
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 05 May 2021
Sec. Heart Failure and Transplantation
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.665797
This article is part of the Research Topic Novelties in the Therapeutic Approaches for Chronic Heart Failure: Cardiovascular targets and beyond View all 12 articles
 Michele Correale1*
Michele Correale1* Lucia Tricarico2Martino Fortunato2
Lucia Tricarico2Martino Fortunato2 Pietro Mazzeo2Savina Nodari3Matteo Di Biase2Natale Daniele Brunetti2
Pietro Mazzeo2Savina Nodari3Matteo Di Biase2Natale Daniele Brunetti2Despite recent advances in chronic heart failure management (either pharmacological or non-pharmacological), the prognosis of heart failure (HF) patients remains poor. This poor prognosis emphasizes the need for developing novel pathways for testing new HF drugs, beyond neurohumoral and hemodynamic modulation approaches. The development of new drugs for HF therapy must thus necessarily focus on novel approaches such as the direct effect on cardiomyocytes, coronary microcirculation, and myocardial interstitium. This review summarizes principal evidence on new possible pharmacological targets for the treatment of HF patients, mainly focusing on microcirculation, cardiomyocyte, and anti-inflammatory therapy.
Despite significant improvements in cardiovascular (CV) mortality over the last decades, CV disease is the main reason for death in several countries. CV therapies improved the survival of patients with CV disease, but, at the same time, increased the number of subjects affected by chronic CV conditions such as heart failure (HF).
Several clinical randomized studies assessed the efficacy of drug therapies in patients with HF. Residual CV mortality, however, remains considerable (1), potentially because novel therapeutic options are too often pursued with an “old” approach, with non-targeted and non-personalized therapies, not based on individual pathophysiology. Each patient affected by HF, instead, may be characterized by different etiologies, clinical characteristics, and comorbidities, each one to be possibly considered (2). Benefits from drugs based on neurohumoral and hemodynamic modulation (3) quickly reach a therapeutic plateau, with very limited possible additional benefit from incremental doses or additional drugs based on the same pathway. Recently, a novel paradigm has been reported in HF with preserved ejection fraction (HFpEF). Some comorbidities (obesity, diabetes mellitus, and chronic obstructive pulmonary disease) drive myocardial dysfunction through coronary microvascular endothelial inflammation in HFpEF. These comorbidities lead to a systemic proinflammatory state causing coronary microvascular endothelial inflammation (4). The development of new drugs for HF therapy must be necessarily focused on additional targets (anatomical or structural as cardiomyocyte and myocardial interstitium, and physiological or functional as microcirculation and inflammation) [(5); Figure 1] above all for HFpEF. Detailed knowledge of the interstitium and of cardiomyocyte biology becomes therefore essential; we thus briefly report principal evidence on new possible pharmacological targets for the treatment of HF patients (Table 1), mainly focusing on microcirculation, cardiomyocyte, and anti-inflammatory therapy.
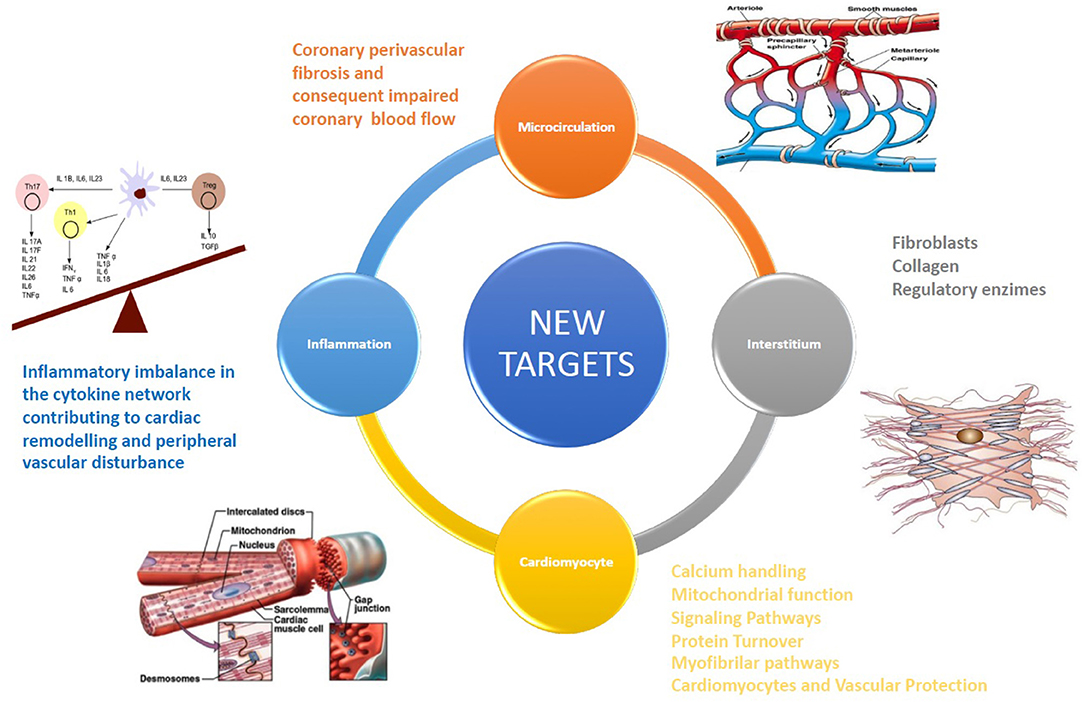
Figure 1. The development of new drugs for HF therapy must be necessarily focused on additional targets, such as cardiomyocytes, coronary microcirculation, and myocardial interstitium.
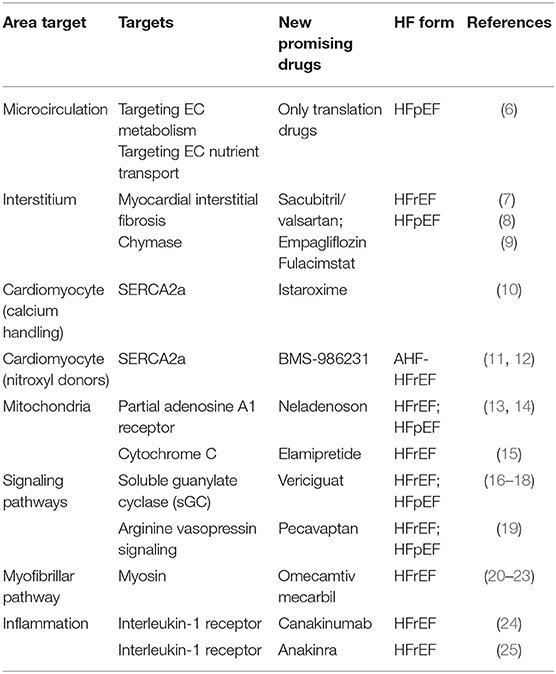
Table 1. New possible pharmacological targets and promising drugs for the treatment of HF patients.
A functional and “healthy” coronary microcirculation is essential for myocardial activity and effectiveness. It plays a pivotal role in the regulation of coronary blood flow in response to cardiac oxygen requirements. Impairment of this mechanism is defined as coronary microvascular dysfunction (CMD) (26). Microcirculatory anomalies can be often found in HF patients (27) and parallel disease progression in ischemic HF (28). CMD is associated with the development of HFpEF. In fact, decreased bio-accessibility of nitric oxide (NO) in endothelial dysfunction plays an important role in HF (29), mainly in HF with (HFpEF) (30). In HFpEF models, coronary microvascular endothelial inflammation reduces nitric oxide bioavailability, cyclic guanosine monophosphate content, and protein kinase G (PKG) activity in adjacent cardiomyocytes (31).
