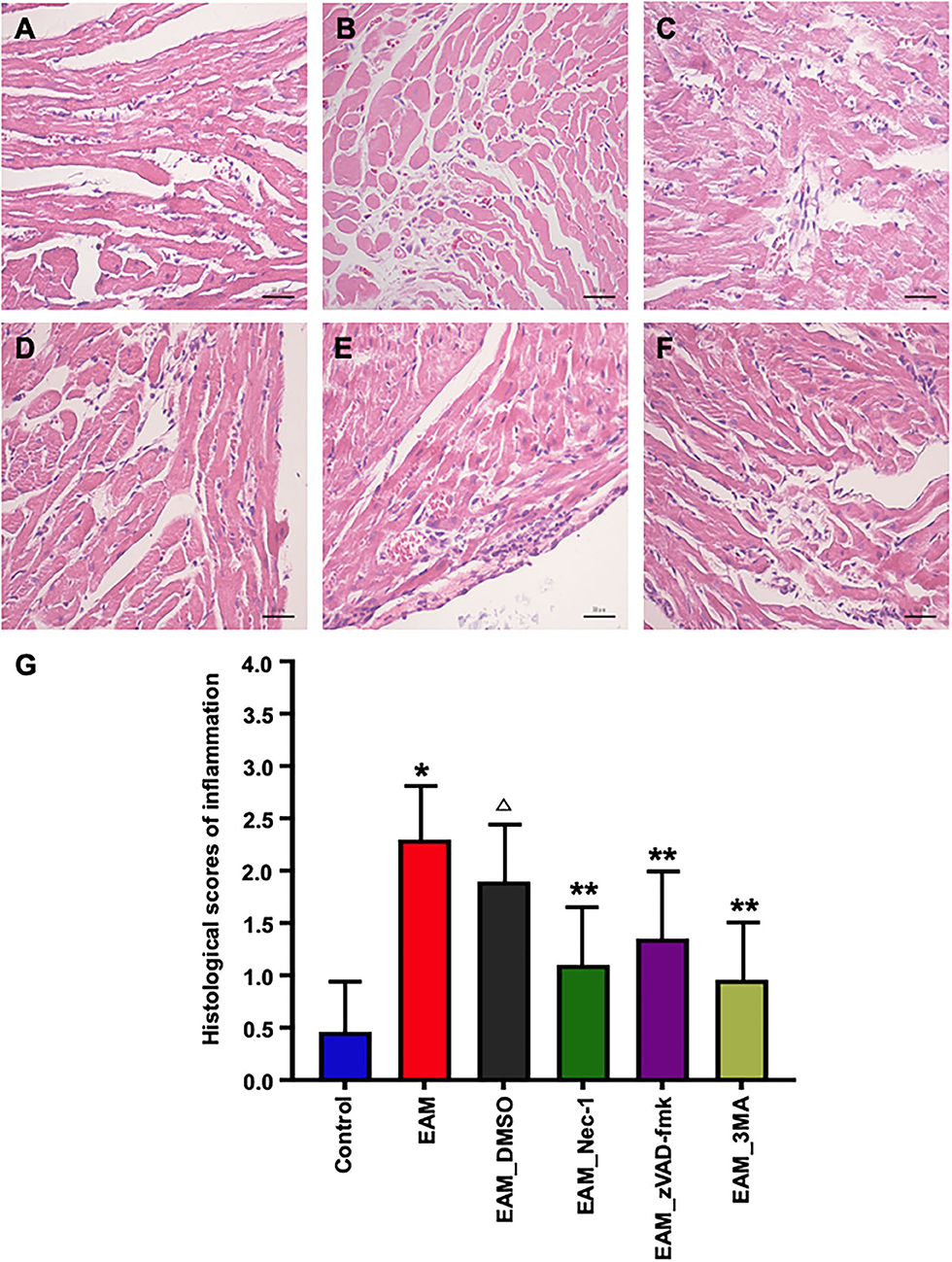
Figure 8. Pathological evaluations of myocardial tissues of various groups by HE staining. (A) In the heart of control group rat, there was no inflammatory infiltrate. (B,C) HE staining showed dense inflammatory infiltrates in the EAM (B) group (3W) and IMM + DMSO group (C). Treating rat with Nec-1, zVAD-fmk, and 3-MA on day 21 attenuated inflammatory infiltrates. Only little scattered inflammatory infiltrates were detected in the EAM + Nec-1 group (D). (G) Quantification of cellular inflammatory infiltration in (A–F). All data are presented as mean ± SEM. A Student's t-test was performed for the control group and IMM group, one-way analysis of variance (ANOVA) followed by the LSD test. *p < 0.05 vs. control group, **p < 0.01 vs. EAM + DMSO group, p < 0.05 vs. EAM. Δ Compared with EAM group, the EAM_DMSO group has no significance.
