 Marie Louise Ndzie Noah†
Marie Louise Ndzie Noah† Gabriel Komla Adzika†
Gabriel Komla Adzika† Richard Mprah
Richard Mprah Adebayo Oluwafemi Adekunle
Adebayo Oluwafemi Adekunle Joseph Adu-Amankwaah
Joseph Adu-Amankwaah Hong Sun*
Hong Sun*- Department of Physiology, Xuzhou Medical University, Xuzhou, China
Cardiovascular diseases (CVDs) characterized by sex–gender differences remain a leading cause of death globally. Hence, it is imperative to understand the underlying mechanisms of CVDs pathogenesis and the possible factors influencing the sex–gender disparities in clinical demographics. Attempts to elucidate the underlying mechanisms over the recent decades have suggested the mechanistic roles of estrogen in modulating cardioprotective and immunoregulatory effect as a factor for the observed differences in the incidence of CVDs among premenopausal and post-menopausal women and men. This review from a pathomechanical perspective aims at illustrating the roles of estrogen (E2) in the modulation of stimuli signaling in the heart during chronic catecholamine stress (CCS). The probable mechanism employed by E2 to decrease the incidence of hypertension, coronary heart disease, and pathological cardiac hypertrophy in premenopausal women are discussed. Initially, signaling via estrogen receptors and β-adrenergic receptors (βARs) during physiological state and CCS were summarized. By reconciling the impact of estrogen deficiency and hyperstimulation of βARs, the discussions were centered on their implications in disruption of nitric oxide synthesis, dysregulation of lipid profiles, and upregulation of nuclear factor of activated T cells, which induces the aforementioned CVDs, respectively. Finally, updates on E2 therapies for maintaining cardiac health during menopause and suggestions for the advancement treatments were highlighted.
Introduction
Cardiovascular diseases (CVDs) remain a leading cause of death globally; hence, it is imperative to understand the underlying mechanisms of its pathogenesis. Studies over the decades and clinical demographics have demonstrated the existence of sex–gender disparity in patients developing CVDs. The cardioprotective role of estrogen has been implicated as a factor causing the sex–gender differences in the occurrence of CVDs. This is owing to the fact that the incidence of hypertension, coronary heart disease (CHD), and pathological cardiac hypertrophy are noticeably less in premenopausal women compared with their age cohort men, until the onset of menopause (1).
Besides catecholamine stress, heredity, ethnic background, aging, and lifestyles (smoking, physical inactivity, and obesity) are the traditional risk factors that broadly influences the pathogenesis of CVDs (2).
Under physiological state, circulating catecholamines stimulate the β-adrenergic receptors (βARs) expressed in the heart to regulate inotropic and chronotropic functions of the heart. However, overstimulation of the βARs during chronic catecholamine stress (CCS) dysregulates the receptors (3, 4). Without any timely treatment intervention to halt the excessive firing of the sympathetic–adrenal medullary system or adaptively modulate effectors post-βARs, the receptor dysregulation cascade initiates the pathogenesis and progression of the aforementioned CVDs. Among the subtypes of βARs, the β2ARs are mostly found mediating catecholamine stimuli during CVDs (4). The loss of estrogen and its residual cardioprotective effect on preventing the dysregulation of the βARs during CCS predisposes post-menopausal women to developing these CVDs just as men of all age cohorts.
This review from a pathomechanical perspective aims at illustrating the roles of E2 in the modulation of stimuli signaling in the heart during CCS and also highlights its implication in causing the obvious sex–gender differences in CVD patients. We initially introduced the subtypes, localizations, and functions of ERs and βARs in the heart briefly. The signaling cascades via ERs and βARs under physiological state and CCS state were summarized. In addition, an overview of how E2 deficiency permits the signaling cascade that dysregulates βARs to initiate and facilitate the progression of CVDs during CCS was discussed. The prior discussions were then reconciled to explain the following: first, how E2 deficiency contributes to the disruption of nitric oxide (NO) synthesis, thereby dysregulating vasoconstriction and vasodilation, which results into hypertension and arrhythmias during CCS; second, dysregulation of lipid profiles due to E2 deficiency, which causes atherosclerosis and progresses into CHD; and lastly, how E2 deficiency permits the upregulation of nuclear factor of activated T cells (NFATs), thereby predisposing post-menopausal women to the occurrence of PCH during CCS. Furthermore, updates on E2 therapies for maintaining cardiac health during menopause and suggestions for the advancement treatments were highlighted.
ERs and βARs Interplay in the Cardiovascular System
ERs could be broadly categorized as non-genomic and genomic, based on both their localization and functions. ERα and ERβ are the classical genomic ERs (Table 1). In the myocardium, they mediate the non-rapid estrogenic activation of transcriptional factors (TFs), such as the signal transducer and activator of transcription (STAT) family, CCAAT-enhancer-binding protein beta (C/EBPβ), CREB, Elk-1, and the nuclear factor kappa B (NF-κB) complex (5, 29–31), while G-protein-coupled estrogen receptor (GPR30) is predominantly cytomembrane bound but also expressed on the endoplasmic reticulum in cardiomyocytes. GPR30 mediates rapidly the non-genomic effects of E2, such as influencing ion channel activities. Besides mediating non-genomic estrogenic effects, GPR30 can indirectly activate TFs via interactions with ERα and ERβ, as reviewed extensively here (5). Intriguingly, inhibition of the non-genomic signaling impeded transcriptional response of genes involved in the cardiovascular function (9). Under physiological state, GPR30 signals to enhanced cardiomyocyte inotropy (rapid response) via Gαs/cAMP/PKA/EPAC pathway, but it switches the signaling via Gαi/PI3K/Akt pathway to exert anti-inotropy and antiapoptosis during CCS (10). By the latter, estrogen confers cardioprotection through GPR30 during CCS. In addition, stimulating protein-1 (Sp-1), found downstream the estrogenic pathway, is a crucial mediator of the indirect genomic signaling that regulates the expression of endothelial nitric oxide synthase (eNOS) and low-density lipoprotein (LDL) receptors (LDLRs) (5). LDLR regulates circulation of cholesterol levels, while NOS facilitates vasodilation, vasoconstriction, and anti-inflammatory responses under normal physiological conditions (32, 33). These are crucial homeostatic roles played by E2 and ERs in ensuring proper cardiovascular function under normal physiological states.
