 Carolyn M. Roos1
Carolyn M. Roos1 Bin Zhang1
Bin Zhang1 Michael A. Hagler1Grace C. Verzosa1
Michael A. Hagler1Grace C. Verzosa1 Runqing Huang1Elise A. Oehler1Arman Arghami1
Runqing Huang1Elise A. Oehler1Arman Arghami1 Jordan D. Miller1,2*
Jordan D. Miller1,2*- 1Department of Surgery, Mayo Clinic, Rochester, MN, United States
- 2Department of Physiology and Biomedical Engineering, Mayo Clinic, Rochester, MN, United States
Background: While a small number of studies suggest that oxidative stress has an influential role in fibrocalcific aortic valve disease (FCAVD), the roles of specific antioxidant enzymes in progression of this disease remain poorly understood. Here, we focused on selectively altering mitochondrial-derived oxidative stress—which has been shown to alter progression of a myriad of age-associated diseases—on the progression of molecular and phenotypic drivers of FCAVD.
Methods: We generated low-density lipoprotein receptor-deficient, Apolipoprotein B100-only mice (LA) that were either haploinsufficient for MnSOD (LA-MnSOD+/−) or genetically overexpressing MnSOD (LA-MnSODTg/0). After 6 months of Western diet feeding, mice underwent echocardiography to assess valvular and cardiac function and tissues were harvested. Quantitative-RT PCR, immunohistochemistry, and histopathology were used to measure changes in molecular pathways related to oxidative stress, calcification, and fibrosis.
Results: While reductions in MnSOD increased oxidative stress, there was not an overt phenotypic effect of MnSOD deficiency on valvular and cardiac function in LA-MnSOD+/− mice. While markers of canonical bone morphogenetic protein signaling tended to increase in valve tissue from LA-MnSOD+/− (e.g., p-SMAD1/5/8 and osterix), we did not observe statistically significant increases in osteogenic signaling. We did, however, observe highly significant reductions in expression of osteopontin, which were associated with significant increases in calcium burden in LA-MnSOD+/− mice. Reciprocally, genetically increasing MnSOD did not preserve valve function in LA-MnSODTg/0, but we did observe slight reductions in p-SMAD1/5/8 levels compared to their non-transgenic littermates. Interestingly, overexpression of MnSOD dramatically increased expression of osteopontin in valve tissue from LA-MnSODTg/0 mice, but was not sufficient to attenuate calcium burden when compared to their LA-MnSOD0/0 littermates.
Conclusions: Collectively, this study demonstrates that maintenance of mitochondrial antioxidant capacity is important in preventing accelerated disease progression in a mouse model of FCAVD, but that effectively altering mitochondrial antioxidant capacity as a monotherapeutic approach to slow key histopathological and molecular drivers of FCAVD remains biologically and therapeutically challenging.
Introduction
Approximately 2–3% of the population over the age of 65 develops fibrocalcific aortic valve disease (FCAVD). While major risk factors for development of FCAVD—such as increasing age and hypercholesterolemia—are frequently shared with atherosclerosis (1), trials of anti-atherosclerotic interventions have yielded largely negative results in patients with FCAVD (2, 3). Consequently, significant gaps remain in our understanding of molecular mechanisms regulating ectopic calcification in the aortic valve.
A large body of work has highlighted the importance of oxidative stress in various pathophysiological processes including tissue remodeling, fibrosis, and calcification. For example, increased oxidative stress contributes to vascular stiffening and remodeling (4, 5) in hypertensive mice, and increases in H2O2 levels augment osteogenic signaling and increase calcification in atherosclerotic blood vessels (6).
In tissue from humans with end-stage FCAVD, increases in oxidative stress and profound reductions in antioxidant enzyme activity in diseased regions of the valve suggested a strong association between increases in oxidative stress and worsening histopathological features of disease (7). While administration of non-selective antioxidants has shown preliminary therapeutic efficacy in preclinical models (8), a causal link between losses in antioxidant capacity in specific subcellular compartments and accelerated ectopic calcification in the valve, however, has been lacking.
Clinically, loss-of-function mutations in mitochondrial antioxidants (such as manganese superoxide dismutase, or MnSOD) are associated with increased risk of vascular calcification. While some reports suggest that experimentally reducing MnSOD worsens vasomotor function (9), atherosclerosis (10), and vascular stiffness (11) in blood vessels from hyperlipidemic mice, data from otherwise unstressed animals suggest that reductions in MnSOD are relatively well-tolerated in most blood vessels. Recently, a report from our laboratory suggested that in chronologically aged mice—with aging being the strongest risk factor for valve calcification in humans—suggested that MnSOD deficiency is not sufficient to induce robust valvular calcification or stenosis (12). It remains unknown, however, if an increase in mitochondrial-derived oxidative stress augments aortic valve calcification and worsens valvular function in mouse models known to develop functional stenosis.
Therefore, the overarching goal of this work was to test the hypothesis that MnSOD is a major regulator of molecular and histopathological drivers of valve disease in a robust mouse model of FCAVD. Our specific hypotheses were 2-fold: (1) Genetic inactivation of one copy of MnSOD will accelerate progression of aortic valve stenosis through augmented osteogenic signaling, increased valvular calcification, and increased tissue remodeling and fibrosis, and (2) genetic overexpression of MnSOD will attenuate osteogenic signaling and valve calcification, reduce tissue remodeling and fibrosis, and slow progression of aortic valve stenosis.
Materials and Methods
Animals
Low-density lipoprotein receptor-deficient (L), apolipoprotein B100/100 (A) mice (referred to as “LA” mice) were used to recapitulate key aspects of FCAVD observed in humans (13–15). In brief, LA mice were crossed with mice that harbored one inactive copy of manganese superoxide dismutase gene (MnSOD+/−). After generating founders that were homozygous for LA and heterozygous for MnSOD, LA-MnSOD+/+ × LA-MnSOD+/− mice intercrosses were used as a breeding strategy so littermate-matched mice could be compared whenever possible (notably MnSOD−/− mice are not viable for postnatal studies due to poor viability) (16). Mice with one copy of an MnSOD transgene (MnSODTg/0) were crossed to the LA-mouse model (17). Once generation of LA-homozygous mice harboring one copy of the MnSOD transgene was achieved, experimental animals were maintained using a LA-MnSOD0/0 × LA-MnSODTg/0 breeding strategy so littermate-matched mice could be compared whenever possible. This resulted in parallel colonies with a minimum of four back-crosses maintained on a mixed background (C57BL/6J and 129S4/SvJae mix), which matches the background strain used for initial characterization of this model (15, 18).
