 Rafael M. Ronsoni1,2*†
Rafael M. Ronsoni1,2*† Marco Aurélio Lumertz Saffi3†
Marco Aurélio Lumertz Saffi3† Marcus Vinicius Magno Gonçalves2†Igor Hidetsu Nakayama2†Tiago Luiz Luz Leiria4†
Marcus Vinicius Magno Gonçalves2†Igor Hidetsu Nakayama2†Tiago Luiz Luz Leiria4†- 1Electrophysiology Department, Instituto de Ritmologia Cardíaca, Joinville, Brazil
- 2Department of Medicine, Universidade da Região de Joinville, Joinville, Brazil
- 3Department of Cardiology, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
- 4Programa de Pós-Graduação em Ciências da Saúde - Instituto de Cardiologia do Rio Grande do Sul/Fundação Universitária de Cardiologia, Porto Alegre, Brazil
Introduction: Current evidence questions the linear sequence traditionally described in atrial fibrillation, blood stasis, intracavitary thrombus, and embolization to the central nervous system. Currently, new perspectives have been described based on questions from the linearly traditional chronology of events; it is within this scope that the article has its objective.
Evidences: The association of the two entities is biologically plausible and supported by different cohorts with a higher risk of developing atrial fibrillation, especially in the cardioembolic form. Concepts (temporal dissociation, biological gradient, etc.) determine the existence of other factors associated with cardioembolism, not exclusively by atrial fibrillation. The entire cascade of events associated with myopathy and atrial remodeling can generate damage to the myocyte and amplify the prothrombotic status. It is important to clarify that atrial myopathy can present itself as atrial fibrillation initially or not, but should always be considered thrombogenic in all the contexts of their clinical presentation. Considering atrial heart disease as a cause of embolic stroke, it could explain that one-third of strokes are considered cryptogenic.
Conclusions: The traditional model exclusively associating the presence of atrial fibrillation in the genesis of thromboembolism is incomplete. The concept of atrial cardiopathy where cardioembolism occurs in a non-atrial fibrillation dependent manner fits better with current data. The future challenge is to effectively detect the various manifestations of atrial heart disease, generating direct implications for the identification of patients at risk of stroke and also for better management after a cardioembolic event.
Introduction
Atrial fibrillation (AF) is considered the most common sustained arrhythmia in clinical practice and with complex pathophysiology, occurring in normal hearts even in the presence of the most varied structural diseases (1–3). Initial results of studies from the Framingham cohort (4) determined the relationship with morbidity and mortality, especially its relationship with stroke (5, 6). Its prevalence has reached epidemic proportions, resulting in a perspective that one in four American or European adults will have AF diagnosed in the near future, without considering a rate between 10 and 25% of asymptomatic cases (2, 3, 7, 8).
Current evidence questions the linear sequence traditionally described in AF, blood stasis, intracavitary thrombus, and embolization to the central nervous system. Currently, new perspectives have been described based on questions from the linearly traditional chronology of events; it is within this scope that the article has its objective (9).
Evidence Between AF and Stroke
Positive Points in the Relationship
The association of the two entities is biologically plausible and supported by different cohorts with a higher risk of developing AF (three to five times), especially in the cardioembolic form (10–12). The association of arrhythmia with the most diverse cardiovascular risk factors associated with stroke is true (age, male gender, diabetes, high blood pressure, heart failure, among others), but despite the possible confounding effect, the association remains independently associated after statistical control of the factors risk (10, 11). And what further strengthens the relationship is that AF remains associated with clinical and neuroimaging stroke patterns related to cardioembolism (13).
Interrogations in the Relationship
However, some doubts are raised, one of which is the association of AF with non-cardioembolic stroke; for example 10% of patients with lacunar stroke have AF and the neurological event associated with the atherosclerosis of large vessels is twice as common in patients with AF. Therefore, the risk of stroke in the context of AF cannot be fully explained by the direct association of AF and stroke, through the cardioembolism pathway, that is, it can occur by other mechanisms (12).
If AF is directly responsible for cardioembolism, it is difficult to understand why elderly patients with cardiovascular risk factors with subclinical AF are associated with twice the risk of stroke as opposed to younger patients with isolated AF who do not have a significant risk of stroke in clinical follow-up. Therefore, these data question the clear biological gradient between stroke and AF; this suggests the occurrence of other associated mechanisms in the elderly population (14, 15).
Another important concept is the temporal dissociation between arrhythmic events and AF. Up to one-third of patients with paroxysmal AF do not show arrhythmic episodes prior to neurological ictus and some will only present an arrhythmic event long after the stroke (16–18). This concept determines the existence of other factors associated with cardioembolism, not exclusively by AF. Another fact that contributes to this question is that the strategies of rhythm control adopted in the treatment of AF were not able to reduce stroke in the clinical follow-up of these patients, despite the apparent success. This has even modified the main trials of rhythm control for AF that have come to value outcomes such as mortality and reduced heart failure, which apparently currently have a stronger relationship with the maintenance of cardiac rhythm (19).
A possible confounding effect is that strokes that affect autonomic centers play a role in triggering AF after neurological stroke. This situation may be a confusing effect in the etiological investigation of stroke, and AF may be unduly responsible for the genesis of the neurological event (20, 21).
Cardioembolic Stroke
Epidemiology of Cardio-Embolic Stroke
Cardioembolic stroke is responsible for about 15–20% of all ischemic events. In developed countries the trend is toward a higher prevalence in relation mainly to atherothrombosis, reaching up to 34.7% (22–25). This is closely linked to better control of atherosclerosis risk factors, such as hypertension and dyslipidemia, generating greater relevance in countries such as Canada. Cardioembolic stroke has tripled its incidence over the past few decades and has a prospect of a further 3× increase by the 2050s in developed countries (26).
Atrial fibrillation is a well-known risk factor for ischemic stroke, causing a five-fold increase in stroke risk, reporting a prevalence of AF in patients with ischemic stroke of about 25–30%. This prevalence is generally underestimated since up to 3 months of follow-up there is an addition of around 20% of new AF diagnoses in previously sinus patients (24).
Atrial fibrillation was most frequently associated with infarcts of the total anterior circulation, occurrence of multiple and bilateral vascular territories accounting for a worse outcome in terms of mortality at 30 days and 1 year, and rate of stroke recurrences in the first year of follow-up (24, 27).
Furthermore, we cannot overlook the possible role of AF in strokes classified as cryptogenic, whose prevalence reaches around one-third of ischemic events. This is explained by the presence of AF in a paroxysmal form that can go undiagnosed during the etiological investigation of the event. Another important fact is that prolonged cardiac monitoring (≥30 days) adds statistically to the diagnoses of subclinical AF in stroke patients initially classified as cryptogenic (28, 29).
