
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 05 March 2021
Sec. Cardiovascular Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.631817
This article is part of the Research Topic New Strategies to Inhibit Cell Death in Myocardial Ischemia-Reperfusion Injury: How to succeed? View all 17 articles
 Wei Wang1
Wei Wang1 Hao Zheng2*
Hao Zheng2*Cardiovascular diseases have been regarded as the leading cause of death around the world, with myocardial infarction (MI) being the most severe form. MI leads to myocardial apoptosis, cardiomyocyte fibrosis, and cardiomyocyte hypertrophy, ultimately leading to heart failure, and death. Micro RNAs (miRNAs) participate in the genesis and progression of myocardial pathology after MI by playing an important regulatory role. This review aims to summarize all available knowledge on the role of miRNAs in the myocardial pathological process after MI to uncover potential major target pathways. In addition, the main therapeutic methods and their latest progress are also reviewed. miRNAs can regulate the main signaling pathways as well as pathological processes. Thus, they have the potential to induce therapeutic effects. Hence, the combination of miRNAs with recently developed exosome nanocomplexes may represent the future direction of therapeutics.
Myocardial infarction (MI) is defined as the death of myocardial cells due to prolonged ischemia and is the most serious manifestation of coronary artery disease (1). However, MI also results in cardiac remodeling, including myocardial fibrosis and cardiac hypertrophy (2). The pathological changes induced by MI can lead to heart failure, cardiac rupture, sudden death, and other adverse events (3). Antithrombotic agents, percutaneous coronary intervention, and bypass surgery are usually applied to treat patients after MI (4, 5). Nonetheless, these approaches only reduce the severity of the coronary artery disease rather than saving the ischemic myocardium and preventing the development of adverse tissue remodeling (6, 7). Therefore, novel therapeutic strategies to reduce myocardial cell death, inhibit adverse remodeling, and/or stimulate heart regeneration are highly needed.
Micro RNAs (miRNAs) are also involved in differential gene expression in the pathophysiology of MI (5, 8). miRNAs originating from DNA sequences are transcribed by RNA polymerase II in the nucleus to form primary products: primary miRNA (pri-miRNA). Pri-miRNA is generally larger than 1000 bPs and is a double-stranded RNA, similar to a long hairpin, consisting of multiple nucleotide fragments. In the nuclear region, endonuclease Drosha (RNAse III) and cofactor Dgcr8 constitute a unique structure-microprocessor. This complex could precisely cut pri-miRNA and degrade it into a 65 bPs secondary product: miRNA precursor (pre-miRNA) (9). Subsequently, these new pre-miRNAs are transported into the cytoplasm through transport complexes that are made of export protein 5 (EXP5), RAN, GTP, and pre-miRNAs (10, 11). Once the complex passes through the nuclear membrane, the RNAse protein (Dicer) clefts the pre-miRNA into about 19-25 bPs miRNAs, and TAR RNA-binding protein (TRBP or PACT) changes the product into double-stranded miRNAs (12, 13). New double-stranded miRNAs are loaded into a specific AGO protein to form a pre-RNA-induced silencing complex (pre-RISC). As one strand of the double helix in the pre-RISC degrades, it is immediately modified to become a mature RISC (14). The complex binds to the 3'UTR region of the target mRNA, resulting in degradation or inhibition of the target mRNA (15) (Figure 1).
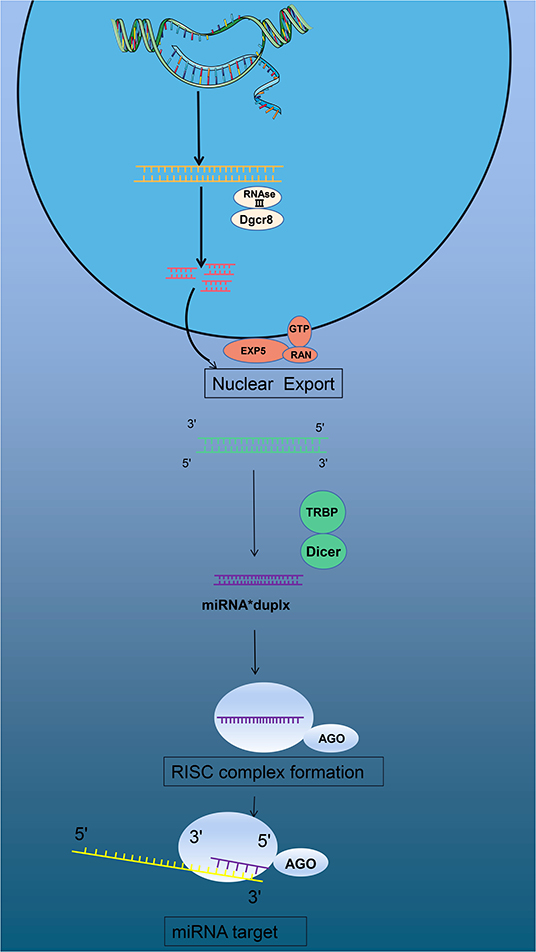
Figure 1. miRNAs form and function.
In the last century, the use of stem cells was thought to be a promising strategy for the treatment of MI (16). As research continues, the exact mechanism of cardiac repair by transplanted cells remains controversial and has yielded inconsistent results. Two main hypotheses exist: (a) direct cardiogenesis/angiogenic differentiation, and (b) indirect stimulation of the regenerative process through paracrine (17, 18). Leda et al. successfully reprogrammed fibroblasts directly into functional cardiomyocytes (19). But low conversion rates and a complex and expensive process have stalled the technology (20). The therapeutic role of exosomes has long been thought to be useful in the treatment of heart injury (21). In combination with nanomaterials, cell transformation is greatly improved (22).
Small non-coding micro RNAs (miRNAs) participate in the pathogenesis and development of myocardial pathology after MI and play an important regulatory role. This study provides a comprehensive overview of miRNAs affecting the pathology after MI and acting on potential targets and access mechanisms. Furthermore, the present therapeutic methods of saving infarct myocardium and latest research progress are summarized. In particular, the challenges and clinical prospects of using miRNA targets for myocardial regenerative therapy are discussed (Table 1 and Figure 2).
