
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 16 March 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.642751
 Xueyu Wang1,2†
Xueyu Wang1,2† Yahong Fu1,2†
Yahong Fu1,2† Zulong Xie3†
Zulong Xie3† Muhua Cao1,2
Muhua Cao1,2 Wenbo Qu1,2
Wenbo Qu1,2 Xiangwen Xi1,2
Xiangwen Xi1,2 Shan Zhong1,2
Shan Zhong1,2 Minghui Piao1,2
Minghui Piao1,2 Xiang Peng1,2
Xiang Peng1,2 Ying Jia1,2
Ying Jia1,2 Lingbo Meng1*
Lingbo Meng1* Jinwei Tian1*
Jinwei Tian1*Background and Aims: Acute coronary syndrome (ACS) is a group of clinical syndromes characterized by rupture or erosion of atherosclerotic unstable plaques. Effective intervention for vulnerable plaques (VP) is of great significance to reduce adverse cardiovascular events.
Methods: Fbn1C1039G+/− mice were crossbred with LDLR−/− mice to obtain a novel model for atherosclerotic VP. After the mice were fed with a high-fat diet (HFD) for 12 or 24 weeks, pathological staining and immunohistochemistry analyses were employed to evaluate atherosclerotic lesions.
Results: Compared to control mice, Fbn1C1039G+/−LDLR−/− mice developed more severe atherosclerotic lesions, and the positive area of oil red O staining in the aortic sinus was significantly increased after 12 weeks (21.7 ± 2.0 vs. 6.3 ± 2.1) and 24 weeks (32.6 ± 2.5 vs. 18.7 ± 2.6) on a HFD. Additional vulnerable plaque characteristics, including significantly larger necrotic cores (280 ± 19 vs. 105 ± 7), thinner fiber caps (14.0 ± 2.8 vs. 32.6 ± 2.7), apparent elastin fiber fragmentation and vessel dilation (3,010 ± 67 vs. 1,465 ± 49), a 2-fold increase in macrophage number (8.5 ± 1.0 vs. 5.0 ± 0.6), obviously decreased smooth muscle cell number (0.6 ± 0.1 vs. 2.1 ± 0.2) and an ~25% decrease in total collagen content (33.6 ± 0.3 vs. 44.9 ± 9.1) were observed in Fbn1C1039G+/−LDLR−/− mice compared with control mice after 24 weeks. Furthermore, spontaneous plaque rupture, neovascularization, and intraplaque hemorrhage were detected in the model mouse plaque regions but not in those of the control mice.
Conclusions: Plaques in Fbn1C1039G+/−LDLR−/− mice fed a HFD show many features of human advanced atherosclerotic unstable plaques. These results suggest that the Fbn1C1039G+/−LDLR−/− mouse is a novel model for investigating the pathological and physiological mechanisms of advanced atherosclerotic unstable plaques.
Atherosclerosis (AS), which results from the accumulation of lipid-rich plaques within artery walls, has become one of the biggest risks to human health. Studies have demonstrated that the growth of atherosclerotic plaques has obvious stages (1). It is widely believed that in the early stage, due to endothelial dysfunction, lipids and lipid-laden macrophages accumulate in the subendothelial area, leading to the formation of foam cells (2). Subsequently, with the continuous progression of plaques and exacerbation of the inflammatory response, a large number of growth factors and inflammatory mediators are secreted by macrophages and other cells, and lipid streaks gradually emerge (3). Moreover, as smooth muscle cells (SMCs) proliferate and migrate to intimal tissue, more elastin and collagen are produced, and lipid streaks gradually change into fibrous plaques. Advanced plaques are enlarged fibrous plaques characterized by narrowed lumen areas, haemodynamic changes, thinner fibrous caps and larger necrotic cores (4). In general, pathological and imaging studies have demonstrated that there are “stable” and “vulnerable” plaques in human coronary arteries (5). Stable plaques, which are accompanied by collagen accumulation, can form a fibrous plaque without a necrotic core. In contrast, vulnerable plaques are characterized by a large necrotic core and a thinner fibrous cap and have a high risk of sudden rupture and thrombosis leading to acute coronary events (5). In recent years, although considerable progress has been made in the prevention and treatment of cardiovascular diseases, atherosclerotic plaque rupture, especially vulnerable plaque rupture, is still the principal pathogenic mechanism of ACS (6, 7). Thus, identifying the specific mechanism of plaque rupture and designing effective prevention and treatment strategies are of great significance (8).
Arteriosclerosis is reported to be a major independent risk factor for cardiovascular diseases in the macro- and microvasculature. As a principal part of extracellular matrices, elastic fibers provide resilience and deformability for arterial vessels. Abnormal changes in the structure and function of elastic fibers can lead to decreased elasticity of vessels, vessel aging and arteriosclerosis. Elastic fibers are composed of two distinct components: insoluble, homogeneous, amorphous elastin, and fibrillin-rich microfibrils (9). Fibrillin-1 (Fbn1), a primary component of microfibrils, provides an architectural scaffold for elastin deposition and crosslinking. Studies have found that mutant fibrillin-1 damages the structure of the vessel wall by rupturing the crosslinking of elastic fibers, resulting in arteriosclerosis (10, 11). Based on this result, the relationship between AS and elastin fragmentation may provide a new perspective in studies of cardiovascular diseases. In previous research, multiple models of AS have been developed with a variety of methods to study the pathological mechanisms of and interventions in atherosclerosis; these methods include using lipid metabolism regulation, physical damage, drug action, and gene knockout techniques to induce atherosclerotic plaque formation. Among these models, apoprotein E-deficient mice (ApoE−/−) and low-density lipoprotein (LDL) receptor-deficient mice (LDLR−/−) have been widely used (12). However, in terms of pathology, these models of atherosclerosis represent some but not all characteristics of human ruptured plaques (13–15). Due to the limitations of human atherosclerotic plaque studies, it is urgent to develop suitable animal models of plaque rupture for basic and clinical research.
In the present study, LDLR−/− mice were crossbred with Fbn1C1039G+/− mice. After feeding the mice a HFD for 12 or 24 weeks, we evaluated the characteristics of plaques in LDLR−/− (the control group) and Fbn1C1039G+/−LDLR−/− (the model group) mice using multiple pathological experiments. We aimed to construct a novel atherosclerosis model for studying the pathological and physiological mechanism of the development of atherosclerotic plaques by introducing a fibrillin-1 heterozygous mutation into LDLR−/− mice via homologous recombination.
