 Calum J. McMullen
Calum J. McMullen Susan Chalmers
Susan Chalmers Rachel Wood
Rachel Wood Margaret R. Cunningham
Margaret R. Cunningham Susan Currie
Susan Currie
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 01 February 2021
Sec. Cardio-Oncology
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.630480
This article is part of the Research TopicAnti-Cancer Drug-Induced CardiotoxicityView all 15 articles
Background: Tyrosine kinase inhibitors (TKIs) have dramatically improved cancer treatment but are known to cause cardiotoxicity. The pathophysiological consequences of TKI therapy are likely to manifest across different cell types of the heart, yet there is little understanding of the differential adverse cellular effects. Cardiac fibroblasts (CFs) play a pivotal role in the repair and remodeling of the heart following insult or injury, yet their involvement in anti-cancer drug induced cardiotoxicity has been largely overlooked. Here, we examine the direct effects of sunitinib malate and imatinib mesylate on adult rat CF viability, Ca2+ handling and mitochondrial function that may contribute to TKI-induced cardiotoxicity. In particular, we investigate whether Ca2+/calmodulin dependent protein kinase II (CaMKII), may be a mediator of TKI-induced effects.
Methods: CF viability in response to chronic treatment with both drugs was assessed using MTT assays and flow cytometry analysis. Calcium mobilization was assessed in CFs loaded with Fluo4-AM and CaMKII activation via oxidation was measured via quantitative immunoblotting. Effects of both drugs on mitochondrial function was determined by live mitochondrial imaging using MitoSOX red.
Results: Treatment of CFs with sunitinib (0.1–10 μM) resulted in concentration-dependent alterations in CF phenotype, with progressively significant cell loss at higher concentrations. Flow cytometry analysis and MTT assays revealed increased cell apoptosis and necrosis with increasing concentrations of sunitinib. In contrast, equivalent concentrations of imatinib resulted in no significant change in cell viability. Both sunitinib and imatinib pre-treatment increased Angiotensin II-induced intracellular Ca2+ mobilization, with only sunitinib resulting in a significant effect and also causing increased CaMKII activation via oxidation. Live cell mitochondrial imaging using MitoSOX red revealed that both sunitinib and imatinib increased mitochondrial superoxide production in a concentration-dependent manner. This effect in response to both drugs was suppressed in the presence of the CaMKII inhibitor KN-93.
Conclusions: Sunitinib and imatinib showed differential effects on CFs, with sunitinib causing marked changes in cell viability at concentrations where imatinib had no effect. Sunitinib caused a significant increase in Angiotensin II-induced intracellular Ca2+ mobilization and both TKIs caused increased mitochondrial superoxide production. Targeted CaMKII inhibition reversed the TKI-induced mitochondrial damage. These findings highlight a new role for CaMKII in TKI-induced cardiotoxicity, particularly at the level of the mitochondria, and confirm differential off-target toxicity in CFs, consistent with the differential selectivity of sunitinib and imatinib.
Cardiotoxicity is a recognized adverse effect of many clinically important drugs and has led to the withdrawal of a number of agents after their introduction to the market (1). Anti-cancer drugs are prevalent among the drug categories that exhibit cardiotoxicity and this can seriously impact upon cancer patient survival. In the long term, patients receiving anti-cancer drugs may be at a greater risk of death from cardiovascular disease than cancer (2). Conventional approaches for assessing cardiotoxic effects are similar to the indices used for assessing heart disease, including measurement of cardiac troponin and natriuretic peptides (3, 4). These markers usually only show significant changes after damage to the heart has occurred. Consequently, there is a real need to identify early onset biomarkers and signaling pathways that are switched on soon after initiating anti-cancer therapy and that correlate with adverse effects on the heart. This will be essential for the development of safer anti-cancer drugs in the future.
A prominent and well-recognized feature of anti-cancer drug-induced cardiotoxicity in cardiac myocytes (CMs) is the accumulation of damaging reactive oxygen species (ROS). Functional effects of ROS accumulation include CM hypertrophy, impaired contraction, apoptosis and autophagy, all of which are key features of cardiotoxicity (5). Importantly though, drug-induced cardiotoxicity does not only affect the contractile cells of the heart. Evidence is emerging that anti-cancer drugs can also have off-target damaging effects on other cells of the cardiovascular system including cardiac fibroblasts (CFs), endothelial cells (ECs), vascular smooth muscle cells (VSMCs), immune cells and cardiac progenitor cells (6). In a healthy heart there is considerable functional interaction between different cell types, particularly between the CMs and CFs. The latter cell type not only secrete chemical mediators that can influence cardiac contractility, but also provide components of extracellular matrix that ensure structural stability (7). In cardiovascular disease, CFs play a pivotal role in cardiac remodeling and in the accompanying contractile dysfunction by exhibiting hyper-proliferation and producing excess collagen. The resulting fibrosis is a key contributor to cardiac dysfunction (8). Recent studies have suggested that anti-cancer therapies can mirror the effects observed in CFs from diseased hearts, including increased oxidative stress and ROS accumulation similar to that seen in CMs but resulting in cardiac fibrosis and potential diastolic dysfunction (9–11).
Among the more novel anti-cancer agents to emerge in recent years are those that prevent cancer cell proliferation and angiogenesis via “selective” targeted tyrosine kinase and vascular endothelial growth factor receptor (VEGFR) inhibition. Unfortunately, these targeted therapies cause cardiotoxicity via both on-target (blocking VEGFR in cardiac cells) and off-target (non-VEGFR) effects (12). Understanding the cellular mechanisms that underpin these cardiotoxic effects will be crucial in improving the safety profile of tyrosine kinase inhibitors (TKIs), especially since these drugs are currently the standard treatment for several types of malignancy. The extent of TKI-induced cardiotoxicity may vary considerably across different members of the TKI drug group. Understanding the fundamental reasons for these differences is essential for tailoring future safer treatments. It is possible that different cardiotoxic responses may reflect an individual drug's capacity for on-target vs. off-target effects (13).
Two well-known TKIs that are currently in clinical use are imatinib and sunitinib. Imatinib mesylate is used for the treatment of chronic myeloid leukemia, gastrointestinal stromal tumors and hypereosinophilic syndrome. Imatinib works by targeting the ABL ATP binding site, stabilizing the inactive form of ABL, preventing tyrosine autophosphorylation and therefore kinase phosphorylation of substrates in cancerous cells, ultimately inducing apoptosis (14). There have been various reports of cardiotoxicity associated with imatinib. At a cellular level the cardiotoxic response has been suggested to include apoptosis as well as necrosis of CMs and this is as a result of mitochondrial dysfunction, ATP depletion and cytochrome C release (15). However, there is speculation over whether the concentrations of imatinib that exert the toxic effects reported on cardiac cells are clinically relevant (16). Despite evidence for cardiotoxicity, overall adverse effects on the heart following imatinib treatment appear relatively uncommon, ranging from 0.5 to 1.7% (17, 18). Sunitinib malate (SUTENT®) on the other hand has been reported to exert a wide range of cardiotoxic effects. Reports of hypertension and congestive heart failure in patients receiving sunitinib therapy ranges from 17 to 43% and 3 to 18% (19) respectively. Sunitinib is commonly used to treat renal cancers and has shown considerable benefit in patients with metastatic renal carcinoma (20). Sunitinib has been suggested to have a number of off-target effects that can adversely affect the heart. Examples of off-target effects include inhibition of AMP-activated protein kinase (21) and activation of calcium/calmodulin dependent protein kinase (CaMKII) (22), both of which, if chronically affected, can result in cardiac dysfunction.
Previous work by our group has highlighted that CaMKII activation occurs in guinea pig hearts following chronic administration of clinically relevant concentrations of either imatinib or sunitinib (22). Increased cardiac CaMKIIδ expression and CaMKII activity were shown to occur in the absence of any overt cardiac dysfunction. We suggested that these changes could reflect early adaptations by the heart to these TKIs which could precede the onset of contractile dysfunction. As such, we suggested that CaMKII might be a useful early onset marker for TKI-induced cardiotoxicity. Our original work comprised in vivo assessment of cardiac function following acute and chronic TKI treatment. CaMKII was assessed in whole cardiac homogenate preparations from TKI-treated hearts. Crucially, there was no investigation of specific cellular responses to either imatinib or sunitinib, evidence that we need to inform us upon more tailored clinical treatments going forward. In order to characterize the cardiotoxic effects of imatinib and sunitinib in more detail, the current study has, for the first time, compared the effects of both TKIs on adult rat CFs across a range of clinically relevant concentrations. Cell phenotype and viability have been assessed along with effects on mitochondrial superoxide production. Here we present novel evidence for distinct differences in the cellular responses to both drugs in CFs. Importantly, we also identify a common role for CaMKII in both imatinib and sunitinib-mediated mitochondrial superoxide production.
