
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
POLICY AND PRACTICE REVIEWS article
Front. Cardiovasc. Med., 04 February 2021
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.604434
This article is part of the Research TopicHighlights in Cardiovascular Biologics and Regenerative Medicine: 2021View all 8 articles
 Paloma Gastelurrutia1,2,3*
Paloma Gastelurrutia1,2,3* Cristina Prat-Vidal1,2,3
Cristina Prat-Vidal1,2,3 Joaquim Vives4,5,6
Joaquim Vives4,5,6 Ruth Coll4
Ruth Coll4 Antoni Bayes-Genis2,3,7,8
Antoni Bayes-Genis2,3,7,8 Carolina Gálvez-Montón2,3
Carolina Gálvez-Montón2,3A systematic and ordered product development program, in compliance with current quality and regulatory standards, increases the likelihood of yielding a successful advanced therapy medicinal product (ATMP) for clinical use as safe and effective therapy. As this is a novel field, little accurate information is available regarding the steps to be followed, and the information to be produced to support the development and use of an ATMP. Notably, successful clinical translation can be somewhat cumbersome for academic researchers. In this article, we have provided a summary of the available information, supported by our experience in Spain throughout the development of an ATMP for myocardial infarction, from the pre-clinical stage to phase I clinical trial approval.
Recent years have brought tremendous developments in cardiac tissue engineering (1). Cell therapy approaches have been attempted for the repair of scars left by myocardial infarction (MI); however, clinical trials have reported only modest and clinically irrelevant improvements in cardiac function (2–4). Thus, the research focus has shifted to cardiac tissue engineering approaches that can deliver cells to the injured myocardium with supportive scaffolds, thus improving cell retention and survival (5). Successful clinical translation of these engineered constructs requires that specific steps be followed from scientific, quality, and regulatory standpoints. First, they have to be conceived and developed in a basic research laboratory. Second, they need to be confirmed as safe and effective in relevant animal models. Third, they must be adapted to current good manufacturing practice (GMP)-compliant procedures for obtaining an advanced therapy medicinal product (ATMP). Finally, the ATMP has to be tested in patients in the context of a clinical trial.
In 2003, the concept of ATMP was defined and introduced in European legislation. ATMP originally comprised gene therapy medicinal products (GTMPs) and somatic cell therapy medicinal products (CTMPs), with the later inclusion of tissue-engineered products (TEPs) in Regulation (EC) No. 1394/2007 (6, 7). The ATMP industry is still emerging, and many therapy proposals in this field are led by independent researchers who have little, if any, process development experience (8).
ATMPs are ground-breaking medicines based on the use of genes, tissues, or cells for the treatment of human disease and injury. They are classified into four main types: GTMPs, CTMPs, TEPs, and combined ATMPs. GTMPs contain genes that have therapeutic, prophylactic, or diagnostic effects. CTMPs include cells or tissues that have been substantially manipulated to change their biological characteristics, physiological functions, or structural properties, or that are intented to be used for a purpose other than their essential functions in the body, with the aim of treating, preventing, or diagnosing a disease. TEPs contain modified cells or tissues of human and/or animal origin (viable or non-viable) that are used to repair, regenerate, or replace human tissue (9). Finally, combined ATMPs contain a medical device as an integral part of the product (10). They may also contain additional substances, such as biomaterials, chemicals, scaffolds, or matrices. The mechanisms of action of ATMPs include pharmacological, immunological, or metabolic activities (10).
Due to the novelty and complexity of ATMPs, there is a lack of accurate information regarding the steps required for their clinical translation, and data that must be produced to achieve regulatory agency approval. This can make it difficult for independent researchers from an academic environment to successfully get an ATMP to the patient population. Investigational medicinal product (IMP) development can be challenging in many aspects, including materializing a research idea, cell/tissue isolation and characterization, preclinical studies to prove safety and efficacy, scaling-up prototypes for human use, defining manufacturing processes in accordance with current GMP, and meeting all ethics and regulatory requirements (10–14). Our group recently developed a pioneering ATMP, named PeriCord, that is presently being tested in humans for cardiac repair after MI. PeriCord comprises a human decellularized pericardial matrix enriched with Wharton's jelly-derived mesenchymal stromal cells (15). Here, we report our experience in creating this IMP in Spain, and through its translation from preclinical stages to use in patients (15).
Basic research laboratories can generate regenerative/repair-driven hypotheses and test them in animal models. To achieve clinical translation, a newly developed ATMP must be proven safe and effective in a planned preclinical program including studies addressing pharmacodynamics (PD), pharmacokinetic (PK), and toxicology (Tox) (16, 17). For a product to be proven safe, the risk-benefit profile must be assessed as acceptable for the target disease or condition, the product must be well-characterized to reduce batch variability and ensure the expected function, and the genetic stability must be confirmed. Accurate characterization of the product is also important for avoiding differences in effectivity, ensuring consistency from dose to dose (11). Thus, as soon as possible, it is important to define the identity, viability, purity, and potency of the ATMP.