Fbn1C1039G+/− mice (C57BL/6 background, purchased from The Jackson Laboratory, Maine, USA) were crossbred with LDLR−/− mice (C57BL/6 background, purchased from Beijing HuaFuKang Bioscience Co. INC, Beijing, China). According to Mendel's genetic laws, we obtained Fbn1C1039G+/−LDLR−/− mice in the second generation. The genotype was identified by polymerase chain reaction, gene sequencing analysis, and agarose gel electrophoresis. The primer sequences were as follows: LDLR-c: CCATATGCATCCCCAGTCTT, LDLR-w: GCGATGGATACACTCAC-TGC, LDLR-m: AATCCATCTTGTTCAATGGCCGATC; C1039G-F: TTGTCCA-TGTGCTTTAAGTAGC, C1039G-R: ACAGAGGTCAGGAGATATGC. To quickly obtain enough Fbn1C1039G+/−LDLR−/− mice, female mice were used for experiments, while males were used for breeding. Eight-week-old female Fbn1C1039G+/−LDLR−/− mice (n = 10) were fed a HFD (Nanjing Junke Bioengineering Co., Ltd., # AAN01, China) containing 10% fat, 2% cholesterol, and 0.5% cholic acid. Female LDLR−/− mice (n = 10) were used as controls. Animals were housed in a temperature-controlled room with a 12-h light/dark cycle and had free access to water and food. They were inspected daily for neurological symptoms (including head tilt, disorientation, and motor disturbances) or sudden death. Body weight was measured every 10 days in the HFD period. All experiments were approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University, China (2015-Yan-043).
To evaluate the plaque characteristics of the two groups in different periods, plaque area and composition in Fbn1C1039G+/−LDLR−/− and LDLR−/− mice were analyzed after 12 or 24 weeks on a HFD. Mice were anesthetized (n = 10 in each group), and blood samples were taken from the retro-orbital plexus for analysis of lipid profiles by a commercially available kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). After fixation and perfusion of the left ventricle with 4% formaldehyde (pH 7.4), tissues (heart, aorta, and brachiocephalic artery) were collected. Subsequently, the heart and arteries were fixed in 4% formaldehyde for 24 h, dehydrated overnight, and embedded in paraffin or Tissue-Tek O.C.T. (SAKURA #4583, USA). Histological analysis was performed on serial paraffin cross sections (4.5 μm) or frozen sections (6 μm).
En face oil red O staining of the aorta was used to quantify the surface area occupied by atherosclerosis. Haematoxylin and eosin (H&E) staining was used to analyze plaque area and necrotic core area. The necrotic core, quantified in three 30 μm-spaced sections per artery, was defined as a hypocellular plaque cavity devoid of collagen and containing necrotic debris and cholesterol clefts. A 3,000 μm2 threshold was implemented to avoid counting very small H&E-negative areas. Fibrous caps were defined as elastin-rich layers covered by overlying plaque. Outward remolding meant that the vessel expanded outward and the lumen area increased. Acute plaque rupture was defined as a visible defect in the cap. Elastin and collagen contents were determined by Movat-Russell staining and Masson staining, respectively.
The percentage of macrophages was determined by anti-Mac-3 immunostaining (BDPharmingen #550292, USA), the percentage of smooth muscle cells was determined by anti-α-SMA staining (Cell Signaling Technology #19245, USA), and the content of matrix metalloproteinase 2 was determined by anti-MMP-2 immunostaining (Wanleibio #WL03224, China). Secondary antibodies were species-appropriate horseradish peroxidase conjugates (Abcam, USA). 3,3′-Diaminobenzidine (KeyGEN BioTECH #KGP1045, China) was used as a chromogen. Intraplaque neovascularization and hemorrhages were examined on slides that were double stained with anti-TER119 (Biolegend #116215, America) and anti-CD31 (Abcam #ab28364, USA) antibodies.
Images were examined and quantified by Image-Pro Plus 6.0 software. All statistical analyses were performed with SPSS software (version 20, IBM Corporation, Armonk, NY, USA) or GraphPad Prism (version 5.0, GraphPad Software, Inc., La Jolla, CA, USA). The results are expressed as the mean ± SEM. Statistical comparisons were carried out using one-way analysis of variance (ANOVA) followed by the LSD test. The Kruskal–Wallis-test was used, followed by the Dunnett-T3 test if the variance was not equal. Differences were considered significant at P < 0.05 (two-sided). The numbers of mice are mentioned in the figure legends.
To determine the effect of the mutation of the fibrillin-1 gene in LDLR−/− mice, we crossbred LDLR−/− mice with Fbn1C1039G+/− mice to generate Fbn1C1039G+/−LDLR−/− mice. As shown in Figures 1A,B, the genotype was identified by polymerase chain reaction, gene sequencing analysis, and agarose gel electrophoresis. Since it is difficult for LDLR−/− mice to develop atherosclerosis by feeding on a chow diet only, we administered a HFD to the two groups of mice. After the mice were fed a HFD for 12 weeks, elastic fiber fracture was observed in Fbn1C1039G+/−LDLR−/− mice, and this phenomenon was more obvious after 24 weeks of HFD, while elastic fibers in LDLR−/− mice were unaffected. As shown in Figure 2, elastic fibers were intact and continuous in LDLR−/− mice, whereas they exhibited progressive fragmentation in Fbn1C1039G+/−LDLR−/− mice, which agrees with previous findings.
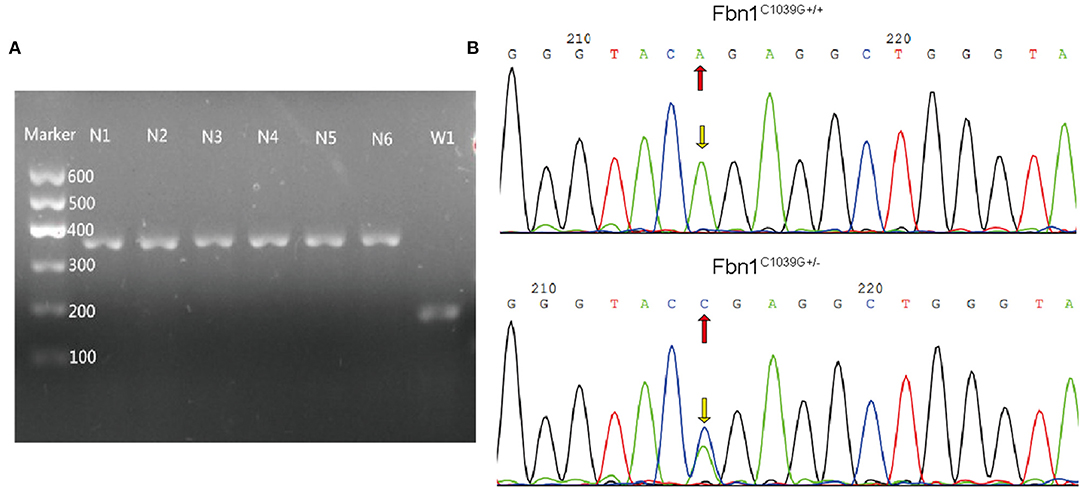
Figure 1. Genotype identification. (A) Agarose gel electrophoresis was used to identify the genotypes of LDLR−/− mice. The length of the LDLR−/− product was 350 bp, while that of the LDLR+/+ product was 167 bp. (B) Gene sequencing analysis showed that Fbn1C1039G+/− had a bimodal waveform, with the nucleotide mutated to C, while Fbn1C1039G+/+ only had a single waveform, and the nucleotide was still A.
