 Aureli Luquero
Aureli Luquero Lina Badimon1,2,3
Lina Badimon1,2,3 Maria Borrell-Pages
Maria Borrell-Pages
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 23 March 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.639727
The relevance of PCSK9 in atherosclerosis progression is demonstrated by the benefits observed in patients that have followed PCSK9-targeted therapies. The impact of these therapies is attributed to the plasma lipid-lowering effect induced when LDLR hepatic expression levels are recovered after the suppression of soluble PCSK9. Different studies show that PCSK9 is involved in other mechanisms that take place at different stages during atherosclerosis development. Indeed, PCSK9 regulates the expression of key receptors expressed in macrophages that contribute to lipid-loading, foam cell formation and atherosclerotic plaque formation. PCSK9 is also a regulator of vascular inflammation and its expression correlates with pro-inflammatory cytokines release, inflammatory cell recruitment and plaque destabilization. Furthermore, anti-PCSK9 approaches have demonstrated that by inhibiting PCSK9 activity, the progression of atherosclerotic disease is diminished. PCSK9 also modulates thrombosis by modifying platelets steady-state, leukocyte recruitment and clot formation. In this review we evaluate recent findings on PCSK9 functions in cardiovascular diseases beyond LDL-cholesterol plasma levels regulation.
Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) is a soluble protein synthesized as a zymogen that undergoes autocatalytic cleavage in the endoplasmic reticulum (1). In 2007, PCSK9 was found to be a ligand for Low Density Lipoprotein Receptor (LDLR) a key cell membrane receptor in cholesterol homeostasis regulation (2). LDLRs bind and internalize low density lipoproteins (LDL) from the bloodstream, clearing the blood from highly-enriched cholesterol lipoproteins. The LDL-LDLR complex is guided to the lysosome where LDLs are digested and LDLRs are recycled to the cell surface to keep clearing LDL particles from the circulation. PCSK9 inhibits LDLR recircularization by promoting its degradation in the lysosomes along with LDLs (3). This effect highly reduces the presence of LDLR at the hepatocyte's cell surface and consequently, there is an increase in LDL particles in the bloodstream.
Although PCSK9 is known since 2003 (4) and was almost immediately associated with hypercholesterolemia (5), the knowledge of its potential role on LDL metabolism regulation and its associated diseases is increasing over the years (6, 7). It was first described that mutations on PCSK9 that lead to gain-of-function variants of the protein were responsible for different cases of human familial hypercholesterolemia [FH; (8, 9)]. FH is an inherited disease where patients have LDL plasma levels above 190 mg/dL, contributing to an elevated risk of atherosclerotic plaque formation and coronary adverse events. Contrarily, PCSK9 loss-of-function mutations are associated with very low levels of LDL in blood reducing the cardiovascular associated risk (10, 11). These data encouraged studies to test if PCSK9 was a good target for clinical trials to treat hypercholesterolemia. Hypercholesterolemia is commonly treated with statins, which are drugs that inhibit HMG-CoA reductase, a key enzyme for cholesterol biosynthesis that reduces cholesterol production and lowers LDL concentration in plasma. However, some patients present statin intolerance which hampers the treatment (12). Both PCSK9 and LDLR gene expression are regulated by SREBP2 [Sterol Regulatory Element-Binding Protein 2; (13–15)]. When intracellular levels of cholesterol are low (as after statin treatments), there is activation of SREBP2 that promotes PCSK9 and LDLR transcription. Therefore, both LDLR and LDLR's inhibitor protein levels increase resulting in an intrinsic loop that limits statin therapy efficacy (16–18). SREBP2 transcriptional activity is regulated upstream by AMPK (AMP-activated protein Kinase). Activation of AMPK leads to SREBP2 phosphorylation and its inability to promote transcription of target genes (19, 20).
Anti-PCSK9 drugs started to be developed as a secondary approach to reduce LDL cholesterol levels in hypercholesterolemic patients. To date, only two monoclonal antibodies targeting PCSK9 are available for treating hypercholesterolemia: evolocumab and alirocumab. They were tested in the OSLER trial (21) and in the ODYSSEY LONG TERM trial (22), respectively (Table 1). Both studies showed a ~60% decrease of LDL particles in blood and a decrease in cardiovascular events including myocardial infarction, unstable angina or stroke (2.18% in placebo and 0.95% in evolocumab-treated patients in the OSLER trial and 5.1% in placebo and 4.6% in alirocumab-treated patients in the ODYSSEY LONG TERM study). However, the cardiovascular events reported during these studies were too low to demonstrate clinical relevance in this area. The FOURIER trial enrolled patients with previous atherosclerotic cardiovascular disease that were on statin therapy (23). Results showed a 59% reduction of LDL content in bloodstream and decreased cardiovascular events including cardiovascular mortality, myocardial infarction, stroke, hospitalization for unstable angina or coronary revascularization by more than 15% in evolocumab treated patients after 26 months follow-up. The ODYSSEY OUTCOMES study in patients with recent acute coronary syndrome at maximum tolerated dose of statins showed that alirocumab administration was associated with a reduced risk of recurrent ischemic cardiovascular events and also with a reduced mortality (24). Finally, the SPIRE 1 and SPIRE 2 trials were randomized trials that compared the efficacy of bococizumab, another anti-PCSK9 antibody, with placebo in patients that suffered previous cardiovascular events. These studies did not show benefits from bococizumab treatment despite showing significant improvements for patients with a high cardiovascular risk. There was reduced clinical efficacy because half of patients receiving bococizumab therapy developed antidrug antibodies probably because bococizumab is a murine humanized antibody containing approximately a 3% of murine sequence. Contrarily, alirocumab and evolocumab are antibodies with full human sequence. The negative results concluded in a premature stop of the trial by the sponsor (25).
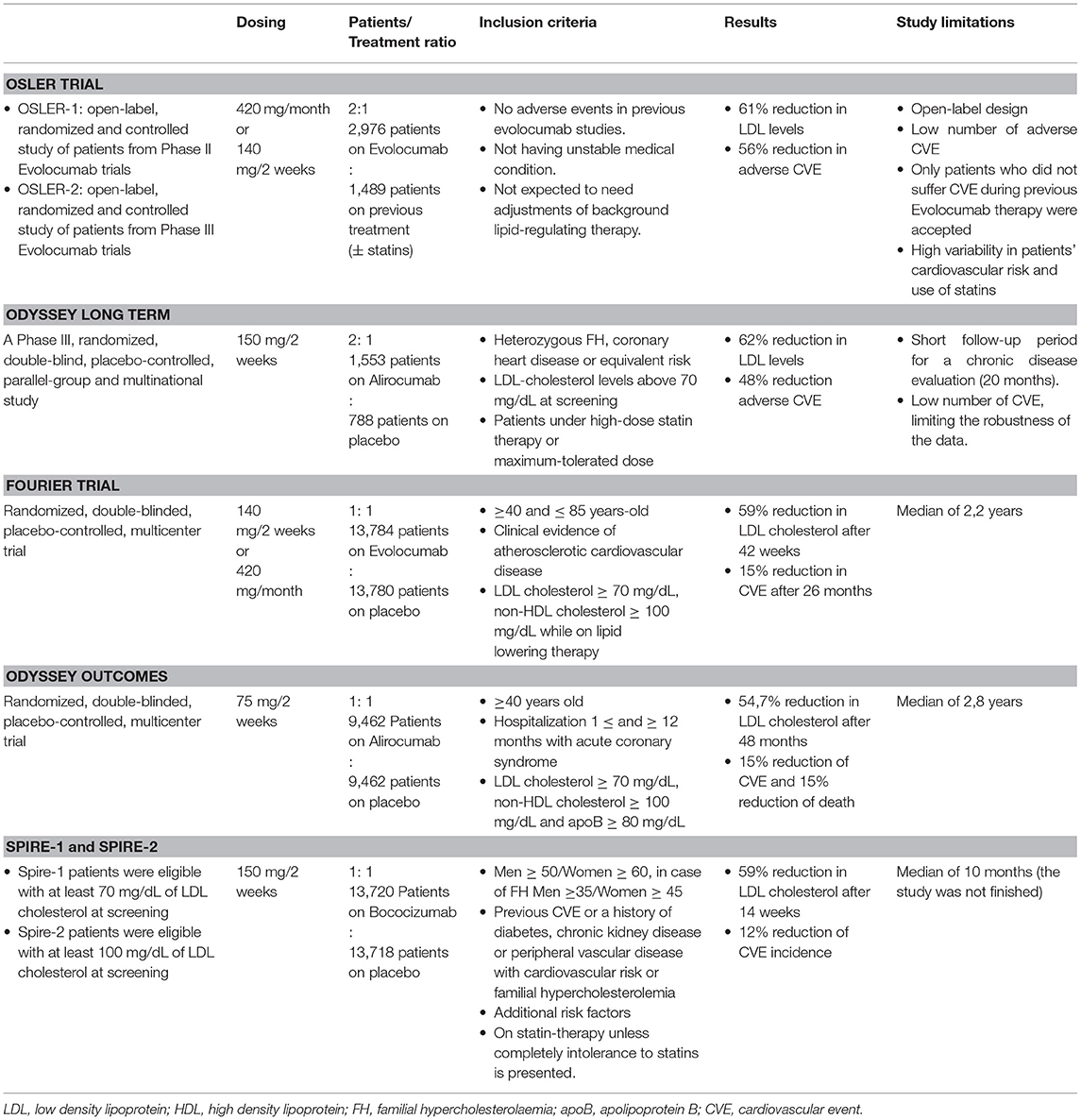
Table 1. Study characteristics and outcomes of clinical trials with monoclonal antibodies against PCSK9.