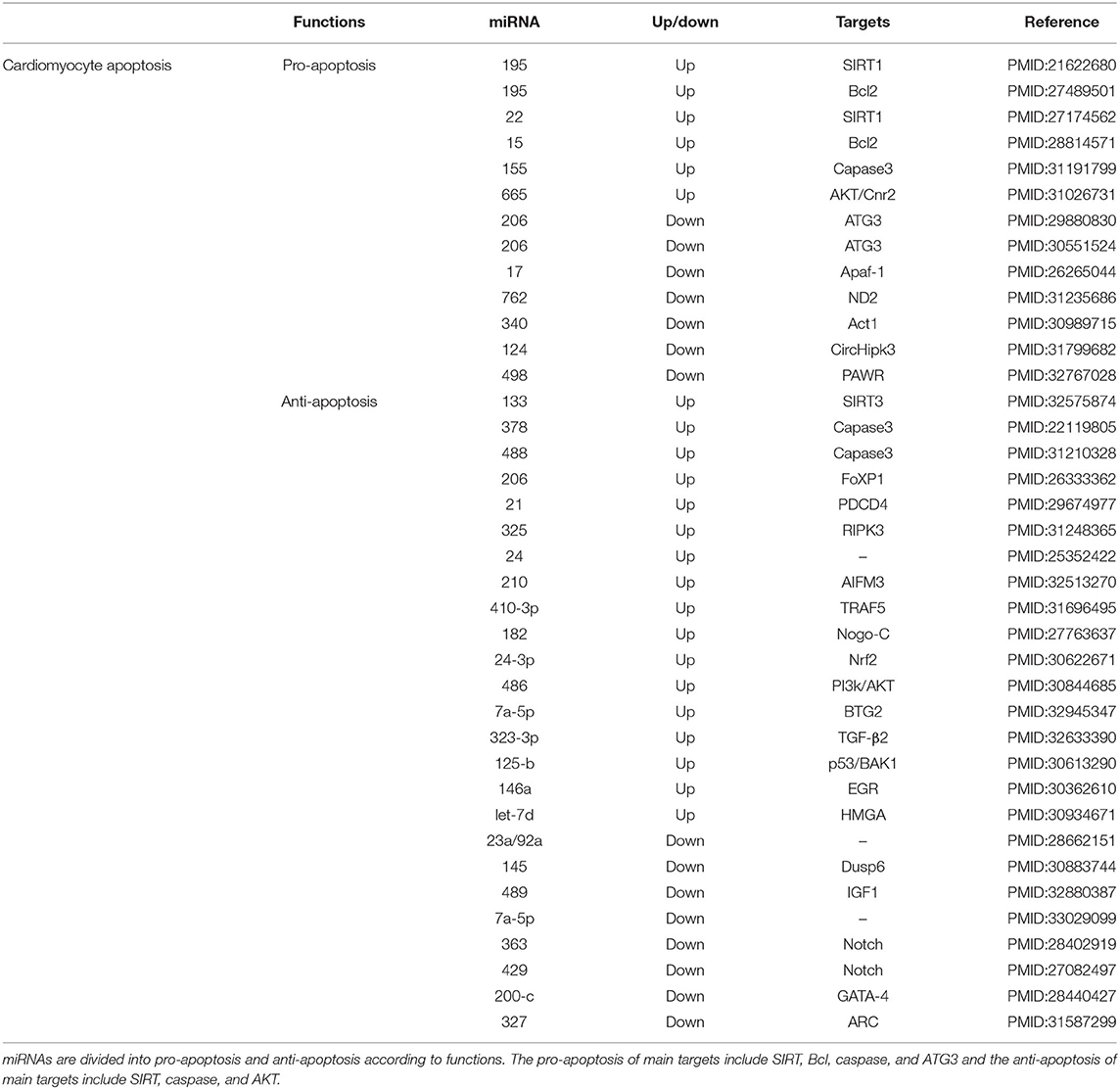
Table 1. Micro RNAs targets and functions in cardiomyocyte apoptosis.
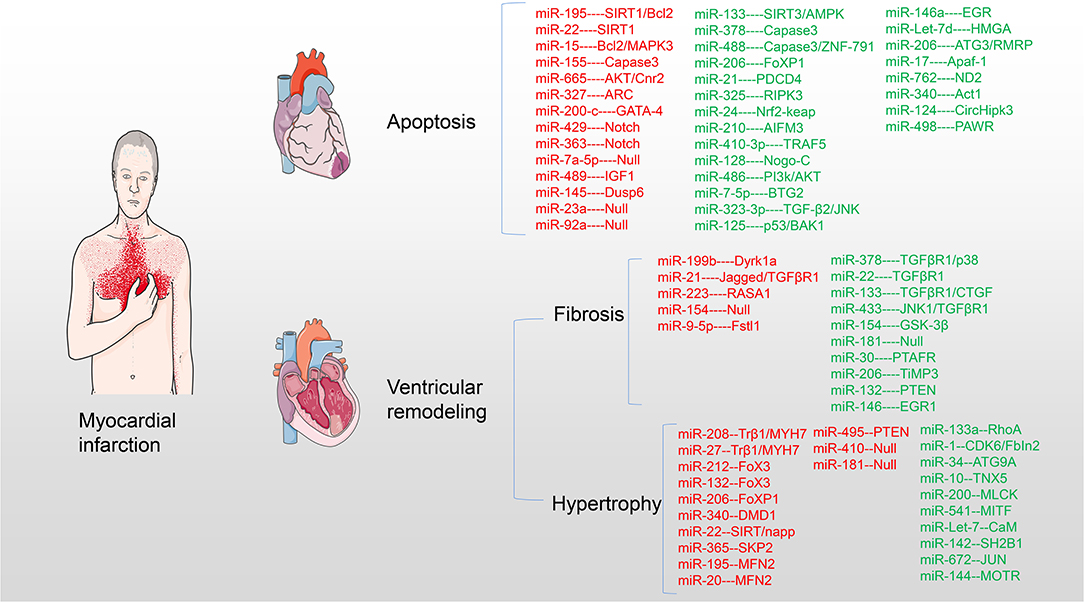
Figure 2. miRNAs target myocardium in pathological process after MI. miRNAs are involved in myocardial cell apoptosis, myocardial fibrosis, and myocardial hypertrophy acting on myocardial targets after MI. Upregulation of red miRNAs promoted the development of pathology, while upregulation of green miRNAs inhibited or even reversed the pathological process.
Apoptosis is a type of programmed cell death promoted by extrinsic and intrinsic pathways through the activation of death receptors and mitochondria, respectively (23–25). The transduction of apoptosis signals is mediated by several pro- and anti-apoptotic factors, including the caspase family, the B cell lymphoma 2 (Bcl-2) family, cytochrome c, and inhibitor of apoptosis proteins (IAP) (26). miRNAs play an important role in myocardial cell apoptosis and heart protection after MI (27–30). Two studies indicated that upregulation of miR-195 and miR-15 in ischemic cardiomyocytes of rats promotes ischemic apoptosis by targeting Bcl-2 (31, 32). In turn, miR-17 can support apoptosis via apoptotic protease activating factor 1 (Apaf-1) which facilitates the formation of apoptosomes containing cytochrome c and deoxyadenosine triphosphate (dATP) (33). However, a previous study suggested that miR-327 inhibits cardiomyocyte apoptosis in vitro and in vivo in rats by targeting the apoptosis repressor with the caspase recruitment domain (ARC) (34). In addition, miR-378 was reported to inhibit caspase-3 expression and attenuate ischemic injury in cardiomyocytes (35), whereas miR-155 did not (36). Overexpression of miRNA-488-3p markedly downregulated the levels of caspase-3 in MI (37). Furthermore, a study revealed that autophagy-related 3 (ATG13) interacts with the fas-associated protein with the death domain to promote the activation of caspase-8 and cell apoptosis (38). ATG13 is also a target of miR-206 and activates the apoptotic factor forkhead box protein 1 (FOXP1) (39, 40). Upregulation of miR-133 can inhibit cardiomyocyte apoptosis, inflammation, and oxidative stress through a mechanism that may be related to the regulation of the SIRT3/AMPK pathway (41). miR-133 is a heart- and muscle-enriched miRNA (42). Sirtuin 1 (Sirt1) also has anti-apoptotic effects that are associated with a reduction in the levels of reactive oxygen species (ROS) (43). In turn, silencing of Sirt1 abolishes the protective effects of miR-22 on hypoxia/reoxygenation-induced mitochondrial dysfunction and cell injury in cardiomyocytes (44). miRNAs also directly suppress the expression of the programmed cell death (PDCD) family, active IAPs, and accelerates cell transcription to inhibit cell apoptosis after MI (45–47). Recently, receptor interacting protein kinase 3 (RIPK3), apoptosis-inducing factor 3 (AIFM3), and tumor necrosis factor receptor-associated factor 5 (TRAF5) were confirmed to be suppressed by miRNAs (48, 49).
Myocardial cells suffer hypoxic damage when MI occurs (50). Two studies confirmed that the activation of protein kinase B (AKT) (51), as well as the ectopic expression of Notch1 (52), inhibited hypoxia-induced apoptosis in culturing human cardiomyocytes under hypoxic conditions (53). Nogo-C is an endoplasmic reticulum protein ubiquitously expressed in tissues including in the heart, which is upregulated in mouse hearts after MI and in cardiomyocytes upon hypoxic treatments (54). Furthermore, knock-down of endogenous NADH dehydrogenase 2 (ND2) significantly decreases intracellular ATP levels and mitochondrial complex-I enzyme activity, whereas it increases ROS levels and apoptotic cell death in cardiomyocytes (55). A study used a H9C2 cardiomyocyte cell line to perform in vitro stimulated ischemia/reperfusion (SI/R) and found a novel function of miR-24-3p in protecting cardiomyocytes from oxidative injury by the activation of the Nrf2/Keap1 pathway (56). Moreover, overexpression of miR-323-3p was also found to reduce oxidative stress and apoptosis of cardiomyocytes via the regulation of the TGF-β2/JNK pathway (57). Additionally, there are still conflicting results regarding miR-7a-5p's protective role on cardiomyocytes upon hypoxic injury (50, 58).