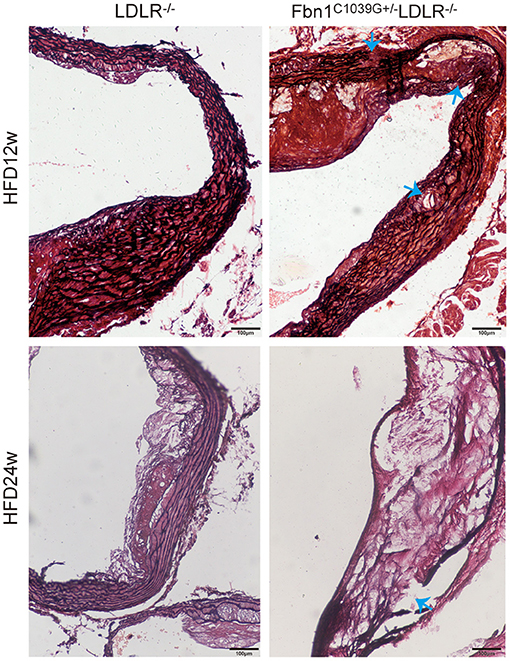
Figure 2. Elastin fragmentation in Fbn1C1039G+/−LDLR−/− mice. The arrow shows Elastin fragmentation in Fbn1C1039G+/−LDLR−/− mice after 12 and 24 w on a HFD, while elastin in LDLR−/− mice was intact. Movat staining: elastin is shown in black.
With the extension of time on the HFD, the degree of the atherosclerotic lesions in the two groups increased. Oil red O staining of the cross-sections of the aortic sinus and en face oil red O staining (the whole aorta from the aortic roots to the bifurcations of the iliac arteries) of Fbn1C1039G+/−LDLR−/− and LDLR−/− mice after 12 or 24 weeks of HFD are shown in Figures 3A,B. The results show that atherosclerotic lesions in Fbn1C1039G+/− LDLR−/− mice were significantly more severe than those in LDLR−/− mice. More specifically, the positive area of oil red O staining in the aortic sinus of Fbn1C1039G+/−LDLR−/− mice was significantly larger than that of LDLR−/− mice after 12 and 24 weeks of HFD (Figure 3A). This phenomenon was also observed in plaques in the full-length aortas of Fbn1C1039G+/−LDLR−/− mice compared with LDLR−/− mice (Figure 3B).
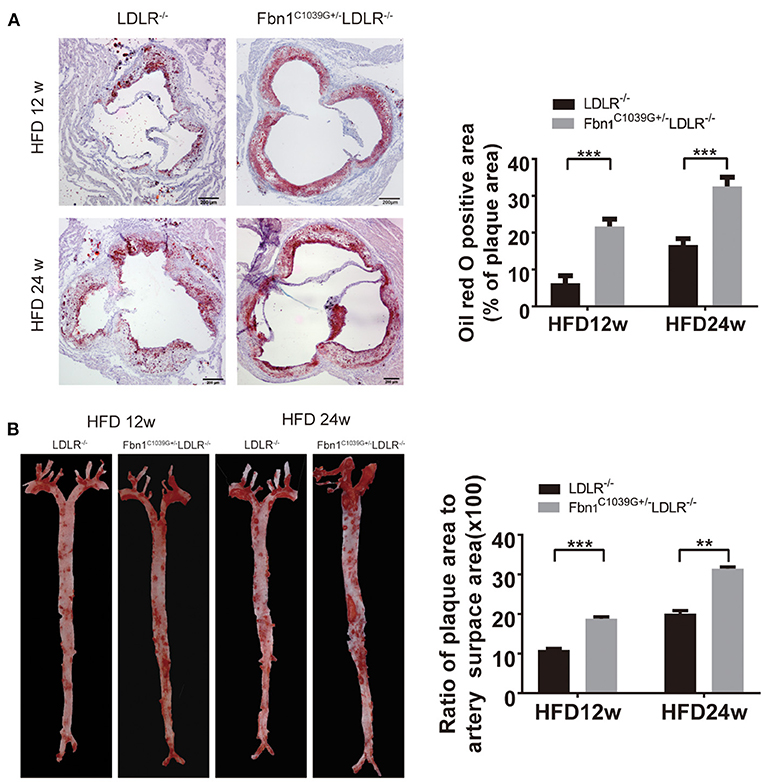
Figure 3. Pathological staining of the aorta and aortic sinus of Fbn1C1039G+/−LDLR−/− and LDLR−/− mice after 12 or 24 weeks on a HFD. (A) Oil red O staining of aortic sinus (n = 5 per group). (B) En face oil red O staining (n = 3 mice in each group). ***P < 0.001, **P < 0.01. The results showed that the Fbn1C1039G+/−LDLR−/− mice exhibited worsened atherosclerosis.
To determine the characteristics of the plaque phenotype in Fbn1C1039G+/−LDLR−/− mice, we performed a comprehensive evaluation of the atherosclerotic lesions of the mice. Cross-sectional analysis of the two groups of mice fed a HFD for 12 and 24 weeks was performed to identify the cellular composition and structural characteristics of atherosclerotic lesions. H&E staining of the aortic sinus was used to quantify atherosclerotic plaque size as well as the area of the necrotic core. The results showed that plaque size in the model mice was significantly higher than that in the control mice after 12 and 24 weeks of HFD feeding (Figure 4A). Accordingly, the area of the necrotic core was significantly larger and the fiber cap was thinner in the model mice than in the control mice. Statistical analysis showed that a necrotic core was one of the features of model mice, and the area was ~3 times that of the control mice (Figure 4B). Masson staining, which is a classic method to visualize collagen fibers, was used to compare the collagen content of atherosclerotic plaques between control and model mice. The results suggested that there were fewer collagen fibers in the model mice, and they were thin and sparsely distributed (Figure 4C). Additionally, compared to control mice, model mice fed a HFD showed abnormal vessel dilation with elastic fiber fragmentation in the arterial wall, and the blood vessel cross-sectional area was significantly larger (Table 1). Immunohistochemical staining of the aortic plaque was used to quantify macrophages, smooth muscle cells and MMP-2 content in plaques. Macrophage content was determined by calculating the percentage of the total plaque area that was positively stained for Mac3. As shown in Figure 4D, compared to that in control mice, the percentage of plaque area that was Mac3-positive was significantly increased in model mice after 12 and 24 weeks of HFD (Figure 4D). Smooth muscle cell content, expressed as the percentage of total plaque area that was positively stained for α-SMA, showed a marked decrease in model mice after both 12 and 24 weeks of HFD (Figure 4E). Immunohistochemistry also showed a significant rise in the percentage of MMP-2-positive plaque area in model mice (Figure 4F). Taken together, the results reveal that the characteristics of the plaque phenotype in Fbn1C1039G+/−LDLR−/− mice are similar to those of human “vulnerable” plaques, including significantly increased vessel area, large plaque size and necrotic core, thin fibrous cap and decreased total collagen. The characteristics are more evident with increasing HFD time.