The inhibitor of apoptosis, such as zVAD-fmk, influences the RIP3-induced programmed necroptosis (9, 14). To observe the influence of apoptosis on necroptosis and autophagy, we use zVAD-fmk, a CASPASE 8 inhibitor of apoptosis, to block the caspase-dependent apoptosis pathway and then monitored the expression level of necroptosis inducing genes, RIP1, RIP3, MLKL, apoptosis specific CASPASE 3, and autophagy specific LC3 I and II. We observed that inhibition of caspase-dependent apoptosis by zVAD-fmk dramatically enhanced necroptosis and autophagy, as assessed by increased expression of RIP1, RIP3, MLKL (Figures 6A–D, 7), and LC3 I and II (Figures 6E,F). Furthermore, EAM rats in the zVAD-fmk group had improved cardiac conditions (Figure 4) with reduced inflammatory infiltration and attenuated myocardial cell injury in comparison to the EAM-DMSO group (Figure 8). These results demonstrate that an apoptosis inhibitor, similar to a necroptosis inhibitor and an autophagy inhibitor, also significantly improves cardiac conditions and reduces inflammatory response in EAM rats. Thus, inhibition of the apoptosis pathway not only shifts myocardial cell death from apoptosis to necroptosis but also triggers autophagy and all the three major types of programmed cell death in EAM rats.
Myocarditis is an inflammatory disease and is one of the leading causes of DCM. DCM is the leading global event for heart transplantation that has an estimated prevalence of ~1:250 people (27). DCM can be detected in patients with progressive heart failure, arrhythmias, thromboembolic disease, or sudden cardiac death (SCD). It is characterized by left ventricular cavity enlargement or dilatation and systolic dysfunction (28). DCM is implicated as the main cause of adult death and can be induced by viral infection and autoimmune and genetic factors (27, 29). Current studies demonstrate that autoimmunity-mediated cardiac damage plays an important role in the pathogenesis of a subset of post-infectious myocarditis, progressive inflammation, fibrosis, and pathological remodeling in DCM (30).
EAM is an effective animal model to study the mechanism of myocardial injury. EAM can be induced in susceptible animals, such as balb/c mouse and Lewis rats, by immunization with PCM or with specific peptides or adoptive transfer of myosin-reactive T cells together with Freud's adjuvant (31). In EAM, CD4+ T cells induce the development of myocarditis, which is characterized by mononuclear macrophage infiltration, necrosis of cardiomyocytes, and proliferation of cardiac fibroblasts (32). To investigate the role of necroptosis, apoptosis, and autophagy, we established a rat model of EAM, which was the best available model to reflect the DCM process, although it might not completely mimic the physiological condition of human DCM.
For many years, apoptosis was considered the classical cell death pathway to maintain homeostasis in cardiovascular disease. Necroptosis, as a caspase-independent cell death pathway, was considered to be a substituted form of cell death when apoptosis was blocked (33). Recently, several studies indicate that necroptosis and autophagy also occur in various cardiovascular pathological conditions with a faster and more effective cell death response than apoptosis (34, 35). Szobi et al. (5) demonstrated that necroptosis may take place in coronary artery disease (5). Nevertheless, little is known whether three forms of cell death take place together in DCM causing sudden cardiac death.
