
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 21 June 2021
Sec. Thrombosis and Haemostasis
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.684920
This article is part of the Research TopicHighlights in Thrombosis: 2021View all 9 articles
 Gaukhar Baidildinova1,2
Gaukhar Baidildinova1,2 Magdolna Nagy1
Magdolna Nagy1 Kerstin Jurk2,3
Kerstin Jurk2,3 Philipp S. Wild2,3,4
Philipp S. Wild2,3,4 Hugo ten Cate1,2,5
Hugo ten Cate1,2,5 Paola E. J. van der Meijden1,5*
Paola E. J. van der Meijden1,5*Platelets are the main players in thrombotic diseases, where activated platelets not only mediate thrombus formation but also are involved in multiple interactions with vascular cells, inflammatory components, and the coagulation system. Although in vitro reactivity of platelets provides information on the function of circulating platelets, it is not a full reflection of the in vivo activation state, which may be relevant for thrombotic risk assessment in various disease conditions. Therefore, studying release markers of activated platelets in plasma is of interest. While this type of study has been done for decades, there are several new discoveries that highlight the need for a critical assessment of the available tests and indications for platelet release products. First, new insights have shown that platelets are not only prominent players in arterial vascular disease, but also in venous thromboembolism and atrial fibrillation. Second, knowledge of the platelet proteome has dramatically expanded over the past years, which contributed to an increasing array of tests for proteins released and shed from platelets upon activation. Identification of changes in the level of plasma biomarkers associated with upcoming thromboembolic events allows timely and individualized adjustment of the treatment strategy to prevent disease aggravation. Therefore, biomarkers of platelet activation may become a valuable instrument for acute event prognosis. In this narrative review based on a systematic search of the literature, we summarize the process of platelet activation and release products, discuss the clinical context in which platelet release products have been measured as well as the potential clinical relevance.
Platelet thrombus formation is a process of crucial importance in hemostasis and thrombosis, starting with platelet activation, adhesion, and aggregation at the vessel wall surface that is damaged by trauma, inflammation, or, in case of atherosclerosis, altered by an atherosclerotic plaque (1, 2). In general, upon vascular damage the platelet membrane glycoprotein (GP) Ib/V/IX complex interacts with von Willebrand factor (vWF) from the damaged endothelium leading to the adhesion of platelets (2, 3). Tethered platelets bind to collagen through their GPVI and integrin α2β1 receptors, which potently trigger platelet activation. The activation process continues toward the release of soluble mediators from activated platelets, an increase of cytosolic Ca2+, and the formation of a platelet thrombus. In parallel, fibrin formation is triggered by the tissue factor-driven-coagulation cascade and amplified by thrombin-mediated feedback reactions as well as the contact activation pathway. Platelet and coagulation activation are highly intertwined with multiple interactions between these two processes. Not only is thrombin a key mediator of platelet activation, platelets also promote coagulation via phosphatidylserine exposure and receptor-mediated binding of coagulation factors (4).
Upon activation, platelets release more than 300 proteins, including P-selectin (CD62P), CD40 ligand (CD40L), platelet factor 4 (PF4), and many others (5). Some of these platelet release markers can reflect the in vivo platelet activation status and hence have already been investigated in clinical studies addressing the involvement of platelet activation in patients with different thrombotic diseases. The soluble platelet biomarkers may provide a better way of assessing the thrombotic risk than the conventional platelet function tests. Impaired in vitro platelet activation based on platelet function tests, may on the one hand point to dysfunctional platelets, but on the other hand to prior activation in the circulation potentially resulting in an exhausted platelet phenotype. Therefore, soluble platelet activation markers reveal the in vivo platelet activation status and provide information on the underlying pathophysiological mechanisms in thrombosis-related disease (6).
The role of platelets in atherothrombotic disease, characterized by arterial thrombus formation as a consequence of atherosclerotic lesion disruption, is well-established. Vascular occlusion underlies the occurrence of ischemia in specific vascular beds, resulting in coronary artery disease (CAD), myocardial infarction (MI), peripheral artery disease (PAD), and ischemic stroke (IS) (7). Although in arterial thromboembolism—as a consequence of atrial fibrillation—and venous thrombosis, coagulation activation is the predominant process, accumulating evidence demonstrates pathogenic roles of platelets herein (8). The conventional treatment strategy for atherothrombotic disease and arterial/venous thromboembolism is based on antiplatelet and anticoagulant drugs, respectively. Especially for patients with atherothrombotic events, the combined antiplatelet and anticoagulant treatment appears beneficial and has recently gained more attention (9).
The active participation of platelets in cardiovascular diseases and the established fact that antiplatelet therapy decreases the risk of (recurrent) thrombotic events underlines the importance of research in platelet pathophysiology (10). In this narrative review based on a systematic search of the literature, we summarized the process of platelet activation and release products, discuss the clinical context in which platelet release products have been measured as well as the potential clinical relevance. Here we focus on soluble platelet biomarkers in patients with arterial thrombosis, venous thrombosis, and atrial fibrillation.
Activated platelets release small biomolecules and more than 300 proteins, which regulate hemostatic, inflammatory, and angiogenic responses of platelets, leukocytes, and vascular cells. Major sources of the platelet protein releasate are granule cargos and proteolytically cleaved/shed membrane-bound proteins such as receptors and platelet-derived extracellular vesicles. Advanced enzyme-linked immunosorbent assay (ELISA)-based assays and mass spectrometry approaches enable the qualitative and quantitative assessment of platelet-released proteins in plasma and of isolated platelets, respectively (11). Platelets contain three major types of granules: α-granules, dense or δ-granules, and lysosomes (12, 13). Rapid granule release can be induced by diverse agonists like thrombin, collagen, and their related peptides (11). The platelet α-granule secretome covers the majority of released platelet proteins, which are synthesized in megakaryocytes or endocytosed from plasma. The α-granules contain large adhesive proteins [vWF, thrombospondin-1 (TSP-1), vitronectin, fibronectin], coagulation factors (factor V, VII, XI, XIII), mitogenic factors [platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), transforming growth factor β (TGF-β)], protease inhibitors [protein C, plasminogen activator inhibitor 1 (PAI-1), tissue factor pathway inhibitor (TFPI)], membrane proteins [P-selectin (CD62P), CD40L], chemokines [β-thromboglobulin (beta-tg), PF4, Regulated upon Activation, Normal T-Cell Expressed and Presumably Secreted (RANTES), stromal cell-derived factor-1α (SDF-1α)], and several other molecules, which are released immediately upon platelet activation (14). Activated platelets also release thromboxane A2 (TxA2), a product of arachidonic acid metabolism (15), and several other eicosanoids (16).
In contrast, dense granules secrete small soluble molecules, such as serotonin, glutamate, adenosine diphosphate (ADP), adenosine triphosphate (ATP), histamine, polyphosphate, Ca2+, and Mg2+ (17, 18). Together with TxA2, they function as positive feedback mediators of platelets to promote platelet aggregation and platelet-based coagulation. Platelet-derived serotonin promotes thrombosis development by inducing vasoconstriction and enhancing platelet activation and thrombus formation. The platelet lysosomes contain enzymes required for extracellular matrix degradation, cell migration, antimicrobial activity, and thrombus remodeling (19, 20). Among these enzymes are cathepsin D and E, β-hexosaminidase, elastase, and heparanase (11). The classical flow cytometry protein for dense granule and lysosomal membrane detection is CD63.
In addition to the release of soluble proteins from granules, proteolytic cleavage of platelet membrane proteins occurs mainly by metalloproteinases (MMP) and the shed fragments can modulate cellular responses. The platelet sheddome, excluding plasma proteins and platelet-derived extracellular vesicles, contains at least 69 membrane proteins (21, 22). Only a fraction of all membrane proteins is cleaved, among these are the externalized surface proteins P-selectin and CD40L, the receptor GPIbα, GPV subunits of the GPIb-IX-V complex, and GPVI (21, 23–25). The ectodomains of the receptors are shed in response to ligand engagement, elevated shear, coagulation, or apoptosis.
Upon activation, platelets secrete beta-tg from α-granules that are derived from the proteolytic cleavage of platelet basic proteins resulting in CTAPIII, CXCL7, and beta-tg. Beta-tg shares significant homology with PF4 (26). Both molecules belong to the chemokine CXC subfamily (27) and are expressed in monocytes, granulocytes, T-cells, and mast cells (7). Yet, platelets have been proposed as the primary and the most rapidly available source of the aforementioned chemokines (28). Beta-tg accounts for almost 10% of the α-granules content and is released into the blood with PF4 and other proteins upon platelet activation (27). The half-life of beta-tg in the blood is 100 min (29), depending on renal clearance (30, 31), while PF4 is rapidly cleared by binding to endothelial cells (32).
Another chemokine secreted by platelet α-granules is SDF-1α or C-X-C motif chemokine ligand 12 (CXCL12), which is involved in inflammatory pathways. SDF-1α is expressed by various cells throughout the body, including immune, stem, and endothelial cells (33), but platelets are thought to be the primary source. Following platelet activation, SDF-1α remains surface-bound and a strong stimulus is required to mediate release. ADP stimulation appears to be most potent in inducing SDF-1α release. Although SDF-1α in the circulation is susceptible to proteolytic degradation, it might be protected in the microenvironment of platelet thrombi (34). There is evidence that SDF-1α via its chemokine receptor CXCR4 induces TxA2 production and dense granule release, which altogether contributes to thrombus formation (35). In addition, ligation of SDF-1α to CXCR4 and CXCR7 regulates monocyte function and macrophage/foam cell differentiation, indicating an important role of SDF-1α in inflammation (36).
TSP-1 is a high-molecular multidomain glycoprotein expressed by various cell types including endothelial cells, monocytes, macrophages, fibroblasts, smooth muscle cells, dendritic cells, and B-cells (37). Similar to the previously mentioned proteins, the main source of TSP-1 is platelets, where it is one of the most abundant granule proteins, synthesized by megakaryocytes. After platelet activation, TSP-1 is released from the α-granules and found either bound to the platelet membrane or in its soluble form in plasma. TSP-1 has multiple functions in hemostasis, angiogenesis, proliferation, migration, endocytosis, immune reactions, and apoptosis. In addition to vWF, TSP-1 has been identified as a high shear substrate for human platelets (38). The TSP-1-CD36 interaction promotes thrombus formation and stabilization under high shear conditions (39). Platelet-originated TSP-1 suppresses the activity of several proteases, amongst others, MMP-2 and−9, plasmin, and cathepsin G. TSP-1-deficient mice models were characterized by improper thrombosis and extended bleeding time (40).
VWF is a multimeric glycoprotein present in platelet α-granules and in Weibel-Palade bodies of endothelial cells (41). Weibel-Palade bodies secrete vWF continuously, but the amount of released vWF can be greatly increased in response to inflammatory stimuli. Since vWF is mainly secreted by endothelial cells, it is a marker for endothelial cell activation rather than platelet activation. After secretion, vWF multimers are cleaved by a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13), which is essential for maintaining normal hemostasis. At sites of vascular damage, collagen-bound vWF binds to the platelet GPIbα-IX-V complex and mediates platelet adhesion, especially under high shear conditions occurring in the arterial system. Furthermore, vWF functions as a carrier protein for coagulation factor VIII in the circulation. There are several reviews discussing the role of vWF in platelet activation and inflammation bringing vWF fame of a risk factor for both arterial and venous thrombosis (42–44).
Activated platelets also produce several eicosanoids, including TxA2, prostaglandin (PG) D2, PGE2, 11-, 12-, and 15-hydroxyeicosatraenoic acid (HETE), through arachidonic acid metabolism by the cyclooxygenase and lipoxygenase pathways (16). TxA2 is synthesized by platelets as well as endothelial cells, macrophages, and neutrophils (45). Via both autocrine and paracrine mechanisms, TxA2 stimulates platelet activation and further aggregation (46, 47). The half-life of TxA2 is about 30 s, therefore it cannot be measured under physiological conditions (46). However, the stable TxA2 metabolite thromboxane B2 (TxB2) has a half-life of 5–7 min and can be assessed by mass spectroscopy, liquid chromatography, and ELISA. A more common approach is to determine the level of TxA2 by measuring the TxB2 urine metabolite 11-dehydrothromboxane B2 (11-DH-TxB2).
PGD2 is mostly released by macrophages, but also to some extent by platelets, and is assumed to be a platelet activation inhibitor. PGE2 is mainly synthesized by endothelial cells and to a lesser extent by platelets. The effect of PGE2 on platelets is concentration-dependent; at low concentrations, it enhances platelet aggregation, while it inhibits aggregation at higher concentrations. 12-HETE is mainly produced by platelets but its effect on platelet activity is not fully investigated (48).
While the platelet secretion markers are rapidly released upon platelet activation, receptor shedding in vitro requires strong platelet stimulation for a prolonged time (6). Shedding results in a soluble shed fragment and a remnant platelet-bound fragment and hence in the loss of receptor-ligand binding function (49). Especially the proteolytic release of GPIbα, GPV, and GPVI has been thoroughly investigated in the last decades.
GPIbα and GPV are part of the GPIb-IX-V complex, expressed exclusively in platelets and megakaryocytes (50) and critical for vWF-dependent platelet adhesion (2). Upon platelet activation, GPIbα shedding is dependent on a disintegrin and metalloproteinase (ADAM)17 activity, whereas GPV can be cleaved by ADAM10/17 and thrombin (51). ADAM17 has a decisive role in GPIbα shedding, determining 90% of the glycocalicin plasma levels, whereas ADAM10 deficiency has no impact on GPIbα shedding. Shear, oxidative stress, serotonin, and GPVI agonists are prominent triggers for ADAM17-mediated GPIbα shedding, resulting in the soluble ectodomain glycocalicin (51, 52). It is hypothesized that glycocalicin is able to trigger hepatic thrombopoietin production in vivo (53), however, the exact role of glycocalicin remains an object of further explorations. In the case of GPV, the main regulator of the shedding process is thrombin and it results in complete elimination of GPV from the platelet surface.
GPVI is another platelet lineage-specific molecule and it functions as a receptor for collagen and fibrin among others (54). The ectodomain shedding is differently regulated by ADAM10 and 17, and the intact receptor is released as a soluble fragment (55). Physiological agonists leading to GPVI shedding are collagen, fibrin, shear stress, antiplatelet autoantibodies, and factor Xa (52). The time frame of GPVI release is dependent on the potency of agonists and, for example, convulxin results in experiments in faster shedding than collagen. The platelet specificity of these receptors makes GPIbα, GPV, and GPVI attractive candidates for identifying platelet activation in vivo.
Contrary to GPVI and the GPIb-IX-V complex, ADAM10/17 are not involved in the shedding of the C-type lectin-like receptor 2 (CLEC-2) upon platelet activation (56). CLEC-2 is abundantly expressed in platelets and megakaryocytes and not in other blood cells (57), albeit a small amount of CLEC-2 is present in liver Kupffer cells (58). Soluble CLEC-2 (sCLEC-2) is shed as a small fragment or could be released bound to other platelet microparticles (56, 59), whereas sGPVI is always shed as a separate fragment. To date, only podoplanin has been recognized as a physiological ligand for CLEC-2 (57). Mouse studies indicate that CLEC-2 has only a minor role as an adhesion receptor in hemostasis, although CLEC-2 maintains vascular integrity at sites of inflammation in the skin. There is accumulating evidence that the CLEC-2-podoplanin interaction plays an important role in thromboinflammation due to the upregulation of podoplanin on tissue-resident macrophages and stromal cells (57). The exact role of CLEC-2 in arterial thrombosis is not completely clear. However, in a mouse model of deep vein thrombosis, comprising inflammatory events, CLEC-2 deficient mice or mice treated with an anti-podoplanin antibody demonstrated substantially decreased thrombus formation (60). In addition, podoplanin can be highly expressed on tumor cells and the platelet CLEC-2/podoplanin axis was shown to promote tumor progression, metastasis, and cancer-induced thrombosis (61).