Risk Factors for Cardioembolic Stroke
Atrial Fibrillation
The mechanisms associated with AF and stroke have already been highlighted. It is closely linked to stroke, increasing its chance by three to five times in the arrhythmia sufferer. Over the next few decades, the number of patients with AF will double and the estimate of arrhythmia-related stroke will triple (26).
Systolic Heart Failure
The main mechanisms associated with intracavitary thrombi are: regional stasis, hypercoagulable state and the association with undiagnosed subclinical AF. This triples the risk of stroke in this population (30).
Patent Foramen Ovale
The supposed mechanism is associated with paradoxical embolism through the venous to arterial system through FOP. The evidence is contradictory in the causal relationship between the two clinical entities, except in stroke patients under 50 years of age (28).
Aortic Arch Atheroma
The presence of enlarged, ulcerated, non-calcified, or movable plaques have been associated with stroke and these characteristics can affect up to 8% of the population over 45 years of age (31). A common problem is that transesophageal echocardiography is not a method used in all services, which ends up underestimating its real prevalence. Currently, the recurrence rate is around 3%, reflecting the better management of atherosclerosis in secondary prevention nowadays (32).
Heart Valve Prosthesis
In the current era of anticoagulation, the risk of stroke in patients with mitral valve is 1.3 and 0.8% in the aortic position, with a lower rate in biological prosthesis than in mechanical ones (33).
Other Causes
Infectious endocarditis (IE); approximately one in every 5 IE has CNS embolization, generating a 20× greater risk of stroke within 30 days of disease progression (26).
New Concept of Atrial Myopathy and Its Relationship With Stroke
Definition of Atrial Myopathy
Atrial myopathy is considered as any structural, architectural, contractile, or electrophysiological change that affects the atria with the potential to produce relevant clinical manifestations (34).
Pathologically, a series of clinical situations (ventricular and valve cardiomyopathies, sleep apnea syndrome, arterial hypertension, diabetes mellitus, obesity, among others) are associated with structural and electrical changes, which are called “atrial remodeling.” Many conditions can coexist and accelerate the process, but the presence of AF greatly stimulates remodeling (35).
It is important to differentiate three fundamental concepts when studying atrial changes; in some cases, they can coexist:
• Atrial remodeling: response of atrial myocytes to electrical, mechanical, or metabolic stressors, such as rapid atrial tachyarrhythmia, volume or pressure overload leading to persistent changes in size, function and electrophysiological properties of the left atrium (35);
• Atrial myopathy: Zipes first described it in 1997. This is the clinical situation that occurs when myocardial diseases are associated with mechanical or electrical dysfunction that usually develop with atrial fibrosis, hypertrophy, or dilation and occur for a variety of causes. Cardiovascular risk factors, such as the clinical factors of the CHA2DS2-VASC score, can cause atrial myopathy even in the absence of AF. Example: isolated atrial fibrosis leading to the worsening of atrial function and the development of symptoms of heart failure and atrial arrhythmias (Figure 1) (35, 36);
• Atrial insufficiency: Any atrial dysfunction causing worsening of cardiac function and symptoms of worsening quality of life or life expectancy, in the absence of significant valve or ventricular diseases. Example: high density of atrial fibrosis causing stroke with isolated AF in a zero-point CHA2DS2-VASC score. Another example: advanced atrial dyssynchrony causing abnormal ventricular filling and symptoms (35). In summary, external aggressions to the atrium (structural heart disease, hypertension, and diabetes) associated or not with the presence of AF induce a slow and progressive remodeling process. It is important to note that structural remodeling, in general, occurs prior to the arrhythmic clinical presentation, and some components are irreversible. Therefore, early treatment of clinical conditions associated with AF is recommended, even before the electrical manifestation in tachyarrhythmia (8, 37). There are already some electrocardiographic risk markers, such as supraventricular ectopies, atrial tachycardia, and left atrial overload associated with stroke regardless of the diagnosis of AF. These markers of atrial dysfunction are specifically associated with cryptogenic and/or embolic stroke and not with the occlusion of small cerebral vessels (38–44).
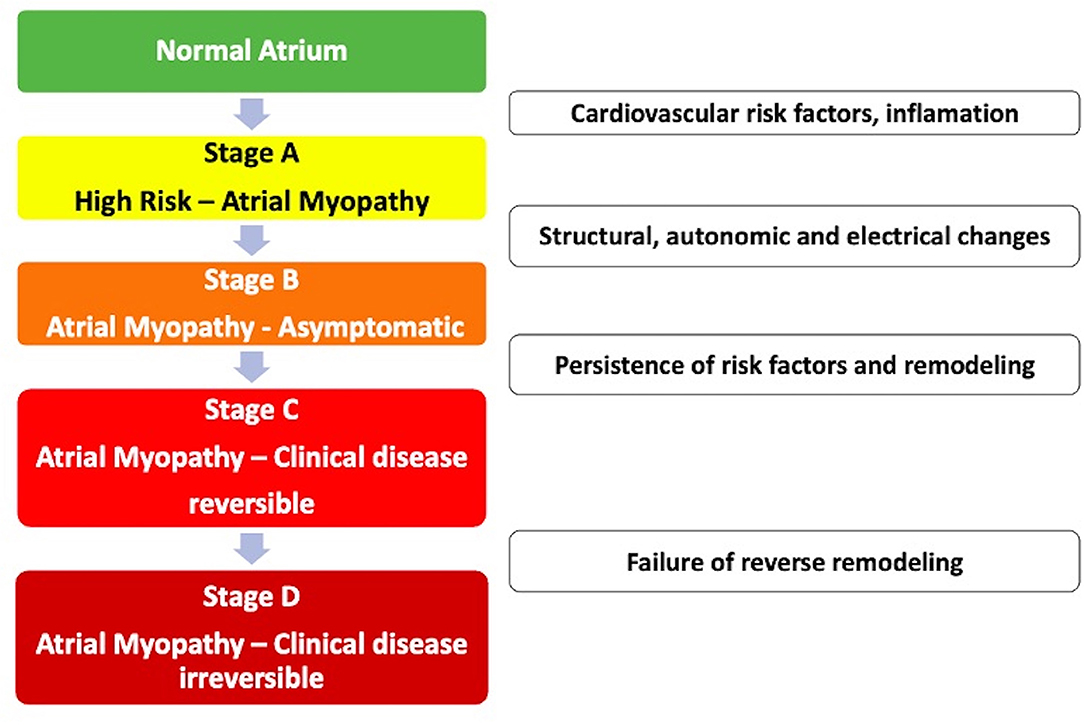
Figure 1. Evolutionary stages of atrial myopathy.