To understand the mechanism of cell death in DCM in EAM, we first assessed cardiac function and myocardial inflammation by echocardiographic examination and HE staining. Our results showed that cardiac conditions as revealed by EF and FS in the EAM group was significantly decreased on 3W with the presence of dense inflammatory infiltrates in cardiomyocytes, suggesting that EAM was successfully established. Increased necroptosis-mediated cardiac cell death was also observed in the viral cardiomyocyte (VMC) mouse model (7). Next, we further assessed necroptosis-, apoptosis-, and autophagy-inducing genes and protein expressions in EAM. Our Western blotting and RT-QPCR results showed that the expression of RIP1, RIP3, MLKL, and LC3 I and II in the EAM group was significantly increased compared to the control group. mRNA expressions of CASPASE3 and BAX were increased, whereas BCL2 expression was decreased, indicating an increase in apoptosis. Immunohistochemical examination showed that brown particles were all distributed in the myocardial cytoplasm, and we also noticed that the level of necroptosis, apoptosis, and autophagy in the 3W rat group was markedly higher than that in the 8W group. These observations confirmed that all three forms of cell death pathway occurred in the EAM “early” stages (3W) more intensely than in the “late” stages (8W), and thus, we selected the “early” stages (3W) of rats to continue to study the relationship between necroptosis, autophagy, and apoptosis.
To understand the relationship and underlying mechanisms among necroptosis, apoptosis, and autophagy, we treated EAM rats with a necroptosis inhibitor (Nec-1), apoptotic inhibitor (zVAD-fmk), and autophagy inhibitor (3-MA). Nec-1 is a necroptosis-specific small-molecule inhibitor, uniquely inhibiting the activation of RIP1 not through the transcriptional regulation but through the blocking of RIP1 kinase activity and the interaction of RIP1–RIP3 (24, 36). First, we observed that Nec-1 treatment decreased the expression of necroptosis-specific genes and proteins, RIP1, RIP3, and MLKL. Second, the expression of apoptosis-inducing genes CASPASE 3 and BAX was increased but BCL2, a protective gene against apoptosis, was decreased. Thus, blockade of necroptosis by Nec-1 not only prevents necrotic cell death but also induces apoptosis in EAM, although the mechanism of this switching is currently unclear. It is demonstrated that the switch from necroptosis to apoptosis was correlated with the RIP1 kinase activity (4, 37). In cylindromatosis (CYLD), RIP1 recruits CASPASE 8 and Fas-associated protein via a death domain (FADD) and TNFR1-associated death domain (TRADD) to trigger apoptosis when CASPASE 8 is activated (37, 38), whereas when CASPASE 8 is blocked, RIP1 and RIP3 interact with each other without degradation and form necrosome to activate necroptosis (25). Our results also indicate that Nec-1 does not have a significant effect on LC3 I and II expression, suggesting that necroptosis inhibition may not affect autophagy. Osborn et al. (39) observed that necroptosis of proliferating FADD-deficient T cells does not depend on hyper-autophagic signaling.
The effect of apoptosis inhibition by zVAD-fmk in EAM induces necroptosis and autophagy. The expression of CASPASE 3 was decreased and RIP1, RIP3, MLKL, and LC3 were markedly increased. Furthermore, in EAM rats, zVAD-fmk improved cardiac conditions, reduced inflammatory infiltration, and attenuated myocardial cell injury compared to the EAM-DMSO group. It is known that apoptosis deficiency can cause a switch from apoptosis to necroptosis, and zVAD-fmk promotes RIP1 phosphorylation by inhibiting CASPASE 8 activation to induce necroptosis (26, 40).