Recent reports suggest that the microcirculation has additional roles in supporting a healthy microenvironment (6). Experimental models have showed that restoring a healthy microcirculation and endothelium could be a possible therapeutic approach to treat HF.
Coronary perivascular fibrosis and the consequently impaired coronary blood flow may represent a new therapeutic target to improve coronary microcirculation (32).
Finally, there is emerging evidence about new translational drugs on the microcirculation (including growth factors and non-coding RNA therapeutics, as well as the targeting of metabolites or metabolic signaling) (6).
Forthcoming trials would better assess coronary microcirculation by cardiac magnetic resonance imaging (cMRI) (33, 34).
The knowledge of interstitium biology is essential for the development of new drugs for HF, providing several potential therapeutic targets in the case of HF: fibroblasts, collagen, and regulatory enzymes regulating collagen synthesis. Myocardial interstitium is not an inert scaffold, but rather an elaborate and active micro-habitat within the myocardium (35). HF fibrotic changes in the interstitium and near capillaries are featured by extracellular matrix (ECM) expansion and myofibroblast secretion of type I collagen. The level of collagen type I crosslinking is related to increased filling pressures in HF patients (36). A new cMRI technique, the T1 mapping (measures the extracellular volume fraction, ECV in human myocardium) permits the distinction of different components of interstitium (cardiomyocytes and connective tissue) and a more precise definition of myocardial fibrosis (37). ECV can be used as a tool in phase II trials to assess the efficacy of novel anti-myocardial fibrosis therapeutics (38).
Myocardial interstitial fibrosis (MIF) is very common in patients with HFpEF and with HFrEF. It is related to cardiac function impairment and poor outcome. It is determined by the changes in the quantity and quality of collagen fibers and in the ECM (39).
Pharmacological drugs formerly utilized with demonstrated safety may also be interesting for the treatment of MIF by means of new mechanisms. The sacubitril/valsartan and the sodium-glucose cotransporter 2 (SGLT2) inhibitor empagliflozin decreased MIF in HF mice with diabetes and improved LV function (7, 8). However, sacubitril/valsartan, in a recent phase III clinical research trial in patients with HFpEF, showed just a marginal reduction of the primary composite endpoint of total hospitalizations for HF and death from CV origin (40). Currently, SGLT2 inhibitors are still under evaluation to determine if they can effectively reduce MIF in humans.
An antifibrotic action was also demonstrated by pirfenidone and tranilast, through the inhibition TGF-β signaling. Prolonged usage of such drugs, however, may cause hepatic toxicity and may culminate in liver failure, therefore further studies are necessary to search for new effective TGF-β pathway targets, but safely too, for MIF reduction (41).
Chymase is a chymotrypsin-like serine protease that is secreted from activated mast cells and other cells, such as cardiomyocytes in the case of tissue damages (42, 43). Chymase is produced after secretion and produces or activates locally profibrotic factors, such as angiotensin (Ang) II, transforming growth factor (TGF) β, and matrix metalloproteinases that take part in adverse remodeling post-MI (44). For these reasons it can be a potential new target for post-MI therapy. Fulacimstat is an orally existing chymase inhibitor which has a multi-functional anti-remodeling effect that reduces LV disfunction after myocardial infarction (9).
Abnormal handling of intracellular Ca2+ in cardiomyocytes plays an important role in impaired cardiac contractility of HF. Ca2+ homeostasis is maintained in cardiomyocytes by ryanodine receptor and Ca-calmodulin kinase IIdelta (CaMKIIdelta), acting separately to manage cardiac Ca2+ handling, which was impaired in cardiac dysfunction (45). Until now there are only translational drugs. However, istaroxime may be a promising drug in the future. Istaroxime is a molecule with a luso-inotropic effect in HF, across the stimulation of SERCA2a ATPase activity and the augmentation of Ca2+ uptake into the sarcoplasmatic reticulum (SR) by mitigating the phospholamban inhibitive effect on SERCA2a in a cAMP/PKA independent way (46). In patients affected by HFrEF during hospitalization for AHF, a 24-h infusion of istaroxime improved the parameters of diastolic and systolic cardiac function without major cardiac adverse effects (10).
SERCA2a is in charge of sequestrating cytosolic Ca2+ back into the sarcoplasmic reticulum, enabling an effective uncoupling of actin-myosin and upcoming ventricular relaxation. Previous studies have proved that the expression of SERCA2a is downregulated in CHF, which later leads to serious systolic and diastolic function impairment.
JTV519 (K201) consolidates the closed state of type-2 ryanodine receptor (RyR2) by improving its affinity for FKBP12.6, which inhibits the leakage of Ca2+. K201 prevents spontaneous diastolic Ca2+ issued during Ca2+ overload by a double inhibitory effect on SR Ca2+-ATPase (SERCA2a) and RyR2, with no antiarrhythmic impact (47).
Another potential target for new HF drugs may be Na+-H+ exchanger-1 (NHE-1). Treatment with cariporide, an NHE-1 inhibitor, induced the reversal of hypertrophy and HF and the attenuation of apoptosis in cardiomyocytes (48), with some antiarrhythmic effects (49).
Nitroxyl (HNO) donors are compounds which improve cardiomyocyte function by direct improvement of SR Ca2+ cycling. BMS-986231 demonstrated a favorable safety and hemodynamic profile in patients hospitalized with advanced HF (11). An ongoing phase II randomized placebo-controlled clinical trial (StandUP-AHF) was designed to provide evidence of tolerability and efficacy of BMS-986231 in patients with HFrEF (12).
Nucleotide-binding oligomerization domain-containing protein 1 (NOD1) is a recent accredited innate immune sensor implicated in CV diseases. The NOD1 pathway was over-expressed in human and murine failing myocardia. Val-Blasco et al. (50) demonstrated that NOD1 may modulate intracellular Ca2+ mishandling in HF, appearing as a novel target for HF therapy.
In HF patients, several mitochondrial anomalies have been found. Impaired mitochondrial electron transport chain activity, enhanced reactive oxygen species (ROS) formation, changed metabolic substrate usage, abnormal mitochondrial dynamics, and modified ion homeostasis are currently being studied (51).
Neladenoson and elamipretide are the most promising drugs affecting mitochondrial function. Neladenoson, the novel partial adenosine A1 agonist, seems to be harmless, with no atrioventricular conduction or neurological side effects in HFrEF patients (13). In particular, the treatment was not related to dose-dependent favorable effects on cardiac function or clinical outcome but was linked with a dose-dependent renal function reduction (14) and no significant change in exercise capacity in patients with HFpEF (52).
Elamipretide is a water-soluble tetrapeptide capable of entering the inner mitochondrial membrane and joining with the phospholipid cardiolipin (53). This association leads to a stabilized cytochrome c conformation in order to promote effective electron transport in mitochondria and improve oxidative phosphorylation (54). LVEF, LV end diastolic pressure, cardiac hypertrophy, myocardial fibrosis, and cardiac ATP synthesis improvement in animal models and humans in treatment with elamipretide was demonstrated (55–57). On the other hand, in the PROGRESS-HF (phase 2) trial, elamipretide did not show a statistically significant change of LV end systolic volume in stable HFrEF patients compared with placebo after 4 weeks (15). More clinical studies of elamipretide in other HF phenotypes are required to assess its potential role in HF. At the same time longer term trials in HFrEF may be necessary too.
