 Celina Madjene1,2,3Alexandre Boutigny1,2,3
Celina Madjene1,2,3Alexandre Boutigny1,2,3 Marie-Christine Bouton1,2,3*
Marie-Christine Bouton1,2,3* Veronique Arocas1,2,3
Veronique Arocas1,2,3 Benjamin Richard1,3,4
Benjamin Richard1,3,4- 1LVTS, INSERM, U1148, Paris, France
- 2Université de Paris, Paris, France
- 3X. Bichat Hospital, Paris, France
- 4Université Sorbonne Paris Nord, Villetaneuse, France
The balance between proteases and protease inhibitors plays a critical role in tissue remodeling during cardiovascular diseases. Different serine protease inhibitors termed serpins, which are expressed in the cardiovascular system, can exert a fine-tuned regulation of protease activities. Among them, protease nexin-1 (PN-1, encoded by SERPINE2) is a very powerful thrombin inhibitor and can also inactivate plasminogen activators and plasmin. Studies have shown that this serpin is expressed by all cell subpopulations in the vascular wall and by circulating cells but is barely detectable in plasma in the free form. PN-1 present in platelet granules and released upon activation has been shown to present strong antithrombotic and antifibrinolytic properties. PN-1 has a broad spectrum of action related to both hemostatic and blood vessel wall protease activities. Different studies showed that PN-1 is not only an important protector of vascular cells against protease activities but also a significant actor in the clearance of the complexes it forms with its targets. In this context, PN-1 overexpression has been observed in the pathophysiology of thoracic aortic aneurysms (TAA) and during the development of atherosclerosis in humans. Similarly, in the heart, PN-1 has been shown to be overexpressed in a mouse model of heart failure and to be involved in cardiac fibrosis. Overall, PN-1 appears to serve as a “hand brake” for protease activities during cardiovascular remodeling. This review will thus highlight the role of PN-1 in the cardiovascular system and deliver a comprehensive assessment of its position among serpins.
Introduction
Protease Nexin-1 (PN-1) is a 50-kDa glycoprotein encoded by the SERPINE2 gene on human chromosome 2 (1). Phylogenetically, it is the closest relative to plasminogen activator inhibitor type-1 (PAI-1 or serpinE1) (2). The serpins comprise a superfamily of proteins that share a conserved tertiary structure. Serpins include inhibitors of serine and papain-like cysteine proteases and non-inhibitory members with other biological functions. PN-1 is a cellular serpin found within diverse organs, such as brain, male and female reproductive systems, kidneys and lungs. PN-1 is also largely expressed in the vessels and the heart (3). This review thus aims to focus on its role in the pathophysiological responses of the cardiovascular system.
Biochemical Properties of PN-1
PN-1 inhibits a broad range of serine proteases explaining its physiological role in various processes ranging from coagulation and fibrinolysis to tissue remodeling and inflammation. In vitro kinetic assays showed that PN-1 reacts rapidly with trypsin and thrombin, with an association rate constant (Ka) of ~2.106 M−1.s−1 (4). The Ka values for the other target proteases including urokinase plasminogen activator (uPA) (4, 5), plasmin and factor XIa (6) are at least 10-fold lower, and 400-fold lower for tissue plasminogen activator (tPA) (4, 5) and activated protein C (7). PN-1 has also been shown to inhibit Factor VII-activating protease (FSAP) (8). As for many other serpins, PN-1 has a high affinity for heparin or heparan sulfate proteoglycans, which targets it to the pericellular space and strongly increases its ability to inhibit thrombin (9–11), thereby making this latter its preferred target. Indeed, unfractionated heparin is responsible for up to a 1,000-fold increase of the Ka value for thrombin, but only a ~10-fold increase for most other proteases and has no impact on the Ka value for plasmin. The crystal structure of the complex between thrombin, PN-1 and heparin demonstrated that heparin acts as a bridge between the serpin and the protease, leading to a ternary complex and enhancing the rate of complex formation (12). The protease-binding site (named the reactive center loop) of PN-1 is situated at the carboxy-terminal end of protein. The reactive site (P1–P′1) represented by the Arg346–Ser347 bond is cleaved by the target serine protease, which results in the formation of a covalent SDS- and heat-stable enzyme–PN-1 complex where both the protease and PN-1 become inactivated (10).