Starting at 2 months of age, experimental mice were fed a Western diet (TD.88137; Harlan Teklad) for 6 months. Mice were housed in a climate-controlled environment with 12-h light/dark cycles and had free access to water and feed. All mice were sacrificed following 6 months of western diet feeding (with all mice consequently being sacrificed at ~8 months of age, see Supplementary Table 1) using an overdose of sodium pentobarbital (≥100 mg/kg), and tissues collected are outlined below. While both sexes were studied in roughly equal proportions, we observed neither substantive nor significant differences in response to genetic alteration of MnSOD between sexes. Consequently, both sexes are reported in aggregated datasets throughout the manuscript. All animal experiments were approved by Mayo Clinic's Institutional Animal Care and Use Committee and comply with National Health Institute Office of Laboratory Animal Welfare's guidelines.
Echocardiography
Prior to sacrifice, all mice underwent high-resolution echocardiography. LA-MnSOD+/− mice were imaged using a GE Vivid 7 ultrasound system, 13-MHz linear probe. LA-MnSODTg/0 mice were imaged using a Vevo VisualSonics, 40-MHz linear probe to evaluate aortic valve and cardiac function via long- and short-axis views, as described previously (12, 19).
Gene Expression
All three aortic valve leaflets were carefully excised from the heart and placed in RNA lysis buffer (Invitrogen)/1% β-mercaptoethanol (Sigma). Isolation of RNA was performed using Invitrogen spin-columns. After conversion of RNA to cDNA (VILO reverse transcriptase, Invitrogen), quantitative real-time PCR was performed on a StepOnePlus RT-PCR machine. TaqMan Gene Expression Assay primers were used to assess mRNA levels for genes of interest. A full table of primers used is provided in the Supplementary Table 2. All genes were normalized by hypoxanthine phosphoribosyltransferase 1 (HPRT1), a commonly used housekeeper gene, and values are expressed via the ΔΔCT method.
Calcification and Fibrosis
The base of the heart was orientated, embedded in OCT, and stored at −80°C until use. Subsequently, samples were sectioned using a Leica CM3050S cryostat to garner 10-micron-thick frozen cross sections of the aortic valve and stored at −80°C until use.
Evaluation of changes in valvular calcification was done using Alizarin Red staining protocol. Adobe Photoshop was utilized to semiquantitatively assess changes in calcium levels of the aortic valve area using well-established methods from our laboratory and others (12, 18, 20). In brief, positive calcium stain (i.e., red punctate areas) was selected and pixel count of stain was determined in Adobe Photoshop. Subsequently, the aortic valve area was measured to normalize to positive calcium stain to report as a calcium ratio. Bright-field microscopy was used to image all sections at 4 × magnification (Olympus).
Evaluation of changes in valvular fibrosis was done using picrosirius red staining. In brief, sections were fixed using 4% paraformaldehyde, rinsed in PBS, and then incubated for 1 h in picrosirius red solution. Following, picrosirius red incubation samples were briefly rinsed in 0.5% acetic acid water, then dehydrated with increasing concentrations of ethanol, then cleared with xylenes, and finally cover slipped with a xylene-containing mounting medium. All sections were imaged using circularized polarizing light at 20 × magnification on a Zeiss ApoTome 2 Axio Imager. Fiji ImageJ (NIH) was utilized to perform analyses of total collagen and collagen thickness as previously described (21).
Immunohistochemistry
Frozen cross sections of aortic valve embedded in OCT were stained for immunofluorescence as previously described (22). Primary antibodies used were as follows: osterix (1:500, Abcam), phospho-SMAD1/5/8 (1:100, Cell Signaling), phospho-SMAD3 (1:100, Abcam), and 3-nitrotyrosine (1:100, Abcam). For all staining assays, negative controls were used to confirm validity and consistency of the staining pattern. All sections were stained with Alexa Fluor 647™ (produced in donkey and specific to the respective species secondary; all used at 1:500 dilution, Invitrogen) and mounted in Prolong Gold Anti-fade with DAPI (Invitrogen). Subsequently, a Zeiss LSM 780 confocal microscope set at 20 × magnification was used to focus and capture images of the base portions of the aortic valve leaflet. Following image acquisition, sections were analyzed using IMAGEJ software (NIH).
Statistical Analyses
All data are expressed in mean ± SEM. Unpaired T-tests were used to detect differences between groups at each time point. Where appropriate, corrections for multiple comparisons between groups were made using the Bonferroni method.
Results
Does Inactivating One Copy of MnSOD Accelerate Valvular and Cardiac Dysfunction?
Using echocardiographic measures of cusp separation distance or transvalvular peak velocity, we found that MnSOD haploinsufficiency did not worsen valvular function (Figures 1A,B). Furthermore, neither ejection fraction nor other measures of left ventricular systolic function differed between genotypes (Supplementary Figures 1A,B), nor did cardiac tissue wet weights differ between groups (Supplementary Figure 1C).
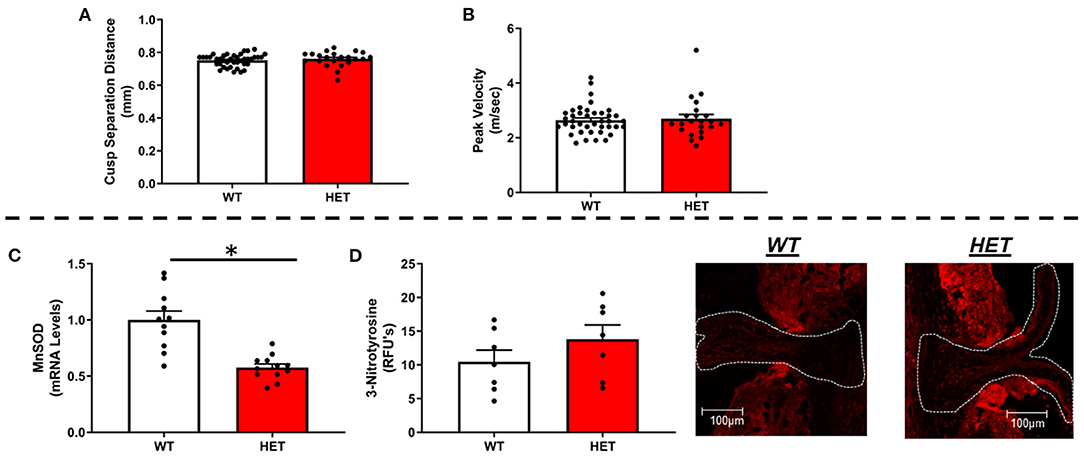
Figure 1. Effects of MnSOD deficiency on valvular function and valvular oxidative stress in hyperlipidemic mice. (A,B) Reductions in mitochondrial antioxidant capacity have no effect on cusp separation distance or transvalvular peak velocity compared to wild-type littermates (n = 40, 22). (C) As expected, MnSOD expression levels were reduced by ~50% in the aortic valve of LA-MnSOD+/− mice compared to their wild-type littermates (n = 11, 12). (D) Losses of mitochondrial antioxidant capacity tended in to increase 3-nitrotyrosine levels—a marker of oxidative stress—compared to wild-type littermates (n = 8, 8). For all panels, * denotes p < 0.05.