Capadenoson, a partial adenosine A1R agonist through inhibition of mPTP opening, might affect mitochondrial function (58). Another potential agonist (VCP746), an atypical A1AR agonist, has been investigated in order to develop ligands that promote A1AR cytoprotection in the absence of adverse hemodynamic effects (59).
Inhibition of mitochondria permeability transition pore (mPTP), in failing cardiomyocytes with cyclosporine A, maintained the expression of cytochrome c oxidase, increasing mitochondrial cytochrome c oxidase-dependent respiration and ATP synthesis (60). Sevoflurane, a volatile anesthetic, increases the threshold of calcium-induced mPTP opening (61).
Mitochondria are an important ROS source, and progress has been achieved to understand the way the matching of energy supply and demand across calcium handling might impact mitochondrial ROS development and removal. In mitochondria, oxidative stress generates a switch toward fission that leads to mitochondrial fragmentation and cell death. Doxycycline defends mitochondria against oxidative stress, and regulates the mitochondrial function causing a change of equilibrium toward fusion, so it might be an innovative therapeutic approach for HF (62).
Ca2+/calmodulin-dependent protein kinase II (CaMKII) has a crucial role in the progression of HF and in the generation of myocardial mitochondrial injury. Inhibition of CaMKII avoids Ca2+ intake toward mitochondria and decreases destruction of these organelles. Therefore, rising evidence supports the targeting of CaMKII and the mPTP as a way to preclude tissue damage. H2S-induced inactivation of CaMKII also may allow for a new therapeutic approach for CV diseases (63).
This approach might provide new possibilities to improve mitochondrial function in HF by fixing cytosolic and mitochondrial ion transporters. So, very specific molecules such as the preclinical CGP-37157, or ranolazine and empagliflozin (64), may be effective. Ranolazine decreases pressure overload-induced cardiac hypertrophy and improves cardiac function by preserving Na+ and Ca2+ handling (65). Empagliflozin decreases CaMKII activity (66). However, mitochondrial biogenesis, the production of ROS, and maintenance of cellular iron homeostasis need further studies to confirm their possible role for novel therapies in HF (67).
The GLP-1 receptor agonist liraglutide might have a therapeutic role in the modulation of cardiac inflammation. Liraglutide improved IL-1β-induced cellular ROS production and NADPH oxidase (NOX)-4 expression. Furthermore, it preserved cardiomyocytes from IL-1β-induced reduced mitochondrial membrane potential and decreased ATP production (68).
Melenovsky et al. demonstrated that, in the presence of profoundly impaired mitochondrial function in HF patients, myocardial iron deficiency may aggravate mitochondrial metabolism and ROS handling. So, the restoration of good levels of myocardial iron might help to ameliorate the bioenergetics of HF (69).
Acetylation of mitochondrial proteins was demonstrated to play an active role in the pathogenesis of cardiac diseases. Proteins are deacetylated by NAD+-dependent deacetylases so-called sirtuins (SIRTs). SIRT3, with the regulation of the activity of enzymes involved in ATP production in the mitochondria, might keep the equilibrium between cardiac function and energy consumption. Downregulation of SIRT3 may lead to the unbalance of mitochondrial bioenergetics related to impaired mitochondrial function. Therefore, the preservation of SIRT3 activity may be a possible approach to avoid pathological consequences of cardiopathies (70).
Myocytes have control mechanisms to maintain functional mitochondria with the removal of compromised mitochondria through specialized autophagy. Huang et al. observed that IGF-IIR signaling impaired expression and circulation of dynamin-related protein (Drp1) and mitofusin (Mfn2) (71). IGF-IIR activates JNK-mediated Bcl-2 phosphorylation to stimulate ULK1/Beclin 1-dependent autophagosome development. Unreasonable mitochondrial fission by Drp1 improved the Rab9-dependent autophagosome identification and swallowing of damaged mitochondria and eventually reduced cardiomyocyte viability. These results showed the link between Rab9-dependent autophagosomes and mitochondrial fission in cardiomyocytes, which gives another potential therapeutic approach.
Reduced NO bioavailability leads to a relative sGC deficiency and a reduction in cGMP synthesis, essential for normal cardiac and vascular function (72–74). Cyclic GMP ameliorates endothelial function and reduces cardiac fibrosis. Direct NO-independent sGC stimulation might provide a new chance to address the relative cGMP deficit in HF. Vericiguat is a novel soluble guanylate cyclase (sGC) stimulator, tested in the SOCRATES PRESERVED study (16), REDUCED study (17), and recently also in the VICTORIA trial (18). It avoids that nitric oxide step and directly binds to induce the sGC. This promising drug raises solid cGMP production and produces the subsequent beneficial effects on the CV system. The VICTORIA trial showed that patients taking vericiguat were 10% less likely to have the primary outcome, a composite of CV death or first hospitalization for HF, than those taking placebo.
Increased arginine vasopressin signaling in either or both the V1a and V2 receptors could contribute to the development of HF. V1a activation could cause vasoconstriction, cardiac hypertrophy, and fibrosis (75) as intracellular signaling pathways are closely related to those for angiotensin II. Renal tubular V2 activation could cause water retention and hyponatremia. A novel dual-balanced vasopressin antagonist (pecavaptan) may lead to decongestion (via V2) and protect heart and vessels (via V1a), improving outcomes. It was shown to be superior to tolvaptan in preclinical models (76). This is the rationale for an ongoing trial (AVANTI) (19) which will enroll HFpEF and HFrEF patients with incomplete decongestion despite standard therapy at day 3–7 after index hospitalization.
All other options are translational drugs. Some receptor-mediated signaling pathways (natriuretic peptides, mediators of glycogen synthase kinase 3 and ERK1/2 pathways, beta adrenergic receptor subtypes, relaxin receptor signaling, TNF/TNF receptor family, TWEAK/Fn14 axis, and micro-RNAs) may represent targets for emerging therapies in HF treatment (77). miRNAs are endogenous non-coding single-stranded RNAs that control gene expression and regulate adaptive and maladaptive cardiac remodeling (78, 79). By blocking the ERK1/2 pathway it is possible to prevent progression of CHF by reducing fibrosis, inflammation, and apoptosis in the myocardium (80).
Recently, more attention has been given to signaling pathways linking cysteine cathepsins. They play a role in synthesis and degradation of the ECM, affecting ventricular remodeling and contractile capacity (81).
The potential therapeutic strategy targeting protein phosphatase 1 (I-1) by restoring the balance of cardiac protein phosphorylation needs further study in order to prove its real clinical benefit (82).
G-protein-coupled receptor (GPCR) kinase-2 (GRK2) is a regulator of GPCRs, in particular beta adrenergic receptors, and has a crucial role in the development and advance of CV disease, like HF. GRK2 inhibition has been examined in several models (83); in a rat model, inhibition by a cyclic peptide C7, improved mitochondrial activity, and improved the contractility (84).
Myosin activators, such as omecamtiv mecarbil and danicamtiv, facilitate the rate-limiting step of the myosin enzymatic cycle and switch the cycle for the benefit of the force-producing state. Myosin activators improve cardiac contractility by increasing the conversion of the actin–myosin complex from a weak to a strong bond, with no variation of intracellular Ca2+ homeostasis, increasing LV systolic function, and without improvement of energy request or arrhythmogenesis (85–87). Omecamtiv mecarbil ameliorates left ventricle systolic ejection time, SV, and CO, without side effects from the inotropic drugs; its bioavailability is high and the drug is safe in HFrEF (88). As shown in the ATOMIC-AHF trial in symptomatic CHF and LVEF lower than 40%, the drug was not associated with improved dyspnea but it did increase systolic EF, decrease the LV end-systolic diameter, and was well-tolerated (20, 21). It also significantly decreased NT-pro-BNP compared with placebo (22); it was also safe for acute HF.