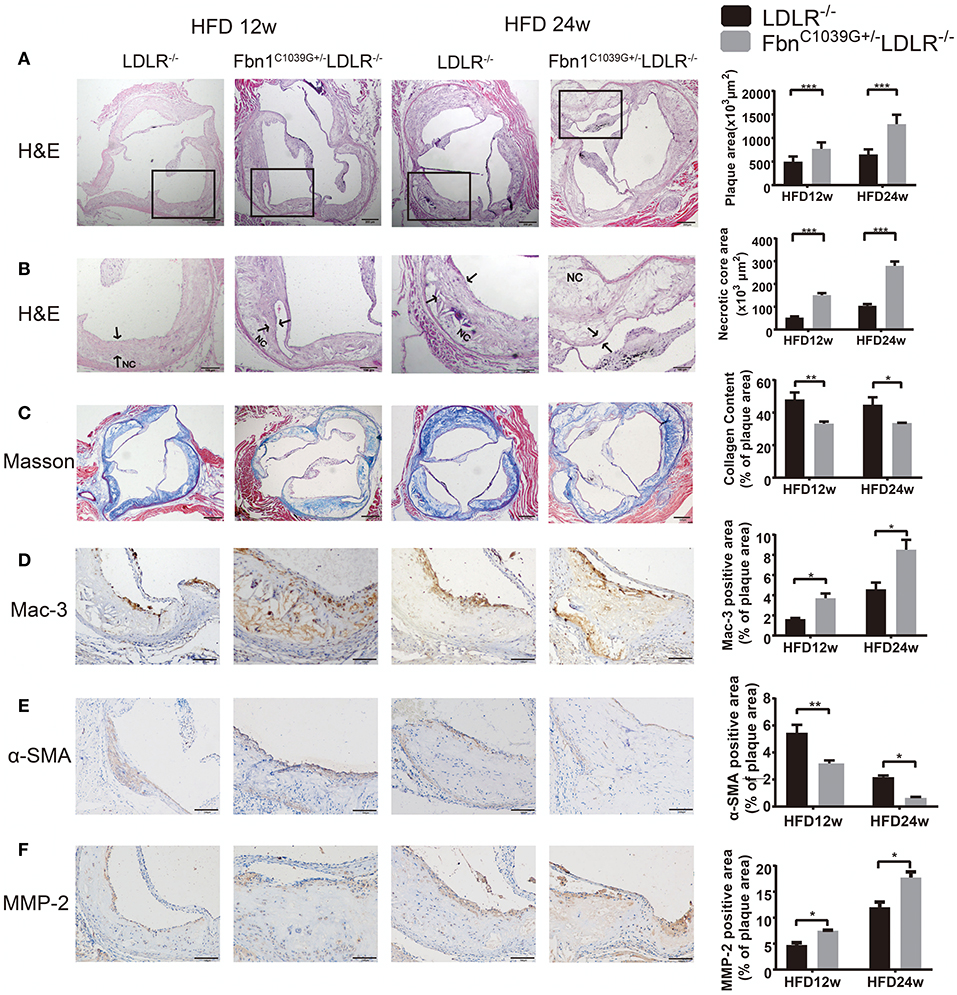
Figure 4. Plaque characteristics of Fbn1C1039G+/−LDLR−/− and LDLR−/− mice after 12 or 24 weeks on a HFD. (A) H and E staining of the aortic sinus of Fbn1C1039G+/−LDLR−/− and LDLR−/− mice after 12 and 24 weeks on a HFD (n = 9). (B) Partial enlargement of (A) showing the necrotic cores (n = 9). (C) Masson staining of the aortic sinus of the two groups of mice after 12 and 24 weeks on a HFD (n = 6). (D) Mac-3 staining of the aortic sinus of Fbn1C1039G+/−LDLR−/− and LDLR−/− mice after 12 and 24 weeks on a HFD (n = 4). (E) α-SMA staining of the aortic sinus of Fbn1C1039G+/−LDLR−/− and LDLR−/− mice after 12 and 24 weeks on a HFD (n = 4). (F) MMP-2 staining of the aortic sinus of Fbn1C1039G+/−LDLR−/− and LDLR−/− mice after 12 and 24 weeks on a HFD (n = 4). The arrows show the fibrous cap. NC, Necrotic core. ***P < 0.001, **P < 0.01, *P < 0.05.

Table 1. Characteristics of Fbn1C1039G+/− LDLR−/− and LDLR−/− mice after 12 or 24 weeks on HFD.
Previous studies have reported that an increase in arterial stiffness aggravated atherosclerotic plaque progression, and the plaques were vulnerable and prone to rupture (11, 16). In the present study, we focused on evaluating the vulnerability of atherosclerotic plaques in the new model. During HFD feeding for 12 and 24 weeks, there was no significant difference in body weight, and no neurological complications or acute cardiovascular events occurred in the two mouse models (Table 1). However, plaque rupture and thrombosis were observed in ~20% of the model mice fed a HFD (n = 10). In addition, we also found ruptured fibrous caps, elastic fiber fragments, and vasa vasorum in Fbn1C1039G+/−LDLR−/− mice, indicating that these typical plaque characteristics bear a strong similarity to those of human advanced AS (Figures 5A,B).
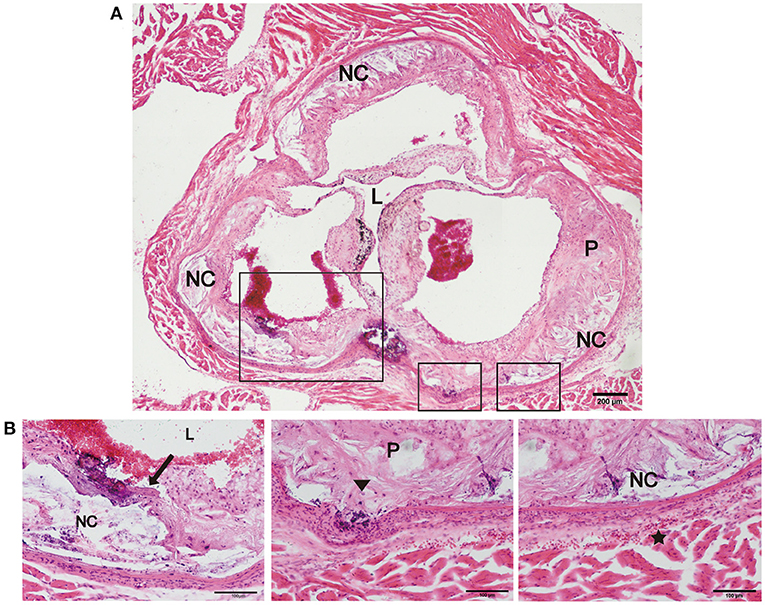
Figure 5. Plaque rupture and vasa vasorum generation in Fbn1C1039G+/−LDLR−/− mice. (A) Plaque rupture and secondary thrombosis in Fbn1C1039G+/−LDLR−/− mice after HFD feeding. (B) Partial enlargements of (A). The arrow shows the ruptured fibrous cap, the triangle shows the fragmentation of elastin, and the star shows the vasa vasorum network. P, plaque; L, lumen; NC, necrotic core.