Moreover, upregulation of miR-340-5p suppresses apoptosis and oxidative stress induced by hypoxia/reoxygenation in H9C2 cells by inhibiting the NF-κB activator 1 (Act1) (59). Lastly, a study suggested that bone marrow mesenchymal stem cell (BM-MSC)-derived vascular endothelial growth factor attenuates cardiac apoptosis via regulation of cardiac miRNA-23a and miRNA-92a in a rat model of multiple sclerosis (60). miRNAs from BM-MSCs can interact with myocardial cells through exosomes (61). Interestingly, exosomes originating from adipose-derived stem cells can also attenuate myocardial damage triggered by acute MI via downregulation of early growth response factor 1 (Egr1) (62).
In summary, to date, more miRNAs with anti-apoptotic activity have been reported than those with pro-apoptotic effects, most of which act on classical pathways such as Bcl-2, caspase, AKT, SIRT, and apoptotic factors.
Myocardial fibrosis is an important feature of most cardiac pathological conditions (63), characterized by alteration of the extracellular matrix (64). Currently, five types of collagen are known to be expressed in the myocardium, among which fibrillar collagen type I (85%) and type III (11%) are commonly expressed in the cardiac extracellular matrix. And, the basement membrane of myocytes and the pericellular space are rarely composed of collagen type IV and V (65). Additionally, fibrillar collagen type VI is related to the adhesion of cellular fibers (66). An MI model was established in SD rats using the LAD ligation method and the study found transforming growth factor-β 1 (TGF-β1) induces the upregulation of miR-21 and downregulation of Jagged1 in cardiac fibroblasts (CFs), which are activated by MI, thereby inducing myofibroblast transformation (67). Additionally, decreased levels of antizyme inhibitor (AZIN1) activate TGF-β1. Furthermore, downregulation of c-Jun N-terminal kinase 1 (JNK1) results in the activation of the extracellular signal-regulated kinase and p38 kinase, leading to Smad3 activation and ultimately cardiac fibrosis (68). miR-133a expression in the infarct border zone of myocardial tissue was found to be significantly decreased after MI. And, the upregulation of miRNA-133a in the myocardial tissue of rats with MI remarkably improved cardiac function and reduced collagen volume fraction (69). Furthermore, the mRNA and protein levels of TGF-β1, connective tissue growth factor, collagen I and III, and α-smooth muscle actin (α-SMA) in myocardial tissue were obviously decreased after miRNA-133a upregulation (70). A study also suggested that miR-223 mimics could enhance cell proliferation and migration, collagen I and III, and α-SMA expression in CFs, which could be mediated via mitogen-activated protein kinase kinase (MEK) 1/2, ERK1/2, and AKT phosphorylation (71). miR-154 has similar functions via glycogen synthase kinase 3 beta (GSK-3β) including reducing the heart and cardiomyocyte size, cardiac fibrosis, lowering the expression of atrial (ANP) and B-type natriuretic peptides (BNP), and of profibrotic markers (72), whereas it increases the expression of p15 (a miR-154 target and cell cycle inhibitor) (73). Furthermore, miR-378 and miR-181a are secreted by cardiomyocytes to act as inhibitors of excessive cardiac fibrosis through a paracrine mechanism (74, 75). Upregulation of miR-132 or phosphatase and tensin homolog (PTEN) silencing activate the PI3K/Akt pathway, thereby repressing cardiomyocyte apoptosis and cardiac fibrosis (76). An earlier study showed that an injection of high mobility group box 1 (HMGB1) into the heart of mice, immediately after MI, had the potential to improve cardiac regeneration and prevent remodeling (77). Recently, a study on CFs isolated from mice hearts upon angiotensin II (Ang II)-induced cardiac fibrosis post-MI revealed that miR-30b-5p and miR-22-3p were downregulated, whereas the platelet activating factor receptor (PTAFR) was upregulated [X. S. (78)]. In addition, miRNAs can directly inhibit myocardial fibrosis and even reverse ventricular remodeling (79, 80). Cardiac CITED4 (CBP/p300-interacting transactivators with E [glutamic acid]/D [aspartic acid]-rich-carboxylterminal domain 4) is sufficient to cause physiological hypertrophy and mitigate adverse ventricular remodeling after MI (81). Although few studies specifically investigated myocardial fibrosis, TGF-β1 is clearly a direct or indirect target underlying this process. Upstream targets of PTAFR and CITED4 have recently been found to be worthy of further exploration (Table 2).

Table 2. Micro RNAs targets and functions in myocardial fibrosis.
Cardiac hypertrophy is an adaptive response when the heart faces various pathological stimuli, such as energy metabolism disorders, increased load, changes in humoral factors, and neuroendocrine activation (82, 83). With myocardial contractility decreasing after MI, ventricular remodeling always occurs with compensatory hypertrophy of the myocardium (84). Although this mechanism has an important role for cardiac function in the early phase of MI, these changes will eventually develop into heart failure and even death (85). Myocardial contractility depends mainly on the expression of two myocardial myosin heavy chain (MHC) genes α and β, called Myh6 and Myh7, respectively (86). Thyroid hormone T3 signaling controls the expression of these two MHC genes by stimulating the expression of Myh6 and inhibiting the expression of Myh7 after birth (87). miR-208 is a heart- and muscle-enriched miRNA (42). Transgenic overexpression of miR-208a in the heart, which is encoded within an intron of Myh7 and regulates the thyroid hormone-associated protein 1 (TRβ1), was shown to be sufficient to induce hypertrophic growth of the heart in mice (88). Another study showed that infarcted hearts have a higher abundance of extracellular vesicular miRNA-27a compared with normal hearts, and that miRNA-27a inhibited PDZ and LIM domain 5 (PDLIM5) translation, leading to cardiomyocyte hypertrophic gene expression (89). Probably, Myh7 is also regulated by the T-box transcription factor 5 (Tbx5) (90). Mice injected with an adeno-associated virus expressing miR-1 showed reduced, and even reversed, myocardial hypertrophy (91). miR-1 is a heart- and muscle-enriched miRNA (42). miR-1 inhibits the expression of cell division protein kinase 6 (CDK6) to inhibit phenylephrine-induced neonatal rat ventricular cardiomyocytes hypertrophy, thereby attenuating the inhibition of the expression of β-MHC and phosphorylated the retinoblastoma protein (92). miR-340 is a pro-eccentric hypertrophy miRNA that targets the cardiomyocyte structure protein dystrophin (93). miR-22 and miR-495 have the opposite effect, with their upregulation significantly increasing cell size and markedly decreasing the expression of Myh6 (94). Moreover, they negatively regulate the PTEN levels in cardiomyocytes (95). Additionally, overexpression of let-7a was found to repress the expression of ANP, BNP, and Myh7, as well as of CaM levels (96). A dual-luciferase reporter assay also showed that let-7a could bind to the 3'–UTR of CAM1 and let-7a possesses a prominent anti-hypertrophic property by targeting CAM genes (97).