All procedures were performed under sterile conditions and conformed to local ethical review committee guidelines as well as to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and Directive 2010/63/EU of the European Parliament. CFs were isolated from male Sprague-Dawley rats weighing between 250 and 350 g. Rats were sacrificed via cervical dislocation and ventricular CFs isolated under sterile conditions via the bulk collagenase digestion method described previously (23). Briefly, isolated hearts were washed in Ca2+ free Krebs solution (120 mM NaCl, 5.4 mM KCl, 0.52 mM NaH2PO4, 20 mM HEPES, 11.1 mM glucose, 3.5 mM MgCl2, 20 mM taurine, 10 mM creatine) supplemented with 1 mM EGTA, 1% (v/v) penicillin/streptomycin; pH 7.4. The aorta and atria were then excised before the remaining ventricular tissue was finely chopped in a digestion buffer containing 0.8 mg/ml collagenase type 1 (Worthington Chemical, UK) and 0.3 mg/ml protease XIV (Sigma-Aldrich) prepared in Ca2+ free Krebs solution. The isolated CFs were resuspended in CF growth medium [DMEM supplemented with 20% (v/v) FBS, 2% (v/v) penicillin/ streptomycin and 1% (v/v) L-glutamine] before being plated in a T75 culture flask and incubated (37°C, 5% CO2). The CF growth media was replenished after 4–5 h of incubation to remove non-adherent cells. The media was replenished 24 h post-isolation to remove any remaining cell debris. Subsequent media changes conducted at 48 h intervals thereafter as per the standard cell culture protocol until 70–80% confluent.
CFs were grown to ~75% confluence in CF growth medium (outlined above) and were treated with sunitinib malate and imatinib mesylate at the indicated concentrations for 18 h in the same CF growth medium containing 20% serum (outlined above). Stock concentrations of drugs were diluted in DMEM supplemented with 2% (v/v) penicillin/streptomycin and 1% (v/v) L-glutamine prior to addition. Working concentrations were based on clinically relevant concentrations previously published (22, 24). Where appropriate, 5 μM KN-93 was added for 2 h pre-treatment prior to the addition of drug for 18 h.
Cell phenotype and growth was monitored using a Nikon Eclipse (TE300) inverted microscope (Nikon, Tokyo, Japan) and a Leica EC3 digital camera affixed to a Leica DM IL LED inverted microscope at 10 × magnification (Leica Biosystems, Wetzlar, Germany).
CFs, human umbilical vein endothelial cells (HUVECs), colonic smooth muscle cells (SMCs) and myofibroblasts (MFs) were seeded in 12-well plates containing 13 mm glass coverslips at a density of 1.5 × 104 cells/ml and cultured until 40–60% confluent. MFs were obtained following passage of CFs to p4 or p5 and were initially identified visually based on dramatic change in cell phenotype where MFs were significantly larger and rounder. Cells were fixed in 3.6% (v/v) formaldehyde in phosphate buffered saline (PBS) for 10 min at room temperature before being permeabilised with 0.25% Triton X-100 in PBS for 10 min at room temperature. Cells were then blocked in 1% (w/v) BSA in PBS for 30 min before incubation with primary antibody diluted in 1% (w/v) BSA in PBS overnight at room temperature [vimentin 1:400; Sigma-Aldrich (V5255)], von Willebrand factor [vWF; 1:100; Abcam (ab6994)] and smooth muscle cell actin [SMCa; 1:200; Abcam (ab7817)]. Following overnight incubation, coverslips were washed three times with PBS before being blocked in 1% (w/v) BSA in PBS for 15 min at room temperature. Coverslips were then incubated (covered) with the corresponding Alexa Fluor™ 488 secondary antibody {1:100; Thermo Fisher, Renfrew, UK [Mouse (A-11001); Rabbit (A-11008)]} in 1% (w/v) BSA in PBS for 1 h at room temperature. Following further washes in PBS, samples were stained with DAPI (1:2,000) for 5 min before being mounted onto glass coverslips with Mowiol® and stored at 4°C until imaged. Samples were imaged using the EVOS™ FL Auto Imaging System (Thermo Fisher) at 20 × magnification.
CFs were seeded in a 96-well plate at a density of 2.5 × 104 cells/well and cultured in growth medium for 24 h prior to drug treatment. Treated samples were incubated with 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide MTT (10 μg/ml) prepared in fresh growth medium for 2 h (37°C, 5% CO2). In living cells, mitochondrial dehydrogenases convert MTT to purple MTT formazan crystals. The purple formazan crystals were resuspended in DMSO and cell viability assessed via optical density at a wavelength of 570 nm. Data was expressed as a percentage of control untreated cells.
CFs were seeded in a 12-well plate at a density of 5 × 104 cells/well and cultured in growth medium until 70% confluent and then treated as per the in vitro drug treatment protocol. CFs were also treated with H2O2 for 18 h to provide single stain controls (600 μM H2O2 annexin V (AnV) control and 1.5 mM H2O2 propidium iodide (PI) control). Once treated, the media from each well was collected into individual FACS tubes. Samples were then dissociated using TrypLE™ Express (37°C) and collected into their respective FACS tubes alongside the previously collected media. Wells were then washed with PBS and added to the corresponding FACS tube to ensure all cells were recovered before being centrifuged at 1,000 g for 5 min. The samples were washed with PBS and 100 μl of AnV binding buffer (BD Biosciences, UK) added to each FACS tube. AnV (BD Biosciences, UK) (5 μl) was then added to all FACS tubes except the control (unstained) and the PI single stain (1.5 mM H2O2 treated) samples. The samples were then covered with aluminum foil to omit light and incubated for 15 min at room temperature. Following incubation, 400 μl of 1:500 PI [Thermo Fisher (P3566)] in AnV binding buffer was added to all FACS tubes except the control (unstained) and annexin V single stain (600μM H2O2 treated) samples. 400μl of AnV binding buffer was added to the control (unstained) and AnV single stain (750μM H2O2 treated) samples. The samples were then analyzed using the BD FACSCanto flow cytometer (BD Biosciences, California, USA) and FlowJo v9 (FlowJo LLC, Oregon, USA).
For assessing Total-CaMKIIδ and Phos-CaMKII expression, CFs were lysed in 150 μl of LDS sample buffer [25% (v/v) 4 × Nu PAGE LDS Sample Buffer (Thermo) and 75 mM DTT] and denatured at 100°C for 5 min. For assessing Ox-CaMKII expression, CFs were lysed in the absence of the reducing agent DTT. Protein lysates were loaded onto 4–20% Mini-PROTEAN TGX Precast Protein Gels and subjected to electrophoresis at 120 V for 110 min in running buffer (3.5 mM SDS, 192 mM glycine, and 25 mM tris base) using a Mini-PROTEAN Tetra chamber (Bio-Rad Laboratories Ltd., UK) at room temperature. Proteins were then transferred to nitrocellulose membranes (0.45 μm pore, GE Healthcare, UK) in transfer buffer [192 mM glycine, 25 mM tris base, 20% (v/v) methanol] using a MiniTrans-Blot Electrophoretic Transfer Cell (Bio-Rad Laboratories Ltd., UK) at a constant current of 280 mA for 110 min at room temperature. Membranes were then blocked in 5% (w/v) BSA in TBS-T [20 mM Tris base, 150 mM NaCl, adjusted to pH = 7.4, and 0.1% (v/v) Tween 20] for 120 min at room temperature. The membranes were then incubated with the appropriate primary antibody in 0.5% (w/v) BSA in TBS-T (0.1%) overnight at 4°C {Ox-CaMKII [1:5,000; Insight biotechnology Limited (GTX36254)]; Phos-CaMKII [1:5,000; Badrilla (A010-50)]; CaMKIIδ [1:5,000; Eurogentec (custom made)]; GAPDH [1;100,000; Abcam (ab8245)]}. Membranes were then washed in TBS-T (0.1% Tween) followed by incubation in the corresponding secondary antibody (1,5,000; Jackson ImmunoResearch, Cambridge, UK [Mouse (715-035-150); Rabbit (111-035-144)] in 0.5% (w/v) BSA in TBS-T (0.1% Tween) for 90 min at room temperature. This was followed by a further wash in TBS-T (0.1% Tween) before being developed via enhanced chemiluminescence and exposure onto X-ray film. For re-probing purposes, membranes were stripped in Tris-based buffer (31 mM Tris base, 35 mM SDS containing 0.7%, pH = 6.7). X-ray film was then scanned on a HP Deskjet 2540 printer scanner and densitometry was carried out using Image J software (25). Blotting data was normalized as indicated in the relevant figure legends and samples processed as percentage of control untreated signal.