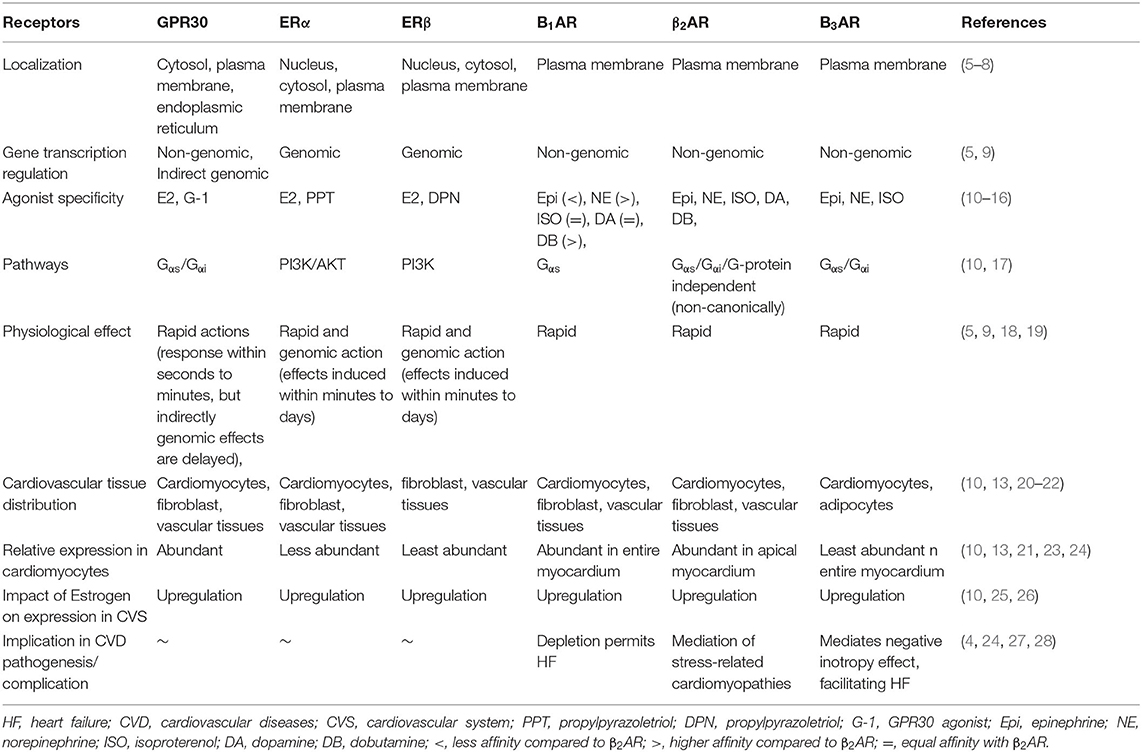
Table 1. Characteristics and activities of estrogen receptors (ERs) and β-adrenergic receptors (βARs).
Just like ERs, βARs are seven transmembrane-spanning receptors; even so, they respond to different agonists and are also well-expressed in the heart (10). βAR subunits β1AR and β2AR are distinctively expressed in the heart. β1AR is widely expressed in all parts of the heart, while β2AR is sparingly expressed in the apical myocardium (Table 1). However, β3AR is mostly expressed in adipocytes and mediates energy metabolism signals (20). In a normal physiological state, stimulation of β1AR and β2AR by circulating catecholamines permits signaling via the classical Gαs/cAMP/PKA, just as E2 does on binding to GPR30 (10). This signaling cascade phosphorylates the L-type Ca2+ channel to ensure the rhythmic contraction of the myocardium. Under stress condition, β2AR, due to its pleiotropic nature, can traffic signaling via Gαi/PI3K/Akt to prevent any cardiac insults. However, during CCS, β2AR functions are dysregulated and are also found to be relatively highly expressed in the myocardium than β1AR (4, 10). This implicates β2AR in the mediation of signaling cascades that initiates the pathogenesis of CVDs and results in heart failures (HFs) (4).
The sharing of similar pathways, Gαs/cAMP/PKA and Gαi/PI3K/Akt by βARs and GPR30 in the cardiovascular system, suggests an interplay between these receptors. A mounting of evidence of the last decades also to prove this (10, 34–36). Besides, even though βARs are not receptors of E2, findings from our recent studies along with others have shown that E2 is able to upregulate the expressions of βARs in cardiomyocytes under stress state and confers cardioprotection by doing so (34, 36). While the underlying mechanisms are still being elucidated, we speculate that E2 via its non-genomic signaling may be modulating activities of G-protein-coupled receptor kinase (GRK) 2 to minimize the internalization of β2ARs during stress.
Estrogen Deficiency and βAR Overstimulation During CCS
E2, being the primary female sex hormones, exerts and modulates several adaptive responses, such as cardioprotection and immunoregulation via its non-genomic and genomic signaling (37). E2 does all these besides its reproductive functions. By the adaptive effects of E2, women have a lower risk of developing CVDs and its associated complications in their reproductive age. The lower risk of CVDs in premenopausal women could be attributed to E2 maintaining the blood pressure (BP) through indirect regulation of lipid profile via LDLR and enhancing NOS activities, preventing cardiac insult during stress via both direct and indirect signaling through Gαi/PI3K/Akt, and conferring vasoprotection and timely regulation of pro- and anti-inflammatory response (37, 38). Unfortunately, male individuals of all age cohorts do not have these estrogenic advantages, as they are predisposed to the occurrence of CVDs. Nonetheless, the deficiency of E2 and the loss of its residual protective effects during menopausal increases the incidence of CVDs just as in men.
At this point, overstimulation of βARs by circulating catecholamines can lead to devastating consequences on the cardiovascular system (CVS). Overstimulation of βARs dysregulates the receptor, usually resulting in the desensitization, internalization, and downregulation of the receptor. This cascade is initiated by PKA negative feedback (heterologous desensitization) or GRK2 phosphorylation—β-arrestins-1 binding at serine/threonine of the receptor (homologous desensitization). These ultimately affects cardiac function and initiates CVD pathogenesis when it prolongs. However, the presence of E2 is able the mitigate the dysregulation of βARs likely via the modulation of GRK2 activities as speculated (39). In addition, E2 signaling via GPR30/Gαi/PI3K/Akt during stress might compliment the β2AR/Gαi/PI3K/Akt signaling to enforce the prevention of any cardiac insult (Figure 1). All these adaptive effects of E2 are lost during its deficiency. As such, the adverse effects of βAR hypersensitization during CCS are aggravated in the absence of E2. This is a probable explanation to why women are resilient to the adverse effects of stress on the CVS until the onset of menopause.