There is a potential link between cardiac hypertrophy and cardiac cell death (98, 99). A study suggested that intravenous miR-144 has a potent effects on cardiac remodeling of rats with MI, which was associated with significant changes in autophagy signaling (100). Cy3-labeled miR-144 was localized to the infarct and border zones and was taken up by cardiomyocytes and macrophages (101). Similarly, knock-down of the autophagy-related protein 9 (ATG9A), which is a direct target of miR-34, downregulated the autophagic activity and cardiomyocyte hypertrophy (102). Furthermore, overexpression of the S-phase kinase-associated protein 2 (Skp2) promoted autophagy and rescued cardiac hypertrophy induced by Ang II. And, Skp2 knock-down further inhibited autophagy and cardiac hypertrophy in mice with MI (103). In contrast, increased miR-206 expression induced cardiac hypertrophy and inhibited cell death in cultured cardiomyocytes. The Yes-associated protein can promote cardiomyocyte growth and survival in postnatal hearts, and increases the abundance of miR-206, which in turn plays an essential role in mediating hypertrophy and survival by silencing FOXP1 in cardiomyocytes (39). miR-133, 541, 200, 624, and 181 can in turn inhibit hypertrophy and improve cardiac function through different mechanisms (104, 105). Recently, a study confirmed that upregulated miR-142-3p could inhibit hypertrophy and mitochondrial SH2B1, a key factor regulating energy metabolism (106). Moreover, miR-195-5p and miRNA-20a-5p can promote cardiac hypertrophy via targeting mitofusin-2 (MFN2), which is a mitochondrial outer membrane fusion protein (107, 108). TRβ1/Myh7, Ang II, and PTEN have been the main targets of research, and MFN2 may be a new major target (Table 3).
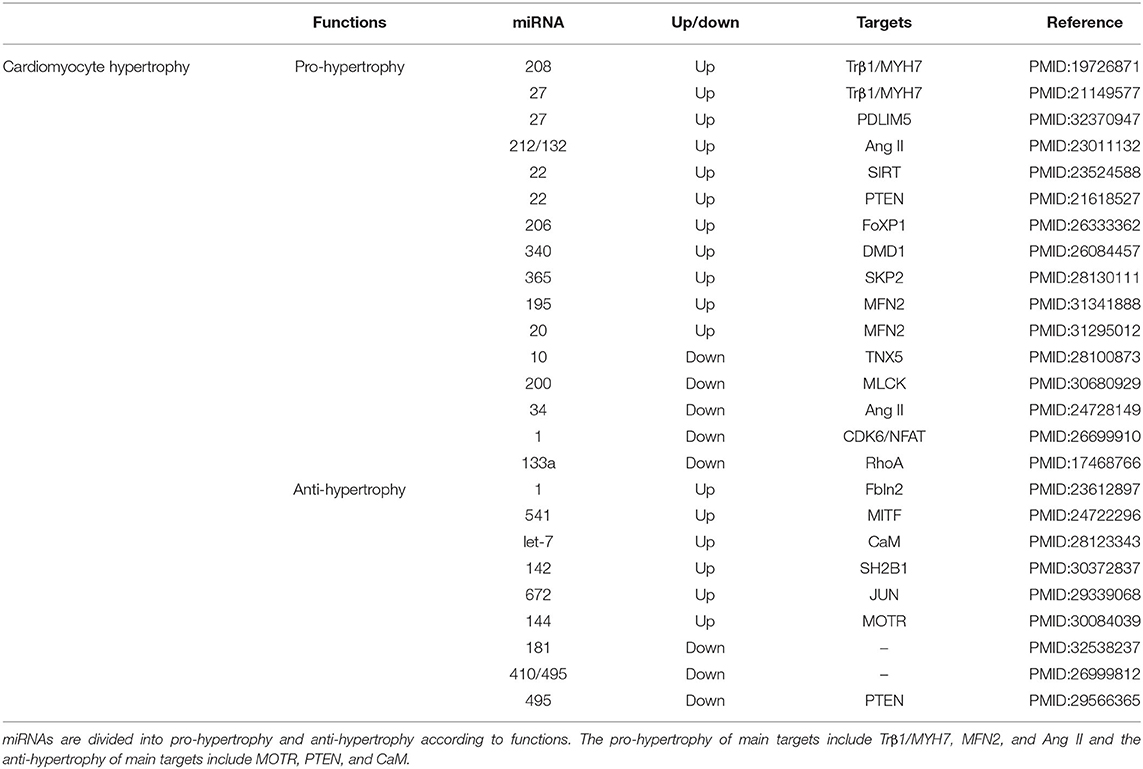
Table 3. Micro RNAs targets and functions in cardiomyocyte hypertrophy.
The majority of patients who survive MI experience a loss of functional cardiomyocytes as a result of the ischemic injury, which leads to ventricular failure with significant alteration of the quality of life and increased risk of mortality (109). Since the proliferation and self-healing capacity of cardiomyocytes in adults is limited, regeneration therapy has emerged as an attractive concept for cardiac repair (110). Compared with traditional interventional stent reperfusion, regenerative therapy can save the myocardium or even regenerate it by promoting angiogenesis, and inhibit, or even avoid adverse cardiac remodeling (111). The main directions of regenerative therapy include stem cell therapy, cardiac fibroblast reprogramming, and exosome therapy.
Scholars have focused on the development of induced pluripotent stem cells, but such treatments have failed to achieve significant benefits in clinical trials (112). This approach has demonstrated limited therapeutic effect mainly due to the risk of immune rejection, genetic instability, tumorigenic potential, low induction efficiency (in the case of induced pluripotent stem cells), and ethical issues (in the case of embryonic stem cells use) (113–115). Leda et al. successfully reprogramed mouse heart and skin fibroblasts into functional induced cardiomyocytes (iCMs) in vitro (19). However, the cardiac fibroblast reprogramming efficiency was extremely low and its requirements are too draconian (116). Furthermore, the iCMs carry other risks such as arrhythmias (117). However, the emergence of exosomes provided an additional tool for myocardial regeneration. Exosomes started to attract attention in 2007, when it was discovered that they have the unique property of transferring miRNAs between cells in vivo, acting as miRNA nanocarriers (118). Recently, mounting evidence has demonstrated the potential of stem cell-derived exosomes, as well as other exosome types, in repairing damage after MI (62, 119). A study confirmed that mesenchymal stem cell (MSC)-derived exosomes electroporated with miR-132 mimics could markedly enhanced the neovascularization in the peri-infarct zone and preserve heart functions (120). Additionally, an injection of exosomes over-expressing miR-21 directly into the infarct zone was found to markedly inhibit cell apoptosis and significantly improve cardiac function in mice (121). MSC-derived exosomes were also found to protect the heart in a porcine model of MI when administered systemically by intravenous injection (122). However, major hurdles remain for the use of exosomes, primarily due to low yields from cell cultures coupled with complicated purification processes (123). Nevertheless, a study reported the self-assembly of a stem cell membrane-camouflaged exosome-mimicking nanocomplex that recapitulated exosome functions, achieving efficient miRNAs delivery and miRNA-mediated myocardial repair (22). Furthermore, a group constructed a functionalized single-walled carbon nanotube bound to siRNA from caspase 3 (F-CNT-siCas3) that demonstrated good water solubility and biocompatibility, but also had a high transfection efficiency of up to 82%, significantly downregulating the expression of the caspase 3 gene and protein in vivo (124). A low molecular weight heparin-encapsulated exosome nanocomplex demonstrated that it could overcome a microvascular obstruction in the infarct, and this structure not only makes myocardial cells uptake miRNAs, which will promote cardiac repair, but will also prevent myocardiocyte apoptosis and attenuate myocardial fibrosis (125). Although the exosome nanocomplex technology is expensive and holds uncertain side effects, it greatly improves the cell conversion rate compared with the previous two regeneration methods, while showing good in vivo results. Thus, an exosome nanocomplex is conducive to further clinical research.