PN-1 in the Vascular System
PN-1 does not circulate in plasma, but is present in blood cells, including platelets (13, 14) and monocytes (14). Active PN-1 is released from platelet α-granules during their activation. Platelet PN-1 displays anti-thrombotic properties via its ability to block thrombin generation and activity (15). This was illustrated by in vivo studies showing an important acceleration of the induction of thrombus formation after vascular injury in PN-1-deficient mice compared to wild-type mice (15). Platelet PN-1 also displays anti-fibrinolytic properties thanks to its ability to block plasmin generation and activity (16), as illustrated in vivo with PN-1-deficient mice that display accelerated and enhanced thrombolysis following treatment with tPA (16). Thus, both PAI-1 and PN-1 may play complementary roles in maintaining the fibrin clot, and therefore largely participate in the resistance of platelet-rich clots to thrombolysis.
The first report of the presence of PN-1 in the vasculature consisted of immunohistochemical studies demonstrating an abundance of PN-1 around cerebral blood vessels (17). Later, PN-1 expression was evidenced in the vascular wall where it is expressed by endothelial cells (18, 19), vascular smooth muscle cells (vSMCs) (20) and fibroblasts (21, 22). Importantly, it is retained at the cell surface of vascular cells and within the extracellular matrix (ECM) of the vessel wall due to its high affinity for heparin sulfate proteoglycans (22) and its ability to bind to the low-density lipoprotein receptor-related protein 1 (LRP1) of the scavenger receptor family (23, 24). PN-1 is expressed by endothelial cells and interacts with thrombomodulin, a high affinity thrombin ligand expressed on the endothelial cell membrane that plays an important role in the regulation of coagulation via the activation of the natural anticoagulant protein C. PN-1-thrombomodulin interaction favors the inhibition of fibrin formation and limits the generation of activated protein C and thrombin activatable fibrinolysis inhibitor (18). Endothelial PN-1 was also shown to protect the endothelial protein C receptor from endogenous shedding, thereby favoring the cytoprotective effects of activated protein C (25). Deficiency of PN-1 in mice does not generate a spontaneous vascular phenotype compromising their survival. However, endothelial PN-1 plays a role in physiological angiogenesis. Indeed, the retina from PN-1-deficient mice displayed increased vascularization with elevated capillary thickness and density, as well as an increased number of veins and arteries, compared to their wild-type littermates (26). Moreover, neovessel formation in Matrigel plug assays in PN-1-deficient mice, as well as the microvascular network sprouting from PN-1-deficient aortic rings, were both largely enhanced compared with their respective controls (27). These data clearly illustrate the important anti-angiogenic potential of vascular PN-1.
PN-1 in Vascular Diseases
PN-1 and Atherosclerosis
Atherosclerosis is a disease characterized by the thickening of the blood vessel wall due to the formation of plaques in the subendothelial intimal space. It involves endothelial cell dysfunction resulting in an alteration of endothelial permeability, allowing the penetration and accumulation of low-density lipoprotein (LDL) particles in the vessel wall where they are susceptible to oxidation. Monocytes are also implicated and transmigrate into the intima where they differentiate into macrophages, becoming foam cells after ingestion of oxidized LDL. VSMC proliferation and migration from the media to the intimal layer, as well as their phenotypic shift into foam cells, are also important features of atherosclerosis development. vSMCs present in the intimal layer form a fibrous cap that contains the plaque. The rupture of the fibrous cap leads to thrombus formation causing blockage of the blood flow (28).
An unbalanced ratio between proteases and their inhibitors is involved throughout the pathophysiology of atherosclerosis. Excessive thrombin, uPA/tPA or plasmin activities are indeed involved in the chronic evolution of the plaque. An important question thus concerns the regulation of these proteases in the vessel wall. In this context, serpins increasingly appear to be critical in regulating protease activity in arterial lesions. Among them, PN-1 has emerged as a key regulator in vascular biology even though its precise mechanism of action remains to be deciphered.
