 Hao Yi-Dan
Hao Yi-Dan Yang Shi-Wei
Yang Shi-Wei- The Key Laboratory of Remodeling-Related Cardiovascular Disease, Ministry of Education, Beijing Institute of Heart, Lung and Blood Vessel Disease, Beijing Anzhen Hospital, Capital Medical University, Beijing, China
The purpose of this review is to bridge the gap between clinical and basic research through providing a comprehensive and concise description of the cellular and molecular aspects of cardioprotective mechanisms and a critical evaluation of the clinical evidence of high-energy phosphates (HEPs) in ischemic heart disease (IHD). According to the well-documented physiological, pathophysiological and pharmacological properties of HEPs, exogenous creatine phosphate (CrP) may be considered as an ideal metabolic regulator. It plays cardioprotection roles from upstream to downstream of myocardial ischemia through multiple complex mechanisms, including but not limited to replenishment of cellular energy. Although exogenous CrP administration has not been shown to improve long-term survival, the beneficial effects on multiple secondary but important outcomes and short-term survival are concordant with its pathophysiological and pharmacological effects. There is urgent need for high-quality multicentre RCTs to confirm long-term survival improvement in the future.
Introduction
The heart is more than a hemodynamic pump. It is also an organ that needs energy from metabolism (1). In fact, altered cardiac metabolism is the primary and upstream pathophysiologic manifestation of myocardial ischemia in humans (2). After coronary blood flow blockage, energy metabolism disorder occurs within a few seconds, followed by mechanical, electrophysiological and structural abnormalities of the myocardium. To date, standard treatments for ischemic heart disease (IHD), including revascularization (thrombolysis, percutaneous coronary intervention, and coronary artery bypass grafting), antithrombotic therapy (antiplatelet and anticoagulant agents), stabilization/reversal of atherosclerosis progression (control of atherosclerotic risk factors), and inhibition of myocardial remodeling (sympathetic and renin-angiotensin-aldosterone system inhibitors), focus on coronary anatomy and on the results of changes in myocardial metabolism rather than on the metabolic changes themselves (2–8). In addition, almost all of the above treatments exert cardioprotection by directly or indirectly affecting heart rate, blood pressure or myocardial perfusion. In contrast, myocardial energy metabolic therapy (MEMT) plays a protective role by regulating the energy synthesis and utilization of myocardial cells without significant impacts on heart rate, blood pressure and perfusion (9, 10). Because of residual cardiovascular risk, MEMT is promisingly emerging as an upstream treatment for IHD (11).
Since the discovery of creatine phosphate (CrP) in 1927 (12) and adenosine triphosphate (ATP) in 1929 (13), the biochemical, physiological, and pharmacological properties of high-energy phosphates (HEPs) have been gradually uncovered. Unlike the single metabolic process of glucose, free fatty acids or amino acids, the pathways and regulations of HEPs biosynthesis and degradation are involved in all metabolic substrates. Moreover, due to the production and consumption of HEPs in different cells and subcellular organelles, the transmembrane transport of HEPs is also a complex process requiring the assistance of many special transporters and catalytic enzymes (14). Therefore, although HEPs have been known for nearly a 100 years, clinicians still have a lot to learn. In recent years, a series of basic and clinical studies have shown potent protection for IHD by exogenous HEPs (15–19). These results have been confirmed in our laboratories (16, 20, 21).
Previous reviews focused either on the cellular and molecular mechanisms of HEPs which is too complex for clinical application (14, 22), or on presenting the clinical evidence which in turn is too simple for clinicians to understand their pathophysiological and pharmacological effects (15, 16). The purpose of this article is to bridge the gap between clinical and basic research.
Overview of High-Energy Phosphates and Their Transformation
It is believed that energy would be concentrated in the chemical bond containing phosphate groups, which yields energy upon hydrolysis (23). Low-energy phosphates are usually linked to phosphoester bonds, which will release 2 and 3 kcal/mol energy. HEPs include a variety of phosphate compounds with energies of hydrolysis higher than 7 kcal/mol (24). ATP and CrP are considered to be the primary HEPs in human body. ATP is the intracellular energy currency, majority of which is not synthesized de novo but generated from adenosine diphosphate (ADP) by oxidative phosphorylation (OP) of mitochondria and cytoplasmic substrate phosphorylation (SP) (Figure 1) (25, 26). Thus, at any given time, the total amount of ATP and ADP remains fairly constant and recycled continuously (27). While, CrP is the storage and transport carrier of energy, which serves to transfer the HEP-bond from the site of ATP production to the site of ATP utilization through “CrP shuttle” (Figure 1) (28–35). Normally the total quantity of ATP in human body is about 0.1 mole (~50 g). However, the energy used by human cells requires the hydrolysis of 100–150 moles (around 50–75 kg) of ATP daily (36). This means that each ATP molecule is recycled 1,000–1,500 times during a single day. The ATP and CrP activity combined, also referred to as the phosphagen system, is the most rapidly available source of energy (37). Unfortunately, the energy available from the store of phosphagen system is limited and can provide energy for a few seconds of maximal activity.
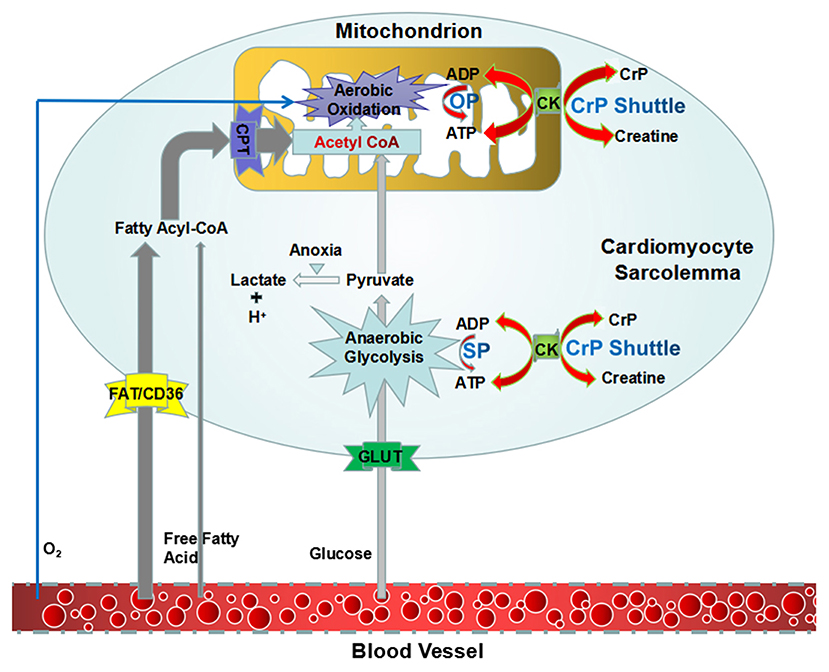
Figure 1. An overview of synthesis of ATP and “CrP shuttle” in cardiomyocyte. ATP is the intracellular energy currency, majority of which is synthesized from ADP by oxidative phosphorylation of mitochondria (predominant) and cytoplasmic substrate phosphorylation (subordinate). CrP is the storage and transport carrier of energy, which serves to transfer the HEP-bond from the site of ATP production to the site of ATP utilization through “CrP shuttle.” ADP, adenosine diphosphate; ATP, adenosine triphosphate; CK, creatine kinase; CrP, creatine phosphate; HEP, high-energy phosphate; OP, oxidative phosphorylation; SP, substrate phosphorylation.
CrP, also known as phosphocreatine or phosphorylated creatine, is a small molecular compound with the formula of C4H10N3O5P, having a molecular weight of 211 daltons. There is one high-energy phosphate bond (N~P) in the chemical structure. As compared, ATP has a relatively more complex molecular structure (C10H16N5O13P3), larger molecular weight (507 daltons), and two high-energy phosphate bonds (O~P). However, the N~P bond of CrP has more energy than either one O~P bond of ATP, 10.3 kcal/mol in comparison with 7.3 kcal/mol (Figure 2) (38). Therefore, CrP can easily provide enough energy and serve as a HEP-bond donor for ATP reconstitution through “CrP shuttle” (28).
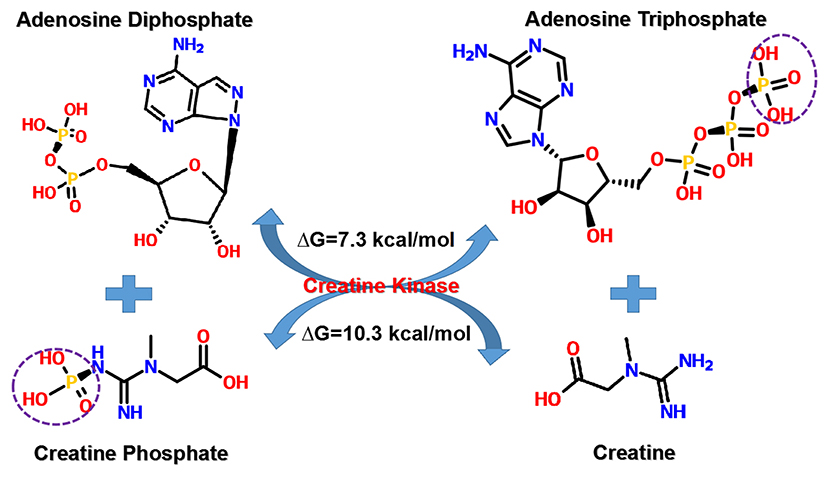
Figure 2. Transfer of HEP-bond through “CrP shuttle.” There is one HEP-bond (N~P) in the chemical structure of CrP. As compared, ATP has a relatively more complex molecular structure and two HEP-bonds (O~P). However, the N~P bond of CrP has more energy than either one O~P bond of ATP, 10.3 kcal/mol in comparison with 7.3 kcal/mol. ATP, adenosine triphosphate; CrP, creatine phosphate; HEP, high-energy phosphate; ΔG, Gibbs free energy change.