Histological and Pathophysiological Classification of Atrial Myopathy
The world's leading cardiac arrhythmia societies have published the only available classification on myopathy, called by the acronym EHRA. It is important to note that it is a classification that is not related to severity or temporal progression (Table 1) (34).
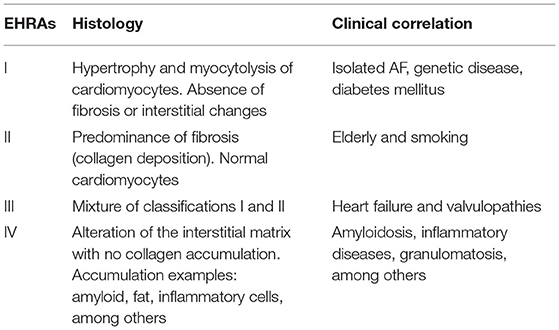
Table 1. Classification on myopathy, called by the acronym EHRA.
The normal characteristics of atrial myocytes are normal below. They have some characteristics of their own such as a prominent sarcoplasmic reticulum (Z tubules) and granules adjacent to the Golgi apparatus that determine natriuretic peptides (e.g. BNP). The others are similar to ventricular myocytes (nucleus, contractile apparatus, cytoskeleton, and organelles). The cellular interstitium is divided into a cellular component (fibroblasts, myofibroblasts, adipocytes, mesenchymal, and inflammatory cells) and extracellular, being mainly of type I collagen fibers. Importantly, collagen is a normal and essential component for the myocytes, being responsible for 5% of normal atrial wall volume. The atrial myocardium is the site of insertion of the post-ganglionic nerve endings of the so-called intrinsic cardiac nervous system, especially in the areas of fat pads. Histological changes to the EHRA classification are shown in Figure 2 (34).
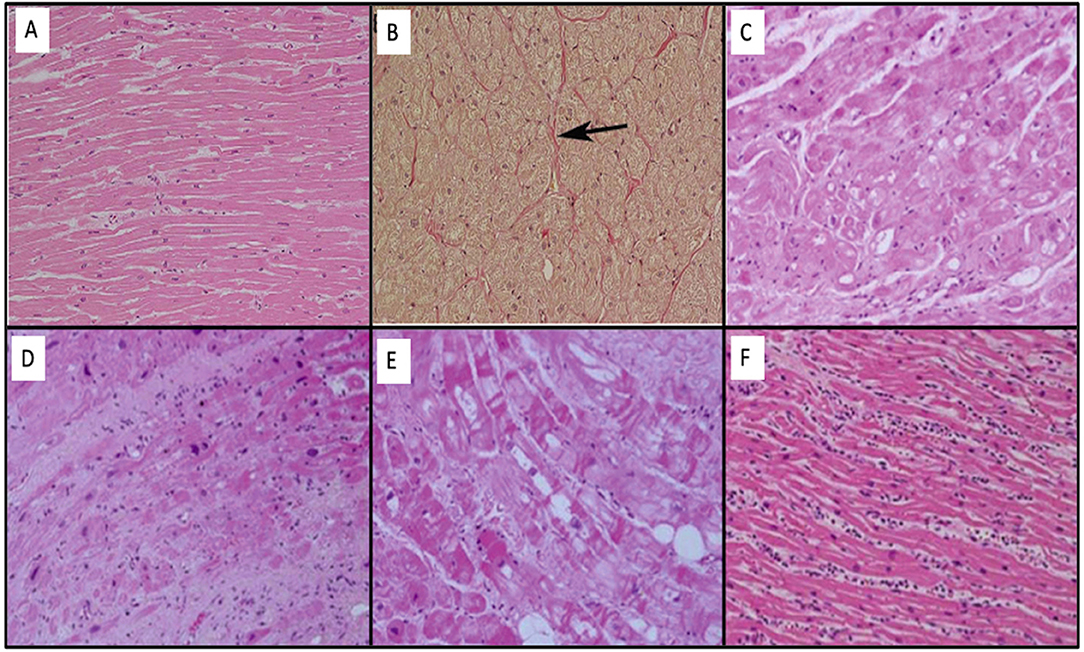
Figure 2. (A) Normal histology of the left atrium with the presence of extensive bands of homogeneous myocytes. (B) Even the atrium in Panel (A) with Van Gieson stain demonstrates that collagen fibers (red color) are identified in the adventitious space of blood vessels (arrow). (C) EHRAs Class I: modifications mainly associated with the myocyte through hypertrophy and myocytolysis. (D) EHRAs Class II: modifications mainly associated with fibrotic changes. (E) EHRAs Class III: mixture of changes in myocyte changes and fibrosis. (F) EHRAs Class IV: neutrophilic myocarditis.
Atrial Remodeling
The maintenance of AF occurs through a series of structural and electrical changes, which are called “atrial remodeling.” A number of clinical situations external to the atrium are associated with slow and progressive remodeling: ventricular and valvular cardiomyopathies, sleep apnea syndrome, arterial hypertension, diabetes mellitus, and obesity, among others. Many conditions can coexist and accelerate the process, but the presence of atrial fibrillation greatly boosts remodeling (35).
The structural basis for the emergence of AF lies in a remodeling process, with proliferation and differentiation of fibroblasts into myofibroblasts; cell hypertrophy, in addition to extracellular matrix deposition, necrosis, and fibrosis; increased cytoplasmic vacuolization, loss of myofibrils, glycogen accumulation, alteration in mitochondrial size, sarcoplasmic reticulum fragmentation, nuclear chromatin dispersion, and connexin alterations, especially connexins 40 and 43 (36, 45).
In association, there are also amyloid, fatty, and myocardial inflammatory infiltration and, in some cases, metabolism alteration, as seen in atrial ischemia due to coronary disease (36, 46). This disorganization of the atrial tissue generates dilation, greater compliance, and lesser contractility, creating an anatomical substrate for the emergence and perpetuation of multiple reentrant and anisotropy circuits, data confirmed in autopsies (36). In the presence of AF, there is an accumulation of intracellular calcium, resulting in adaptive and inflammatory responses, generating apoptosis and, consequently, atrial fibrosis (36).