Experiments in animal models should be conducted to evaluate the biodistribution of an ATMP in principal organs (e.g., brain, liver, spleen, lungs, kidneys, and gonads), to identify target organs and tissues (18), and to assess local effects. Studies must review and confirm the expected mechanism of action of the product's active ingredient, i.e., the biologically active ingredient that exerts the expected therapeutic effect. Animal experiments should also reveal an absence of tumorigenic activity and an acceptable safety profile. In terms of effectiveness, animal models may prove efficacy by confirming the hypothesis at the preclinical level. In some cases, no animal model is available for the specific pathology under investigation. In this case, a new model can be designed if the basic research group has sufficient experience in the pre-clinical large animal setting, and can fine-tune a model to mimic a human pathological scenario. However, sometimes a disease cannot be reproduced due to its high complexity or mortality. In this scenario, pre-clinical studies can be performed using different animal models that present the different symptoms separately.
Prior to administration to a patient, the product must comply with the above-described requirements, as well as others.
We refer as the translational stage, the phase during which the researchers have a proof of concept and are ready to start working to develop a product for clinical use. Several considerations must be taken into account for products intended for human use:
1. The preclinical product must be adapted to fulfill the regulatory and legal requirements for human use. To this end, the ATMP must be manufactured following procedures in compliance with current GMP standards and in GMP-accredited facilities. If the preclinical product was not originally produced using GMP procedures and/or clinical grade reagents, it should be modified. Due to the potential impact on the final product's safety and quality, ATMP developers must make available the quality attributes and composition of all raw materials used in the manufacturing process. Importantly, this information is required for the regulatory submission, especially when animal-derived components are used. Any raw materials and excipients must be suitable for clinical use, and some may have to be substituted for others with similar characteristics upon appropriate risk analysis and experimental testing (e.g., xeno-free, clinical grade ingredients) (19), which may be more expensive. Additionally, sometimes extensive ex vivo expansion is required to yield sufficient cell numbers for clinical use, and scale-up processes should be defined and incorporated within the manufacturing procedure to adapt doses and/or final product dimensions to human scale (20).
2. For autologous cell products, “scaling-out” usually requires the ability to conduct multiple parallel bioprocesses by using traditional planar 2D cultures for the production of larger amounts of finished product (20, 21). For unmatched allogeneic products, “scaling-up” involves the use of expansion platforms, such as bioreactors, to entail reproducible and robust manufacturing bioprocess, therefore offering major advantages with respect to GMP implementation.
Some specific documents must be generated to collect all of the information required by the competent authorities for their evaluation and approval of the IMP, and their approval of a clinical trial together with the local ethics committee. These aspects will be detailed in the section ATMP approval for clinical trials.
Several issues must be addressed prior to the actual production of an ATMP.
1. GMP compliance: The ATMP manufacturing facilities must comply with current GMP, thus guaranteeing the quality and safety of the manufactured products. This accreditation is granted by the competent authorities, e.g., the AEMPS in Spain. GMP is a quality management system to ensure that products are consistently processed and controlled according to quality standards. This system guarantees traceability at all times, covering every aspect of manufacturing and distribution. GMP covers all aspects of production, from the starting materials, premises, and equipment to staff training and personal hygiene. It is essential to have detailed written procedures (standard operating procedures; SOP) for each process that could affect the finished product quality, which are followed each time a product is manufactured (22). Quality control documents are required to register all utilized processes and materials, including batch records and expiration dates. The products commonly used in preclinical studies are often unsuitable for clinical use. For some such ingredients, excipients, or culture reagents, there is an equivalent, generally more expensive, GMP-compliant alternative. However, other materials may be more difficult to replace. For example, bovine serum has been widely used in cell culture for over a century (23), but the current trend is to switch from the use of serum or platelet lysate to serum-free chemically defined media formulations to alleviate concerns regarding safety (i.e., xenoviral transfer and contamination), efficacy (batch-to-batch variation), and scarcity (11, 20, 23).
2. Reproducibility, safety, and consistency: There are several critical process stages for assuring consistency and meeting the established specifications of the finished drug product (DP)—including tissue harvesting, cell isolation and expansion, cell and finished product storage, lot size (number of cells per production batch), and methods. The manufacturing bioprocess is designed to minimize the risks of deviating from specifications along the production process, such that there is no need to solely rely on testing of the finished product. This is known as “quality by design (QbD),” an approach that aims to ensure product quality by combining statistical, analytical, and risk-management methodologies into the design, development, and manufacturing processes (24– 26). Acceptance and release criteria must be determined to ensure the safety and consistency of the final DP, with regards to microbiological growth (negative serological and microbiological testing), physiochemical properties, dose range, cell viability, and endotoxin levels, among other concerns.