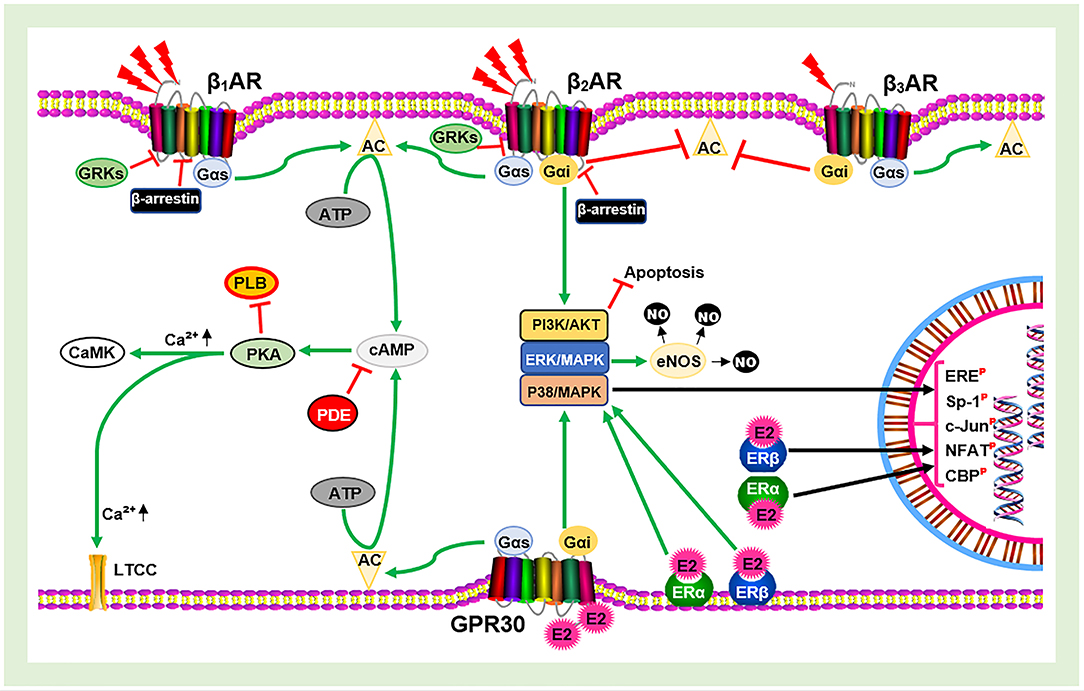
Figure 1. Schematic illustration of estrogen receptors (ERs) and β-adrenergic receptors (βARs) signaling. AC, Adenylyl cyclase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; GRKs, G-protein-coupled receptor kinases; E2, estrogen; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; PDE, phosphodiesterase; PLB, phospholamban; LTCC, L-type calcium channel; CaMK, calmodulin-dependent kinase; ERE, estrogen receptor element; Sp-1, stimulating protein-1; NFAT, nuclear factor of activated T cells;  , Phosphorylated;
, Phosphorylated;  , Inhibit;
, Inhibit;  , Upregulate;
, Upregulate;  , Catecholamine.
, Catecholamine.
Mechanistic Roles of Estrogen in Catecholamine Stress-Related Sex-Gender Disparities in CVDs
Herein, we discuss the probable signaling cascades facilitated by E2 to prevent the occurrence of hypertension, coronary heart disease, and pathological cardiac hypertrophy. Although there are many other CVDs occurring due to CCS and have sex–gender disparities, the focus of this review will be centered on these CVDs.
Hypertension
Hypertension is a constant increase in BP that causes damage to several organs. The pathophysiology of hypertension comprises arrhythmias, elevated systemic vascular resistance, stiffness, and receptivity to stimuli, which can result in other CVS-related pathological conditions (40). Vasodilation and vasoconstriction are the opposing physiological functions that regulate the tension of blood in circulation. They are essential for maintaining homeostatic function as they retain body heat, minimize blood loss during injuries, and regulate of arterial pressure. Vasodilation is facilitated by the activities of nitric oxide (NO); as such, the bioavailability of NO is crucial for the regulation of the tension/pressure of blood in circulation. The eNOS produces the NO explicitly in the vascular endothelium to induce the vasorelaxation (41). The impairment of eNOS and deficiency of NO leads to endothelial dysfunction, prolonged vasoconstriction, and an increase in BP (41). Prolongation of these vascular dysfunctions ultimately results in hypertension.
In a Ca2+-independent manner, E2 and shear stress (circulating catecholamine) are the known agonists that activate eNOS. By signaling via GPR30/Gαi, E2 induces Akt, ERK1/2, or CaMK-II to phosphorylate eNOS at Ser1177, thereby activating eNOS and enhancing NO production for normal vascular function (42, 43). In addition, stimulation of β2AR by catecholamines induces NO production via Gαi/PI3K/Akt phosphorylation of eNOS (44). However, during CCS, β2AR dysregulation and dysfunction may impede its mediation of the activation of eNOS, as such affects the NO bioavailability and causes endothelial dysfunction. In premenopausal women, the inability of β2AR to facilitate NO production might be masked by E2-induced activation of eNOS–NO production. Therefore, the occurrence of catecholamine-stress-induced endothelial dysfunction, which progresses in hypertension, may be averted by E2 in such age cohort of female individuals. Besides, E2 also prevents the dysregulation of β2AR; hence, the adverse effect of CCS on NO production may be mitigated by it. Nonetheless, the deficiency E2 and the loss of its residual protective effects (typically during menopause) permit endothelial dysfunction, and its follow-up cascades that result in hypertension and HF. Unfortunately, male individuals of all age cohort are pre-disposed to developing HF resulting from hypertension during CCS (45).
Furthermore, it has been suggested that E2 exerts the following adaptive effects on the renin–angiotensin system (RAS) pathway to prevent hypertension: (1) E2 enhances signaling of angiotensin II (Ang II) via angiotensin II receptor type 2 (AT2R) to facilitate vasodilation (2, 46) E2 upregulates angiotensin-converting enzymes (ACE) 2 expression to enhance the conversion of Ang II to Ang-(1–4, 29–31) for signaling via Mas receptor (MasR) to facilitate vasodilation (47). Detailed mechanistic role of E2 in preventing hypertension via the RAS pathway has been demonstrated elsewhere (48, 49).
Coronary Heart Disease
CHD is the ultimate result of coronary artery disease (CAD), although these CVD terms are used interchangeably. CAD is characterized by a reduction in blood circulation to the myocardium caused by obstructive atherosclerotic (fibro-fatty plaque buildups) vasoconstriction in the epicardial coronary arteries (50, 51). The prolonged reduction in blood flow to the myocardium (ischemia) causes heart attacks or induce cardiomyocytes death and myocardial infarction, which elicits inflammatory responses and consequently marked fibrosis (51). The fundamental pathomechanism of CHD initiation and progression has to do with lipid accumulation in the CVS. Moreover, unhealthy lifestyles, such as smoking and obesity [high low-density lipoprotein (LDL) cholesterol, low high-density lipoprotein (HDL) cholesterol], aging, hypertension, family history of premature atherosclerosis, hyperlipidemia, diabetes, chronic stress, and menopause are its known risk factors (45). Among these traditional risk factors, chronic stress and E2-deficiency-related factor will be the focus of this study.
NO made available due to shear stress signaling via β2AR contributes to the retardation of atherosclerosis, besides regulating blood pressure. However, CCS is implicated in the pathogenesis of CHD because it dysregulates β2AR (44) and induces dysfunction of eNOS, thereby expediting the occurrence of atherosclerosis. In addition, compared to premenopausal women, post-menopausal women are at an increased risk of developing CAD/CHD due to loss of the pre-discussed vasodilation and vasoprotective roles E2 (42, 43).
Nonetheless, since CHD pathogenesis is involved in lipid accumulation in the intimal layer of arteries, this suggests that besides vasoregulation, E2 and ERs may be using multiple mechanisms to regulate lipid profile levels in circulation and impeding LDL depositions to minimize the occurrence of CHD premenopausal women. Efforts to elucidate the mechanisms employed by E2 and ERs to result in the sex–gender differences in the development of CHD have suggested the following.