P-selectin, also known as CD62P, GMP-140, PADGEM (platelet activation-dependent granule external membrane protein), is a transmembrane single-chain glycoprotein (62) and the largest among the selectin family (63). Platelet P-selectin is embedded on the membrane of α-granules and also stored in Weibel-Palade bodies of vascular endothelial cells. Upon platelet activation, the membrane of α-granules merge with the platelet membrane via exocytosis, leading to P-selectin translocation to the platelet surface where it is rapidly cleaved off or slowly internalized, resulting in the release of soluble P-selectin (sP-selectin), whereas endothelial surface P-selectin is internalized within 30 min (21, 32, 64). The platelet surface P-selectin is usually referred to as CD62P and can be measured by flow cytometry in contrast to plasma released sP-selectin. The shedding mechanism remains unknown (21). It was shown that binding of platelet P-selectin to P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes leading to leukocyte rolling (65) and endothelial cells is required for P-selectin shedding, but the protease responsible for this is not discovered yet (66). Platelet-leukocyte aggregates (PLA) can be detected in blood and recognized as one of the most reliable markers for platelet activation (67). There is evidence from mice studies that rather dimeric than monomeric sP-selectin contributes to activation of leukocytes, thereby promoting vascular leukocyte recruitment and microvesicle formation (68).
Several studies acknowledged that the plasma level of sP-selectin originates predominantly from platelets, even though it may also be an indicator of endothelial cell activation, hence plasma levels of sP-selectin have been recognized as a biomarker of activated and degranulated platelets (64, 69, 70). This was also supported by the positive correlation between the level of sP-selectin and platelet count. SP-selectin activates leukocytes and promotes their adhesion to platelets (68).
Similar to P-selectin, CD40L (CD154 or GP39) is another externalized surface protein, which has potent pro-inflammatory properties (71) and belongs to the cytokine tumor necrosis factor (TNF) family (72). CD40L is detected on the surface of various cells including hematopoietic cells, like platelets, basophils, monocytes, macrophages, and non-hematopoietic cells such as mast, endothelial, and smooth muscle cells (72), suggesting a broad range of CD40L functions in vivo (73). Upon platelet activation by collagen or thrombin, CD40L, also located within the α-granule membrane, is mobilized to the platelet surface (21) and is enzymatically cleaved by MMP-2 and MMP-9 within a period of minutes to hours to generate soluble CD40L (sCD40L) (74). Despite the numerous sources of CD40L mentioned above, it was estimated that more than 95% of plasma sCD40L is derived from activated platelets and therefore might reflect the platelet activation status (75). SCD40L increases thrombus stability and promotes the expression of tissue factor, chemokines, and pro-inflammatory biological response modifier molecules (76).
The shedding of receptor ectodomains represents an efficient mechanism for the irreversible downregulation of receptor expression on the platelet surface, resulting in decreased ligand binding. This leads to an essential and tight control of platelet responsiveness in primary hemostasis and coagulation but also in inflammatory processes where activated platelets modulate the activation state of leukocytes and vascular cells through direct receptor/glycoprotein-mediated interactions. The physiological functions of released factor from platelets are summarized in Figure 1.
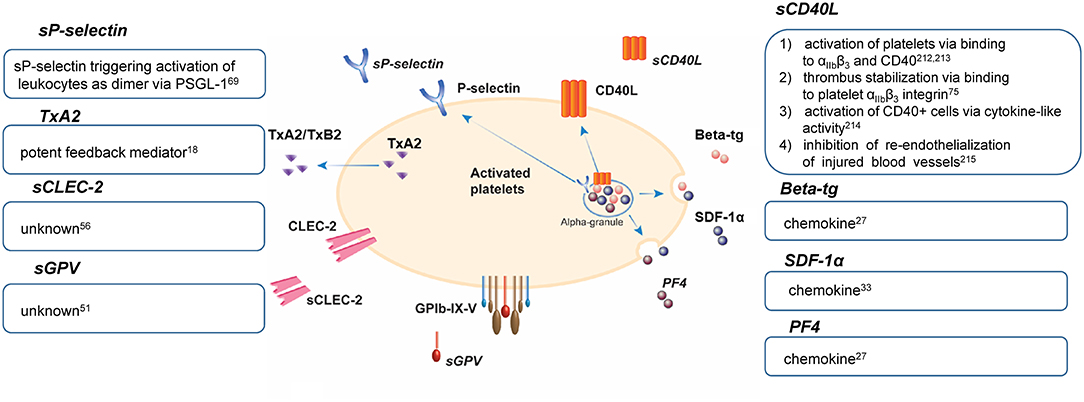
Figure 1. Physiological functions of platelet release factors (77–80). beta-tg, β-thromboglobulin; CD40L, CD40 ligand; CLEC-2, C-type lectin-like receptor 2; GP, glycoprotein; PF4, platelet factor 4; PSGL-1, P-selectin glycoprotein ligand-1; SDF-1α, stromal cell-derived factor-1α; TxA2, thromboxane A2; TxB2, thromboxane B2; s, soluble.
Atherosclerosis is a systemic chronic disease resulting from lipid accumulation in the intima of arteries and chronic inflammation accompanied by platelet activation (81, 82). Coronary artery disease (CAD), defined by the presence of significant atherosclerosis within one or more major coronary arteries, is prone to trigger atherothrombosis on ruptured or eroded atherosclerotic plaques (83), a process in which platelets play a dominant role.
Several studies reported increased levels of platelet biomarkers in patients with CAD, demonstrated mainly by elevated levels of sP-selectin (84–87), sCD40L (86, 88), and sGPV (84, 86) compared to healthy individuals or non-CAD patients (Table 1). Lindmark et al. (85) reported elevated levels of platelet-monocyte (PMA) and platelet-neutrophil aggregates (PNA) measured by flow cytometry. The PMA and PNA levels were significantly higher in patients with unstable CAD vs. stable CAD, who in turn were characterized by slightly but not significantly higher levels compared to controls. Details of these studies are presented in Supplementary Table 1. The levels of sP-selectin were shown to be comparable between South Asian and white European CAD patients on antiplatelet drugs (89).
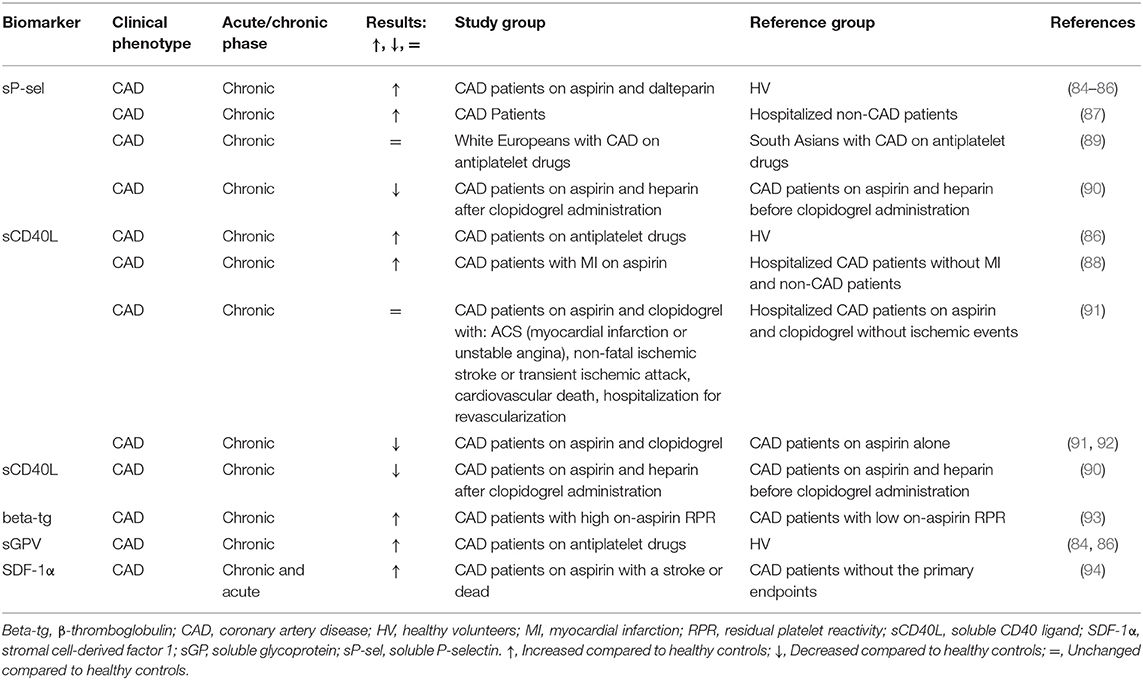
Table 1. Soluble biomarkers of platelet activation in coronary artery disease (CAD) patients.
Various research groups have suggested that the administration of antiplatelet drugs leads to a decline in the level of platelet activation biomarkers. The level of sP-selectin was lower in patients with stable CAD receiving aspirin compared to aspirin-naïve patients (90), while 5-day aspirin administration did not influence the level of sP-selectin in another study (84). The association between on-aspirin platelet reactivity and the level of beta-tg was studied by Pettersen et al. (93) who demonstrated that CAD patients with high residual platelet reactivity (RPR) had a higher level of beta-tg. At the same time, these patients were not characterized by hypercoagulability based on thrombin generation, and hence the authors speculated that the high on-aspirin RPR would rather depend on increased endothelial cell and platelet activation. However, no clinical outcomes were investigated for further exploration.
There is evidence that clopidogrel administration to aspirin-treated patients with CAD significantly reduced the levels of sP-selectin and sCD40L (90–92). In the study of Kaufman et al. (90), the decrease in sP-selectin and sCD40L after administration of a loading dose of clopidogrel did not correlate with platelet reactivity, indicating that the decline in soluble protein levels was likely due to initially elevated levels as a consequence of the percutaneous coronary intervention (PCI) procedure. The level of sP-selectin correlated moderately with sCD40L levels and platelet aggregation in response to arachidonic acid, ADP, and collagen, revealing a link between platelet activity and platelet aggregability (90).
The elevation of sCD40L is particularly evident in patients with recent MI who had higher levels of sCD40L than patients with non-MI CAD, or no CAD-patients (88). Higher sCD40L was accompanied by increased platelet activation as evidenced by increased PMA, PNA, and platelet-surface activated αIIbβ3, determined by flow cytometry in whole blood. The extent of platelet activation was related to CAD stability, with the highest platelet activation in recent-MI patients. However, platelet CD62P did not differ between the groups. SCD40L was associated with female gender, hematocrit, and C-reactive protein and inversely associated with hypertension (91). However, no associations between sCD40L and clinical outcomes were noted in this study.
SGPV levels have been found increased in CAD patients (84, 86), one study appraised sGPV as a relevant biomarker for atherosclerotic patients (84). The second study indicated that platelet activation probably better correlates with intima-media thickness than with angiographic severity of CAD or may reflect thrombogenic abnormalities (86).
Ghasemzadeh et al. (94) reported in a study population of 599 patients that higher plasma SDF-1α level was associated with a nearly 5- and 6-fold increase in the risk of MI and cardiovascular death, respectively, providing a potentially powerful prognostic tool for patients with CAD.
Peripheral artery disease (PAD) is a severe systemic manifestation of atherosclerosis that typically becomes symptomatic in the legs (claudication) but also carries a high risk for MI and ischemic stroke. Similar to symptomatic CAD, patients with confirmed PAD who are treated with antiplatelet and possible anticoagulant drugs, are characterized by increased levels of sP-selectin (95–99), sCD40L (98), and sGPV (84) compared to healthy volunteers (Table 2, Supplementary Table 1).
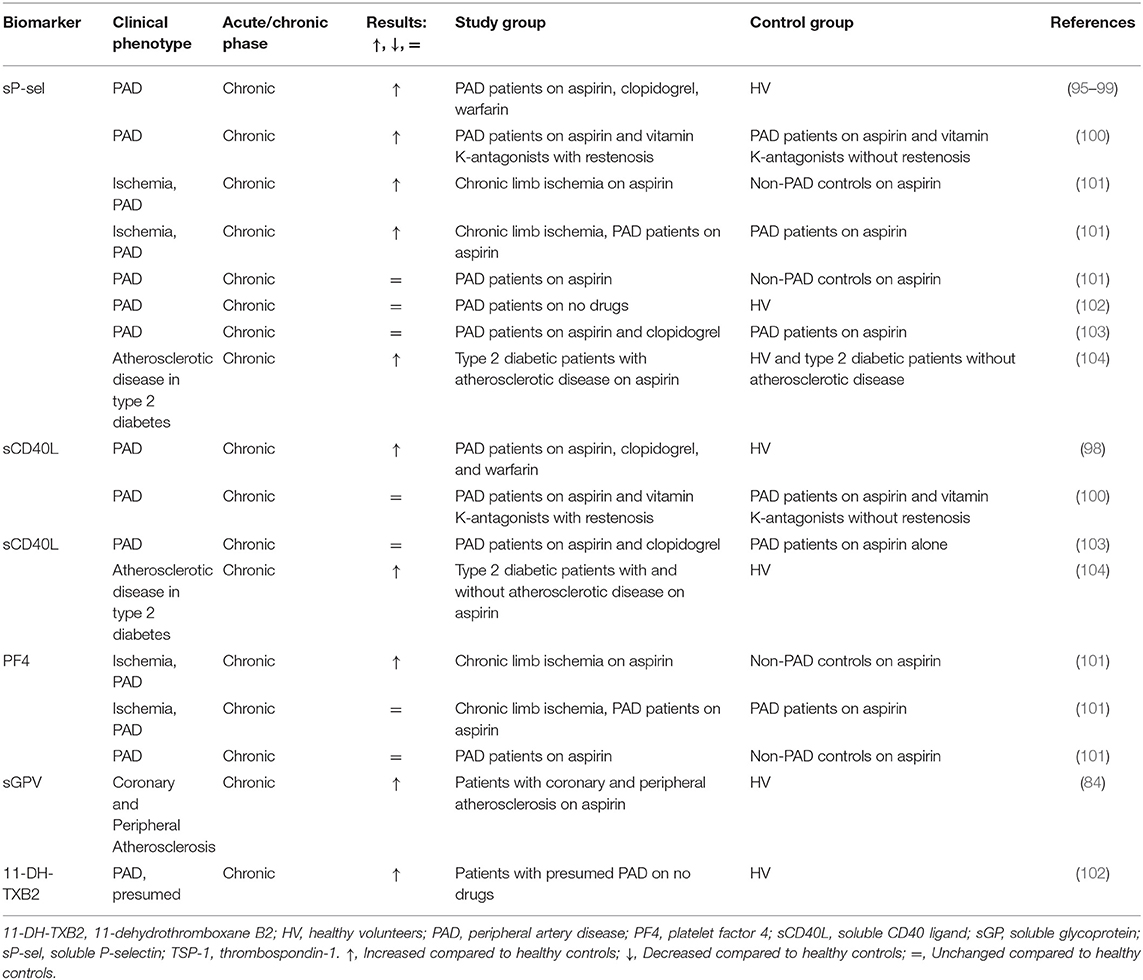
Table 2. Soluble biomarkers of platelet activation in peripheral artery disease (PAD) and atherosclerotic patients.
The level of sP-selectin was also higher compared to healthy volunteers in a study that included patients with presumed PAD, as well as those in whom the diagnosis was confirmed (102). SP-selectin correlated with the severity of PAD (99). This is also confirmed by Zamzam et al. (101), who demonstrated sP-selectin and PF4 were significantly higher in a group with chronic limb-threatening ischemia compared with non-PAD controls but did not differ between PAD and non-PAD groups. The levels of sP-selectin were significantly higher in type 2 diabetic patients with the atherosclerotic disease compared to patients with type 2 diabetes only or healthy subjects, while sCD40L levels were significantly elevated in diabetes patients compared to control subjects, with no difference between two diabetic subgroups (104).
Platelet surface CD62P and CD63 (a dense granule and lysosome membrane glycoprotein), as well as sP-selectin, were higher in the patients compared to the control group (98). CD62P well (r = 0.525) and mildly (r = 0.314) but significantly correlated with CD63 and sP-selectin, respectively. Tsakiris found no correlation between CD62P and sP-selectin (100). SCD40L failed to correlate with any of platelet activation markers both in the study of Blann et al. (98) and Tan et al. (104).