In particular, miRNAs play an important role in the pathology of myocardial apoptosis, fibrosis, and hypertrophy after MI. Targets of miRNAs have significant therapeutic potential, although there are still some conflicting data. The majority of miRNAs and their targets have consistent actions. In particular, SIRT, Bcl-2, Bax, caspase, TGF-β1, TRβ1/Myh7, and MFN2 are believed to play a more significantly prominent role than other targets. In addition, with the development of exosome therapy in combination with nanomaterials, some of the limitations of stem cell therapy (such as low conversion rates and poor cardiac absorption) can be overcome. Exosome nanocomplexes cannot only carry myocardium-friendly miRNAs, but can also directly deliver analogs of important targets into the myocardium in the future. Whether exosome nanocomplexes can treat infarcted myocardium by acting as vectors for the main targets of miRNAs, similar to cocktail therapy, may be the next major direction of exploration. Exosome nanocomplexes with miRNAs are more likely to be successfully taken forward into clinical evaluation than other experimental strategies; however, they also have several limitations. First, the up- and down-stream relationship with the target needs further verification and improvement. There are still conflicting effects of miRNAs (such as miR-Let-7 and miR-154) and more experimental studies are needed. Second, miRNAs act on multiple targets and are involved in several mechanisms; thus, it is necessary to weigh the advantages and disadvantages of their activities. Lastly, treatment with miRNAs is complicated and expensive, and more clinical studies are needed to confirm their therapeutic potential. With the perfectly targeted mechanism and the continuous improvement of exosome therapeutic materials, we believe that mature technologies and drugs based on miRNAs used to save the infarcted myocardium will soon be available to all.
HZ supervised the writing of the manuscript. WW and HZ prepared the manuscript and wrote the draft together. WW prepared the figures. All authors have read and agreed to the published version of the manuscript.
This work was supported by the Natural Science Foundation of Zhejiang Province (LY18H020004).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thanks all of the individuals who participated in the investigations.
1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics−2015 update: a report from the American Heart Association. Circulation. (2015) 131:e29–322. doi: 10.1161/CIR.0000000000000152
2. Kumar D, Jugdutt BI. Apoptosis and oxidants in the heart. J Lab Clin Med. (2003) 142:288–97. doi: 10.1016/S0022-2143(03)00148-3
3. Rizzacasa B, Amati F, Romeo F, Novelli G, Mehta JL. Epigenetic modification in coronary atherosclerosis: JACC review topic of the week. J Am Coll Cardiol. (2019) 74:1352–65. doi: 10.1016/j.jacc.2019.07.043
4. Weintraub WS, Daniels SR, Burke LE, Franklin BA, Goff DC. Value of primordial and primary prevention for cardiovascular disease: a policy statement from the American Heart Association. Circulation. (2011) 124:967–90. doi: 10.1161/CIR.0b013e3182285a81
5. Martinez SR, Gay MS, Zhang L. Epigenetic mechanisms in heart development and disease. Drug Discov Today. (2015) 20:799–811. doi: 10.1016/j.drudis.2014.12.018
6. Nichols M, Townsend N, Scarborough P, Rayner M. Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J. (2014) 35:2950–9. doi: 10.1093/eurheartj/ehu299
7. Taylor J. Third universal definition of myocardial infarction. Eur Heart J. (2012) 33:2506–7. doi: 10.1093/eurheartj/ehs184
8. Wojciechowska A, Braniewska A, Kozar-Kaminska K. MicroRNA in cardiovascular biology and disease. Adv Clin Exp Med. (2017) 26:865–74. doi: 10.17219/acem/62915
9. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014) 15:509–24. doi: 10.1038/nrm3838
10. Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. (2003) 17:3011–6. doi: 10.1101/gad.1158803
11. Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. (2004) 303:95–8. doi: 10.1126/science.1090599
12. Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. (2001) 293:834–8. doi: 10.1126/science.1062961
13. Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. (2001) 409:363–6. doi: 10.1038/35053110
14. Cao DD, Li L, Chan WY. MicroRNAs: key regulators in the central nervous system and their implication in neurological diseases. Int J Mol Sci. (2016) 17:842. doi: 10.3390/ijms17060842
15. Rorbach G, Unold O, Konopka BM. Distinguishing mirtrons from canonical miRNAs with data exploration and machine learning methods. Sci Rep. (2018) 8:7560. doi: 10.1038/s41598-018-25578-3
16. Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, et al. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. (1998) 98:687–98. doi: 10.1161/01.CIR.98.7.687
17. Johnson T, Zhao L, Manuel G, Taylor H, Liu D. Approaches to therapeutic angiogenesis for ischemic heart disease. J Mol Med. (2019) 97:141–51. doi: 10.1007/s00109-018-1729-3
18. Huang K, Hu S, Cheng K. A new era of cardiac cell therapy: opportunities and challenges. Adv Healthc Mater. (2019) 8:e1801011. doi: 10.1002/adhm.201801011
19. Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. (2010) 142:375–86. doi: 10.1016/j.cell.2010.07.002
20. Sadahiro T, Yamanaka S, Ieda M. Direct cardiac reprogramming: progress and challenges in basic biology and clinical applications. Circ Res. (2015) 116:1378–91. doi: 10.1161/CIRCRESAHA.116.305374
21. Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. (2010) 4:214–22. doi: 10.1016/j.scr.2009.12.003
22. Yao C, Wu W, Tang H, Jia X, Tang J, Ruan X, et al. Self-assembly of stem cell membrane-camouflaged nanocomplex for microRNA-mediated repair of myocardial infarction injury. Biomaterials. (2020) 257:120256. doi: 10.1016/j.biomaterials.2020.120256
23. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. (2004) 5:897–907. doi: 10.1038/nrm1496
24. Djiadeu P, Kotra LP, Sweezey N, Palaniyar N. Surfactant protein D delays Fas- and TRAIL-mediated extrinsic pathway of apoptosis in T cells. Apoptosis. (2017) 22:730–40. doi: 10.1007/s10495-017-1348-4
25. Sim WK, Park JH, Kim KY, Chung IS. Robustaflavone induces G0/G1 cell cycle arrest and apoptosis in human umbilical vein endothelial cells and exhibits anti-angiogenic effects in vivo. Sci Rep. (2020) 10:11070. doi: 10.1038/s41598-020-67993-5
26. Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. (2005) 17:393–403. doi: 10.1016/j.molcel.2004.12.030
27. Wong LL, Saw EL, Lim JY, Zhou Y, Richards AM, Wang P. MicroRNA Let-7d-3p contributes to cardiac protection via targeting HMGA2. Int J Mol Sci. (2019) 20:E1522. doi: 10.3390/ijms20071522
28. Bai M, Pan CL, Jiang GX, Zhang YM, Zhang Z. CircHIPK3 aggravates myocardial ischemia-reperfusion injury by binding to miRNA-124–3p. Eur Rev Med Pharmacol Sci. (2019) 23:10107–14. doi: 10.26355/eurrev_201911_19580
29. Wu G, Tan J, Li J, Sun X, Du L, Tao S. miRNA-145–5p induces apoptosis after ischemia-reperfusion by targeting dual specificity phosphatase 6. J Cell Physiol. (2019). doi: 10.1002/jcp.28291. [Epub ahead of print].