To further determine the relationship between autophagy and necroptosis, we treated EAM rats with 3-MA, an inhibitor of autophagy. Our data showed that the levels of RIP1, RIP3, and MLKL were upregulated in the 3-MA group and thus necroptosis can also be triggered when autophagy is blocked in EAM. We also observed that blockage of autophagy can also improve cardiac conditions and attenuate inflammatory response. Thus, inhibition of autophagy by 3-MA not only switches cell death from autophagy to necroptosis but also protects cardiac conditions from myocarditis. Many studies demonstrated the extensive crosstalk between necroptosis and autophagy as autophagy is capable of inducing, suppressing, or having no effect on necroptosis and apoptosis (39, 41–44). In a cardiac ischemia–reperfusion injury (IRI) mouse model, RIP3K (RIP3 kinase) is shown to decrease mitophagy and promote mitochondria-mediated apoptosis, further implying switching between autophagy and apoptosis (45). In osteosarcoma, autophagy and necroptosis affect the proliferation and invasion of osteosarcoma cells (46). Both cancer and normal cells can induce autophagy and necroptosis in response to nutrient or serum starvation (47). Oberst (48) illustrated that the recruitment of FADD to autophagosomes directly activates RIP1 and RIP3. In contrast, recent studies have also demonstrated that autophagy could inhibit necroptosis under some special conditions, such as stimulation by TNF-α, certain antigen encounters, or starvation (44). We observe that inhibition of necroptosis pathway by Nec-1 may not have much effect on autophagy, whereas inhibition of autophagy increases necroptosis. However, none of the inhibitors reversed LVEF to a normal level. A cell death pathway of necroptosis, or apoptosis, or autophagy cannot be inhibited entirely, but it can activate cells to choose an alternative pathway to participate in cell death (40). These three programmed cell death pathways are not completely isolated, and they are somewhat interrelated. It appears that, in our system, necroptosis and apoptosis are interchangeable and complimentary as blockage of either pathway activates the other. Similar conclusion is suggested by Murphy and Silke (33). In contrast, necroptosis blockage does not have any effect on autophagy, but autophagy blockage increases necroptosis. Thus, taken together, our results suggest that autophagy is an independent cell death pathway that acts upstream of the necroptosis signaling pathway, and it can suppress other types of cell death such as necroptosis. However, it appears that the mechanism of the autophagy pathway is a more complex process.
We established an EAM model of rats where all three pathways of cell death necroptosis, apoptosis, and autophagy take place with deterioration of cardiac conditions and inflammatory infiltration in cardiomyocytes. Blockage of each cell death pathway, with their respective inhibitor, independently alleviates cardiac conditions with reduced inflammatory infiltration. Each inhibitor not only blocks the specific pathway but also induces alternative pathways to promote cell death in EAM rats. Nevertheless, the interplay between autophagy, apoptosis, and necroptosis is evident in EAM (Figure 9), but the functional role and the interrelated molecular mechanisms of these three cell death pathways remain a subject of further investigations.
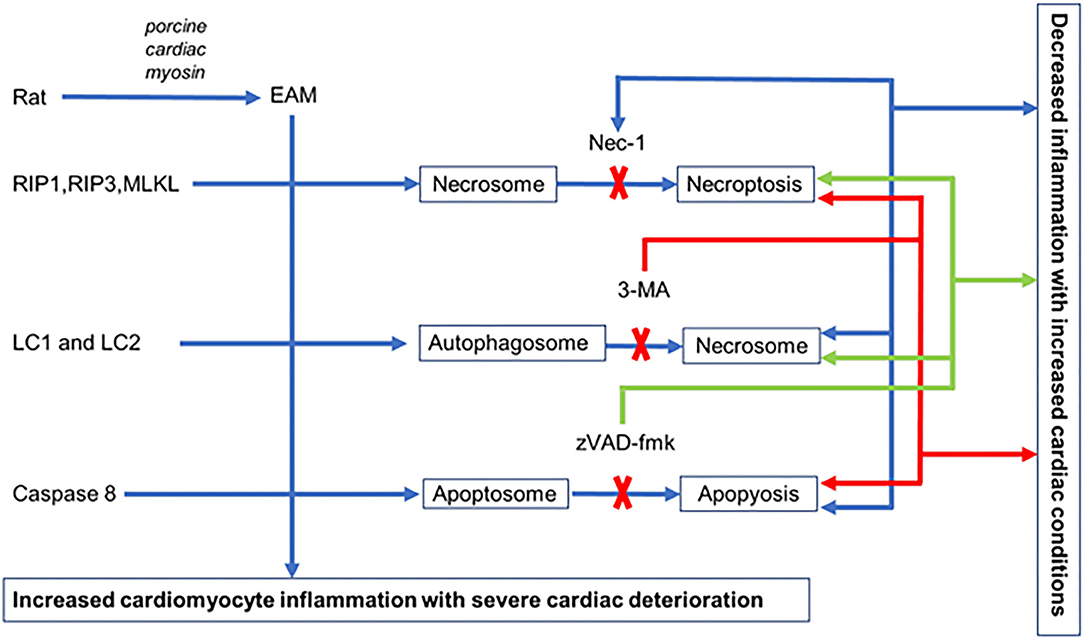
Figure 9. Hypothetical illustration of cell death in EAM model. In EAM, all three processes of cell death take place, but when necroptosis is blocked by Nec-1, apoptosis increases but there is no effect on autophagy. When autophagy is blocked by 3-MA, both apoptosis and necroptosis are increased. When apoptosis is blocked by zVAD-fmk, it increases both necroptosis and autophagy.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by Institutional Review Board of the First Affiliated Hospital of Nanchang University.