Tsakiris et al. (100) investigated the association between sP-selectin and sCD40L levels in relation to the development of restenosis within 6 months after peripheral angioplasty in patients with PAD. While sP-selectin was associated with outcome (restenosis), no such association was found for sCD40L. SCD40L was suggested to be more linked to endothelial activation due to its correlation with other endothelial activation markers (100). Eikelboom et al. (103) addressed the additive effect of clopidogrel-mediated platelet inhibition on top of aspirin treatment, showing inhibition of ADP- and collagen-induced platelet aggregation, but no reduction in sP-selectin and sCD40L.
Since sCD40L did not correlate with any other markers of platelet activation (sP-selectin, CD62P, CD63) (98, 104), sP-selectin may be a more reliable biomarker for atherosclerotic patient stratification than sCD40L. Burdess et al. (105) criticized the use of sP-selectin and sCD40L measured by ELISA due to the lack of consistency of measured levels in the same group of patients within 1 day and between days and found poor and no correlation with flow cytometry results confirming results of Tsakiris et al. (100), Blann et al. (98), and Tan et al. (104).
Platelet biomarkers in relation to atherosclerotic risk factors were addressed in a population-based study with nearly 3,000 participants; no significant associations were found between sCD40L level and the risk factors (106). The authors also claimed that sCD40L is not a useful tool to screen for subclinical atherosclerosis. Another study included more than 300 patients with atherosclerosis after PCI with endovascular stent implantation (107) and found a strong correlation of PF4, TSP-1, and sCD40L with each other as well as with peak thrombin generation and endogenous thrombin potential, while sP-selectin only correlated weakly with TSP-1. This was explained by assuming that PF4, TSP-1, and sCD40L are mostly of platelet origin, whereas sP-selectin is primarily released by endothelial cells in patients with advanced atherosclerosis.
SGPV was increased in both PAD and CAD patients compared to healthy subjects but was insensitive to 5 days of aspirin treatment (84). Since we found only two studies that measured sGPV in PAD and CAD patients (84, 86), current data on this biomarker are still limited.
Acute coronary syndrome (ACS) describes the predominant situation of symptomatic CAD due to ischemia of the heart, oftentimes in response to atherothrombotic occlusion (108). CAD is the most common cause of arterial thrombosis in ACS and MI. Platelets are the main culprits in the development of ACS and subsequent cardiovascular events (109).
While many studies, as discussed above, show evidence of increased platelet activity in CAD, studies in ACS focus more on the dynamics of platelet release markers in response to pharmacological intervention. Studies investigating the effect of αIIbβ3 antagonists on the release of platelet biomarkers showed reduced sCD40L release from activated platelets (110, 111) (Table 3, Supplementary Table 1). In the study by Ray et al. (111), sCD40L was associated with coronary thrombosis and three different treatments were compared: bivalirudin alone, bivalirudin with αIIbβ3 inhibitors, and unfractionated heparin (UFH) with αIIbβ3 inhibitors. Bivalirudin, the direct thrombin inhibitor, in large-scale randomized trials has been demonstrated to reduce bleeding and thrombocytopenia compared to heparin plus αIIbβ3 inhibitors, while ischemia rates in patients after PCI were similar (112). Ray et al. (111) described levels of sCD40L to be significantly lower in the UFH group compared with the other two groups, indicating that UFH combined with αIIbβ3 inhibitors reduced sCD40L release more strongly than bivalirudin with or without αIIbβ3 inhibitors.
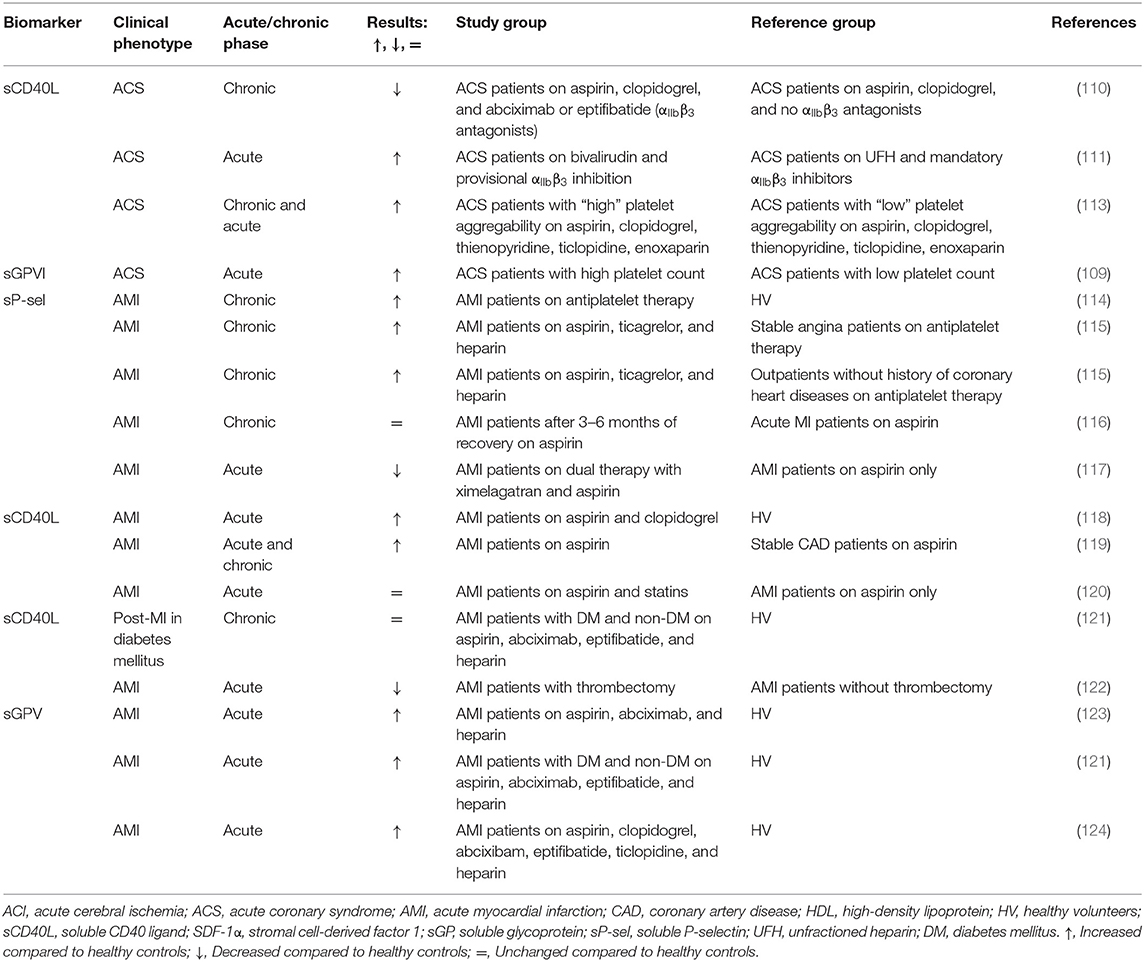
Table 3. Soluble biomarkers of platelet activation in acute coronary syndrome (ACS) and acute myocardial infarction (AMI) patients.
The second study described the reducing effect of treatment with two different αIIbβ3 antagonists (eptifibatide or abciximab) on plasma sCD40L levels after PCI compared to pre-PCI (110). This reduction was not observed in patients without αIIbβ3 antagonists (control); baseline levels were comparable between the different treatment groups. For control patients not treated with clopidogrel before the PCI, clopidogrel administration at the end of the procedure reduced plasma sCD40L significantly 18–24 h after PCI. PMA followed a similar pattern, however, the correlation between the two markers was not assessed.
In another study, ADP-induced platelet aggregation was measured in ACS patients and the patients were subsequently divided into the “high aggregation” (above median) or “low aggregation” (below median) group (113). Elevated sCD40L and sP-selectin levels were found in ACS patients with relatively high platelet aggregability in response to ADP. The authors speculated that CD40L-related enhancement of inflammation and coagulation theoretically might increase the risk of restenosis and in-stent thrombosis in CAD patients and, as a prove, referred to several studies that found associations between restenosis and the CD40 system (125–127).
The study in ACS patients addressing sGPVI demonstrated an inverse correlation between plasma sGPVI levels and platelet count; comparable results were found for platelet surface-expressed GPVI levels. This suggests that patients with lower platelet counts have a higher platelet activation state and, in line with this, these patients were prone to have poorer clinical outcomes (composite of MI, stroke, cardiovascular death) (109). So far, only one study addressed sGPVI levels in relation to platelet count in ACS patients (109). Other correlations with the severity of the disease or clinical outcomes were not described.
In patients with acute MI (AMI) there is evidence for elevated plasma levels of sP-selectin (128), sCD40L (118), and sGPV (121, 123, 124) compared to healthy individuals. A constant elevation of sP-selectin in patients with AMI over the period of 3–6 months was presented by Christersson et al. (117) and Järemo et al. (116). Patients with AMI had significantly higher sP-selectin levels compared with stable angina patients and outpatients without a history of coronary heart disease (115). In the study of Christersson et al. (117), aspirin-treated patients randomized to higher doses of a direct thrombin inhibitor showed lower sP-selectin levels than patients on lower doses or placebo. Another study reported no difference in the sP-selectin level in AMI patients at 3–6 h after infarction compared to 1 day after infarction (116). In the study of Huisse et al. (123), elevated plasma levels of sGPV were found in combination with increased flow cytometry assessed CD62P and activated αIIbβ3 presentation ex vivo in AMI patients compared to healthy volunteers, confirming platelet activation by different tests.
Following up on the topic of drug-related effects on platelet release factors, the addition of statins to conventional aspirin therapy was not beneficial in AMI patients (120). Several articles addressed sCD40L in AMI patients in comparison to other thrombotic diseases. An elevated sCD40L level was observed in patients with AMI compared to age/sex-matched controls with stable CAD (119). In another study, sCD40L levels were higher in patients with MI and diabetes than in subjects with MI alone or those with diabetes alone. However, the difference was not statistically significant which might be explained by the low number of study subjects in each group (121). Interestingly, the levels of sCD40L distinguished not only patients with thrombus formation vs. control subjects but also patients with high-burden thrombus formation in the infarct-related artery vs. low-burden thrombus formation (118).
Together with sP-selectin and sCD40L, sGPV was also elevated in AMI and ACS patients (121, 124) and was recognized as a more sensitive marker of thrombus-induced platelet activation than platelet-derived microparticles (123). Similar to CAD, SDF-1α was also studied in AMI patients, showing that increased SDF-1α levels were associated with the risk factors older age, lower levels of high-density lipoprotein (HDL) cholesterol, and smoking. After adjustment for these factors, SDF-1α correlated with incident heart failure and all-cause mortality (129).
Ischemic stroke, not based on cardiac embolism, is predominantly a consequence of atherothrombosis in the carotid and other cranial arteries (130). Platelet activation as measured by release markers during an acute ischemic stroke (AIS) has been demonstrated in many studies, showing elevated plasma levels of sP-selectin (131–138), sCD40L (134), sGPVI (139), and sCLEC2 (140, 141) in comparison to healthy volunteers (Table 4, Supplementary Table 1).
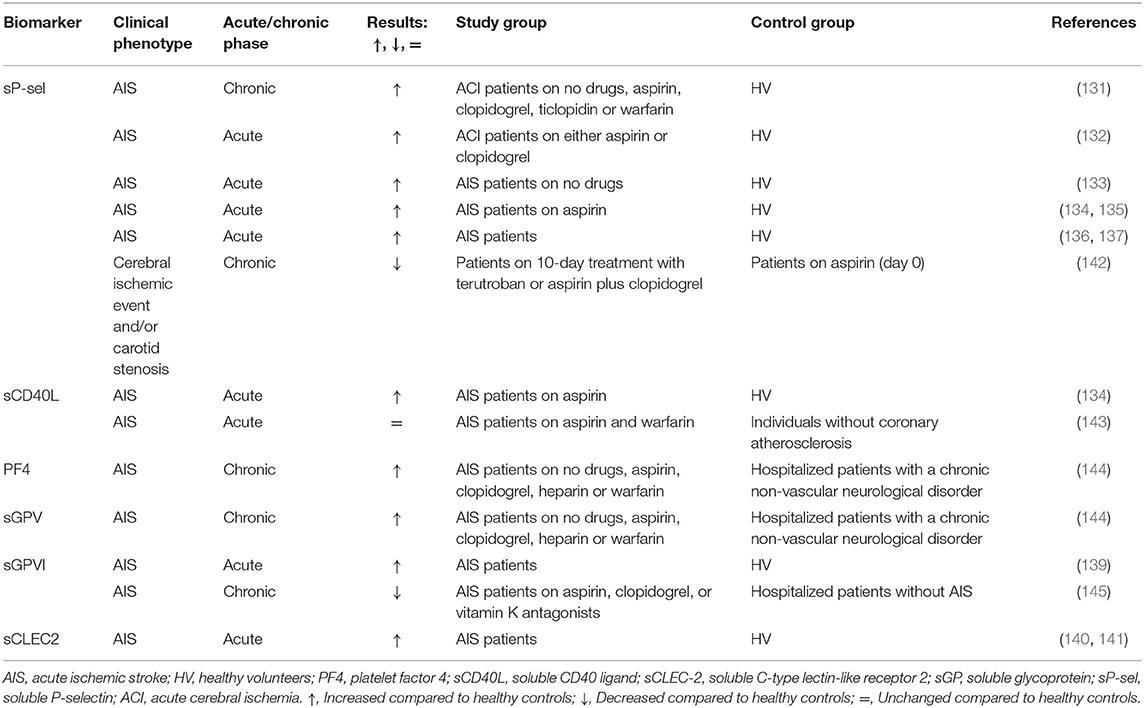
Table 4. Soluble biomarkers of platelet activation in acute ischemic stroke (AIS) patients.
In several studies, sP-selectin again was an indicator of platelet activation and increased levels were found in patients with AIS, independent of treatment with antithrombotics (133–137). This increase was also reported for CD62P (134). In a randomized study with patients at high risk of recurrent IS, treatment groups with different antiplatelet therapies (APT) were compared. A significant reduction in sP-selectin was demonstrated 10 days after treatment with terutroban or clopidogrel plus aspirin, while a decreasing trend was reported after treatment with aspirin or terutroban plus aspirin (142). Spontaneous and arachidonic acid-induced platelet aggregation was either low or decreased both at baseline (day 0) and day 10. The impaired aggregation response is an expected observation since patients were on aspirin during the run-in period. In that case, sP-selectin might be recognized as a more sensitive marker for assessing the antiplatelet drug effect. Both aspirin and clopidogrel lowered the sP-selectin level in patients with acute cerebral infarction (132). SP-selectin in this study positively correlated with flow cytometry detected PMA (r = 0.454, P < 0.05). Additionally, the prognostic value of sP-selectin levels was underlined by its strong correlation with the onset time of progressive IS (137).
In one study, plasma levels of sCD40L and platelet CD62P were found to be similar in AIS patients compared to controls (143). However, platelet surface CD40L expression and PMA levels were higher in patients compared to controls as assessed with flow cytometry. The control group included individuals without coronary atherosclerosis but with similar treatment and risk factors for cardiovascular diseases, which might explain the lack of difference in the level of sCD40L. The lack of significance might also be due to the small sample size of 41 patients vs. 10 controls.
SGPV was elevated in patients with AIS compared to control patients without vascular complications and antithrombotic treatment; this sGPV increase was not influenced by antithrombotic treatment (144). Multivariate analysis demonstrated a correlation between sGPV and stroke, platelet, and leukocyte counts, but not with cardiovascular risk factors. Interestingly, sGPV positively correlated with the PF4 level.
There are two studies where sGPVI was measured; elevated sGPVI levels were found in IS patients compared to healthy volunteers (139), while reduced sGPVI levels were seen in comparison to patients with non-ischemic events (145). In the latter study, the control group consisted of patients with other cerebral disorders, which might distort the interpretation of the sGPVI level in IS patients. Additionally, Wurster et al. (145) evaluated GPVI levels in chronic IS patients whereas Al-Tamimi et al. (139) investigated acute phase patients. Interestingly, Wurster et al. (145) did report increased levels of platelet-surface GPVI in IS patients. Inconsistency between soluble and platelet-surface expressed GPVI levels might be explained by the method used to measure sGPVI, since a newly developed ELISA assay was applied.