30. Chai Q, Zheng M, Wang L, Wei M, Yin Y, Ma F, et al. Circ_0068655 promotes cardiomyocyte apoptosis via miR-498/PAWR Axis. Tissue Eng Regen Med. (2020) 17:659–70. doi: 10.1007/s13770-020-00270-8
31. Hang P, Sun C, Guo J, Zhao J, Du Z. BDNF-mediates down-regulation of MicroRNA-195 inhibits ischemic cardiac apoptosis in rats. Int J Biol Sci. (2016) 12:979–89. doi: 10.7150/ijbs.15071
32. Liu Y, Yang L, Yin J, Su D, Pan Z, Li P, et al. MicroRNA-15b deteriorates hypoxia/reoxygenation-induced cardiomyocyte apoptosis by downregulating Bcl-2 and MAPK3. J Investig Med. (2018) 66:39–45. doi: 10.1136/jim-2017-000485
33. Song S, Seo HH, Lee SY, Lee CY, Lee J, Yoo KJ, et al. MicroRNA-17-mediated down-regulation of apoptotic protease activating factor 1 attenuates apoptosome formation and subsequent apoptosis of cardiomyocytes. Biochem Biophys Res Commun. (2015) 465:299–304. doi: 10.1016/j.bbrc.2015.08.028
34. Li Q, Yang J, Zhang J, Liu XW, Yang CJ, Fan ZX, et al. Inhibition of microRNA-327 ameliorates ischemia/reperfusion injury-induced cardiomyocytes apoptosis through targeting apoptosis repressor with caspase recruitment domain. J Cell Physiol. (2020) 235:3753–67. doi: 10.1002/jcp.29270
35. Fang J, Song XW, Tian J, Chen HY, Li DF, Wang JF, et al. Overexpression of microRNA-378 attenuates ischemia-induced apoptosis by inhibiting caspase-3 expression in cardiac myocytes. Apoptosis. (2012) 17:410–23. doi: 10.1007/s10495-011-0683-0
36. Guo J, Liu HB, Sun C, Yan XQ, Hu J, Yu J, et al. MicroRNA-155 promotes myocardial infarction-induced apoptosis by targeting rna-binding protein QKI. Oxid Med Cell Longev. (2019) 2019:4579806. doi: 10.1155/2019/4579806
37. Zheng HF, Sun J, Zou ZY, Zhang Y, Hou GY. MiRNA-488–3p suppresses acute myocardial infarction-induced cardiomyocyte apoptosis via targeting ZNF791. Eur Rev Med Pharmacol Sci. (2019) 23:4932–9. doi: 10.26355/eurrev_201906_18083
38. Ding S, Abudupataer M, Zhou Z, Chen J, Li H, Xu L, et al. Histamine deficiency aggravates cardiac injury through miR-206/216b-Atg13 axis-mediated autophagic-dependant apoptosis. Cell Death Dis. (2018) 9:694. doi: 10.1038/s41419-018-0723-6
39. Yang Y, Del Re DP, Nakano N, Sciarretta S, Zhai P, Park J, et al. miR-206 mediates YAP-induced cardiac hypertrophy and survival. Circ Res. (2015) 117:891–904. doi: 10.1161/CIRCRESAHA.115.306624
40. Kong F, Jin J, Lv X, Han Y, Liang X, Gao Y, et al. Long noncoding RNA RMRP upregulation aggravates myocardial ischemia-reperfusion injury by sponging miR-206 to target ATG3 expression. Biomed Pharmacother. (2019) 109:716–25. doi: 10.1016/j.biopha.2018.10.079
41. Sun B, Liu S, Hao R, Dong X, Fu L, Han B. RGD-PEG-PLA Delivers MiR-133 to infarct lesions of acute myocardial infarction model rats for cardiac protection. Pharmaceutics. (2020) 12:575. doi: 10.3390/pharmaceutics12060575
42. Zhang L, Ding H, Zhang Y, Wang Y, Zhu W, Li P. Circulating MicroRNAs: biogenesis and clinical significance in acute myocardial infarction. Front Physiol. (2020) 11:1088. doi: 10.3389/fphys.2020.01088
43. Zhu H, Yang Y, Wang Y, Li J, Schiller PW, Peng T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc Res. (2011) 92:75–84. doi: 10.1093/cvr/cvr145
44. Du JK, Cong BH, Yu Q, Wang H, Wang L, Wang CN, et al. Upregulation of microRNA-22 contributes to myocardial ischemia-reperfusion injury by interfering with the mitochondrial function. Free Radic Biol Med. (2016) 96:406–17. doi: 10.1016/j.freeradbiomed.2016.05.006
45. Guo C, Deng Y, Liu J, Qian L. Cardiomyocyte-specific role of miR-24 in promoting cell survival. J Cell Mol Med. (2015) 19:103–112. doi: 10.1111/jcmm.12393
46. Gu H, Liu Z, Li Y, Xie Y, Yao J, Zhu Y, et al. Serum-derived extracellular vesicles protect against acute myocardial infarction by regulating miR-21/PDCD4 signaling pathway. Front Physiol. (2018) 9:348. doi: 10.3389/fphys.2018.00348
47. Zhang DY, Wang BJ, Ma M, Yu K, Zhang Q, Zhang XW. MicroRNA-325–3p protects the heart after myocardial infarction by inhibiting RIPK3 and programmed necrosis in mice. BMC Mol Biol. (2019) 20:17. doi: 10.1186/s12867-019-0136-9
48. Teng YL, Ren F, Xu H, Song HJ. Overexpression of miRNA-410–3p protects hypoxia-induced cardiomyocyte injury via targeting TRAF5. Eur Rev Med Pharmacol Sci. (2019) 23:9050–7. doi: 10.26355/eurrev_201910_19307
49. Cheng H, Chang S, Xu R, Chen L, Song X, Wu J, et al. Hypoxia-challenged MSC-derived exosomes deliver miR-210 to attenuate post-infarction cardiac apoptosis. Stem Cell Res Ther. (2020) 11:224. doi: 10.1186/s13287-020-01737-0
50. Yu J, Yang W, Wang W, Wang Z, Pu Y, Chen H, et al. Involvement of miR-665 in protection effect of dexmedetomidine against Oxidative Stress Injury in myocardial cells via CB2 and CK1. Biomed Pharmacother. (2019) 115:108894. doi: 10.1016/j.biopha.2019.108894
51. Sun XH, Wang X, Zhang Y, Hui J. Exosomes of bone-marrow stromal cells inhibit cardiomyocyte apoptosis under ischemic and hypoxic conditions via miR-486–5p targeting the PTEN/PI3K/AKT signaling pathway. Thromb Res. (2019) 177:23–32. doi: 10.1016/j.thromres.2019.02.002
52. Xu H, Jin L, Chen Y, Li J. Downregulation of microRNA-429 protects cardiomyocytes against hypoxia-induced apoptosis by increasing Notch1 expression. Int J Mol Med. (2016) 37:1677–85. doi: 10.3892/ijmm.2016.2558
53. Meng X, Ji Y, Wan Z, Zhao B, Feng C, Zhao J, et al. Inhibition of miR-363 protects cardiomyocytes against hypoxia-induced apoptosis through regulation of Notch signaling. Biomed Pharmacother. (2017) 90:509–16. doi: 10.1016/j.biopha.2017.03.080
54. Jia S, Qiao X, Ye J, Fang X, Xu C, Cao Y, et al. Nogo-C regulates cardiomyocyte apoptosis during mouse myocardial infarction. Cell Death Dis. (2016) 7:e2432. doi: 10.1038/cddis.2016.331
55. Yan K, An T, Zhai M, Huang Y, Wang Q, Wang Y, et al. Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. (2019) 10:500. doi: 10.1038/s41419-019-1734-7
56. Xiao X, Lu Z, Lin V, May A, Shaw DH, Wang Z, et al. MicroRNA miR-24–3p reduces apoptosis and regulates keap1-Nrf2 pathway in mouse cardiomyocytes responding to ischemia/reperfusion injury. Oxid Med Cell Longev. (2018) 2018:7042105. doi: 10.1155/2018/7042105
57. Shi CC, Pan LY, Zhao YQ, Li Q, Li JG. MicroRNA-323–3p inhibits oxidative stress and apoptosis after myocardial infarction by targeting TGF-beta2/JNK pathway. Eur Rev Med Pharmacol Sci. (2020) 24:6961–70. doi: 10.26355/eurrev_202006_21688
58. Liang D, Jin Y, Lin M, Xia X, Chen X, Huang A. Down-regulation of Xist and Mir-7a-5p improves LPS-induced myocardial injury. Int J Med Sci. (2020) 17:2570–7. doi: 10.7150/ijms.45408
59. Li D, Zhou J, Yang B, Yu Y. microRNA-340–5p inhibits hypoxia/reoxygenation-induced apoptosis and oxidative stress in cardiomyocytes by regulating the Act1/NF-kappaB pathway. J Cell Biochem. (2019) 120:14618–27. doi: 10.1002/jcb.28723
60. Song YS, Joo HW, Park IH, Shen GY, Lee Y, Shin JH, et al. Bone marrow mesenchymal stem cell-derived vascular endothelial growth factor attenuates cardiac apoptosis via regulation of cardiac miRNA-23a and miRNA-92a in a rat model of myocardial infarction. PLoS ONE. (2017) 12:e0179972. doi: 10.1371/journal.pone.0179972
61. Zhu LP, Tian T, Wang JY, He JN, Chen T, Pan M, et al. Hypoxia-elicited mesenchymal stem cell-derived exosomes facilitates cardiac repair through miR-125b-mediated prevention of cell death in myocardial infarction. Theranostics. (2018) 8:6163–77. doi: 10.7150/thno.28021
62. Pan J, Alimujiang M, Chen Q, Shi H, Luo X. Exosomes derived from miR-146a-modified adipose-derived stem cells attenuate acute myocardial infarction-induced myocardial damage via downregulation of early growth response factor 1. J Cell Biochem. (2019) 120:4433–43. doi: 10.1002/jcb.27731
63. Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. (2007) 117:568–75. doi: 10.1172/JCI31044
64. Weber KT, Janicki JS, Shroff SG, Pick R, Chen RM, Bashey RI. Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ Res. (1988) 62:757–65. doi: 10.1161/01.RES.62.4.757
65. Pezel T, Viallon M, Croisille P, Sebbag L, Bochaton T, Garot J, et al. Imaging interstitial fibrosis, left ventricular remodeling, and function in stage A and B heart failure. JACC Cardiovasc Imaging. (2020). doi: 10.1016/j.jcmg.2020.05.036. [Epub ahead of print].