As barrier cells, endothelial cells have an important role in the occurrence and development of atherosclerosis. Endothelial dysfunction can lead to inflammatory cell invasion and lipid deposition in the subendothelial area, which can exacerbate atherosclerosis. With the increase in plaque size and the stenosis of the arterial lumen, haemodynamic changes often occur in vessels, exacerbating endothelial injury and promoting neovascularization (17). Due to the immature neovasculature, vessel walls are particularly vulnerable to rupture and leakage, resulting in intraplaque hemorrhage and plaque rupture (18). Because another prominent characteristic of human vulnerable plaques is the presence of intraplaque neovascularization, we further observed whether this feature was also present in our new model. Using immunofluorescence staining, the internal structural features of the plaques in the two models were observed after 24 weeks of HFD feeding. Compared to control mice, Fbn1C1039G+/−LDLR−/− mice showed inner surface structural discontinuity and endothelial barrier impairment, as shown in Figures 6A,B. Furthermore, neovascularization and intraplaque hemorrhage were detected in model mice, but no signs of neovascularization or intraplaque hemorrhage were observed in control mice (Figures 6A,B). According to statistical analysis, there was no difference in the percentages of TER119- or CD31-positive plaque areas in control and model mice (Figures 6C,D). However, there was a significant difference in the percentage of TER119-positive areas in the adventitia and media between the two groups (Figure 6E).
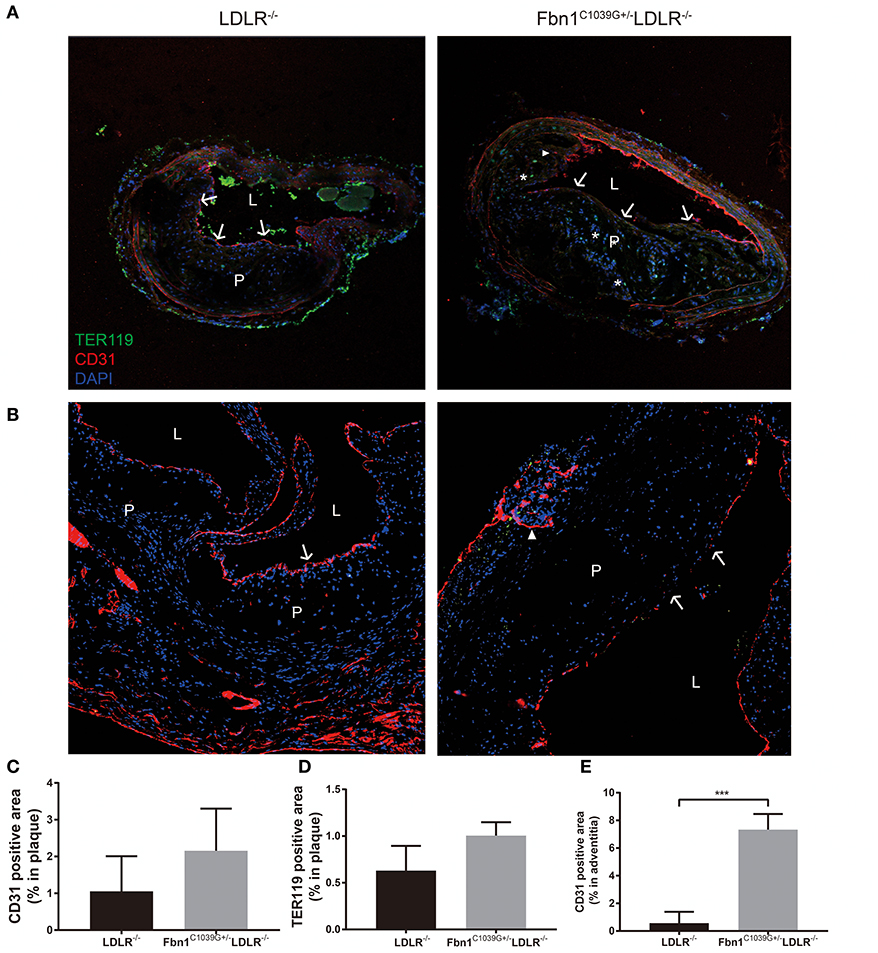
Figure 6. Damaged endothelium, intraplaque hemorrhage and neovascularization. (A) Immunofluorescence staining of the brachial artery in Fbn1C1039G+/−LDLR−/− mice and LDLR−/− mice after 24 weeks on a HFD. (B) Immunofluorescence staining of the aortic sinus in Fbn1C1039G+/−LDLR−/− mice and LDLR−/− mice after 24 weeks on a HFD. (C) Immunofluorescence staining quantification of the CD31-positive area (% of plaque) in Fbn1C1039G+/−LDLR−/− mice and LDLR−/− mice after 24 weeks on a HFD. (D) Immunofluorescence staining statistics of the TER119-positive area (% of plaque) in Fbn1C1039G+/−LDLR−/− mice and LDLR−/− mice after 24 weeks on a HFD. (E) Quantification of the CD31-positive area (% of media and adventitia) in Fbn1C1039G+/−LDLR−/− mice and LDLR−/− mice after 24 weeks on a HFD (n = 5 in Fbn1C1039G+/−LDLR−/− mice, n = 3 in LDLR−/− mice), ***p < 0.001. Magnification: ×100. TER119 shows red blood cells, CD31 shows endothelial cells, and DAPI shows nuclei. The arrow indicates the endothelial barrier, the triangle indicates neovascularization, and *indicates intraplaque hemorrhage. P, plaque; L, lumen.
ACS is known to be a major cause of death from coronary heart disease (CHD) that has a high disability rate and poor prognosis. Although it has been demonstrated that the existing prevention and treatment strategies have a certain therapeutic effect against CHD, the incidence of ACS and complications caused by VP rupture is still high and has become an urgent clinical problem to be solved. With the development of basic medicine, the pathophysiological mechanism of CHD has been extensively studied. However, due to the lack of an ideal animal model for VP, it is currently difficult to transform preliminary basic research into clinical applications. Arterial stiffness is caused by aging or risk factors and mainly manifests as the degradation of extracellular matrix (ECM), which is characterized by collagen deposition and elastic fiber fracture and is a degenerative process of elastic arteries. Research has found that in terms of pathophysiology, arteriosclerosis and AS are synergistic processes that potentiate each other to exacerbate vascular damage (19). On the one hand, in stiff arteries, diastolic blood pressure (DBP) falls sharply, leading to insufficient diastolic coronary perfusion, which may exacerbate myocardial ischaemia under AS (20). On the other hand, the development and progression of AS, especially plaques, can promote the destruction of elastic fibers and outward vascular remodeling, resulting in the enlargement of the luminal area and worsening the severity of arterial stiffness (21).