Cells were seeded into black sided, clear bottomed 96-well plate at a density of 2.5 × 104 cells/well and cultured for 24 h before treatment with sunitinib and imatinib as described previously. Culture medium was aspirated and the cells washed twice with HBSS (136.9 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 0.4 mM MgSO4·7H2O, 0.5 mM MgCl2.6H2O, 0.3 mM Na2HPO4.2H2O, 0.4 mM KH2PO4, 5.6 mM glucose, 4.2 mM NaHCO3). Cells were then incubated with 2 μM Fluo4-AM Ca2+ indicator dye in Fluo4 buffer [1 mM MgCl, 1.5 mM CaCl, 0.3 mM probenecid, and 0.1% (w/v) BSA in HBSS] for 2 h (37°C, 5% CO2). Cells were then washed twice with Fluo4 buffer before adding 80 μl of Fluo4 buffer to each well. A compound plate was prepared using the ligands of interest, diluted to 3 × the final concentration in Fluo4 buffer. The plate reader transfers 40 μl of compound to each well, diluting the compound 1:3 to reach a 1 × desired final concentration for each sample. Fluorescence excitation was then measured using a Flexstation 3 microplate reader (Molecular Devices, Wokingham, UK) at Ex:494/Em:525 nm and SoftMax® Pro software, version 5.4.3.
MitoSOX fluorescence was recorded on a Nikon TiE inverted epifluorescence microscope with 40 × 1.3 NA oil immersion objective, illuminated with 515 nm excitation light (Pe4000 multi-LED; CoolLED, Andover, UK) and emitted light >550 nm selected by dichroic mirror and captured on an ORCA Flash 4.0 CMOS camera (Hamamatsu, Welwyn Garden City, UK). Cell imaging chambers were placed inside a stage-top incubator (H301; OKO labs, Naples, Italy) and maintained at 37°C throughout the imaging process. CFs were seeded into the 8-well chambers at a density of 2.5 × 104 cells/well, cultured in CF growth medium for 24 h and then treated as per the in vitro drug treatment protocol. Antimycin A (10 μM) was used as a positive control. The treated samples were then washed with HBSS before being incubated with 3 μM MitoSOX Red for 5 min at room temperature in the absence of light. CFs were recorded for 5 min using WinFluor V4 0.8 live cell imaging software. Signals were normalized by selecting 10 cells from each sample and measuring fluorescence intensity for each cell. A blinded process was used for cell selection. Fluorescence intensity was then measured using ImageJ software (25).
Data are presented as mean values ± S.E.M of n observations, where n represents the number of samples. Comparisons were assessed by one-way ANOVA with post-hoc Dunnett's-test using GraphPad Prism (version 7.0a). P-values < 0.05 were considered significant.
The phenotype of healthy CFs was first established via immunohistochemistry and brightfield imaging. Immunohistological staining showed a strong positive stain for vimentin in untreated isolated CFs, consistent with healthy, undifferentiated CFs. To exclude the possibility of contamination with SMCs or ECs, CFs were also stained for smooth muscle cell alpha actin (SMCa) (a marker of smooth muscle cell phenotype) and von Willebrand Factor (vWF) (a marker of endothelial cell phenotype). Staining was compared across CFs, myofibroblasts (MFs), SMCs and ECs. Although a positive stain for both SMCa and vWF was obtained in CFs, staining was significantly less than that obtained in the positive controls for SMCs and of a very different pattern than that seen in the positive controls for HUVECs. Staining was also less than that obtained in the phenotypically transformed MFs (Figure 1A).
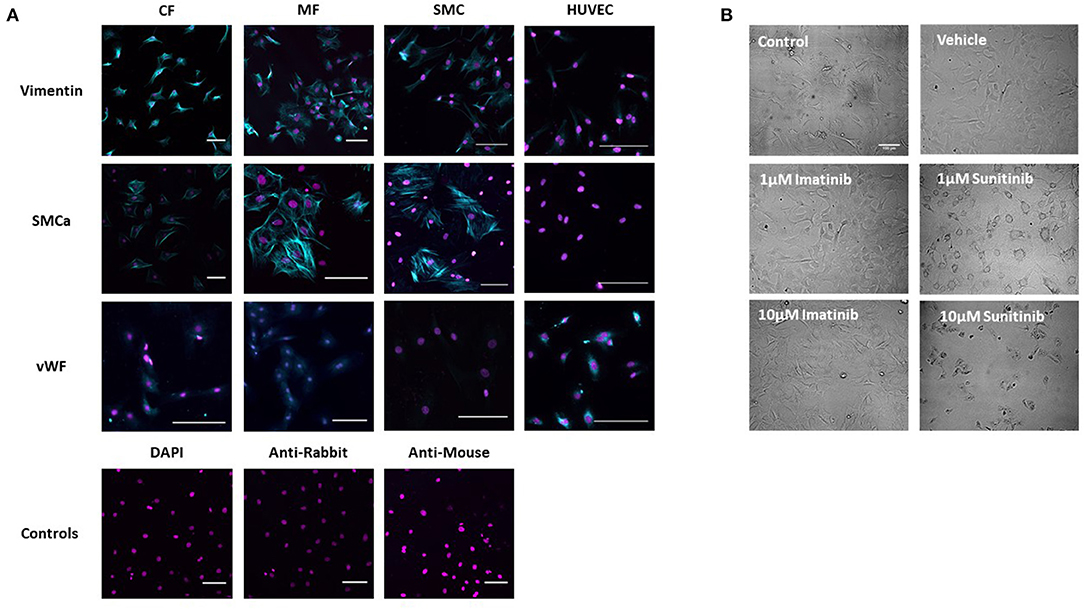
Figure 1. Assessment of Cardiac Fibroblast phenotype. (A) CF phenotype was confirmed via immunohistochemistry by staining for vimentin, smooth muscle cell alpha actin (SMCa) and von Willebrand Factor (vWF). Staining was compared between CFs and myofibroblasts (MF). Smooth muscle cells (SMC) and human umbilical vein endothelial cells (HUVECs) were used as positive controls. Cells treated with DAPI alone and secondary antibodies alone were used as negative controls. (B) Brightfield images showing CF treated with sunitinib and imatinib (0.1–10 μM) for 18 h in the presence of serum. Scale bar 100 μm. Images are from one experiment, representative of two others.
In order to assess the effects of imatinib and sunitinib on CF phenotype, cells were treated with increasing concentrations of drug (1–10 μM) for 18 h and then subjected to brightfield imaging (Figure 1B). Sunitinib-induced changes in CF phenotype were evident at 1 μM sunitinib and became more apparent as concentrations increased. Sunitinib-treated CFs appeared larger and more transparent, with the formation of vacuole-like structures evident in the main cell body. Obvious cell loss was apparent at 10 μM sunitinib. The observed changes in CF phenotype were associated with subtle changes in cell marker expression determined via quantitative immunoblotting, although these changes were not statistically significant (data not shown). Imatinib treatment did not have any discernible effect on CF phenotype until much higher concentrations.
To determine whether the changes in phenotype correlated with reduced cell viability, MTT assays and FACS analysis were performed. MTT assays revealed that sunitinib, but not imatinib, reduced CF viability (Figure 2). CF viability was significantly reduced at 3 and 10μM sunitinib, consistent with the cell loss observed in brightfield imaging.