Does Inactivating One Copy of MnSOD Effectively Alter Redox State and Impact Expression of Other Anti- or Pro-oxidant Enzymes in FCAVD?
As expected, there was an observed ~50% reduction in gene expression of MnSOD in heterozygous mice compared to their wild-type littermates (Figure 1C), which was associated with a tendency toward increases in 3-nitrotyrosine levels in the valve cusp (Figure 1D). Consistent with previous reports using this model, expression of CuZnSOD—the other major intracellular SOD isoform—did not differ between genotypes (Supplementary Figure 2A). Extracellular SOD, however, was significantly but modestly reduced in LA-MnSOD+/− mice compared to their wild-type littermates (Supplementary Figure 2B). Changes in mRNA levels of other pro-oxidant markers known to play significant roles in cardiovascular tissues (e.g., NOX1, NOX2, and NOX4) were unremarkable and not significant (Supplementary Figures 2C–E).
Do Reductions in Mitochondrial Antioxidant Capacity Upregulate Pro-osteogenic Pathways and Increase Calcium Burden in FCAVD?
Interestingly, MnSOD haploinsufficiency did not alter mRNA levels of bone-morphogenetic protein-2 (BMP2) (Figure 2A) or BMP4 (Supplementary Figure 3A), although we did observe a tendency toward higher levels of p-smad1/5/8 protein levels in LA-MnSOD+/− mice compared to their LA-MnSOD+/+ littermates (Figure 2E). While expression of other direct BMP target genes was not significantly altered in LA-MnSOD+/− mice compared to their LA-MnSOD+/+ littermates (Figure 2B) and Supplementary Figure 3B), protein levels of osterix—a late-stage marker of osteoblastogenesis—were modestly increased in LA-MnSOD+/− mice (Supplementary Figure 3C). Interestingly, while we did observe noticeable and paradoxically unexpected reductions in mRNA levels of the osteogenic transcription factor Runx2 (Figure 2C) in LA-MnSOD+/− mice, we also observed substantial and significant reductions in mRNA levels of osteopontin (Figure 2D). Ultimately, these molecular changes resulted in significant increases of calcium burden in LA-MnSOD+/− mice compared to their LA-MnSOD+/+ littermates (assessed using Alizarin red staining in Figure 2F).
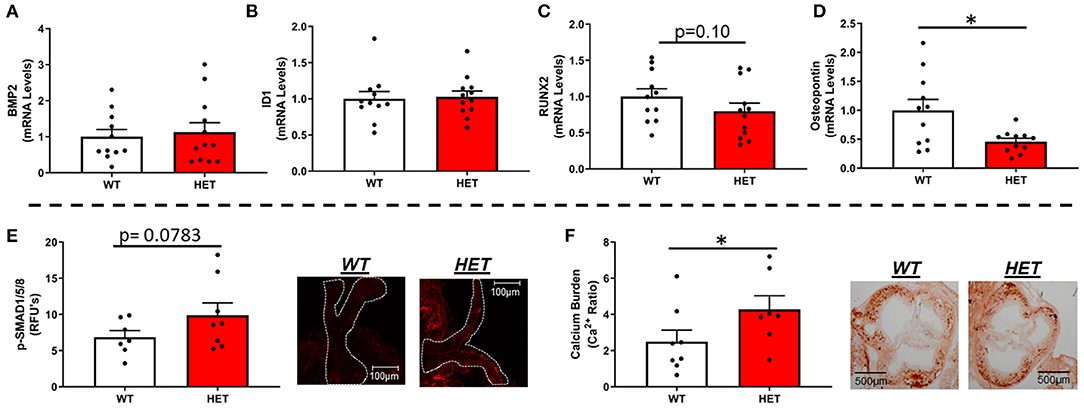
Figure 2. Effects of MnSOD deficiency on valvular osteogenic signaling and calcium burden in hyperlipidemic mice. (A) Bone morphogenetic protein-2 (BMP2) expression levels remained unchanged by reductions in mitochondrial antioxidant capacity (n = 11, 12). (B) Expression levels of ID1—a canonical BMP signaling target—did not change between LA-MnSOD+/+ and LA-MnSOD+/− mice (n = 11, 12). (C) Expression levels of RUNX2, a BMP-related transcription factor, was modestly reduced in LA-MnSOD+/− compared to their LA-MnSOD+/+ littermates (n = 11, 12). (D) Interestingly, osteopontin mRNA levels were significantly reduced in aortic valves from LA-MnSOD+/− mice compared to LA-MnSOD+/+ littermates (n = 11, 12). (E) Immunohistochemical assessment of p-SMAD1/5/8 suggested moderately increased canonical BMP signaling in LA-MnSOD+/− mice compared to LA-MnSOD+/+ mice (n = 8, 8). (F) Histological evaluations of calcium burden using Alizarin Red staining showed significant increases in valvular calcium burden in LA-MnSOD+/− compared to their wild-type littermates (higher magnification images are available in the Supplementary Figure 4) (n = 8, 8). For all panels, * denotes p < 0.05.
Do Reductions in Mitochondrial Antioxidant Capacity Increase Matrix Remodeling and Fibrogenic Processes in FCAVD?
In contrast to our hypothesis, mRNA levels of TGFβ-1—a major regulator of fibrogenesis and matrix remodeling—were unchanged by loss of one copy of MnSOD (Figure 3A). Furthermore, assessment of phospho-SMAD3 proteins levels via immunofluorescent staining showed no significant differences between the two genotypes (Figure 3B). Other genes related to fibrosis were largely unchanged as a result of losses in mitochondrial antioxidant capacity in LA-MnSOD+/− mice (Supplementary Figures 3D,E). Using picrosirius red staining to assess fibrotic burden and collagen fiber thickness, we found that total fibrotic burden tended to be reduced in LA-MnSOD+/− mice compared to wild-type littermates (Figure 3C, p = n.s.). There were no observed changes in collagen fiber thickness between genotypes (Figures 3D,E).