3. Scalability: Process scalability is a major factor to consider when starting from a preclinical product and aiming to develop its corresponding IMP format. For translational allogeneic cell therapy that requires cells, reagents, and culture media, large numbers of cells are needed to achieve a certain dose ready for use in several patients, thus affecting product cost and quality (17). The cells must retain their key quality attributes of identity, potency, purity, and safety, and there must be no changes in quality with regards to cell differentiation capability (11). Quality by design strategies and classical experimental design approaches can be applied to optimize cell culture media, reagents, and process parameters to create a robust, reproducible, and scalable process (11, 27).
4. Stability: The DP stability period must be determined based on cell stability and the product life cycle (28). Cells can multiply, but do not have to multiply continuously, only when needed for their normal cycle. When they multiply, they must maintain a stable karyotype and stable expression of cytokine markers and telomerase. The product life cycle is the process a product goes through once it is released until it is removed upon reaching its expiration date. This shelf-life is determined based on changes in the product characteristics, and is assessed in the preclinical stage and during product development, and might be reassessed during the clinical trial phase according to the product stability protocols.
All of these factors must be considered to manufacture a clinically appropriate product adapted from the preclinical version. The end results must be a comparable product in terms of identity, purity, potency, and safety (29).
After the preclinical and translational stages, and upon achieving ATMP manufacturing and validation, all required information must be presented to the competent authority and final approval obtained to commence a clinical trial. Here we summarize all of the documentation required for presentation to the Spanish Agency of Medicines and Medical Devices (AEMPS):
1. Target Product Profile (TPP): This is not a mandatory document in Spain, but rather a summary of the necessary information about the product development process. A TPP is created with the aim of ensuring that the drug development process is efficient and provides all of the required medical, technical, and scientific information about a product (30). It can be worthwhile to start the TPP at an early stage (preclinical stage) to provide a clear vision of the whole process.
2. Investigational Medicinal Product Dossier (IMPD): The IMPD, including modules 1 to 5 (described below), is one of the main IMP-related documents required for clinical trial approval in one or more European Union Member States. The IMPD includes regional administrative information and summaries of the data related to the quality, manufacture, and control of any IMP (including both reference product and placebo), as well as data from non-clinical and clinical studies (Figure 1).
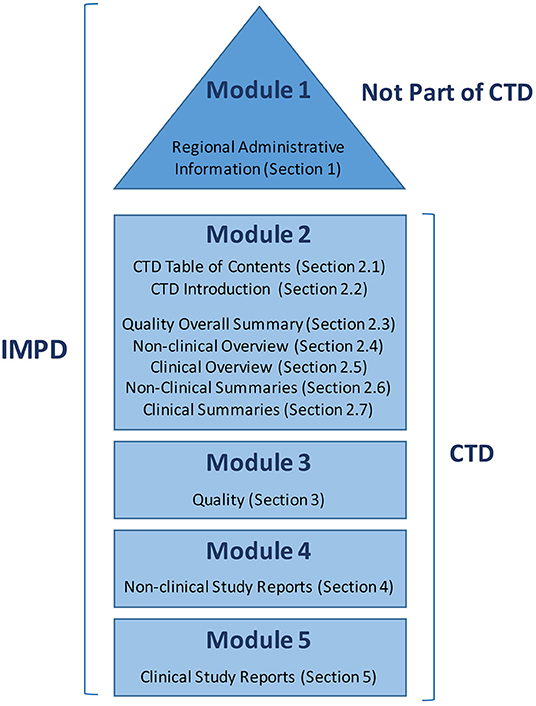
Figure 1. Parts of the Investigational Medicinal Product Dossier (IMPD) as collected in the Notice to Applicants prepared by the European Commission.
The AEMPS does not offer guidance on writing an IMPD, but suggests following the European Guidance from the European Medicines Agency (EMA) (31). Figure 1 shows the parts of the IMPD as summarized in a Notice to Applicants (32) prepared by the European Commission in consultation with competent authorities of the member states, the EMA, and other interested parties as a guideline for fulfilling the obligations raised by Article 6 of Regulation (EC) No. 726/2004, and with respect to Annex I of Directive 2001/83/EC (European Commission Notice to Applicants). Based on this Notice to Applicants, we adapted the index for our IMP, as shown in Table 1.