Besides monoclonal antibodies, other approaches that target PCSK9 have been developed. Inclisiran (a small interference RNA) and statin administration in patients with atherosclerotic cardiovascular disease or heterozygous hypercholesterolemia patients reduced PCSK9 levels in blood and LDL-cholesterol levels in ORION-9 and ORION-10/ORION-11 phase 3 clinical trials (26, 27).
Regulation of cholesterol-rich LDL blood levels is not the only role that PCSK9 drives in atherosclerosis pathogenesis. There is a strong background suggesting alternative roles for PCSK9 in the development of atherosclerosis (28, 29). Indeed, a prospective cohort of 4.232 sixty-year-old men and women living in Stockholm County showed that independently of LDL plasma levels, PCSK9 levels correlate with elevated probability of future cardiovascular events (30). In another study that included 643 participants, high plasma PCSK9 levels correlated with enhanced atherosclerosis progression independently of LDL, as measured by carotid plaque formation and total plaque area (31). However, prospective studies have failed to demonstrate a relationship between PCSK9 expression levels and future risk of cardiovascular events despite revealing correlations between PCSK9 plasma levels and atherosclerotic markers including LDL-cholesterol, blood triglycerides or insulin (32, 33).
This review will discuss the role of PCSK9 in modulating the activity of different cell lineages involved in atherosclerosis progression including macrophages, vascular smooth muscle cells (VSMC), endothelial cells (EC), lymphocytes and platelets and its associated cardiovascular risk. We will comment on PCSK9 effector roles showing that they extend far beyond the regulation of LDL particles and reveal new insights by which PCSK9 inhibitors may lower the incidence of atherosclerosis progression.
Atherosclerosis is commonly described as a chronic inflammatory disease that starts with an excess of cholesterol accumulation in the vascular wall triggering inflammation (34). Macrophages are inflammatory cells that play a key role in lipid uptake and atherosclerosis progression. In 2012, it was shown that LDLR expressed in the surface of human macrophages were downregulated by PCSK9 produced by VSMC, reducing the ability of macrophages to internalize native LDL molecules and avoiding the formation of foam cells, indicating that PCSK9-stimulated macrophages reduce foam cells formation and hence, reduce atherosclerosis progression (35). However, native LDL molecules are not the major source of cholesterol accumulation in macrophages. Upon vascular extravasation, LDL molecules undergo several modifications including aggregation and oxidation. LDL aggregation and oxidation occur after extracellular matrix components such as glycosaminoglycans (36) or chondroitin sulfate proteoglycans (37) retain native LDL particles and facilitate their modification by several secreted enzymes including secretory phospholipase A2, sphingomyelinase, lipoxygenase or myeloperoxidase (38–40). Modified LDL particles generate aggregated (agLDL) and oxidized (oxLDL) LDLs, which are the major source of cholesterol ester accumulation in macrophages and VSMCs [Figure 1; (41–44)]. Macrophages do not internalize agLDL or oxLDL through LDLR but through a different group of receptors called scavenger receptors (45) and LDLR related proteins [LRPs; (46–48)].
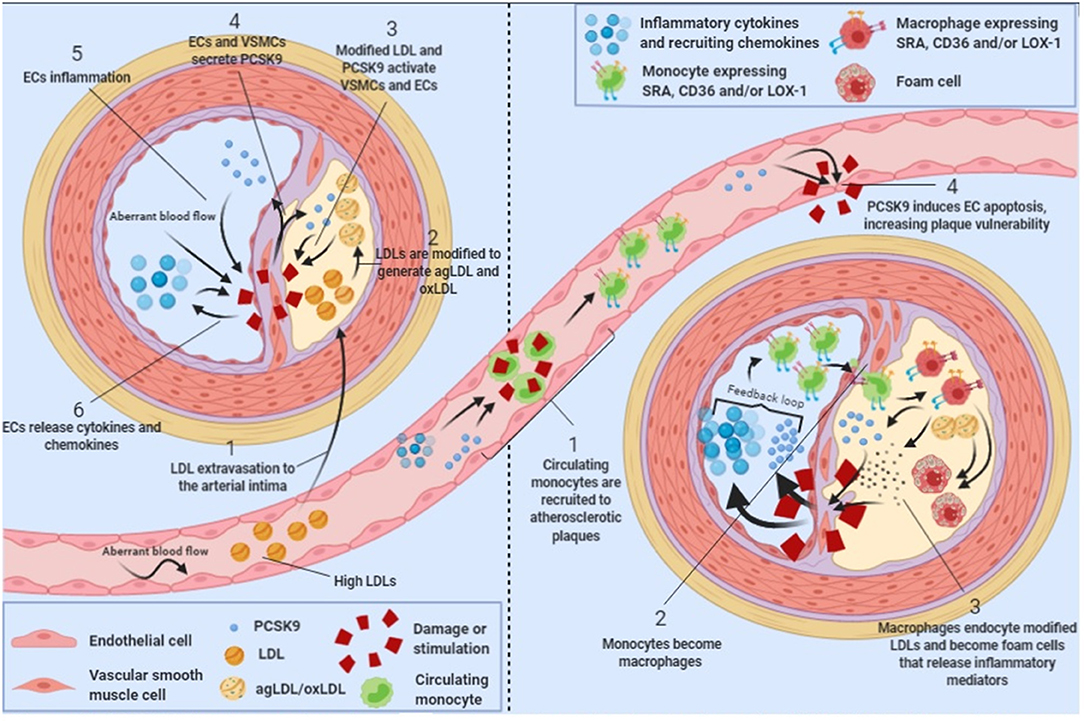
Figure 1. PCSK9 in atherosclerosis progression. Schematic showing the role of PCSK9 in different stages of atherosclerosis progression.
Scavenger receptors including scavenger receptor A (SRA), cluster of differentiation 36 (CD36) and lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) promote the endocytosis of oxLDL particles in monocytes and macrophages and their expression is highly increased under different inflammatory stimulus including lipopolysaccharide (LPS) or tumor necrosis factor-α [TNFα; Figure 1; (49, 50)]. Main features of scavenger receptors are summarized in Table 2. LPS are major components of the outer membrane of Gram-negative bacteria that are recognized by Toll-Like Receptor 4 (TLR4) expressed in macrophage's cell surface. Binding of LPS to TLR4 triggers an intracellular response that activates both MAPK and NFκB pathways triggering inflammation. PCSK9 expression levels are increased in mouse macrophages after LPS stimulation as a result of the activation of the NLRP3 (NOD-Like Receptor Protein 3) inflammasome. Indeed, NLRP3 and its downstream signals IL-1β, IL-18, and caspase 1 all participate in PCSK9 secretion as confirmed by specific gene deletion experiments (51).
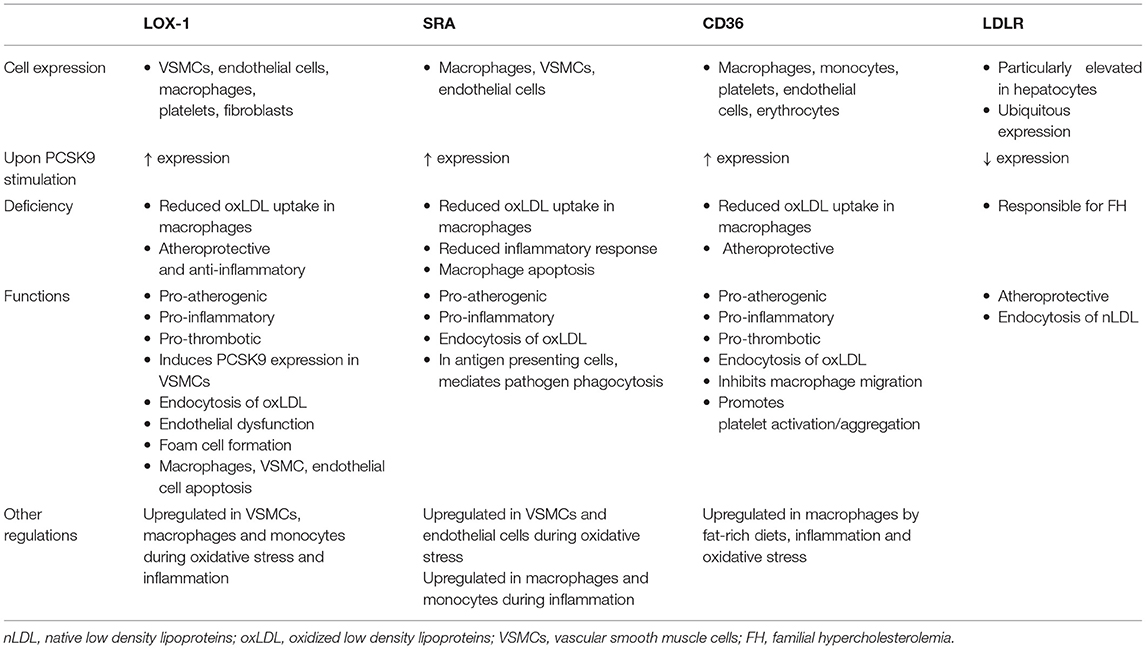
Table 2. Scavenger Receptors and LDLR main features.