First, physiological levels E2 signaling via ERα upregulates the expression of LDLR and downregulates scavenger receptor class B type 1 (SR-B1), the HDL receptor (52). By this, E2 enhances rapidly the uptake of LDL (bad cholesterol) in circulation while keeping circulating levels of HDL (good cholesterol) high, thus keeping the lipid profile in a healthy balance. Unlike HDL, which gets to the liver and helps with the removal of other forms of cholesterol from the bloodstream, LDL only gets deposited in arterial walls (atherosclerosis); thus, they are given the names good and bad cholesterol, respectively. In short, these studies have demonstrated that E2 via ERα upregulated LDLR expressions to decrease bloodstream levels as well as the deposition of LDL to prevents CAD/CHD (52, 53).
In addition, Fu et al. recently demonstrated that by non-genomic signaling via GPR30-phospholipase C-γ (PLCγ), E2 preserves the expression of LDLR (54). Proprotein convertase subtilisin/kexin type 9 (PCSK9), a negative modulator of lipid metabolism, has been shown to mediate the degradation of LDLR. PCSK9 does this by binding to the LDLR extracellular domain and then facilitating LDLR endocytosis and degradation by lysosomes (55). By degrading LDLR, PCSK9 elevates circulating LDL levels and increases the risk of CAD/CHD. However, E2 via GPR30 was shown to inhibit the PCSK9-mediated cascade that degrades LDLR, thereby preserving LDLR expressions and keeping bloodstream LDL levels and the risk of CAD/CHD low (54).
Furthermore, it has been suggested that besides downregulating SR-B1 to keep bloodstream HDL levels high via ERα signaling, E2 also inhibits LDL transcytosis via GPR30 and SR-B1 (56). Transcytosis is an integral vesicular that helps transport macromolecules (e.g., LDL) across the interior of a cell by endocytosis of the molecule at the apical endothelial surface and exocytosis at the basal membrane. Intriguingly, SR-B1 was identified as one of the receptors that can transport LDL through endothelial cells of the human coronary artery (57). However, the finding of Ghaffari et al. demonstrated that instead of ERα, GPR30 was specifically found mediating the downregulation of SR-B1 and the inhibition of LDL transcytosis to lower the rate of atherosclerosis (56) and ultimately ischemia and CAD/CHD.
Lastly, based on the well-known adaptive immunoregulatory roles of E2 and its receptors (38), it can be speculated that in cases where there are sudden ruptures of atherosclerotic plaque, E2 signaling via ERs may lessen the platelet aggregation and thrombosis while modulating timely pro- and anti-inflammatory responses to mitigate the possible fatal outcomes. Conversely, androgen has been demonstrated to facilitate maladaptive and immunosuppressive responses (58–63). This is evident, as data over the decades have suggested that more men and post-menopausal women die from atherosclerosis/CAD/CHD than premenopausal women (37, 38). In summary, all these findings have shown that deficiency of E2 and dysregulation of ERs (both genomic and non-genomic) distort healthy balance in lipid profiles and permits LDL depositions into the intimal layer of the coronary arteries, which progresses into CHD.
Pathological Cardiac Hypertrophy
The chronic upregulation of levels of circulating catecholamines overstimulates β2AR, inducing a non-canonical signaling that is facilitated by GRK5 phosphorylation—β-arrestins-1 binding at serine/threonine of the receptor (17, 64). In addition, because GRK5 has a nuclear localization sequence (NLS) identical to homeobox-containing transcription factors, it can translocate to the nuclei where it activates the TFs: myocyte enhancer factor 2 (MEF2), GATA4, Csx/Nkx2–5, and NF-kB (17, 65). An irreversible pathological cardiac hypertrophied (PCH) heart is the result of these cascades if there are no timely intervention to subdue the adverse remodeling. PCH patients suffer heart malfunction resulting from markedly thickened ventricular walls and a decrease in the left ventricular chamber. As such, it becomes impossible for the heart to replenish sufficient blood rapidly for the next ejection (66).
In the recent decade, considerable efforts have been devoted to elucidating the pathomechanism underlying the occurrence of PCH during CCS. Besides the maladaptive roles played by GRK5, the nuclear factor of activated T cells, the cytoplasm (NFATc) is at the center of the signaling cascades maladaptively remodeling the heart. Our earlier studies suggested that PCH maybe the result of synergies of maladaptive signal cascades occurring in cardiomyocytes and immune cells; intriguingly, NFATs play essential roles in these cells (4). Of the four intracellular Ca2+-dependent isoforms of NFAT (NFATc1–NFATc4) reviewed elsewhere (67), NFATc3 has been shown to be the isoform implicated in facilitating the exacerbation of PCH (17). Under physiological state, NFATs are cytosolic due to being heavily phosphorylated. However, activation of intercellular calcium regulating receptors (e.g., ERs) upregulated Ca2+-calmodulin-activated calcineurin, which dephosphorylates the NFATs, enabling them to translocate in the nuclei and regulate gene transcription adaptively (68). Hullmann et al. demonstrated that during chronic stress (pressure overload), GRK5 activated NFATc3 in a kinase-independent manner, which exacerbated PCH (17).
As well-demonstrated, NFATc3 plays an essential maladaptive role in the adverse remodeling of the heart; nonetheless, E2 and ERs have been shown to modulate their activation and signaling activities adaptively to exert antihypertrophic effects. First, E2 has been demonstrated to suppress nuclear localization of GRK5 (69), thereby impeding its activation of NFATc3. In addition, the overexpression of ERα and ERβ were found to have repressed the transcriptional activities of NFATc3, while the contrast was observed when endogenous ERα and ERβ were knocked down. Furthermore, an agonist of ERα (delphinidin) was found to inhibit NFAT activation and also inhibited histone deacetylase (HDAC), which also has the ability to participate in inducing PCH (70, 71). This suggests that even though E2 increases intracellular Ca2+ concentrations to modulate NFATc3 activities via non-genomic signaling (GPR30) adaptively, it might also have the ability to repress or normalize intracellular Ca2+ concentration via genomic signaling (ERα and ERβ) to inhibit NFATc3 activities and attenuate the occurrence of PCH during CCS. Finally, maladaptive inflammatory responses are also implicated in the pathogenesis and exacerbation of PCH (4); however, the adaptive immunoregulatory roles of E2 might mitigate the extent of adverse cardiac remodeling, while androgen does the contrary. As such, the deficiency of E2 and/or depletion in ER expression predisposes an individual to the occurrence of PCH during CCS.
Altogether, these might also explain why premenopausal women have a low tendency of developing PCH, while male individuals of all age cohorts and menopausal women are likely to have their hearts pathological remodeled when having CCS.
Conclusions and Perspectives on E2 Replacement Therapy
Sex–gender disparities in the clinical demographics of hypertension, CAD/CHD, and PCH have been demonstrated to be likely due to the diverse protective mechanistic roles of E2 and ERs in modulating signaling cascades even during CCS, to circumvent adverse outcomes in premenopausal women (Figure 2). In contrast, male individuals of all age cohorts are disadvantaged due to lack of physiological levels of E2. The incidence rates of these CVDs are higher in male individuals, and their lack of estrogen expedites the progression into heart failure and death.