Its effects on CV outcomes were tested in the GALACTIC-HF trial (23). Among patients with HFrEF, those who received this drug revealed a significant relative risk reduction in a composite of a HF event or CV death, compared with placebo. A phase III study (METEORIC-HF) is currently ongoing to assess its effects on exercise capacity1. This new drug seems to be a promising new approach for patients affected by HFrEF.
The giant protein titin is anchored at the Z-disc; changes in titin stiffness occur in HF through a switch in the expression fraction of the two major titin isoforms in cardiac sarcomeres, N2BA (compliant) and N2B (stiffer). In HF patients, increased passive stiffness can be detected because of a titin phosphorylation deficit (89), thus representing another potential drug target in HF.
To the best of our knowledge, there are not sufficiently valid data (anti-TNF-α drugs have not shown benefits in CHF) about anti-inflammatory drugs in HF. Previous studies showed that inflammatory mediators may be relevant in the CHF, contributing to adverse remodeling and peripheral vascular anomalies. Inflammatory mechanisms are also hypothesized in HFpEF, with pro-inflammatory/pro-fibrotic or immunological alterations (90).
Previous research showed increased values of inflammatory cytokines such as TNF-α, IL-1β, and IL-6 in HF. In HF patients, this improvement in inflammatory mediators does not appear to be joined by an equivalent increase in anti-inflammatory cytokines such as IL-10 and TGF-β; thus resulting in an inflammatory imbalance in the cytokine system (91). Interleukin-1β (IL-1β) is known to depress cardiac function. A small secondary analysis of the CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) trial (24) confirmed IL-1 as a potential therapeutic target in HF. Instead, in patients with LVEF <50% and administration of anakinra (IL-1 receptor antagonist) was only associated with exercise capacity improvement after 12 weeks of therapy (25).
Recently in some drugs already used in HF therapy, such as glycosides (92) and ivabradine (93), anti-inflammatory effects have been demonstrated.
Others authors have proved the possible advantage of the combination of methotrexate with conventional therapy, through the interaction between the activated immune and inflammatory mediator's system (94).
Pentoxyfylline, intravenous immunoglobulin, thalidomide, and statins, have demonstrated encouraging results in smaller studies (95), although these have not been verified in multicenter randomized studies with relevant endpoints (96–101). In a meta-analysis, lipophilic statins were superior to hydrophilic statins in terms of follow-up LVEF, BNP, C-reactive protein, and IL-6 in HF (102).
Some recently discovered therapeutic targets (peroxisome proliferator-activated receptor gamma activators, Rho-kinase, p38 mitogen-activated protein kinase, nuclear transcription factor NF-kappaB) modulating parasympathetic tone, macrophage inhibitors, and chemokine receptor antagonists, have already been tested.
Oleuropein mitigates the progression of HF, presumably by anti-oxidative and anti-inflammatory effects (103). Also oral taurine supplementation in HF with LVEF <50% has anti-atherogenic and anti-inflammatory effects (104).
Another possible strategy can be represented by resveratrol. The resveratrol treatment decreased the galectin-3 level, which is secreted by macrophages, and plays a role in mediating cardiac fibrosis and inflammation. This leads to the reduction of IL-1 and IL-6 levels. The reduced activity of leukocytes may be a significant effect of resveratrol, and it can lead to heart function improvement in HFrEF (105).
The most encouraging results for near future developments are given by gliflozins.
Encouraging results in type 2 diabetic patients with prior CV events from the EMPAREG-HF study (empagliflozin) (106), from the CANVAS program (canagliflozin) (107, 108), and from the DECLARE-TIMI 58 trial (dapagliflozin) (109) induced researchers to test SGLT2i in people with HF regardless of the presence of diabetes mellitus. Results from the DAPA-HF trial (110) and from the EMPEROR-Reduced trial (111) in both diabetics and non-diabetics showed an important reduction in CV death and HF hospitalization. The mechanisms underlying the cardiac effects of gliflozins have not yet been fully defined, although possible effects of these drugs on cardiomyocytes could be supposed. Ongoing clinical trials will clarify the effects and mechanisms of gliflozins in HFrEF and HFpEF patients with and without T2DM (112, 113). New results have been derived from the studies on endothelial function in CHF (114), especially interesting results have been obtained in HFpEF in treatment with empagliflozin (115, 116).
Other possible approaches for the future include RyR2 and its associated accessory proteins, which might be potential new drug targets. RyR2 placed on the sarcoplasmic reticulum produces systolic Ca2+ transients within cardiomyocytes. Appropriate functioning of RyR2 is therefore pivotal to the timing and force produced by cardiomyocytes. Impaired intracellular Ca2+ handing secondary to impaired function of RyR2 may be related to HF (117).
Normal cardiac Ca2+ handling is also due to striated muscle preferably expressed protein kinase (SPEG), a member of the myosin light chain kinase family. SPEG has been causally linked to HF and atrial fibrillation, so it can be taken into consideration (118).
Gene therapy is still far from being clinically applicable (119). The gene cysteine-rich secretory protein LCCL domain containing 1 (CRISPLD1) is overexpressed in HF. The downregulation of some signaling pathways upon CRISPLD1-KO implicates a role in adverse remodeling. These discoveries offer novel candidate genes with encouraging potential roles for therapeutic interventions (120).
The cardiac bridging integrator 1 gene (cBIN1) therapy stabilizes the subcellular membrane within cardiomyocytes, preserving intracellular distribution of calcium (121), and potentially could be a new way forward to find new drugs.
Possible new HF drugs, above all for HFpEF, might target myocytes, mitochondria, microvascular circulation, and interstitium. Further studies are needed to transfer evidence derived from pre-clinical evidence to real-life with new possible therapeutic approaches aimed at new therapeutic targets in HF.
MC and SN: substantial conception. LT, MF, and PM: acquisition of data. MD: drafting of the article for important intellectual content. NB: revising the article for final approval of the submitted paper. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^ClinicalTrials.gov Identifier: NCT03759392.
1. Butler J, Fonarow GC, Gheorghiade M. Strategies and opportunities for drug development in heart failure. JAMA. (2013) 309:1593–4. doi: 10.1001/jama.2013.1063
2. Correale M, Monaco I, Brunetti ND, Di Biase M, Metra M, Nodari S, et al. Redefining biomarkers in heart failure. Heart Fail Rev. (2018) 23:237–53. doi: 10.1007/s10741-018-9683-2
3. Muiesan ML, Paini A, Agabiti Rosei C. Current pharmacological therapies in heart failure patients. High Blood Press Cardiovasc Prev. (2017) 24:107–14. doi: 10.1007/s40292-017-0194-3
4. Correale M, Paolillo S, Mercurio V, Limongelli G, Barillà F, Ruocco G, et al. Comorbidities in chronic heart failure: an update from Italian Society of Cardiology (SIC) working group on heart failure. Eur J Intern Med. (2020) 71:23–31. doi: 10.1016/j.ejim.2019.10.008
5. Quarta G, Gori M, Iorio A, D'Elia E, Moon JC, Iacovoni A, et al. Cardiac magnetic resonance in heart failure with preserved ejection fraction: myocyte, interstitium, microvascular, and metabolic abnormalities. Eur J Heart Fail. (2020) 22:1065–75. doi: 10.1002/ejhf.1961
6. Luxán G, Dimmeler S. The vasculature: a therapeutic target in heart failure? Cardiovasc Res. (2021). doi: 10.1093/cvr/cvab047. [Epub ahead of print].