ZZ and HW: conceptualization and funding acquisition. YW, ZZ, XC, AA, IA, and HW: methodology. YW, ZZ, and XC: validation. YW, ZZ, AM, and HW: formal analysis. ZZ, AM, QY, VN, AA, IA, and HW: writing—review and editing. QY and VN participated in the revision and editing of the paper and the revision of English grammar. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the National Nature Science Foundation of China (Nos. 81660067 and 81260044), the Technology Support project of Jiangxi province (No. 2010BSA12000) to ZZ, and AHA SDG Grant 17SDG33410868 to HW. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.696362/full#supplementary-material
CFA, Complete Freund's adjuvant; CHF, congestive heart failure; CYLD, cylindromatosis; DCM, dilated cardiomyopathy; EAM, experimental autoimmune myocarditis model; EF, ejection fraction; FADD, Fas-associated protein via death domain; LVEDd, left ventricular end-diastolic diameter; LVESd, left ventricular end-systolic diameter; 3-MA, 3-Methyladenine; MLKL, mixed lineage kinase domain-like; Nec-1, necrostatin-1; PCM, porcine cardiac myosin; RIP, receptor-interacting protein kinase; TNFRT, tumor necrosis factor receptor tumor; VMC, viral myocarditis.
1. Rose NR. Viral myocarditis. Curr Opin Rheumatol. (2016) 28:383–9. doi: 10.1097/BOR.0000000000000303
2. Reddy J, Massilamany C, Buskiewicz I, Huber AS. Autoimmunity in viral myocarditis. Curr Opin Rheumatol. (2013) 25:502–8. doi: 10.1097/BOR.0b013e3283620036
3. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. (2015) 14:48. doi: 10.1186/s12943-015-0321-5
4. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. (2010) 11:700–14. doi: 10.1038/nrm2970
5. Szobi A, Gonçalvesová E, Varga ZV, Leszek P, Kuśmierczyk M, Hulman M, et al. Analysis of necroptotic proteins in failing human hearts. J Transl Med. (2017) 15:86. doi: 10.1186/s12967-017-1189-5
6. Zhe-Wei S, Li-Sha G, Yue-Chun L. The role of necroptosis in cardiovascular disease. Front Pharmacol. (2018) 9:721. doi: 10.3389/fphar.2018.00721
7. Zhou F, Jiang X, Teng L, Yang J, Ding J, He C. Necroptosis may be a novel mechanism for cardiomyocyte death in acute myocarditis. Mol Cell Biochem. (2018) 442:11–8. doi: 10.1007/s11010-017-3188-5
8. Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev. (2012) 249:205–17. doi: 10.1111/j.1600-065X.2012.01147.x
9. Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. (2011) 471:368–72. doi: 10.1038/nature09857
10. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
11. Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA. (2014) 111:15072–7. doi: 10.1073/pnas.1408987111
12. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. (2014) 24:105–21. doi: 10.1038/cr.2013.171
13. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
14. Zhou W, Yuan J. SnapShot: necroptosis. Cell. (2014) 158:464–464.e461. doi: 10.1016/j.cell.2014.06.041