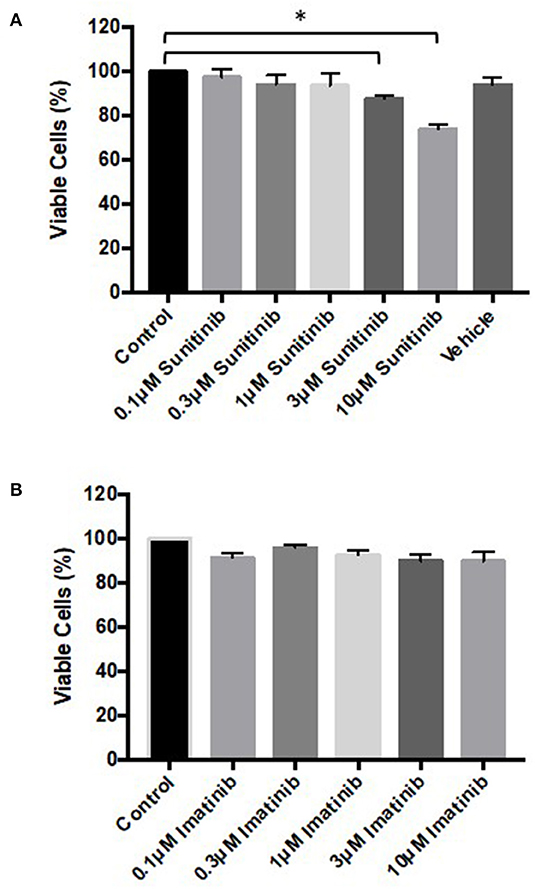
Figure 2. Sunitinib treatment reduces cardiac fibroblast viability. Cells were treated with sunitinib or imatinib (0.1–10 μM) in the presence of serum for 18 h. Cell proliferation was assessed via MTT assays. (A) Sunitinib treated CF (B) imatinib treated CF. Results are expressed as mean ± S.E.M and are normalized to control (n = 3, *p < 0.05).
Further analysis via FACS confirmed these findings. Sunitinib treatment caused a reduction in the number of healthy cells (Figure 3B) with a concomitant increase in the number of early and late apoptotic cells at 3 and 10 μM treatments, as well as an increase in necrotic cells at 3μM reaching significance for necrosis at 10 μM sunitinib (Figure 3B). Imatinib treatment had no significant effect on CF viability (Figure 4).
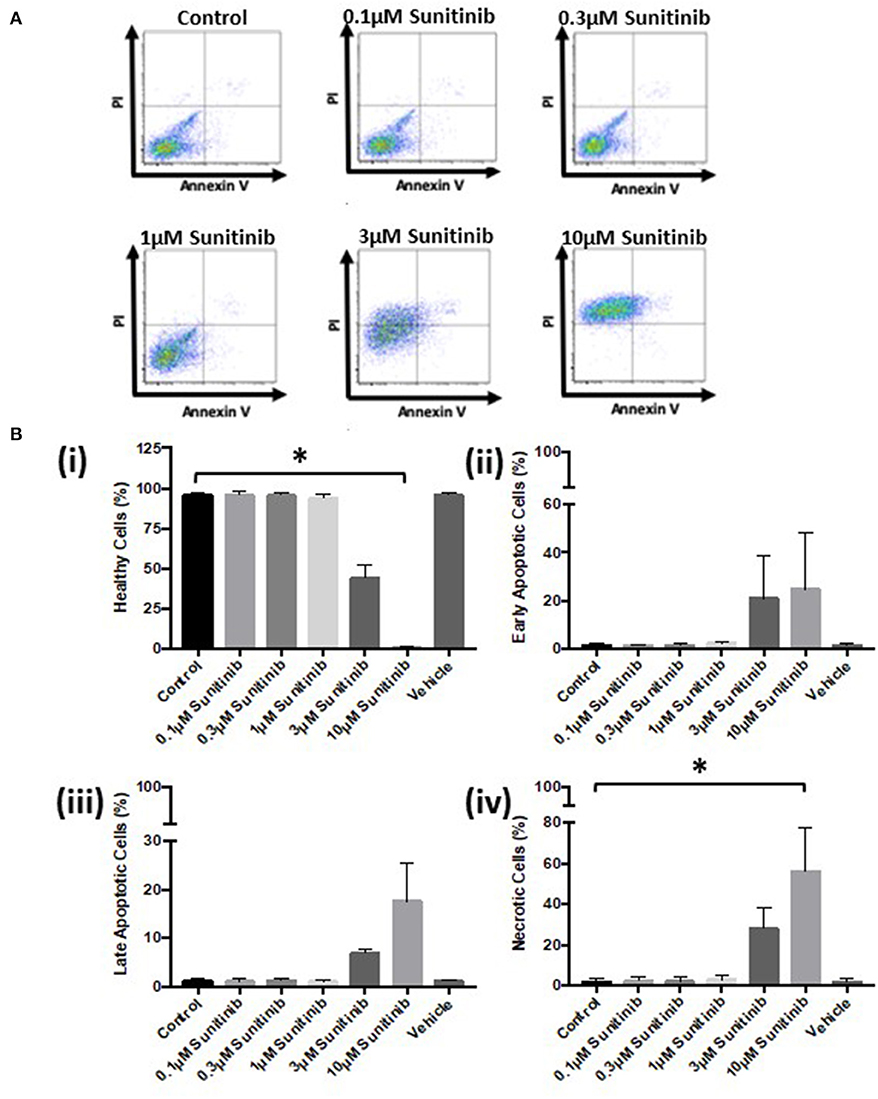
Figure 3. Sunitinib treatment causes increased necrosis in cardiac fibroblasts. Cells were treated with sunitinib (0.1–10 μM) in the presence of serum for 18 h. Cell viability was determined by PI and AnV staining. (A) Representative flow cytometry dot plots with double Annexin V-APC/PI staining for cells treated with sunitinib. (B) Histograms detailing the percentage of (i) healthy (ii) early apoptotic (iii) late apoptotic and (iv) necrotic cells following sunitinib treatment. Results are expressed as mean ± S.E.M (n = 3, *p < 0.05).
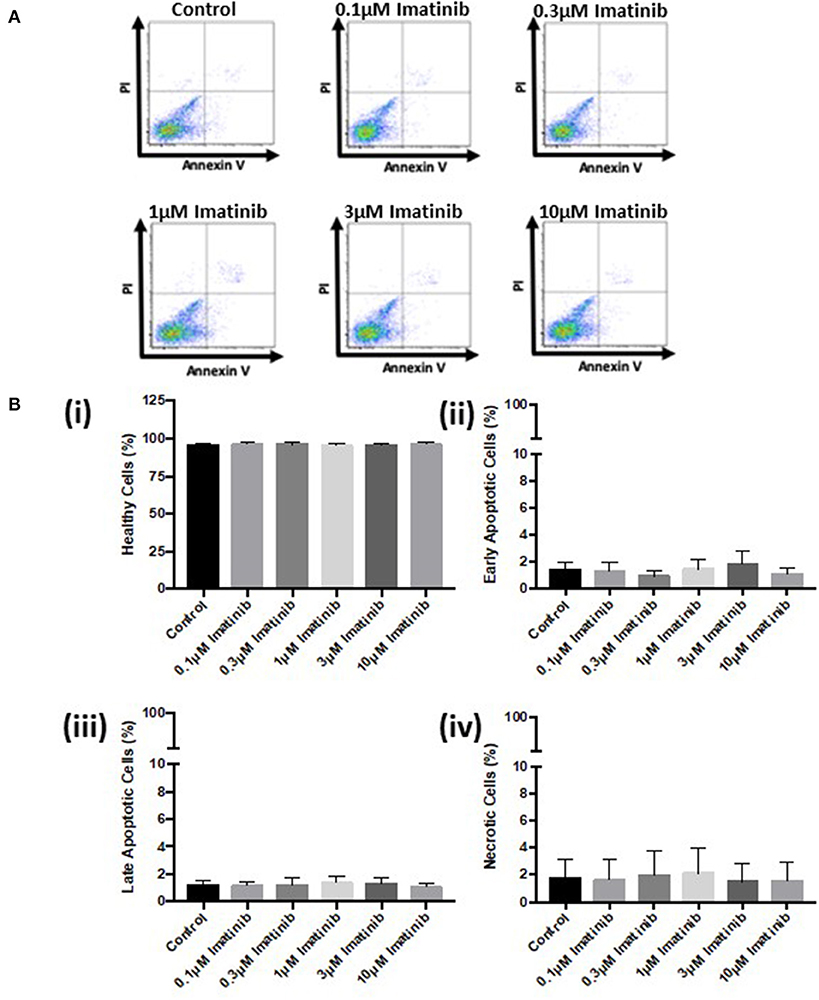
Figure 4. Imatinib treatment does not cause apoptosis or necrosis in cardiac fibroblasts. Cells were treated with imatinib (0.1–10 μM) in the presence of serum for 18 h. Cell viability was then determined by PI and AnV staining. (A) Representative flow cytometry dot plots with double Annexin V-APC/PI staining for cells treated with sunitinib. (B) Histograms detailing the percentage of (i) healthy (ii) early apoptotic (iii) late apoptotic and (iv) necrotic cells following imatinib treatment. Results are expressed as mean ± S.E.M (n = 3).