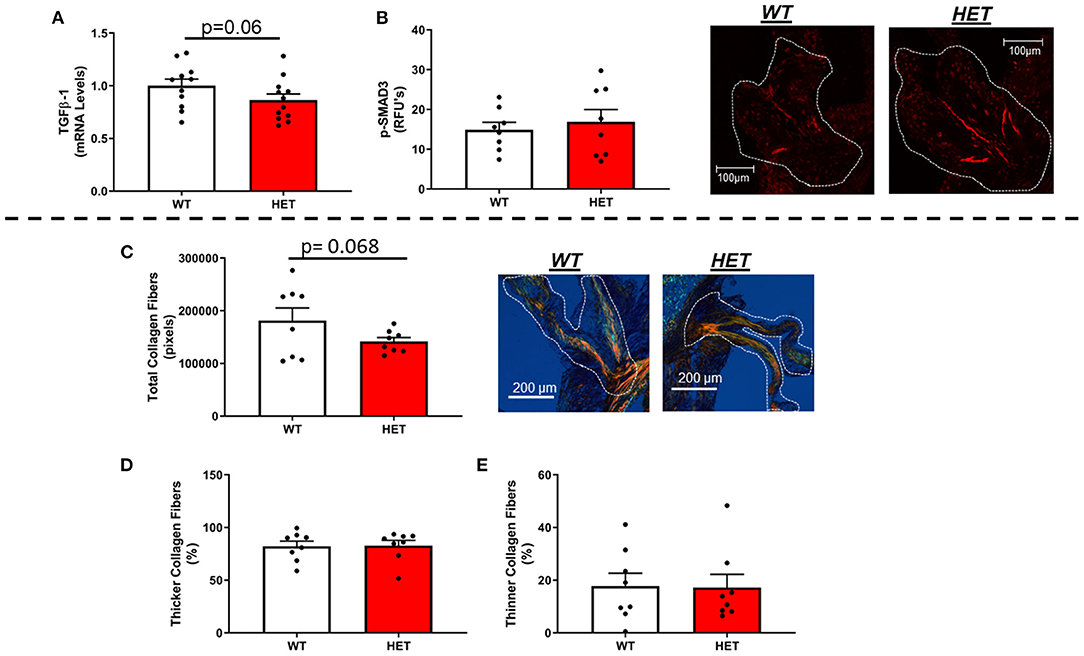
Figure 3. Effects of MnSOD deficiency on valvular fibrogenic signaling, tissue fibrosis, and collagen fiber thickness in hyperlipidemic mice. (A) TGFβ-1 expression levels tended to decrease in aortic valves from mice with MnSOD haploinsufficiency compared to their wild-type counterparts (n = 11, 12). (B) Immunofluorescence of phospho-SMAD3 protein was unchanged with reductions in MnSOD (n = 8, 8). (C–E) Picrosirius red staining was used to evaluate changes in total collagen burden. (C). Losses of MnSOD in LA-MnSOD+/− mice tended to reduce total collagen fibers compared to their LA-MnSOD+/+ littermates. (D,E) Genetic reductions in MnSOD did not alter collagen fiber composition in LA-MnSOD+/− mice compared to their LA-MnSOD+/+ littermates (n = 8, 8).
Interim Summary of Effects of Reducing MnSOD on FCAVD
Collectively, genetic inactivation of MnSOD had no effect on valve function, ventricular function, or fibrogenic signaling in this model. Interestingly, despite accelerating accrual of valvular calcium, we observed modest and paradoxically reduced expression of the osteogenic markers Runx2 and Osteopontin in valves from mice with MnSOD haploinsufficiency.
Does Increasing Mitochondrial Antioxidant Capacity Protect Against Valvular and Cardiac Dysfunction in This Mouse Model of FCAVS?
In contrast to our working hypothesis, transgenic overexpression of MnSOD did not prevent reductions in cusp separation distance, nor did it have an effect on transvalvular peak velocity in LA-MnSODTg/0 mice compared to their LA-MnSOD0/0 littermates (Figures 4A,B). Secondly, increasing mitochondrial antioxidant capacity did not have an overt impact on ejection fraction or left ventricular systolic function (Supplementary Figures 5A,B), nor did cardiac tissue wet weights differ between groups (Supplementary Figure 5C).

Figure 4. Effects of MnSOD overexpression on valvular function and valvular oxidative stress in hyperlipidemic mice. (A,B) Increasing mitochondrial antioxidant capacity had no impact on cusp separation distance and peak velocity compared to LA-MnSOD0/0 mice (n = 17, 15). (C) mRNA levels for MnSOD were increased by three-fold in LA-MnSODTg/0 mice compared to their wild-type littermates (n = 12, 10). (D) Interestingly, increasing mitochondrial antioxidant capacity did not reduce 3-nitrotyrosine levels in valves from LA-MnSODTg/0 mice compared to their LA-MnSOD0/0 littermates (n = 10, 10). For all panels, * denotes p < 0.05.
Does Genetically Increasing MnSOD Alter Redox State and Impact Expression of Other Anti- or Pro-oxidant Enzymes in FCAVD?
As expected, MnSOD expression levels were increased approximately three-fold compared to their wild-type littermates (Figure 4C). Surprisingly, levels of 3-nitrotyrosine—indicator of oxidative stress—did not show reductions in valve tissue from mice that carried the MnSOD transgene (Figure 4D). Consistent with prior reports, genetically increasing MnSOD did not result in altered expression of other SOD isoforms in the aortic valve (Supplementary Figures 6A,B), nor did it influence mRNA levels of other pro-oxidant enzymes in aortic valve tissue (Supplementary Figure 6C).
Does Overexpression of MnSOD Reduce Pro-osteogenic Signaling and Slow Calcium Burden in Hyperlipidemic Mice?
Consistent with our data in LA-MnSOD+/− mice, increasing MnSOD expression in LA-MnSODTg/0 mice did not alter mRNA levels of BMP2 or BMP4 (Figure 5A and Supplementary Figure 7A) and only tended to reduce p-SMAD1/5/8 levels in aortic valve tissue (Figure 5E). While expressions of some biomarkers of osteoblastogenesis (such as MSX2) were unchanged (Supplementary Figure 7B), we were surprised to observe significant increases in mRNA levels of the direct p-SMAD1/5/8 target ID1 (Figure 5B) as well as mRNA levels of the osteogenic transcription factor RUNX2 in LA-MnSODTg/0 compared to their LA-MnSOD0/0 littermates (Figure 5C). Interestingly, we again observed a profound effect of MnSOD on mRNA levels of osteopontin, which were now increased in LA-MnSODTg/0 mice (Figure 5D). Collectively, these apparently opposing molecular changes resulted in no net impact on calcium burden in LA-MnSODTg/0 mice when compared to their LA-MnSOD0/0 littermates (Figure 5F).