Two articles originating from the same cohort of AIS patients consisting of 323 individuals with a follow-up of 1 year showed that sCLEC-2 might be used as a predictor for AIS; the elevated level of the biomarker was significantly correlated with stroke progression and death. Patients with the highest sCLEC-2 level had an 8-fold higher risk of progressive stroke or death compared to the patients in the lowest quartile (140, 141).
Although atrial fibrillation (AF) is currently considered a condition that in the vast majority of cases requires oral anticoagulation to prevent thromboembolic stroke, research from past decades also considered the role of platelets in this setting (146–148). For this reason, there is quite some literature on the involvement of activated platelets in AF-related hypercoagulability. Many studies reported elevated levels of sP-selectin (149–156) and sCD40L (154, 157, 158) in patients with AF compared to healthy subjects. In addition, increased concentrations of plasma beta-tg (84, 151, 159, 160) and sGPV (160) were documented (Table 5, Supplementary Table 1). Choudhury et al. (153) additionally measured platelet surface CD62P and CD63 by flow cytometry in whole blood. Both markers were elevated in AF patients as well as sP-selectin compared to healthy people. However, CD62P strongly correlated with CD63 (r = 0.6; p < 0.001), but not with sP-selectin.
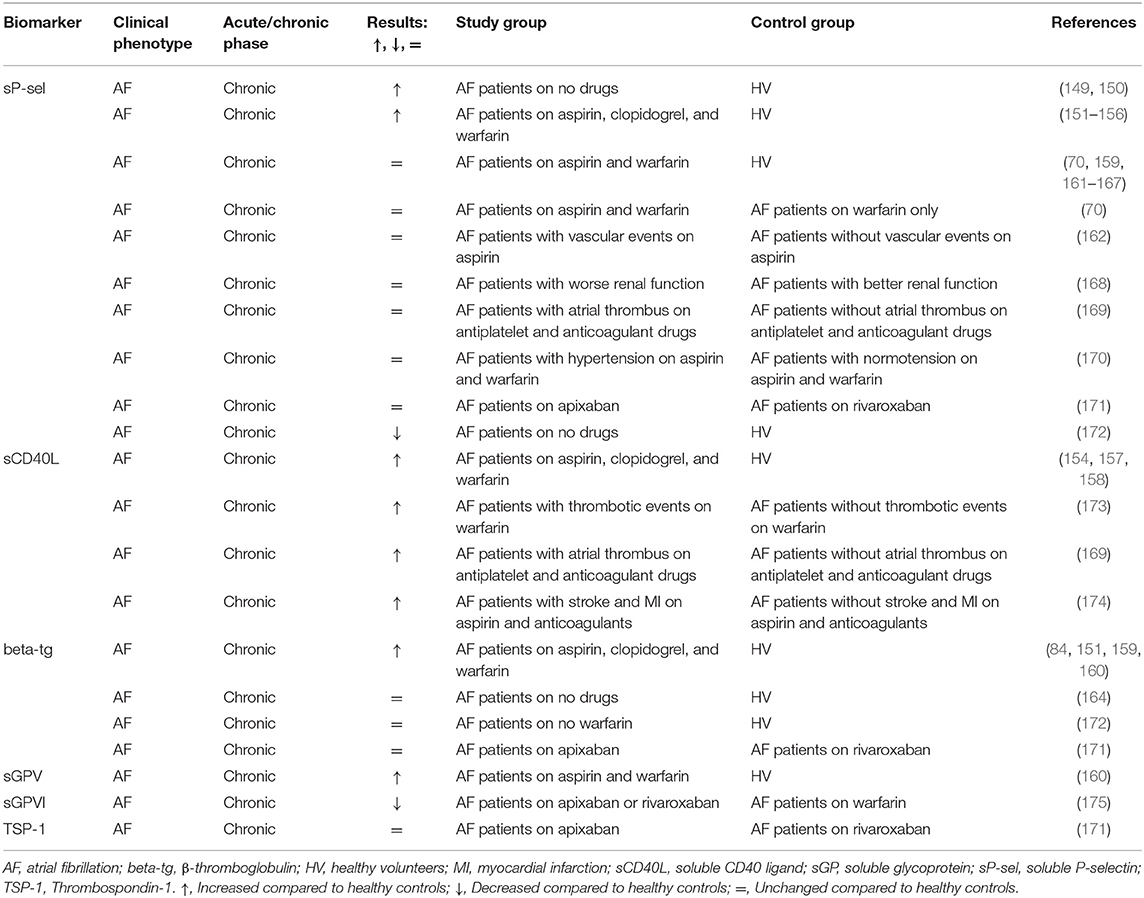
Table 5. Soluble biomarkers of platelet activation in atrial fibrillation (AF) patients.
However, some studies found no difference or even a decrease in biomarker levels when comparing patients with AF and controls. This was observed most strikingly for sP-selectin. Yet the majority of articles demonstrating no difference between the groups adjusted the association between sP-selectin levels and AF severity or prognosis for confounding factors. For example, sP-selectin in AF patients correlated with diabetes but not with other recognized AF risk factors such as increasing age, recent heart failure, and prior cerebral ischemia (70). The absence of an association between sP-selectin levels and AF or cardiovascular risk was again claimed by this group a year later (161, 162). In one of their studies, Conway et al. (70) pointed out that the lack or absence of adequate adjustment for cardiovascular diseases may falsely link changes in sP-selectin levels to AF.
The level of sP-selectin was found to be unrelated to clinical outcomes (IS, MI, or vascular death) (162) and left atrial thrombus formation (169) in AF patients. Similar sP-selectin levels were reported when comparing different treatment groups; patients on warfarin plus aspirin vs. warfarin alone (70) or patients on rivaroxaban vs. apixaban (171). In the study by Steppich et al., rivaroxaban and apixaban did not influence levels of beta-tg and TSP-1. However, these direct oral anticoagulants were found to be more effective than warfarin in suppressing sGPVI measurements (175).
SCD40L was elevated in AF patients with embolic events, atrial thrombus formation (169, 173), stroke, and MI (174) compared to AF patients without these conditions. Other studies provide evidence that sCD40L is inversely related to stroke risk (176). In one of the largest studies including 880 subjects, Lip et al. reported that patients at the highest risk of stroke as determined by increased age and blood pressure, impaired left ventricular function, and previous thromboembolism, had lower levels of sCD40L than people without any of these factors. SCD40L, in contrast to sP-selectin and beta-tg, was a prognostic biomarker for vascular events in AF patients (173, 174). No correlation was found between sCD40L and sP-selectin in the study of Choudhury et al. (154).
Beta-tg levels were higher in patients with AF and similar to sP-selectin indifferent to aspirin (159, 160), warfarin (160, 172), rivaroxaban, and apixaban (171) administration. No relation was found between platelet aggregation induced by ADP, collagen, epinephrine, and thrombin and the plasma platelet activation markers sP-selectin (151), beta-tg (151, 160), and sGPV (160).
One of the unsolved questions in AF research is whether any of the observed changes in platelet biomarkers reflect the arrhythmia per se, or the comorbidity (152–154). An effect of AF was postulated based on two studies showing that lone AF was associated with elevated sP-selectin compared to age-matched controls. Lone AF patients had also enhanced sGPV levels (160) further supporting a role of platelet activation in AF since sGPV comes exclusively from platelets.
Despite the fact that venous thrombosis is traditionally not regarded a condition that is dependent on platelet activation, clinical studies have clearly shown a protective effect of low-dose aspirin on recurrent venous thromboembolism (VTE). This effect is most likely explained by the inhibition of platelets as the low dose of aspirin does not have any demonstrable anti-inflammatory effects in humans (185). Although the effect of oral anticoagulation is clinically more relevant than APT to prevent recurrent VTE, the involvement of platelets in venous thrombosis remains of interest, particularly for settings in which the addition of APT may be considered, like in acute VTE or periprocedural, in case of venous stenting.
One of the most thorough explorations on platelet biomarkers in VTE was done by Riedl et al. (23), who studied several biomarkers and their mutual associations. The researchers compared sP-selectin, sCD40L, PF4, and TSP-1 among three groups: cancer patients with VTE, cancer patients without VTE, and healthy subjects. Interestingly, only sP-selectin was elevated among all biomarkers in cancer patients with VTE, compared to the other groups which were not different from each other, indicating that VTE rather than cancer was responsible for the sP-selectin increase (Table 6, Supplementary Table 1). Although TSP-1 was increased in both cancer groups compared to healthy volunteers, it was not affected by the presence of VTE. The authors concluded that sCD40L, PF4, and TSP-1 cannot predict VTE development, while sP-selectin, on the contrary, could have predictive potential (23). SCD40L, PF4, and TSP-1 mutually correlated with each other and weakly with sP-selectin which is released not only by platelets but also by endothelial cells. Based on this, Riedl et al. (23) suggested that VTE is more associated with endothelial rather than platelet activation.
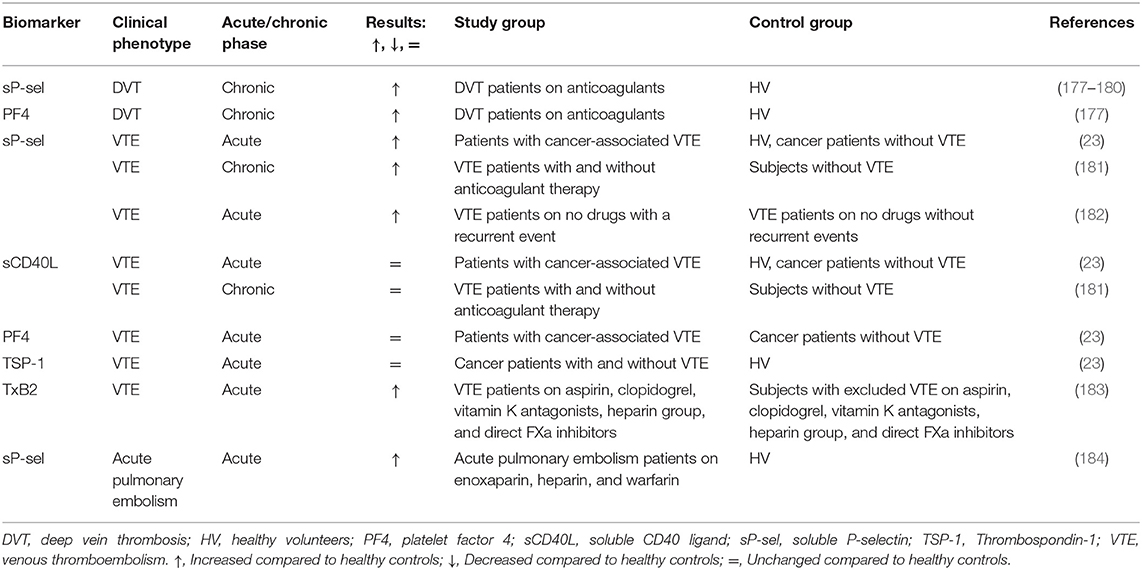
Table 6. Soluble biomarkers of platelet activation in patients with venous thromboembolism (VTE).
This conclusion is also supported by Migliacci et al. (181) who measured the level of sP-selectin and sCD40L in VTE patients and compared them to controls. However, it is important to mention that the control group included subjects with AF and valve prosthesis together with healthy volunteers. In accordance with the findings of Riedl et al., the level of sP-selectin was significantly higher in the patient group in contrast to sCD40L, which was similar between the patients and controls. In this study, also plasma vWF level was measured, which was higher in patients and correlated weakly but significantly with sP-selectin. The fact that vWF reflects endothelial activation and is stored together with sP-selectin in endothelial cells (186), supports the suggestion that endothelial activation is more pronounced in VTE patients compared to platelet activation and is responsible for the elevation of sP-selectin level in plasma.
In contrast to the above-mentioned study (23), Furio et al. (177) observed a significant increase in PF4 levels in deep vein thrombosis (DVT) patients compared to healthy volunteers. However, it should be considered that Riedl et al. (23) studied cancer patients with VTE in the acute phase, while Furio et al. (177) included patients with DVT in a chronic phase that, in addition to possible effects of anticancer treatment, may explain differences in results for this biomarker.
TxB2 was significantly elevated in patients with confirmed VTE diagnosis, independent of aspirin intake, in contrast to patients with excluded VTE (183). Non-aspirin VTE cases presented significantly shorter closure times with collagen/ADP and collagen/epinephrine in the platelet function analyzer compared to controls. Within the group of non-aspirin users, platelet aggregability in response to ADP or collagen was lower in VTE-cases compared to patients with excluded VTE. Patients with VTE showed higher platelet CD63 surface presentation ex vivo and lower platelet-dependent thrombin generation triggered by tissue factor, independent of therapy.
Other studies mostly concentrated on sP-selectin unanimously observing elevated levels of this biomarker in patients compared to healthy subjects, regardless of the location of venous thrombosis. SP-selectin was higher in patients with DVT (177–180) and acute pulmonary embolism (184). In line with elevated sP-selectin plasma levels in patients with DVT, Furio et al. (177) observed increased platelet CD62P presentation. In contrast, Chung et al. (184) reported unaltered platelet CD62P expression and PLA, but increased activated integrin αIIbβ3 in patients with acute pulmonary embolism compared to controls. In the study of Kyrle et al. (182), sP-selectin appeared to be predictive for VTE recurrence, i.e., individuals with VTE and higher sP-selectin levels were more likely to have a second VTE event.
However, some studies questioned if sP-selectin reflects platelet and not endothelial activation since the levels of sP-selectin did not correlate with sCD40L (23, 181) and TSP-1 levels (23). Another study provided evidence that the elevated sP-selectin level and enhanced urinary 11-DH-TxB2 excretion in DVT patients was due to increased platelet activation (179). Therefore, we conclude that the question about the presence of platelet activation in DVT patients remains open. However, sP-selectin might be used as a prognostic tool for the recurrent VTE or incidence of VTE in cancer patients.
Platelets are important contributors to the development of arterial thrombotic events. They are involved in atherosclerotic plaque formation and plaque rupture can lead to ischemia or infarction (187). Platelets are involved in thrombosis not only as the first violins of the blood coagulation process, but also as promoters of inflammation (188). The identification of changes in the level of plasma biomarkers associated with upcoming thromboembolic events could allow timely adjustment of the treatment strategy in order to prevent the disease aggravation. Therefore, biomarkers of platelet activation may become a valuable instrument for the prognosis of acute events.
An ideal biomarker should be specific, accurate, reproducible by a simple technique, independent from pre-analytical artifacts, cost-effective, and acceptable to patients (26, 32, 188). In contrast to plaque material, platelets are accessible through routine venipuncture and are easily counted within a minute by a standard cell counter. This allows serial sampling and long-term monitoring.
However, despite numerous clinical studies evaluating platelet biomarkers, data remain inconclusive. Several biomarkers were suggested but none of these can be recognized as a robust diagnostic marker. Interindividual variability and inconsistencies in cutoff values impede the implementation of biomarkers of platelet activation in the wide clinical practice. Besides this, the ex vivo manipulations like the blood drawing procedure and centrifugation can pre-activate platelets and distort real numbers of biomarkers level (189–192). The measurements can be also influenced by the type of anticoagulation, storage, and thawing procedures (32).
A common problem in the summarized clinical studies may be limited power due to small sample sizes and confounding due to incomplete or absent adjustment for risk factors for thrombosis, as identified as a problem in assessing sP-selectin levels in AF. There are many molecules expressed by platelets. Some of them are exclusive for platelets such as GPIbα, GPV, and GPVI, whereas others are also synthesized by other cells, e.g., P-selectin, CD40L, and SDF-1α. Our review demonstrates that the limelight of clinicians' attention was obviously mainly focused on sP-selectin and sCD40L. Currently, sP-selectin is recognized as an important marker of platelet activation and was found to be elevated in a broad range of conditions including various types of cardiovascular diseases (unstable angina, thrombocytopenia, arterial hypertension, stroke, AMI, congestive heart failure) as discussed, but also in other conditions, including autoimmune disorders (Sjogren's syndrome, systemic lupus erythematosus), diabetes, or psychiatric disorders (64, 193–200).