PCSK9 expression in macrophages after TNFα stimulation relies on the generation of reactive oxygen species (ROS). ROS inhibitors diphenyleneiodonium (DPI) and apocynin reduce PCSK9 expression while ROS inducers pyocyanin and antimycin A increase PCSK9 release showing that PCSK9 is expressed during macrophage proinflammatory procedures (50). ROS production is dependent on NADPH oxidase. Upon TNFα-stimulation lack of different NADPH oxidase complex subunits reduces the amount of scavenger receptors in the surface of macrophages (50). Recombinant PCSK9 administration increases SRA, CD36, and LOX-1 both at gene and protein levels in cultured mouse macrophages. Concomitantly, oxLDL uptake is increased (Figure 1). This increased lipid uptake is abolished in macrophages that lack SRA, CD36, or LOX-1 suggesting that all three receptors are involved in oxLDL uptake and consequently, in the generation of foam cells in atherosclerosis (50). Table 2 summarizes the involvement and regulation of scavenger receptors and LDLR in different processes associated with atherosclerosis.
Other cell surface receptors expressed in macrophages modulated by PCSK9 are LRP1, LRP5, and LRP8. These receptors belong to the LRP subfamily of the LDLR superfamily of receptors and conserve the characteristic EGF domain that allows PCSK9 binding (52). LRP1 surface levels, together with LDLR, are downregulated by human PCSK9 in atherosclerotic mouse macrophages inducing increased gene expression of the proinflammatory markers TNFα and IL-1β and decreased gene expression of the anti-inflammatory markers IL-10 and arginase-1 indicating enhanced macrophage polarization toward a pro-inflammatory phenotype (53). LRP8 (aka apoER2) a receptor known for recognizing ApoE protein is also downregulated upon recombinant PCSK9 binding in different cell lines including HEK293, 3T3 fibroblasts, CHO, NeuroA2 and HuH7 (54). We have recently described that LRP5 is required for lipid internalization in human macrophages as in the absence of PCSK9 and/or LRP5, macrophages show reduced cholesterol ester accumulation (55). Both proteins form a complex at the perinuclear area of human macrophages that immunoprecipitate together. Their interaction is stronger in lipid loaded macrophages (55). In addition, macrophages silenced for LRP5 show reduced release of PCSK9, indicating that LRP5 is involved in soluble PCSK9 release, probably by participating in the intracellular transport of PCSK9 to the plasma membrane (55). Furthermore, we also show that the complex LRP5-PCSK9 up-regulates TLR4/NFκB signaling to favor macrophage inflammation. Interestingly, LRP5 surface levels remain unaltered by secreted PCSK9 (55).
Finally, VLDLR, a receptor that also belongs to the LDLR superfamily and displays a similar structure to that of LDLR is also downregulated by PCSK9 binding. Indeed, treatment of HEK293 cells or 3T3 fibroblasts cells with human recombinant PCSK9 shows a downregulation of VLDLR expression levels (54). Both VLDLR and LRP8 are known to generate anti-inflammatory signaling in macrophages (56).
PCSK9 is mainly produced in liver, kidney and small intestine (4). However, it is also expressed in vascular cells including endothelial cells (ECs) and vascular smooth muscle cells [VSMCs; (35, 57, 58)]. Vascular cells are affected by hemodynamic factors like blood flow that, by inducing wall shear stress, play a critical role in atherosclerosis development and progression (59). Human ECs and VSMCs under low-blood flow have higher PCSK9 protein expression than cells under high blood flow, an effect conserved even after LPS stimulation. Indeed, aortas from Wt mice showed significantly higher PCSK9 expression in high shear stress regions, an effect further potentiated by LPS administration (60). Also, in rabbits fed at high-fat diet, low-flow aortic regions had higher PCSK9 expression while regions with high flow such the aortic arch showed lower vascular PCSK9 expression [Figure 1; (61)]. Therefore, there is a negative correlation between PCSK9 vascular expression levels and blood flow.
PCSK9 has been shown to promote vascular inflammation. Binding of PCSK9 to the inflammatory receptor TLR4 was first hypothesized by the structural homology of the C-terminal domain of PCSK9 and the TLR4 ligand resistin in in silico simulations (62). TLRs are cell receptors that recognize pathogens and regulate the expression of pro-inflammatory cytokines and also the early immune responses to infection (63). Among TLRs, TLR4 acts as a receptor for LPS and activates NF-κB to promote an inflammatory response (64). PCSK9 expression in ECs and VSMCs is dependent on the TLR4/NFκB signaling pathway as inhibition of different components of the activation cascade show that PCSK9 expression relies on the TLR4-MyD88-NFkB axis and is independent of the TLR4/TRIF signaling, postulating the MyD88 pathway as a possible target for future therapies to prevent excessive PCSK9 production in the vasculature (61). Hence, PCSK9 synthesis is regulated by the TLR4 receptor signaling pathway through MyD88 and NFκB activation, and soluble PCSK9 can act as an inflammatory mediator by TLR4 binding and recognition as demonstrated in ApoE knockout mice (65).
Vascular stability depends on cellular apoptosis. PCSK9 modulates the expression of the apoptosis inducer Bax and the apoptosis inhibitor Bcl-2. The balance between these two proteins is key to prevent or trigger apoptosis (66, 67). Lipid loaded endothelial cells show increased Bax protein levels and decreased Bcl-2 levels that lead to caspase 3 and caspase 9 activation inducing cell apoptosis (68). PCSK9 silencing by siRNA, inhibits apoptosis as silenced PCSK9 cannot phosphorylate p38 and JNK (both members of the MAPK signaling pathway) allowing the activation of the apoptosis inhibitor Bcl-2 (69–72). Interestingly, p38 and JNK are also responsible of Bax and Bad phosphorylation that activate programmed cell death (73, 74). Hence, PCSK9 may be promoting MAPK signaling cascade activation and endothelial cell apoptosis [Figure 1], a mechanism that has already been described in cancer cells (75).
Plaque formation is a complex process that includes lipoprotein retention, inflammatory cells recruitment, VSMC proliferation, matrix synthesis, apoptosis, and necrosis (76). Several lines of evidence sustain that PCSK9 promotes plaque formation in mice and human (29, 67, 77). Indeed, PCSK9 increases LDL uptake by macrophages scavenger receptors contributing to cell foam formation (50); it favors inflammation at the atherosclerotic vascular wall by inducing the expression of adhesion molecules, chemoattractants and inflammatory cytokines (78) and it induces ECs apoptosis reducing vessel stability (69). Furthermore, increased PCSK9 expression levels are associated to low shear stress (60, 61). Therefore, PCSK9 is an efficient target for the development therapies toward the prevention and treatment of atherosclerotic plaque formation.
Anti-PCSK9 therapy in mice reduced by half the plaque area in the aortic root, and the infiltration of pro-inflammatory macrophages in the atherosclerotic plaque was decreased (79). Serum levels of CXCL1, CXCL3, and CXCL10 (known chemoattractants for leukocytes), mainly produced by ECs and VSMCs, were reduced (79). Also, Pcsk9 knockout mice show reduced expression of vascular cell adhesion molecule 1 (VCAM-1), a protein needed for immune cell adhesion to the vascular wall (57).
Anti-PCSK9 vaccination is an alternative to monoclonal antibody therapy. Vaccination involves the conjugation of a peptide (8–13 amino acids) that mimics the N-terminal domain of mature PCSK9 to a carrier protein that confers immunogenic properties to activate the immune system. Syntheses of host specific antibodies against PCSK9 generating long-term inhibition are obtained. Vaccination therapy aims to overcome monoclonal antibody therapy disadvantages including short in vivo half-lives, frequent dosage administration and high costs (80). In atherosclerosis mice models, inhibition of PCSK9 activity through vaccination decreased the expression of intercellular adhesion molecule 1 (ICAM-1) in the diseased aortic root and consequently there was a reduction in monocyte adhesion and migration to the endothelium that contributed to a reduction in atherosclerotic lesions (81). The anti-PCSK9 vaccine AT04A, which generates persistent humoral immune response against PCSK9 for 1 year in mice, reduced LDL content by more than 50% (81). It also reduced NLRP3 inflammasome expression in macrophages (81), a powerful inducer for PCSK9 expression and secretion in macrophages needed for the formation and progression of atherosclerotic plaques (51).