15. Korkusuz H, Esters P, Huebner F, Bug R, Ackermann H, Vogl JT. Accuracy of cardiovascular magnetic resonance in myocarditis: comparison of MR and histological findings in an animal model. J Cardiovasc Magn Reson. (2010) 12:49. doi: 10.1186/1532-429X-12-49
16. Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. (2013) 27:1640–9. doi: 10.1101/gad.223321.113
17. Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA. et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. (2012) 107:270. doi: 10.1007/s00395-012-0270-8
18. Koudstaal S, Oerlemans MI, Van der Spoel TI, Janssen AW, Hoefer IE, Doevendans PA, et al. Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur J Clin Invest. (2015) 45:150–9. doi: 10.1111/eci.12391
19. Tian F, Yao J, Yan M, Sun X, Wang W, Gao W, et al. 5-aminolevulinic acid-mediated sonodynamic therapy inhibits RIPK1/RIPK3-dependent necroptosis in THP-1-derived foam cells. Sci Rep. (2016) 6:21992. doi: 10.1038/srep21992
20. Watson LE, Sheth M, Denyer RF, Dostal ED. Baseline echocardiographic values for adult male rats. J Am Soc Echocardiogr. (2004) 17:161–7. doi: 10.1016/j.echo.2003.10.010
21. Herman E, Eldridge S. Spontaneously occurring cardiovascular lesions in commonly used laboratory animals. Cardiooncology. (2019) 5:6. doi: 10.1186/s40959-019-0040-y
22. Zheng Z, Yu Y, Potla R, Wu Y, Wu H. Fibrinogen-like protein-2 causes deterioration in cardiac function in experimental autoimmune myocarditis rats through regulation of programmed death-1 and inflammatory cytokines. Immunology. (2018) 153:246–52. doi: 10.1111/imm.12837
23. Liu W, Li WM, Gao C, Sun LN. Effects of atorvastatin on the Th1/Th2 polarization of ongoing experimental autoimmune myocarditis in Lewis rats. J Autoimmun. (2005) 25:258–63. doi: 10.1016/j.jaut.2005.06.005
24. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
25. Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, et al. Structural basis of RIP1 inhibition by necrostatins. Structure. (2013) 21:493–9. doi: 10.1016/j.str.2013.01.016
26. Dong K, Zhu ZC, Wang FH, Ke GJ, Yu Z, Xu X. Activation of autophagy in photoreceptor necroptosis after experimental retinal detachment. Int J Ophthalmol. (2014) 7:745–52. doi: 10.3980/j.issn.2222-3959.2014.05.01
27. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. (2013) 10:531–47. doi: 10.1038/nrcardio.2013.105
28. Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. (2013) 309:896–908. doi: 10.1001/jama.2013.1363
29. Tayal U, Prasad S, Cook AS. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. (2017) 9:20. doi: 10.1186/s13073-017-0410-8
30. Hoetzenecker K, Zimmermann M, Hoetzenecker W, Schweiger T, Kollmann D, Mildner M, et al. Mononuclear cell secretome protects from experimental autoimmune myocarditis. Eur Heart J. (2015) 36:676–85. doi: 10.1093/eurheartj/ehs459
31. Liu J, Wu P, Wang Y, Du Y, Liu AS, Zhang Y, et al. Ad-HGF improves the cardiac remodeling of rat following myocardial infarction by upregulating autophagy and necroptosis and inhibiting apoptosis. Am J Transl Res. (2016) 8:4605–4627.