7. Suematsu Y, Miura S, Goto M, Matsuo Y, Arimura T, Kuwano T, et al. LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur J Heart Fail. (2016) 18:386–93. doi: 10.1002/ejhf.474
8. Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M, et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol. (2017) 16:9. doi: 10.1186/s12933-016-0489-z
9. Düngen HD, Kober L, Nodari S, Schou M, Otto C, Becka M, et al. Safety and tolerability of the chymase inhibitor fulacimstat in patients with left ventricular dysfunction after myocardial infarction-results of the CHIARA MIA 1 trial. Clin Pharmacol Drug Dev. (2019) 8:942–51. doi: 10.1002/cpdd.633
10. Carubelli V, Zhang Y, Metra M, Lombardi C, Felker GM, Filippatos G, et al. Treatment with 24 hour istaroxime infusion in patients hospitalised for acute heart failure: a randomised, placebo-controlled trial. Eur J Heart Fail. (2020) 22:1684–93. doi: 10.1002/ejhf.1743
11. Tita C, Gilbert EM, Van Bakel AB, Grzybowski J, Haas GJ, Jarrah M, et al. A Phase 2a dose-escalation study of the safety, tolerability, pharmacokinetics and haemodynamic effects of BMS-986231 in hospitalized patients with heart failure with reduced ejection fraction. Eur J Heart Fail. (2017) 19:1321–32. doi: 10.1002/ejhf.897
12. Felker GM, Borentain M, Cleland JG, DeSouza MM, Kessler PD, O'Connor CM, et al. Rationale and design for the development of a novel nitroxyl donor in patients with acute heart failure. Eur J Heart Fail. (2019) 21:1022–31. doi: 10.1002/ejhf.1504
13. Voors AA, Düngen HD, Senni M, Nodari S, Agostoni P, Ponikowski P, et al. Safety and tolerability of neladenoson bialanate, a novel oral partial adenosine A1 receptor agonist, in patients with chronic heart failure. J Clin Pharmacol. (2017) 57:440–51. doi: 10.1002/jcph.828
14. Voors AA, Bax JJ, Hernandez AF, Wirtz AB, Pap AF, Ferreira AC, et al. Safety and efficacy of the partial adenosine A1 receptor agonist neladenoson bialanate in patients with chronic heart failure with reduced ejection fraction: a phase IIb, randomized, double-blind, placebo-controlled trial. Eur J Heart Fail. (2019) 21:1426–33. doi: 10.1002/ejhf.1591
15. Butler J, Khan MS, Anker SD, Fonarow GC, Kim RJ, Nodari S, et al. Effects of elamipretide on left ventricular function in patients with heart failure with reduced ejection fraction: the PROGRESS-HF phase 2 trial. J Card Fail. (2020) 26:429–37. doi: 10.1016/j.cardfail.2020.02.001
16. Filippatos G, Maggioni AP, Lam CSP, Pieske-Kraigher E, Butler J, Spertus J, et al. Patient-reported outcomes in the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED ejection fraction (SOCRATES-PRESERVED) study. Eur J Heart Fail. (2017) 19:782–91. doi: 10.1002/ejhf.800
17. Gheorghiade M, Greene SJ, Butler J, Filippatos G, Lam CS, Maggioni AP, et al. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: the SOCRATES-REDUCED randomized trial. JAMA. (2015) 314:2251–62. doi: 10.1001/jama.2015.15734
18. Armstrong PW, Pieske B, Anstrom KJ, Ezekowitz J, Hernandez AF, Butler J, et al. Vericiguat in patients with heart failure and reduced ejection fraction. N Engl J Med. (2020) 382:1883–93. doi: 10.1056/NEJMoa1915928
19. Goldsmith SR, Burkhoff D, Gustafsson F, Voors A, Zannad F, Kolkhof P, et al. Dual vasopressin receptor antagonism to improve congestion in patients with acute heart failure: design of the AVANTI trial. J Card Fail. (2021) 27:233–41. doi: 10.1016/j.cardfail.2020.10.007
20. Teerlink JR, Felker GM, McMurray JJV, Ponikowski P, Metra M, Filippatos GS, et al. Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: the ATOMIC-AHF study. J Am Coll Cardiol. (2016) 67:1444–55. doi: 10.1016/j.jacc.2016.01.031
21. Swenson AM, Tang W, Blair CA, Fetrow CM, Unrath WC, Previs MJ, et al. Omecamtiv mecarbil enhances the duty ratio of human beta-cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. J Biol Chem. (2017) 292:3768–78. doi: 10.1074/jbc.M116.748780
22. Kaplinsky E, Mallarkey G. Cardiac myosin activators for heart failure therapy: focus on omecamtiv mecarbil. Drugs Context. (2018) 7:212518. doi: 10.7573/dic.212518
23. Teerlink JR, Diaz R, Felker GM, McMurray JJV, Metra M, Solomon SD, et al. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N Engl J Med. (2020) 384:105–16. doi: 10.1056/NEJMoa2025797
24. Trankle CR, Canada JM, Cei L, Abouzaki N, Oddi-Erdle C, Kadariya D, et al. Usefulness of canakinumab to improve exercise capacity in patients with long-term systolic heart failure and elevated c-reactive protein. Am J Cardiol. (2018) 122:1366–70. doi: 10.1016/j.amjcard.2018.07.002
25. Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, et al. Interleukin-1 blockade in recently decompensated systolic heart failure: results From REDHART (recently decompensated heart failure anakinra response trial). Circ Heart Fail. (2017) 10:e004373. doi: 10.1161/CIRCHEARTFAILURE.117.004373
26. Vancheri F, Longo G, Vancheri S, Henein M. Coronary microvascular dysfunction. J Clin Med. (2020) 9:2880. doi: 10.3390/jcm9092880
27. Herrmann J, Kaski JC, Lerman A. Coronary microvascular dysfunction in the clinical setting: from mystery to reality. Eur Heart J. (2012) 33:2771–82. doi: 10.1093/eurheartj/ehs246
28. Gil KE, Pawlak A, Frontczak-Baniewicz M, Gil RJ, Nasierowska-Guttmejer A. The proposed new classification of coronary microcirculation as the predictor of the heart failure progression in idiopathic dilated cardiomyopathy. Cardiovasc Pathol. (2015) 24:351–8. doi: 10.1016/j.carpath.2015.08.001
29. Quitter F, Figulla HR, Ferrari M, Pernow J, Jung C. Increased arginase levels in heart failure represent a therapeutic target to rescue microvascular perfusion. Clin Hemorheol Microcirc. (2013) 54:75–85. doi: 10.3233/CH-2012-1617
30. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschöpe C, et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. (2016) 4:312–24. doi: 10.1016/j.jchf.2015.10.007
31. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