In humans, PCSK9 inhibitors therapy added to statin therapy is capable of increasing fibrous cap thickening in acute coronary syndrome patients, reducing plaque vulnerability (82). However, the LDL-cholesterol lowering capacities of both PCSK9 inhibitors and statin treatment cannot solely explain the increased fibrous cap thickness suggesting that an unknown pleiotropic effect such as an anti-inflammatory effect independent of lowering LDL-cholesterol may be involved (82). PCSK9 inhibitor treatment in an atherogenic mouse model increased the number of circulating endothelial progenitor cells and circulating angiogenic cells, markers of endothelial and vascular health associated with positive outcomes as reduced occurrence of cardiovascular events and death associated to cardiovascular causes (83).
The role of PCSK9 during atherosclerosis progression in the adaptive immune system has been studied. Dendritic cells (DCs) and T lymphocytes are localized in the atherosclerotic plaque, usually at sites prone to rupture (84). DCs mainly work as antigen presenting cells to T lymphocytes. They phagocyte antigens and present them to T lymphocytes in a process that involves MHC and TCR complexes (in DCs and T lymphocytes, respectively). In atheroma plaques, DCs present oxLDL fragments to T-lymphocytes that are then activated (85). The importance of T cell activation in atherosclerosis was demonstrated because ApoE knockout and immunodeficient (severe combined immunodeficiency mice without functional B and T lymphocytes) mice had less atherosclerotic lesions than ApoE knockout mice alone. Furthermore, CD4 T lymphocytes from ApoE knockout mice transferred to immunodeficient ApoE knockout mice induce the generation of atherosclerotic lesions (86). PCSK9 is induced by oxLDL in DCs and enhances the expression of proteins involved in T cell activation including CD80, CD83, CD86, and HLA-DR and the production of pro-inflammatory cytokines including TNFα, IL-1β, and IL-6. The expression of all these proteins was reduced when PCSK9 was silenced, and TGFβ and IL-10 expression levels were increased (87). T cells activated by oxLDL-stimulated DCs produced mainly IFNγ and IL-17, indicating a polarization toward an anti-inflammatory Th1/Th17 phenotype. These anti-inflammatory T regulatory cells inhibit foam cell formation and reverse the pro-inflammatory phenotype of macrophages reducing atherosclerosis progression (88). PCSK9 is a key molecule in Th17 response as atherosclerotic Pcsk9/Ldlr/Apobec (apolipoprotein B mRNA-editing catalytic polypeptide-1) triple knockout mice had significant lower Th17 production in comparison with atherosclerotic Ldlr/Apobec double knockout mice. This was associated with changes in the different cellular sources of Th17 (Th17 lymphocytes or γδTCR+ T cells). Indeed, mice lacking PCSK9 had a reduced number of Th17 lymphocytes as well as a reduced expression of RORγT, the transcription factor needed for Th17 lymphocyte differentiation (89).
A very recent work shows that PCSK9 downregulates the expression of MHC class I proteins in tumor cells by promoting its internalization and degradation in lysosomes (in a similar manner to that of PCSK9 with LDLR). Therefore, PCSK9 decreases the cytotoxic T lymphocyte response against the tumor (90). In an atherosclerotic context it seems plausible that modified LDLs could stimulate antigen presenting cells such as DCs or B cells to produce a variety of cytokines that would guide T lymphocytes differentiation toward a particular inflammatory subtype.
In 2003, after the discovery of PCSK9 gain-of-function mutations in FH patients the first monoclonal antibodies against PCSK9 were tested in preclinical and clinical studies (including ODYSSEY LONG TERM, ODYSSEY OUTCOMES, and FOURIER) demonstrating efficacy in reducing LDL cholesterol plasma levels in patients (91). In 2020 a recent sub analysis of the FOURIER and ODYSSEY OUTCOMES trials revealed that PCSK9 inhibition in patients with stable atherosclerosis and hyperlipidemia on statin therapy significantly reduces the risk of venous thromboembolism supporting a protective role for antiPCSK9 antibodies in human cardiovascular diseases (92). Monoclonal antibodies against PCSK9 are also capable of reversing the pro-inflammatory phenotype of atherogenic macrophages in patients with FH. PCSK9 inhibitors reduced CCR2, CX3CR1, and integrins CD11b and CD18 expression in circulating monocytes suggesting a lower infiltrating and chemoattractant capacity. PCSK9 antibody treatment reduced the production of TNFα by monocytes while the production of anti-inflammatory cytokine IL-10 was enhanced (93). In fact, circulating monocytes from FH patients were enriched with lipid droplets despite non-detectable LDLR expression but increased expression of CD36 and SRA [Figure 1; (93)]. Also, ABCA1 protein, a protein responsible for cholesterol efflux in macrophages, was inhibited upon PCSK9 expression (94). Taken together, these results suggest that circulating monocytes are pre-conditioned in FH patients due to PCSK9 activity, which enhances their infiltrating capacity, lipid accumulation and pro-inflammatory activity. Indeed, a prospective study with heterozygous FH patients under standard statin therapy revealed a positive correlation between circulating levels of PCSK9 and adverse cardiovascular events (95).
Several risk factors associated with cardiovascular disease, including hyperlipidaemia, induce endothelial dysfunction and lead to arterial or venous thrombosis (96). In arteries with ongoing atherosclerosis progression, atherosclerotic plaque rupture is the main cause for thrombosis (97).
Pcsk9 knockout mice show reduced carotid artery thrombosis induced by FeCl3 (a technique to rapidly and accurately induce thrombi formation) in different sized arteries and veins (98). Upon FeCl3 stimulation, 70% of Pcsk9 knockout mice developed non-occlusive non-stable thrombi after 30 min while 57% of Wt mice showed total artery occlusion before 15 min after FeCl3 administration suggesting a role for PCSK9 in platelet reactivity (98). Furthermore, platelets from Pcsk9 knockout mice show a significant reduction in glycoprotein IIB/IIIA expression levels, P-selectin expression levels and in circulating platelet-leukocyte aggregates in comparison with Wt mice indicating lower platelet activation in Pcsk9 knockout mice (98). Similarly, Pcsk9 knockout mice also show reduced thrombi formation after inferior vena cava ligation in comparison to Wt mice (99). Thrombi generated by inferior vena cava ligation in Pcsk9 knockout mice have less leukocyte attachment as leukocyte recruitment is dependent on P-selectin and CXCL1, which are downregulated in Pcsk9 knockout mice (100). However, it is unknown whether this inflammatory cell recruitment is downregulated because of PCSK9's role in lipid uptake or because PCSK9 has lipid-independent functions on platelet's steady-state (Figure 2 illustrates some of the mechanisms by which PCSK9 induces thrombosis).
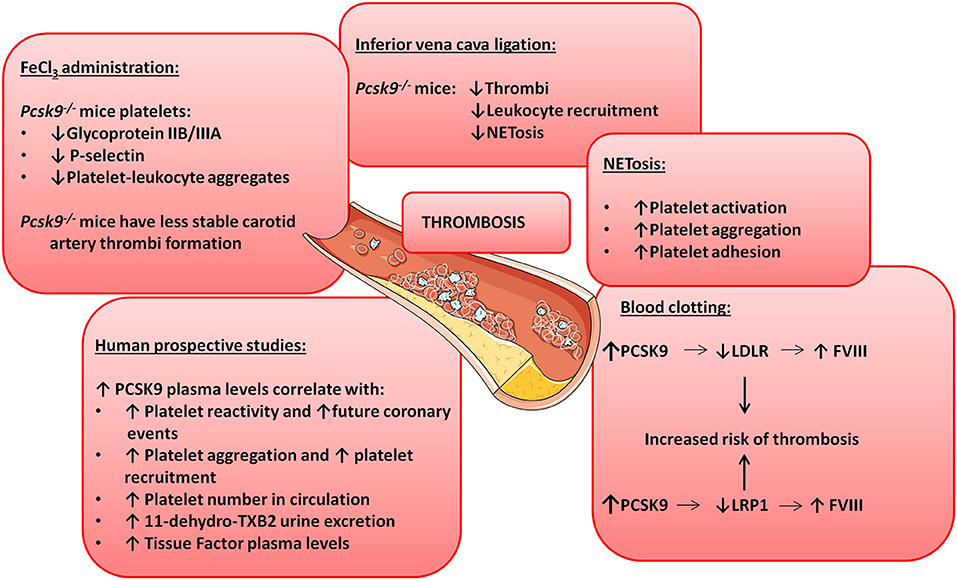
Figure 2. PCSK9 aggravates thrombosis. Schematic showing the involvement of PCSK9 in different thrombotic processes.
NETosis is the process by which neutrophils release their nuclei content composed of DNA and antimicrobial proteins including neutrophil elastases and histones, creating networks of extracellular fibers that trap and facilitate the killing of pathogens (101). NETosis is linked to thrombosis because it causes platelet activation, aggregation and adhesion (102) and promotes the initiation of the coagulation cascade (100). In Pcsk9 knockout mice NETosis is significantly reduced, despite that the total number of blood neutrophils and leukocytes are increased suggesting that PCSK9 can induce thrombosis by stimulating NETosis [Figure 2; (99)].