Fibrillin is the primary component of microfibrils and is encoded by the Fbn1 gene. Mutation of the Fbn1 gene impairs elastic fiber cross-linking, reduces the elasticity of vessels, and affects the structure of the aortic wall. In the cardiovascular system, rupture of elastic fibers can cause arteriosclerosis and dilation of the ascending aorta, which may complicate aortic dissection or aneurysm (11). In this study, we introduced a fibrillin-1 mutation into LDLR−/− mice via homologous recombination to construct a novel AS model. After feeding the mice a HFD for 12 or 24 weeks, the characteristics of the model were evaluated using multiple pathological experiments. The results showed that elastic fiber fracture was observed in Fbn1C1039G+/−LDLR−/− mice (Figure 2). Although Fbn1C1039G+/−LDLR−/− mice showed no significant difference in lipid level compared with LDLR−/− mice, the positive areas of oil red O staining in the aortic sinus and full-length aorta increased significantly (Figures 3A,B). The results showed that atherosclerotic lesions of Fbn1C1039G+/−LDLR−/− mice were more severe than those of LDLR−/− mice, indicating that arteriosclerosis with fiber fracture can exacerbate the development of AS. Further analysis of the plaque phenotype established that the plaques of model mice were more unstable and had characteristics similar to those of human VP. Fbn1C1039G+/−LDLR−/− mice fed a HFD were observed to have larger necrotic cores, thinner fibrous caps, more inflammatory cells, increased MMP-2, and significantly fewer collagen fibers and SMCs than control mice (Figures 4A–F). A thin fibrous cap mainly consisting of SMCs, ECM and collagen fibers is recognized as a precursor to atherosclerotic plaque rupture. In the process of AS development, with endothelial dysfunction and SMC phenotype switching, SMCs proliferate and migrate around the fibrous cap to stabilize the plaque. In turn, the SMCs in the fibrous cap migrate into the plaque core. Then, due to stimulation by inflammatory factors, many SMCs undergo apoptosis, resulting in a decrease in SMC content (Figure 4E) and thinning of fiber caps, increasing the likelihood of plaque rupture (22, 23). Therefore, in the present study, the thinning of the fiber cap in the model mice might be associated with the surge of inflammatory factors and the apoptosis of SMCs. Further experiments are still needed to explore the specific mechanism.
Moreover, compared to that of LDLR−/− mice, the percentage of Mac3-positive area was significantly increased in the plaques of Fbn1C1039G+/−LDLR−/− mice after 12 and 24 weeks of HFD (Figure 4D), and the total number of macrophages was closely related to the progression and severity of plaques. It is well-known that macrophage accumulation in vessel walls is a hallmark of the development of atherosclerosis (24, 25). Stimulated by multiple environmental factors, macrophages can switch to many kinds of functional phenotypes that play essential roles in plaque phenotype and stability (26, 27). Studies have revealed that M1 macrophages are mainly located in unstable areas of plaques and are associated with plaque progression. In contrast, M2 polarization markers are mainly expressed in the stable zones and are associated with plaque regression (26, 28). Proinflammatory M1 macrophages can secrete multiple inflammatory cytokines, such as TNF, IL-6, and IL-1β, which activate endothelial cells, promote SMC proliferation, reduce collagen synthesis and matrix degradation and finally aggravate VP progression (24, 29). Matrix metalloproteinases (MMPs), which are widely produced by monocytes/macrophages, SMCs, endothelial cells, fibroblasts, and neoplastic cells, play a crucial role in protein degradation (30). Studies have demonstrated that MMP dysregulation may be involved in vascular and cardiac remodeling and atherogenesis (31, 32). In addition, similar to M1 macrophages, MMPs are highly expressed in unstable areas of atherosclerotic plaques and may have the ability to promote the destabilization of plaque lesions (33). In this study, we detected the content of MMP-2 in plaques and observed a significant increase in model mice (Figure 4F); this may be an important factor leading to plaque instability. However, more work is needed to observe the spatial distribution of different phenotypic macrophages and the regulatory effect of MMPs on atherosclerotic lesions in model mice.
The LDL receptor is a membrane receptor that mediates the plasma level of cholesterol-rich LDL. Studies have found that LDLR deficiency significantly affects the uptake and clearance of LDL as well as other lipoproteins, resulting in abnormal plasma lipid metabolism (34, 35). Pathological studies have demonstrated that LDLR−/− mice fed with HFD have been widely used as an atherosclerotic model for basic research. Nevertheless, LDLR−/− murine models develop relatively less severe coronary atherosclerosis but require a long-term HFD, which places certain limitations on mechanistic research of VP (36). Apolipoprotein E (ApoE) is a lipoprotein that exerts antiatherogenic effects by affecting lipoprotein metabolism directly and indirectly during the development of atherosclerosis. ApoE is a high-affinity ligand of a majority of lipoprotein particles, including LDL-, LDLR- and LDLR-related proteins, and plays an important role in lipoprotein remnant clearance (37). ApoE−/− mice spontaneously develop hyperlipidaemia and extensive atherosclerosis even on a normal chow diet. However, neither ApoE−/− nor LDLR−/− mice can fully represent the characteristics of atherosclerotic plaque rupture in humans. Van der Donckt et al. found that mice with the Fbn1C1039G+/− mutation show a series of pathophysiological changes, such as elevated pulse pressure, progressive aortic dilation and decreased contractility of cardiomyocytes, resulting in arterial stiffness and cardiac dysfunction (11). ApoE−/−Fbn1C1039G+/− mice can be a perfect model of human end-stage atherosclerosis for exploring the specific mechanism of plaque destabilization and therapeutic targets for advanced atherosclerosis (38). However, sudden death and typical neurological complications can be observed in ApoE−/−Fbn1C1039G+/− mice during HFD. Moreover, there was a 50% mortality rate after 20 weeks of HFD (39). In the present study, during the whole period of high-fat diet feeding, all of the mice were in good condition, and no neurological complications or acute cardiovascular events were recorded, which is in contrast to a previous study using ApoE−/−Fbn1C1039G+/− mice (11). Asymptomatic plaque rupture of this model mouse might be related to less vascular stenosis and lesion thrombosis (40). This may also have occurred because the infarction vessel was not located in an important position; thus, no corresponding symptoms occurred. These hypotheses need to be verified by further pathological tests of the heart and brain. This characteristic enables the model mice to avoid the problem of high mortality observed in ApoE−/− Fbn1C1039G+/− mice and may reduce the interference of high mortality in subsequent studies.
Furthermore, elastic fiber fragments, plaque rupture, and thrombosis were observed in Fbn1C1039G+/−LDLR−/− mice, indicating a highly unstable plaque phenotype that bears a strong similarity to that of human advanced atherosclerosis (Figures 5A,B). In addition, compared with LDLR−/− mice, inner surface structural discontinuity and endothelial barrier impairment appeared in Fbn1C1039G+/−LDLR−/− mice. Neovascularization and intraplaque hemorrhage were observed within the plaque regions of the model mice (Figure 6). Neovascularization and intraplaque hemorrhage are prominent characteristics of human vulnerable plaques and are closely related to future ischaemic events (41). In general, the vasa vasorum is generated when the vasculature is damaged and provides oxygen and nutrients for tissue repair (42). However, the vasa vasorum, influenced by lipid deposition, endothelial injury, and a hypoxic inflammatory microenvironment, plays an intricate role in the progression of plaques (43). With progressive intimal thickening and luminal narrowing, the hypoxic area further expands, causing vasa vasorum to grow across the vessel wall to the lumen, thereby supplying oxygen and nutrients to the inner layers (44–46). Since ~80% of the new vessels are immature, vessel walls are particularly vulnerable and easily rupture and leak, leading to intraplaque hemorrhage and plaque breakage (18). Erythrocyte exudation can exacerbate the local inflammatory response, accelerating the progression of plaques (45).