32. Dai Z, Aoki T, Fukumoto Y, Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol. (2012) 60:416–21. doi: 10.1016/j.jjcc.2012.06.009
33. Lee DC, Simonetti OP, Harris KR, Holly TA, Judd RM, Wu E, et al. Magnetic resonance versus radionuclide pharmacological stress perfusion imaging for flow-limiting stenoses of varying severity. Circulation. (2004) 110:58–65. doi: 10.1161/01.CIR.0000133389.48487.B6
34. Nakamori S, Onishi K, Nakajima H, Yoon YE, Nagata M, Kurita T, et al. Impaired myocardial perfusion reserve in patients with fatty liver disease assessed by quantitative myocardial perfusion magnetic resonance imaging. Circ J. (2012) 76:2234–40. doi: 10.1253/circj.CJ-11-1487
35. Eckhouse SR, Spinale FG. Changes in the myocardial interstitium and contribution to the progression of heart failure. Heart Fail Clin. (2012) 8:7–20. doi: 10.1016/j.hfc.2011.08.012
36. López B, Querejeta R, González A, Larman M, Díez J. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension. (2012) 60:677–83. doi: 10.1161/HYPERTENSIONAHA.112.196113
37. Schelbert EB, Piehler KM, Zareba KM, Moon JC, Ugander M, Messroghli DR, et al. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc. (2015) 4:e002613. doi: 10.1161/JAHA.115.002613
38. Webber M, Jackson SP, Moon JC, Captur G. Myocardial fibrosis in heart failure: anti-fibrotic therapies and the role of cardiovascular magnetic resonance in drug trials. Cardiol Ther. (2020) 9:363–76. doi: 10.1007/s40119-020-00199-y
39. González A, López B, Ravassa S, San José G, Díez J. The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim Biophys Acta Mol Cell Res. (2019) 1866:1421–32. doi: 10.1016/j.bbamcr.2019.06.001
40. Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. (2019) 381:1609–20. doi: 10.1056/NEJMoa1908655
41. Díez J, González A, Kovacic JC. Myocardial interstitial fibrosis in nonischemic heart disease, Part 3/4: JACC focus seminar. J Am Coll Cardiol. (2020) 75:2204–18. doi: 10.1016/j.jacc.2020.03.019
42. Takai S, Jin D. Improvement of cardiovascular remodelling by chymase inhibitor. Clin Exp Pharmacol Physiol. (2016) 43:387–93. doi: 10.1111/1440-1681.12549
43. Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS ONE. (2011) 6:e28501. doi: 10.1371/journal.pone.0028501
44. Dell'Italia LJ, Collawn JF, Ferrario CM. Multifunctional role of chymase in acute and chronic tissue injury and remodeling. Circ Res. (2018) 122:319–36. doi: 10.1161/CIRCRESAHA.117.310978
45. Currie S, Elliott EB, Smith GL, Loughrey CM. Two candidates at the heart of dysfunction: the ryanodine receptor and calcium/calmodulin protein kinase II as potential targets for therapeutic intervention-An in vivo perspective. Pharmacol Ther. (2011) 131:204–20. doi: 10.1016/j.pharmthera.2011.02.006
46. Ferrandi M, Barassi P, Tadini-Buoninsegni F, Bartolommei G, Molinari I, Tripodi MG, et al. Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br J Pharmacol. (2013) 169:1849–61. doi: 10.1111/bph.12278
47. Patel C, Deoghare S. Heart failure: novel therapeutic approaches. J Postgrad Med. (2015) 61:101–8. doi: 10.4103/0022-3859.153104
48. Kim JO, Kwon EJ, Song DW, Lee JS, Kim DH. MiR-185 inhibits endoplasmic reticulum stress-induced apoptosis by targeting Na+/H+ exchanger-1 in the heart. BMB Rep. (2016) 49:208–13. doi: 10.5483/BMBRep.2016.49.4.193
49. Baartscheer A, Hardziyenka M, Schumacher CA, Belterman CN, van Borren MM, Verkerk AO, et al. Chronic inhibition of the Na+/H+ - exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodelling. Br J Pharmacol. (2008) 154:1266–75. doi: 10.1038/bjp.2008.189
50. Val-Blasco A, Piedras MJGM, Ruiz-Hurtado G, Suarez N, Prieto P, Gonzalez-Ramos S, et al. Role of NOD1 in heart failure progression via regulation of Ca2+ handling. J Am Coll Cardiol. (2017) 69:423–33. doi: 10.1016/j.jacc.2016.10.073
51. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. (2017) 14:238–50. doi: 10.1038/nrcardio.2016.203
52. Shah SJ, Voors AA, McMurray JJV, Kitzman DW, Viethen T, Bomfim Wirtz A, et al. Effect of neladenoson bialanate on exercise capacity among patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. (2019) 321:2101–12. doi: 10.1001/jama.2019.6717
53. Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. (2014) 96:672–83. doi: 10.1038/clpt.2014.174
54. Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. (2014) 171:2017–28. doi: 10.1111/bph.12468
55. Sabbah HN, Gupta RC, Kohli S, Wang M, Hachem S, Zhang K. Chronic therapy with elamipretide (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ Heart Fail. (2016) 9:e002206. doi: 10.1161/CIRCHEARTFAILURE.115.002206
56. Shi J, Dai W, Hale SL, Brown DA, Wang M, Han X, et al. Bendavia restores mitochondrial energy metabolism gene expression and suppresses cardiac fibrosis in the border zone of the infarcted heart. Life Sci. (2015) 141:170–8. doi: 10.1016/j.lfs.2015.09.022
57. Daubert MA, Yow E, Dunn G, Marchev S, Barnhart H, Douglas PS, et al. Novel mitochondria-targeting peptide in heart failure treatment: a randomized, placebo-controlled trial of elamipretide. Circ Heart Fail. (2017) 10:e004389. doi: 10.1161/CIRCHEARTFAILURE.117.004389
58. Nell PG, Albrecht-Kupper B. The adenosine A1 receptor and its ligands. Prog Med Chem. (2009) 47:163–201. doi: 10.1016/S0079-6468(08)00204-X
59. Vecchio EA, Chuo CH, Baltos JA, Ford L, Scammells PJ, Wang BH, et al. The hybrid molecule, VCP746, is a potent adenosine A2B receptor agonist that stimulates anti-fibrotic signalling. Biochem Pharmacol. (2016) 117:46–56. doi: 10.1016/j.bcp.2016.08.007
60. Sharov VG, Todor AV, Imai M, Sabbah HN. Inhibition of mitochondrial permeability transition pores by cyclosporine A improves cytochrome C oxidase function and increases rate of ATP synthesis in failing cardiomyocytes. Heart Fail Rev. (2005) 10:305–10. doi: 10.1007/s10741-005-7545-1