Increased values of sCD40L have been found in cardiovascular diseases including PAD (128), CAD (86), AF (154, 157, 158, 173, 174), AIS (134), and also in patients with diabetes (201). Similarly, beta-tg and PF4 were reported to be altered in other diseases too, including cancer (202–205), ischemic heart disease (206), and AF (207). SGPVI reflected activation of platelets in patients with AIS (139), microangiopathy (208), rheumatoid arthritis (59, 209), and Alzheimer's disease (210). Thus, it can be concluded that the above biomarkers do not specifically reflect thrombosis but probably also reflect diverse other processes some of which may in part be associated with platelet activation. At this stage, there are no data indicating distinct platelet activation profiles related to specific diseases, or predicting diseases (21).
Platelet activation is a well-known contributor to the pathogenesis of arterial thrombosis and leads to increased levels of platelet activation biomarkers. As discussed, some markers are platelet specific, whereas others may be increased due to activation of other cell types including endothelial cells. So far, it remains challenging to distinguish the exact input of platelet activation and vessel wall pathology into the increase of sP-selectin or sCD40L based on the data provided. Similarly, elevations of biomarkers and risk associations may vanish upon adjustment for confounding factors, as mentioned for AF.
Evidence for using platelet biomarkers as a prognostic and stratifying tool in DVT is still scarce. Interestingly, a recent study described platelet-related parameters in patients with confirmed VTE compared to patients with suspected but unconfirmed VTE, independent of the underlying cardiovascular profile (183). Herein VTE patients were characterized with elevated expression of platelet activation markers in combination with lower platelet-dependent thrombin generation in vitro. These findings clearly underscore the role of platelets in VTE. A current overview of platelet released activation biomarkers in arterial and venous cardiovascular diseases determined by ELISA-based tests is presented in Figure 2.
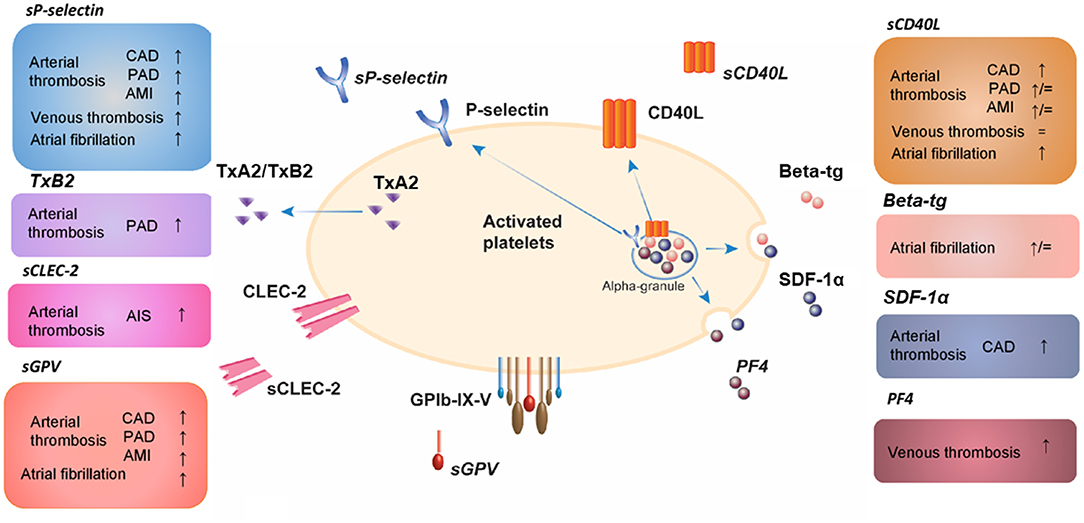
Figure 2. Platelet released activation biomarkers in arterial and venous cardiovascular diseases determined by ELISA-based tests. AIS, acute ischemic stroke; AMI, acute myocardial infarction; beta-tg, β-thromboglobulin; CAD, coronary artery disease; CD40L, CD40 ligand; CLEC-2, C-type lectin-like receptor 2; GP, glycoprotein; PF4, platelet factor 4; SDF-1α, stromal cell-derived factor-1α; TxA2, thromboxane A2; TxB2, thromboxane B2; PAD, peripheral artery disease; s, soluble; ↑, increased compared to healthy controls; ↓, decreased compared to healthy controls; =, unchanged compared to healthy controls.
Several authors recommend implementing a combination of several biomarkers, which allows a more objective assessment of a patient's current state since the pathogenesis of thrombosis is a complex process involving the interplay between inflammation, coagulation, and cellular activation (211–214). It is also worthwhile to link biomarker assays to platelet function tests and platelet surface markers to obtain a more comprehensive understanding of the disease state.
It becomes obvious that there is a need for larger clinical trials to investigate the diagnostic potential of the biomarkers discussed in the thrombosis setting (32). The application of machine learning for the identification of signatures of platelet biomarkers could better reflect the biological complexity and multifactorial processes and overcome the high interindividual variability and limitations due to the scatter of measurement results. The newly available high-throughput protein technologies open up possibilities here that could lead to new insights.
Inclusion of newer, less well-studied plasma markers of platelet activation, such as sGPVI, sGPIbα, SDF-1α, sGPV, and sCLEC2, in clinical studies might be valuable in the search for reliable thrombotic biomarkers. For interpretation and comparison, future studies measuring biomarkers should ideally report detailed information on clinical parameters, pre-analytical and analytical variables. This information should be stratified and analyzed to determine its influence on the association between disease severity and biomarker level.
GB, MN, PM, and HC drafted the manuscript. All authors have seen and approved the final version of the manuscript, participated in the interpretation of the findings, reviewed the manuscript, and revised it critically before submission.
GB was supported by a PhD fellowship from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement No. 813409.
PW has received research funding outside the present study from Boehringer Ingelheim, Sanofi-Aventis, Bayer Healthcare, Daiichi Sankyo Europe, and Novartis, and received outside the present study honoraria for lectures or consulting from Boehringer Ingelheim, Bayer HealthCare, Evonik, AstraZeneca, and Sanofi-Aventis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.684920/full#supplementary-material
11-DH-TxB2, 11-dehydrothromboxane B2; ACS, Acute coronary syndrome; ADAM, A disintegrin and metalloproteinase family of proteinases; ADAMTS, A disintegrin and metalloproteinase with a thrombospondin type 1 motif; ADP, Adenosine diphosphate; AF, Atrial fibrillation; AIS, Acute ischemic stroke; AMI, Acute myocardial infarction; APT, Antiplatelet therapy; ATP, Adenosine triphosphate; Beta-tg, β-thromboglobulin; CAD, Coronary artery disease; CD40L, CD40 ligand; CLEC-2, C-type lectin-like receptor 2; CXCL, CXC ligand; DVT, Deep vein thrombosis; ELISA, Enzyme-linked immunosorbent assay; GP, Glycoprotein; HDL, High-density lipoprotein; HETE, Hydroxyeicosatetraenoic acid; IS, Ischemic stroke; MI, Myocardial infarction; MMP, Metalloproteinase(s); PAD, Peripheral artery disease; PADGEM, Platelet activation-dependent granule external membrane protein; PAI-1, Plasminogen activator inhibitor 1; PCI, Percutaneous coronary intervention; PDGF, Platelet-derived growth factor; PF4, Platelet factor 4; PG, Prostaglandin; PLA, Platelet-leukocyte aggregates; PMA, Platelet-monocyte aggregates; PNA, Platelet-neutrophil aggregates; PSGL-1, P-selectin glycoprotein ligand-1; RANTES, Regulated upon activation, normal T-cell expressed and presumably secreted; RPR, Residual platelet reactivity; sCD40L, Soluble CD40 ligand; sCLEC-2, Soluble C-type lectin-like receptor 2; SDF-1α, Stromal cell-derived factor-1α; sP-selectin, Soluble P-selectin; TFPI, Tissue factor pathway inhibitor; TGF-β, Transforming growth factor β; TNF, Tumor necrosis factor; TSP-1, Thrombospondin-1; TxA2, Thromboxane A2; TxB2, Thromboxane B2; UFH, Unfractionated heparin; VEGF, Vascular endothelial growth factor; VTE, Venous thromboembolism; VWF, Von Willebrand factor.
1. Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. (2007) 357:2482–94. doi: 10.1056/NEJMra071014
2. Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med. (2011) 89:109–21. doi: 10.1007/s00109-010-0691-5
3. Andrews RK, Shen Y, Gardiner EE, Dong JF, López JA, Berndt MC. The glycoprotein Ib-IX-V complex in platelet adhesion and signaling. Thromb Haemost. (1999) 82:357–64. doi: 10.1055/s-0037-1615854
4. Swieringa F, Spronk HMH, Heemskerk JWM, van der Meijden PEJ. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. (2018) 2:450–60. doi: 10.1002/rth2.12107
5. Coppinger JA, Maguire PB. Insights into the platelet releasate. Curr Pharm Des. (2007) 13:2640–6. doi: 10.2174/138161207781662885
6. Baaten CCFMJ, Swieringa F, Misztal T, Mastenbroek TG, Feijge MAH, Bock PE, et al. Platelet heterogeneity in activation-induced glycoprotein shedding: functional effects. Blood Adv. (2018) 2:2320–31. doi: 10.1182/bloodadvances.2017011544
7. Iida Y, Doi T, Tokuda H, Matsushima-Nishiwaki R, Tsujimoto M, Kuroyanagi G, et al. Rho-kinase regulates human platelet activation induced by thromboxane A2 independently of p38 MAP kinase. Prostaglandins Leukot Essent Fat Acids. (2015) 94:73–81. doi: 10.1016/j.plefa.2014.11.006
8. Montoro-García S, Schindewolf M, Stanford S, Larsen OH, Thiele T. The role of platelets in venous thromboembolism. Semin Thromb Hemost. (2016) 42:242–51. doi: 10.1055/s-0035-1570079
9. Chan NC, Weitz JI. Antithrombotic agents. Circ Res. (2019) 124:426–36. doi: 10.1161/CIRCRESAHA.118.313155
10. Kamath S, Blann A, Lip G. Platelet activation: assessment and quantification. Eur Heart J. (2001) 22:1561–71. doi: 10.1053/euhj.2000.2515
11. Pagel O, Walter E, Jurk K, Zahedi RP, Pagel O, Walter E, et al. Taking the stock of granule cargo : platelet releasate proteomics. Platelets. (2017) 28:119–28. doi: 10.1080/09537104.2016.1254762
12. Heijnen H, van der Sluijs P. Platelet secretory behaviour: as diverse as the granules or not? J Thromb Haemost. (2015) 13:2141–51. doi: 10.1111/jth.13147
13. Flaumenhaft R, Sharda A. The life cycle of platelet granules. F1000Research. (2018) 7:1–12. doi: 10.12688/f1000research.13283.1
14. Jurk K, Kehrel BE. Platelets : physiology and biochemistry. Semin Thromb Hemost. (2005) 1:381–92. doi: 10.1055/s-2005-916671
15. Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. (2006) 99:1293–304. doi: 10.1161/01.RES.0000251742.71301.16
16. Crescente M, Menke L, Chan MV, Armstrong PC, Warner TD. Eicosanoids in platelets and the effect of their modulation by aspirin in the cardiovascular system (and beyond). Br J Pharmacol. (2019) 176:988–99. doi: 10.1111/bph.14196
17. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. (2014) 123:2759–67. doi: 10.1182/blood-2013-11-462432
18. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. (2017) 38:785–91. doi: 10.1093/eurheartj/ehw550
19. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. (2018) 122:337–51. doi: 10.1161/CIRCRESAHA.117.310795
20. Jurk K. Platelet granules - secretory and secretive. Hamostaseologie. (2017) 37:208–10. doi: 10.5482/HAMO-16-07-0023
21. Au AE, Josefsson EC. Regulation of platelet membrane protein shedding in health and disease. Platelets. (2017) 28:342–53. doi: 10.1080/09537104.2016.1203401
22. Wu J, Heemskerk JWM, Baaten CCFMJ. Platelet membrane receptor proteolysis : implications for platelet function. Front Cardiovasc Med. (2021) 7:1–13. doi: 10.3389/fcvm.2020.608391
23. Riedl J, Hell L, Kaider A, Koder S, Marosi C, Zielinski C, et al. Association of platelet activation markers with cancer-associated venous thromboembolism. Platelets. (2016) 27:80–5. doi: 10.3109/09537104.2015.1041901
24. Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, Von Andrian UH, et al. Tumor necrosis factor-α-converting enzyme (ADAM17) mediates GPIbα shedding from platelets in vitro and in vivo. Circ Res. (2004) 95:677–83. doi: 10.1161/01.RES.0000143899.73453.11
25. Gardiner EE, Arthur JF, Kahn ML, Berndt MC, Andrews RK. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood. (2004) 104:3611–7. doi: 10.1182/blood-2004-04-1549
26. Gurney D, Lip GYH, Blann AD. A reliable plasma marker of platelet activation: does it exist? Am J Hematol. (2002) 70:139–44. doi: 10.1002/ajh.10097
27. Brandt E, Petersen F, Ludwig A, Ehlert JE, Bock L, Flad HD. The β-thromboglobulins and platelet factor 4: blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J Leukoc Biol. (2000) 67:471–8. doi: 10.1002/jlb.67.4.471
28. Shi G, Field DJ, Long X, Mickelsen D, Ko K, Ture S, et al. Platelet factor 4 mediates vascular smooth muscle cell injury responses. Blood. (2013) 121:4417–27. doi: 10.1182/blood-2012-09-454710
29. van Wyk V, Heyns AD, de Wet JI, Kotzé HF, Lótter MG. A formula for correcting for the in vitro release of platelet beta- thromboglobulin. Thromb Res. (1987) 46:659–68. doi: 10.1016/0049-3848(87)90267-2
30. Dawes J, Smith RC, Pepper DS. The release, distribution, and clearance of human β-thromboglobulin and platelet factor 4. Thromb Res. (1978) 12:851–61. doi: 10.1016/0049-3848(78)90279-7
31. Minar E, Ehringer H. Influence of acetylsalicylic acid (1.0 g/day) on platelet survival time, β-thromboglobulin and platelet factor 4 in patients with peripheral arterial occlusive disease. Thromb Res. (1987) 45:791–802. doi: 10.1016/0049-3848(87)90089-2
32. Ferroni P, Riondino S, Vazzana N, Santoro N, Guadagni F, Davì G. Biomarkers of platelet activation in acute coronary syndromes. Thromb Haemost. (2012) 108:1109–23. doi: 10.1160/TH12-08-0550
33. Liepelt A, Tacke F. Stromal cell-derived factor-1 (SDF-1) as a target in liver diseases. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G203–9. doi: 10.1152/ajpgi.00193.2016
34. Chatterjee M, Gawaz M. Platelet-derived CXCL12 (SDF-1α): basic mechanisms and clinical implications. J Thromb Haemost. (2013) 11:1954–67. doi: 10.1111/jth.12404
35. Walsh TG, Harper MT, Poole AW. SDF-1α is a novel autocrine activator of platelets operating through its receptor CXCR4. Cell Signal. (2015) 27:37–46. doi: 10.1016/j.cellsig.2014.09.021
36. Chatterjee M, Von Ungern-Sternberg SNI, Seizer P, Schlegel F, Büttcher M, Sindhu NA, et al. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. (2015) 6:1–16. doi: 10.1038/cddis.2015.233
37. Binsker U, Kohler TP, Hammerschmidt S. Contribution of human thrombospondin-1 to the pathogenesis of gram-positive bacteria. J Innate Immun. (2019) 11:303–15. doi: 10.1159/000496033
38. Jurk K, Clemetson KJ, de Groot PG, Brodde MF, Steiner M, Savion N, et al. Thrombospondin-1 mediates platelet adhesion at high shear via glycoprotein Ib (GPIb): an alternative/backup mechanism to von Willebrand factor. FASEB J. (2003) 17:1490–2. doi: 10.1096/fj.02-0830fje
39. Kuijpers MJE, De Witt S, Nergiz-Unal R, Van Kruchten R, Korporaal SJA, Verhamme P, et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler Thromb Vasc Biol. (2014) 34:1187–92. doi: 10.1161/ATVBAHA.113.302917
40. Aburima A, Berger M, Spurgeon BEJ, Webb BA, Wraith KS, Febbraio M, et al. Thrombospondin-1 promotes hemostasis through modulation of cAMP signaling in blood platelets. Blood. (2021) 137:678–89. doi: 10.1182/blood.2020005382
41. Ruggeri ZM, Mendolicchio GL. Interaction of von willebrand factor with platelets and the vessel wall. Hamostaseologie. (2015) 35:211–24. doi: 10.5482/HAMO-14-12-0081
42. Calabrò P, Gragnano F, Golia E, Grove EL. Von Willebrand factor and venous thromboembolism: pathogenic link and therapeutic implications. Semin Thromb Hemost. (2018) 44:249–60. doi: 10.1055/s-0037-1605564
43. Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. (2017) 15:1285–94. doi: 10.1111/jth.13696
44. Denorme F, Vanhoorelbeke K, De Meyer SF. von Willebrand factor and platelet glycoprotein Ib: a thromboinflammatory axis in stroke. Front Immunol. (2019) 10:2884. doi: 10.3389/fimmu.2019.02884
45. Rucker D, Dhamoon AS. Physiology, Thromboxane A2. StatPearls Publishing (2019). Available online at: http://www.ncbi.nlm.nih.gov/pubmed/30969639 (accessed August 6, 2020).