The PCSK9-REACT study is an observational, prospective study where patients with recent acute coronary syndromes underwent coronary intervention and received P2Y12 inhibitors (103). P2Y12 is a chemoreceptor for adenosine diphosphate (ADP) involved in platelet aggregation (104) and a target for thromboembolism treatments using antagonists as ticagrelor or prasugrel (105). The study revealed a strong correlation between PCSK9 blood levels and platelet reactivity (103). It also showed that elevated PCSK9 plasma levels are associated with future coronary events as 22% of patients with the highest PCSK9 plasma levels suffered coronary events while only 2% of the patients in the lower tertile experienced coronary events. In line with these results, human recombinant PCSK9 added to healthy human plasma was capable of significantly increasing platelet aggregation and reducing aggregation lag time when platelets were stimulated with epinephrine (98). The platelet enhancing capacity is because addition of PCSK9 increased the total number of platelets that express the activation marker glycoprotein IIB/IIIA by 36% [Figure 2; (98)].
A relationship between PCSK9 plasma levels and total number of circulating platelets has also been shown in patients with stable coronary artery disease (106). Similarly, atrial fibrillation patients show a strong correlation between PCSK9 plasma levels and platelet reactivity as elevated PCSK9 levels positively correlate with elevated risk for this cardiovascular event (107). These patients also have higher rate of platelet aggregation and recruitment coincidentally with higher expression levels of thromboxane B2 (TxB2, a platelet activation marker), higher release of P-selectin and enhanced ROS formation (108). Correlation between elevated PCSK9 plasma levels and elevated urine excretion of TxB2 was also found (108). Taken together, these results show that not only platelet number but also platelet reactivity is enhanced when PCSK9 plasma levels are elevated (Figure 2).
Cholesterol incorporation into platelet membranes induces platelet reactivity while cholesterol depletion from membranes is associated with platelet stability (109). Thus, PCSK9 inhibition would decrease plasma LDL levels reducing platelet reactivity. As a matter of fact, statins treatment in hypercholesterolemic patients, is able to reduce platelet membrane cholesterol (110). It remains a matter of discussion whether PCSK9 exerts a direct effect on platelets or the effects depend on the dyslipidaemia generated by PCSK9 binding to LDLR. Dyslipidaemia induces the generation of oxLDL and agLDL, which in turn, facilitate platelet activation by binding scavenger receptors on platelet's surface including LOX-1 and CD36 (111–113). Once activated, platelets are capable of oxidizing LDLs, generating a positive feedback of platelet activation (114). PCSK9 inhibition also downregulates lipoprotein (a) [Lp(a)] serum levels in patients with inherited dyslipidemias (115). Since Lp(a) enhances platelet activation and thrombosis (116–119), PCSK9 may prevent thrombosis by lowering Lp(a) levels. A direct effect of PCSK9 on platelets independent of its effects on dyslipidaemia has been recently shown as PCSK9 inhibitors can enhance oxidative stress (as a result of the activation of the Nox2 and cPLA2 signaling cascades) and block platelet activation in Wt human platelets (108).
PCSK9 inhibitors are being tested to modulate platelet activation in humans. Patients with primary hypercholesterolemia with previous statin treatment were treated for 12 months with alirocumab or evolocumab and after only 2 months treatment a significant decrease in the platelet activation marker CD62P was found (120). Soluble CD40, soluble P-selectin and platelet factor 4 plasma levels were also reduced after 12 months of statin and PCSK9 inhibitor treatment. The study also shows that hypercholesterolemic patients with additional acetylsalicylic acid administration to statins and PCSK9 inhibitors have decreased platelet aggregation (120). A trend in the reduction of platelet aggregation in patients without acetylsalicylic acid administration was observed but there were no significant differences because of the low number of patients that followed this treatment (120).
PCSK9 is also involved in blood clotting. Clotting formation is a complex chemical process were circulating blood clotting factors will sequentially induce protein cleavages to generate thrombin and fibrin (121). A correlation between elevated blood clotting Factor VIII (FVIII) plasma levels and arterial thrombosis has been shown in both animal and human studies (122–124). FVIII synthesis and clearance (and therefore FVIII plasma levels) are regulated by the liver. Indeed, LDLR and LRP1 expressed in hepatocytes promote FVIII endocytosis and degradation (125–128). Although not demonstrated yet, a connection between PCSK9 and FVIII seems plausible. Indeed, downregulation of LDLR expression in hepatocytes cell surface regulated by PCSK9 induces an increase in FVIII plasma levels and therefore an increased risk of thrombosis and cardiovascular events (127). PCSK9 can also reduce LRP1 cell surface expression further increasing FVIII plasma levels (53, 129). Finally, in patients that produce anti-phospholipidic antibodies, polymorphisms in PCSK9 and LDLR genes are associated with thrombosis progression supporting a role in clotting formation for the PCSK9-LDLR axis [Figure 2; (130)].
Since PCSK9 was first described as the inducer of some FH pathologies, a lot of interest has been placed in the achievement of an effective inhibitory treatment. PCSK9 inhibitors administered to patients revealed a key role of PCSK9 in atherosclerotic disease as its inhibition reduced plasma LDL-cholesterol levels with improved clinical cardiovascular outcomes demonstrating a multifactorial and pathophysiological role for PCSK9 in atherosclerosis progression. Interestingly, PCSK9 functions are far from only regulating LDL-cholesterol plasma levels by reducing hepatic LDLR expression. Indeed, recent findings demonstrate that PCSK9 is also actively modulating inflammation, plaque formation and thrombosis. Hence, the benefits observed from PCSK9 inhibitory therapies may not only be induced by its plasma lipid-lowering capacities but also by reducing the impact of several other mechanisms in which PCSK9 is involved that are actively promoting atherosclerosis. Unfortunately, the information on PCSK9 interactome is still limited and further investigations on the role of PCSK9's activity on different signaling pathways are still needed to generate a clear vision of PCSK9 full potential during atherosclerosis progression. Despite PCSK9 has been studied mostly in cardiovascular diseases, it also participates in general mechanisms shared by many other diseases, and hence it is conceivable that PCSK9 is involved in the initiation and progression of other pathologies with powerful inflammatory or thrombotic components.
AL: research performance, draft manuscript preparation, writing and review. MB-P: conceptualization, funding acquisition, draft manuscript preparation, writing, review, and editing. LB: conceptualization and funding acquisition. All authors contributed to the article and approved the submitted version.
This work was supported by the Spanish Ministry of Science and Innovation and FEDER funds (PID2019-107160RB-I00 to LB); the Instituto de Salud Carlos III (CIBERCV CB16/11/00411 to LB, TERCEL RD16/0011/018 to LB and FIS2020-01282 to MB-P); the Generalitat of Catalunya-Secretaria d'Universitats i Recerca del Departament d'Economia i Coneixement de la Generalitat (2017SGR1480 to LB); the Spanish Society of Cardiology (FEC 2019 to MB-P); and the Fundación Investigación Cardiovascular-Fundación Jesus Serra for their continuous support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank S. Huertas and M. A. Velasco for their continuous support to the group.