Therefore, as a VP mouse model with spontaneous plaque rupture, this model is of great significance to drug development, equipment upgrading, and further exploration of the mechanism of atherosclerosis. Moreover, compared with pig or dog models, mouse models have a shorter breeding cycle, lower feeding costs and are easier to use. Compared to models built by balloon injury or artery ligation, this model better simulates the pathological physiological process of human vulnerable plaques. In addition, mouse models with the C57BL/6 background have high repeatability, which is beneficial to researchers exploring the characteristics and intervention means of vulnerable plaques.
However, there are still some limitations in our study: (1) we neglected to compare the liver conditions, such as inflammation, fibrosis or steatosis, of the two groups; (2) only a small number of mice were used for some staining experiments; and (3) additional methods, such as RT-PCR or WB, should be used to evaluate plaque vulnerability. Future study to address these problems is needed.
In conclusion, the Fbn1C1039G+/−LDLR−/− mouse is a suitable model for the study of vulnerable plaques. Compared to the LDLR−/− mice, the model mice exhibited a prominent increase in plaque destabilization. The comprehensive analysis revealed that the plaques in the model had the following characteristics of vulnerable plaques: (1) larger necrotic cores, thinner fibrous caps, and significantly fewer collagen fibers and smooth muscle cells; (2) more inflammatory cells and MMP-2 content; (3) spontaneous plaque rupture and secondary thrombosis; and (4) neovascularization and intraplaque hemorrhage.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Ethics Committee of the Second Affiliated Hospital of Harbin Medical University.
XW and YF performed the animal experiments and wrote the paper. ZX and MC raised the animals and collected the tissues. WQ and XX conducted the pathological experiments. SZ and MP designed the primers and conducted the PCR assays. XP and YJ analyzed the data. LM and JT conceived of, designed, and supervised the study. All authors read and approved the final manuscript.
This work was supported by grants from the National Natural Science Foundation of China (81971715, 91739113 to JT, 81801812 to ZX and 81827806 to Bo Yu), the National Key R&D Program of China (2016YFC1301100 to Bo Yu), Fok Ying-Tong Education Foundation for Young Teachers (171032 to JT), the Foundation of Guangxi Key Laboratory of Diabetic Systems Medicine (GKLCDSM-20200101-01 to JT), the Natural Science Foundation of Heilongjiang Province of China (H2018022 to JT), the Key Laboratory of Myocardial Ischemia, Harbin Medical University, the Ministry of Education (KF201820 to YF), and the Postgraduate Research and Practice Innovation Program of Harbin Medical University (YJSSJCX2018-64HYD to XW and YJSSJCX2019-38HYD to YF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Bo Yu and Jiangtian Tian for the financial supports.
ACS, acute coronary syndrome; VP, vulnerable plaque; HFD, high-fat diet; AS, atherosclerosis; SMC, smooth muscle cells; Fbn1, Fibrillin1; ApoE−/−, apoprotein E–deficient mice; LDLR−/−, low-density lipoprotein (LDL) receptor-deficient mice; H&E, Haematoxylin-eosin; CHD, coronary heart disease; ECM, extracellular matrix; DBP, diastolic blood pressure; MMP, matrix metalloproteinase.
1. Yokoya K, Takatsu H, Suzuki T, Hosokawa H, Ojio S, Matsubara T, et al. Process of progression of coronary artery lesions from mild or moderate stenosis to moderate or severe stenosis: a study based on four serial coronary arteriograms per year. Circulation. (1999) 100:903–9. doi: 10.1161/01.CIR.100.9.903
2. Nakagami H, Kaneda Y, Ogihara T, Morishita R. Endothelial dysfunction in hyperglycemia as a trigger of atherosclerosis. Curr Diabetes Rev. (2005) 1:59–63. doi: 10.2174/1573399052952550
3. Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. (1999) 138:S419–20. doi: 10.1016/S0002-8703(99)70266-8
4. Ahmadi A, Leipsic J, Blankstein R, Taylor C, Hecht H, Stone GW, et al. Do plaques rapidly progress prior to myocardial infarction? The interplay between plaque vulnerability and progression. Circ Res. (2015) 117:99–104. doi: 10.1161/CIRCRESAHA.117.305637
5. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. (2006) 47(Suppl. 8):C13–8. doi: 10.1016/j.jacc.2005.10.065
6. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Circulation. (2003) 108:1664–72. doi: 10.1161/01.CIR.0000087480.94275.97
7. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. (2000) 20:1262–75. doi: 10.1161/01.ATV.20.5.1262
8. Yla-Herttuala S, Bentzon JF, Daemen M, Falk E, Garcia-Garcia HM, Herrmann J, et al. Stabilisation of atherosclerotic plaques. Position paper of the European Society of Cardiology (ESC) Working Group on atherosclerosis and vascular biology. Thromb Haemost. (2011) 106:1–19. doi: 10.1160/TH10-12-0784
10. Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. (2004) 114:172–81. doi: 10.1172/JCI200420641
11. Van der Donckt C, Van Herck JL, Schrijvers DM, Vanhoutte G, Verhoye M, Blockx I, et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur Heart J. (2015) 36:1049–58. doi: 10.1093/eurheartj/ehu041
12. Getz GS, Reardon CA. Do the Apoe-/- and Ldlr-/- mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol. (2016) 36:1734–41. doi: 10.1161/ATVBAHA.116.306874
13. Ni M, Chen WQ, Zhang Y. Animal models and potential mechanisms of plaque destabilisation and disruption. Heart. (2009) 95:1393–8. doi: 10.1136/hrt.2008.143461
14. Rosenfeld ME, Carson KG, Johnson JL, Williams H, Jackson CL, Schwartz SM. Animal models of spontaneous plaque rupture: the holy grail of experimental atherosclerosis research. Curr Atheroscler Rep. (2002) 4:238–42. doi: 10.1007/s11883-002-0025-3
15. Cullen P, Baetta R, Bellosta S, Bernini F, Chinetti G, Cignarella A, et al. Rupture of the atherosclerotic plaque: does a good animal model exist? Arterioscler Thromb Vasc Biol. (2003) 23:535–42. doi: 10.1161/01.ATV.0000060200.73623.F8