66. Heumuller SE, Talantikite M, Napoli M, Armengaud J, Morgelin M, Hartmann U, et al. C-terminal proteolysis of the collagen VI alpha3 chain by BMP-1 and proprotein convertase(s) releases endotrophin in fragments of different sizes. J Biol Chem. (2019) 294:13769–80. doi: 10.1074/jbc.RA119.008641
67. Zhou XL, Xu H, Liu ZB, Wu QC, Zhu RR, Liu JC. miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1. J Cell Mol Med. (2018) 22:3816–24. doi: 10.1111/jcmm.13654
68. Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H, et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. (2016) 6:2068–83. doi: 10.7150/thno.15007
69. Yu BT, Yu N, Wang Y, Zhang H, Wan K, Sun X, et al. Role of miR-133a in regulating TGF-beta1 signaling pathway in myocardial fibrosis after acute myocardial infarction in rats. Eur Rev Med Pharmacol Sci. (2019) 23:8588–97. doi: 10.26355/eurrev_201910_19175
70. Hong Y, Cao H, Wang Q, Ye J, Sui L, Feng J, et al. MiR-22 may suppress fibrogenesis by targeting TGFbetaR I in cardiac fibroblasts. Cell Physiol Biochem. (2016) 40:1345–53. doi: 10.1159/000453187
71. Liu X, Xu Y, Deng Y, Li H. MicroRNA-223 Regulates cardiac fibrosis after myocardial infarction by targeting RASA1. Cell Physiol Biochem. (2018) 46:1439–54. doi: 10.1159/000489185
72. Dong P, Liu WJ, Wang ZH. MiR-154 promotes myocardial fibrosis through beta-catenin signaling pathway. Eur Rev Med Pharmacol Sci. (2018) 22:2052–60. doi: 10.26355/eurrev_201804_14735
73. Bernardo BC, Nguyen SS, Gao XM, Tham YK, Ooi JY, Patterson NL, et al. Inhibition of miR-154 protects against cardiac dysfunction and fibrosis in a mouse model of pressure overload. Sci Rep. (2016) 6:22442. doi: 10.1038/srep22442
74. Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan L, et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics. (2018) 8:2565–82. doi: 10.7150/thno.22878
75. Vaskova E, Ikeda G, Tada Y, Wahlquist C, Mercola M, Yang PC. Sacubitril/valsartan improves cardiac function and decreases myocardial fibrosis via downregulation of exosomal miR-181a in a rodent chronic myocardial infarction model. J Am Heart Assoc. (2020) 9:e015640. doi: 10.1161/JAHA.119.015640
76. Zhang CJ, Huang Y, Lu JD, Lin J, Ge ZR, Huang H. Upregulated microRNA-132 rescues cardiac fibrosis and restores cardiocyte proliferation in dilated cardiomyopathy through the phosphatase and tensin homolog-mediated PI3K/Akt signal transduction pathway. J Cell Biochem. (2018). doi: 10.1002/jcb.27081
77. Limana F, Esposito G, D'Arcangelo D, Di Carlo A, Romani S, Melillo G, et al. HMGB1 attenuates cardiac remodelling in the failing heart via enhanced cardiac regeneration and miR-206-mediated inhibition of TIMP-3. PLoS ONE. (2011) 6:e19845. doi: 10.1371/journal.pone.0019845
78. Zhao XS, Ren Y, Wu Y, Ren HK, Chen H. MiR-30b-5p and miR-22–3p restrain the fibrogenesis of post-myocardial infarction in mice via targeting PTAFR. Eur Rev Med Pharmacol Sci. (2020) 24:3993–4004. doi: 10.26355/eurrev_202004_20869
79. da Costa Martins PA, Salic K, Gladka MM, Armand AS, Leptidis S, el Azzouzi H, et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol. (2010) 12:1220–7. doi: 10.1038/ncb2126
80. Xiao Y, Zhang Y, Chen Y, Li J, Zhang Z, Sun Y, et al. Inhibition of microRNA-9–5p protects against cardiac remodeling following myocardial infarction in mice. Hum Gene Ther. (2019) 30:286–301. doi: 10.1089/hum.2018.059
81. Lerchenmuller C, Rabolli CP, Yeri A, Kitchen R, Salvador AM, Liu LX, et al. CITED4 protects against adverse remodeling in response to physiological and pathological stress. Circ Res. (2020) 127:631–46. doi: 10.1161/CIRCRESAHA.119.315881
82. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. (2016) 97:245–62. doi: 10.1016/j.yjmcc.2016.06.001
83. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. (2018) 15:387–407. doi: 10.1038/s41569-018-0007-y
84. Guimaraes DA, Dos Passos MA, Rizzi E, Pinheiro LC, Amaral JH, Gerlach RF, et al. Nitrite exerts antioxidant effects, inhibits the mTOR pathway and reverses hypertension-induced cardiac hypertrophy. Free Radic Biol Med. (2018) 120:25–32. doi: 10.1016/j.freeradbiomed.2018.03.006
85. Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. (2015) 89:1401–1438. doi: 10.1007/s00204-015-1477-x
86. Weiss A, Leinwand LA. The mammalian myosin heavy chain gene family. Annu Rev Cell Dev Biol. (1996) 12:417–39. doi: 10.1146/annurev.cellbio.12.1.417
87. Morkin E. Control of cardiac myosin heavy chain gene expression. Microsc Res Tech. (2000) 50:522–31. doi: 10.1002/1097-0029(20000915)50:6andlt;522::AID-JEMT9andgt;3.0.CO;2-U.3.0.CO;2-U
88. Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. (2009) 119:2772–86. doi: 10.1172/JCI36154
89. Tian C, Hu G, Gao L, Hackfort BT, Zucker IH. Extracellular vesicular MicroRNA-27a* contributes to cardiac hypertrophy in chronic heart failure. J Mol Cell Cardiol. (2020) 143:120–31. doi: 10.1016/j.yjmcc.2020.04.032
90. Nishi H, Ono K, Horie T, Nagao K, Kinoshita M, Kuwabara Y, et al. MicroRNA-27a regulates beta cardiac myosin heavy chain gene expression by targeting thyroid hormone receptor beta1 in neonatal rat ventricular myocytes. Mol Cell Biol. (2011) 31:744–55. doi: 10.1128/MCB.00581-10
91. Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, et al. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. (2013) 2:e000078. doi: 10.1161/JAHA.113.000078
92. Yuan W, Tang C, Zhu W, Zhu J, Lin Q, Fu Y, et al. CDK6 mediates the effect of attenuation of miR-1 on provoking cardiomyocyte hypertrophy. Mol Cell Biochem. (2016) 412:289–96. doi: 10.1007/s11010-015-2635-4
93. Zhou J, Gao J, Zhang X, Liu Y, Gu S, Zhang X, et al. microRNA-340–5p functions downstream of cardiotrophin-1 to regulate cardiac eccentric hypertrophy and heart failure via target gene dystrophin. Int Heart J. (2015) 56:454–8. doi: 10.1536/ihj.14-386
94. Huang ZP, Chen J, Seok HY, Zhang Z, Kataoka M, Hu X, et al. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res. (2013) 112:1234–43. doi: 10.1161/CIRCRESAHA.112.300682
95. Fu J, Chen Y, Li F. Attenuation of MicroRNA-495 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. Cardiology. (2018) 139:245–54. doi: 10.1159/000487044