61. Onishi A, Miyamae M, Kaneda K, Kotani J, Figueredo VM. Direct evidence for inhibition of mitochondrial permeability transition pore opening by sevoflurane preconditioning in cardiomyocytes: comparison with cyclosporine A. Eur J Pharmacol. (2012) 675:40–6. doi: 10.1016/j.ejphar.2011.11.040
62. Riba A, Deres L, Eros K, Szabo A, Magyar K, Sumegi B, et al. Doxycycline protects against ROS-induced mitochondrial fragmentation and ISO-induced heart failure. PLoS ONE. (2017) 12:e0175195. doi: 10.1371/journal.pone.0175195
63. Wu D, Hu Q, Tan B, Rose P, Zhu D, Zhu YZ. Amelioration of mitochondrial dysfunction in heart failure through S-sulfhydration of Ca2+/calmodulin-dependent protein kinase II. Redox Biol. (2018) 19:250–62. doi: 10.1016/j.redox.2018.08.008
64. Makrecka-Kuka M, Korzh S, Videja M, Vilks K, Cirule H, Kuka J, et al. Empagliflozin protects cardiac mitochondrial fatty acid metabolism in a mouse model of diet-induced lipid overload. Cardiovasc Drugs Ther. (2020) 34:791–7. doi: 10.1007/s10557-020-06989-9
65. Nie J, Duan Q, He M, Li X, Wang B, Zhou C, et al. Ranolazine prevents pressure overload-induced cardiac hypertrophy and heart failure by restoring aberrant Na+ and Ca2+ handling. J Cell Physiol. (2019) 234:11587–601. doi: 10.1002/jcp.27791
66. Mustroph J, Wagemann O, Lücht CM, Trum M, Hammer KP, Sag CM, et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. (2018) 5:642–8. doi: 10.1002/ehf2.12336
67. Dietl A, Maack C. Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr Heart Fail Rep. (2017) 14:338–49. doi: 10.1007/s11897-017-0347-7
68. Zhang L, Tian J, Diao S, Zhang G, Xiao M, Chang D. GLP-1 receptor agonist liraglutide protects cardiomyocytes from IL-1β-induced metabolic disturbance and mitochondrial dysfunction. Chem Biol Interact. (2020) 332:109252. doi: 10.1016/j.cbi.2020.109252
69. Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, et al. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail. (2017) 19:522–30. doi: 10.1002/ejhf.640
70. Parodi-Rullán RM, Chapa-Dubocq XR, Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Front Physiol. (2018) 9:1094. doi: 10.3389/fphys.2018.01094
71. Huang CY, Lai CH, Kuo CH, Chiang SF, Pai PY, Lin JY, et al. Inhibition of ERK-Drp1 signaling and mitochondria fragmentation alleviates IGF-IIR-induced mitochondria dysfunction during heart failure. J Mol Cell Cardiol. (2018) 122:58–68. doi: 10.1016/j.yjmcc.2018.08.006
72. Gheorghiade M, Marti CN, Sabbah HN, Roessig L, Greene SJ, Böhm M, et al. Soluble guanylate cyclase: a potential therapeutic target for heart failure. Heart Fail Rev. (2013) 18:123–34. doi: 10.1007/s10741-012-9323-1
73. Buys ES, Sips P, Vermeersch P, Raher MJ, Rogge E, Ichinose F, et al. Gender-specific hypertension and responsiveness to nitric oxide in sGCalpha1 knockout mice. Cardiovasc Res. (2008) 79:179–86. doi: 10.1093/cvr/cvn068
74. van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. (2012) 126:830–9. doi: 10.1161/CIRCULATIONAHA.111.076075
75. Kolkhof P, Pook E, Pavkovic M, Kretschmer A, Buchmüller A, Tinel H, et al. Vascular protection and decongestion without renin-angiotensin-aldosterone system stimulation mediated by a novel dual-acting vasopressin V1a/V2 receptor antagonist. J Cardiovasc Pharmacol. (2019) 74:44–52. doi: 10.1097/FJC.0000000000000677
76. Mondritzki T, Mai TA, Vogel J, Pook E, Wasnaire P, Schmeck C, et al. Cardiac output improvement by pecavaptan: a novel dual-acting vasopressin V1a/V2 receptor antagonist in experimental heart failure. Eur J Heart Fail. (2020). doi: 10.1002/ejhf.2001. [Epub ahead of print].
77. Tuttolomondo A, Simonetta I, Pinto A. MicroRNA and receptor mediated signaling pathways as potential therapeutic targets in heart failure. Expert Opin Ther Targets. (2016) 20:1287–300. doi: 10.1080/14728222.2016.1212017
78. Koeck I, Burkhard FC, Monastyrskaya K. Activation of common signaling pathways during remodeling of the heart and the bladder. Biochem Pharmacol. (2016) 102:7–19. doi: 10.1016/j.bcp.2015.09.012
79. Zhao Y, Ponnusamy M, Dong Y, Zhang L, Wang K, Li P. Effects of miRNAs on myocardial apoptosis by modulating mitochondria related proteins. Clin Exp Pharmacol Physiol. (2017) 44:431–40. doi: 10.1111/1440-1681.12720
80. Huby AC, Turdi S, James J, Towbin JA, Purevjav E. FasL expression in cardiomyocytes activates the ERK1/2 pathway, leading to dilated cardiomyopathy and advanced heart failure. Clin Sci. (2016) 130:289–99. doi: 10.1042/CS20150624
81. O'Toole D, Zaeri AAI, Nicklin SA, French AT, Loughrey CM, Martin TP. Signalling pathways linking cysteine cathepsins to adverse cardiac remodelling. Cell Signal. (2020) 76:109770. doi: 10.1016/j.cellsig.2020.109770
82. Weber S, Meyer-Roxlau S, El-Armouche A. Role of protein phosphatase inhibitor-1 in cardiac beta adrenergic pathway. J Mol Cell Cardiol. (2016) 101:116–26. doi: 10.1016/j.yjmcc.2016.09.007
83. Cannavo A, Komici K, Bencivenga L, D'amico ML, Gambino G, Liccardo D, et al. GRK2 as a therapeutic target for heart failure. Expert Opin Ther Targets. (2018) 22:75–83. doi: 10.1080/14728222.2018.1406925
84. Ciccarelli M, Sorriento D, Fiordelisi A, Gambardella J, Franco A, Del Giudice C, et al. Pharmacological inhibition of GRK2 improves cardiac metabolism and function in experimental heart failure. ESC Heart Fail. (2020) 7:1571–84. doi: 10.1002/ehf2.12706
85. Shen YT, Malik FI, Zhao X, Depre C, Dhar SK, Abarzúa P, et al. Improvement of cardiac function by a cardiac Myosin activator in conscious dogs with systolic heart failure. Circ Heart Fail. (2010) 3:522–7. doi: 10.1161/CIRCHEARTFAILURE.109.930321
86. Psotka MA, Teerlink JR. Direct myosin activation by omecamtiv mecarbil for heart failure with reduced ejection fraction. Handb Exp Pharmacol. (2017) 243:465–90. doi: 10.1007/164_2017_13
87. Szentandrassy N, Horvath B, Vaczi K, Kistamas K, Masuda L, Magyar J, et al. Dose-dependent electrophysiological effects of the myosin activator omecamtiv mecarbil in canine ventricular cardiomyocytes. J Physiol Pharmacol. (2016) 67:483–9.