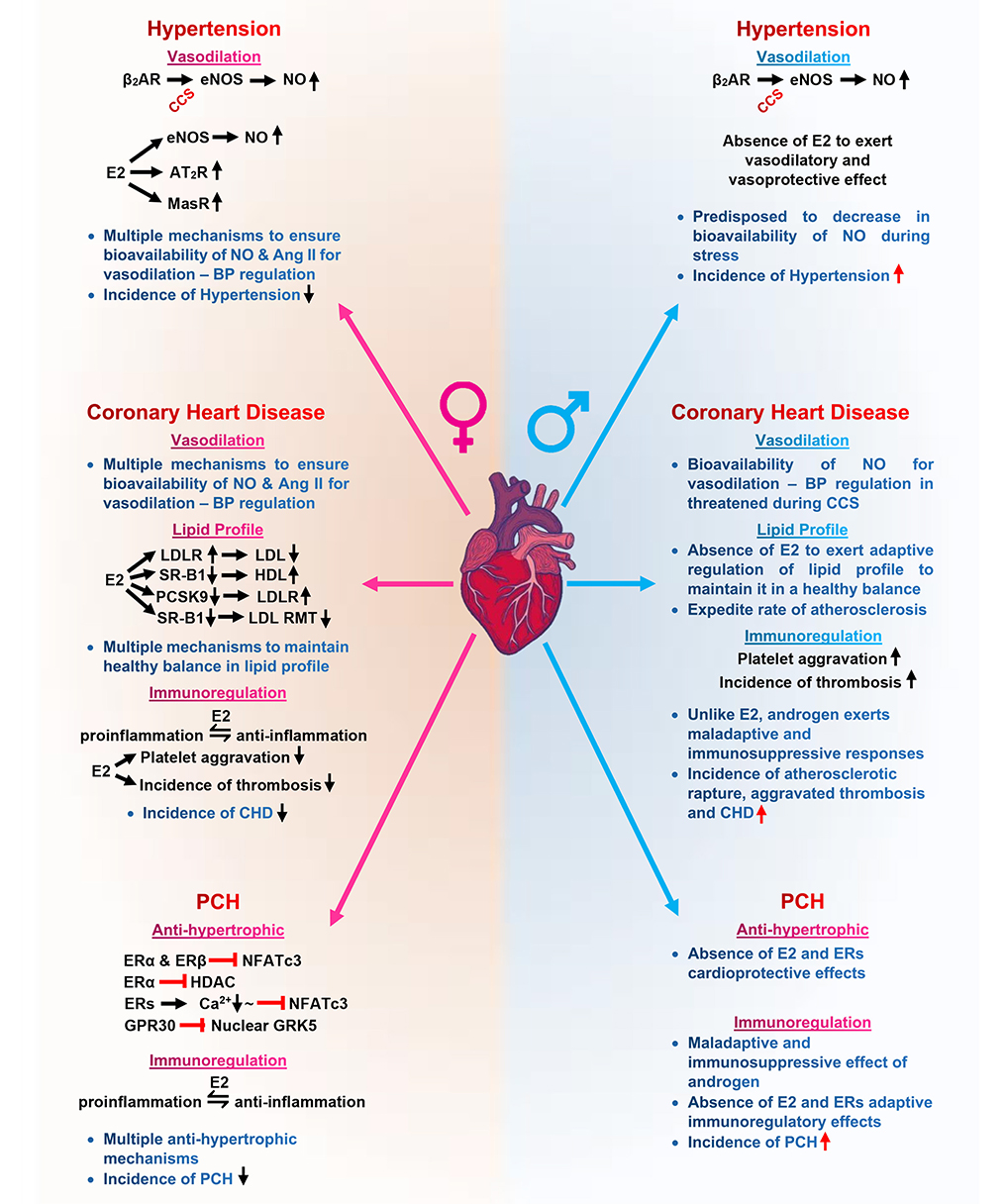
Figure 2. Graphical abstract of sex–gender disparities in cardiovascular diseases mediated by the mechanistic role of estrogen on eNOS, lipid profile, and NFATs. eNOS, endothelial nitric oxide synthase; NO, nitric oxide; Ang II, angiotensin II; AT2R, angiotensin II receptor type 2; MasR, Mas receptor; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; RMT, receptor-mediated transcytosis; SR-B1, scavenger receptor class B type 1; PCSK9, proprotein convertase subtilisin/kexin type 9; GRK5, G protein-coupled receptor kinases 5; HDAC, histone deacetylase;  , signaling;
, signaling;  , upregulate;
, upregulate;  , downregulate;
, downregulate;  , inhibit;
, inhibit;  , normalize.
, normalize.
E2 replacement therapy (E2RT) is used to control menopausal symptoms and minimize the incidence of cardiovascular disease in post-menopausal women (53, 72, 73). However, the usage of E2RT decreased significantly, following reports from the randomized controlled trial by the Women's Health Initiative (WHI), which concluded that an increased risk of breast cancer was associated with using menopausal hormone therapy (MHT) (74–79). Attempts to explain the finding of WHI revealed that the type and regimen of MHT used during the randomized controlled trial might have influenced the increased risk of breast cancer. In summary, the combination of conjugated equine estrogen plus medroxyprogesterone acetate has been advised against (80–82). In addition, it has been suggested that the MHT should be initiated early within the critical window (5–6 years from menopausal) to circumvent the reported side effects of MHT (83, 84). The recent findings of Shao et al. and Hodia et al. have provided evidence to support the need to initiate MHT within 5–6 years after menopause (83, 85). It was demonstrated that individuals who started oral E2RT within 6 years after menopause had less progression of atherosclerosis than those who started 10 years or more after menopause (83).
Furthermore, it has also been demonstrated that by targeting GPR30, post-menopausal coronary atherosclerosis and its accompanying maladaptive inflammatory responses were attenuated without any side effect. It was explained that the agonist (G-1) via GPR30 did not have any uterotrophic effects; hence, no malignancies and hyperplasia were observed (86). Based on this evidence, it is proposed that by initiating E2RT within 5–6 years after menopausal or by exclusively targeting GPR30, the incidence of hypertension, CHD, and PCH may be minimized in post-menopausal women with fewer side effects.
Author Contributions
The review idea was conceived by MN. GA, MN, and RM drafted and wrote the manuscript. With the supervision of HS, GA, MN, RM, AA, and JA-A revised and proofread the manuscript. All authors accepted the final version of the manuscript.