1. Poirier S, Mayer G. The biology of PCSK9 from the endoplasmic reticulum to lysosomes: new and emerging therapeutics to control low-density lipoprotein cholesterol. Drug Des Devel Ther. (2013) 7:1135–48. doi: 10.2147/DDDT.S36984
2. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. (2007) 282:18602–12. doi: 10.1074/jbc.M702027200
3. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. (2004) 279:48865–75. doi: 10.1074/jbc.M409699200
4. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Bélanger Jasmin S, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. (2003) 100:928–33. doi: 10.1073/pnas.0335507100
5. Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. (2003) 34:154–6. doi: 10.1038/ng1161
6. Soutar AK. Unexpected roles for PCSK9 in lipid metabolism. Curr Opin Lipidol. (2011) 22:192–6. doi: 10.1097/MOL.0b013e32834622b5
7. Seidah NG, Abifadel M, Prost S, Boileau C, Prat A. The proprotein convertases in hypercholesterolemia and cardiovascular diseases: emphasis on proprotein convertase subtilisin/Kexin 9. Pharmacol Rev. (2017) 69:33–52. doi: 10.1124/pr.116.012989
8. Timms KM, Wagner S, Samuels ME, Forbey K, Goldfine H, Jammalapati S, et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet. (2004) 114:349–53. doi: 10.1007/s00439-003-1071-9
9. Fasano T, Sun XM, Patel DD, Soutar AK. Degradation of LDLR protein mediated by “gain of function” PCSK9 mutants in normal and ARH cells. Atherosclerosis. (2009) 203:166–71. doi: 10.1016/j.atherosclerosis.2008.10.027
10. Cariou B, Ouguerram K, Zaïr Y, Guerois R, Langhi C, Kourimate S, et al. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. (2009) 29:2191–7. doi: 10.1161/ATVBAHA.109.194191
11. Benjannet S, Hamelin J, Chrétien M, Seidah NG. Loss- and gain-of-function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J Biol Chem. (2012) 287:33745–55. doi: 10.1074/jbc.M112.399725
12. Reiner Z. Resistance and intolerance to statins. Nutr Metab Cardiovasc Dis. (2014) 24:1057–66. doi: 10.1016/j.numecd.2014.05.009
13. Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. (2004) 24:1454–9. doi: 10.1161/01.ATV.0000134621.14315.43
14. Costet P, Cariou B, Lambert G, Lalanne F, Lardeux B, Jarnoux AL, et al. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J Biol Chem. (2006) 281:6211–8. doi: 10.1074/jbc.M508582200
15. Miserez AR, Muller PY, Barella L, Barella S, Staehelin HB, Leitersdorf E, et al. Sterol-regulatory element-binding protein (SREBP)-2 contributes to polygenic hypercholesterolaemia. Atherosclerosis. (2002) 164:15–26. doi: 10.1016/S0021-9150(01)00762-6
16. Mayne J, Dewpura T, Raymond A, Cousins M, Chaplin A, Lahey KA, et al. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. (2008) 7:22. doi: 10.1186/1476-511X-7-22
17. Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. (2008) 49:394–8. doi: 10.1194/jlr.M700437-JLR200
18. Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, et al. Strong induction of PCSK9 gene expression through HNF1α and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. (2010) 51:1486–95. doi: 10.1194/jlr.M003566
19. Sui GG, Xiao HB, Lu XY, Sun ZL. Naringin activates AMPK resulting in altered expression of SREBPs, PCSK9, and LDLR to reduce body weight in obese C57BL/6J mice. J Agric Food Chem. (2018) 66:8983–90. doi: 10.1021/acs.jafc.8b02696
20. Li X, Hu X, Pan T, Dong L, Ding L, Wang Z, et al. Kanglexin, a new anthraquinone compound, attenuates lipid accumulation by activating the AMPK/SREBP-2/PCSK9/LDLR signalling pathway. Biomed Pharmacother. (2021) 133:110802. doi: 10.1016/j.biopha.2020.110802
21. Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. (2015) 372:1500–9. doi: 10.1056/NEJMoa1500858
22. Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. (2015) 372:1489–99. doi: 10.1056/NEJMoa1501031
23. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. (2017) 376:1713–22. doi: 10.1056/NEJMoa1615664
24. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. (2018) 379:2097–107. doi: 10.1056/NEJMoa1801174
25. Ridker PM, Revkin J, Amarenco P, Brunell R, Curto M, Civeira F, et al. Cardiovascular efficacy and safety of bococizumab in high-risk patients. N Engl J Med. (2017) 376:1527–39. doi: 10.1056/NEJMoa1701488
26. Raal FJ, Kallend D, Ray KK, Turner T, Koenig W, Wright RS, et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N Engl J Med. (2020) 382:1520–30. doi: 10.1056/NEJMoa1913805
27. Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N Engl J Med. (2020) 382:1507–19. doi: 10.1056/NEJMoa1912387
28. Cariou B, Ding Z, Mehta JL. PCSK9 and atherosclerosis: beyond LDL-cholesterol lowering. Atherosclerosis. (2016) 253:275–7. doi: 10.1016/j.atherosclerosis.2016.08.007
29. Shapiro MD, Fazio S. PCSK9 and atherosclerosis - lipids and beyond. J Atheroscler Thromb. (2017) 24:462–72. doi: 10.5551/jat.RV17003
30. Leander K, Mälarstig A, Van'T Hooft FM, Hyde C, Hellénius ML, Troutt JS, et al. Circulating proprotein convertase subtilisin/kexin type 9 (PCSK9) predicts future risk of cardiovascular events independently of established risk factors. Circulation. (2016) 133:1230–9. doi: 10.1161/CIRCULATIONAHA.115.018531
31. Xie W, Liu J, Wang W, Wang M, Qi Y, Zhao F, et al. Association between plasma PCSK9 levels and 10-year progression of carotid atherosclerosis beyond LDL-C: a cohort study. Int J Cardiol. (2016) 215:293–8. doi: 10.1016/j.ijcard.2016.04.103
32. Ridker PM, Rifai N, Bradwin G, Rose L. Plasma proprotein convertase subtilisin/kexin type 9 levels and the risk of first cardiovascular events. Eur Heart J. (2016) 37:554–60. doi: 10.1093/eurheartj/ehv568
33. Zhu YM, Anderson TJ, Sikdar K, Fung M, McQueen MJ, Lonn EM, et al. Association of proprotein convertase subtilisin/kexin type 9 (PCSK9) with cardiovascular risk in primary prevention. Arterioscler Thromb Vasc Biol. (2015) 35:2254–9. doi: 10.1161/ATVBAHA.115.306172
34. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. (1993) 362:801–9. doi: 10.1038/362801a0
35. Ferri N, Tibolla G, Pirillo A, Cipollone F, Mezzetti A, Pacia S, et al. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis. (2012) 220:381–6. doi: 10.1016/j.atherosclerosis.2011.11.026
36. Hurt-Camejo E, Camejo G, Rosengren B, López F, Ahlström C, Fager G, et al. Effect of arterial proteoglycans and glycosaminoglycans on low density lipoprotein oxidation and its uptake by human macrophages and arterial smooth muscle cells. Arterioscler Thromb Vasc Biol. (1992) 12:569–83. doi: 10.1161/01.ATV.12.5.569
37. Williams KJ. Arterial wall chondroitin sulfate proteoglycans: diverse molecules with distinct roles in lipoprotein retention and atherogenesis. Curr Opin Lipidol. (2001) 12:477–87. doi: 10.1097/00041433-200110000-00002
38. Deevska GM, Sunkara M, Morris AJ, Nikolova-Karakashian MN. Characterization of secretory sphingomyelinase activity, lipoprotein sphingolipid content and LDL aggregation in ldlr-/- mice fed on a high-fat diet. Biosci Rep. (2012) 32(Pt 5):479–90. doi: 10.1042/BSR20120036
39. Leitinger N, Watson AD, Hama SY, Ivandic B, Qiao JH, Huber J, et al. Role of group II secretory phospholipase A2 in atherosclerosis: 2. Potential involvement of biologically active oxidized phospholipids. Arterioscler Thromb Vasc Biol. (1999) 19:1291–8. doi: 10.1161/01.ATV.19.5.1291
40. Yoshida H, Kisugi R. Mechanisms of LDL oxidation. Clin Chim Acta. (2010) 411:1875–82. doi: 10.1016/j.cca.2010.08.038
41. Llorente-Cortés V, Otero-Viñas M, Camino-López S, Llampayas O, Badimon L. Aggregated low-density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation. (2004) 110:452–9. doi: 10.1161/01.CIR.0000136032.40666.3D
42. Costales P, Fuentes-Prior P, Castellano J, Revuelta-Lopez E, Corral-Rodríguez MÁ, Nasarre L, et al. K domain CR9 of low density lipoprotein (LDL) receptor-related protein 1 (LRP1) is critical for aggregated LDL-induced foam cell formation from human vascular smooth muscle cells. J Biol Chem. (2015) 290:14852–65. doi: 10.1074/jbc.M115.638361
43. Quinn MT, Parthasarathy S, Fong LG, Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc Natl Acad Sci USA. (1987) 84:2995–8. doi: 10.1073/pnas.84.9.2995
44. Parthasarathy S, Quinn MT, Steinberg D. Is oxidized low density lipoprotein involved in the recruitment and retention of monocyte/macrophages in the artery wall during the initiation of atherosclerosis? Basic Life Sci. (1988) 49:375–80. doi: 10.1007/978-1-4684-5568-7_58
45. Beppu M, Ohishi K, Kikugawa K. Modification of delipidated apoprotein B of low density lipoprotein by lipid oxidation products in relation to macrophage scavenger receptor binding. Biol Pharm Bull. (1994) 17:51–7. doi: 10.1248/bpb.17.51
46. Hu L, Boesten LSM, May P, Herz J, Bovenschen N, Huisman MV, et al. Macrophage low-density lipoprotein receptor-related protein deficiency enhances atherosclerosis in apoE/LDLR double knockout mice. Arterioscler Thromb Vasc Biol. (2006) 26:2710–5. doi: 10.1161/01.ATV.0000249641.96896.e6
47. Borrell-Pages M, Romero JC, Juan-Babot O, Badimon L. Wnt pathway activation, cell migration, and lipid uptake is regulated by low-density lipoprotein receptor-related protein 5 in human macrophages. Eur Heart J. (2011) 32:2841–50. doi: 10.1093/eurheartj/ehr062
48. Waltmann MD, Basford JE, Konaniah ES, Weintraub NL, Hui DY. Apolipoprotein E receptor-2 deficiency enhances macrophage susceptibility to lipid accumulation and cell death to augment atherosclerotic plaque progression and necrosis. Biochim Biophys Acta Mol Basis Dis. (2014) 1842:1395–405. doi: 10.1016/j.bbadis.2014.05.009
49. Hossain E, Ota A, Karnan S, Takahashi M, Mannan SB, Konishi H, et al. Lipopolysaccharide augments the uptake of oxidized LDL by up-regulating lectin-like oxidized LDL receptor-1 in macrophages. Mol Cell Biochem. (2015) 400:29–40. doi: 10.1007/s11010-014-2259-0
50. Ding Z, Liu S, Wang X, Theus S, Deng X, Fan Y, et al. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res. (2018) 114:1145–53. doi: 10.1093/cvr/cvy079
51. Ding Z, Wang X, Liu S, Zhou S, Kore RA, Mu S, et al. NLRP3 inflammasome via IL-1β regulates PCSK9 secretion. Theranostics. (2020) 10:7100–10. doi: 10.7150/thno.45939
52. Go GW, Mani A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J Biol Med. (2012) 85:19–28.
53. Giunzioni I, Tavori H, Covarrubias R, Major AS, Ding L, Zhang Y, et al. Local effects of human PCSK9 on the atherosclerotic lesion. J Pathol. (2016) 238:52–62. doi: 10.1002/path.4630
54. Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. (2008) 283:2363–72. doi: 10.1074/jbc.M708098200
55. Badimon L, Luquero A, Crespo J, Peña E, Borrell-Pages M. PCSK9 and LRP5 in macrophage lipid internalization and inflammation. Cardiovasc Res. (2020) cvaa254. doi: 10.1093/cvr/cvaa254. [Epub ahead of print].