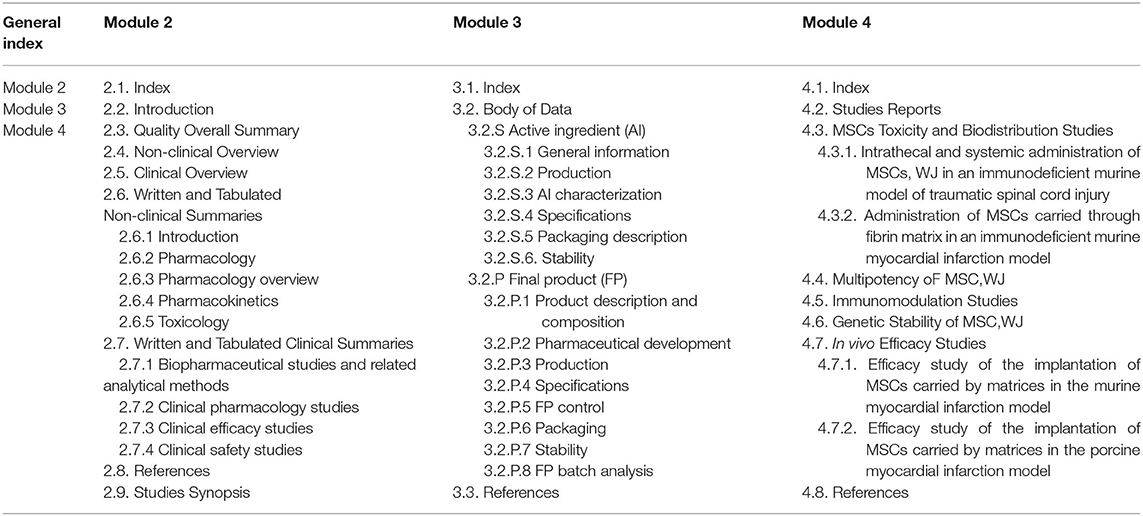
Table 1. Investigational medicinal product dossier (IMPD) index adapted from the Notice to applicants by the European Commission.
The IMPD includes the following modules:
- Module 1: This section is not a part of the Common Technical Document (CTD), and includes all regional administrative information required by the AEMPS.
- Module 2: This module provides an overview of all of the information, divided into several sections, including a table of contents (2.1), introduction (2.2), review of the quality information (2.3), review of the non-clinical data (2.4), review of the clinical data (2.5), and two tabulated summaries: one for the non-clinical data (2.6) and one for the clinical data (2.7).
- Module 3: This section is a compendium of the available quality information, which will also be sent as a discrete document in Block I of the required documentation for the Ethics Committee and AEMPS evaluation (see below). The document includes data regarding the characterization, fabrication, specifications, stability, packaging, and quality control analysis of the active ingredient and the finished product (i.e., the ATMP).
- Module 4: This module constitutes a compendium of the available non-clinical data, which includes toxicity and biodistribution studies, mechanism of action, and safety data. For TEPs, this includes information concerning the genetic stability of the active cells, and in vivo efficacy studies.
- Module 5: This non-compulsory module includes clinical study reports, when available.
This structure indicates the necessary experiments for gathering all of the required data in terms of quality and fabrication (Module 2, section Quality overall summary, and Module 3), as well as the pharmacological mechanism of action, pharmacokinetics, toxicity, biodistribution, and safety (Module 4). The IMPD serves as an excellent tool for defining all necessary preclinical studies, and for aligning manufacturing process requirements with product specifications (11).
3. Common Technical Document (CTD): The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) has the mission of bringing together regulatory authorities and pharmaceutical industries worldwide to ensure that safe, effective, and high-quality medicines are developed and registered in the most resource-efficient manner (33). Toward this aim, they created a common format, the CTD (Figure 1), to gather information regarding quality, safety, and efficacy. Module 1 is region-specific, while Modules 2, 3, 4, and 5 are intended to be common among all regions—as has been mandatory since July 2003 in the EU and Japan, and is the strongly recommended format of choice in the United States.
4. Protocol: The protocol describes how the clinical trial will be conducted, and is the most important document for clinical trial execution. The protocol must include a title; a protocol number, version number, and date; the study objective and justification, containing all relevant previous clinical and preclinical data; the study design; candidate inclusion and exclusion criteria; intervention or treatment; risks and benefits, including adverse reaction definition and reporting; expected trial start and end dates; study withdrawal and blind-opening, if applicable; data management, and statistical analysis; ethical and regulatory considerations; document archive and font documents; quality control; funding; publication policy; references; and annexes. It is highly recommended that this document include a figure explaining the study design and a table detailing the scheduled study visits.
5. Informed consent form: Informed consent is the procedure guaranteeing that a subject gives consent to participate in a research study, having and understanding all of the information related to the trial objectives, its possible benefits, the risks and inconveniences, the available alternatives, and his/her rights and responsibilities. The candidate subject must have time to think and ask any necessary questions. All of the provided information must be collected in a document with a version number updated with current legislation, and two printed copies must be signed, dated, and kept by the investigator and the patient.
6. Investigator's Brochure (IB): This document is addressed to the researchers involved in the trial, providing a compilation of the clinical and non-clinical data regarding the product, and the reference safety information. The IB includes all of the IMP-related information from the IMPD that the investigator will need to correctly manage the IMP and the included subjects, ensuring their understanding of the study rationale, IMP dose, dose range, administration, and safety monitoring parameters. The IB should include a title page; summary; table of contents; introduction; section describing the product's physical, chemical, and pharmaceutical properties; non-clinical studies (pharmacology pharmacokinetics, and toxicology); effects in humans (pharmacokinetics, safety, and efficacy); and a summary for the investigator. If an event that is not listed in this document occurs during the trial, the investigator must consider it a non-expected adverse event, and must notate it as such. The contents are detailed in the guideline EMA/CHMP/ICH/135/1995 and the required information is obtained from the IMPD (34).