88. Teerlink JR, Felker GM, McMurray JJ, Solomon SD, Adams KF Jr., Cleland JG, et al. Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF): a phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet. (2016) 388:2895–903. doi: 10.1016/S0140-6736(16)32049-9
89. Kruger M, Linke WA. Titin-based mechanical signalling in normal and failing myocardium. J Mol Cell Cardiol. (2009) 46:490–8. doi: 10.1016/j.yjmcc.2009.01.004
90. Tschope C, Van Linthout S, Kherad B. Heart failure with preserved ejection fraction and future pharmacological strategies: a glance in the crystal ball. Curr Cardiol Rep. (2017) 19:70. doi: 10.1007/s11886-017-0874-6
91. Gullestad L, Kjekshus J, Damås JK, Ueland T, Yndestad A, Aukrust P. Agents targeting inflammation in heart failure. Expert Opin Investig Drugs. (2005) 14:557–66. doi: 10.1517/13543784.14.5.557
92. Furst R, Zundorf I, Dingermann T. New knowledge about old drugs: the anti-inflammatory properties of cardiac glycosides. Planta Med. (2017) 83:977–84. doi: 10.1055/s-0043-105390
93. Rohm I, Kretzschmar D, Pistulli R, Franz M, Schulze PC, Stumpf C, et al. Impact of ivabradine on inflammatory markers in chronic heart failure. J Immunol Res. (2016) 2016:6949320. doi: 10.1155/2016/6949320
94. Gong KZ, Song G, Spiers JP, Kelso EJ, Zhang ZG. Activation of immune and inflammatory systems in chronic heart failure: novel therapeutic approaches. Int J Clin Pract. (2007) 61:611–21. doi: 10.1111/j.1742-1241.2007.01295.x
95. Van Linthout S, Riad A, Dhayat N, Spillmann F, Du J, Dhayat S, et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia. (2007) 50:1977–86. doi: 10.1007/s00125-007-0719-8
96. Feinstein MJ, Jhund P, Kang J, Ning H, Maggioni A, Wikstrand J, et al. Do statins reduce the risk of myocardial infarction in patients with heart failure? A pooled individual-level reanalysis of CORONA and GISSI-HF. Eur J Heart Fail. (2015) 17:434–41. doi: 10.1002/ejhf.247
97. Tavazzi L, Tognoni G, Franzosi MG, Latini R, Maggioni AP, Marchioli R, et al. Rationale and design of the GISSI heart failure trial: a large trial to assess the effects of n-3 polyunsaturated fatty acids and rosuvastatin in symptomatic congestive heart failure. Eur J Heart Fail. (2004) 6:635–41. doi: 10.1016/j.ejheart.2004.03.001
98. Perrone-Filardi P, Savarese G, Scarano M, Cavazzina R, Trimarco B, Minneci S, et al. Prognostic impact of metabolic syndrome in patients with chronic heart failure: data from GISSI-HF trial. Int J Cardiol. (2015) 178:85–90. doi: 10.1016/j.ijcard.2014.10.094
99. Latini R, Gullestad L, Masson S, Nymo SH, Ueland T, Cuccovillo I, et al. Pentraxin-3 in chronic heart failure: the CORONA and GISSI-HF trials. Eur J Heart Fail. (2012) 14:992–9. doi: 10.1093/eurjhf/hfs092
100. Sanders-van Wijk S, Masson S, Milani V, Rickenbacher P, Gorini M, Tavazzi LT, et al. Interaction of galectin-3 concentrations with the treatment effects of beta-blockers RAS blockade in patients with systolic heart failure: a derivation-validation study from TIME-CHF GISSI-HF. Clin Chem. (2016) 62:605–16. doi: 10.1373/clinchem.2015.246850
101. Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, Latini R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. (2008) 372:1231–9. doi: 10.1016/S0140-6736(08)61240-4
102. Bonsu KO, Reidpath DD, Kadirvelu A. Effects of statin treatment on inflammation and cardiac function in heart failure: an adjusted indirect comparison meta-analysis of randomized trials. Cardiovasc Ther. (2015) 33:338–46. doi: 10.1111/1755-5922.12150
103. Janahmadi Z, Nekooeian AA, Moaref AR, Emamghoreishi M. Oleuropein attenuates the progression of heart failure in rats by antioxidant and antiinflammatory effects. Naunyn Schmiedebergs Arch Pharmacol. (2017) 390:245–52. doi: 10.1007/s00210-016-1323-6
104. Ahmadian M, Roshan VD, Aslani E, Stannard SR. Taurine supplementation has anti-atherogenic and anti-inflammatory effects before and after incremental exercise in heart failure. Ther Adv Cardiovasc Dis. (2017) 11:185–94. doi: 10.1177/1753944717711138
105. Gal R, Deres L, Horvath O, Eros K, Sandor B, Urban P, et al. Resveratrol improves heart function by moderating inflammatory processes in patients with systolic heart failure. Antioxidants. (2020) 9:1108. doi: 10.3390/antiox9111108
106. Zinman B, Inzucchi SE, Lachin JM, Wanner C, Ferrari R, Fitchett D, et al. Rationale, design, and baseline characteristics of a randomized, placebocontrolled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME). Cardiovasc Diabetol. (2014) 13:102. doi: 10.1186/1475-2840-13-102
107. Neal B, Perkovic V, Matthews DR, Mahaffey KW, Fulcher G, Meininger G, et al. Rationale, design and baseline characteristics of the CANagliflozin cardioVascular Assessment Study-Renal (CANVAS-R): a randomized, placebo-controlled trial. Diabetes Obes Metab. (2017) 19:387–93. doi: 10.1111/dom.12829
108. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
109. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. The design and rationale for the Dapagliflozin Effect on Cardiovascular Events (DECLARE)-TIMI 58 trial. Am Heart J. (2018) 200:83–9. doi: 10.1016/j.ahj.2018.01.012
110. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
111. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383:1413–24. doi: 10.1056/NEJMoa2022190
112. Santos-Gallego CG, Garcia-Ropero A, Mancini D, Pinney SP, Contreras JP, Fergus I, et al. Rationale and design of the EMPA-TROPISM Trial (ATRU-4): are the “cardiac benefits” of empagliflozin independent of its hypoglycemic activity? Cardiovasc Drugs Ther. (2019) 33:87–95. doi: 10.1007/s10557-018-06850-0
113. Jensen J, Omar M, Kistorp C, Poulsen MK, Tuxen C, Gustafsson I, et al. Empagliflozin in heart failure patients with reduced ejection fraction: a randomized clinical trial (Empire HF). Trials. (2019) 20:374. doi: 10.1186/s13063-019-3474-5
114. Correale M, Leopizzi A, Mallardi A, Ranieri A, Suriano MP, D'Alessandro D, et al. Switch to direct anticoagulants and improved endothelial function in patients with chronic heart failure and atrial fibrillation. Thromb Res. (2020) 195:16–20. doi: 10.1016/j.thromres.2020.06.046
115. Kolijn D, Pabel S, Tian Y, Lódi M, Herwig M, Carrizzo A, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. (2021)117:495–507. doi: 10.1093/cvr/cvaa123
116. Juni RP, Kuster DWD, Goebel M, Helmes M, Musters RJP, van der Velden J, et al. Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl Sci. (2019) 4:575–91. doi: 10.1016/j.jacbts.2019.04.003
117. Connell P, Word TA, Wehrens XHT. Targeting pathological leak of ryanodine receptors: preclinical progress and the potential impact on treatments for cardiac arrhythmias and heart failure. Expert Opin Ther Targets. (2020) 24:25–36. doi: 10.1080/14728222.2020.1708326
118. Campbell H, Aguilar-Sanchez Y, Quick AP, Dobrev D, Wehrens X. SPEG: a key regulator of cardiac calcium homeostasis. Cardiovasc Res. (2020). doi: 10.1093/cvr/cvaa290. [Epub ahead of print].
119. Fargnoli AS, Katz MG, Bridges CR, Hajjar RJ. Gene therapy in heart failure. Handb Exp Pharmacol. (2017) 243:395–21. doi: 10.1007/164_2016_81
120. Khadjeh S, Hindmarsh V, Weber F, Cyganek L, Vidal RO, Torkieh S, et al. CRISPLD1: a novel conserved target in the transition to human heart failure. Basic Res Cardiol. (2020) 115:27. doi: 10.1007/s00395-020-0784-4
Keywords: drug therapy, therapy targets, heart failure, inflammation, micro-circulation, interstitium
Citation: Correale M, Tricarico L, Fortunato M, Mazzeo P, Nodari S, Di Biase M and Brunetti ND (2021) New Targets in Heart Failure Drug Therapy. Front. Cardiovasc. Med. 8:665797. doi: 10.3389/fcvm.2021.665797
Received: 08 February 2021; Accepted: 19 March 2021;
Published: 05 May 2021.
Edited by:
Antonio Sorgente, EpiCURA, BelgiumReviewed by:
Alberto Aimo, Sant'Anna School of Advanced Studies, ItalyCopyright © 2021 Correale, Tricarico, Fortunato, Mazzeo, Nodari, Di Biase and Brunetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele Correale, b3BzZmNvQHRpbi5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.