Funding
This study was supported by National Natural Science Foundation of China (Nos. 81461138036 and 81370329), Post-graduate Research & Practice Innovation Program of Jiangsu Province, China (KYCX17-1712), and Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Boese AC, Kim SC, Yin KJ, Lee JP, Hamblin MH. Sex differences in vascular physiology and pathophysiology: estrogen and androgen signaling in health and disease. Am J Physiol Heart Circ Physiol. (2017) 313:H524–45. doi: 10.1152/ajpheart.00217.2016
2. Flora GD, Nayak MK. A brief review of cardiovascular diseases, associated risk factors and current treatment regimes. Curr Pharm Des. (2019) 25:4063–84. doi: 10.2174/1381612825666190925163827
3. Huang CK, Lee SO, Chang E, Pang H, Chang C. Androgen receptor (AR) in cardiovascular diseases. J Endocrinol. (2016) 229:R1–16. doi: 10.1530/joe-15-0518
4. Adzika GK, Machuki JO, Shang W, Hou H, Ma T, Wu L, et al. Pathological cardiac hypertrophy: the synergy of adenylyl cyclases inhibition in cardiac and immune cells during chronic catecholamine stress. J Mol Med (Berl). (2019) 97:897–907. doi: 10.1007/s00109-019-01790-0
5. Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
6. Reuter H. Localization of beta adrenergic receptors, and effects of noradrenaline and cyclic nucleotides on action potentials, ionic currents and tension in mammalian cardiac muscle. J Physiol. (1974) 242:429–51. doi: 10.1113/jphysiol.1974.sp010716
7. Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. (2002) 415:206–12. doi: 10.1038/415206a
8. Wachter SB, Gilbert EM. Beta-adrenergic receptors, from their discovery and characterization through their manipulation to beneficial clinical application. Cardiology. (2012) 122:104–12. doi: 10.1159/000339271
9. Puglisi R, Mattia G, Carè A, Marano G, Malorni W, Matarrese P. Non-genomic effects of estrogen on cell homeostasis and remodeling with special focus on cardiac ischemia/reperfusion injury. Front Endocrinol (Lausanne). (2019) 10:733. doi: 10.3389/fendo.2019.00733
10. Machuki JO, Zhang HY, Harding SE, Sun H. Molecular pathways of oestrogen receptors and β-adrenergic receptors in cardiac cells: recognition of their similarities, interactions and therapeutic value. Acta Physiol (Oxf). (2018) 222:e12978. doi: 10.1111/apha.12978
11. Williams RS, Bishop T. Selectivity of dobutamine for adrenergic receptor subtypes: in vitro analysis by radioligand binding. J Clin Invest. (1981) 67:1703–11. doi: 10.1172/jci110208
12. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. (2006) 2:207–12. doi: 10.1038/nchembio775
13. Paur H, Wright PT, Sikkel MB, Tranter MH, Mansfield C, O'Gara P, et al. High levels of circulating epinephrine trigger apical cardiodepression in a β2-adrenergic receptor/Gi-dependent manner: a new model of Takotsubo cardiomyopathy. Circulation. (2012) 126:697–706. doi: 10.1161/circulationaha.112.111591
14. Dietrichs ES, Sager G, Tveita T. Altered pharmacological effects of adrenergic agonists during hypothermia. Scand J Trauma Resusc Emerg Med. (2016) 24:143. doi: 10.1186/s13049-016-0339-8
15. Evans NJ, Bayliss AL, Reale V, Evans PD. Characterisation of signalling by the endogenous GPER1 (GPR30) receptor in an embryonic mouse hippocampal cell line (mHippoE-18). PLoS ONE. (2016) 11:e0152138. doi: 10.1371/journal.pone.0152138
17. Hullmann JE, Grisanti LA, Makarewich CA, Gao E, Gold JI, Chuprun JK, et al. GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ Res. (2014) 115:976–85. doi: 10.1161/circresaha.116.304475
18. Lefkowitz RJ, Williams LT. Catecholamine binding to the beta-adrenergic receptor. Proc Natl Acad Sci USA. (1977) 74:515–9. doi: 10.1073/pnas.74.2.515
19. Sampson KJ, Kass RS. Molecular mechanisms of adrenergic stimulation in the heart. Heart Rhythm. (2010) 7:1151–3. doi: 10.1016/j.hrthm.2010.02.013
20. Wu JL, Liu WZ, Liu JH, Qiao LY, Yuan YN. Distribution and quantification of β-3 adrenergic receptor in tissues of sheep. Animal. (2011) 5:88–93. doi: 10.1017/s1751731110001564
21. Pugach EK, Blenck CL, Dragavon JM, Langer SJ, Leinwand LA. Estrogen receptor profiling and activity in cardiac myocytes. Mol Cell Endocrinol. (2016) 431:62–70. doi: 10.1016/j.mce.2016.05.004
22. Gourdy P, Guillaume M, Fontaine C, Adlanmerini M, Montagner A, Laurell H, et al. Estrogen receptor subcellular localization and cardiometabolism. Mol Metab. (2018) 15:56–69. doi: 10.1016/j.molmet.2018.05.009
23. Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3β pathway after ischemia/reperfusion. PLoS ONE. (2015) 10:e0135988. doi: 10.1371/journal.pone.0135988
24. de Lucia C, Eguchi A, Koch WJ. New insights in cardiac β-adrenergic signaling during heart failure and aging. Front Pharmacol. (2018) 9:904. doi: 10.3389/fphar.2018.00904
25. Monjo M, Pujol E, Roca P. alpha2- to beta3-Adrenoceptor switch in 3T3-L1 preadipocytes and adipocytes: modulation by testosterone, 17beta-estradiol, and progesterone. Am J Physiol Endocrinol Metab. (2005) 289:E145–50. doi: 10.1152/ajpendo.00563.2004
26. Riedel K, Deussen AJ, Tolkmitt J, Weber S, Schlinkert P, Zatschler B, et al. Estrogen determines sex differences in adrenergic vessel tone by regulation of endothelial β-adrenoceptor expression. Am J Physiol Heart Circ Physiol. (2019) 317:H243–54. doi: 10.1152/ajpheart.00456.2018
27. Bhadada SV, Patel BM, Mehta AA, Goyal RK. β(3) receptors: role in cardiometabolic disorders. Ther Adv Endocrinol Metab. (2011) 2:65–79. doi: 10.1177/2042018810390259
28. Yang LK, Tao YX. Physiology and pathophysiology of the β(3)-adrenergic receptor. Prog Mol Biol Transl Sci. (2019) 161:91–112. doi: 10.1016/bs.pmbts.2018.09.003
29. Fox EM, Andrade J, Shupnik MA. Novel actions of estrogen to promote proliferation: integration of cytoplasmic and nuclear pathways. Steroids. (2009) 74:622–7. doi: 10.1016/j.steroids.2008.10.014
30. Furth PA. STAT signaling in different breast cancer sub-types. Mol Cell Endocrinol. (2014) 382:612–5. doi: 10.1016/j.mce.2013.03.023