96. Zhou X, Sun F, Luo S, Zhao W, Yang T, Zhang G, et al. Let-7a is an antihypertrophic regulator in the heart via targeting calmodulin. Int J Biol Sci. (2017) 13:22–31. doi: 10.7150/ijbs.16298
97. Wang D, Zhai G, Ji Y, Jing H. microRNA-10a Targets T-box 5 to inhibit the development of cardiac hypertrophy. Int Heart J. (2017) 58:100–6. doi: 10.1536/ihj.16-020
98. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. (2007) 13:613–8. doi: 10.1038/nm1582
99. Liu F, Li N, Long B, Fan YY, Liu CY, Zhou QY, et al. Cardiac hypertrophy is negatively regulated by miR-541. Cell Death Dis. (2014) 5:e1171. doi: 10.1038/cddis.2014.141
100. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. (2012) 3:1078. doi: 10.1038/ncomms2090
101. Li J, Cai SX, He Q, Zhang H, Friedberg D, Wang F, et al. Intravenous miR-144 reduces left ventricular remodeling after myocardial infarction. Basic Res Cardiol. (2018) 113:36. doi: 10.1007/s00395-018-0694-x
102. Huang J, Sun W, Huang H, Ye J, Pan W, Zhong Y, et al. miR-34a modulates angiotensin II-induced myocardial hypertrophy by direct inhibition of ATG9A expression and autophagic activity. PLoS ONE. (2014) 9:e94382. doi: 10.1371/journal.pone.0094382
103. Wu H, Wang Y, Wang X, Li R, Yin D. MicroRNA-365 accelerates cardiac hypertrophy by inhibiting autophagy via the modulation of Skp2 expression. Biochem Biophys Res Commun. (2017) 484:304–310. doi: 10.1016/j.bbrc.2017.01.108
104. Lu Y, Wu F. A new miRNA regulator, miR-672, reduces cardiac hypertrophy by inhibiting JUN expression. Gene. (2018) 648:21–30. doi: 10.1016/j.gene.2018.01.047
105. Hu S, Cheng M, Guo X, Wang S, Liu B, Jiang H, et al. Down-regulation of miR-200c attenuates AngII-induced cardiac hypertrophy via targeting the MLCK-mediated pathway. J Cell Mol Med. (2019) 23:2505–16. doi: 10.1111/jcmm.14135
106. Liu BL, Cheng M, Hu S, Wang S, Wang L, Tu X, et al. Overexpression of miR-142–3p improves mitochondrial function in cardiac hypertrophy. Biomed Pharmacother. (2018) 108:1347–56. doi: 10.1016/j.biopha.2018.09.146
107. Wang L, Qin D, Shi H, Zhang Y, Li H, Han Q. MiR-195–5p promotes cardiomyocyte hypertrophy by targeting MFN2 and FBXW7. Biomed Res Int. (2019) 2019:1580982. doi: 10.1155/2019/1580982
108. Sun D, Li C, Liu J, Wang Z, Liu Y, Luo C, et al. Expression profile of microRNAs in hypertrophic cardiomyopathy and effects of microRNA-20 in inducing cardiomyocyte hypertrophy through regulating gene MFN2. DNA Cell Biol. (2019) 38:796–807. doi: 10.1089/dna.2019.4731
109. Angert D, Houser SR. Stem cell therapy for heart failure. Curr Treat Options Cardiovasc Med. (2009) 11:316–327. doi: 10.1007/s11936-009-0032-6
110. Muller P, Lemcke H, David R. Stem cell therapy in heart diseases - cell types, mechanisms and improvement strategies. Cell Physiol Biochem. (2018) 48:2607–55. doi: 10.1159/000492704
111. Xiong YY, Gong ZT, Tang RJ, Yang YJ. The pivotal roles of exosomes derived from endogenous immune cells and exogenous stem cells in myocardial repair after acute myocardial infarction. Theranostics. (2021) 11:1046–58. doi: 10.7150/thno.53326
112. Johnston KJ, Wen H, Kotwal A, Joynt Maddox KE. Comparing preventable acute care use of rural versus urban americans: an observational study of national rates during 2008–2017. J Gen Intern Med. (2021) 24:3993–4004. doi: 10.1007/s11606-020-06532-4
113. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
114. Gerber MA, Baltimore RS, Eaton CB, Gewitz M, Rowley AH, Shulman ST, et al. Prevention of rheumatic fever and diagnosis and treatment of acute Streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. (2009) 119:1541–51. doi: 10.1161/CIRCULATIONAHA.109.191959
115. Machiraju P, Greenway SC. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J Stem Cells. (2019) 11:33–43. doi: 10.4252/wjsc.v11.i1.33
116. Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. (2012) 485:599–604. doi: 10.1038/nature11139
117. Nam YJ, Lubczyk C, Bhakta M, Zang T, Fernandez-Perez A, McAnally J, et al. Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development. (2014) 141:4267–78. doi: 10.1242/dev.114025
118. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
119. Zhao J, Li X, Hu J, Chen F, Qiao S, Sun X, et al. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc Res. (2019) 115:1205–16. doi: 10.1093/cvr/cvz040
120. Ma T, Chen Y, Chen Y, Meng Q, Sun J, Shao L, et al. MicroRNA-132, Delivered by mesenchymal stem cell-derived exosomes, promote angiogenesis in myocardial infarction. Stem Cells Int. (2018) 2018:3290372. doi: 10.1155/2018/3290372
121. Song Y, Zhang C, Zhang J, Jiao Z, Dong N, Wang G, et al. Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics. (2019) 9:2346–60. doi: 10.7150/thno.29945
122. Charles CJ, Li RR, Yeung T, Mazlan SMI, Lai RC, de Kleijn DPV, et al. Systemic mesenchymal stem cell-derived exosomes reduce myocardial infarct size: characterization with mri in a porcine model. Front Cardiovasc Med. (2020) 7:601990. doi: 10.3389/fcvm.2020.601990
123. Lazar E, Benedek T, Korodi S, Rat N, Lo J, Benedek I. Stem cell-derived exosomes - an emerging tool for myocardial regeneration. World J Stem Cells. (2018) 10:106–115. doi: 10.4252/wjsc.v10.i8.106
124. Li Y, Yu H, Zhao L, Zhu Y, Bai R, Jin Z, et al. Effects of carbon nanotube-mediated Caspase3 gene silencing on cardiomyocyte apoptosis and cardiac function during early acute myocardial infarction. Nanoscale. (2020) 12:21599–604. doi: 10.1039/D0NR05032F
Keywords: MI, apoptosis, hypertrophy, fibrosis, miRNA
Citation: Wang W and Zheng H (2021) Myocardial Infarction: The Protective Role of MiRNAs in Myocardium Pathology. Front. Cardiovasc. Med. 8:631817. doi: 10.3389/fcvm.2021.631817
Received: 21 November 2020; Accepted: 08 February 2021;
Published: 05 March 2021.
Edited by:
Sarawut Kumphune, Chiang Mai University, ThailandReviewed by:
Junjie Yang, University of Alabama at Birmingham, United StatesCopyright © 2021 Wang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Zheng, emhlbmdoYW82MTY5QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.