Inflammation is closely linked to AF, as suggested by higher concentrations of inflammatory markers identified in the persistent form than paroxysmal in sinus rhythm. Advancing age is considered a risk factor for cardiovascular diseases, such as AF, as it causes a decline in cardiac structure and function; the mechanisms are not fully elucidated. Inflammatory mediators are also associated in elderly people with AF, which may be a possible explanation for the correlation of both entities. Inflammatory markers already identified in high concentrations are C-reactive protein; interleukin types 1B, 2, 6, and 8; fibrinogen; and tumor necrosis factor (36).
Activation of the renin-angiotensin-aldosterone system is also implicated in the amplification of atrial remodeling by several pathways, increasing the susceptibility to atrial fibrillation (46). The system promotes extracellular matrix fibrosis, leading to atrial repolarization heterogeneities and predisposition to the development of AF. Angiotensin II can increase the activity of cardiomyocytes in the pulmonary veins, which can trigger AF (2).
After the onset of arrhythmia, the electrical and mechanical disorganization imposed on the atria intensifies the atrial remodeling process, leading to the perpetuation of the arrhythmia (47).
The studies of Wijffels et al. (48) and Morillo et al. (49) identified a significant decrease and dispersion of the atrial refractory period and a loss or inversion of the adaptation property of the refractory period in relation to the stimulation frequency in experimental models of AF. These changes are likely in the ionic environment and are mediated mainly by slow Ca (L-Ca2) channels and electrical remodeling affects calcium and potassium channels (Ito e Ikur) (36, 50, 51).
Both studies demonstrated an increased susceptibility to the induction and sustainability of AF in relation to the time of induced tachycardia, proving that AF leads to AF (“AF begets AF”) (48, 49). Another mechanism involved was the decrease in wavelength; the induction of multiple wave-fronts allowed the activation of smaller atrial zones not reached by longer waves due to the presence of conduction blockade zones (51, 52).
Situations associated with oxidative stress generate an increase in intracellular calcium, by modifying type 2 ryanodine and intracellular calcium receptors, causing cellular calcium accumulation and facilitating the generation of triggered activity, cell hypertrophy, cell apoptosis, and fibrosis (36).
Atrial fibrillation results in both electrophysiological and autonomic remodeling of the atrium. Remodeling was characterized by increased atrial sympathetic innervation and heterogeneity of this innervation. This conclusion was confirmed by images of the atrial sympathetic nerve terminals of dogs by positron emission tomography, as well as by the increase in tissue norepinephrine content (53).
Obesity and obstructive sleep apnea syndrome cause autonomic remodeling by sympathetic activation, and this interferes with calcium metabolism, precipitating activity triggered mainly in muscle invaginations of pulmonary veins rich in innervation of the autonomic nervous system (36).
It is important to emphasize that structural remodeling, in general, occurs prior to the arrhythmic clinical presentation and some components are irreversible. Therefore, early treatment of clinical situations associated with atrial fibrillation is recommended, even before the electrical manifestation in tachyarrhythmia (37).
Progression from paroxysmal to persistent AF peaks in the first year, between 4 and 9% depending on the patient's treatment center; then there is a continuous growth, reaching 18–25% in 5 years of follow-up, a situation influenced by the clinical profile of the patient (8).
Biomarkers
In recent years, the adoption of clinical tools with the objective of stratifying the risk of stroke, the most used being the CHA2DS2VASC scheme (age, sex, heart failure, or previous neurological vascular event parameters) (54). Although widely accepted by cardiology societies, its ability to predict the event is suboptimal [C statistic, 0.64; 95% confidence interval (CI), 0.58–0.70] (55).
In this context, biomarkers try to fill this gap by adding the advantage in trying to express the severity and duration of the disease or the individual response to the studied insult. In relation to AF, it could generate data with greater precision in relation to thrombogenicity and the propensity to develop it in the future (56). Some types of biomarkers have been described related to stress or myocardial injury (troponins and natriuretic peptides), coagulopathies (D-dimer, plasminogen activator inhibitor, tissue factor, and P-selectin), endothelial damage (thrombomodulin, E-selectin, and von Willebrand factor), inflammatory (C-reactive protein, interleukin-6, and tumor necrosis factor-alpha), fibrosis and extracellular matrix turnover (transforming growth factor-b, myeloperoxidase, and metallopeptidases and their inhibitor), or genetic factors (micro-RNA and single-nucleotide polymorphisms) (57).
A balance is needed between complex laboratory tests in relation to clinical tools that are easy and quick to use in the wards. Currently, there is a greater value in detecting and confirming the patient really at low risk of thromboembolic phenomena, so biomarkers with high negative predictive value may have a value in the future of daily clinical practice (57).
Justifications for the Association of Atrial Myopathy and Stroke
It is well known that AF is related to atrial remodeling, only that these changes can occur with or without arrhythmia, so is it possible that myocardiopathy can cause stroke, even occasionally in the absence of AF? (43, 44, 58–63).
It was identified that episodes of AF with 6 min are related to an increased risk of stroke, except that atrial remodeling takes a few weeks for its complete institution, that is, another inconsistency in the direct role of AF and the neurological event appears (16, 64). This suggests that in this group of patients, atrial structural changes occur prior to the diagnosis of AF and the neurological event.
An important data in this context of cardioembolic stroke physiopathology is temporal dissociation. It was described based on data that demonstrated that cerebrovascular events and the presence of episodes of AF were not linear, that is, in many cases the robust temporal correlation between the two clinical events was lacking (35).
The ASSERT study of 2,580 patients with pacemaker and stroke demonstrated that only 8% of patients had a record of AF 30 days before the stroke and 16% had the first arrhythmic event detected only after clinical ictus (36). This suggests that arrhythmia may be a marker of atrial cardiomyopathy, excluding the previous concept that AF was the direct cause of cardioembolism (35, 36).
Contributing to the hypothesis, we have identified the inability of rhythm control in AF strategies in stroke prevention in clinical follow-up after maintenance at a steady pace sinus (36). It is interesting that, following this concept, the amount of atrial fibrosis, captured by magnetic resonance, in cases of cryptogenic stroke is similar to the cases related to manifest AF and greater than that of the cases associated with other etiologies (35).
Prothrombotic Status
The entire cascade of events associated with cardiomyopathy and atrial remodeling can generate damage to the myocytes and also an expression of prothrombotic factors on the atrial endothelial surface associated with platelet activation and inflammatory cells amplifying the prothrombotic status, mainly in the left atrial appendage (35, 37).