31. Cavalcanti FN, Lucas TF, Lazari MF, Porto CS. Estrogen receptor ESR1 mediates activation of ERK1/2, CREB, and ELK1 in the corpus of the epididymis. J Mol Endocrinol. (2015) 54:339–49. doi: 10.1530/jme-15-0086
32. Sharma JN, Al-Omran A, Parvathy SS. Role of nitric oxide in inflammatory diseases. Inflammopharmacology. (2007) 15:252–9. doi: 10.1007/s10787-007-0013-x
33. Salerno AG, van Solingen C, Scotti E, Wanschel A, Afonso MS, Oldebeken SR, et al. LDL receptor pathway regulation by miR-224 and miR-520d. Front Cardiovasc Med. (2020) 7:81. doi: 10.3389/fcvm.2020.00081
34. Hou H, Zhao Z, Machuki JO, Zhang L, Zhang Y, Fu L, et al. Estrogen deficiency compromised the β(2)AR-Gs/Gi coupling: implications for arrhythmia and cardiac injury. Pflugers Arch. (2018) 470:559–70. doi: 10.1007/s00424-017-2098-4
35. Möller C, Stiermaier T, Brabant G, Graf T, Thiele H, Eitel I. Comprehensive assessment of sex hormones in Takotsubo syndrome. Int J Cardiol. (2018) 250:11–5. doi: 10.1016/j.ijcard.2017.10.047
36. Machuki JO, Zhang HY, Geng J, Fu L, Adzika GK, Wu L, et al. Estrogen regulation of cardiac cAMP-L-type Ca(2+) channel pathway modulates sex differences in basal contraction and responses to β(2)AR-mediated stress in left ventricular apical myocytes. Cell Commun Signal. (2019) 17:34. doi: 10.1186/s12964-019-0346-2
37. Fairweather D. Sex differences in inflammation during atherosclerosis. Clin Med Insights Cardiol. (2014) 8:49–59. doi: 10.4137/cmc.s17068
38. Arnal JF, Douin-Echinard V, Brouchet L, Tremollières F, Laurell H, Lenfant F, et al. Understanding the oestrogen action in experimental and clinical atherosclerosis. Fundam Clin Pharmacol. (2006) 20:539–48. doi: 10.1111/j.1472-8206.2006.00445.x
39. Abraham AD, Schattauer SS, Reichard KL. Estrogen regulation of GRK2 inactivates kappa opioid receptor signaling mediating analgesia, but not aversion. J Neurosci. (2018) 38:8031–43. doi: 10.1523/jneurosci.0653-18.2018
40. Lu J, Lu Y, Wang X, Li X, Linderman GC, Wu C, et al. Prevalence, awareness, treatment, and control of hypertension in China: data from 1.7 million adults in a population-based screening study (China PEACE Million Persons Project). Lancet. (2017) 390:2549–58. doi: 10.1016/s0140-6736(17)32478-9
41. Li Q, Youn JY, Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J Hypertens. (2015) 33:1128–36. doi: 10.1097/hjh.0000000000000587
42. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. (1999) 399:597–601. doi: 10.1038/21218
43. Kolluru GK, Siamwala JH, Chatterjee S. eNOS phosphorylation in health and disease. Biochimie. (2010) 92:1186–98. doi: 10.1016/j.biochi.2010.03.020
44. Hu Q, Zhang T, Li Y, Feng J, Nie R, Wang X, et al. β2AR-dependent signaling contributes to in-vivo reendothelialization capacity of endothelial progenitor cells by shear stress. J Hypertens. (2020) 38:82–94. doi: 10.1097/hjh.0000000000002203
45. Steptoe A, Kivimäki M. Stress and cardiovascular disease: an update on current knowledge. Annu Rev Public Health. (2013) 34:337–54. doi: 10.1146/annurev-publhealth-031912-114452
46. Mishra JS, Te Riele GM, Qi QR, Lechuga TJ, Gopalakrishnan K, Chen DB, et al. Estrogen receptor-β mediates estradiol-induced pregnancy-specific uterine artery endothelial cell angiotensin type-2 receptor expression. Hypertension. (2019) 74:967–74. doi: 10.1161/hypertensionaha.119.13429
47. Xue B, Zhang Z, Johnson RF, Johnson AK. Sensitization of slow pressor angiotensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment. Hypertension. (2012) 59:459–66. doi: 10.1161/hypertensionaha.111.185116
48. Maric-Bilkan C, Manigrasso MB. Sex differences in hypertension: contribution of the renin-angiotensin system. Gend Med. (2012) 9:287–91. doi: 10.1016/j.genm.2012.06.005
49. Xue B, Zhang Z, Beltz TG, Guo F, Hay M, Johnson AK. Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am J Physiol Heart Circ Physiol. (2014) 307:H191–8. doi: 10.1152/ajpheart.01012.2013
50. Marzilli M, Merz CN, Boden WE, Bonow RO, Capozza PG, Chilian WM, et al. Obstructive coronary atherosclerosis and ischemic heart disease: an elusive link! J Am Coll Cardiol. (2012) 60:951–6. doi: 10.1016/j.jacc.2012.02.082
51. Taqueti VR, Di Carli MF. Clinical significance of noninvasive coronary flow reserve assessment in patients with ischemic heart disease. Curr Opin Cardiol. (2016) 31:662–9. doi: 10.1097/hco.0000000000000339
52. Lee JY, Hyun HS, Park HG, Seo JH, Lee EY, Lee JS, et al. Effects of hormone therapy on serum lipid levels in postmenopausal Korean women. J Menopausal Med. (2015) 21:104–11. doi: 10.6118/jmm.2015.21.2.104
53. Roelfsema F, Yang RJ, Veldhuis JD. Differential effects of estradiol and progesterone on cardiovascular risk factors in postmenopausal women. J Endocr Soc. (2018) 2:794–805. doi: 10.1210/js.2018-00073
54. Fu W, Gao XP, Zhang S, Dai YP, Zou WJ, Yue LM. 17β-Estradiol inhibits PCSK9-mediated LDLR degradation through GPER/PLC activation in HepG2 cells. Front Endocrinol (Lausanne). (2019) 10:930. doi: 10.3389/fendo.2019.00930
55. Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. (2009) 50:S172–7. doi: 10.1194/jlr.R800091-JLR200
56. Ghaffari S, Naderi Nabi F, Sugiyama MG, Lee WL. Estrogen inhibits LDL (low-density lipoprotein) transcytosis by human coronary artery endothelial cells via GPER (G-protein-coupled estrogen receptor) and SR-BI (scavenger receptor class B type 1). Arterioscler Thromb Vasc Biol. (2018) 38:2283–94. doi: 10.1161/atvbaha.118.310792
57. Armstrong SM, Sugiyama MG, Fung KY, Gao Y, Wang C, Levy AS, et al. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc Res. (2015) 108:268–77. doi: 10.1093/cvr/cvv218
58. Olsen NJ, Viselli SM, Fan J, Kovacs WJ. Androgens accelerate thymocyte apoptosis. Endocrinology. (1998) 139:748–52. doi: 10.1210/endo.139.2.5729
59. Friedl R, Brunner M, Moeslinger T, Spieckermann PG. Testosterone inhibits expression of inducible nitric oxide synthase in murine macrophages. Life Sci. (2000) 68:417–29. doi: 10.1016/s0024-3205(00)00953-x
60. Dulos GJ, Bagchus WM. Androgens indirectly accelerate thymocyte apoptosis. Int Immunopharmacol. (2001) 1:321–8. doi: 10.1016/s1567-5769(00)00029-1