Immunohistochemical studies demonstrated the presence of PN-1 in the healthy vascular wall and particularly in vSMCs (20). PN-1 has also been shown to be associated with vSMCs in advanced carotid atherosclerotic lesions, but also with macrophages and platelets (14, 29). Accumulation of PN-1 was detectable in very early lesions and was increased in complicated plaques: globally, PN-1 was present in the cap, in the necrotic core and in the mural thrombus (14, 30). In fact, the biological activity of PN-1 appears to be involved in the different stages of atherosclerotic plaque progression. During the early stage, PN-1 may be involved in endothelial dysfunction. Indeed, at the endothelial level, PN-1 has been shown to interact with thrombomodulin, a glycoprotein that transforms thrombin from a pro- to an anticoagulant protein (18). Thrombomodulin interaction with PN-1 accentuates the ability of the latter to inhibit thrombin. In advanced atherosclerotic plaques, PN-1 is largely expressed by platelets and inflammatory cells including monocytes/macrophages. In agreement with this observation, PN-1 has been shown to be up-regulated in lipopolysaccharide-activated monocytes and degraded in macrophages (14). Because monocytes/macrophages are exposed to an inflammatory environment in atherothrombotic lesions, PN-1 overexpression may represent a cell defense reaction against proteases present in the atherosclerotic plaque. Indeed, vSMCs synthesize and secrete tPA that is able to drive the conversion of plasminogen into plasmin at the cell surface, leading to matrix degradation, cell detachment, and death (31). However, PN-1 is also overexpressed by vSMCs in the advanced plaque where it is able to form covalent complexes with plasmin (30). Both endocytic LRP-1 and PN-1 are highly expressed in human atheroma, making PN-1 a crucial actor in plasmin internalization by vSMCs, via LRP-1 (30). PN-1 has also been shown to form covalent complexes with FSAP (8), a circulating protease found in human atherosclerotic plaques and supposed to play a regulatory role in their progression and vulnerability (32). The fibrous cap plays a crucial role in the development of atherosclerosis because its thickness is tightly related to the vulnerability of atherosclerotic plaques. PN-1 may also influence the thickness of the fibrous cap, by acting on the migration of vSMCs. Indeed, overexpression of PN-1 by vSMCs has been shown to significantly reduce their adhesion, spreading and migration on vitronectin, an adhesive protein found in atherosclerotic plaques (33). This effect is related to the high affinity of PN-1 to vitronectin, shown by direct-binding in vitro assays (11). Moreover, PN-1 can limit thrombin-induced vSMC proliferation (20) and (i) prevents the pro-apoptotic effect of high thrombin concentrations (34), (ii) inhibits plasminogen activation in the peri-cellular environment, and (iii) prevents plasmin-induced cell detachment (34). Taken together, these data raise the possibility that PN-1 overexpression during atherosclerosis could significantly influence the stability of the plaque. At the most complicated stage of atherosclerosis, rupture of the plaque can trigger localized, often occlusive, thrombus formation. PN-1 can thus also accumulate within thrombi generated during atherothrombosis since platelets are a reservoir of this serpin. Via its ability to inhibit plasmin generation and activity within the thrombus, platelet PN-1 is assumed to contribute to thrombus stabilization and is therefore also a non-negligible contributor to thrombus resistance to lysis (16).
Given its ubiquitous expression in the atheromatous lesions and its inhibitory activity against numerous deleterious proteases present in the atheroma, PN-1 can undoubtedly regulate the characteristics of the atherosclerotic plaque at different stages of development.
PN-1 and Aneurysms
Aortic aneurysms are also diseases characterized by intense remodeling due to an imbalance in favor of proteolytic degradation of the vascular wall ECM, leading to progressive dilation and eventually to rupture. Despite various possible etiologies, all thoracic aneurysms of the ascending aorta (TAA) share common pathophysiological features leading to structural deterioration of the aortic wall. VSMCs apoptosis and the degradation of collagen and elastic fibers are the two principal modifications occurring within the medial layer characterizing TAA. The relevance of the antiprotease activity of PN-1 expressed by vSMCs has been emphasized by its ability to regulate in vitro pericellular plasminogen activation (35) and therefore cell resistance to proteolytic aggression, as observed during atherosclerosis. In human biopsies, PN-1 expression was found to be increased in the medial layer of TAA compared with the aortic medial layer from healthy donors and the protein colocalized with vSMCs. Interestingly, cultured vSMCs from TAA continued to display an increased level of PN-1 mRNA expression compared with control vSMCs (36). This was found to be due to the permanent epigenetic activation of the smad2 pathway in vivo in the arterial wall of TAA, an activation which persisted in cultures of vSMCs of TAA origin. Hence, human cultured vSMCs from TAA had a limited capacity to convert plasminogen into plasmin, and were therefore protected against apoptosis-induced detachment after plasminogen or plasmin treatment (36). Indeed, PN-1 overexpression was shown to be associated with aneurysmal dilatation, whereas the absence of PN-1 overexpression was associated with aortic dissections (36). Together, these data show that overproduction of PN-1 by vSMCs in vivo during TAA development may participate in the increased ability of the cells to resist the proteolytic environment.
The clearance of PN-1/plasmin complexes has also been addressed specifically in the TAA context. PN-1, LRP-1 and plasmin were shown to colocalize in the media of human TAA where PN-1 amounts correlated with plasmin activity (37). The uptake of PN-1/plasmin complexes was shown to be partly mediated by LRP-1 in vSMCs. These results strongly suggest that PN-1 might play a protective role in vivo during TAA development, as discussed for atherosclerosis, but further experimental animal models are required to fully understand its impact on TAA pathophysiology.