The AEMPS is the national regulatory agency in Spain. There are several different ways of corresponding with the AEMPS. First, for preliminary specific questions related to the proposed study, enquiry e-mails can be sent to the Support Office for Independent Clinical Research, Clinical Trials Division, Pharmaceutical Medicines for Human Use Department.
Second, in the early stages, when considering moving toward the clinical phase, scientific advice can be sought. Scientific advice meetings are face-to-face sessions at AEMPS facilities, at which researchers present their intentions and the questions that they want to discuss with experts from the regulatory agency. These professionals act as assessors for the investigational team, and include representatives from different areas, such as the Division of Biological Products, Advanced Therapies and Biotechnology, and Department of Medical Devices. After the meeting, correspondence occurs between the experts and the Quality Research Department, Pharmacology and Clinical Assessment Division, Medicines for Human Use Department, to whom minutes of the meeting must be sent. Figure 2 presents a template for summarizing meeting minutes. This document is revised and accepted by the AEMPS, and will be part of the collected documentation required to obtain regulatory approval for the clinical trial.

Figure 2. Template to write minutes after obtaining scientific advice from the Spanish Agency of Medicines and Medical Devices (AEMPS).
Third, the documentation is submitted via the AEMPS platform (https://ecm.aemps.es/). Figure 3 shows the list of documents that must be uploaded to the platform (35). The evaluation is separated into two main blocks. Block I includes the documents that must be sent to the AEMPS and to the local ethics committee. Block II comprises documents to be sent exclusively for local ethics committee evaluation. The AEMPS must validate Block I, and the local ethics committee must validate Blocks I and II within a maximum of 10 natural days (Figure 4) (36). Both organizations can request rectifications and/or clarifications and the process can be extended over time. Upon approval by the AEMPS and the local ethics committee, the clinical trial must be publicly registered in the Spanish Clinical Trial Registry (REEC, from Registro Español de Estudios Clínicos; https://reec.aemps.es/).

Figure 3. List of documents to upload to the Spanish Agency of Medicines and Medical Devices (AEMPS) platform for investigational medicinal product (IMP) approval.
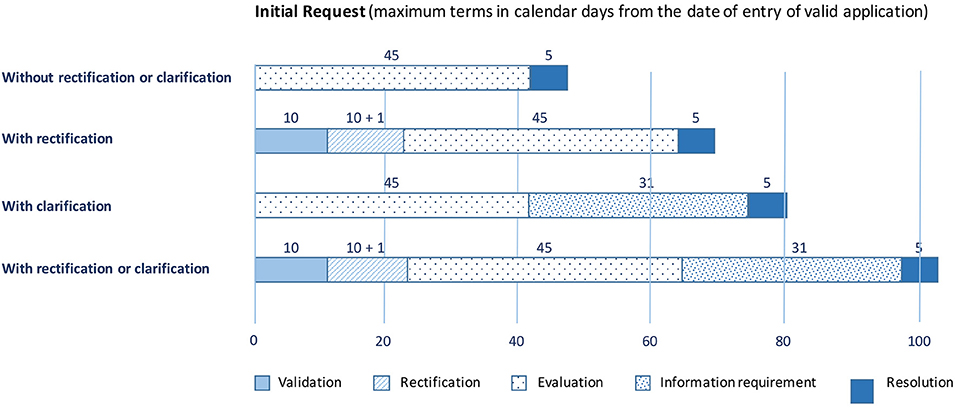
Figure 4. Due dates after Spanish Agency of Medicines and Medical Devices (AEMPS) document submission.
The European Regulation on ATMPs has introduced a classification procedure that gives developers the opportunity to verify whether their product can be considered an ATMP. This procedure is optional, free of charge, and may occur at any stage of ATMP development prior to applying for marketing authorization. There is also the possibility of briefing meetings for preparing ATMP classification, which are organized by the EMA Innovation Task Force and represent a starting point for interactions between the European agency and the ATMP developers (6), enabling the EMA to provide scientific advice. However, AEMPS decisions are harmonized with decision by the EMA, and it is not necessary to seek both.
After local regulatory approval, the clinical trial should be registered in the European clinical trials database (EudraCT, https://eudract.ema.europa.eu/eudract-web/index.faces) to allow public consultation of updated study-related information.
Our group is a multidisciplinary team—including basic researchers (biologists, biochemists, and veterinarians) and clinical researchers (physicians and pharmacists). This diversity may not be common in basic research groups, which may need to look outside of their group for a clinical perspective early in the translation process. Notably, despite being a quite large multidisciplinary group, we still had to seek a partner with broad experience in production and product quality. This is why we created a consortium with a center that had the knowledge that we were lacking, had GMP procedures established in their daily activity, and possessed all of the facilities needed for product production.