Situations and clinical signs associated with inflammation amplify the prothrombotic process because they are associated with the increased concentration of interleukin 6 and the activation of tissue factor, factor VII, and von Willebrand factor. This leads to endothelial damage, platelet activation, and greater sensitivity to thrombin, generating a vicious cycle between pro-thrombotic and inflammatory status (36) (Figure 3). It is important to clarify that atrial myopathy can present itself as AF initially but should always be considered thrombogenic in all the contexts of their clinical presentation (36). On the other hand, the burden of AF is important, since patients persistently and permanently have a higher stroke rate than found in paroxysmal; the arrhythmic load may be associated with a greater impairment atrial disease due to myopathy and, consequently, greater prothrombotic status (36).
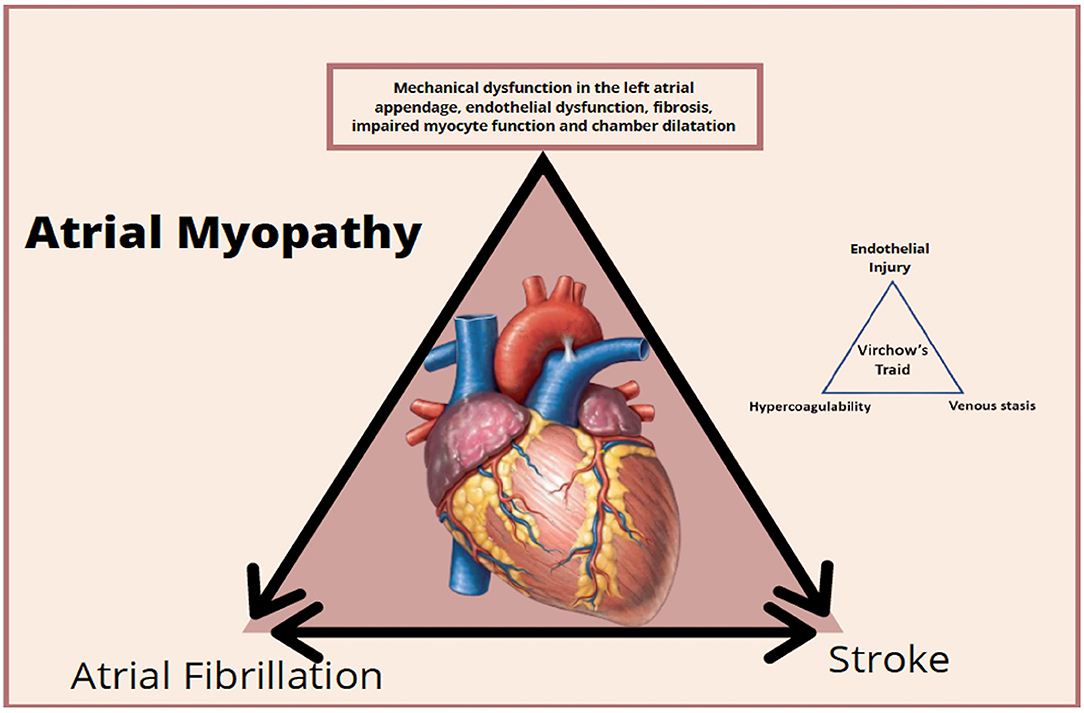
Figure 3. Clinical stages and prothrombotic status of atrial myopathy.
What probably occurs in the presence of AF is that thrombogenic mechanisms are amplified through contractile dysfunction and blood stasis. This ends up meeting the results of clinical trials where a control of the rhythm of AF was not associated with reduced stroke, probably because it did not reduce the evolution of atrial heart disease (19).
Clinical Implications
Anticoagulation in Sinus Rhythm
Traditionally, the use of anticoagulants in patients with AF reduces the rate of stroke in the clinical follow-up by around 50–60% (65). In the WARCEF study, warfarin reduced the incidence of ischemic stroke in patients with HFREF compared to aspirin (HR 0.52, 95% CI 0.33–0.88, p = 0.0055) (66). Coumarins also reduce the chance of stroke in other types of heart disease, such as in patients with mechanical cardiac prostheses and with post-infarction ventricular thrombus (33). This pooling of evidence indirectly suggests that anticoagulation could prevent the risk of ischemic stroke in other cardiomyopathies (67).
The WARSS study was a multicenter randomized clinical trial that tested anticoagulation with warfarin versus aspirin in the secondary prevention of stroke in patients with a non-cardioembolic mechanism. Although the primary endpoint showed no benefit, post-hoc analysis showed that there was evidence of a benefit among those with NT-proBNP elevations above 750 pg/ml (HR 0.30, 95% CI 0.12–0.84; p = 0.021) (68). This constitutes a hypothesis that justified the performance of a clinical trial testing anticoagulation vs. antiplatelet therapy among patients with embolic stroke of undetermined source (ESUS) and atrial heart disease (67).
The NAVIGATE ESUS and RE-SPECT ESUS trials tested anticoagulation in ESUS, concluding that oral anticoagulation is not associated with reduced stroke recurrence rates compared to aspirin (69, 70). One of the reasons for the lack of benefit is the presence of overlapping potential embolic sources, characteristic of this population where more than 65% have more than one source. Certain sources (such as atrial heart disease, left ventricular disease, FOP, and cancer) are likely to benefit from anticoagulant therapy, whereas aortic plaques, cervical, or intracranial atherosclerosis may respond better to aspirin (71). Perhaps in the near future the ARCADIA trial will be able to answer this question; this study will include 1,100 patients with ESUS and with evidence of atrial heart disease in groups comparing apixaban and aspirin. The definition of heart disease will use at least one of the three selected biomarkers (PTFV1 >5,000 μV/ms on 12-lead ECG; Serum NT-proBNP >250 pg/ml; left atrial diameter index ≥3 cm/m2 on echocardiogram) (72). Although the optimal choice of biomarkers is not yet clear, the attempt to select patients with ESUS and heart disease is commendable in an attempt to demonstrate the role of anticoagulation in this subtype of stroke.
Suggestion for Clinical Practice
Considering atrial heart disease (with or without the presence of AF) as a cause of embolic stroke, it could explain that one-third of strokes are considered cryptogenic (Figure 2) (73). Many are suspected of cardiomegaly clinically and through imaging, but only in one-third of patients an episode of AF is diagnosed even after 3 years of continuous cardiac monitoring (28). Therefore, the classification of cryptogenic may be overestimated, considering this association.
This sequence of facts implies the role of continuous monitoring to detect AF after an event considered initially cryptogenic. The absence of AF detection during the monitoring period cannot be the sole and exclusive reason for discontinuing anticoagulant therapy in association with other signs of atrial heart disease and the presence of clinical suspicion and stroke image (19).