In contrast to TAA, the role of PN-1 in abdominal aortic aneurysms (AAA) has not yet been addressed. Previous reports have shown that the enzymes of the fibrinolytic system are also involved in AAA progression (38, 39) and local overexpression of PAI-1 in the mouse was accordingly reported to prevent the development of the disease (40). The role of the plasminergic system remains nevertheless incompletely understood (41) and the study of PN-1 in this context could provide new insights into the understanding of how proteases and their counter-regulators participate in the evolution of AAA.
PN-1 in Cardiac Fibrosis
Myocardial fibrosis is an important pathophysiological process defined as an excessive accumulation of matrix proteins and is a well-established morbi-mortality marker. It increases myocardial stiffness, alters systolic function and contributes to malignant arrhythmias (42).
PN-1 in the heart has received less attention although it has been reported to be present in mouse heart (43). Moreover, in rats a high overexpression of PN-1 was described in in vivo heart failure models (44). Li et al. were the first to assess the role of PN-1 in cardiac fibrosis (45). They showed that both cardiomyocytes and myocardial fibroblasts express PN-1, even though the level of PN-1 expression in the former was only half that in the latter. They also found, in an in vivo mouse model of cardiac fibrosis induced by transverse aortic constriction (TAC), that collagen deposition was increased after 4 weeks, associated with a slight increase in PN-1 expression in the heart (45). Moreover, they showed that pro-fibrotic mediators like angiotensin II and transforming growth factor-β (TGF-β) could induce, in myocardial fibroblasts, an increased expression of collagen associated with PN-1 overexpression, at both the messenger and protein levels. Such an up-regulation of PN-1 induced by TGF-β has also been observed in vitro in human pulmonary fibroblasts (46). Reciprocally, the knockdown of PN-1 appears to partially attenuate cardiac fibrosis (45). However, cardiac expression of PN-1 is only partially impaired and these data do not allow us to draw clear conclusions as to the role of PN-1 in cardiac injury.
PN-1 appears to be importantly involved in fibrotic processes. Interestingly, depending on the affected tissue, PN-1 displays either anti-fibrotic properties as described in pulmonary fibrosis (47) or in contrast, pro-fibrotic properties as described here in cardiac fibrosis or as reported in scleroderma, a disease also characterized by ECM accumulation in skin and visceral tissue (48). The link between PN-1 and cardiac fibrosis can also be mediated, at least in part, by its antiprotease inhibitor activity, in particular by its ability to inhibit thrombin and uPA. Indeed, the direct inhibition of thrombin with dabigatran was shown to attenuate cardiac fibrosis and improve global cardiac function in a TAC murine model (49). The importance of the uPA/plasmin/matrix-metalloproteinase (MMP) system in collagen degradation has been well-characterized (50). PAI-1, a serpin close to PN-1, has also been shown to exert pro-or anti-fibrotic effects in different organs. The inhibition by PAI-1 of uPA- and tPA-mediated conversion of plasminogen to plasmin was shown to decrease plasmin-mediated MMP activation, and consequently to increase matrix accumulation and fibrosis in different tissues including lung, liver and kidney (51). In contrast, in the heart, PAI-1 protects mice from hypertension-induced cardiac fibrosis (52). Indeed, although PAI-1 is upregulated by TGF-β in numerous cell types (53), in the myocardium, PAI-1 was shown to inhibit TGF-β production specifically in cardiomyocytes (51).
More detailed studies are required to decipher the role of PN-1 in cardiac fibrosis. Indeed, in pathological conditions, such as pressure overload models or myocardial infarction, inflammation plays an important role in adaptative and inadaptive responses, where monocytes and macrophages are key components of the inflammatory pathophysiology (54). Because PN-1 is expressed by inflammatory cells and has been shown to be closely related to the inflammatory reaction in lung fibrosis, we can hypothesize that PN-1 can also participate in cardiac inflammation and consequently, in cardiac fibrosis.
Conclusions
The close relationships between PN-1 and proteases of the coagulation and fibrinolytic systems, as well as between PN-1 and the endocytic receptor LRP1, explain the impact of this serpin in the cardiovascular system (Table 1). Essentially, PN-1 participates in maintaining the homeostatic function of the arterial wall and the cardiac tissue, as illustrated by its overexpression in the different cardiovascular pathologies mentioned in this review (Figure 1).

Table 1. Expected effect of PN-1 in cardiovascular diseases depending on its targets or partners.