The contents of HEPs vary significantly in different tissues. The highest levels of HEPs are found in muscle, heart, brain, spermatozoa, and retina (14). The concentration and distribution of HEPs in vivo can be determined non-invasively by 31P-magnetic resonance spectroscopy (MRS) (39, 40). The myocardial CrP/ATP ratio measured by 31P-MRS reflects the viability and energy metabolic status of cardiomyocytes (41). Over a wide range of cardiac workloads, the CrP/ATP ratio is essentially invariant and consistent with a constant free ADP concentration (42, 43). The cutoff point for CrP/ATP ratio (>1.60 and <1.60), which was established retrospectively and need to be evaluated prospectively, is a stronger predictor of cardiovascular death (44). The ratio is decreased upon myocardial ischemia (45, 46).
The Biosynthesis, Degradation and Turnover of Endogenous Creatine Phosphate
The biosynthesis of CrP begins by formation of creatine from three essential amino acids: arginine, glycine, and methionine (Figure 3) (14). The entire glycine molecule is incorporated whereas arginine furnishes its amidino group to yield guanidinoacetic acid (GAA), which then methylated at the amidino group to give creatine. It is postulated, but largely accepted, that the main route of creatine synthesis involves formation of guanidinoacetate in kidney, and methylation in liver (47–49). These reactions are respectively catalyzed by two rate-limiting enzymes, i.e., L-arginine:glycine amidinotransferase (AGAT) and S-adenosyl-L-methionine:N-guanidinoacetate methyltransferase (GAMT) (47–50). To complete the phosphorylation process, creatine is then transported to tissues such as muscle, heart, and brain by a specific Na+- and Cl−-dependent plasma membrane transporter (51). CrP production is catalyzed by creatine kinase (CK), which is a dimer of M and B (M = muscle, B = brain) subunits produced by different structural genes. Three isozymes are possible: BB, MB, and MM. Cardiac muscle contains significant amounts of CK-MB (25–46% of total CK activity, as opposed to <5% in skeletal muscle), so that in myocardial infarction the rise in serum total CK activity is accompanied by a parallel rise in that of CK-MB (14, 52, 53).
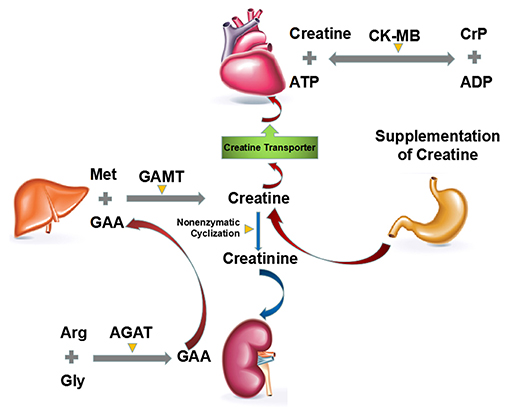
Figure 3. Major routes of biosynthesis, degradation and turnover of endogenous CrP. The biosynthesis of CrP begins by formation of creatine from three essential amino acids: arginine, glycine, and methionine. The main route of creatine synthesis involves formation of GAA in kidney, and methylation in liver. These reactions are, respectively, catalyzed by AGAT and GAMT. Then creatine is transported to heart by a specific Na+- and Cl−-dependent plasma membrane transporter. The degradation of creatine and CrP is an irreversible, non-enzymatic cyclization to creatinine, which should be supplemented by diet or de novo biosynthesis. ADP, adenosine diphosphate; AGAT, L-arginine:glycine amidinotransferase; ATP, adenosine triphosphate; CK, creatine kinase; CrP, creatine phosphate; GAA, guanidinoacetic acid; GAMT, S-adenosyl-L-methionine:N-guanidinoacetate methyltransferase.
Unlike the biosynthesis, the degradation of creatine and CrP is an irreversible, non-enzymatic cyclization to creatinine (Figure 3) (54, 55). Almost constant fraction of the body creatine (1.1%/day) and CrP (2.6%/day) is converted into creatinine, giving an overall conversion rate for total creatine pool (creatine + CrP) of ~1.7%/day (56).
For example, in a 70 kg man containing around 120 g of creatine pool, roughly 2 g/day are converted into creatinine and have to be replaced by creatine or CrP supplementation or from de novo biosynthesis (14).
Myocardial Metabolic Changes During Ischemia/Reperfusion: Substrates, Pathways, Metabolites, and Purine Nucleotide Cycle
Within a few seconds after coronary blood flow blockage, the oxygenated hemoglobin in ischemic zone rapidly depletes. The main pathway used to generate energy in myocardium changes from aerobic oxidation of mitochondria to cytoplasmic anaerobic glycolysis (Table 1) (57, 58). And the primary substrate of myocardial energy metabolism also changes from free fatty acids to glucose (Table 1) (57–61). However, the HEPs synthesized by glycolysis are far from meeting the energy requirements of heart. Under such condition, the ischemic myocardium preferentially utilizes the energy contained in endogenous CrP, followed by ATP, ADP, and adenosine monophosphate (AMP) (Figure 4) (62–67). AMP can also be decomposed into adenosine, hypoxanthine, etc. under the action of 5'-nucleotidase (Figure 4) (62, 68). The above reaction ultimately leads to a decrease in intracellular adenine nucleotide pool (ATP + ADP + AMP), resulting in a significant reduction in high-energy phosphate precursors. If the myocardium recover aerobic oxidation in a short period of time, AMP can be reoxidized to ADP and ATP to replenish energy. If not, it is no longer possible to reoxidize AMP to ADP or ATP. Furthermore, the lactic acid and other intermediate products produced by glycolysis accumulate in cardiomyocytes (Figure 4) (57, 58). After 10 min of ischemia, the intracellular pH will drop to 5.8–6.0 (69, 70). The rate of ADP rephosphorylation to ATP by anaerobic glycolysis is slowed down by acidosis (71).

Table 1. Myocardial energy metabolism: source, process and site of ATP production.
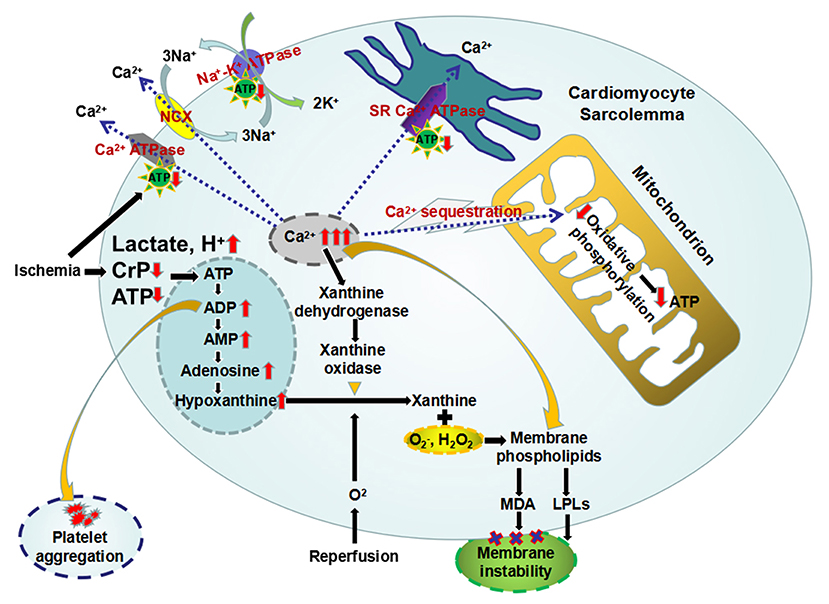
Figure 4. The primary metabolic changes and the secondary celluar injuries during myocardial ischemia/reperfusion. Ischemic myocardium preferentially utilizes the energy contained in CrP, followed by ATP, ADP, and AMP. And AMP can be further decomposed into adenosine and hypoxanthine, which leads to a decrease in intracellular adenine nucleotide pool. Furthermore, the lactic acid produced by glycolysis accumulate in cardiomyocytes, resulting intracellular acidosis. The loss of HEPs eliminates three of the four mechanisms of cellular calcium homeostasis, leading intracellular Ca2+ overload. Mitochondrial sequestration, the remaining mechanism, causes overloading of the mitochondria with Ca2+ and diminished capacity for oxidative phosphorylation. And overloaded intracellular Ca2+ induces the conversion of xanthine dehydrogenase to xanthine oxidase. The latter can produce oxygen free radicals, which in turn oxidize the membrane phospholipids and produce MDA, causing the membrane instability. In addition, intracellular accumulation of metabolic intermediates, including AMP, lactic acid, Ca2+, and H+, etc, may activate membrane phospholipase to make cell membrane degrade to LPLs, which also contribute to myocardial membrane instability. Increased ADP can induce platelet adhesion and aggregation. ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; CrP, creatine phosphate; HEPs, high-energy phosphates; LPLs, lysophospholipids; MDA, malondialdehyde.