We had studied cardiac regeneration as a basic research group for almost 10 years. Then we spent over a year to adapt our product and our processes to GMP and, together with our partner center, we generated the IMPD. The optimum approach is to start planning the full process from the very beginning, when a research idea is generated, with the involvement of all experts. Considering the development of an investigational product as a project enables early planning of a scheme that includes preclinical and in-vivo animal models and quality aspects, along with bioprocess aspects, as well as compliance with the GMP and legal framework requirements (16). This approach is important to ensure appropriate design of the non-clinical package required for a clinical trial—helping to avoid skipping process steps; setting overly optimistic deadlines; underestimating the financial, time, and personnel requirements; and performing studies without proper documentation (16). Figure 5 depicts the whole process for clinical translation. We performed a lot of work without having considered all GMP requirements, which resulted in a longer overall process. We are now aware of these issues for future projects that we hope to be developing together as a consortium.
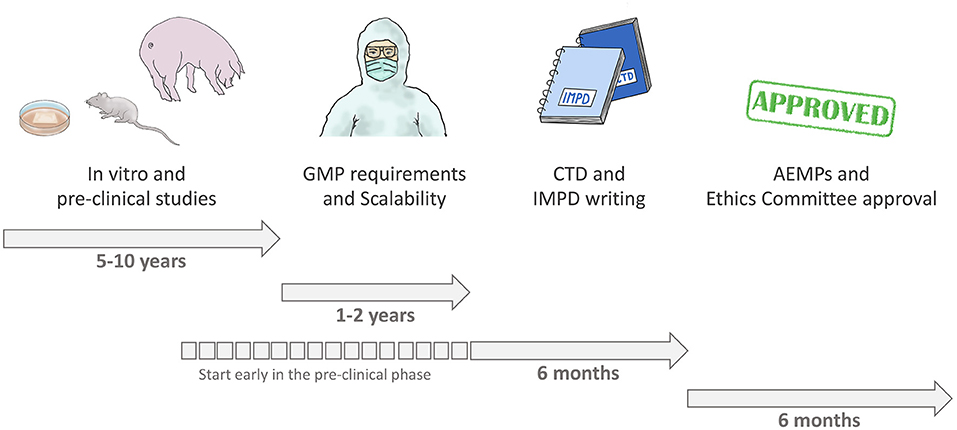
Figure 5. Process of clinical translation.
In conclusion, basic research laboratories can generate innovative regenerative/repair-driven hypotheses; however, preclinical products must go through a complex process of translational adaptation before reaching patients. Intentions should be discussed with the competent authorities as early as possible, as they may guide researchers to avoid possible errant pathways that would result in great losses of time and money. When intending to assess an ATMP in a clinical trial, the IMPD is the most complex and important document to develop. Its structure provides researchers with guidance regarding the preclinical data needed to generate a safe and effective IMP. Key steps along the process include scalability and GMP manufacturing, along with animal models to perform PD, PK, and Tox studies.
PG and CG-M conceived the presented idea. PG wrote the manuscript with support from CG-M, CP-V, JV, and RC. CG-M, JV, and AB-G performed a critical revision of the manuscript. All authors gave final approval of the version to be published.
This work was supported by the Spanish Cell Therapy Network (TerCel) [RD16/0011/0006 and RD16/0011/0028]; and CIBER Cardiovascular [CB16/11/00403] projects, as a part of the National R&D&I Plan, and was co-funded by the ISCIII-Sub-Directorate General for Research Assessment and Promotion and the European Regional Development Fund (ERDF); the Government of Catalonia: Agency for Management of University and Research Grants (AGAUR) [2017-SGR-483 and 2017-SGR-719], PERIS Program (Health Department) [SLT002/16/00234 and SLT002/16/00209] and CERCA Program; La Caixa Banking Foundation; grants from the Spanish Ministry of Science, Innovation and Universities (MICINN) [SAF2017-84324-C2-1-R]; Institute of Health Carlos III (ISCIII) [PI18/00256, PIC18/00014, ICI19/00039, and PI19/01788], and cofunded by European Union (ERDF/ESF) - A way to build Europe. This work has also been funded by the Sociedad Española de Cardiología and by the Call for Expression of Interest (EoI) for Collaborative Projects on Regenerative Medicine 2019 of Center for Regenerative Medicine of Barcelona (CMR[B]). The funders of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to acknowledge members of both the Cell Therapy Service and Barcelona Tissue Bank (Banc de Sang i Teixits, Barcelona), and members of the ICREC Research Lab for their support. We would also like to acknowledge the invaluable help and support of the AEMPS.