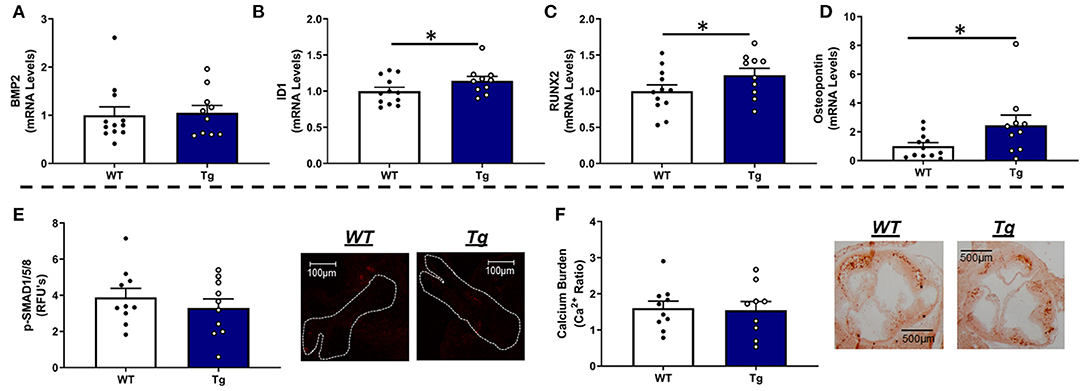
Figure 5. Effects of MnSOD overexpression on osteogenic signaling and calcium burden in valve tissue from hyperlipidemic mice. (A) Increasing mitochondrial antioxidant capacity levels did not alter BMP2 expression levels in the aortic valve compared to wild-type littermates (n = 12, 10). (B) Expression levels of ID1 were significantly increased in LA-MnSODTg/0 compared to LA-MnSOD0/0 littermates (n = 12, 10). (C) Expression levels of RUNX2 were significantly increased in LA-MnSODTg/0 compared to the LA-MnSOD0/0 littermates (n = 12, 10) (D) Osteopontin expression levels were significantly increased in LA-MnSODTg/0 mice compared to LA-MnSOD0/0 littermates (n = 12, 10). (E) Increasing MnSOD tended to reduce protein levels of phospho-SMAD1/5/8 in the aortic valve but failed to reach significance (n = 10, 10). (F) Interestingly, calcium deposition in the aortic valve was unchanged between genotypes despite dramatic increases in MnSOD expression in LA-MnSODTg/0 mice (higher-magnification images are available in the Supplementary Figure 8) (n = 10, 10). For all panels, * denotes p < 0.05.
Does Increasing Mitochondrial Antioxidant Capacity Increase Matrix Remodeling and Fibrogenic Processes in FCAVD?
In LA mice carrying the MnSOD transgene, mRNA levels of TGFβ-1 were modestly increased (Figure 6A) and associated with parallel changes in COL1A1 mRNA levels (Supplementary Figure 7C). Immunofluorescence-based evaluation of phospho-SMAD3 protein levels in the aortic valve showed no significant differences between LA-MnSODTg/0 and LA-MnSOD0/0 mice (Figure 6B). Histological assessment of tissue fibrosis using picrosirius red staining showed that increasing mitochondrial antioxidant capacity slightly augmented total collagen burden in the aortic valve tissue from LA-MnSODTg/0 mice compared to LA-MnSOD0/0 (Figure 6C). Furthermore, composition analyses of the collagen fibers showed modest reductions in the proportion of thinner collagen fibers in mice overexpressing MnSOD, while thicker collagen fibers were slightly higher in LA-MnSODTg/0 mice compared to their LA-MnSOD0/0 littermates (Figures 6D,E).
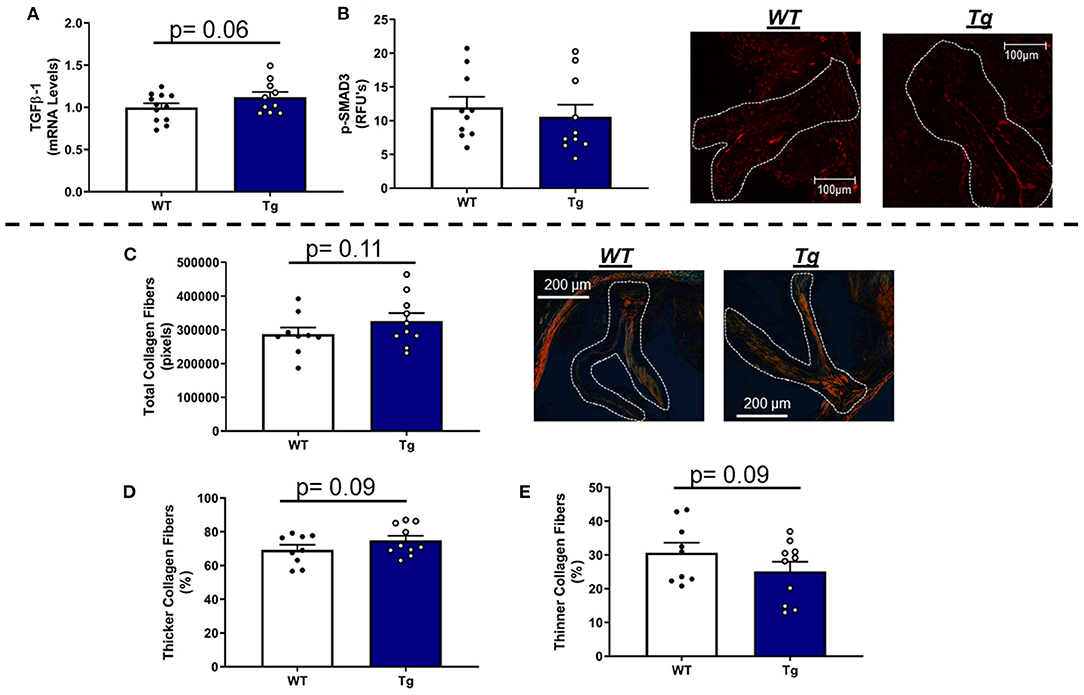
Figure 6. Effects of MnSOD overexpression on fibrogenic signaling and collagen deposition in aortic valves from hyperlipidemic mice. (A) Overexpression of MnSOD modestly increased TGFβ-1 expression levels in the aortic valve compared to wild-type mice (n = 12, 10). (B) Immunofluorescence of phospho-SMAD3 protein was unchanged between LA-MnSOD0/0 and LA-MnSODTg/0 mice (n = 10, 10). (C) Picrosirius red staining to evaluate changes in collagen burden resulted in modest increases in LA-MnSODTg/0 mice compared to wild-type mice (n = 10, 10). (D,E) The increase in total collagen fibers in LA-MnSODTg/0 mice was associated with a tendency toward greater collagen fiber thickness compared to their LA-MnSODTg/0 littermates (n = 10, 10).