Secondary to the metabolic changes, myocardial ischemia/reperfusion injuries occur as follows: intracellular Ca2+ overload, accumulation of arrhythmogenic intermediates and oxygen free radicals, myocardial membrane instability, electrophysiological changes in cardiomyocytes, mitochondrial damage, and platelet aggregation, etc (Figure 4).
Pathophysiological and Pharmacological Effects of Exogenous Creatine Phosphate on Myocardial Ischemia
The clinical effects of ATP in patients with cardiovascular disorders have been evaluated in early studies (72–74). Intravenous administration of ATP can interrupt the reentry pathways through the atrial ventricular node and restore normal sinus rhythm accompanied by relatively high incidences of advanced atrioventricular block and other adverse reactions, which makes paroxysmal supraventricular tachycardia the primary cardiovascular indication (75). And it seems quite paradoxical that oral administration of ATP may lead to a progressive diminution of plasma ATP level (76). Furthermore, exogenous ATP is a charged molecule containing three negative charges that is not freely permeable through cell membranes (77–79). In addition, there are enzymes that decompose ATP on the surface of cell membrane, including ATPase, adenylate kinase and AMP deaminase, which can split ATP into ADP, AMP, adenosine, and inorganic phosphate (80, 81). Since the first publication by Parrat and Marshall (82), CrP has been substantially demonstrated to be effective in protection of ischemic myocardium. The following we will focus on the pathophysiological and pharmacological effects of exogenous CrP, including but not limited to supplementing cellular energy.
Replenishment of Intracelluar ATP
It has been observed that the exogenous CrP could be incorporated into intracellular ATP molecules and increase the tissue level of ATP (83). Although exogenous CrP uptake was 3–4 orders of magnitude lower than ATP conversion in the case of normal cardiac work, it may be important in maintaining subsarcolemmal pools of CrP or ATP (Figure 4) (35, 83). The exogenous CrP uptake rate can be markedly increased in hypokinetic segments of ischemic myocardium (35, 83–86). Low-dose CrP may promote intracellular ATP synthesis mainly through substrate. After reaching a certain concentration of 10 mmol/L, it can significantly inhibit 5'-nucleotidase and AMP deaminase, thereby maintaining the nucleotide pool level, indicating that CrP does not only act as a energy substrate but also a regulator able to bind to the active sites of the enzymes and change their activity (62, 68, 87–90).
Attenuation of Intracellular Ca2+ Overload in Cardiomyocytes
Normally, extracellular fluid has a concentration of Ca2+ 10,000 times higher than intracellular fluid (91). Furthermore, there is an electrical force driving Ca2+ into the cell because of the negative resting membrane potential (91, 92). However, there is little leakage of Ca2+ into the cardiomyocyte except during the action potential. Even the Ca2+ that enters the cell during action potentials must be removed from the cell otherwise an accumulation of Ca2+ would lead to cellular dysfunction (92). Main mechanisms maintaining the intracellular to extracellular concentration and charge gradients include: (1) pumping Ca2+ out of the cytoplasm by the plasma membrane Ca2+ ATPase (93), (2) exchange of Ca2+ for Na+ driven by the intracellular to extracellular concentration gradient of Na+ as a result of the plasma membrane Na+-K+ ATPase (94), (3) sequestration of cytoplasm Ca2+ in sarcoplasmic reticulum (SR) by the SR Ca2+ ATPase (95), and (4) accumulation of intracellular Ca2+ by oxidation-dependent calcium sequestration inside the mitochondria (96). The loss of HEPs during ischemia eliminates three of the four mechanisms of cellular calcium homeostasis (Figure 4). Mitochondrial sequestration, the remaining mechanism, causes overloading of the mitochondria with Ca2+ and diminished capacity for oxidative phosphorylation (Figure 4) (97). Furthermore, activation of phospholipases and protein kinases (98), production of arachidonic acid (99, 100), and oxygen free radicals (101) are all involved in the destruction of membrane integrity. This, in turn, causes a massive and rapid influx of Ca2+ into the cell.
Several studies have shown that intracellular Ca2+ overload is a major cause of myocardial cell damage and cardiac dysfunction in IHD. CrP can reduce Ca2+ influx by providing energy to ATP-dependent Ca2+ ATPase and Na+-K+ ATPase on the plasma membrane (102, 103). At the same time, the Ca2+ ATPase activity on the sarcoplasmic reticulum is restored, and Ca2+ enter the sarcoplasmic reticulum to avoid the myocardial stiffness contracture (104). Furthermore, CrP binds to membrane phospholipids through zwitterionic interaction, which can enhance membrane stability (105, 106). In addition, CrP can also provide energy for the sliding of actin-myosin filaments, promoting the rapid recovery of myocardial contractility (107).
Protection of Heart From Oxidative Stress-Induced Myocardial Injury
And overloaded intracellular Ca2+ induces the conversion of xanthine dehydrogenase to xanthine oxidase (108–111). The latter can produce superoxide and xanthine from hypoxanthine upon reperfusion (Figure 4) (112). Furthermore, more damaging free radicals could be produced by the metal catalyzed Haber-Weiss reaction (113–115). The large amount of oxygen free radicals generated by the above reactions can in turn oxidize the membrane phospholipids and produce malondialdehyde (MDA), causing the membrane instability (Figure 4) (116). Zucchi et al. (117) found that supplementation of exogenous CrP could reduce the product of phospholipid peroxidation, MDA, by inhibiting ADP/AMP degradation and Ca2+ accumulation in cardiomyocytes. Myocardial peroxidation damage is alleviated through all of the above mechanisms.
Stabilization of Membrane Structure
Maintaining the integrity of the phospholipid bilayer membrane is a basic requirement for preserving overall cell viability. Myocardial membrane instability due to the decrease of ATP production and accumulation of acid metabolites plays a key role in the pathogenesis of ischemia-reperfusion injury, especially the electrophysiological manifestation of ischemia (118). The possibility that lysophospholipids (LPLs) contribute to myocardial membrane instability was first reported by Hajdu (119). Normally their concentration is maintained very low, but LPLs in sufficient quantities are potent detergents, which can alter general properties of the membrane such as fluidity and permeability (120). Furthermore, LPLs have been shown to affect the activities of plasma membrane Na+-K+ ATPase (121). Upon myocardial ischemia, intracellular accumulation of metabolic intermediates, including AMP, lactic acid, Ca2+, and H+, etc, may activate membrane phospholipase to make cell membrane degrade to LPLs (Figure 4). At 8 min after ischemia, a 60% increase in LPLs levels occurred, which could either be reacylated or transacylated to form precursor phospholipids or further degraded, depending on the energy state of the cell (121–123). Supplementation of exogenous CrP can provide energy to ATP-dependent Ca2+ ATPase and Na+-K+ ATPase on the plasma membrane and reduce the activation of anaerobic glycolysis, which blocks the process of phospholipids degradation and stabilizes the cell membrane. In addition, the integrity of the mitochondrial structure during ischemia is the basis for oxidative phosphorylation to synthesize ATP after reperfusion. CrP also has protective effects on the mitochondrial membrane and its oxidative phosphorylation function (124–126).
Broad Spectrum Antiarrhythmic Effects
Normally, the electrophysiological properties of cardiomyocytes require cell membrane integrity and maintaining of intracellular to extracellular concentration and charge gradients. Metabolic changes after myocardial ischemia, including the decrease of ATP production and accumulation of acid metabolites, lead to decreased activity of ATP-dependent transport systems. ATP-sensitive K+ channels (KATP), inactivated by normal cellular ATP levels, will open and permit K+ to leave the cell upon ischemia (127, 128). Furthermore, decreased activity of Na+/K+-ATPase leads to extracellular accumulation of K+ and inactivation of fast Na+ channels that are responsible for the rapid depolarization (129). These mechanisms lead to a series of electrophysiological changes in cardiomyocytes, including: (1) the resting membrane potential and the action potential amplitude are significantly decreased; (2) the depolarization speed is slowed down; (3) the action potential duration (APD) is shortened; (4) the distance from the resting membrane potential to the K+ equilibrium potential is increased; (5) the conduction velocity rate is slowed down (130). All of the above changes ultimately can contribute to arrhythmias.