46. Helgadóttir H, Ólafsson Í, Andersen K, Gizurarson S. Stability of thromboxane in blood samples. Vasc Health Risk Manag. (2019) 15:143–7. doi: 10.2147/VHRM.S204925
47. Jobe SM. Failure to Release and Aspirin-Like Defects. 3rd ed. New York, NY: Elsevier Inc. (2019).
48. Maskrey BH, Rushworth GF, Law MH, Treweeke AT, Wei J, Leslie SJ, et al. 12-hydroxyeicosatetraenoic acid is associated with variability in aspirin-induced platelet inhibition. J Inflamm. (2014) 11:1–7. doi: 10.1186/s12950-014-0033-4
49. Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang HY, et al. Deciphering the human platelet sheddome. Blood. (2011) 117:15–27. doi: 10.1182/blood-2010-05-283838
50. Li R, Emsley J. The organizing principle of the platelet glycoprotein Ib-IX-V complex. J Thromb Haemost. (2013) 11:605–14. doi: 10.1111/jth.12144
51. Bender M, Stegner D, Nieswandt B. Model systems for platelet receptor shedding. Platelets. (2017) 28:325–32. doi: 10.1080/09537104.2016.1195491
52. Montague SJ, Andrews RK, Gardiner EE. Mechanisms of receptor shedding in platelets. Blood. (2018) 132:2535–45. doi: 10.1182/blood-2018-03-742668
53. Hoffmeister KM. The role of lectins and glycans in platelet clearance. J Thromb Haemost. (2011) 9:35–43. doi: 10.1111/j.1538-7836.2011.04276.x
54. Onselaer MB, Hardy AT, Wilson C, Sanchez X, Babar AK, Miller JLC, et al. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv. (2017) 1:1495–504. doi: 10.1182/bloodadvances.2017007732
55. Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by ADAM family metalloproteinases. J Thromb Haemost. (2007) 5:1530–7. doi: 10.1111/j.1538-7836.2007.02590.x
56. Inoue O, Osada M, Nakamura J, Kazama F, Shirai T, Tsukiji N, et al. Soluble CLEC-2 is generated independently of ADAM10 and is increased in plasma in acute coronary syndrome: comparison with soluble GPVI. Int J Hematol. (2019) 110:285–94. doi: 10.1007/s12185-019-02680-4
57. Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. (2019) 129:12–23. doi: 10.1172/JCI122955
58. Tang T, Li L, Tang J, Li Y, Lin WY, Martin F, et al. A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol. (2010) 28:749–55. doi: 10.1038/nbt.1644
59. Gitz E, Pollitt AY, Gitz-Francois JJ, Alshehri O, Mori J, Montague S, et al. CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood. (2014) 124:2262–70. doi: 10.1182/blood-2014-05-572818
60. Payne H, Ponomaryov T, Watson SP, Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood. (2017) 129:2013–20. doi: 10.1182/blood-2016-09-742999
61. Quintanilla M, Montero LM, Renart J, Villar EM. Podoplanin in inflammation and cancer. Int J Mol Sci. (2019) 20:1–38. doi: 10.3390/ijms20030707
62. Blann AD, Nadar SK, Lip GYH. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. (2003) 24:2166–79. doi: 10.1016/j.ehj.2003.08.021
63. Watson ML, Kingsmore SF, Johnston GI, Siegelman MH, Le Beau MM, Lemons RS, et al. Genomic organization of the selectin family of leukocyte adhesion molecules on human and mouse chromosome 1. J Exp Med. (1990) 172:263–72. doi: 10.1084/jem.172.1.263
64. Kappelmayer J, Nagy B, Miszti-Blasius K, Hevessy Z, Setiadi H. The emerging value of P-selection as a disease marker. Clin Chem Lab Med. (2004) 42:475–86. doi: 10.1515/CCLM.2004.082
65. Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL, Xu T, et al. P-selectin primes leukocyte integrin activation during inflammation. Nat Immunol. (2007) 8:882–92. doi: 10.1038/ni1491
66. Dole VS, Bergmeier W, Patten IS, Hirahashi J, Mayadas TN, Wagner DD. PSGL-1 regulates platelet P-selectin-mediated endothelial activation and shedding of P-selectin from activated platelets. Thromb Haemost. (2007) 98:806–12. doi: 10.1160/TH07-03-0207
67. Weyrich AS. Platelets: more than a sack of glue. Hematology. (2014) 2014:400–3. doi: 10.1182/asheducation-2014.1.400
68. Panicker SR, Mehta-D'souza P, Zhang N, Klopocki AG, Shao B, McEver RP. Circulating soluble P-selectin must dimerize to promote inflammation and coagulation in mice. Blood. (2017) 130:181–91. doi: 10.1182/blood-2017-02-770479
69. André P, Hartwell D, Hrachovinová I, Saffaripour S, Wagner DD. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci USA. (2000) 97:13835–40. doi: 10.1073/pnas.250475997
70. Conway DSG, Pearce LA, Chin BSP, Hart RG, Lip GYH. Plasma von Willebrand factor and soluble P-selectin as indices of endothelial damage and platelet activation in 1321 patients with nonvalvular atrial fibrillation: relationship to stroke risk factors. Circulation. (2002) 106:1962–7. doi: 10.1161/01.CIR.0000033220.97592.9A
71. Chandler AB, Earhart AD, Speich HE, Kueter TJ, Hansen J, White MM, et al. Regulation of CD40L (CD154) and CD62P (p-selectin) surface expression upon GPIIb-IIIa blockade of platelets from stable coronary artery disease patients. Thromb Res. (2010) 125:44–52. doi: 10.1016/j.thromres.2009.04.017
72. Lam FW, Vijayan KV, Rumbaut RE. Platelets and their interactions with other immune cells. Compr Physiol. (2015) 5:1265–80. doi: 10.1002/cphy.c140074
73. Henn V, Slupsky JR, Gräfe M, Anagnostopoulos I, Förster R, Müller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. (1998) 391:591–4. doi: 10.1038/35393
74. André P, Srinivasa Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, et al. CD40L stabilizes arterial thrombi by a β3 integrin-dependent mechanism. Nat Med. (2002) 8:247–52. doi: 10.1038/nm0302-247
75. Szmitko PE, Wang CH, Weisel RD, De Almeida JR, Anderson TJ, Verma S. New markers of Inflammation and endothelial cell activation part I. Circulation. (2003) 108:1917–23. doi: 10.1161/01.CIR.0000089190.95415.9F
76. Aloui C, Prigent A, Sut C, Tariket S, Hamzeh-Cognasse H, Pozzetto B, et al. The signaling role of cd40 ligand in platelet biology and in platelet component transfusion. Int J Mol Sci. (2014) 15:22342–64. doi: 10.3390/ijms151222342
77. Prasad KSS, Andre P, Yan Y, Phillips DR. The platelet CD40L/GP IIb-IIIa axis in atherothrombotic disease. Curr Opin Hematol. (2003) 10:356–61. doi: 10.1097/00062752-200309000-00006
78. Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol. (2005) 25:2428–34. doi: 10.1161/01.ATV.0000184765.59207.f3
79. May AE, Kälsch T, Massberg S, Herouy Y, Schmidt R, Gawaz M. Engagement of glycoprotein IIb/IIIa (α IIb β 3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation. (2002) 106:2111–7. doi: 10.1161/01.CIR.0000033597.45947.0F
80. Furman MI, Krueger LA, Linden MD, Barnard MR, Frelinger AL, Michelson AD. Release of soluble CD40L from platelets is regulated by glycoprotein IIb/IIIa and actin polymerization. J Am Coll Cardiol. (2004) 43:2319–25. doi: 10.1016/j.jacc.2003.12.055
81. Lindemann S, Kramer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost. (2007) 5:203–11. doi: 10.1111/j.1538-7836.2007.02517.x
82. Fuentes EQ, Fuentes FQ, Andrés V, Pello OM, De Mora JF, Palomo IG. Role of platelets as mediators that link inflammation and thrombosis in atherosclerosis. Platelets. (2013) 24:255–62. doi: 10.3109/09537104.2012.690113
83. Libby P, Pasterkamp G, Crea F, Jang IK. Reassessing the mechanisms of acute coronary syndromes: the “vulnerable plaque” and superficial erosion. Circ Res. (2019) 124:150–60. doi: 10.1161/CIRCRESAHA.118.311098
84. Blann AD, Lanza F, Galajda P, Gurney D, Moog S, Cazenave JP, et al. Increased platelet glycoprotein V levels in patients with coronary and peripheral atherosclerosis: the influence of aspirin and cigarette smoking. Thromb Haemost. (2001) 86:777–83. doi: 10.1055/s-0037-1616131
85. Lindmark E, Wallentin L, Siegbahn A. Blood cell activation, coagulation, and inflammation in men and women with coronary artery disease. Thromb Res. (2001) 103:249–59. doi: 10.1016/S0049-3848(01)00313-9
86. Tan K, Tayebjee M, MacFadyen R, Lip G. Relation of platelet activation to coronary angiographic severity and collateralization. Am J Cardiol. (2005) 96:208–10. doi: 10.1016/j.amjcard.2005.03.045
87. Warzok F, Steiner M, Blann AD, Weber F, Urbaszek W, Schuff-Werner P. Immediate and late effects of coronary angiography on soluble endothelial cell markers and P-selectin in patients with and without coronary artery disease. Blood Coagul Fibrinolysis. (1999) 10:381–8. doi: 10.1097/00001721-199909000-00009
88. Linden MD, Furman MI, Freilinger AL, Fox ML, Barnard MR, Li Y, et al. Indices of platelet activation and the stability of coronary artery disease. J Thromb Haemost. (2007) 5:761–5. doi: 10.1111/j.1538-7836.2007.02462.x
89. Jaumdally RJ, Varma C, Blann AD, MacFadyen RJ, Lip GYH. Indices of angiogenesis, platelet activation, and endothelial damage/dysfunction in relation to ethnicity and coronary artery disease: differences in central versus peripheral levels. Ann Med. (2007) 39:628–33. doi: 10.1080/07853890701636265
90. Kaufmann J, Wellnhofer E, Kappert K, Urban D, Meyborg H, Hauptmann T, et al. Soluble P-selectin level correlates with acetylsalicylic acid but not with clopidogrel response in patients with stable coronary artery disease after a percutaneous coronary intervention. Coron Artery Dis. (2013) 24:312–20. doi: 10.1097/MCA.0b013e328360efd3
91. Gremmel T, Frelinger AL, Michelson AD. Soluble CD40 ligand in aspirin-treated patients undergoing cardiac catheterization. PLoS ONE. (2015) 10:e0134599. doi: 10.1371/journal.pone.0134599
92. Azar RR, Kassab R, Zoghbi A, Aboujaoudé S, El-Osta H, Ghorra P, et al. Effects of clopidogrel on soluble CD40 ligand and on high-sensitivity C-reactive protein in patients with stable coronary artery disease. Am Heart J. (2006) 151:521.e1–4. doi: 10.1016/j.ahj.2005.10.021
93. Pettersen AÅR, Arnesen H, Opstad TB, Bratseth V, Seljeflot I. Markers of endothelial and platelet activation are associated with high on-aspirin platelet reactivity in patients with stable coronary artery disease. Thromb Res. (2012) 130:424–8. doi: 10.1016/j.thromres.2012.06.016
94. Ghasemzadeh N, Hritani AW, De Staercke C, Eapen DJ, Veledar E, Al Kassem H, et al. Plasma stromal cell-derived factor 1α/CXCL12 level predicts long-term adverse cardiovascular outcomes in patients with coronary artery disease. Atherosclerosis. (2015) 238:113–8. doi: 10.1016/j.atherosclerosis.2014.10.094
95. Makin AJ, Chung NAY, Silverman SH, Lip GYH. Alterations of thrombogenesis, endothelial damage and oxidative stress with reperfusion during femoral artery bypass surgery for peripheral vascular disease. Pathophysiol Haemost Thromb. (2002) 32:158–64. doi: 10.1159/000070421
96. Makin AJ, Chung NAY, Silverman SH, Lip GYH. Thrombogenesis and endothelial damage/dysfunction in peripheral artery disease: relationship to ethnicity and disease severity. Thromb Res. (2003) 111:221–6. doi: 10.1016/j.thromres.2003.09.012
97. Makin AJ, Chung NAY, Silverman SH, Moss MS, Lip GYH. Indices of thrombogenesis, endothelial damage and platelet function following percutaneous peripheral artery angiography and angioplasty for peripheral vascular disease. Pathophysiol Haemost Thromb. (2003) 33:102–8. doi: 10.1159/000073854
98. Blann A, Tan K, Tayebjee M, Davagnanam I, Moss M, Lip G. Soluble CD40L in peripheral artery disease: relationship with disease severity, platelet markers and the effects of angioplasty. Thromb Haemost. (2005) 93:578–83. doi: 10.1160/TH04-09-0586
99. Tan K, Tayebjee M, Lynd C, Blann A, Lip G. Platelet microparticles and soluble P selectin in peripheral artery disease: relationship to extent of disease and platelet activation markers. Ann Med. (2005) 37:61–6. doi: 10.1080/07853890410018943
100. Tsakiris D, Tschoepl M, Wolf K, Labs K, Jager K, Marbet G. Platelets and cytokines in concert with endothelial activation in patients with peripheral arterial occlusive disease. Blood Coagul Fibrinolysis. (2000) 11:165–73. doi: 10.1097/00001721-200003000-00008
101. Zamzam A, Syed MH, Rand ML, Singh K, Hussain MA, Jain S, et al. Altered coagulation profile in peripheral artery disease patients. Vascular. (2020) 28:368–77. doi: 10.1177/1708538120915997
102. Blann AD, Adams R, Ashleigh R, Naser S, Kirkpatrick U, McCollum CN. Changes in endothelial, leucocyte and platelet markers following contrast medium injection during angiography in patients with peripheral artery disease. Br J Radiol. (2001) 74:811–7. doi: 10.1259/bjr.74.885.740811
103. Eikelboom JW, Hankey GJ, Thom J, Claxton A, Yi Q, Gilmore G, et al. Enhanced antiplatelet effect of clopidogrel in patients whose platelets are least inhibited by aspirin: a randomized crossover trial. J Thromb Haemost. (2005) 3:2649–55. doi: 10.1111/j.1538-7836.2005.01640.x
104. Tan K, Tayebjee M, Lim H, Lip G. Clinically apparent atherosclerotic disease in diabetes is associated with an increase in platelet microparticle levels. Diabet Med. (2005) 22:1657–62. doi: 10.1111/j.1464-5491.2005.01707.x
105. Burdess A, Michelsen AE, Brosstad F, Fox KAA, Newby DE, Nimmo AF. Platelet activation in patients with peripheral vascular disease: reproducibility and comparability of platelet markers. Thromb Res. (2012) 129:50–5. doi: 10.1016/j.thromres.2011.08.015
106. De Lemos JA, Zirlik A, Schönbeck U, Varo N, Murphy SA, Khera A, et al. Associations between soluble CD40 ligand, atherosclerosis risk factors, and subclinical atherosclerosis: results from the Dallas Heart Study. Arterioscler Thromb Vasc Biol. (2005) 25:2192–6. doi: 10.1161/01.ATV.0000182904.08513.60
107. Gremmel T, Ay C, Riedl J, Kopp CW, Eichelberger B, Koppensteiner R, et al. Platelet-specific markers are associated with monocyte-platelet aggregate formation and thrombin generation potential in advanced atherosclerosis. Thromb Haemost. (2016) 115:615–21. doi: 10.1160/th15-07-0598