56. Baitsch D, Bock HH, Engel T, Telgmann R, Müller-Tidow C, Varga G, et al. Apolipoprotein e induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. (2011) 31:1160–8. doi: 10.1161/ATVBAHA.111.222745
57. Ding Z, Liu S, Wang X, Deng X, Fan Y, Shahanawaz J, et al. Cross-Talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc Res. (2015) 107:556–67. doi: 10.1093/cvr/cvv178
58. Ding Z, Liu S, Wang X, Mathur P, Dai Y, Theus S, et al. Cross-Talk between PCSK9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxidants Redox Signal. (2016) 25:997–1008. doi: 10.1089/ars.2016.6631
59. Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling. molecular, cellular, and vascular behavior. J Am Coll Cardiol. (2007) 49:2379–93. doi: 10.1016/j.jacc.2007.02.059
60. Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun C, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxidants Redox Signal. (2015) 22:760–71. doi: 10.1089/ars.2014.6054
61. Liu S, Deng X, Zhang P, Wang X, Fan Y, Zhou S, et al. Blood flow patterns regulate PCSK9 secretion via MyD88-mediated pro-inflammatory cytokines. Cardiovasc Res. (2019) 116:1721–32. doi: 10.1093/cvr/cvz262
62. Hampton EN, Knuth MW, Li J, Harris JL, Lesley SA, Spraggon G. The self-inhibited structure of full-length PCSK9 at 1.9 Å reveals structural homology with resistin within the C-terminal domain. Proc Natl Acad Sci USA. (2007) 104:14604–9. doi: 10.1073/pnas.0703402104
63. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. (2005) 17:1–14. doi: 10.1093/intimm/dxh186
64. Guijarro-Muñoz I, Compte M, Álvarez-Cienfuegos A, Álvarez-Vallina L, Sanz L. Lipopolysaccharide activates toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway and proinflammatory response in human pericytes. J Biol Chem. (2014) 289:2457–68. doi: 10.1074/jbc.M113.521161
65. Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. (2017) 262:113–22. doi: 10.1016/j.atherosclerosis.2017.04.023
66. Molostvov G, Morris A, Rose P, Basu S. Modulation of Bcl-2 family proteins in primary endothelial cells during apoptosis. Pathophysiol Haemost Thromb. (2002) 32:85–91. doi: 10.1159/000065081
67. Yurtseven E, Ural D, Baysal K, Tokgözoglu L. An update on the role of PCSK9 in atherosclerosis. J Atheroscler Thromb. (2020) 27:909–18. doi: 10.5551/jat.55400
68. Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS, Liu LS. PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax-caspase9-caspase3 pathway. Mol Cell Biochem. (2012) 359:347–58. doi: 10.1007/s11010-011-1028-6
69. Li J, Liang X, Wang Y, Xu Z, Li G. Investigation of highly expressed PCSK9 in atherosclerotic plaques and ox-LDL-induced endothelial cell apoptosis. Mol Med Rep. (2017) 16:1817–25. doi: 10.3892/mmr.2017.6803
70. De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, et al. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. (2006) 281:21353–61. doi: 10.1074/jbc.M511052200
71. Willis SN, Fletcher JI, Kaufmann T, Van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. (2007) 315:856–9. doi: 10.1126/science.1133289
72. Dhanasekaran DN, Premkumar Reddy E. JNK-signaling: a multiplexing hub in programmed cell death. Genes Cancer. (2017) 8:682–94. doi: 10.18632/genesandcancer.155
73. Donovan N, Becker EBE, Konishi Y, Bonni A. JNK phosphorylation and activation of bad couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. (2002) 277:40944–9. doi: 10.1074/jbc.M206113200
74. Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, et al. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. (2004) 23:1889–99. doi: 10.1038/sj.emboj.7600194
75. Xu B, Li S, Fang Y, Zou Y, Song D, Zhang S, et al. proprotein convertase subtilisin/kexin type 9 promotes gastric cancer metastasis and suppresses apoptosis by facilitating MAPK signaling pathway through HSP70 up-regulation. Front Oncol. (2021) 10:609663. doi: 10.3389/fonc.2020.609663
76. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114:1852–66. doi: 10.1161/CIRCRESAHA.114.302721
77. Ruscica M, Tokgözoglu L, Corsini A, Sirtori CR. PCSK9 inhibition and inflammation: a narrative review. Atherosclerosis. (2019) 288:146–55. doi: 10.1016/j.atherosclerosis.2019.07.015
78. Kühnast S, Van Der Hoorn JWA, Pieterman EJ, Van Den Hoek AM, Sasiela WJ, Gusarova V, et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res. (2014) 55:2103–12. doi: 10.1194/jlr.M051326
79. Schuster S, Rubil S, Endres M, Princen HMG, Boeckel JN, Winter K, et al. Anti-PCSK9 antibodies inhibit pro-atherogenic mechanisms in APOE*3Leiden.CETP mice. Sci Rep. (2019) 9:11079. doi: 10.1038/s41598-019-47242-0
80. Galabova G, Brunner S, Winsauer G, Juno C, Wanko B, Mairhofer A, et al. Peptide-based anti-PCSK9 vaccines-an approach for long-term LDLc management. PLoS ONE. (2014) 9:e114469. doi: 10.1371/journal.pone.0114469
81. Landlinger C, Pouwer MG, Juno C, Van Der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur Heart J. (2017) 38:2499–507. doi: 10.1093/eurheartj/ehx260
82. Yano H, Horinaka S, Ishimitsu T. Effect of evolocumab therapy on coronary fibrous cap thickness assessed by optical coherence tomography in patients with acute coronary syndrome. J Cardiol. (2020) 75:289–95. doi: 10.1016/j.jjcc.2019.08.002
83. Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. (2005) 353:999–1007. doi: 10.1056/NEJMoa043814
84. Bobryshev YV, Watanabe T. Ultrastructural evidence for association of vascular dendritic cells with T-lymphocytes and with B-cells in human atherosclerosis. J Submicrosc Cytol Pathol. (1997) 29:209–21.