Studies have shown that in myocardial ischemia and reperfusion, CrP can play a broad spectrum antiarrhythmic effects through several electrophysiological mechanisms, including but not limited to ATP replenishment (131). Firstly, by providing energy to ATP-dependent KATP channels and Na+/K+-ATPase, exogenous CrP can reduce extracellular accumulation of K+ and reactivate the fast Na+ channels, suggesting a Class I antiarrhythmic role (132). Secondly, by prolonging ventricular myocardium APD and effective refractory period (ERP) under normoxic but not ischemic conditions, exogenous CrP can prevent reentrant circuits forming between the ischemic and non-ischemic zone and play a class III antiarrhythmic role (132, 133). Thirdly, by attenuating intracellular Ca2+ overload, exogenous CrP can inhibit Ca2+-mediated activation of inward current channels and triggered activity, exerting a class IV antiarrhythmic role (134, 135). Furthermore, exogenous CrP can also play an antiarrhythmic role by reducing the accumulation of arrhythmogenic lysophosphoglycerides and increasing the threshold of ventricular fibrillation (136–138).
Inhibiting Platelet Aggregation and Improving Microvascular Function
It is known that ADP can not only induce platelet adhesion and aggregation, but also amplify the aggregation effects of collagen, thrombin and other inducers (Figure 4) (139, 140). ADP may still affect the platelets when the arachidonate pathway is blocked (141). Exogenous CrP can inhibit platelet aggregation and then improve the microvascular function by rapid removal of ADP and formation of ATP, which is an inhibitor of ADP-induced platelet aggregation (19, 142).
Clinical Application of Exogenous Creatine Phosphate in Ischemic Heart Disease: Evidence and Evaluation
As mentioned above, energy metabolic abnormalities are the upstream and primary pathophysiologic manifestation of myocardial ischemia. Whereas, hemodynamic, electrophysiological, morphological, clinical, biochemical and imaging changes are the downstream, and secondary consequence of myocardial energy metabolic abnormalities. The depletion of HEPs is involved in both upstream and downstream changes in myocardial ischemia. As demonstrated in vitro and animal experiments, CrP was suggested to be potentially beneficial in patients with acute and chronic myocardial ischaemic injury through multiple mechanisms, including but not limited to ATP replenishment. In fact, results from a large number of clinical studies substantially support that supplementation of exogenous CrP is associated with improved short-term survival (143, 144), enhancement of cardiac systolic and diastolic function (145–147), lower peak CK-MB/troponin release (20, 148–152), reduction in the incidence of major arrhythmias (144, 151, 153–156), etc. There is still uncertainty, however, whether the administration of exogenous CrP can improve long-term outcomes, rather than just the secondary endpoints or pathophysiological process of IHD.
Limitations and Perspectives
According to a meta-analysis performed by Landoni et al. (16), although more than 4,000 articles were screened, only 12 studies comparing CrP with placebo or standard treatment in patients with IHD met the design requirements for controlled or case-matched clinical trials. Unfortunately, there is insufficient statistical power to obtain results on long-term survival due to the common limitations, including: (1) single center trial; (2) small sample size; (3) short-term follow-up; (4) secondary end-points; (5) choice of standard treatment rather than placebo as the comparator; (6) administration routes and doses of CrP varying significantly among the studies; (7) inadequate baseline information or baseline bias (20, 143, 144, 146, 150, 151, 153, 156). In addition, majority of the studies were published before the “era of revascularization” and patients were recruited from those undergoing non-revascularization therapy or mixed, significantly different from the current practice (143, 144, 146, 150, 153, 156).
At first glance, it is surprising that exogenous CrP has not been shown to improve long-term survival in clinical studies. In fact, there are two sides to the same issue. On one side, CrP may plays extensive roles in every physiological and pathophysiological process from upstream to downstream of myocardial ischemia. On the other side, the myocardial intracellular actions of CrP lack target and pathway specificity. Furthermore, the uptake and distribution of exogenous CrP in vivo lack of tissue and cell specificity. Such non-specificities lead to uncertainties in the dominant pharmacological mechanism, optimal administration route and dose, as well as treatment window of exogenous CrP in individualized patients with IHD. Moreover, the cardioprotection of exogenous CrP may be limited by endogenous CrP levels. However, owing to the physiological, pathophysiological, and pharmacological plausibility of its effects and to the concordance of the beneficial effects of exogenous CrP on multiple secondary but important outcomes and short-term survival, there is urgent need for high-quality multicentre randomized controlled trials (RCTs) to confirm long-term survival improvement. In addition, further studies are needed to investigate the causality between changes in endogenous/exogenous CrP levels and IHD progression and prognosis (157).
To better understand the pathophysiological and pharmacological effects, we specified the context for all cited researches as cell study (19, 23–27, 29–35, 48–54, 69, 70, 91–103, 118, 128), animal study (12, 41, 42, 45, 46, 65, 66, 68, 71–74, 76–78, 81–90, 99, 104, 111, 116–119, 129, 133–135) and human study (15–18, 20, 21, 37–40, 44, 58, 108–110, 143–156). Furthermore, we detailed the indications, contraindications, side effects, and application instructions of CrP supplement in Table 2.
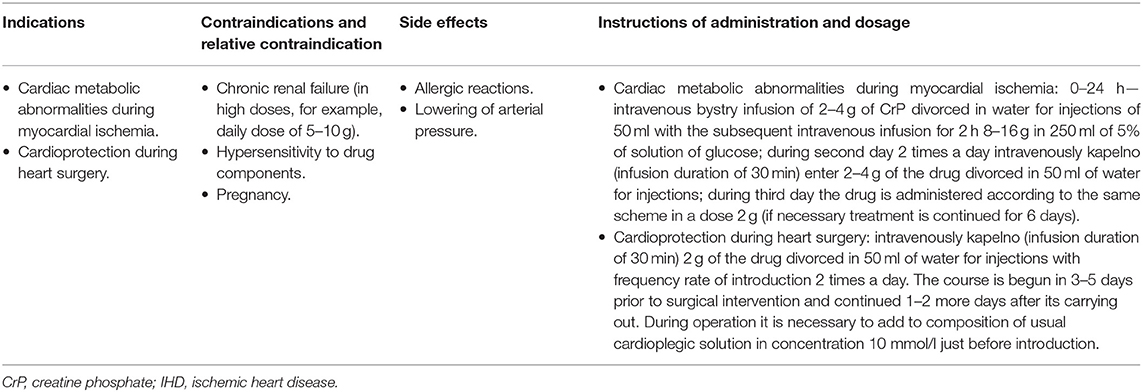
Table 2. The indications, contraindications, side effects, and application instructions of CrP supplement for IHD.
Conclusions
The purpose of this article is to provide a comprehensive and concise description of the cellular and molecular aspects of cardioprotective mechanisms and a critical evaluation of the clinical evidence of HEPs in IHD. According to the well-documented physiological, pathophysiological and pharmacological properties of HEPs, exogenous CrP may be considered as an ideal metabolic regulator. It plays cardioprotection roles from upstream to downstream of myocardial ischemia through multiple complex mechanisms, including but not limited to replenishment of cellular energy. Although exogenous CrP administration has not been shown to improve long-term survival, the beneficial effects on multiple secondary but important outcomes and short-term survival are concordant with its pathophysiological and pharmacological effects. There is urgent need for high-quality multicentre RCTs to confirm long-term survival improvement in the future.
Author Contributions
HY-D, ZY-X, and YS-W contributed toward drafting and critically reviewing the document and agree to be accountable for all aspects of the work. YS-W and ZY-J provided his views and comments on the manuscript, made the final decision about the journal selection as well as approved the submission of the manuscript to the journal. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ADP, adenosine diphosphate; AGAT, L-arginine:glycine amidinotransferase; AMP, adenosine monophosphate; APD, action potential duration; ATP, adenosine triphosphate; CK, creatine kinase; CrP, creatine phosphate; ERP, effective refractory period; GAA, guanidinoacetic acid; GAMT, S-adenosyl-L-methionine:N-guanidinoacetate methyltransferase; HEPs, high-energy phosphates; IHD, ischemic heart disease; KATP, ATP-sensitive K+ channels; LPLs, lysophospholipids; MDA, malondialdehyde; MEMT, myocardial energy metabolic therapy; MRS, magnetic resonance spectroscopy; OP, oxidative phosphorylation; RCTs, randomized controlled trials; SP, substrate phosphorylation.