61. Torcia MG, Nencioni L, Clemente AM, Civitelli L, Celestino I, Limongi D, et al. Sex differences in the response to viral infections: TLR8 and TLR9 ligand stimulation induce higher IL10 production in males. PLoS ONE. (2012) 7:e39853. doi: 10.1371/journal.pone.0039853
62. Kissick HT, Sanda MG, Dunn LK, Pellegrini KL, On ST, Noel JK, et al. Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc Natl Acad Sci USA. (2014) 111:9887–92. doi: 10.1073/pnas.1402468111
63. Schurz H, Salie M, Tromp G, Hoal EG, Kinnear CJ, Möller M. The X chromosome and sex-specific effects in infectious disease susceptibility. Hum Genomics. (2019) 13:2. doi: 10.1186/s40246-018-0185-z
64. Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, et al. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci USA. (2005) 102:1442–7. doi: 10.1073/pnas.0409532102
65. Patial S, Shahi S, Saini Y, Lee T, Packiriswamy N, Appledorn DM, et al. G-protein coupled receptor kinase 5 mediates lipopolysaccharide-induced NFκB activation in primary macrophages and modulates inflammation in vivo in mice. J Cell Physiol. (2011) 226:1323–33. doi: 10.1002/jcp.22460
66. Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. (2013) 128:388–400. doi: 10.1161/circulationaha.113.001878
67. Kar P, Mirams GR, Christian HC, Parekh AB. Control of NFAT isoform activation and NFAT-dependent gene expression through two coincident and spatially segregated intracellular Ca(2+) signals. Mol Cell. (2016) 64:746–59. doi: 10.1016/j.molcel.2016.11.011
68. Park YJ, Yoo SA, Kim M, Kim WU. The role of calcium-calcineurin-NFAT signaling pathway in health and autoimmune diseases. Front Immunol. (2020) 11:195. doi: 10.3389/fimmu.2020.00195
69. Goncalves GK, Scalzo S, Alves AP, Agero U, Guatimosim S, Reis AM. Neonatal cardiomyocyte hypertrophy induced by endothelin-1 is blocked by estradiol acting on GPER. Am J Physiol Cell Physiol. (2018) 314:C310–22. doi: 10.1152/ajpcell.00060.2017
70. Kee HJ, Kook H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J Biomed Biotechnol. (2011) 2011:928326. doi: 10.1155/2011/928326
71. Dayoub O, Le Lay S. Estrogen receptor α/HDAC/NFAT axis for delphinidin effects on proliferation and differentiation of T lymphocytes from patients with cardiovascular risks. Sci Rep. (2017) 7:9378. doi: 10.1038/s41598-017-09933-4
72. Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-beta to inhibit calcineurin. Endocrinology. (2008) 149:3361–9. doi: 10.1210/en.2008-0133
73. Boardman HM, Hartley L, Eisinga A, Main C, Roquéi Figuls M, Bonfill Cosp X, et al. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst Rev. (2015) 3:CD002229. doi: 10.1002/14651858.CD002229.pub4
74. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA. (2002) 288:321–33. doi: 10.1001/jama.288.3.321
75. Modena MG, Sismondi P, Mueck AO, Kuttenn F, Lignières B, Verhaeghe J, et al. New evidence regarding hormone replacement therapies is urgently required transdermal postmenopausal hormone therapy differs from oral hormone therapy in risks and benefits. Maturitas. (2005) 52:1–10. doi: 10.1016/j.maturitas.2005.05.003
76. Schierbeck LL, Rejnmark L, Tofteng CL, Stilgren L, Eiken P, Mosekilde L, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ. (2012) 345:e6409. doi: 10.1136/bmj.e6409
77. Antoine C, Ameye L, Paesmans M, de Azambuja E, Rozenberg S. Menopausal hormone therapy use in relation to breast cancer incidence in 11 European countries. Maturitas. (2016) 84:81–8. doi: 10.1016/j.maturitas.2015.11.010
78. Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ. (2017) 8:33. doi: 10.1186/s13293-017-0152-8
79. Kohn GE, Rodriguez KM, Hotaling J, Pastuszak AW. The history of estrogen therapy. Sex Med Rev. (2019) 7:416–21. doi: 10.1016/j.sxmr.2019.03.006
80. LaCroix AZ, Chlebowski RT, Manson JE, Aragaki AK, Johnson KC, Martin L, et al. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. JAMA. (2011) 305:1305–14. doi: 10.1001/jama.2011.382
81. Hickey M, McNamara HC, Mishra GD. Menopausal estrogen therapy and breast cancer mortality. JAMA. (2018) 319:193. doi: 10.1001/jama.2017.18308
82. Shufelt C, Bairey Merz CN, Pettinger MB, Choi L, Chlebowski R, Crandall CJ, et al. Estrogen-alone therapy and invasive breast cancer incidence by dose, formulation, and route of delivery: findings from the WHI observational study. Menopause. (2018) 25:985–91. doi: 10.1097/gme.0000000000001115
83. Shao H, Breitner JC, Whitmer RA, Wang J, Hayden K, Wengreen H, et al. Hormone therapy and Alzheimer disease dementia: new findings from the Cache County Study. Neurology. (2012) 79:1846–52. doi: 10.1212/WNL.0b013e318271f823
84. Modena MG. “Estrogens and the Heart: Do they Help or Hurt?” How estrogen impacts the cardiovascular system. SOJ Gynecol Obstet Womens Health. (2016) 2:8. doi: 10.15226/2381-2915/2/1/00108
85. Hodis HN, Mack WJ, Henderson VW, Shoupe D, Budoff MJ, Hwang-Levine J, et al. Vascular effects of early versus late postmenopausal treatment with estradiol. N Engl J Med. (2016) 374:1221–31. doi: 10.1056/NEJMoa1505241
Keywords: catecholamine stress, β-adrenergic receptors, estrogen, eNOS, lipid profile, NFATs, cardiovascular diseases
Citation: Ndzie Noah ML, Adzika GK, Mprah R, Adekunle AO, Adu-Amankwaah J and Sun H (2021) Sex–Gender Disparities in Cardiovascular Diseases: The Effects of Estrogen on eNOS, Lipid Profile, and NFATs During Catecholamine Stress. Front. Cardiovasc. Med. 8:639946. doi: 10.3389/fcvm.2021.639946
Received: 10 December 2020; Accepted: 06 January 2021;
Published: 12 February 2021.
Edited by:
Angela H. E. M. Maas, Radboud University Nijmegen Medical Centre, NetherlandsReviewed by:
Tharmarajan Ramprasath, Georgia State University, United StatesMaria Grazia Modena, University of Modena and Reggio Emilia, Italy
Copyright © 2021 Ndzie Noah, Adzika, Mprah, Adekunle, Adu-Amankwaah and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Sun, c3VuaCYjeDAwMDQwO3h6aG11LmVkdS5jbg==
†These authors have contributed equally to this work