As the institution of the concept of atrial heart disease is likely to control the concomitant risk factors that are associated with AF and atrial heart disease, they can be beneficial in reducing thromboembolic risk and not just restoring the atrial rhythm alone (74).
In the same way, sometimes AF can be considered as isolated, when it occurs in the absence of abnormal atrial substrate markers and without vascular risk factors, where the risk of stroke is minimized or even canceled (75).
Conclusions
The traditional model exclusively associating the presence of AF in the genesis of thromboembolism is incomplete. The concept of atrial cardiopathy where cardioembolism occurs in a non-AF dependent manner fits better with current data. The future challenge is to effectively detect the various manifestations of atrial heart disease, generating direct implications for the identification of patients at risk of stroke and also for better management after a cardioembolic event.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Raviele A, Natale A, Calkins H, Camm JA, Cappato R, Ann Chen S, et al. Venice chart international consensus document on atrial fibrillation ablation: 2011 update. J Cardiovasc Electrophysiol. (2012) 23:890–923. doi: 10.1111/j.1540-8167.2012.02381.x
2. Morin DP, Bernard ML, Madias C, Rogers PA, Thihalolipavan S, Estes NA III. The state of the art: atrial fibrillation epidemiology, prevention, and treatment. Mayo Clin Proc. (2016) 91:1778–810. doi: 10.1016/j.mayocp.2016.08.022
3. Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation-executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the (2001). Guidelines for the management of patients with atrial fibrillation). Eur Heart J. (2006) 27:1979–2030. doi: 10.1093/eurheartj/ehm315
4. Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med. (1982) 306:1018–22. doi: 10.1056/NEJM198204293061703
5. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke. (1991) 22:983–8. doi: 10.1161/01.STR.22.8.983
6. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham study. Arch Int Med. (1987) 147:1561–4. doi: 10.1001/archinte.147.9.1561
7. Zulkifly H, Lip GYH, Lane DA. Epidemiology of atrial fibrillation. Int J Clin Pract. (2018) 72:e13070. doi: 10.1111/ijcp.13070
8. Zoni-Berisso M, Lercari F, Carazza T, Domenicucci S. Epidemiology of atrial fibrillation: European perspective. Clin Epidemiol. (2014) 6:213–20. doi: 10.2147/CLEP.S47385
9. Kamel H, Okin PM, Elkind MS, Iadecola C. Atrial fibrillation and mechanisms of stroke: time for a new model. Stroke. (2016) 47:895–900. doi: 10.1161/STROKEAHA.115.012004
10. Wolf PA, Dawber TR, Thomas HE Jr, Kannel WB. Epidemiologic assessment of chronic atrial fibrillation and risk of stroke: the Framingham study. Neurology. (1978) 28:973–7. doi: 10.1212/WNL.28.10.973
11. Manolio TA, Kronmal RA, Burke GL, O'Leary DH, Price TR. Short-term predictors of incident stroke in older adults. Cardiovascular health study. Stroke. (1996) 27:1479–86. doi: 10.1161/01.STR.27.9.1479
12. Lodder J, Bamford JM, Sandercock PA, Jones LN, Warlow CP. Are hypertension or cardiac embolism likely causes of lacunar infarction? Stroke. (1990) 21:375–81. doi: 10.1161/01.STR.21.3.375
13. Boiten J, Lodder J. Lacunar infarcts. Pathogenesis and validity of the clinical syndromes. Stroke. (1991) 22:1374–8. doi: 10.1161/01.STR.22.11.1374
14. Chao TF, Liu CJ, Chen SJ, Wang KL, Lin YJ, Chang SL, et al. Atrial fibrillation and the risk of ischemic stroke: does it still matter in patients with a CHA2DS2-VASc score of 0 or 1? Stroke. (2012) 43:2551–5. doi: 10.1161/STROKEAHA.112.667865
15. Healey JS, Connolly SJ, Gold MR, Israel CW, Van Gelder IC, Capucci A, et al. Subclinical atrial fibrillation and the risk of stroke. N Engl J Med. (2012) 366:120–9. doi: 10.1056/NEJMoa1105575
16. Turakhia MP, Ziegler PD, Schmitt SK, Chang Y, Fan J, Than CT, et al. Atrial fibrillation burden and short-term risk of stroke: case-crossover analysis of continuously recorded heart rhythm from cardiac electronic implanted devices. Circ Arrhyth Electrophysiol. (2015) 8:1040–7. doi: 10.1161/CIRCEP.114.003057
17. Brambatti M, Connolly SJ, Gold MR, Morillo CA, Capucci A, Muto C, et al. Temporal relationship between subclinical atrial fibrillation and embolic events. Circulation. (2014) 129:2094–9. doi: 10.1161/CIRCULATIONAHA.113.007825
18. Martin DT, Bersohn MM, Waldo AL, Wathen MS, Choucair WK, Lip GY, et al. Randomized trial of atrial arrhythmia monitoring to guide anticoagulation in patients with implanted defibrillator and cardiac resynchronization devices. Eur Heart J. (2015) 36:1660–8. doi: 10.1093/eurheartj/ehv115
19. Al-Khatib SM, Allen LaPointe NM, Chatterjee R, Crowley MJ, Dupre ME, Kong DF, et al. Rate- and rhythm-control therapies in patients with atrial fibrillation: a systematic review. Ann Intern Med. (2014) 160:760–73. doi: 10.7326/M13-1467
20. Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. (2014) 114:1500–15. doi: 10.1161/CIRCRESAHA.114.303772
21. Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, et al. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. (2001) 104:2886–91. doi: 10.1161/hc4901.101760
22. Bogousslavsky J, Cachin C, Regli F, Despland PA, Van Melle G, Kappenberger L. Cardiac sources of embolism and cerebral infarction–clinical consequences and vascular concomitants: the Lausanne Stroke Registry. Neurology. (1991) 41:855–9. doi: 10.1212/WNL.41.6.855
23. Palacio S, Hart RG. Neurologic manifestations of cardiogenic embolism: an update. Neurol Clin. (2002). 20:179–93. doi: 10.1016/S0733-8619(03)00058-6
24. Marini C, De Santis F, Sacco S, Russo T, Olivieri L, Totaro R, et al. Contribution of atrial fibrillation to incidence and outcome of ischemic stroke: results from a population-based study. Stroke. (2005) 36:1115–9. doi: 10.1161/01.STR.0000166053.83476.4a
25. Engdahl J, Andersson L, Mirskaya M, Rosenqvist M. Stepwise screening of atrial fibrillation in a 75-year-old population: implications for stroke prevention. Circulation. (2013) 127:930–7. doi: 10.1161/CIRCULATIONAHA.112.126656
26. Kamel H, Healey JS. Cardioembolic stroke. Circ Res. (2017) 120:514–26. doi: 10.1161/CIRCRESAHA.116.308407
27. Arquizan C, Lamy C, Mas JL. [Simultaneous supratentorial multiple cerebral infarctions]. Rev Neurol (Paris). (1997) 153:748–53.