16. Van Herck JL, De Meyer GR, Martinet W, Van Hove CE, Foubert K, Theunis MH, et al. Impaired fibrillin-1 function promotes features of plaque instability in apolipoprotein E-deficient mice. Circulation. (2009) 120:2478–87. doi: 10.1161/CIRCULATIONAHA.109.872663
17. Li X, Yang Q, Wang Z, Wei D. Shear stress in atherosclerotic plaque determination. DNA Cell Biol. (2014) 33:830–8. doi: 10.1089/dna.2014.2480
18. Sluimer JC, Kolodgie FD, Bijnens AP, Maxfield K, Pacheco E, Kutys B, et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol. (2009) 53:1517–27. doi: 10.1016/j.jacc.2008.12.056
19. Palombo C, Kozakova M. Arterial stiffness, atherosclerosis and cardiovascular risk: Pathophysiologic mechanisms and emerging clinical indications. Vascul Pharmacol. (2016) 77:1–7. doi: 10.1016/j.vph.2015.11.083
20. Ikonomidis I, Makavos G, Lekakis J. Arterial stiffness and coronary artery disease. Curr Opin Cardiol. (2015) 30:422–31. doi: 10.1097/HCO.0000000000000179
21. Jankowski P, Bilo G, Kawecka-Jaszcz K. The pulsatile component of blood pressure: its role in the pathogenesis of atherosclerosis. Blood Press. (2007) 16:238–45. doi: 10.1080/08037050701428166
22. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. (2014) 115:662–7. doi: 10.1161/CIRCRESAHA.115.304634
23. Hermus L, Lefrandt JD, Tio RA, Breek JC, Zeebregts CJ. Carotid plaque formation and serum biomarkers. Atherosclerosis. (2010) 213:21–9. doi: 10.1016/j.atherosclerosis.2010.05.013
24. Weissberg PL, Bennett MR. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:1928–9. doi: 10.1056/NEJM199906173402418
25. Qin F, Rong T, Ruiyan Z, Xiaoxiang Y. Research progress on the role of inflammatory response in vulnerable plaque and its mechanism. Chin J Arterioscler. (2019) 27:301–6. doi: 10.3969/j.issn.1007-3949.2019.04.006
26. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. (2015) 12:10–7. doi: 10.1038/nrcardio.2014.173
27. Shibata MA, Harada-Shiba M, Shibata E, Tosa H, Matoba Y, Hamaoka H, et al. Crude alpha-mangostin suppresses the development of atherosclerotic lesions in apoe-deficient mice by a possible M2 macrophage-mediated mechanism. Int J Mol Sci. (2019) 20:1722. doi: 10.3390/ijms20071722
28. Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque instability and rupture. Arterioscler Thromb Vasc Biol. (2007) 27:15–26. doi: 10.1161/01.ATV.0000251503.35795.4f
29. Mosser DM. The many faces of macrophage activation. J Leukoc Biol. (2003) 73:209–12. doi: 10.1189/jlb.0602325
30. Jones CB, Sane DC, Herrington DM. Matrix metalloproteinases: a review of their structure and role in acute coronary syndrome. Cardiovasc Res. (2003) 59:812–23. doi: 10.1016/S0008-6363(03)00516-9
31. Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. (2002) 90:251–62. doi: 10.1161/res.90.3.251
32. Kampoli AM, Tousoulis D, Papageorgiou N, Antoniades C, Androulakis E, Tsiamis E, et al. Matrix metalloproteinases in acute coronary syndromes: current perspectives. Curr Top Med Chem. (2012) 12:1192–205. doi: 10.2174/1568026611208011192
33. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. (1994) 94:2493–503. doi: 10.1172/JCI117619
34. Teupser D, Persky AD, Breslow JL. Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement). Arterioscler Thromb Vasc Biol. (2003) 23:1907–13. doi: 10.1161/01.ATV.0000090126.34881.B1
35. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. (1993) 92:883–93. doi: 10.1172/JCI116663
36. Ma Y, Wang W, Zhang J, Lu Y, Wu W, Yan H, et al. Hyperlipidemia and atherosclerotic lesion development in Ldlr-deficient mice on a long-term high-fat diet. PLoS ONE. (2012) 7:e35835. doi: 10.1371/journal.pone.0035835
37. Greenow K, Pearce NJ, Ramji DP. The key role of apolipoprotein E in atherosclerosis. J Mol Med (Berl). (2005) 83:329–42. doi: 10.1007/s00109-004-0631-3
38. Gargiulo S, Gramanzini M, Mancini M. Molecular imaging of vulnerable atherosclerotic plaques in animal models. Int J Mol Sci. (2016) 17:1511. doi: 10.3390/ijms17091511
39. Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, et al. Animal models of atherosclerosis. Eur J Pharmacol. (2017) 816:3–13. doi: 10.1016/j.ejphar.2017.05.010
40. Lee KY, Chang K. Understanding vulnerable plaques: current status and future directions. Korean Circ J. (2019) 49:1115–22. doi: 10.4070/kcj.2019.0211
41. Turc G, Oppenheim C, Naggara O, Eker OF, Calvet D, Lacour J-C. Relationships between recent intraplaque hemorrhage and stroke risk factors in patients with carotid stenosis: the HIRISC study. Arterioscler Thromb Vasc Biol. (2012) 32:492–9. doi: 10.1161/ATVBAHA.111.239335
42. Xu J, Lu X, Shi GP. Vasa vasorum in atherosclerosis and clinical significance. Int J Mol Sci. (2015) 16:11574–608. doi: 10.3390/ijms160511574
43. Qi ZW, Li M, Zhu K, Zhang JP. Research advance of outer membrane vasa vasorum in atherosclerotic vulnerable plaque. Chin J Arterioscler. (2019) 2019:542–6.
44. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Role of lipids and intraplaque hypoxia in the formation of neovascularization in atherosclerosis. Ann Med. (2017) 49:661–77. doi: 10.1080/07853890.2017.1366041
45. Parma L, Peters HAB, Baganha F, Sluimer JC, de Vries MR, Quax PHA. Prolonged hyperoxygenation treatment improves vein graft patency and decreases macrophage content in atherosclerotic lesions in ApoE3*Leiden mice. Cells. (2020) 9:336. doi: 10.3390/cells9020336
Keywords: atherosclerosis, vulnerable plaque, animal model, plaque rupture, Fbn1C1039G +/–LDLR–/– mice
Citation: Wang X, Fu Y, Xie Z, Cao M, Qu W, Xi X, Zhong S, Piao M, Peng X, Jia Y, Meng L and Tian J (2021) Establishment of a Novel Mouse Model for Atherosclerotic Vulnerable Plaque. Front. Cardiovasc. Med. 8:642751. doi: 10.3389/fcvm.2021.642751
Received: 16 December 2020; Accepted: 23 February 2021;
Published: 16 March 2021.
Edited by:
Tetsuro Miyazaki, Juntendo University Urayasu Hospital, JapanReviewed by:
Marcello Mancini, National Research Council (CNR), ItalyCopyright © 2021 Wang, Fu, Xie, Cao, Qu, Xi, Zhong, Piao, Peng, Jia, Meng and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingbo Meng, bWVuZ2xpbmdibzUwQDE2My5jb20=; Jinwei Tian, dGlhbmppbndlaWRyMjAwOUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.