References
1. Opie LH. Proof that glucose-insulin-potassium provides metabolic protection of ischaemic myocardium. Lancet. (1999) 353:768–9. doi: 10.1016/S0140-6736(98)00385-7
2. Weiss RG, Bottomley PA, Hardy CJ, Gerstenblith G. Regional myocardial metabolism of high-energy phosphates during isometric exercise in patients with coronary artery disease. N Engl J Med. (1990) 323:1593–600. doi: 10.1056/NEJM199012063232304
3. Fihn SD, Blankenship JC, Alexander KP, Bittl JA, Byrne JG, Fletcher BJ, et al. 2014 ACC/AHA/AATS/PCNA/SCAI/STS focused update of the guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines, and the American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. (2014) 64:1929–49. doi: 10.1016/j.jacc.2014.07.017
4. Amsterdam EA, Wenger NK, Brindis RG, Casey DE, Ganiats TG, Holmes DR, et al. 2014 AHA/ACC Guideline for the Management of Patients with Non-ST-Elevation Acute Coronary Syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. (2014) 64:e139–228. doi: 10.1016/j.jacc.2014.09.016
5. Levine GN, Bates ER, Blankenship JC, Bailey SR, Bittl JA, Cercek B, et al. 2015 ACC/AHA/SCAI Focused Update on Primary Percutaneous Coronary Intervention for Patients With ST-Elevation Myocardial Infarction: an Update of the 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention and the 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction. J Am Coll Cardiol. (2016) 67:1235–50. doi: 10.1016/j.jacc.2015.10.005
6. Knuuti J, Wijns W, Saraste A, Capodanno D, Barbato E, Funck-Brentano C, et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur Heart J. (2020) 41:407–77. doi: 10.1093/eurheartj/ehz425
7. Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. (2018) 39:119–77. doi: 10.1093/eurheartj/ehx393
8. Roffi M, Patrono C, Collet JP, Mueller C, Valgimigli M, Andreotti F, et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J. (2016) 37:267–315. doi: 10.1093/eurheartj/ehv320
9. Kolwicz SC, Tian R. Metabolic therapy at the crossroad: how to optimize myocardial substrate utilization. Trends Cardiovasc Med. (2009) 19:201–7. doi: 10.1016/j.tcm.2009.12.005
10. Stanley WC. Myocardial energy metabolism during ischemia and the mechanisms of metabolic therapies. J Cardiovasc Pharmacol Ther. (2004) 9(Suppl. 1):S31–45. doi: 10.1177/107424840400900104
11. Fruchart JC, Davignon J, Hermans MP, Al-Rubeaan K, Amarenco P, Assmann G, et al. Residual macrovascular risk in 2013: what have we learned. Cardiovasc Diabetol. (2014) 13:26. doi: 10.1186/1475-2840-13-26
12. Eggleton P, Eggleton GP. The inorganic phosphate and a labile form of organic phosphate in the gastrocnemius of the frog. Biochem J. (1927) 21:190–5. doi: 10.1042/bj0210190
13. Ennor AH, Morrison JF. Biochemistry of the phosphagens and related guanidines. Physiol Rev. (1958) 38:631–74. doi: 10.1152/physrev.1958.38.4.631
14. Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. (2000) 80:1107–213. doi: 10.1152/physrev.2000.80.3.1107
15. Mingxing F, Landoni G, Zangrillo A, Monaco F, Lomivorotov VV, Hui C, et al. Phosphocreatine in cardiac surgery patients: a meta-analysis of randomized controlled trials. J Cardiothorac Vasc Anesth. (2018) 32:762–70. doi: 10.1053/j.jvca.2017.07.024
16. Landoni G, Zangrillo A, Lomivorotov VV, Likhvantsev V, Ma J, De Simone F, et al. Cardiac protection with phosphocreatine: a meta-analysis. Interact Cardiovasc Thorac Surg. (2016) 23:637–46. doi: 10.1093/icvts/ivw171
17. Sharov VG, Saks VA, Kupriyanov VV, Lakomkin VL, Kapelko VI, AYa S, et al. Protection of ischemic myocardium by exogenous phosphocreatine. I. Morphologic and phosphorus 31-nuclear magnetic resonance studies. J Thorac Cardiovasc Surg. (1987) 94:749–61. doi: 10.1016/S0022-5223(19)36191-4
18. Semenovsky ML, Shumakov VI, Sharov VG, Mogilevsky GM, Asmolovsky AV, Makhotina LA, et al. Protection of ischemic myocardium by exogenous phosphocreatine. II. Clinical, ultrastructural, and biochemical evaluations. J Thorac Cardiovasc Surg. (1987) 94:762–9. doi: 10.1016/S0022-5223(19)36192-6
19. Sharov VG, Afonskaya NI, Ruda MY, Cherpachenko NM, EYa P, Markosyan RA, et al. Protection of ischemic myocardium by exogenous phosphocreatine (neoton): pharmacokinetics of phosphocreatine, reduction of infarct size, stabilization of sarcolemma of ischemic cardiomyocytes, and antithrombotic action. Biochem Med Metab Biol. (1986) 35:101–14. doi: 10.1016/0885-4505(86)90064-2
20. Ke-Wu D, Xu-Bo S, Ying-Xin Z, Shi-Wei Y, Yu-Jie Z, Dong-Mei S, et al. The effect of exogenous creatine phosphate on myocardial injury after percutaneous coronary intervention. Angiology. (2015) 66:163–8. doi: 10.1177/0003319713515996
21. Yang SW, Park KH, Zhou YJ. The impact of hypoglycemia on the cardiovascular system: physiology and pathophysiology. Angiology. (2016) 67:802–9. doi: 10.1177/0003319715623400
22. Strumia E, Pelliccia F, D'Ambrosio G. Creatine phosphate: pharmacological and clinical perspectives. Adv Ther. (2012) 29:99–123. doi: 10.1007/s12325-011-0091-4
23. Fiske CH, Subbarow Y. The nature of the “inorganic phosphate” in voluntary muscle. Science. (1927) 65:401–3. doi: 10.1126/science.65.1686.401
24. de Meis L. How enzymes handle the energy derived from the cleavage of high-energy phosphate compounds. J Biol Chem. (2012) 287:16987–7005. doi: 10.1074/jbc.X112.363200
25. Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. (2010) 88:40–50. doi: 10.1093/cvr/cvq240
26. Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. (2008) 1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011
27. D'Alessandro M, Melandri BA. ATP hydrolysis in ATP synthases can be differently coupled to proton transport and modulated by ADP and phosphate: a structure based model of the mechanism. Biochim Biophys Acta. (2010) 1797:755–62. doi: 10.1016/j.bbabio.2010.03.007
28. Langen P, Hucho F. Karl Lohmann and the discovery of ATP. Angew Chem Int Ed Engl. (2008) 47:1824–7. doi: 10.1002/anie.200702929
29. Erickson-Viitanen S, Geiger P, Yang WC, Bessman SP. The creatine-creatine phosphate shuttle for energy transport-compartmentation of creatine phosphokinase in muscle. Adv Exp Med Biol. (1982) 151:115–25. doi: 10.1007/978-1-4684-4259-5_17
30. McClellan G, Weisberg A, Winegrad S. Energy transport from mitochondria to myofibril by a creatine phosphate shuttle in cardiac cells. Am J Physiol. (1983) 245:C423–7. doi: 10.1152/ajpcell.1983.245.5.C423
31. Savabi F, Geiger PJ, Bessman SP. Myofibrillar end of the creatine phosphate energy shuttle. Am J Physiol. (1984) 247:C424–32. doi: 10.1152/ajpcell.1984.247.5.C424
32. Bessman SP, Carpenter CL. The creatine-creatine phosphate energy shuttle. Annu Rev Biochem. (1985) 54:831–62. doi: 10.1146/annurev.bi.54.070185.004151
33. Bessman SP. The creatine phosphate energy shuttle–the molecular asymmetry of a “pool”. Anal Biochem. (1987) 161:519–23. doi: 10.1016/0003-2697(87)90483-0
34. Saks VA, Rosenshtraukh LV, Smirnov VN, Chazov EI. Role of creatine phosphokinase in cellular function and metabolism. Can J Physiol Pharmacol. (1978) 56:691–706. doi: 10.1139/y78-113
35. Jacobus WE. Respiratory control and the integration of heart high-energy phosphate metabolism by mitochondrial creatine kinase. Annu Rev Physiol. (1985) 47:707–25. doi: 10.1146/annurev.ph.47.030185.003423
37. Kerksick CM, Roberts MD, Dalbo VJ, Sunderland KL. Intramuscular phosphagen status and the relationship to muscle performance across the age spectrum. Eur J Appl Physiol. (2016) 116:115–27. doi: 10.1007/s00421-015-3246-1
39. Solaiyappan M, Weiss RG, Bottomley PA. Neural-network classification of cardiac disease from 31P cardiovascular magnetic resonance spectroscopy measures of creatine kinase energy metabolism. J Cardiovasc Magn Reson. (2019) 21:49. doi: 10.1186/s12968-019-0560-5
40. Melenovsky V, Hlavata K, Sedivy P, Dezortova M, Borlaug BA, Petrak J, et al. Skeletal muscle abnormalities and iron deficiency in chronic heart failurean exercise 31P magnetic resonance spectroscopy study of calf muscle. Circ Heart Fail. (2018) 11:e004800. doi: 10.1161/CIRCHEARTFAILURE.117.004800