Altered Ca2+ handling is known to be associated with cell injury and death. Given the effects of TKI treatment, particularly sunitinib, on CF phenotype and viability (Figures 1–4), the possibility that either drug may affect Ca2+ mobilization in CFs was investigated. The effects of TKI pre-treatment on agonist-induced Ca2+ mobilization were examined. We have previously used Angiotensin II (AngII) to induce intracellular Ca2+ release in CFs (26) so this was applied to CFs in the current study to elicit responses.
AngII was first applied to CFs across a range of concentrations (0.01–10 μM) to determine a suitable concentration that would elicit a sub-maximal Ca2+ release in CFs (Figures 5A,B). Ang II at 0.3 μM elicited a rapid release of Ca2+ with a peak of ~80% of maximum. Using this sub-maximal concentration ensured that any subsequent effects of TKI pre-treatment (stimulatory or inhibitory) could be determined. CFs were pre-treated with a range of concentrations of either sunitinib (0.001–1 μM) or imatinib (0.01–1 μM) for 18 h and were then subjected to stimulation with 0.3 μM Ang II to induce Ca2+ release. Responses were compared with AngII responses in untreated cells. Cells treated with sunitinib only did not evoke intracellular calcium release (Figures 5C,F). Sunitinib pre-treatment resulted in a dose-dependent increase in AngII-evoked Ca2+ release (Figure 5C) with significance observed at 1 μM sunitinib (Figures 5D,F). Imatinib pre-treatment, even at higher concentrations, did not result in any significant effect on AngII-evoked Ca2+release (Figures 5E,F).
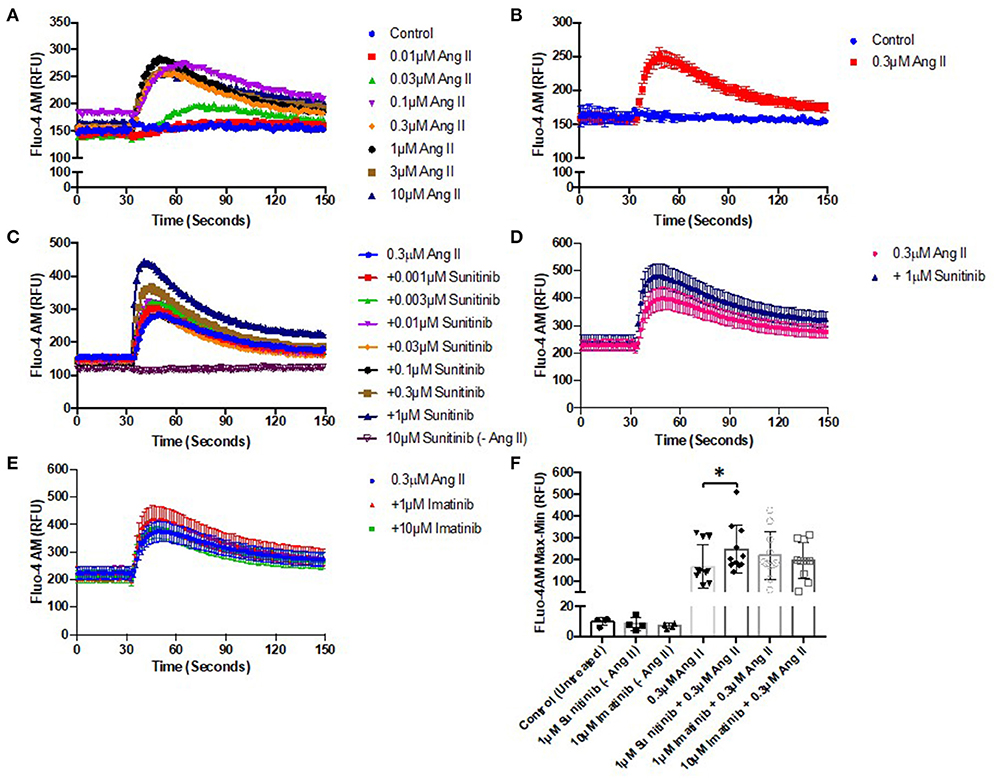
Figure 5. Sunitinib increases Angiotensin II—induced Ca2+ mobilization in cardiac fibroblasts. (A) Effect of increased AngII concentrations (0.01–10 μM) on intracellular Ca2+ mobilization (B) Representative trace of Ca2+ mobilization in response to 0.3 μM AngII stimulation (C) Effects of increasing concentrations of sunitinib (0.001–1 μM) on AngII induced Ca2+ mobilization. Sunitinib in the absence of Ang II is also shown. Results shown are mean data from one experiment, representative of five other experiments. (D) Comparison of maximal (1 μM) sunitinib Ang II-induced Ca2+ mobilization vs. control. (E) Effects of increasing concentrations of imatinib (1–10 μM) on AngII induced Ca2+ mobilization. (F) Effects of TKI-treatment on peak agonist-induced Ca2+ mobilization. Calcium mobilization was determined by measuring fluorescence at Ex:494/Em:525 nm. Results are expressed as mean ± S.E.M of 6 biological replicates, *p < 0.05.
The effects of TKI treatment on agonist-induced intracellular Ca2+ mobilization, combined with the effects on cell viability, raised the possibility that TKIs may affect Ca2+ dependent processes within CFs and, in particular Ca2+-dependent protein activation. Previous work has shown that the Ca2+ dependent protein kinase, CaMKII, is activated and expression levels of the delta isoform (CaMKIIδ) are significantly increased in guinea pig cardiac homogenates following in vivo infusion of sunitinib and imatinib (22). We therefore examined the effects of chronic (18 h) treatment of CFs with both TKIs (at 1 and 10 μM) on CaMKIIδ expression and CaMKII activation (via either phosphorylation or oxidation). TKI treatment of CFs did not alter CaMKIIδ expression (Figure 6A) nor was phosphorylation of CaMKII affected. Interestingly though, sunitinib treatment did increase CaMKII activation via oxidation at 1 μM sunitinib. There was a subsequent decrease in oxidation at 10 μM sunitinib likely due to cell death as indicated by the lower signal for GAPDH (Figures 6C,D). Imatinib treatment had no discernible effect on CaMKII expression or activation.
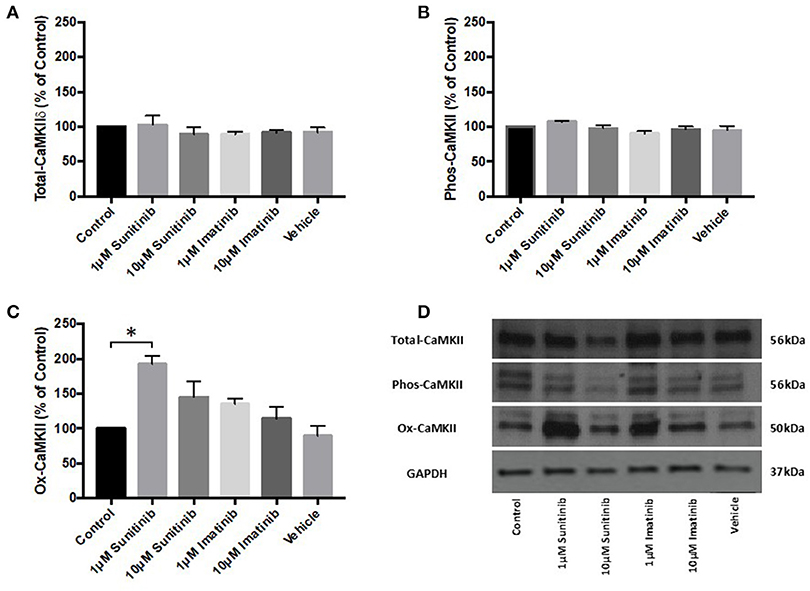
Figure 6. Sunitinib treatment increases ox-CaMKII expression in cardiac fibroblasts. Following the indicated treatments with either sunitinib or imatinib, CaMKIIδ expression or CaMKII activation via phosphorylation or oxidation was determined via quantitative immunoblotting (A) CaMKIIδ expression, normalized to GAPDH (B) Phos-CaMKII expression, normalized to CaMKIIδ (C) Ox-CaMKII expression, normalized to CaMKIIδ (D) Representative immunoblot showing CaMKIIδ expression and CaMKII activation following TKI treatment. Results are expressed as mean ratios protein: GAPDH ± S.E.M and are normalized to control (n = 3, *p < 0.05).