Interim Summary of Effects of Overexpressing MnSOD on FCAVD
Collectively, genetic overexpression of MnSOD was largely ineffective at reducing biomarkers of oxidative stress in this model and had no beneficial effect on valve function or ventricular function following 6 months of Western diet feeding. While overexpression of MnSOD failed to convey therapeutic benefit with regard to slowing accrual of valvular calcium, we again observed paradoxically increases in expression of the osteogenic markers Runx2 and Osteopontin in valves from mice overexpressing MnSOD and did not observe notable or significant changes in molecular or structural components of extracellular matrix integrity or fibrosis.
Discussion
The results from this study provide four major novel findings: (1) genetically decreasing or increasing MnSOD did not alter valvular and cardiac function in hyperlipidemic mice; (2) altering MnSOD levels appears to modulate several key molecular changes implicated in progression of FCAVD, although these changes appear to often occur in a conflicting, non-orchestrated, and context-dependent manner; (3) the effects of MnSOD on valvular calcification appear to be largely unidirectional, with reductions in MnSOD accelerating valvular calcification but overexpression of MnSOD failing to exert protection against valvular calcification; and (4) ultimately, therapeutic approaches to altering MnSOD levels as a monotherapy remain elusive for the treatment of FCAVD.
Lifelong Alterations in Mitochondrial Antioxidant Capacity Do Not Affect Aortic Valve Function in Mild-To-Moderate FCAVD
A major finding of this study was that increasing or decreasing expression of MnSOD—over a roughly four-fold range (from deficiency to overexpression)—did not have a detectable impact on aortic valve function this model of FCAVD. This observation is largely consistent with previous reports from our laboratory, suggesting that reductions of MnSOD in aging mice are not sufficient to induce overt valvular stenosis (12), and chronological aging without incremental pathophysiological insults is unlikely to provide a sufficient biological context (e.g., activation of osteogenic signaling) for MnSOD haploinsufficiency to trigger overt calcific valvular disease (12, 23). Importantly, most other models (including hyperlipidemic murine (24), lagomorph (8), or porcine models (25)) do not develop hemodynamically significant aortic valve dysfunction, which consequently precludes the assessment of true therapeutic benefit of antioxidant interventions. As discussed in subsequent sections, however, there is a growing body of literature suggesting that targeting other mechanisms influencing redox state, other specific reactive oxygen species, or developing more “holistic” approaches to improving mitochondrial function and/or redox state may be more effective in modulating pathophysiological cardiovascular tissue calcification and fibrosis and may consequently be more viable to pursue.
Changes in Mitochondrial Antioxidant Capacity Fail to Orchestrate Changes in Osteogenic Signaling That Are Consistent With Therapeutic Viability
In the current study, we found that p-SMAD1/5/8 levels were coarsely inversely associated with mRNA levels of MnSOD. Our initial focus on BMP signaling stemmed from the seminal reports from Demer and others showing that this signaling cascade is commonly upregulated in cardiovascular lesions and sufficient to result in overt tissue calcification (26). While previous work suggests that BMP signaling can be significantly influenced by reactive oxygen species (and Nox4 activity in particular) (27, 28), our data suggest that the interplay between MnSOD and canonical BMP signaling is unlikely to play a major role in driving key features of FCAVD.
After observing small effect sizes on BMP signaling, we quickly shifted our focus to other major regulators of ectopic osteogenesis such as Runx2, Msx2, and other mechanisms which Chen, Towler, and others have clearly demonstrated is modulated by hydrogen peroxide levels in vascular smooth muscle (6, 29). Here, we observed a unique finding where expression of Runx2 is inversely associated with mRNA levels of MnSOD, and Msx2 is largely unaffected by alterations in mitochondrial redox state. While we were initially surprised by this finding—particularly given the previous work of Liberman demonstrating that systemic administration of non-specific antioxidants reduces osteogenic signaling in valve tissue (8)—the changes we observed in Runx2 are entirely consistent with previous reports in orthotopic calcification where increasing MnSOD levels induce Runx2 expression and activity and promote bone ossification (30). Also, the observations from Lai and colleagues stemmed from interventions in cells derived from the aorta (29), where NADPH oxidases are thought to be a larger contributor to increased oxidative stress compared to the diseased valve (7). Collectively, from the perspective of therapeutic utility, one could arrive at the conclusion that MnSOD may function as a “downstream” regulator of a subset of osteogenic mechanisms in valve tissue that is conserved with bone, and would consequently likely to be of limited therapeutic utility.
To determine whether there were other molecular mechanisms that influenced cell and phenotypic fate in FCAVD, we measured the expression of osteopontin and found that it was significantly downregulated in valves from LA-MnSOD+/− mice and significantly increased in valves from LA-MnSODTg/0 mice. Numerous reports suggest that osteopontin is near-ubiquitously associated with cardiovascular calcification in a variety of models, and the groundbreaking work by Giachelli et al. (31–33) strongly suggests that upregulation of osteopontin is an adaptive and protective response. Importantly, while this may serve to protect against calcific mass expansion in the aortic valve, numerous reports also suggest that upregulation of osteopontin can independently contribute to mitochondrial dysfunction, tissue fibrosis, and cellular apoptosis (34, 35). Ultimately, while it seems biologically plausible that reductions in osteopontin are a primary, permissive event enabling accelerated valvular calcification in MnSOD-deficient mice (or reciprocally, a primary protective mechanism in MnSOD-transgenic mice), such a complicated molecular interplay warrants further investigation.
Altering MnSOD Expression Has Little Effect on Fibrogenic Signaling or Fibrosis in FCAVD
Emerging data from our group and others strongly suggest that tissue fibrosis may play a significant role in the pathogenesis of FCAVD in both humans and experimental animal models of valve disease. Clinically, women have been shown to have less valvular calcification compared to men at any given severity of stenosis (even after normalizing for body size), suggesting that other factors such as fibrosis may disproportionately contribute to valvular dysfunction in some patient populations (36). Experimentally, Weiss and colleagues have shown that valvular fibrosis can be a primary driver of valvular dysfunction in hyperlipidemic mice with hyperactivation of the renin–angiotensin system (37).
However, regardless of the relative contributions of cusp fibrosis to valvular dysfunction, our data generally suggest that gain or loss of function of MnSOD does not have substantive effects on overall fibrotic burden or collagen fiber thickness in valve tissue from mice in this model system. Importantly—and as noted above—this lack of an effect was observed despite changes in expression of osteopontin and the well-documented impact of osteopontin on fibrosis and matrix remodeling in other tissues (38, 39). Our findings differ from several investigations of the role of MnSOD in the regulation of matrix deposition and remodeling in other tissues (e.g., heart or vasculature), where activity of MnSOD appears to inversely associate with cardiac tissue fibrosis (40) or indices of fibrous cap stability in atherosclerotic plaques (41). Thus, further investigation is warranted into the potential differential unique and context-dependent roles osteopontin may play in FCAVD.