32. Su Z, Zhang P, Yu Y, Lu H, Liu Y, Ni P, et al. HMGB1 facilitated macrophage reprogramming towards a proinflammatory M1-like phenotype in experimental autoimmune myocarditis development. Sci Rep. (2016) 6:21884. doi: 10.1038/srep21884
33. Murphy JM, Silke J. Ars Moriendi; the art of dying well - new insights into the molecular pathways of necroptotic cell death. EMBO Rep. (2014) 15:155–64. doi: 10.1002/embr.201337970
34. Myers JM, Fairweather D, Huber SA, Cunningham WM. Autoimmune myocarditis, valvulitis, and cardiomyopathy. Curr Protoc Immunol Chapter. (2013) 15:Unit 15.14.11–51. doi: 10.1002/0471142735.im1514s101
35. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. (2014) 24:69–79. doi: 10.1038/cr.2013.161
36. Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. (2013) 5:70–8. doi: 10.1016/j.celrep.2013.08.044
37. Moquin DM, McQuade T, Chan KF. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE. (2013) 8:e76841. doi: 10.1371/journal.pone.0076841
38. Newton K, Dugger DL, Wickliffe KE N, Kapoor, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. (2014) 343:1357–60. doi: 10.1126/science.1249361
39. Osborn SL, Diehl G, Han SJ, Xue L, Kurd N, Hsieh K, et al. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci USA. (2010) 107:13034–9. doi: 10.1073/pnas.1005997107
40. Saeed WK, Jun DW. Necroptosis: an emerging type of cell death in liver diseases. World J Gastroenterol. (2014) 20:12526–32. doi: 10.3748/wjg.v20.i35.12526
41. Martyniszyn L, Szulc-Dabrowska L, Boratyńska-Jasińska A, Struzik J, Winnicka A, Niemiałtowski M. Crosstalk between autophagy and apoptosis in RAW 264.7 macrophages infected with ectromelia orthopoxvirus. Viral Immunol. (2013) 26:322–35. doi: 10.1089/vim.2013.0003
42. Besirli CG, Chinskey ND, Zheng QD, Zacks ND. Autophagy activation in the injured photoreceptor inhibits fas-mediated apoptosis. Invest Ophthalmol Vis Sci. (2011) 52:4193–9. doi: 10.1167/iovs.10-7090
43. Cai N, Zhao X, Jing Y, Sun K, Jiao S, Chen X, et al. Autophagy protects against palmitate-induced apoptosis in hepatocytes. Cell Biosci. (2014) 4:28. doi: 10.1186/2045-3701-4-28
44. Kim HJ, Hwang KE, Park DS, Oh SH, Jun HY, Yoon KH, et al. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J Transl Med. (2017) 15:123. doi: 10.1186/s12967-017-1223-7
45. Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. (2017) 13:498–507. doi: 10.1016/j.redox.2017.07.007
46. Li J, Yang Z, Li Y, Xia J, Li D, Li H, et al. Cell apoptosis, autophagy and necroptosis in osteosarcoma treatment. Oncotarget. (2016) 7:44763–78. doi: 10.18632/oncotarget.8206
47. Jung S, Li C, Duan J, Lee S, Kim K, Park Y, et al. TRIP-Br1 oncoprotein inhibits autophagy, apoptosis, and necroptosis under nutrient/serum-deprived condition. Oncotarget. (2015) 6:29060–75. doi: 10.18632/oncotarget.5072
Keywords: experimental autoimmune myocarditis, necroptosis, RIP1/RIP3/MLKL pathway, autophagy, necrostatin-1
Citation: Wu Y, Zheng Z, Cao X, Yang Q, Norton V, Adini A, Maiti AK, Adini I and Wu H (2021) RIP1/RIP3/MLKL Mediates Myocardial Function Through Necroptosis in Experimental Autoimmune Myocarditis. Front. Cardiovasc. Med. 8:696362. doi: 10.3389/fcvm.2021.696362
Received: 16 April 2021; Accepted: 22 July 2021;
Published: 23 August 2021.
Edited by:
Christian Schulte, University Heart and Vascular Center Hamburg (UHZ), GermanyReviewed by:
Jun Ren, University of Washington, United StatesCopyright © 2021 Wu, Zheng, Cao, Yang, Norton, Adini, Maiti, Adini and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenzhong Zheng, Z3JlYXRlZnVsQDE2My5jb20=; Hao Wu, aGFvLnd1M0BjaGlsZHJlbnMuaGFydmFyZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.