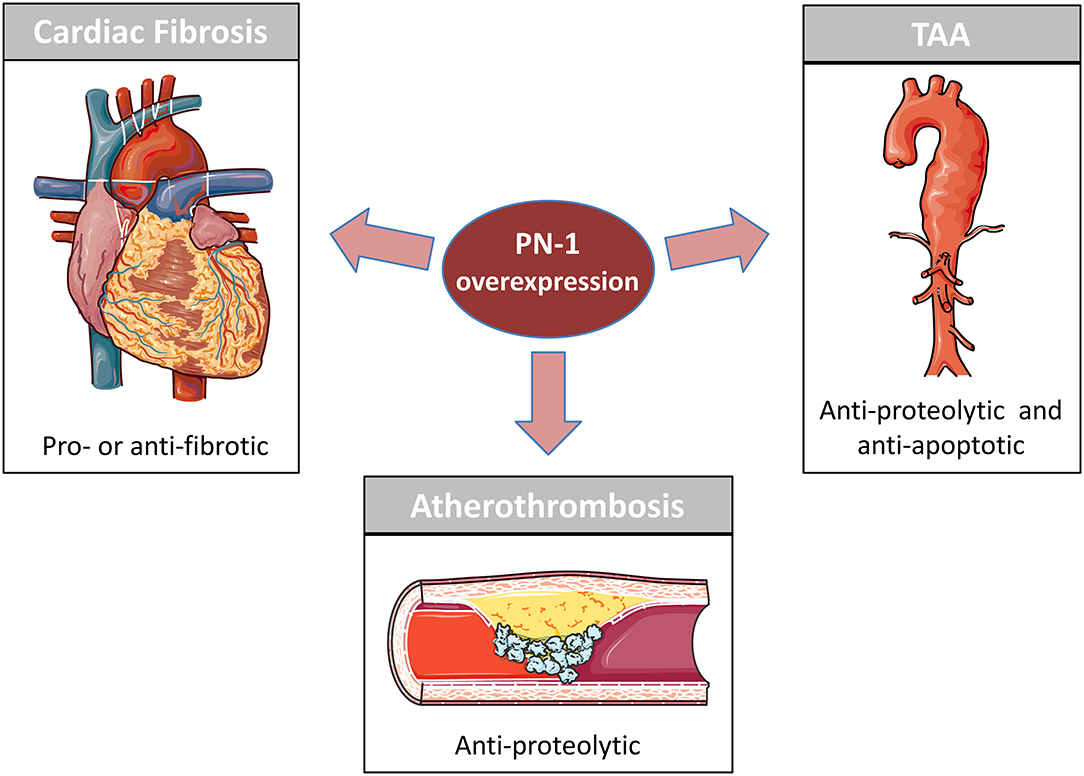
Figure 1. PN-1 in cardiovascular disease. PN-1 is overexpressed in Atherosclerosis, Cardiac Fibrosis and Thoracic Aortic Aneurysm (TAA). Previous studies have shown PN-1 to be an important protective actor in atherosclerosis and TAA by reducing the impact of the proteolytic environment on the vascular cells. PN-1 is also involved in cardiac fibrosis but can be either anti-fibrotic and protective or pro-fibrotic and deleterious depending on its targets (see Table 1).
Author Contributions
CM and BR generated the figure and the table. VA and M-CB provided critical feedback and edited the review. All authors contributed to the review.
Funding
This work was supported by INSERM, Université de Paris, Université Sorbonne Paris Nord, Fondation de France (n°86496 and 96226), and the French Federation of Cardiology.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Carter RE, Cerosaletti KM, Burkin DJ, Fournier RE, Jones C, Greenberg BD, et al. The gene for the serpin thrombin inhibitor (PI7), protease nexin I, is located on human chromosome 2q33-q35 and on syntenic regions in the mouse and sheep genomes. Genomics. (1995) 27:196–9. doi: 10.1006/geno.1995.1025
2. Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. (2000) 10:1845–64. doi: 10.1101/gr.GR-1478R
3. Bouton MC, Boulaftali Y, Richard B, Arocas V, Michel JB, Jandrot-Perrus M. Emerging role of serpinE2/protease nexin-1 in hemostasis and vascular biology. Blood. (2012) 119:2452–7. doi: 10.1182/blood-2011-10-387464
4. Scott RW, Bergman BL, Bajpai A, Hersh RT, Rodriguez H, Jones BN, et al. Protease nexin. Properties and a modified purification procedure. J Biol Chem. (1985) 260:7029–34. doi: 10.1016/S0021-9258(18)88883-4
5. Eaton DL, Scott RW, Baker JB. Purification of human fibroblast urokinase proenzyme and analysis of its regulation by proteases and protease nexin. J Biol Chem. (1984) 259:6241–7. doi: 10.1016/S0021-9258(20)82132-2
6. Knauer DJ, Majumdar D, Fong PC, Knauer MF. SERPIN regulation of factor XIa. The novel observation that protease nexin 1 in the presence of heparin is a more potent inhibitor of factor XIa than C1 inhibitor. J Biol Chem. (2000) 275:37340–6. doi: 10.1074/jbc.M003909200
7. Hermans JM, Stone SR. Interaction of activated protein C with serpins. Biochem J. (1993) 295 (Pt 1):239–45. doi: 10.1042/bj2950239
8. Muhl L, Nykjaer A, Wygrecka M, Monard D, Preissner KT, Kanse SM. Inhibition of PDGF-BB by Factor VII-activating protease (FSAP) is neutralized by protease nexin-1, and the FSAP-inhibitor complexes are internalized via LRP. Biochem J. (2007) 404:191–6. doi: 10.1042/BJ20061630
9. Cunningham DD. Regulation of neuronal cells and astrocytes by protease nexin-1 and thrombin. Ann N Y Acad Sci. (1992) 674:228–36. doi: 10.1111/j.1749-6632.1992.tb27491.x
10. Evans DL, McGrogan M, Scott RW, Carrell RW. Protease specificity and heparin binding and activation of recombinant protease nexin I. J Biol Chem. (1991) 266:22307–12. doi: 10.1016/S0021-9258(18)54571-3
11. Rovelli G, Stone SR, Guidolin A, Sommer J, Monard D. Characterization of the heparin-binding site of glia-derived nexin/protease nexin-1. Biochemistry. (1992) 31:3542–9. doi: 10.1021/bi00128a031
12. Li W, Huntington JA. Crystal structures of protease nexin-1 in complex with heparin and thrombin suggest a 2-step recognition mechanism. Blood. (2012) 120:459–67. doi: 10.1182/blood-2012-03-415869
13. Gronke RS, Knauer DJ, Veeraraghavan S, Baker JB. A form of protease nexin I is expressed on the platelet surface during platelet activation. Blood. (1989) 73:472–8. doi: 10.1182/blood.V73.2.472.472