Limitations
One notable limitation of this study is that overexpression of MnSOD did fail to significantly reduce cellular markers of oxidative stress in LA-MnSODTg/0 mice compared to their LA-MnSOD0/0 littermates. While there is a potential for technical reasons for this observation [e.g., 3-NT staining is not specific to the mitochondria (42)], there are ample data demonstrating that MnSOD can undergo a number of posttranslational modifications in disease states [e.g., deacetylation by sirtuin 3 is essential for optimal MnSOD activity (43)]. Furthermore, it is possible that MnSOD expression is already dramatically suppressed in diseased valve tissue (7), which would serve to minimize the observed biological effect size of reducing or increasing MnSOD expression by moderate genetic alterations. Consequently, future investigation into mechanisms regulating “optimal” mitochondrial enzymatic function—both from a reductionist perspective and from a holistic organelle perspective—will be essential for any successful therapeutic intervention targeting mitochondria.
Summary and Translational Perspectives
Collectively, our data support the intriguing hypothesis that, in FCAVD, mitochondrial antioxidant capacity is endogenously “titrated” to an optimal level to minimize progression of valvular calcification. Data in support of this hypothesis show that reductions of MnSOD accelerates valvular calcification but that overexpression of MnSOD fails to reduce valvular calcification and results in paradoxical increases in key regulators of osteoblastogenesis such as Runx2. While this latter finding echoes the challenges faced by numerous previous investigators aiming to leverage antioxidant therapies to treat cardiovascular diseases (44, 45), it is possible that specific subtypes of patients [e.g., those with loss-of-function mutations in MnSOD (46)] may benefit significantly from targeted antioxidant therapy. Ultimately, we believe a more nuanced and multipronged approach accounting for the highly context-dependent effects of mitochondrial antioxidant capacity will be essential to the development of novel therapeutics aimed at slowing progression of FCAVD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Mayo Clinic Institutional Animal Care and Use Committee.
Author Contributions
CR: conceptualization, methodology, data curation (histology and immunohistochemistry), interpretation, writing—original drafts, and writing—reviewing and editing. BZ: data curation (histology and immunohistochemistry) and writing—reviewing and editing. MH: data curation (qPCR). GV, EO, and RH: data curation (echocardiography). AA: data curation (histology). JM: conceptualization, methodology, funding, interpretation, writing—original drafts, writing—reviewing and editing, and supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Heart, Lung, and Blood Institute (R00-HL092235, R01-HL111121), the Robert and Arlene Kogod Center on Aging at Mayo Clinic, and the Center for Regenerative Medicine at Mayo Clinic.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to express sincere gratitude to Dr. Arlan Richardson (University of Oklahoma Health Sciences Center) for providing the MnSODTg/0 mice, which were used to generate the LA-MnSODTg/0 mice for this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.694881/full#supplementary-material
References
1. Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular health study. J Am Coll Cardiol. (1997) 29:630–4. doi: 10.1016/s0735-1097(96)00563-3
2. Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. (2005) 352:2389–97. doi: 10.1056/NEJMoa043876
3. Hermans H, Herijgers P, Holvoet P, Verbeken E, Meuris B, Flameng W, et al. Statins for calcific aortic valve stenosis: into oblivion after saltire and seas? an extensive review from bench to bedside. Curr Probl Cardiol. (2010) 35:284–306. doi: 10.1016/j.cpcardiol.2010.02.002
4. Forstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
5. Singh U, Jialal I. Oxidative stress and atherosclerosis. Pathophysiology. (2006) 13:129–42. doi: 10.1016/j.pathophys.2006.05.002
6. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. (2008) 283:15319–27. doi: 10.1074/jbc.M800021200
7. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. (2008) 52:843–50. doi: 10.1016/j.jacc.2008.05.043
8. Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J Jr, Pomerantzeff PM, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. (2008) 28:463–70. doi: 10.1161/ATVBAHA.107.156745
9. Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. (2007) 27:1941–6. doi: 10.1161/ATVBAHA.107.146852
10. Ohashi M, Runge MS, Faraci FM, Heistad DD. MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. (2006) 26:2331–6. doi: 10.1161/01.ATV.0000238347.77590.c9
11. Zhou RH, Vendrov AE, Tchivilev I, Niu XL, Molnar KC, Rojas M, et al. Mitochondrial oxidative stress in aortic stiffening with age: the role of smooth muscle cell function. Arterioscler Thromb Vasc Biol. (2012) 32:745–55. doi: 10.1161/ATVBAHA.111.243121
12. Roos CM, Hagler M, Zhang B, Oehler EA, Arghami A, Miller JD. Transcriptional and phenotypic changes in aorta and aortic valve with aging and MnSOD deficiency in mice. Am J Physiol Heart Circ Physiol. (2013) 305:H1428–39. doi: 10.1152/ajpheart.00735.2012
13. Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res. (2011) 108:1392–412. doi: 10.1161/CIRCRESAHA.110.234138
14. Weiss RM, Miller JD, Heistad DD. Fibrocalcific aortic valve disease: opportunity to understand disease mechanisms using mouse models. Circ Res. (2013) 113:209–22. doi: 10.1161/CIRCRESAHA.113.300153
15. Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. (2006) 114:2065–9. doi: 10.1161/CIRCULATIONAHA.106.634139
16. Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. (1996) 93:9782–7. doi: 10.1073/pnas.93.18.9782
17. Jang YC, Perez VI, Song W, Lustgarten MS, Salmon AB, Mele J, et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J Gerontol A Biol Sci Med Sci. (2009) 64:1114–25. doi: 10.1093/gerona/glp100
18. Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K, et al. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. (2010) 30:2482–6. doi: 10.1161/ATVBAHA.110.211029
19. Casaclang-Verzosa G, Enriquez-Sarano M, Villaraga HR, Miller JD. Echocardiographic approaches and protocols for comprehensive phenotypic characterization of valvular heart disease in mice. J Vis Exp. (2017) 2017:54110. doi: 10.3791/54110
20. Mosch J, Gleissner CA, Body S, Aikawa E. Histopathological assessment of calcification and inflammation of calcific aortic valves from patients with and without diabetes mellitus. Histol Histopathol. (2017) 32:293–306. doi: 10.14670/HH-11-797
21. Rich L, Whittaker P. Collagen and picrosirius red staining: a polarized light assessment of fibrillar hue and spatial distribution. J morphol sci. (2017) 22:97–104.