Since sunitinib can cause activation of CaMKII via oxidation in CFs, we next explored whether TKI treatment may result in elevated levels of reactive oxygen species (ROS) in these cells. Furthermore, we were intrigued to explore the possibility that any TKI-mediated increase in ROS may be via CaMKII-mediated effects at the level of the mitochondria. To investigate the possible role of mitochondrial ROS in mediating the cardiotoxic mechanism of TKIs, the mitochondrial superoxide indicator, MitoSOX Red, was used to determine changes in mitochondrial superoxide production.
Live cell imaging revealed that both sunitinib and imatinib significantly increased mitochondrial superoxide production in CFs. This is evident at all concentrations tested and the fold-changes observed following TKI treatment were markedly higher than that observed with the mitochondrial complex III inhibitor Antimycin A, which was used as a positive control for increased superoxide production (Figures 7A(i),(ii)). Sunitinib resulted in considerably higher fluorescence than imatinib (Figures 7B(i) vs. 7C(i)), again suggesting the increased potency and cardiotoxic potential of sunitinib when compared with imatinib. Interestingly, when CFs were pre-treated with the CaMKII inhibitor KN-93, sunitinib-mediated superoxide production was significantly reduced (Figure 8A). Moreover, in cells treated with 10 μM imatinib, KN-93 pre-treatment completely abolished imatinib-induced mitochondrial superoxide production (Figure 8B).
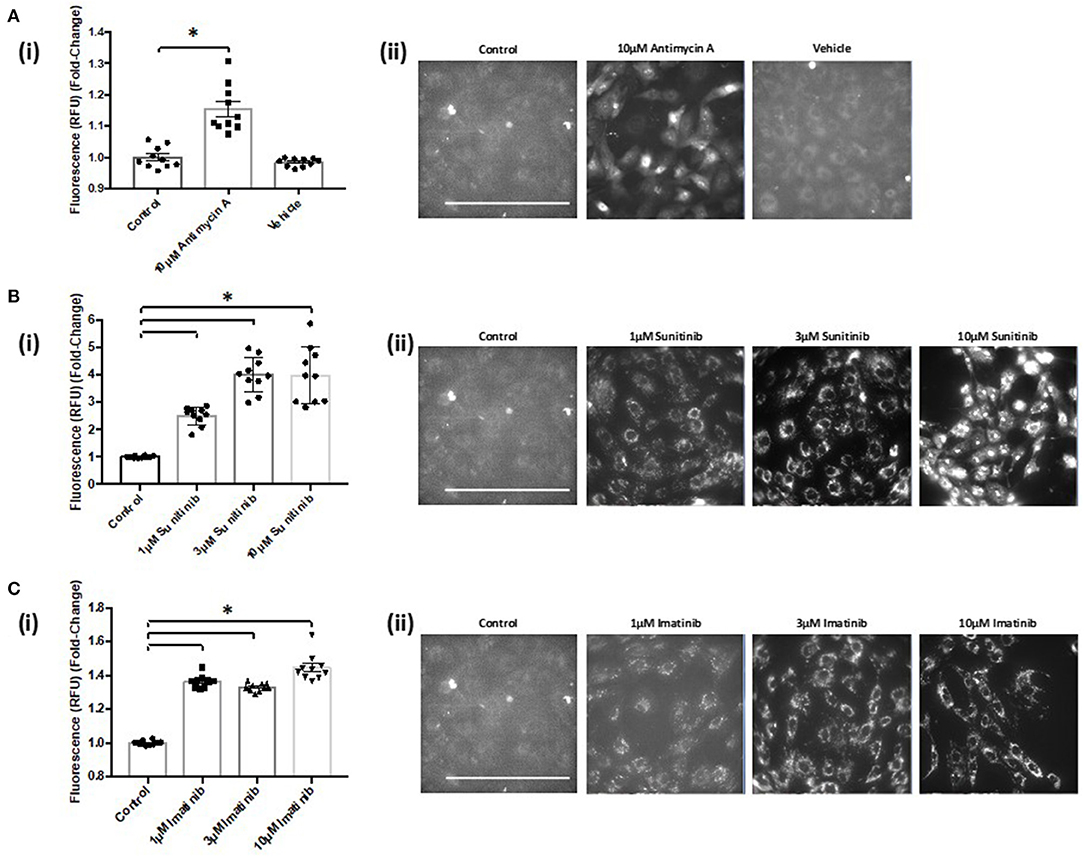
Figure 7. Sunitinib and imatinib treatments both cause increased mitochondrial superoxide production in cardiac fibroblasts. (Ai) Histogram showing superoxide production assessed via fluorescence intensity in antimycin A and vehicle treated CFs (Aii) Representative images of MitoSOX Red analysis of antimycin A and vehicle treated samples. (Bi) Superoxide production assessed via fluorescence intensity in sunitinib treated CFs (Bii) Representative images of MitoSOX Red analysis of sunitinib treated samples. (Ci) Superoxide production assessed via fluorescence intensity in imatinib treated CFs (Cii) Representative images of MitoSOX Red analysis of imatinib treated samples. Scale bar 100 μm. Results shown are mean data from one experiment (n = 10), representative of two other experiments (*p < 0.05).
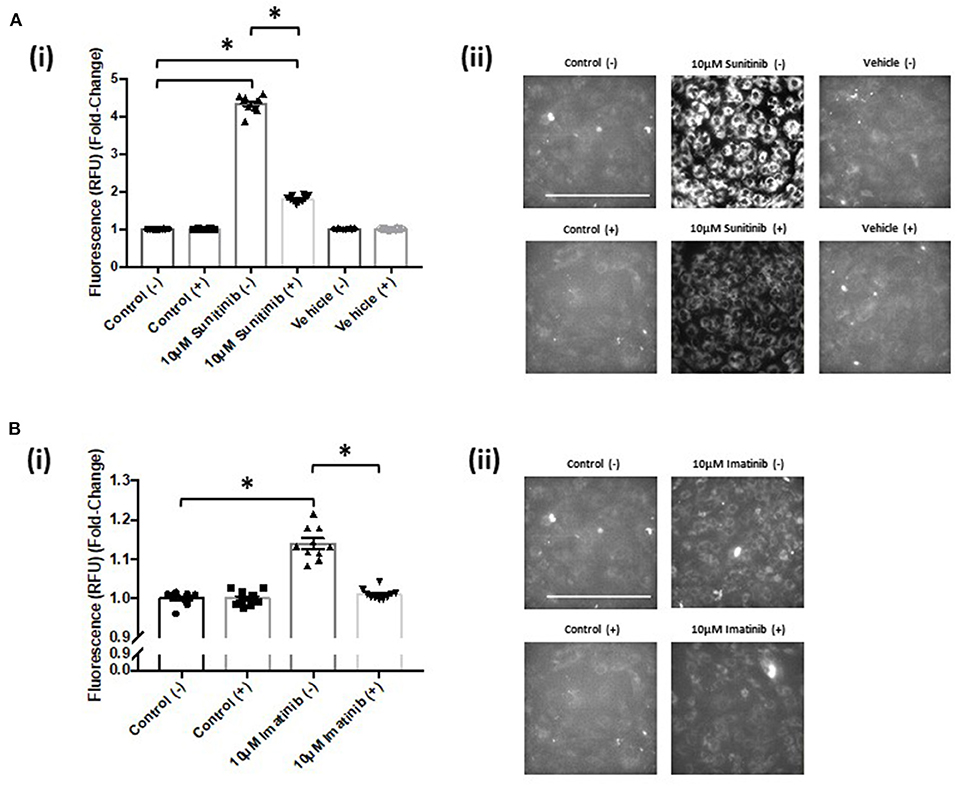
Figure 8. KN-93 reduces mitochondrial superoxide production in both sunitinib and imatinib treated cardiac fibroblasts. (Ai) Superoxide production assessed via fluorescence intensity in sunitinib treated CF in the presence (+) or absence (−) of 5 μM KN-93. (Aii) Representative images of MitoSOX Red analysis of sunitinib treated samples in the presence (+) or absence (−) of KN-93. (Bi) Superoxide production assessed via fluorescence intensity in imatinib treated CF in the presence (+) or absence (−) of KN-93. (Bii) Representative images of MitoSOX Red analysis of imatinib treated samples in the presence (+) or absence (−) of KN-93. Scale bar 100 μm. Results shown are mean data and images from one experiment where the same control (−) presented previously for non-KN 93 treatment has been used as a reference comparison for control (+) (n = 10). Imatinib and sunitinib treatments from the same experiment have been presented separately for clarity and are representative of two other experiments (*p < 0.05).