108. Kumar A, Cannon CP. Acute coronary syndromes: diagnosis and management, part I. Mayo Clin Proc. (2009) 84:917–38. doi: 10.4065/84.10.917
109. Bigalke B, Stellos K, Stakos D, Joos T, Pötz O, Geisler T, et al. Influence of platelet count on the expression of platelet collagen receptor glycoprotein VI (GPVI) in patients with acute coronary syndrome. Thromb Haemost. (2009) 101:911–5. doi: 10.1160/TH08-06-0399
110. Furman MI, Krueger LA, Linden MD, Fox ML, Ball SP, Barnard MR, et al. GPIIb-IIIa antagonists reduce thromboinflammatory processes in patients with acute coronary syndromes undergoing percutaneous coronary intervention. J Thromb Haemost. (2005) 3:312–20. doi: 10.1111/j.1538-7836.2005.01124.x
111. Ray MJ, Juneja M, Bett N, Walters DL. A comparison of anticoagulation with bivalirudin and provisional GPIIb/IIIa inhibition with unfractionated heparin and mandatory GPIIb/IIIa inhibition during percutaneous coronary intervention in relation to platelet activation and the inhibition of coagu. EuroIntervention. (2009) 5:330–5. doi: 10.4244/V5I3A52
112. Stone GW, Witzenbichler B, Guagliumi G, Peruga JZ, Brodie BR, Dudek D, et al. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med. (2008) 358:2218–30. doi: 10.1056/NEJMoa0708191
113. Obradovic SD, Antovic JP, Antonijevic NM, Ratkovic NG, Vojvodic DV, Subota VS, et al. Elevations in soluble CD40 ligand in patients with high platelet aggregability undergoing percutaneous coronary intervention. Blood Coagul Fibrinolysis. (2009) 20:283–9. doi: 10.1097/MBC.0b013e328329f28c
114. Lee K, Blann A, Lip G. Effects of omega-3 polyunsaturated fatty acids on plasma indices of thrombogenesis and inflammation in patients post-myocardial infarction. Thromb Res. (2006) 118:305–312. doi: 10.1016/j.thromres.2005.07.018
115. Carnevale R, Sciarretta S, Valenti V, Nonno F, Calvieri C, Nocella C, et al. Low-grade endotoxaemia enhances artery thrombus growth via Toll-like receptor 4 : implication for myocardial infarction. Eur Heart J. (2020) 41:1–10. doi: 10.1093/eurheartj/ehz893
116. Järemo P, Hansson G, Nilsson O. Elevated inflammatory parameters are associated with lower platelet density in acute myocardial infarctions with ST-elevation. Thromb Res. (2000) 100:471–8. doi: 10.1016/S0049-3848(00)00366-2
117. Christersson C, Oldgren J, Wallentin L, Siegbahn A. Treatment with an oral direct thrombin inhibitor decreases platelet activity but increases markers of inflammation in patients with myocardial infarction. J Intern Med. (2011) 270:215–23. doi: 10.1111/j.1365-2796.2011.02354.x
118. Youssef AA, Chang LT, Sheu JJ, Lee FY, Chua S, Yeh KH, et al. Association between circulating level of CD40 ligand and angiographic morphologic features indicating high-burden thrombus formation in patients with acute myocardial infarction undergoing primary coronary intervention. Circ J. (2007) 71:1857–61. doi: 10.1253/circj.71.1857
119. Undas A, Szułdrzyński K, Brummel-Ziedins KE, Tracz W, Zmudka K, Mann KG. Systemic blood coagulation activation in acute coronary syndromes. Blood. (2009) 113:2070–8. doi: 10.1182/blood-2008-07-167411
120. Pastuszczak M, Kotlarz A, Mostowik M, Zalewski J, Zmudka K, Undas A. Prior simvastatin treatment is associated with reduced thrombin generation and platelet activation in patients with acute ST-segment elevation myocardial infarction. Thromb Res. (2010) 125:382–6. doi: 10.1016/j.thromres.2009.06.021
121. Morel O, Hugel B, Jesel L, Lanza F, Douchet MP, Zupan M, et al. Sustained elevated amounts of circulating procoagulant membrane microparticles and soluble GPV after acute myocardial infarction in diabetes mellitus. Thromb Haemost. (2004) 91:345–53. doi: 10.1160/TH03-05-0294
122. Ohashi Y, Kawashima S, Mori T, Terashima M, Ichikawa S, Ejiri J, et al. Soluble CD40 ligand and interleukin-6 in the coronary circulation after acute myocardial infarction. Int J Cardiol. (2006) 112:52–8. doi: 10.1016/j.ijcard.2005.09.051
123. Huisse M-G, Lanoy E, Tcheche D, Feldman L, Bezeaud A, Anglès-Cano E, et al. Prothrombotic markers and early spontaneous recanalization in ST-segment elevation myocardial infarction. Thromb Haemost. (2007) 98:420–6. doi: 10.1160/TH06-11-0621
124. Morel O, Hugel B, Jesel L, Mallat Z, Lanza F, Douchet MP, et al. Circulating procoagulant microparticles and soluble GPV in myocardial infarction treated by primary percutaneous transluminal coronary angioplasty. A possible role for GPIIb-IIIa antagonists. J Thromb Haemost. (2004) 2:1118–1126. doi: 10.1111/j.1538-7836.2004.00805.x
125. Yan JC, Ma GS, Zhu J, Feng Y, Luo D, Wu ZG, et al. The clinical implications of increased coexpression of CD40-CD40 ligand system and C-reactive protein in patients after percutaneous coronary intervention. Clin Chim Acta. (2006) 374:140–1. doi: 10.1016/j.cca.2006.05.025
126. Yan JC, Ding S, Liang Y, Ma GS, Zhu J, Feng Y, et al. Relationship between upregulation of CD40 system and restenosis in patients after percutaneous coronary intervention. Acta Pharmacol Sin. (2007) 28:339–43. doi: 10.1111/j.1745-7254.2007.00520.x
127. Türker S, Güneri S, Akdeniz B, Özcan MA, Baris N, Badak Ö, Kirimli Ö, et al. Usefulness of preprocedural soluble CD40 ligand for predicting restenosis after percutaneous coronary intervention in patients with stable coronary artery disease. Am J Cardiol. (2006) 97:198–202. doi: 10.1016/j.amjcard.2005.08.024
128. Lee WJ, Sheu WHH, Chen YT, Liu TJ, Liang KW, Ting CT, et al. Circulating CD40 ligand is elevated only in patients with more advanced symptomatic peripheral arterial diseases. Thromb Res. (2006) 118:619–26. doi: 10.1016/j.thromres.2005.10.012
129. Subramanian S, Liu C, Aviv A, Ho JE, Courchesne P, Muntendam P, et al. Stromal cell-derived factor 1 as a biomarker of heart failure and mortality risk. Arterioscler Thromb Vasc Biol. (2014) 34:2100–5. doi: 10.1161/ATVBAHA.114.303579
130. Cherian P, Hankey GJ, Eikelboom JW, Thom J, Baker RI, McQuillan A, et al. Endothelial and platelet activation in acute ischemic stroke and its etiological subtypes. Stroke. (2003) 34:2132–7. doi: 10.1161/01.STR.0000086466.32421.F4
131. AnŽej S, BoŽič M, Antovič A, Peternel P, Gašperšič N, Rot U, et al. Evidence of hypercoagulability and inflammation in young patients long after acute cerebral ischaemia. Thromb Res. (2007) 120:39–46. doi: 10.1016/j.thromres.2006.08.005
132. Cao YJ, Wang YM, Zhang J, Zeng YJ, Liu CF. The effects of antiplatelet agents on platelet-leukocyte aggregations in patients with acute cerebral infarction. J Thromb Thrombolysis. (2009) 27:233–8. doi: 10.1007/s11239-007-0190-x
133. Lip GYH, Blann AD, Farooqi IS, Zarifis J, Sagar G, Beevers DG. Sequential alterations in haemorheology, endothelial dysfunction, platelet activation and thrombogenesis in relation to prognosis following acute stroke: the West Birmingham Stroke Project. Blood Coagul Fibrinolysis. (2002) 13:339–47. doi: 10.1097/00001721-200206000-00010
134. Lukasik M, Dworacki G, Michalak S, Kufel-Grabowska J, Golanski J, Watala C, et al. Aspirin treatment influences platelet-related inflammatory biomarkers in healthy individuals but not in acute stroke patients. Thromb Res. (2011) 128:e73–80. doi: 10.1016/j.thromres.2011.06.016
135. Nadar SK, Lip GYH, Blann AD. Platelet morphology, soluble P selectin and platelet P-selectin in acute ischaemic stroke. The West Birmingham stroke project. Thromb Haemost. (2004) 92:1342–8. doi: 10.1160/TH04-07-0433
136. Kozuka K, Kohriyama T, Ikeda J, Nakamura S, Nomura E, Kajikawa H. Endothelial markers and adhesion molecules in acute ischemic stroke-Sequential change and differences in stroke subtype. Atherosclerosis. (2002) 161:161–8. doi: 10.1016/S0021-9150(01)00635-9
137. Wang Q, Zhao W, Bai S. Association between plasma soluble P-selectin elements and progressive ischemic stroke. Exp Ther Med. (2013) 5:1427–33. doi: 10.3892/etm.2013.985
138. Jurk K, Jahn U, Aken H, Van Schriek C, Droste DW, Ritter MA, et al. Platelets and Blood Cells Platelets in patients with acute ischemic stroke are exhausted and refractory to thrombin, due to cleavage of the seven-transmembrane thrombin receptor (PAR-1). Thromb Haemost. (2004) 91:334–44. doi: 10.1160/TH03-01-0044
139. Al-Tamimi M, Gardiner EE, Thom JY, Shen Y, Cooper MN, Hankey GJ, et al. Soluble glycoprotein VI is raised in the plasma of patients with acute ischemic stroke. Stroke. (2011) 42:498–500. doi: 10.1161/STROKEAHA.110.602532
140. Wu X, Zhang W, Li H, You S, Shi J, Zhang C, et al. Plasma C-type lectin-like receptor 2 as a predictor of death and vascular events in patients with acute ischemic stroke. Eur J Neurol. (2019) 26:1334–40. doi: 10.1111/ene.13984
141. Zhang X, Zhang W, Wu X, Li H, Zhang C, Huang Z, et al. Prognostic significance of plasma CLEC-2 (C-type lectin-like receptor 2) in patients with acute ischemic stroke. Stroke. (2019) 50:45–52. doi: 10.1161/STROKEAHA.118.022563
142. Bal Dit Sollier C, Crassard I, Simoneau G, Bergmann JF, Bousser MG, Drouet L. Effect of the thromboxane prostaglandin receptor antagonist terutroban on arterial thrombogenesis after repeated administration in patients treated for the prevention of ischemic stroke. Cerebrovasc Dis. (2009) 28:505–13. doi: 10.1159/000236915
143. Oberheiden T, Nguyen XD, Fatar M, Elmas E, Blahak C, Morper N, et al. Platelet and monocyte activation in acute ischemic stroke-is there a correlation with stroke etiology? Clin Appl Thromb. (2012) 18:87–91. doi: 10.1177/1076029611412359
144. Wolff V, Aleil B, Giroud M, Lorenzini JL, Meyer N, Wiesel ML, et al. Soluble platelet glycoprotein V is a marker of thrombosis in patients with ischemic stroke. Stroke. (2005) 36:17–9. doi: 10.1161/01.STR.0000155738.02753.4d
145. Wurster T, Poetz O, Stellos K, Kremmer E, Melms A, Schuster A, et al. Plasma levels of soluble glycoprotein VI (sGPVI) are associated with ischemic stroke. Platelets. (2013) 24:560–5. doi: 10.3109/09537104.2012.746455
146. Masunaga N, Abe M, Ogawa H, Aono Y, Ikeda S, Doi K, et al. Current status, time trends and outcomes of combination therapy with oral anticoagulant and antiplatelet drug in patients with atrial fibrillation — the fushimi AF registry. Circ J. (2018) 82:2983–91. doi: 10.1253/circj.CJ-18-0872
147. Matsumura-Nakano Y, Shizuta S, Komasa A, Morimoto T, Masuda H, Shiomi H, et al. Open-label randomized trial comparing oral anticoagulation with and without single antiplatelet therapy in patients with atrial fibrillation and stable coronary artery disease beyond 1 year after coronary stent implantation. Circulation. (2019) 139:604–16. doi: 10.1161/CIRCULATIONAHA.118.036768
148. So CH, Eckman MH. Combined aspirin and anticoagulant therapy in patients with atrial fibrillation. J Thromb Thrombolysis. (2017) 43:7–17. doi: 10.1007/s11239-016-1425-5
149. Blann A, Li-Saw-Hee F, Lip G. Increased membrane and soluble p-selectin in atrial fibrillation. Circulation. (1999) 100:e86–7. doi: 10.1161/01.CIR.100.17.e86
150. Li-Saw-Hee FL, Blann AD, Lip GYH. A cross-sectional and diurnal study of thrombogenesis among patients with chronic atrial fibrillation. J Am Coll Cardiol. (2000) 35:1926–31. doi: 10.1016/S0735-1097(00)00627-6
151. Kamath S, Blann A, Chin B, Lip G. A prospective randomized trial of aspirin-clopidogrel combination therapy and dose-adjusted warfarin on indices of thrombogenesis and platelet activation in atrial fibrillation. J Am Coll Cardiol. (2002) 40:484–90. doi: 10.1016/S0735-1097(02)01984-8
152. Choudhury A, Chung I, Blann AD, Lip GYH. Elevated platelet microparticle levels in nonvalvular atrial fibrillation: relationship to P-selectin and antithrombotic therapy. Chest. (2007) 131:809–15. doi: 10.1378/chest.06-2039
153. Choudhury A, Chung I, Blann AD, Lip GYH. Platelet Surface CD62P and CD63, mean platelet volume, and soluble/platelet p-selectin as indexes of platelet function in atrial fibrillation. A comparison of “healthy control subjects” and “disease control subjects” in sinus rhythm. J Am Coll Cardiol. (2007) 49:1957–1964. doi: 10.1016/j.jacc.2007.02.038
154. Choudhury A, Chung I, Panja N, Patel J, Lip GYH. Soluble CD40 ligand, platelet surface CD40 ligand, and total platelet CD40 ligand in atrial fibrillation: relationship to soluble p-selectin, stroke risk factors, and risk factor intervention. Chest. (2008) 134:574–81. doi: 10.1378/chest.07-2745
155. Fu R, Wu S, Wu P, Qiu J. A Study of blood soluble P-selectin, fibrinogen, and von Willebrand factor levels in idiopathic and lone atrial fibrillation. Europace. (2011) 13:31–6. doi: 10.1093/europace/euq346
156. Wysokinski WE, Cohoon KP, Melduni RM, Mazur M, Ammash N, Munger T, et al. Association between P-selectin levels and left atrial blood stasis in patients with nonvalvular atrial fibrillation. Thromb Res. (2018) 172:4–8. doi: 10.1016/j.thromres.2018.10.009
157. Choudhury A, Freestone B, Patel J, Lip GYH. Relationship of soluble CD40 ligand to vascular endothelial growth factor, angiopoietins, and tissue factor in atrial fibrillation: a link among platelet activation, angiogenesis, and thrombosis? Chest. (2007) 132:1913–9. doi: 10.1378/chest.07-1565
158. Lim HS, Willoughby SR, Schultz C, Gan C, Alasady M, Lau DH, et al. Effect of atrial fibrillation on atrial thrombogenesis in humans: impact of rate and rhythm. J Am Coll Cardiol. (2013) 61:852–60. doi: 10.1016/j.jacc.2012.11.046
159. Kamath S, Chin B, Blann A, Lip G. A study of platelet activation in paroxysmal, persistent and permanent atrial fibrillation. Blood Coagul Fibrinolysis. (2002) 13:627–36. doi: 10.1097/00001721-200210000-00008