AEMPS, Spanish Agency of Medicines and Medical Devices; AEMPS, Agencia Española del Medicamento y Producto Sanitario; ATMP, Advanced Therapy Medicinal Product; MTA, Medicamento de Terapia Avanzada; CTD, Common Technical Document; IMP, Investigational Medicinal Product; PEI, Producto En fase de Investigación; IMPD, Investigational Medicinal Product Dossier; EPEI, Expediente de Producto En fase de Investigación; TPP, Target Product Profile; FDP, Ficha de Desarrollo del Producto.
1. Jarrell DK, Vanderslice EJ, VeDepo MC, Jacot JG. Engineering myocardium for heart regeneration-advancements, considerations, and future directions. Front Cardiovasc Med. (2020) 7:586261. doi: 10.3389/fcvm.2020.586261
2. Gyöngyösi M, Haller PM, Blake DJ, Martin Rendon E. meta-analysis of cell therapy studies in heart failure and acute myocardial infarction. Circ Res. (2018) 123:301–08. doi: 10.1161/CIRCRESAHA.117.311302
3. Fish KM, Ishikawa K, Hajjar RJ. Stem cell therapy for acute myocardial infarction: on the horizon or still a dream? Coron Artery Dis. (2018) 29:89–91. doi: 10.1097/MCA.0000000000000589
4. Nguyen PK, Rhee JW, Wu JC. Adult stem cell therapy and heart failure, 2000 to 2016: a systematic review. JAMA Cardiol. (2016) 1:831–41. doi: 10.1001/jamacardio.2016.2225
5. Gálvez-Montón C, Prat-Vidal C, Roura S, Soler-Botija C, Bayes-Genis A. Update: Innovation in cardiology (IV). Cardiac tissue engineering and the bioartificial heart. Rev Esp Cardiol. (2013) 66:391–9. doi: 10.1016/j.recesp.2012.11.013
6. Voltz-Girolt C1, Celis P, Boucaumont M, D'Apote L, Pinheiro MH, Papaluca-Amati M. The advanced therapy classification procedure. Overview of experience gained so far. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. (2011) 54:811–5. doi: 10.1007/s00103-011-1309-y
7. The European Parliament and the Council of the European Union (2007). Regulation. (EC) No. 1394/2007. OJ L324, 121–37.
8. Cuende N, Rasko JEJ, Bckoh M, Dominici M, Konomou L. Cell, tissue and gene products with marketing authorization in 2018 worldwide. Cytotherapy. (2018) 20:1401–3. doi: 10.1016/j.jcyt.2018.09.010
9. Advanced Therapy Medicinal Products: Overview. Available online at: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview (accessed July 15, 2019).
10. Adopted Reflection Paper on Classification of ATMPs Rev.1 (08/06/2015). Available online at: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-classification-advanced-therapy-medicinal-products_en-0.pdf (accessed July 15, 2019).
11. Campbell A, Brieva T, Raviv L, Rowley J, Niss K, Brandwein H, et al. Concise review: process development considerations for cell therapy. Stem Cells Transl Med. (2015) 4:1155–63. doi: 10.5966/sctm.2014-0294
12. Borán T, Menezes-Ferreira M, Reischl I, Celis P, Ferry N, Gänsbacher B, et al. Clinical development and commercialization of advanced therapy medicinal products in the European Union: how are the product pipeline and regulatory framework evolving? Hum Gene Ther Clin Dev. (2017) 28:126–35. doi: 10.1089/humc.2016.193
13. Schneider CK, Salmikangas P, Jilma P, Flamion B, Todorova LR, Paphitou A, et al. Committee for advanced therapies (cat). Challenges with advanced therapy medicinal products and how to meet them. Nat Rev Drug Discov. (2010) 9:195–201. doi: 10.1038/nrd3052
14. Shukla V, Seoane-Vazquez E, Fawaz S, Brown L, Rodriguez-Monguio R. The landscape of cellular and gene therapy products: authorization, discontinuations, and cost. Hum Gene Ther Clin Dev. (2019) 30:102–13. doi: 10.1089/humc.2018.201
15. Prat-Vidal C, Rodríguez-Gómez L, Aylagas M, Nieto-Nicolau N, Gastelurrutia P, Agustí E, et al. First-in-human PeriCord cardiac bioimplant: scalability and GMP manufacturing of an allogeneic engineered tissue graft. EBioMed. (2020) 15:102729. doi: 10.1016/j.ebiom.2020.102729
17. Vives J, Oliver-Vila I, Pla A, Quality compliance in the shift from cell transplantation to cell therapy in non-pharma environments. Cytotherapy. (2015) 17:1009–14. doi: 10.1016/j.jcyt.2015.02.002
18. Reyes-Moreno B, Coca M, Codinach M, López-Lucas Md, Del Mazo A, Oliver-Vila I, et al. Assessment of biodistribution using mesenchymal stromal cells: algorithm for study design and challenges in detection methodologies. Cytotherapy. (2017) 19:1060–9. doi: 10.1016/j.jcyt.2017.06.004
19. Vives J, Blanco M, Caminal M, Coca M, Codinach M, Coll R, et al. Development of an advanced cell therapy product indicated for the treatment of gonarthrosis. BMC Proc. (2015) 9(Suppl 9):O9. doi: 10.1186/1753-6561-9-S9-O9
20. García-Fernández C, Lopez-Fernandez A, Borrós S, Lecina M, Vives J. Strategies for large-scale expansion of clinical-grade human multipotent mesenchymal stromal cells. Biochem Eng J. (2020) 159:107601. doi: 10.1016/j.bej.2020.107601
21. Cherian DS, Bhuvan T, Meagher L, Heng TSP. Biological considerations in scaling up therapeutic cell manufacturing. Front Pharmacol. (2020) 11:654. doi: 10.3389/fphar.2020.00654
22. International Society for Pharmaceutical Engineering. Good Manufacturing Practice (GMP) Resources. Available online at: https://ispe.org/initiatives/regulatory-resources/gmp (accesed March 19, 2020).