14. Mansilla S, Boulaftali Y, Venisse L, Arocas V, Meilhac O, Michel JB, et al. Macrophages and platelets are the major source of protease nexin-1 in human atherosclerotic plaque. Arterioscler Thromb Vasc Biol. (2008) 28:1844–50. doi: 10.1161/ATVBAHA.108.171389
15. Boulaftali Y, Adam F, Venisse L, Ollivier V, Richard B, Taieb S, et al. Anticoagulant and antithrombotic properties of platelet protease nexin-1. Blood. (2010) 115:97–106. doi: 10.1182/blood-2009-04-217240
16. Boulaftali Y, Ho-Tin-Noe B, Pena A, Loyau S, Venisse L, Francois D, et al. Platelet protease nexin-1, a serpin that strongly influences fibrinolysis and thrombolysis. Circulation. (2011) 123:1326–34. doi: 10.1161/CIRCULATIONAHA.110.000885
17. Choi BH, Suzuki M, Kim T, Wagner SL, Cunningham DD. Protease nexin-1. Localization in the human brain suggests a protective role against extravasated serine proteases. Am J Pathol. (1990) 137:741–7.
18. Bouton MC, Venisse L, Richard B, Pouzet C, Arocas V, Jandrot-Perrus M. Protease nexin-1 interacts with thrombomodulin and modulates its anticoagulant effect. Circ Res. (2007) 100:1174–81. doi: 10.1161/01.RES.0000265066.92923.ee
19. Leroy-Viard K, Jandrot-Perrus M, Tobelem G, Guillin MC. Covalent binding of human thrombin to a human endothelial cell-associated protein. Exp Cell Res. (1989) 181:1–10. doi: 10.1016/0014-4827(89)90177-8
20. Bouton MC, Richard B, Rossignol P, Philippe M, Guillin MC, Michel JB, et al. The serpin protease-nexin 1 is present in rat aortic smooth muscle cells and is upregulated in L-NAME hypertensive rats. Arterioscler Thromb Vasc Biol. (2003) 23:142–7. doi: 10.1161/01.ATV.0000047867.98019.2D
21. Baker JB, Low DA, Simmer RL, Cunningham DD. Protease-nexin: a cellular component that links thrombin and plasminogen activator and mediates their binding to cells. Cell. (1980) 21:37–45. doi: 10.1016/0092-8674(80)90112-9
22. Farrell DH, Wagner SL, Yuan RH, Cunningham DD. Localization of protease nexin-1 on the fibroblast extracellular matrix. J Cell Physiol. (1988) 134:179–88. doi: 10.1002/jcp.1041340203
23. Crisp RJ, Knauer DJ, Knauer MF. Roles of the heparin and low density lipid receptor-related protein-binding sites of protease nexin 1 (PN1) in urokinase-PN1 complex catabolism. The PN1 heparin-binding site mediates complex retention and degradation but not cell surface binding or internalization. J Biol Chem. (2000) 275:19628–37. doi: 10.1074/jbc.M909172199
24. Knauer MF, Kridel SJ, Hawley SB, Knauer DJ. The efficient catabolism of thrombin-protease nexin 1 complexes is a synergistic mechanism that requires both the LDL receptor-related protein and cell surface heparins. J Biol Chem. (1997) 272:29039–45. doi: 10.1074/jbc.272.46.29039
25. Boulaftali Y, Francois D, Venisse L, Jandrot-Perrus M, Arocas V, Bouton MC. Endothelial protease nexin-1 is a novel regulator of A disintegrin and metalloproteinase 17 maturation and endothelial protein C receptor shedding via furin inhibition. Arterioscler Thromb Vasc Biol. (2013) 33:1647–54. doi: 10.1161/ATVBAHA.113.301494
26. Selbonne S, Francois D, Raoul W, Boulaftali Y, Sennlaub F, Jandrot-Perrus M, et al. Protease nexin-1 regulates retinal vascular development. Cell Mol Life Sci. (2015) 72:3999–4011. doi: 10.1007/s00018-015-1972-5
27. Selbonne S, Azibani F, Iatmanen S, Boulaftali Y, Richard B, Jandrot-Perrus M, et al. In vitro and in vivo antiangiogenic properties of the serpin protease nexin-1. Mol Cell Biol. (2012) 32:1496–505. doi: 10.1128/MCB.06554-11
28. Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