160. Kamath S, Blann A, Chin B, Lanza F, Aleil B, Cazenave JP, et al. A study of platelet activation in atrial fibrillation and the effects of antithrombotic therapy. Eur Heart J. (2002) 23:1788–95. doi: 10.1053/euhj.2002.3259
161. Conway DSG, Heeringa J, Van Der Kuip DAM, Chin BSP, Hofman A, Witteman JCM, et al. Atrial fibrillation and the prothrombotic state in the elderly: the Rotterdam study. Stroke. (2003) 34:413–7. doi: 10.1161/01.STR.0000051728.85133.32
162. Conway DSG, Pearce LA, Chin BSP, Hart RG, Lip GYH. Prognostic value of plasma von Willebrand factor and soluble P-selectin as indices of endothelial damage and platelet activation in 994 patients with nonvalvular atrial fibrillation. Circulation. (2003) 107:3141–5. doi: 10.1161/01.CIR.0000077912.12202.FC
163. Goldsmith IRA, Li-Saw-Hee FL, Blann AD, Lip GYH. Increased platelet activation and endothelial dysfunction in patients with atrial fibrillation immediately following percutaneous balloon mitral valvuloplasty. Clin Cardiol. (2000) 23:587–90. doi: 10.1002/clc.4960230808
164. Kamath S, Blann AD, Caine GJ, Gurney D, Chin BSP, Lip GYH. Platelet P-selectin levels in relation to plasma soluble P-selectin and β-thromboglobulin levels in atrial fibrillation. Stroke. (2002) 33:1237–42. doi: 10.1161/01.STR.0000013739.82306.7F
165. Conway DSG, Buggins P, Hughes E, Lip GYH. Predictive value of indexes of inflammation and hypercoagulability on success of cardioversion of persistent atrial fibrillation. Am J Cardiol. (2004) 94:508–10. doi: 10.1016/j.amjcard.2004.04.070
166. Conway DSG, Buggins P, Hughes E, Lip GYH. Relation of interleukin-6, C-reactive protein, and the prothrombotic state to transesophageal echocardiographic findings in atrial fibrillation. Am J Cardiol. (2004) 93:1368–73. doi: 10.1016/j.amjcard.2004.02.032
167. Conway DSG, Buggins P, Hughes E, Lip GYH. Relationship of interleukin-6 and C-reactive protein to the prothrombotic state in chronic atrial fibrillation. J Am Coll Cardiol. (2004) 43:2075–82. doi: 10.1016/j.jacc.2003.11.062
168. Lau YC, Xiong Q, Blann AD, Lip GYH. Relationship between renal function and circulating microparticles, soluble P-selectin and E-selectin levels in atrial fibrillation. J Thromb Thrombolysis. (2017) 43:18–23. doi: 10.1007/s11239-016-1427-3
169. Tarnowski D, Poitz DM, Plichta L, Heidrich FM, Wiedemann S, Ruf T, et al. Comparison of diverse platelet activation markers as indicators for left atrial thrombus in atrial fibrillation. Platelets. (2018) 29:41–7. doi: 10.1080/09537104.2017.1293805
170. Varughese GI, Patel JV, Tomson J, Lip GYH. Effects of blood pressure on the prothrombotic risk in 1235 patients with non-valvular atrial fibrillation. Heart. (2007) 93:495–9. doi: 10.1136/hrt.2006.099374
171. Steppich B, Dobler F, Brendel LC, Hessling G, Braun SL, Steinsiek AL, et al. Effect of the FXa inhibitors Rivaroxaban and Apixaban on platelet activation in patients with atrial fibrillation. J Thromb Thrombolysis. (2017) 43:490–7. doi: 10.1007/s11239-017-1495-z
172. Li-Saw-Hee FL, Blann AD, Goldsmith I, Lip GYH. Indexes of hypercoagulability measured in peripheral blood reflect levels in intracardiac blood in patients with atrial fibrillation secondary to mitral stenosis. Am J Cardiol. (1999) 83:1206–9. doi: 10.1016/S0002-9149(99)00060-0
173. Duygu H, Barisik V, Kurt H, Turk U, Ercan E, Kose S. Prognostic value of plasma soluble CD40 ligand in patients with chronic non-valvular atrial fibrillation. Europace. (2008) 10:210–4. doi: 10.1093/europace/eum284
174. Ferro D, Loffredo L, Polimeni L, Fimognari F, Villari P, Pignatelli P, et al. Soluble CD40 ligand predicts ischemic stroke and myocardial infarction in patients with nonvalvular atrial fibrillation. Arterioscler Thromb Vasc Biol. (2007) 27:2763–8. doi: 10.1161/ATVBAHA.107.152777
175. Pignatelli P, Pastori D, Bartimoccia S, Menichelli D, Vicario T, Nocella C, et al. Anti Xa oral anticoagulants inhibit in vivo platelet activation by modulating glycoprotein VI shedding. Pharmacol Res. (2016) 113:484–9. doi: 10.1016/j.phrs.2016.09.035
176. Lip GYH, Patel JV, Hughes E, Hart RG. High-sensitivity C-reactive protein and soluble CD40 ligand as indices of inflammation and platelet activation in 880 patients with nonvalvular atrial fibrillation: relationship to stroke risk factors, stroke risk stratification schema, and prognosis. Stroke. (2007) 38:1229–37. doi: 10.1161/01.STR.0000260090.90508.3e
177. Furio E, García-Fuster MJ, Redon J, Marques P, Ortega R, Sanz MJ, et al. CX 3 CR1/CX 3 CL1 axis mediates platelet-leukocyte adhesion to arterial endothelium in younger patients with a history of idiopathic deep vein thrombosis. Thromb Haemost. (2018) 118:562–71. doi: 10.1055/s-0038-1629897
178. Blann A, Noteboom W, Rosendaal F. Increased soluble P-selectin levels following deep venous thrombosis: cause or effect? Br J Haematol. (2000) 108:191–3. doi: 10.1046/j.1365-2141.2000.01813.x
179. Patrignani P, Di Febbo C, Tacconelli S, Douville K, Guglielmi MD, Horvath RJ, et al. Differential association between human prostacyclin receptor polymorphisms and the development of venous thrombosis and intimal hyperplasia: a clinical biomarker study. Pharmacogenet Genomics. (2008) 18:611–20. doi: 10.1097/FPC.0b013e328301a774
180. Rectenwald JE, Myers DD, Hawley AE, Longo C, Henke PK, Guire KE, et al. D-dimer, P-selectin, and microparticles: novel markers to predict deep venous thrombosis. Thromb Haemost. (2005) 94:1312–7. doi: 10.1160/TH05-06-0426
181. Migliacci R, Becattini C, Pesavento R, Davi G, Vedovati MC, Guglielmini G, et al. Endothelial dysfunction in patients with spontaneous venous thromboembolism. Haematologica. (2007) 92:812–8. doi: 10.3324/haematol.10872
182. Kyrle PA, Hron G, Eichinger S, Wagner O. Circulating P-selectin and the risk of recurrent venous thromboembolism. Thromb Haemost. (2007) 97:880–3. doi: 10.1160/TH07-02-0115
183. Panova-Noeva M, Wagner B, Nagler M, Koeck T, ten Cate V, Prochaska JH, et al. Comprehensive platelet phenotyping supports the role of platelets in the pathogenesis of acute venous thromboembolism – results from clinical observation studies. EBioMedicine. (2020) 60:102978. doi: 10.1016/j.ebiom.2020.102978
184. Chung T, Connor D, Joseph J, Emmett L, Mansberg R, Peters M, et al. Platelet activation in acute pulmonary embolism. J Thromb Haemost. (2007) 5:918–24. doi: 10.1111/j.1538-7836.2007.02461.x
185. Simes J, Becattini C, Agnelli G, Eikelboom JW, Kirby AC, Mister R, et al. Aspirin for the prevention of recurrent venous thromboembolism the INSPIRE collaboration. Circulation. (2014) 130:1062–71. doi: 10.1161/CIRCULATIONAHA.114.008828
186. Ballermann BJ. Endothelial cell activation. Kidney Int. (1998) 53:1810–26. doi: 10.1046/j.1523-1755.1998.00943.x
187. Badimon L, Padró T, Vilahur G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur Hear J Acute Cardiovasc Care. (2012) 1:60–74. doi: 10.1177/2048872612441582
188. Pasalic L, Wang SSY, Chen VMY. Platelets as biomarkers of coronary artery disease. Semin Thromb Hemost. (2016) 42:223–33. doi: 10.1055/s-0036-1572328
189. Gurbel PA, Becker RC, Mann KG, Steinhubl SR, Michelson AD. Platelet function monitoring in patients with coronary artery disease. J Am Coll Cardiol. (2007) 50:1822–34. doi: 10.1016/j.jacc.2007.07.051
190. Weber M, Rabenau B, Stanisch M, Elsaesser A, Mitrovic V, Heeschen C, et al. Influence of sample type and storage conditions on soluble CD40 ligand assessment. Clin Chem. (2006) 52:888–91. doi: 10.1373/clinchem.2005.062083
191. Varo N, Nuzzo R, Natal C, Libby P, Schönbeck U. Influence of pre-analytical and analytical factors on soluble CD40L measurements. Clin Sci. (2006) 111:341–7. doi: 10.1042/CS20060047
192. Riondino S, Martini F, La Farina F, Spila A, Guadagni F, Ferroni P. Increased plasma levels of soluble CD40 ligand correlate with platelet activation markers and underline the need for standardized pre-analytical conditions. Clin Biochem. (2010) 43:666–70. doi: 10.1016/j.clinbiochem.2009.12.021
193. Amin HM, Ahmad S, Walenga JM, Hoppensteadt DA, Leitz H, Fareed J. Soluble P-selectin in human plasma: effect of anticoagulant matrix and its levels in patients with cardiovascular disorders. Clin Appl Thromb. (2000) 6:71–6. doi: 10.1177/107602960000600204
194. Becs G, Hudák R, Fejes Z, Debreceni IB, Bhattoa HP, Balla J, et al. Haemodiafiltration elicits less platelet activation compared to haemodialysis. BMC Nephrol. (2016) 17:1–10. doi: 10.1186/s12882-016-0364-x
195. Bunescu A, Seideman P, Lenkei R, Levin K, Egberg N. Enhanced Fcγ receptor I, α Mβ 2 integrin receptor expression by monocytes and neutrophils in rheumatoid arthritis: interaction with platelets. J Rheumatol. (2004) 31:2347–55.
196. Nomura S, Kanazawa S, Fukuhara S. Effects of efonidipine on platelet and monocyte activation markers in hypertensive patients with and without type 2 diabetes mellitus. J Hum Hypertens. (2002) 16:539–47. doi: 10.1038/sj.jhh.1001447
197. Sakamaki F, Kyotani S, Nagaya N, Sato N, Oya H, Satoh T, et al. Increased plasma P-selectin and decreased thrombomodulin in pulmonary arterial hypertension were improved by continuous prostacyclin therapy. Circulation. (2000) 102:2720–5. doi: 10.1161/01.CIR.102.22.2720
198. Sellam J, Proulle V, Jüngel A, Ittah M, Miceli Richard C, Gottenberg JE, et al. Increased levels of circulating microparticles in primary Sjögren's syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res Ther. (2009) 11:1–11. doi: 10.1186/ar2833
199. Takeda I, Kaise S, Nishimaki T, Kasukawa R. Soluble P-selectin in the plasma of patients with connective tissue diseases. Int Arch Allergy Immunol. (1994) 105:128–34. doi: 10.1159/000236814
200. Wang F, Xing T, Wang N, Liu L. Clinical significance of plasma CD146 and P-selectin in patients with type 2 diabetic nephropathy. Cytokine. (2012) 57:127–9. doi: 10.1016/j.cyto.2011.10.010
201. Lim HS, Blann AD, Lip GYH. Soluble CD40 ligand, soluble P-selectin, interleukin-6, and tissue factor in diabetes mellitus: relationsips to cardiovascular disease and risk factor intervention. Circulation. (2004) 109:2524–8. doi: 10.1161/01.CIR.0000129773.70647.94
202. Levine SP, Suarez AJ, Sorenson RR, Raymond NM, Knieriem LK. Platelet factor 4 release during exercise in patients with coronary artery disease. Am J Hematol. (1984) 17:117–27. doi: 10.1002/ajh.2830170204
203. Voisin PJ, Rousselle D. Reduction of beta-thromhoglobulin by artificial. Metabolism. (1983) 138:138–41. doi: 10.1016/0026-0495(83)90218-4
204. Walz DA. Platelet-released proteins as molecular markers for the activation process. Semin Thromb Hemost. (1984) 10:270–9. doi: 10.1055/s-2007-1004432
205. Poruk KE, Firpo MA, Huerter LM, Scaife CL, Emerson LL, Boucher KM, et al. Serum platelet factor 4 is an independent predictor of survival and venous thromboembolism in patients with pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev. (2010) 19:2605–10. doi: 10.1158/1055-9965.EPI-10-0178
206. Kim MH, Huo SH, Kim KS, Kim MS, Song JS. Study on the platelet factor and β-thromboglobulin in the patients with ischemic heart disease. Korean J Intern Med. (1986) 1:1–6. doi: 10.3904/kjim.1986.1.1.1
207. Wu N, Tong S, Xiang Y, Wu L, Xu B, Zhang Y, et al. Association of hemostatic markers with atrial fibrillation: a meta-analysis and meta-regression. PLoS ONE. (2015) 10:e0124716. doi: 10.1371/journal.pone.0124716
208. Yamashita Y, Naitoh K, Wada H, Ikejiri M, Mastumoto T, Ohishi K, et al. Elevated plasma levels of soluble platelet glycoprotein VI (GPVI) in patients with thrombotic microangiopathy. Thromb Res. (2014) 133:440–4. doi: 10.1016/j.thromres.2013.11.023
209. Stack JR, Madigan A, Helbert L, Dunne E, Gardiner EE, Andrews RK, et al. Soluble glycoprotein VI, a specific marker of platelet activation is increased in the plasma of subjects with seropositive rheumatoid arthritis. PLoS ONE. (2017) 12:e0188027. doi: 10.1371/journal.pone.0188027
210. Laske C, Leyhe T, Stransky E, Eschweiler GW, Bueltmann A, Langer H, et al. Association of platelet-derived soluble glycoprotein VI in plasma with Alzheimer's disease. J Psychiatr Res. (2008) 42:746–51. doi: 10.1016/j.jpsychires.2007.07.017
211. deFilippi CR, Seliger SL. Biomarkers for prognostication after acute coronary syndromes. New times and statistics. J Am Coll Cardiol. (2009) 54:365–7. doi: 10.1016/j.jacc.2009.04.031
212. Eapen DJ, Ghasemzadeh N, MacNamara JP, Quyyumi A. The evaluation of novel biomarkers and the multiple biomarker approach in the prediction of cardiovascular disease. Curr Cardiovasc Risk Rep. (2014) 8:1–4. doi: 10.1007/s12170-014-0408-3
213. Mayeux R. Biomarkers: potential uses and limitations. NeuroRx. (2004) 1:182–8. doi: 10.1602/neurorx.1.2.182
Keywords: platelets, biomarkers, thrombosis, venous thromboembolism, atrial fibrillation, arterial thrombosis
Citation: Baidildinova G, Nagy M, Jurk K, Wild PS, ten Cate H and van der Meijden PEJ (2021) Soluble Platelet Release Factors as Biomarkers for Cardiovascular Disease. Front. Cardiovasc. Med. 8:684920. doi: 10.3389/fcvm.2021.684920
Received: 24 March 2021; Accepted: 24 May 2021;
Published: 21 June 2021.
Edited by:
Colin E. Evans, Northwestern University, United StatesReviewed by:
Dianne E. Van Der Wal, Australian Red Cross Blood Service, AustraliaCopyright © 2021 Baidildinova, Nagy, Jurk, Wild, ten Cate and van der Meijden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola E. J. van der Meijden, cC52YW5kZXJtZWlqZGVuQG1hYXN0cmljaHR1bml2ZXJzaXR5Lm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.