In the current study we have evaluated the effects of sunitinib and imatinib on both phenotype and function of adult rat CFs to gain insight into possible mechanisms underlying the cardiotoxic profiles of both drugs in the non-contractile fibroblasts of the heart. Effects on cell phenotype, viability, intracellular Ca2+ release and mitochondrial superoxide production were assessed and, for the first time, a potential role for CaMKII activation via oxidation as an underlying mechanism of action of sunitinib in CFs was highlighted. Interestingly, but unsurprisingly, sunitinib and imatinib exerted differential effects on CFs and this may provide an indication of the broader cardiotoxic potential of each drug.
Initial characterization of CFs was performed using immunocytochemistry and this revealed distinct vimentin staining of CFs with low levels of SMCa and altered distribution of vWF confirming results from previous studies (27, 28). Importantly, the distinction between CFs and MFs was highlighted with a clear difference in phenotype and SMCa staining across the two (Figure 1A). Treatment of CFs with sunitinib and imatinib at relatively low concentrations (1–10 μM) and in the presence of serum enabled a realistic assessment of the potential for these drugs to affect CF phenotype and performance in the clinical setting. Previous investigations have often used much higher concentrations of drugs which may not be clinically relevant (29) and very few studies have assessed the effects on CFs (11). Sunitinib induced clear alterations in cell phenotype and viability at concentrations as low as 1 μM whereas imatinib effects were less pronounced and phenotypic changes only started to become evident at a 10-fold higher concentration. This pattern was maintained when the effects of both drugs on cell viability were studied in more detail. Using MTT assays and FACS analysis, a sunitinib-induced reduction in viability was observed at 3 and 10 μM with significant levels of necrosis occurring at 10 μM (Figures 2, 3). There were no significant effects of imatinib at any of the concentrations tested (Figures 2, 4). These observations fit with the differences observed in cardiotoxicity reported for these drugs in the clinic (29–31). Differences in % viability were detected across MTT and flow cytometry and this reflects the differences in sensitivity between both assays. Importantly, imatinib did not induce any measurable change in viability assessed via either assay.
Previous work by our group has highlighted that acute and chronic treatment of guinea pigs with sunitinib and imatinib can alter the expression and/or activity of CaMKII measured in ventricular cardiac homogenates (22). CaMKII is well-established as a pivotal molecule in cardiac physiology regulating cellular processes such as excitation-contraction coupling, cellular mechanics and energetics (32, 33). CaMKII is also recognized as an important mediator of pathological signaling and remodeling in disease (34, 35). As well as a recognized role in cardiomyocyte physiology and pathophysiology, a role for CaMKII in CF dysfunction has also been highlighted (27). The possibility that sunitinib and/or imatinib might alter CaMKII activity in CFs has not previously been explored but this could be a central factor contributing to the underlying mechanism of cardiotoxicity. In particular, given the contribution that CFs have to extracellular matrix deposition and to cardiac myocyte function, detrimental effects on CF viability by TKIs could impact directly upon cardiac contractility leading in particular to impaired diastolic function. Interestingly, sunitinib was able to increase AngII-mediated intracellular Ca2+ release and this trend was evident at all concentrations of sunitinib tested (0.001–1 μM), becoming significant at 1 μM. Lower concentrations were used in these experiments to closely mirror plasma concentrations that are seen in patients (24, 36) and to investigate whether intracellular signaling (in the form of Ca2+ release) may be more sensitive to drug-induced effects than those effects observed phenotypically and functionally. Although imatinib appeared to increase intracellular Ca2+, this effect was not significant even up to 10 μM imatinib. This raises the potential that sunitinib (and possibly imatinib) may have for exacerbating Ca2+-activated responses. The contribution that excess Ca2+ release has toward apoptosis and necrosis is well-documented (37, 38). Sustained CaMKII activation is a central feature of these pathological processes and the enzyme is initially activated in response to increased intracellular [Ca2+]. CaMKII can also be activated by post-translational modifications downstream of Ca2+/calmodulin binding. This can include autophosphorylation, oxidation, S-nitrosylation and O-GlcNAcylation. All of these modifications can occur during sustained and pathological activation (39). Here, for the first time, we have investigated the ability of both sunitinib and imatinib to induce either phosphorylation or oxidation of CaMKII. As well as the possibility that both drugs may activate CaMKII via elevations in intracellular Ca2+, data shown here suggest that sunitinib can also cause oxidation of CaMKII in CFs (Figure 6). Oxidation of CaMKII is able to alter the Ca2+ sensitivity of the enzyme enabling activation at low intracellular [Ca2+] and sustaining activity in the absence of Ca2+/calmodulin (40, 41) and this may be a means by which the cardiotoxic effects of CaMKII are harnessed by certain anti-cancer drugs. This mode of activation of CaMKII may be a distinguishing feature in differences in mechanism of action between the two TKIs although this would require further investigation. Interestingly, CaMKII has been linked to activation of a regulated form of necrosis, termed necroptosis (33). Necroptosis can be triggered via activation of receptor-interacting protein (RIP3) which has CaMKII as a substrate, and it has been shown that disruption of either RIP3 or CaMKII can significantly reduce cell death (42). Here we have shown that sunitinib can induce necrosis (we have not assessed necroptosis) and can activate CaMKII. It is possible that the two processes are linked in the mechanism of action of sunitinib-induced cardiotoxicity in CFs.
Since increased oxidation of CaMKII was evident following sunitinib treatment, we investigated whether either sunitinib or imatinib treatment could result in increased levels of mitochondrial superoxide. Although the role of ROS in cardiac pathophysiology remains unclear, ROS have been implicated in a variety of processes that affect cardiac function, both contractile and non-contractile processes (43, 44). While ROS have been implicated in the pathogenesis of anthracycline induced cardiotoxicity, there is less evidence of the effects of ROS in TKI-induced cardiotoxicity, particularly relating to effects on non-contractile cells of the heart. Importantly, current evidence suggests that anti-oxidant therapy does not offer cardioprotection against anthracycline-induced cardiotoxicity (5). Therefore, a better understanding of how ROS production may be mediated by anti-cancer agents and relate to cardiotoxic effects is required. Here we have shown that both sunitinib and imatinib can induce significant increases in mitochondrial superoxide in CFs following 18 h treatment and this occurs across all concentrations tested (1–10 μM) (Figure 7). Interestingly, when we pre-treat CFs with a CaMKII inhibitor (KN-93) and repeat TKI treatment at the highest concentrations of TKI tested (10 μM), the effects of both sunitinib and imatinib on mitochondrial superoxide production are abrogated (Figure 8). It is likely that KN-93 will inhibit global CaMKII in the CF and this may explain why we see an effect on imatinib induced superoxide production in the absence of any quantifiable CaMKII activation in this study. These results highlight that interventions interfering with CaMKII targeting to the mitochondria or with CaMKII oxidation are worthy of further study. Methionine sulfoxide reductase A (MsrA) can reduce oxidized CaMKII and could be a promising target to limit TKI-induced oxidative stress (40). It is possible that sunitinib-induced effects on CaMKII oxidation and superoxide production are not mutually exclusive and one may exacerbate the other driving toxicity (Figure 9). These mitochondrial events are already known to be a central feature of myocardial disease (45).