41. Weiss RG, Chatham JC, Georgakopolous D, Charron MJ, Wallimann T, Kay L, et al. An increase in the myocardial PCr/ATP ratio in GLUT4 null mice. FASEB J. (2002) 16:613–5. doi: 10.1096/fj.01-0462fje
42. Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science. (1986) 232:1121–3. doi: 10.1126/science.3704638
43. Zhang J, Duncker DJ, Xu Y, Zhang Y, Path G, Merkle H, et al. Transmural bioenergetic responses of normal myocardium to high workstates. Am J Physiol. (1995) 268:H1891–905. doi: 10.1152/ajpheart.1995.268.5.H1891
44. Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. (1997) 96:2190–6. doi: 10.1161/01.CIR.96.7.2190
45. McDonald KM, Yoshiyama M, Francis GS, Ugurbil K, Cohn JN, Zhang J. Myocardial bioenergetic abnormalities in a canine model of left ventricular dysfunction. J Am Coll Cardiol. (1994) 23:786–93. doi: 10.1016/0735-1097(94)90769-2
46. Liao R, Nascimben L, Friedrich J, Gwathmey JK, Ingwall JS. Decreased energy reserve in an animal model of dilated cardiomyopathy. Relationship to contractile performance. Circ Res. (1996) 78:893–902. doi: 10.1161/01.RES.78.5.893
47. Kreider RB, Kalman DS, Antonio J, Ziegenfuss TN, Wildman R, Collins R, et al. International Society of Sports Nutrition position stand: safety and efficacy of creatine supplementation in exercise, sport, and medicine. J Int Soc Sports Nutr. (2017) 14:18. doi: 10.1186/s12970-017-0173-z
48. Popolo A, Adesso S, Pinto A, Autore G, Marzocco S. L-Arginine and its metabolites in kidney and cardiovascular disease. Amino Acids. (2014) 46:2271–86. doi: 10.1007/s00726-014-1825-9
49. Barcelos RP, Stefanello ST, Mauriz JL, Gonzalez-Gallego J, Soares FA. Creatine and the liver: metabolism and possible interactions. Mini Rev Med Chem. (2016) 16:12–8. doi: 10.2174/1389557515666150722102613
50. Iqbal F, Hoeger H, Lubec G, Bodamer O. Biochemical and behavioral phenotype of AGAT and GAMT deficient mice following long-term creatine monohydrate supplementation. Metab Brain Dis. (2017) 32:1951–61. doi: 10.1007/s11011-017-0092-3
51. Sora I, Richman J, Santoro G, Wei H, Wang Y, Vanderah T, et al. The cloning and expression of a human creatine transporter. Biochem Biophys Res Commun. (1994) 204:419–27. doi: 10.1006/bbrc.1994.2475
52. Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the 'phosphocreatine circuit' for cellular energy homeostasis. Biochem J. (1992) 281 (Pt 1):21–40. doi: 10.1042/bj2810021
53. Saks VA, Ventura-Clapier R, Aliev MK. Metabolic control and metabolic capacity: two aspects of creatine kinase functioning in the cells. Biochim Biophys Acta. (1996) 1274:81–8. doi: 10.1016/0005-2728(96)00011-4
54. Brosnan ME, Brosnan JT. Renal arginine metabolism. J Nutr. (2004) 134:2791S−5S; discussion 2796S−2797S. doi: 10.1093/jn/134.10.2791S
55. Kashani K, Rosner MH, Ostermann M. Creatinine: from physiology to clinical application. Eur J Intern Med. (2020) 72:9–14. doi: 10.1016/j.ejim.2019.10.025
56. Walker JB. Creatine: biosynthesis, regulation, and function. Adv Enzymol Relat Areas Mol Biol. (1979) 50:177–242. doi: 10.1002/9780470122952.ch4
57. Mallet RT, Manukhina EB, Ruelas SS, Caffrey JL, Downey HF. Cardioprotection by intermittent hypoxia conditioning: evidence, mechanisms, and therapeutic potential. Am J Physiol Heart Circ Physiol. (2018) 315:H216–32. doi: 10.1152/ajpheart.00060.2018
58. Ait-Aissa K, Blaszak SC, Beutner G, Tsaih SW, Morgan G, Santos JH, et al. Mitochondrial oxidative phosphorylation defect in the heart of subjects with coronary artery disease. Sci Rep. (2019) 9:7623. doi: 10.1038/s41598-019-43761-y
59. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
60. Lopaschuk GD, Collins-Nakai RL, Itoi T. Developmental changes in energy substrate use by the heart. Cardiovasc Res. (1992) 26:1172–80. doi: 10.1093/cvr/26.12.1172
61. Kodde IF, van der Stok J, Smolenski RT, de Jong JW. Metabolic and genetic regulation of cardiac energy substrate preference. Comp Biochem Physiol A Mol Integr Physiol. (2007) 146:26–39. doi: 10.1016/j.cbpa.2006.09.014
62. Scolletta S, Biagioli B. Energetic myocardial metabolism and oxidative stress: let's make them our friends in the fight against heart failure. Biomed Pharmacother. (2010) 64:203–7. doi: 10.1016/j.biopha.2009.10.002
63. Heather LC, Clarke K. Metabolism, hypoxia and the diabetic heart. J Mol Cell Cardiol. (2011) 50:598–605. doi: 10.1016/j.yjmcc.2011.01.007
64. Sano M, Fukuda K. [Metabolic remodeling in the ischemic and non-ischemic failing heart]. Nihon Rinsho. (2011) 69(Suppl. 7):60–8.
65. Braasch W, Gudbjarnason S, Puri PS, Ravens KG, Bing RJ. Early changes in energy metabolism in the myocardium following acute coronary artery occlusion in anesthetized dogs. Circ Res. (1968) 23:429–38. doi: 10.1161/01.RES.23.3.429
66. Neely JR, Rovetto MJ, Whitmer JT, Morgan HE. Effects of ischemia on function and metabolism of the isolated working rat heart. Am J Physiol. (1973) 225:651–8. doi: 10.1152/ajplegacy.1973.225.3.651
67. Jennings RB, Murry CE, Steenbergen C, Reimer KA. Development of cell injury in sustained acute ischemia. Circulation. (1990) 82:II2.
68. Headrick JP, Willis RJ. 5'-Nucleotidase activity and adenosine formation in stimulated, hypoxic and underperfused rat heart. Biochem J. (1989) 261:541–50. doi: 10.1042/bj2610541
69. Vanheel B, Van de Voorde J. Differential influence of extracellular and intracellular pH on K+ accumulation in ischaemic mammalian cardiac tissue. J Mol Cell Cardiol. (1995) 27:1443–55. doi: 10.1006/jmcc.1995.0136
70. Beauloye C, Bertrand L, Krause U, Marsin AS, Dresselaers T, Vanstapel F, et al. No-flow ischemia inhibits insulin signaling in heart by decreasing intracellular pH. Circ Res. (2001) 88:513–9. doi: 10.1161/01.RES.88.5.513
71. Lancaster MK, Harrison SM. Changes in contraction, cytosolic Ca2+ and pH during metabolic inhibition and upon restoration of mitochondrial respiration in rat ventricular myocytes. Exp Physiol. (1998) 83:349–60. doi: 10.1113/expphysiol.1998.sp004118
72. Kopf GS, Chaudry I, Condos S, Baue AE. Reperfusion with ATP-MgCl2 following prolonged ischemia improves myocardial performance. J Surg Res. (1987) 43:114–7. doi: 10.1016/0022-4804(87)90152-1
73. Thelin S, Hultman J, Ronquist G, Hansson HE. Myocardial high-energy phosphates, lactate and pyruvate during moderate or severe normothermic ischemia in rat hearts perfused with phosphoenolpyruvate and ATP in cardioplegic solution. Scand J Thorac Cardiovasc Surg. (1987) 21:245–9. doi: 10.3109/14017438709106033
74. Martinesi L, Belli C. [Experimental study on the effects of polarizing solutions with and without ATP on the electrocardiographic pattern of myocardial ischemia caused by pitressin]. G Clin Med. (1967) 48:720–46.
75. Camm AJ, Garratt CJ. Adenosine and supraventricular tachycardia. N Engl J Med. (1991) 325:1621–9. doi: 10.1056/NEJM199112053252306
76. Kichenin K, Seman M. Chronic oral administration of ATP modulates nucleoside transport and purine metabolism in rats. J Pharmacol Exp Ther. (2000) 294:126–33.
77. Glynn IM. Membrane adenosine triphosphatase and cation transport. Br Med Bull. (1968) 24:165–9. doi: 10.1093/oxfordjournals.bmb.a070620
78. Dieterle Y, Ody C, Ehrensberger A, Stalder H, Junod AF. Metabolism and uptake of adenosine triphosphate and adenosine by porcine aortic and pulmonary endothelial cells and fibroblasts in culture. Circ Res. (1978) 42:869–76. doi: 10.1161/01.RES.42.6.869
80. Dunkley CR, Manery JF, Dryden EE. The conversion of ATP to IMP by muscle surface enzymes. J Cell Physiol. (1966) 68:241–7. doi: 10.1002/jcp.1040680305
81. Fedelesová M, Ziegelhöffer A, Krause EG, Wollenberger A. Effect of exogenous adenosine triphosphate on the metabolic state of the excised hypothermic dog heart. Circ Res. (1969) 24:617–27. doi: 10.1161/01.RES.24.5.617
82. Parratt JR, Marshall RJ. The response of isolated cardiac muscle to acute anoxia: protective effect of adenosine triphosphate and creatine phosphate. J Pharm Pharmacol. (1974) 26:427–33. doi: 10.1111/j.2042-7158.1974.tb09308.x
83. Down WH, Chasseaud LF, Ballard SA. The effect of intravenously administered phosphocreatine on ATP and phosphocreatine concentrations in the cardiac muscle of the rat. Arzneimittelforschung. (1983) 33:552–4.