22. Thalji NM, Hagler MA, Zhang H, Casaclang-Verzosa G, Nair AA, Suri RM, et al. Nonbiased molecular screening identifies novel molecular regulators of fibrogenic and proliferative signaling in myxomatous mitral valve disease. Circ Cardiovasc Genet. (2015) 8:516–28. doi: 10.1161/CIRCGENETICS.114.000921
23. Hinton RB Jr, Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, et al. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart Circ Physiol. (2008) 294:H2480–8. doi: 10.1152/ajpheart.91431.2007
24. Tanaka K, Sata M, Fukuda D, Suematsu Y, Motomura N, Takamoto S, et al. Age-associated aortic stenosis in apolipoprotein E-deficient mice. J Am Coll Cardiol. (2005) 46:134–41. doi: 10.1016/j.jacc.2005.03.058
25. Porras AM, Shanmuganayagam D, Meudt JJ, Krueger CG, Hacker TA, Rahko PS, et al. Development of aortic valve disease in familial hypercholesterolemic swine: implications for elucidating disease etiology. J Am Heart Assoc. (2015) 4:e002254. doi: 10.1161/JAHA.115.002254
26. Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. (1993) 91:1800–9. doi: 10.1172/JCI116391
27. Mandal CC, Ganapathy S, Gorin Y, Mahadev K, Block K, Abboud HE, et al. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem J. (2011) 433:393–402. doi: 10.1042/BJ20100357
28. Simone S, Cosola C, Loverre A, Cariello M, Sallustio F, Rascio F, et al. BMP-2 induces a profibrotic phenotype in adult renal progenitor cells through Nox4 activation. Am J Physiol Renal Physiol. (2012) 303:F23–34. doi: 10.1152/ajprenal.00328.2011
29. Lai CF, Shao JS, Behrmann A, Krchma K, Cheng SL, Towler DA. TNFR1-activated reactive oxidative species signals up-regulate osteogenic Msx2 programs in aortic myofibroblasts. Endocrinology. (2012) 153:3897–910. doi: 10.1210/en.2012-1216
30. Gao J, Feng Z, Wang X, Zeng M, Liu J, Han S, et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. (2018) 25:229–40. doi: 10.1038/cdd.2017.144
31. Speer MY, McKee MD, Guldberg RE, Liaw L, Yang HY, Tung E, et al. Inactivation of the osteopontin gene enhances vascular calcification of matrix gla protein-deficient mice: evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J Exp Med. (2002) 196:1047–55. doi: 10.1084/jem.20020911
32. Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H, et al. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol. (2002) 161:2035–46. doi: 10.1016/S0002-9440(10)64482-3
33. Worcester EM, Blumenthal SS, Beshensky AM, Lewand DL. The calcium oxalate crystal growth inhibitor protein produced by mouse kidney cortical cells in culture is osteopontin. J Bone Miner Res. (1992) 7:1029–36. doi: 10.1002/jbmr.5650070905
34. Yousefi K, Irion CI, Takeuchi LM, Ding W, Lambert G, Eisenberg T, et al. Osteopontin promotes left ventricular diastolic dysfunction through a mitochondrial pathway. J Am Coll Cardiol. (2019) 73:2705–18. doi: 10.1016/j.jacc.2019.02.074
35. Dalal S, Zha Q, Singh M, Singh K. Osteopontin-stimulated apoptosis in cardiac myocytes involves oxidative stress and mitochondrial death pathway: role of a pro-apoptotic protein BIK. Mol Cell Biochem. (2016) 418:1–11. doi: 10.1007/s11010-016-2725-y
36. Sritharen Y, Enriquez-Sarano M, Schaff HV, Casaclang-Verzosa G, Miller JD. Pathophysiology of aortic valve stenosis: is it both fibrocalcific and sex specific? Physiology. (2017) 32:182–96. doi: 10.1152/physiol.00025.2016
37. Chu Y, Lund DD, Doshi H, Keen HL, Knudtson KL, Funk ND, et al. Fibrotic aortic valve stenosis in hypercholesterolemic/hypertensive mice. Arterioscler Thromb Vasc Biol. (2016) 36:466–74. doi: 10.1161/ATVBAHA.115.306912
38. Matsui Y, Jia N, Okamoto H, Kon S, Onozuka H, Akino M, et al. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension. (2004) 43:1195–201. doi: 10.1161/01.HYP.0000128621.68160.dd
39. Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. (2005) 2:e251. doi: 10.1371/journal.pmed.0020251
40. Kwak HB, Lee Y, Kim JH, Van Remmen H, Richardson AG, Lawler JM. MnSOD overexpression reduces fibrosis and pro-apoptotic signaling in the aging mouse heart. J Gerontol A Biol Sci Med Sci. (2015) 70:533–44. doi: 10.1093/gerona/glu090
41. Vendrov AE, Stevenson MD, Alahari S, Pan H, Wickline SA, Madamanchi NR, et al. Attenuated superoxide dismutase 2 activity induces atherosclerotic plaque instability during aging in hyperlipidemic mice. J Am Heart Assoc. (2017) 6:6775. doi: 10.1161/JAHA.117.006775
42. Herce-Pagliai C, Kotecha S, Shuker DE. Analytical methods for 3-nitrotyrosine as a marker of exposure to reactive nitrogen species: a review. Nitric Oxide. (1998) 2:324–36. doi: 10.1006/niox.1998.0192
43. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. (2010) 40:893–904. doi: 10.1016/j.molcel.2010.12.013
44. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. (2017) 19:42. doi: 10.1007/s11883-017-0678-6
45. Thomson MJ, Puntmann V, Kaski JC. Atherosclerosis and oxidant stress: the end of the road for antioxidant vitamin treatment? Cardiovasc Drugs Ther. (2007) 21:195–210. doi: 10.1007/s10557-007-6027-1
Keywords: MnSOD, mitochondria, fibrocalcific aortic valve disease, calcification, oxidative stress
Citation: Roos CM, Zhang B, Hagler MA, Verzosa GC, Huang R, Oehler EA, Arghami A and Miller JD (2021) Effects of Altering Mitochondrial Antioxidant Capacity on Molecular and Phenotypic Drivers of Fibrocalcific Aortic Valve Stenosis. Front. Cardiovasc. Med. 8:694881. doi: 10.3389/fcvm.2021.694881
Received: 13 April 2021; Accepted: 17 May 2021;
Published: 24 June 2021.
Edited by:
Cynthia St. Hilaire, University of Pittsburgh, United StatesReviewed by:
Joshua D. Hutcheson, Florida International University, United StatesJoy Lincoln, Medical College of Wisconsin, United States
Copyright © 2021 Roos, Zhang, Hagler, Verzosa, Huang, Oehler, Arghami and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordan D. Miller, bWlsbGVyLmpvcmRhbkBtYXlvLmVkdQ==