85. Frostegård J, Zhang Y, Sun J, Yan K, Liu A. Oxidized low-density lipoprotein (OxLDL)-treated dendritic cells promote activation of T cells in human atherosclerotic plaque and blood, which is repressed by statins: microRNA let-7c is integral to the effect. J Am Heart Assoc. (2016) 5:e003976. doi: 10.1161/JAHA.116.003976
86. Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4+ T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. (2000) 102:2919–22. doi: 10.1161/01.CIR.102.24.2919
87. Liu A, Frostegård J. PCSK9 plays a novel immunological role in oxidized LDL-induced dendritic cell maturation and activation of T cells from human blood and atherosclerotic plaque. J Intern Med. (2018) 284:193–210. doi: 10.1111/joim.12758
88. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. (2015) 35:280–7. doi: 10.1161/ATVBAHA.114.303568
89. Kim YU, Kee P, Danila D, Teng BB. A critical role of PCSK9 in mediating il-17-producing T cell responses in hyperlipidemia. Immune Netw. (2019) 19:e41. doi: 10.4110/in.2019.19.e41
90. Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. (2020) 588:693–8. doi: 10.1038/s41586-020-2911-7
91. El Khoury P, Elbitar S, Ghaleb Y, Khalil YA, Varret M, Boileau C, et al. PCSK9 mutations in familial hypercholesterolemia: from a groundbreaking discovery to anti-PCSK9 therapies. Curr Atheroscler Rep. (2017) 19:49. doi: 10.1007/s11883-017-0684-8
92. Marston NA, Gurmu Y, Melloni GEM, Bonaca M, Gencer B, Sever PS, et al. The effect of PCSK9 inhibition on the risk of venous thromboembolism. Circulation. (2020) 141:1600–7. doi: 10.1161/CIRCULATIONAHA.120.046397
93. Bernelot Moens SJ, Neele AE, Kroon J, Van Der Valk FM, Van Den Bossche J, Hoeksema MA, et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J. (2017) 38:1584–93. doi: 10.1093/eurheartj/ehx002
94. Adorni MP, Cipollari E, Favari E, Zanotti I, Zimetti F, Corsini A, et al. Inhibitory effect of PCSK9 on Abca1 protein expression and cholesterol efflux in macrophages. Atherosclerosis. (2017) 256:1–6. doi: 10.1016/j.atherosclerosis.2016.11.019
95. Cao YX, Jin JL, Sun D, Liu HH, Guo YL, Wu NQ, et al. Circulating PCSK9 and cardiovascular events in FH patients with standard lipid-lowering therapy. J Transl Med. (2019) 17:367. doi: 10.1186/s12967-019-2123-9
96. Gresele P, Momi S, Migliacci R. Endothelium, venous thromboembolism and ischaemic cardiovascular events. Thromb Haemost. (2010) 103:56–61. doi: 10.1160/TH09-08-0562
97. Santos-Gallego CG, Picatoste B, Badimón JJ. Pathophysiology of acute coronary syndrome. Curr Atheroscler Rep. (2014) 16:401. doi: 10.1007/s11883-014-0401-9
98. Camera M, Rossetti L, Barbieri SS, Zanotti I, Canciani B, Trabattoni D, et al. PCSK9 as a positive modulator of platelet activation. J Am Coll Cardiol. (2018) 71:952–4. doi: 10.1016/j.jacc.2017.11.069
99. Wang H, Wang Q, Wang J, Guo C, Kleiman K, Meng H, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) deficiency is protective against venous thrombosis in mice. Sci Rep. (2017) 30:14360. doi: 10.1038/s41598-017-14307-x
100. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
101. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
102. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
103. Navarese EP, Kolodziejczak M, Winter MP, Alimohammadi A, Lang IM, Buffon A, et al. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: the PCSK9-REACT study. Int J Cardiol. (2017) 227:644–9. doi: 10.1016/j.ijcard.2016.10.084
104. Dorsam RT, Kunapuli SP. Central role of the P2Y12 receptor in platelet activation. J Clin Invest. (2004) 113:340–5. doi: 10.1172/JCI20986
105. Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. (2001) 409:202–7. doi: 10.1038/35051599
106. Li S, Zhu CG, Guo YL, Xu RX, Zhang Y, Sun J, et al. The relationship between the plasma PCSK9 levels and platelet indices in patients with stable coronary artery disease. J Atheroscler Thromb. (2015) 22:76–84. doi: 10.5551/jat.25841
107. Pastori D, Nocella C, Farcomeni A, Bartimoccia S, Santulli M, Vasaturo F, et al. Relationship of PCSK9 and urinary thromboxane excretion to cardiovascular events in patients with atrial fibrillation. J Am Coll Cardiol. (2017) 70:1455–62. doi: 10.1016/j.jacc.2017.07.743
108. Cammisotto V, Pastori D, Nocella C, Bartimoccia S, Castellani V, Marchese C, et al. PCSK9 regulates Nox2-mediated platelet activation via CD36 receptor in patients with atrial fibrillation. Antioxidants. (2020) 9:296. doi: 10.3390/antiox9040296
109. Shattil SJ, Anaya-Galindo R, Bennett J, Colman RW, Cooper RA. Platelet hypersensitivity induced by cholesterol incorporation. J Clin Invest. (1975) 55:636–43. doi: 10.1172/JCI107971
110. Panes O, González C, Hidalgo P, Valderas JP, Acevedo M, Contreras S, et al. Platelet tissue factor activity and membrane cholesterol are increased in hypercholesterolemia and normalized by rosuvastatin, but not by atorvastatin. Atherosclerosis. (2017) 257:164–71. doi: 10.1016/j.atherosclerosis.2016.12.019
111. Chen K, Febbraio M, Li W, Silverstein RL. A specific cd36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res. (2008) 102:1512–9. doi: 10.1161/CIRCRESAHA.108.172064
112. Hofmann A, Brunssen C, Morawietz H. Contribution of lectin-like oxidized low-density lipoprotein receptor-1 and LOX-1 modulating compounds to vascular diseases. Vascul Pharmacol. (2017). doi: 10.1016/j.vph.2017.10.002. [Epub ahead of print].
113. Shen MY, Chen FY, Hsu JF, Fu RH, Chang CM, Chang CT, et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood. (2016) 127:1336–45. doi: 10.1182/blood-2015-05-646117
114. Carnevale R, Bartimoccia S, Nocella C, Di Santo S, Loffredo L, Illuminati G, et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis. (2014) 237:108–16. doi: 10.1016/j.atherosclerosis.2014.08.041
115. Sahebkar A, Watts GF. New therapies targeting apoB metabolism for high-risk patients with inherited dyslipidaemias: what can the clinician expect? Cardiovasc Drugs Ther. (2013) 27:559–67. doi: 10.1007/s10557-013-6479-4
116. Rand ML, Sangrar W, Hancock MA, Taylor DM, Marcovina SM, Packham MA, et al. Apolipoprotein(a) enhances platelet responses to the thrombin receptor-activating peptide SFLLRN. Arterioscler Thromb Vasc Biol. (1998) 18:1393–9. doi: 10.1161/01.ATV.18.9.1393
117. Martínez C, Rivera J, Loyau S, Corral J, González-Conejero R, Lozano ML, et al. Binding of recombinant apolipoprotein(a) to human platelets and effect on platelet aggregation. Thromb Haemost. (2001) 85:686–93. doi: 10.1055/s-0037-1615654
118. Dentali F, Gessi V, Marcucci R, Gianni M, Grandi AM, Franchini M. Lipoprotein(a) as a risk factor for venous thromboembolism: a systematic review and meta-analysis of the literature. Semin Thromb Hemost. (2017) 43:614–20. doi: 10.1055/s-0036-1598002
119. Nguyen S, Ilano L, Oluoha N, Pakbaz Z. Lipoprotein(a) a risk factor for venous thrombosis and pulmonary embolism in patients younger than 50 years of age. Blood. (2018) 132(Suppl. 1):5055. doi: 10.1182/blood-2018-99-113975
120. Barale C, Bonomo K, Frascaroli C, Morotti A, Guerrasio A, Cavalot F, et al. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: a 12-month follow-up. Nutr Metab Cardiovasc Dis. (2020) 30:282–91. doi: 10.1016/j.numecd.2019.09.012
121. Smith SA, Travers RJ, Morrissey JH. How it all starts: initiation of the clotting cascade. Crit Rev Biochem Mol Biol. (2015) 50:326–36. doi: 10.3109/10409238.2015.1050550
122. Bank I, Libourel EJ, Middeldorp S, Hamulyák K, Van Pampus ECM, Koopman MMW, et al. Elevated levels of FVIII:C within families are associated with an increased risk for venous and arterial thrombosis. J Thromb Haemost. (2005) 3:79–84. doi: 10.1111/j.1538-7836.2004.01033.x
123. Milgrom A, Lee K, Rothschild M, Makadia F, Duhon G, Min S, et al. Thrombophilia in 153 Patients With Premature Cardiovascular Disease ≤ Age 45. Clin Appl Thromb. (2018) 24:295–302. doi: 10.1177/1076029617703481
124. Machlus KR, Lin FC, Wolberg AS. Procoagulant activity induced by vascular injury determines contribution of elevated factor VIII to thrombosis and thrombus stability in mice. Blood. (2011) 118:3960–8. doi: 10.1182/blood-2011-06-362814
125. Saenko EL, Yakhyaev AV, Mikhailenko I, Strickland DK, Sarafanov AG. Role of the low density lipoprotein-related protein receptor in mediation of factor VIII catabolism. J Biol Chem. (1999) 274:37685–92. doi: 10.1074/jbc.274.53.37685
126. Bovenschen N, Herz J, Grimbergen JM, Lenting PJ, Havekes LM, Mertens K, et al. Elevated plasma factor VIII in a mouse model of low-density lipoprotein receptor-related protein deficiency. Blood. (2003) 101:3933–9. doi: 10.1182/blood-2002-07-2081
127. Bovenschen N, Mertens K, Hu L, Havekes LM, Van Vlijmen BJM. LDL receptor cooperates with LDL receptor-related protein in regulating plasma levels of coagulation factor VIII in vivo. Blood. (2005) 106:906–12. doi: 10.1182/blood-2004-11-4230
128. Paciullo F, Gresele P. Effect of statins on measures of coagulation: potential role of low-density lipoprotein receptors. Eur Heart J. (2019) 40:392. doi: 10.1093/eurheartj/ehy680
129. Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS ONE. (2013) 8:e64145. doi: 10.1371/journal.pone.0064145
Keywords: atherosclerosis, PCSK9 (proprotein convertase subtilisin kexin type 9), lipoprotein receptors, inflammation, lipid loading, LDL—cholesterol
Citation: Luquero A, Badimon L and Borrell-Pages M (2021) PCSK9 Functions in Atherosclerosis Are Not Limited to Plasmatic LDL-Cholesterol Regulation. Front. Cardiovasc. Med. 8:639727. doi: 10.3389/fcvm.2021.639727
Received: 10 December 2020; Accepted: 01 March 2021;
Published: 23 March 2021.
Edited by:
Gabrielle Fredman, Albany Medical College, United StatesReviewed by:
Marit Westerterp, University of Groningen, NetherlandsCopyright © 2021 Luquero, Badimon and Borrell-Pages. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Borrell-Pages, bWJvcnJlbGxwYUBzYW50cGF1LmNhdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.