84. Thelin S, Hultman J, Ronquist G, Juhlin C, Hansson HE, Lindgren PG. Improved myocardial protection by creatine phosphate in cardioplegic solution. An in vivo study in the pig during normothermic ischemia. Thorac Cardiovasc Surg. (1987) 35:137–42. doi: 10.1055/s-2007-1020217
85. Thelin S, Hultman J, Ronquist G, Hansson HE. Metabolic and functional effects of creatine phosphate in cardioplegic solution. Studies on rat hearts during and after normothermic ischemia. Scand J Thorac Cardiovasc Surg. (1987) 21:39–45. doi: 10.3109/14017438709116917
86. Rosenshtraukh LV, Saks VA, Yuriavichus IA, Nesterenko VV, Undrovinas AI, Smirnov VN, et al. Effect of creatine phosphate on the slow inward calcium current, action potential, and contractile force of frog atrium and bentricle. Biochem Med. (1979) 21:1–5. doi: 10.1016/0006-2944(79)90049-8
87. Schopf G, Rumpold H, Müller MM. Alterations of purine salvage pathways during differentiation of rat heart myoblasts towards myocytes. Biochim Biophys Acta. (1986) 884:319–25. doi: 10.1016/0304-4165(86)90180-7
88. Lewandowski ED, White LT. Pyruvate dehydrogenase influences postischemic heart function. Circulation. (1995) 91:2071–9. doi: 10.1161/01.CIR.91.7.2071
89. Snaith CD, Wright G, Lofkin M. The effects of aspartate and 2-oxoglutarate upon glycolytic energy metabolites and mechanical recovery following global ischaemia in isolated rat hearts. J Mol Cell Cardiol. (1992) 24:305–15. doi: 10.1016/0022-2828(92)93167-I
90. Ronca-Testoni S, Raggi A, Ronca G. Muscle AMP aminohydrolase. 3. A comparative study on the regulatory properties of skeletal muscle enzyme from various species. Biochim Biophys Acta. (1970) 198:101–12. doi: 10.1016/0005-2744(70)90038-0
91. Peters T. Basic mechanisms of cellular calcium homeostasis. Acta Otolaryngol Suppl. (1988) 460:7–12. doi: 10.3109/00016488809125129
93. Carafoli E, Malmström K, Sigel E, Crompton M. The regulation of intracellular calcium. Clin Endocrinol. (1976) 5(Suppl.):49S−59S. doi: 10.1111/j.1365-2265.1976.tb03815.x
94. Carafoli E. The regulation of intracellular calcium. Adv Exp Med Biol. (1982) 151:461–72. doi: 10.1007/978-1-4684-4259-5_51
95. Blaustein MP, Ratzlaff RW, Kendrick NK. The regulation of intracellular calcium in presynaptic nerve terminals. Ann N Y Acad Sci. (1978) 307:195–212. doi: 10.1111/j.1749-6632.1978.tb41943.x
96. Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. (1967) 213:137–9. doi: 10.1038/213137a0
98. Golfman LS, Haughey NJ, Wong JT, Jiang JY, Lee D, Geiger JD, et al. Lysophosphatidylcholine induces arachidonic acid release and calcium overload in cardiac myoblastic H9c2 cells. J Lipid Res. (1999) 40:1818–26. doi: 10.1016/S0022-2275(20)34898-7
99. Hoffmann P, Richards D, Heinroth-Hoffmann I, Mathias P, Wey H, Toraason M. Arachidonic acid disrupts calcium dynamics in neonatal rat cardiac myocytes. Cardiovasc Res. (1995) 30:889–98. doi: 10.1016/S0008-6363(95)00133-6
100. Oe H, Kuzuya T, Hoshida S, Nishida M, Hori M, Kamada T, et al. Calcium overload and cardiac myocyte cell damage induced by arachidonate lipoxygenation. Am J Physiol. (1994) 267:H1396–402. doi: 10.1152/ajpheart.1994.267.4.H1396
101. Chang JC, Lien CF, Lee WS, Chang HR, Hsu YC, Luo YP, et al. Intermittent hypoxia prevents myocardial mitochondrial Ca2+ overload and cell death during ischemia/reperfusion: the role of reactive oxygen species. Cells. (2019) 8:564. doi: 10.3390/cells8060564
102. Semb SO, Sejersted OM. Calcium induced contracture stimulates Na,K-pump rate in isolated sheep cardiac Purkinje fibers. J Mol Cell Cardiol. (1997) 29:2197–212. doi: 10.1006/jmcc.1997.0455
103. Dhalla NS, Singh JN, McNamara DB, Bernatsky A, Singh A, Harrow JA. Energy production and utilization in contractile failure due to intracellular calcium overload. Adv Exp Med Biol. (1983) 161:305–16. doi: 10.1007/978-1-4684-4472-8_16
104. Tani M, Hasegawa H, Suganuma Y, Shinmura K, Kayashi Y, Nakamura Y. Protection of ischemic myocardium by inhibition of contracture in isolated rat heart. Am J Physiol. (1996) 271:H2515–9. doi: 10.1152/ajpheart.1996.271.6.H2515
105. Tokarska-Schlattner M, Epand RF, Meiler F, Zandomeneghi G, Neumann D, Widmer HR, et al. Phosphocreatine interacts with phospholipids, affects membrane properties and exerts membrane-protective effects. PLoS ONE. (2012) 7:e43178. doi: 10.1371/journal.pone.0043178
106. Saks VA, Dzhaliashvili IV, Konorev EA, Strumia E. [Molecular and cellular aspects of the cardioprotective mechanism of phosphocreatine]. Biokhimiia. (1992) 57:1763–84.
107. Günther J, Oddoy A, Schubert E. [Mechanical characteristics of isotonic work and high-energy phosphates in frog ventricle after 1-fluoro-2,4-dinitrobenzene]. Acta Biol Med Ger. (1978) 37:613–21.
108. Srivastava SK, Ansari NH, Liu S, Izban A, Das B, Szabo G, et al. The effect of oxidants on biomembranes and cellular metabolism. Mol Cell Biochem. (1989) 91:149–57. doi: 10.1007/BF00228090
109. Malis CD, Bonventre JV. Mechanism of calcium potentiation of oxygen free radical injury to renal mitochondria. A model for post-ischemic and toxic mitochondrial damage. J Biol Chem. (1986) 261:14201–8. doi: 10.1016/S0021-9258(18)67004-8
110. Kock R, Delvoux B, Sigmund M, Greiling H. A comparative study of the concentrations of hypoxanthine, xanthine, uric acid and allantoin in the peripheral blood of normals and patients with acute myocardial infarction and other ischaemic diseases. Eur J Clin Chem Clin Biochem. (1994) 32:837–42. doi: 10.1515/cclm.1994.32.11.837
111. Gneushev ET, Naumova VV, Bogoslovskii VA. [Hypoxanthine content of peripheral venous blood in infarct and ischemia of the myocardium]. Ter Arkh. (1978) 50:20–4.
112. McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. (1985) 312:159–63. doi: 10.1056/NEJM198501173120305
113. Fridovich I. Superoxide radical: an endogenous toxicant. Annu Rev Pharmacol Toxicol. (1983) 23:239–57. doi: 10.1146/annurev.pa.23.040183.001323
114. McCord JM. The superoxide free radical: its biochemistry and pathophysiology. Surgery. (1983) 94:412–4.
115. Tien M, Svingen BA, Aust SD. An investigation into the role of hydroxyl radical in xanthine oxidase-dependent lipid peroxidation. Arch Biochem Biophys. (1982) 216:142–51. doi: 10.1016/0003-9861(82)90198-9
116. Ballagi-Pordány G, Richter J, Koltai M, Aranyi Z, Pogátsa G, Schaper W. Is malondialdehyde a marker of the effect of oxygen free radicals in rat heart tissue. Basic Res Cardiol. (1991) 86:266–72. doi: 10.1007/BF02190606
117. Zucchi R, Poddighe R, Limbruno U, Mariani M, Ronca-Testoni S, Ronca G. Protection of isolated rat heart from oxidative stress by exogenous creatine phosphate. J Mol Cell Cardiol. (1989) 21:67–73. doi: 10.1016/0022-2828(89)91494-6
118. Houang EM, Bartos J, Hackel BJ, Lodge TP, Yannopoulos D, Bates FS, et al. Cardiac muscle membrane stabilization in myocardial reperfusion injury. JACC Basic Transl Sci. (2019) 4:275–87. doi: 10.1016/j.jacbts.2019.01.009
119. Hajdu S, Weiss H, Titus E. The isolation of a cardiac active principle from mammalian tissue. J Pharmacol Exp Ther. (1957) 120:99–113.