29. Kanse SM, Chavakis T, Al-Fakhri N, Hersemeyer K, Monard D, Preissner KT. Reciprocal regulation of urokinase receptor (CD87)-mediated cell adhesion by plasminogen activator inhibitor-1 and protease nexin-1. J Cell Sci. (2004) 117(Pt 3):477–85. doi: 10.1242/jcs.00861
30. Boukais K, Bayles R, Borges Lde F, Louedec L, Boulaftali Y, Ho-Tin-Noe B, et al. Uptake of plasmin-PN-1 complexes in early human atheroma. Front Physiol. (2016) 7:273. doi: 10.3389/fphys.2016.00273
31. Meilhac O, Ho-Tin-Noe B, Houard X, Philippe M, Michel JB, Angles-Cano E. Pericellular plasmin induces smooth muscle cell anoikis. FASEB J. (2003) 17:1301–3. doi: 10.1096/fj.02-0687fje
32. Kanse SM, Parahuleva M, Muhl L, Kemkes-Matthes B, Sedding D, Preissner KT. Factor VII-activating protease (FSAP): vascular functions and role in atherosclerosis. Thromb Haemost. (2008) 99:286–9. doi: 10.1160/TH07-10-0640
33. Richard B, Pichon S, Arocas V, Venisse L, Berrou E, Bryckaert M, et al. The serpin protease nexin-1 regulates vascular smooth muscle cell adhesion, spreading, migration and response to thrombin. J Thromb Haemost. (2006) 4:322–8. doi: 10.1111/j.1538-7836.2006.01710.x
34. Rossignol P, Ho-Tin-Noe B, Vranckx R, Bouton MC, Meilhac O, Lijnen HR, et al. Protease nexin-1 inhibits plasminogen activation-induced apoptosis of adherent cells. J Biol Chem. (2004) 279:10346–56. doi: 10.1074/jbc.M310964200
35. Richard B, Arocas V, Guillin MC, Michel JB, Jandrot-Perrus M, Bouton MC. Protease nexin-1: a cellular serpin down-regulated by thrombin in rat aortic smooth muscle cells. J Cell Physiol. (2004) 201:138–45. doi: 10.1002/jcp.20103
36. Gomez D, Kessler K, Borges LF, Richard B, Touat Z, Ollivier V, et al. Smad2-dependent protease nexin-1 overexpression differentiates chronic aneurysms from acute dissections of human ascending aorta. Arterioscler Thromb Vasc Biol. (2013) 33:2222–32. doi: 10.1161/ATVBAHA.113.301327
37. Boukais K, Borges LF, Venisse L, Touat Z, Francois D, Arocas V, et al. Clearance of plasmin-PN-1 complexes by vascular smooth muscle cells in human aneurysm of the ascending aorta. Cardiovasc Pathol. (2018) 32:15–25. doi: 10.1016/j.carpath.2017.10.002
38. Schneiderman J, Bordin GM, Engelberg I, Adar R, Seiffert D, Thinnes T, et al. Expression of fibrinolytic genes in atherosclerotic abdominal aortic aneurysm wall. A possible mechanism for aneurysm expansion. J Clin Invest. (1995) 96:639–45. doi: 10.1172/JCI118079
39. Wang YX, Martin-McNulty B, Freay AD, Sukovich DA, Halks-Miller M, Li WW, et al. Angiotensin II increases urokinase-type plasminogen activator expression and induces aneurysm in the abdominal aorta of apolipoprotein E-deficient mice. Am J Pathol. (2001) 159:1455–64. doi: 10.1016/S0002-9440(10)62532-1
40. Qian HS, Gu JM, Liu P, Kauser K, Halks-Miller M, Vergona R, et al. Overexpression of PAI-1 prevents the development of abdominal aortic aneurysm in mice. Gene Ther. (2008) 15:224–32. doi: 10.1038/sj.gt.3303069
41. Rein CM, Cardenas JC, Church FC. The controversial role of the urokinase system in abdominal aortic aneurysm formation and rupture. Arterioscler Thromb Vasc Biol. (2011) 31:2769–71. doi: 10.1161/ATVBAHA.111.237123
42. Centurion OA, Scavenius KE, Garcia LB, Torales JM, Mino LM. Potential mechanisms of cardiac injury and common pathways of inflammation in patients with COVID-19. Crit Pathw Cardiol. (2021) 20:44–52. doi: 10.1097/HPC.0000000000000227
43. Mansuy IM, van der Putten H, Schmid P, Meins M, Botteri FM, Monard D. Variable and multiple expression of Protease Nexin-1 during mouse organogenesis and nervous system development. Development. (1993) 119:1119–34.