23. Brindley DA, Davie NL, Culme-Seymour EJ, Mason C. Peak serum: implications of serum supply for cell therapy manufacturing. Regen Med. (2012) 7:7–13. doi: 10.2217/rme.11.112
24. Rathore AS. Roadmap for implementation of quality by design (QbD) for biotechnology products. Trends Biotechnol. (2009) 27:546–53. doi: 10.1016/j.tibtech.2009.06.006
25. Rathore AS, Winkle H. Quality by design for biopharmaceuticals. Nat Biotechnol. (2009) 27:26–34. doi: 10.1038/nbt0109-26
26. Del Mazo-Barbara A, Nieto V, Mirabel C, Reyes B, García-López J, Oliver-Vila I, Vives J. Streamlining the qualification of computerized systems in GxP-compliant academic cell therapy facilities. Cytotherapy. (2016) 18:1237–9. doi: 10.1016/j.jcyt.2016.06.003
27. Agency European Medicines. Ich Guideline Q8 (R2) on Pharmaceutical Development. Vol. EMA/CHMP/ICH/167068/2004. London (2017).
28. Mirabel C, Puente-Massaguer E, Del Mazo-Barbara A, Reyes B, Morton P, Gòdia F, Vives J. Stability enhancement of clinical grade multipotent mesenchymal stromal cell-based products. J Transl Med. (2018) 16:291. doi: 10.1186/s12967-018-1659-4
29. Demonstration of Comparability of Human Biological Products Including Therapeutic Biotechnology-Derived Products. Available online at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/demonstration-comparability-human-biological-products-including-therapeutic-biotechnology-derived (accessed August 1, 2019).
30. Curry S, Brown R. The Target Product Profile as a Planning Tool in Drug Discovery Research. Business Briefing: PharmaTech 2003, World Markets Research Centre (2003), p. 67–71.
31. European Investigational Medicinal Product Dossiers. Available online at: http://www.imp-dossier.eu/ (accessed July 3, 2019).
32. European Commission Notice to Applicants for Medicinal products for human use. Volume 2B. Presentation and Format of the Dossier Common Technical Document (CTD). Available online at: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf (accessed August 1, 2019).
33. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Available online at: https://www.ich.org/page/mission (accessed March 19, 2020).
35. Anexo I. Documentación del ensayo e identificación de los documentos al cargarlos en el Portal ECM – Aemps. Available online at: https://www.aemps.gob.es/investigacionClinica/medicamentos/docs/anexo1-Ins-AEMPS-EC.pdf (accessed April 20, 2020).
36. Documento de instrucciones de la Agencia Española de Medicamentos y Productos Sanitarios para la realización de ensayos clínicos en España Versión 10 de 17 de diciembre de 2018. Available online at: https://www.aemps.gob.es/investigacionClinica/medicamentos/docs/Instrucciones-realizacion-ensayos-clinicos.pdf (accesed August 2, 2019).
Keywords: advanced therapy medicinal product, investigational medicinal product, investigational medicinal product dossier (IMPD), translational (regulation), regulatory agency
Citation: Gastelurrutia P, Prat-Vidal C, Vives J, Coll R, Bayes-Genis A and Gálvez-Montón C (2021) Transitioning From Preclinical Evidence to Advanced Therapy Medicinal Product: A Spanish Experience. Front. Cardiovasc. Med. 8:604434. doi: 10.3389/fcvm.2021.604434
Received: 09 September 2020; Accepted: 04 January 2021;
Published: 04 February 2021.
Edited by:
Kim Connelly, St. Michael's Hospital, United KingdomReviewed by:
Manuel M. Mazo, University of Navarra, SpainCopyright © 2021 Gastelurrutia, Prat-Vidal, Vives, Coll, Bayes-Genis and Gálvez-Montón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paloma Gastelurrutia, cGdhc3RlbHVycnV0aWFAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.