28. Sanna T, Diener HC, Passman RS, Di Lazzaro V, Bernstein RA, Morillo CA, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. (2014) 370:2478–86. doi: 10.1056/NEJMoa1313600
29. Gladstone DJ, Spring M, Dorian P, Panzov V, Thorpe KE, Hall J, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. (2014) 370:2467–77. doi: 10.1056/NEJMoa1311376
30. Witt BJ, Brown RD Jr, Jacobsen SJ, Weston SA, Ballman KV, Meverden RA, et al. Ischemic stroke after heart failure: a community-based study. Am Heart J. (2006) 152:102–9. doi: 10.1016/j.ahj.2005.10.018
31. Meissner I, Khandheria BK, Sheps SG, Schwartz GL, Wiebers DO, Whisnant JP, et al. Atherosclerosis of the aorta: risk factor, risk marker, or innocent bystander? A prospective population-based transesophageal echocardiography study. J Am Coll Cardiol. (2004) 44:1018–24. doi: 10.1016/j.jacc.2004.05.075
32. Amarenco P, Davis S, Jones EF, Cohen AA, Heiss WD, Kaste M, et al. Clopidogrel plus aspirin versus warfarin in patients with stroke and aortic arch plaques. Stroke. (2014) 45:1248–57. doi: 10.1161/STROKEAHA.113.004251
33. Cannegieter SC, Rosendaal FR, Briet E. Thromboembolic and bleeding complications in patients with mechanical heart valve prostheses. Circulation. (1994) 89:635–41. doi: 10.1161/01.CIR.89.2.635
34. Goette A, Kalman JM, Aguinaga L, Akar J, Cabrera JA, Chen SA, et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: definition, characterization, and clinical implication. Heart Rhythm. (2017) 14:e3–40. doi: 10.1016/j.hrthm.2016.05.028
35. Bisbal F, Baranchuk A, Braunwald E, Bayes de. Luna A, Bayes-Genis A. Atrial failure as a clinical entity: JACC review topic of the week. J Am Coll Cardiol. (2020) 75:222–32. doi: 10.1016/j.jacc.2019.11.013
36. Shen MJ, Arora R, Jalife J. Atrial myopathy. JACC Basic Transl Sci. (2019) 4:640–54. doi: 10.1016/j.jacbts.2019.05.005
37. Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. (2016) 37:2893–962. doi: 10.1093/eurheartj/ehw210
38. Larsen BS, Kumarathurai P, Falkenberg J, Nielsen OW, Sajadieh A. Excessive atrial ectopy and short atrial runs increase the risk of stroke beyond incident atrial fibrillation. J Am Coll Cardiol. (2015) 66:232–41. doi: 10.1016/j.jacc.2015.05.018
39. Kamel H, O'Neal WT, Okin PM, Loehr LR, Alonso A, Soliman EZ. Electrocardiographic left atrial abnormality and stroke subtype in the atherosclerosis risk in communities study. Ann Neurol. (2015) 78:670–8. doi: 10.1002/ana.24482
40. Kamel H, Soliman EZ, Heckbert SR, Kronmal RA, Longstreth WT Jr, Nazarian S, et al. P-wave morphology and the risk of incident ischemic stroke in the Multi-Ethnic Study of Atherosclerosis. Stroke. (2014) 45:2786–8. doi: 10.1161/STROKEAHA.114.006364
41. Kamel H, Hunter M, Moon YP, Yaghi S, Cheung K, Di Tullio MR, et al. Electrocardiographic left atrial abnormality and risk of stroke: Northern Manhattan study. Stroke. (2015) 46:3208–12. doi: 10.1161/STROKEAHA.115.009989
42. Benjamin EJ, D'Agostino RB, Belanger AJ, Wolf PA, Levy D. Left atrial size and the risk of stroke and death. The Framingham heart study. Circulation. (1995) 92:835–41. doi: 10.1161/01.CIR.92.4.835
43. Yaghi S, Moon YP, Mora-McLaughlin C, Willey JZ, Cheung K, Di Tullio MR, et al. Left atrial enlargement and stroke recurrence: the Northern Manhattan Stroke Study. Stroke. (2015) 46:1488–93. doi: 10.1161/STROKEAHA.115.008711
44. Di Tullio MR, Sacco RL, Sciacca RR, Homma S. Left atrial size and the risk of ischemic stroke in an ethnically mixed population. Stroke. (1999) 30:2019–24. doi: 10.1161/01.STR.30.10.2019
45. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. (2008) 51:802–9. doi: 10.1016/j.jacc.2007.09.064
46. January CT, Wann LS, Calkins H, Chen LY, Cigarroa JE, Cleveland JC Jr, et al. 2019 AHA/ACC/HRS focused update of the 2014. AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines and the heart rhythm society. J Am Coll Cardiol. (2019). 74:104–32. doi: 10.1016/j.jacc.2019.01.011
47. Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. (2014) 114:1483–99. doi: 10.1161/CIRCRESAHA.114.302226
48. Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. (1995) 92:1954–68. doi: 10.1161/01.CIR.92.7.1954
49. Morillo CA, Klein GJ, Jones DL, Guiraudon CM. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation. (1995) 91:1588–95. doi: 10.1161/01.CIR.91.5.1588
50. van der Velden HMW, van der Zee L, Wijffels MC, van Leuven C, Dorland R, Vos MA, et al. Atrial fibrillation in the goat induces changes in monophasic action potential and mRNA expression of ion channels involved in repolarization. J Cardiovasc Electrophysiol. (2000) 11:1262–9. doi: 10.1046/j.1540-8167.2000.01262.x
51. Konings KT, Kirchhof CJ, Smeets JR, Wellens HJ, Penn OC, Allessie MA. High-density mapping of electrically induced atrial fibrillation in humans. Circulation. (1994) 89:1665–80. doi: 10.1161/01.CIR.89.4.1665
52. Allessie MA. Atrial electrophysiologic remodeling: another vicious circle? J Cardiovasc Electrophysiol. (1998) 9:1378–93. doi: 10.1111/j.1540-8167.1998.tb00114.x
53. Olgin JE, Sih HJ, Hanish S, Jayachandran JV, Wu J, Zheng QH, et al. Heterogeneous atrial denervation creates substrate for sustained atrial fibrillation. Circulation. (1998) 98:2608–14. doi: 10.1161/01.CIR.98.23.2608
54. Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the euro heart survey on atrial fibrillation. Chest. (2010) 137:263–72. doi: 10.1378/chest.09-1584
55. Sanders GD, Lowenstern A, Borre E, Chatterjee R, Goode A, Sharan L, et al. Stroke Prevention in Patients with Atrial Fibrillation: A Systematic Review Update. Rockville, MD: Agency for Healthcare Research and Quality (US). Report No.: 18-EHC018-EFReport No.: 2018-SR-04 (2018). doi: 10.23970/AHRQEPCCER214
56. Hylek EM. Biomarkers for prediction of stroke and bleeds in atrial fibrillation. Circulation. (2019) 139:772–4. doi: 10.1161/CIRCULATIONAHA.118.038635
57. Szymanski FM, Lip GY, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Opolski G. Stroke risk factors beyond the CHA(2)DS(2)-VASc score: can we improve our identification of “high stroke risk” patients with atrial fibrillation? Am J Cardiol. (2015) 116:1781–8. doi: 10.1016/j.amjcard.2015.08.049
58. Cai H, Li Z, Goette A, Mera F, Honeycutt C, Feterik K, et al. Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation. (2002) 106:2854–8. doi: 10.1161/01.CIR.0000039327.11661.16
59. Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. (1997) 96:1180–4. doi: 10.1161/01.CIR.96.4.1180
60. Mihm MJ Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, et al. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation. (2001) 104:174–80. doi: 10.1161/01.CIR.104.2.174
61. Vaziri SM, Larson MG, Benjamin EJ, Levy D. Echocardiographic predictors of nonrheumatic atrial fibrillation. The Framingham Heart Study. Circulation. (1994) 89:724–30. doi: 10.1161/01.CIR.89.2.724
62. Warraich HJ, Gandhavadi M, Manning WJ. Mechanical discordance of the left atrium and appendage: a novel mechanism of stroke in paroxysmal atrial fibrillation. Stroke. (2014) 45:1481–4. doi: 10.1161/STROKEAHA.114.004800
63. Stollberger C, Chnupa P, Kronik G, Brainin M, Finsterer J, Schneider B, et al. Transesophageal echocardiography to assess embolic risk in patients with atrial fibrillation. ELAT study group embolism in left atrial thrombi. Ann Intern Med. (1998) 128:630–8. doi: 10.7326/0003-4819-128-8-199804150-00004
64. De Jong AM, Maass AH, Oberdorf-Maass SU, Van Veldhuisen DJ, Van Gilst WH, Van Gelder IC. Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc Res. (2011) 89:754–65. doi: 10.1093/cvr/cvq357
65. van Walraven C, Hart RG, Singer DE, Laupacis A, Connolly S, Petersen P, et al. Oral anticoagulants vs aspirin in nonvalvular atrial fibrillation: an individual patient meta-analysis. Jama. (2002) 288:2441–8. doi: 10.1001/jama.288.19.2441
66. Homma S, Thompson JL, Pullicino PM, Levin B, Freudenberger RS, Teerlink JR, et al. Warfarin and aspirin in patients with heart failure and sinus rhythm. N Engl J Med. (2012) 366:1859–69. doi: 10.1056/NEJMoa1202299
67. Elkind MSV. Atrial cardiopathy and stroke prevention. Curr Cardiol Rep. (2018) 20:103. doi: 10.1007/s11886-018-1053-0
68. Longstreth WT Jr, Kronmal RA, Thompson JL, Christenson RH, Levine SR, Gross R, et al. Amino terminal pro-B-type natriuretic peptide, secondary stroke prevention, and choice of antithrombotic therapy. Stroke. (2013) 44:714–9. doi: 10.1161/STROKEAHA.112.675942
69. Hart RG, Connolly SJ, Mundl H. Rivaroxaban for stroke prevention after embolic stroke of undetermined source. N Engl J Med. (2018) 379:987. doi: 10.1056/NEJMc1809065
70. Diener HC, Sacco RL, Easton JD, Granger CB, Bernstein RA, Uchiyama S, et al. Dabigatran for prevention of stroke after embolic stroke of undetermined source. N Engl J Med. (2019) 380:1906–17. doi: 10.1056/NEJMoa1813959
71. Ntaios G. Embolic Stroke of Undetermined source: JACC review topic of the week. J Am Coll Cardiol. (2020) 75:333–40. doi: 10.1016/j.jacc.2019.11.024
72. Kamel H, Longstreth WT Jr, Tirschwell DL, Kronmal RA, Broderick JP, Palesch YY, et al. The AtRial cardiopathy and antithrombotic drugs in prevention after cryptogenic stroke randomized trial: rationale and methods. Int J Stroke. (2019) 14:207–14. doi: 10.1177/1747493018799981
73. Marnane M, Duggan CA, Sheehan OC, Merwick A, Hannon N, Curtin D, et al. Stroke subtype classification to mechanism-specific and undetermined categories by TOAST, A-S-C-O, and causative classification system: direct comparison in the North Dublin population stroke study. Stroke. (2010) 41:1579–86. doi: 10.1161/STROKEAHA.109.575373
74. Magnani JW, Lopez FL, Soliman EZ, Maclehose RF, Crow RS, Alonso A, et al. wave indices, obesity, and the metabolic syndrome: the atherosclerosis risk in communities study. Obesity (Silver Spring). (2012) 20:666–72. doi: 10.1038/oby.2011.53
Keywords: atrial fibrillation, stroke, atrial myopathy, cardioembolic stroke, review
Citation: Ronsoni RM, Saffi MAL, Gonçalves MVM, Nakayama IH and Luz Leiria TL (2021) A New Vision at the Interface of Atrial Fibrillation and Stroke. Front. Cardiovasc. Med. 8:689313. doi: 10.3389/fcvm.2021.689313
Received: 02 April 2021; Accepted: 12 July 2021;
Published: 09 August 2021.
Edited by:
Nicola Mumoli, ASST Ovest Milanese, ItalyReviewed by:
Carmine Morisco, University of Naples Federico II, ItalyAndre Rodrigues Duraes, Federal University of Bahia, Brazil
Copyright © 2021 Ronsoni, Saffi, Gonçalves, Nakayama and Luz Leiria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael M. Ronsoni, cmFmYWVscm9uc29uaSYjeDAwMDQwO2dtYWlsLmNvbQ==
†These authors have contributed equally to this work