44. Lu B, Yu H, Zwartbol M, Ruifrok WP, van Gilst WH, de Boer RA, et al. Identification of hypertrophy- and heart failure-associated genes by combining in vitro and in vivo models. Physiol Genomics. (2012) 44:443–54. doi: 10.1152/physiolgenomics.00148.2011
45. Li X, Zhao D, Guo Z, Li T, Qili M, Xu B, et al. Overexpression of serpinE2/protease nexin-1 contribute to pathological cardiac fibrosis via increasing collagen deposition. Sci Rep. (2016) 6:37635. doi: 10.1038/srep37635
46. Francois D, Venisse L, Marchal-Somme J, Jandrot-Perrus M, Crestani B, Arocas V, et al. Increased expression of protease nexin-1 in fibroblasts during idiopathic pulmonary fibrosis regulates thrombin activity and fibronectin expression. Lab Invest. (2014) 94:1237–46. doi: 10.1038/labinvest.2014.111
47. Francois D, Arocas V, Venisse L, Aymonnier K, Idir L, Martos R, et al. Hematopoietic protease nexin-1 protects against lung injury by preventing thrombin signaling in mice. Blood Adv. (2018) 2:2389–99. doi: 10.1182/bloodadvances.2018018283
48. Strehlow D, Jelaska A, Strehlow K, Korn JH. A potential role for protease nexin 1 overexpression in the pathogenesis of scleroderma. J Clin Invest. (1999) 103:1179–90. doi: 10.1172/JCI1918
49. Dong A, Mueller P, Yang F, Yang L, Morris A, Smyth SS. Direct thrombin inhibition with dabigatran attenuates pressure overload-induced cardiac fibrosis and dysfunction in mice. Thromb Res. (2017) 159:58–64. doi: 10.1016/j.thromres.2017.09.016
50. Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost. (2001) 86:324–33. doi: 10.1055/s-0037-1616230
51. Flevaris P, Vaughan D. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin Thromb Hemost. (2017) 43:169–77. doi: 10.1055/s-0036-1586228
52. Gupta KK, Donahue DL, Sandoval-Cooper MJ, Castellino FJ, Ploplis VA. Plasminogen activator inhibitor-1 protects mice against cardiac fibrosis by inhibiting urokinase-type plasminogen activator-mediated plasminogen activation. Sci Rep. (2017) 7:365. doi: 10.1038/s41598-017-00418-y
53. Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. (2012) 227:493–507. doi: 10.1002/jcp.22783
Keywords: PN-1, atherosclerosis, aneurysm, fibrosis, serpinE2, heart failure, smooth muscle cell
Citation: Madjene C, Boutigny A, Bouton M-C, Arocas V and Richard B (2021) Protease Nexin-1 in the Cardiovascular System: Wherefore Art Thou? Front. Cardiovasc. Med. 8:652852. doi: 10.3389/fcvm.2021.652852
Received: 13 January 2021; Accepted: 10 March 2021;
Published: 31 March 2021.
Edited by:
Rory R. Koenen, Maastricht University, NetherlandsReviewed by:
Lucas Tirloni, National Institutes of Health (NIH), United StatesJames Huntington, University of Cambridge, United Kingdom
Copyright © 2021 Madjene, Boutigny, Bouton, Arocas and Richard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie-Christine Bouton, bWFyaWUtY2hyaXN0aW5lLmJvdXRvbkBpbnNlcm0uZnI=