120. Weltzien HU. Cytolytic and membrane-perturbing properties of lysophosphatidylcholine. Biochim Biophys Acta. (1979) 559:259–87. doi: 10.1016/0304-4157(79)90004-2
121. Shaikh NA, Downar E. Time course of changes in porcine myocardial phospholipid levels during ischemia. A reassessment of the lysolipid hypothesis. Circ Res. (1981) 49:316–25. doi: 10.1161/01.RES.49.2.316
122. van den Bosch H. Phosphoglyceride metabolism. Annu Rev Biochem. (1974) 43:243–77. doi: 10.1146/annurev.bi.43.070174.001331
123. Sobel BE, Corr PB. Biochemical mechanisms potentially responsible for lethal arrhythmias induced by ischemia: the lysolipid hypothesis. Adv Cardiol. (1979) 26:76–85. doi: 10.1159/000402391
124. Scott ID, Nicholls DG. Energy transduction in intact synaptosomes. Influence of plasma-membrane depolarization on the respiration and membrane potential of internal mitochondria determined in situ. Biochem J. (1980) 186:21–33. doi: 10.1042/bj1860021
125. Nakahara T, Takeo S. Irreversible changes in oxidative phosphorylation activity of the mitochondrial membrane from hearts subjected to hypoxia and reoxygenation. Can J Cardiol. (1986) 2:24–33.
126. Berkich DA, Salama G, LaNoue KF. Mitochondrial membrane potentials in ischemic hearts. Arch Biochem Biophys. (2003) 420:279–86. doi: 10.1016/j.abb.2003.09.021
127. Grant AO. Cardiac ion channels. Circ Arrhythm Electrophysiol. (2009) 2:185–94. doi: 10.1161/CIRCEP.108.789081
128. Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia: a theoretical study of altered cell excitability and action potential duration. Cardiovasc Res. (1997) 35:256–72. doi: 10.1016/S0008-6363(97)00093-X
129. Kléber AG. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ Res. (1983) 52:442–50. doi: 10.1161/01.RES.52.4.442
130. Klabunde RE. Cardiac electrophysiology: normal and ischemic ionic currents and the ECG. Adv Physiol Educ. (2017) 41:29–37. doi: 10.1152/advan.00105.2016
131. Hoffman BF, Rosen MR. Cellular mechanisms for cardiac arrhythmias. Circ Res. (1981) 49:1–15. doi: 10.1161/01.RES.49.1.1
132. Rosenshtraukh LV, Witt RC, Nance PN, Rozanski GJ. Electrophysiologic effects of exogenous phosphocreatine in cardiac tissue: potential antiarrhythmic actions. Am Heart J. (1990) 120:1111–9. doi: 10.1016/0002-8703(90)90124-G
133. Singh BN, Nademanee K. Control of cardiac arrhythmias by selective lengthening of repolarization: theoretic considerations and clinical observations. Am Heart J. (1985) 109:421–30. doi: 10.1016/0002-8703(85)90629-5
134. El-Sherif N, Gough WB, Zeiler RH, Mehra R. Triggered ventricular rhythms in 1-day-old myocardial infarction in the dog. Circ Res. (1983) 52:566–79. doi: 10.1161/01.RES.52.5.566
135. Ferrier GR, Moffat MP, Lukas A. Possible mechanisms of ventricular arrhythmias elicited by ischemia followed by reperfusion. Studies on isolated canine ventricular tissues. Circ Res. (1985) 56:184–94. doi: 10.1161/01.RES.56.2.184
136. Kryzhanovskii SA, Kacharava VG, Marko R, Kelemen K, Kaverina NV, Sakhs VA. [Electrophysiologic study of the anti-arrhythmic mechanism of action of phosphocreatine in acute myocardial ischemia and reperfusion]. Kardiologiia. (1991) 31:66–9.
137. Anyukhovsky EP, Javadov SA, Preobrazhensky AN, Beloshapko GG, Rosenshtraukh LV, Saks VA. Effect of phosphocreatine and related compounds on the phospholipid metabolism of ischemic heart. Biochem Med Metab Biol. (1986) 35:327–34. doi: 10.1016/0885-4505(86)90090-3
138. Robinson LA, Braimbridge MV, Hearse DJ. Creatine phosphate: an additive myocardial protective and antiarrhythmic agent in cardioplegia. J Thorac Cardiovasc Surg. (1984) 87:190–200. doi: 10.1016/S0022-5223(19)37413-6
139. Niewiarowski S, Thomas DP. Platelet aggregation by ADP and thrombin. Nature. (1966) 212:1544–7. doi: 10.1038/2121544a0
140. Packham MA, Guccione MA, Chang PL, Mustard JF. Platelet aggregation and release: effects of low concentrations of thrombin or collagen. Am J Physiol. (1973) 225:38–47. doi: 10.1152/ajplegacy.1973.225.1.38
141. Zucker MB, Peterson J. Effect of acetylsalicylic acid, other nonsteroidal anti-inflammatory agents, and dipyridamole on human blood platelets. J Lab Clin Med. (1970) 76:66–75.
142. Macfarlane DE, Mills DC. The effects of ATP on platelets: evidence against the central role of released ADP in primary aggregation. Blood. (1975) 46:309–20. doi: 10.1182/blood.V46.3.309.309
143. Golikov AP, Riabinin VA. [Neoton in the treatment of myocardial infarct and unstable stenocardia]. Kardiologiia. (1993) 33:15–7, 3–4.
144. Samarenko MB. [Effect of phosphocreatine on the incidence of ventricular heart rhythm disorders in the 1st 24 hours of myocardial infarct]. Kardiologiia. (1986) 26:47–50.
145. Du XH, Liang FY, Zhao XW. [Effects of phosphocreatine on plasma brain natriuretic peptide level in elderly patients with chronic congestive heart failure]. Nan Fang Yi Ke Da Xue Xue Bao. (2009) 29:154–55, 159.
146. Perepech NB, Nedoshivin AO, Nesterova IV. [Neoton and thrombolytic therapy of myocardial infarction]. Ter Arkh. (2001) 73:50–55.
147. Perepech NB, Nedoshivin AO, Kutuzova AE. [Exogenous phosphocreatine in the prevention and treatment of cardiac insufficiency in patients with myocardial infarction]. Klin Med. (1993) 71:19–22.
148. Chambers DJ, Braimbridge MV, Kosker S, Yamada M, Jupp RA, Crowther A. Creatine phosphate (Neoton) as an additive to St. Thomas' Hospital cardioplegic solution (Plegisol). Results of a clinical study. Eur J Cardiothorac Surg. (1991) 5:74–81. doi: 10.1016/1010-7940(91)90004-4
149. Cheng SX, Hu QH. [Cardioprotective effect of exogenous phosphocreatine in patients undergoing open heart surgery]. Hunan Yi Ke Da Xue Xue Bao. (2001) 26:353–5.
150. Cisowski M, Bochenek A, Kucewicz E, Wnuk-Wojnar AM, Morawski W, Skalski J, et al. The use of exogenous creatine phosphate for myocardial protection in patients undergoing coronary artery bypass surgery. J Cardiovasc Surg. (1996) 37:75–80.
151. Guo-han C, Jian-hua G, Xuan H, Jinyi W, Rong L, Zhong-min L. Role of creatine phosphate as a myoprotective agent during coronary artery bypass graft in elderly patients. Coron Artery Dis. (2013) 24:48–53. doi: 10.1097/MCA.0b013e32835aab95
152. Pagani L, Musiani A. [The use of systemic phosphocreatine in heart surgery]. Minerva Anestesiol. (1992) 58:199–205.
153. Cerný J, Nemec P, Bucek J, Cerný E, Papousek F, Lojek A. [The effect of creatine phosphate in patients after surgery in ischemic heart disease]. Vnitr Lek. (1993) 39:153–9.
154. Chambers DJ, Haire K, Morley N, Fairbanks L, Strumia E, Young CP, et al. St. Thomas' Hospital cardioplegia: enhanced protection with exogenous creatine phosphate. Ann Thorac Surg. (1996) 61:67–75. doi: 10.1016/0003-4975(95)00819-5
155. Zhidkov IL, Ivanov VA, Kozhevnikov VA, Charnaia MA, Mukhamedzianova AR, Trekova NA. [Intraoperative myocardial protection with extracellular cardioplegic solutions in patients with cardiac valve diseases]. Anesteziol Reanimatol. (2007) 2:38–42.
156. MYa R, Samarenko MB, Afonskaya NI, Saks VA. Reduction of ventricular arrhythmias by phosphocreatine (Neoton) in patients with acute myocardial infarction. Am Heart J. (1988) 116:393–7. doi: 10.1016/0002-8703(88)90611-4
Keywords: high-energy phosphates, creatine phosphate, energy metabolism, ischemic heart disease, cardioprotection
Citation: Yi-Dan H, Ying-Xin Z, Shi-Wei Y and Yu-Jie Z (2021) High-Energy Phosphates and Ischemic Heart Disease: From Bench to Bedside. Front. Cardiovasc. Med. 8:675608. doi: 10.3389/fcvm.2021.675608
Received: 03 March 2021; Accepted: 17 June 2021;
Published: 28 July 2021.
Edited by:
Zhong Wang, University of Michigan, United StatesReviewed by:
Xu Chen, University of Mississippi Medical Center, United StatesZhihua Wang, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2021 Yi-Dan, Ying-Xin, Shi-Wei and Yu-Jie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Shi-Wei, eWFuZy5zaGl3ZWlAY2NtdS5lZHUuY24=; Zhou Yu-Jie, YXp6eWpfMTJAMTYzLmNvbQ==