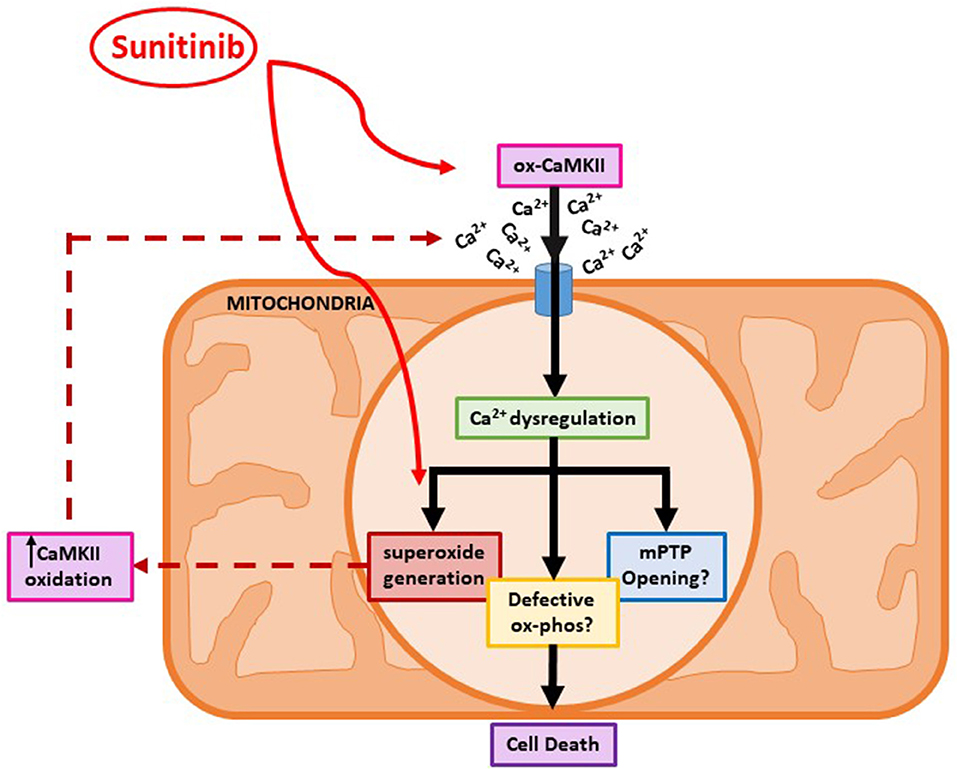
Figure 9. A proposed mechanism for sunitinib-induced mitochondrial dysfunction in adult cardiac fibroblasts. Schematic showing proposed sunitinib-induced events at the level of the mitochondria in CFs. Sunitinib treatment may exert direct effects on mitochondrial superoxide production that can then result in CaMKII oxidation. Sunitinib-induced increases in CaMKII oxidation and activation can then go on to cause elevated superoxide production so a positive feedback loop may exist exacerbating the cardiotoxic effects. The combination of elevated CaMKII oxidation and increased superoxide production ultimately causes toxicity at the level of the mitochondria and can contribute to CF cell death.
Recent work using transgenic mice and computational modeling has shown that myocardial infarction surgery causes significant elevation of mitochondrial CaMKII along with left ventricular dilatation. Pathological remodeling is reversed in mice with genetic mitochondrial CaMKII inhibition (46). The significance of mitochondrial targeted CaMKII activation and inhibition in the switch between normal function and pathological consequences is striking and is pertinent to the results presented in the current study.
For the first time we have demonstrated differential cardiotoxic effects in adult CFs in response to sunitinib and imatinib at low, clinically relevant concentrations of each drug. We have shown that sunitinib is capable of mediating an increase in agonist-induced [Ca2+]i and can activate CaMKII via oxidation. Both drugs lead to significant elevation of mitochondrial superoxide production and this is reversed in the presence of CaMKII inhibition. The implications of these results suggest that TKI-mediated cardiotoxicity could be reduced or reversed via targeted CaMKII inhibition. Development of CaMKII inhibitors to target heart disease is already ongoing. The use of more targeted CaMKII inhibitors in the treatment of anti-cancer drug cardiotoxicity could be an additional niche area for the pharmaceutical industry and fundamentally, could represent a new and important therapeutic approach in the field of cardio-oncology.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
SCu and MC worked on the conception and organization of the research project. Experimental work was conducted predominantly by CM. Data analysis was conducted by CM, SCh, RW, and MC. SCu and CM wrote the first draft of the manuscript. MC and SCh reviewed the manuscript. All authors read and approved the final version of the manuscript.
This work was supported by a Princess Royal Scholarship (Tenovus Scotland) to CM (S170866).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Augusto Montezano and Dr. Livia De Lucca Canargo, University of Glasgow, for the gift of primary smooth muscle cells for our study. We would also like to thank Graeme Mackenzie and Margaret MacDonald for their technical assistance with this work. Thanks also to Brian Leiper, Carol Whitehouse, Lee Wheeler, and Peter Conte for their assistance with the animal husbandry.
1. Curigliano G, Cardinale D, Dent S, Criscitiello C, Aseyev O, Lenihan D, et al. Cardiotoxicity of anti-cancer treatments: epidemiology, detection and management. CA Cancer J Clin. (2016) 66:309–25. doi: 10.3322/caac.21341
2. Carver JR, Shapiro CL, Ng A, Jacobs L, Schwartz C, Virgo KS, et al. American society of clinical oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol. (2007) 25:3991–4008. doi: 10.1200/JCO.2007.10.9777
3. Henri C, Heinonen T, Tardif JC. The role of biomarkers in decreasing risk of cardiac toxicity after cancer therapy. Biomarkers Cancer. (2016) 8:39–45. doi: 10.4137/BIC.S31798
4. Kilickap S, Barista I, Akgul E. cTnT can be a useful marker for early detection of anthracycline cardiotoxicity. Ann Oncol. (2005) 16:798–804. doi: 10.1093/annonc/mdi152
5. Angsutararux P, Luanpitpong S, Issaragrisil S. Chemotherapy-induced cardiotoxicity: overview of the roles of oxidative stress. Oxid Med Cell Longev. (2015) 2015:795602. doi: 10.1155/2015/795602
6. Gambardella J, Trimarco B, Iaccarino G, Sorriento D. Cardiac nonmyocyte cell functions and crosstalks in response to cardiotoxic drugs. Oxid Med Cell Longev. (2017) 2017:1089359. doi: 10.1155/2017/1089359
7. Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblast. Cardiovasc Res. (2005) 65:40–51. doi: 10.1016/j.cardiores.2004.08.020
8. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodelling. Pharmacol Therap. (2009) 123:255–78. doi: 10.1016/j.pharmthera.2009.05.002
9. Narikawa M, Umemura M, Tanaka R, Hikichi M, Nagasako A, Fujita T, et al. Doxorubicin induces trans-differentiation and MMP1 expression in cardiac fibroblasts via cell death-independent pathways. PLoS ONE. (2019) 14:e0221940. doi: 10.1371/journal.pone.0221940
10. CJ McMullen, C McCluskey, SJ Kim, Laovitthayanggoon S, MacDonald M, Safar M, et al. Anti-cancer tyrosine kinase inhibitors increase oxidative stress in primary cardiac fibroblasts. Heart J. (2018) 104:4.SCF22. doi: 10.1136/heartjnl-2018-SCF.22
11. Burke MJ, Walmsley R, Munsey TS, Smith AJ. Receptor tyrosine kinase inhibitors cause dysfunction in adult rat cardiac fibroblasts in vitro. Toxicol In Vitro. (2019) 58:178–86. doi: 10.1016/j.tiv.2019.03.026
12. Chen MH, Kerkela R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapies. Circulation. (2008) 118:84–95. doi: 10.1161/CIRCULATIONAHA.108.776831
13. Cheng H, Force T. Why do kinase inhibitors cause cardiotoxicity and what can be done about it? Circ Res. (2010) 106:21–34. doi: 10.1016/j.pcad.2010.06.006
14. Beininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukaemia. Blood. (2005) 105:2640–53. doi: 10.1182/blood-2004-08-3097
15. Kerkela R, Grazette L, Yacobi R, Illiescu C, Patten R, Beahm C., et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. (2006) 12:908–16. doi: 10.1038/nm1446
16. Wenyue Hu, Lu S, McAlpine I, Jamieson JD, Lee DU, Marroquin L, et al. Mechanistic investigation of imatinib-induced cardiac toxicity and the involvement of c-Abl kinase. Toxicol Sci. (2012) 129:188–99. doi: 10.1093/toxsci/kfs192
17. Atallah E, Durand JB, Kantarjian H, Cortes J. Congestive heart failure is a rare event in patients receiving imatinib therapy. Blood. (2007) 110:1233–7. doi: 10.1182/blood-2007-01-070144
18. Hatfield A, Owen S, Pilot PR. In reply to cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. (2007) 13:15–6. doi: 10.1038/nm0107-13a
19. Chen Z, Ai D. Cardiotoxicity associated with targeted anti-cancer therapies. Mol Clin Oncol. (2016) 4:675–81. doi: 10.3892/mco.2016.800
20. Rock EP, Goodman V, Jiang JX, Mahjoob K, Verbois SL, Morse D, et al. Food and drug administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist. (2007) 12:107–13. doi: 10.1634/theoncologist.12-1-107
21. Kerkela R, Woulfe KC, Durand JB, Vagnozzi R, Kramer D, Chu TF, et al. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin Transl Sci. (2009) 2:15–25. doi: 10.1111/j.1752-8062.2008.00090.x
22. Mooney L, Skinner M, Coker SJ, Currie S. Effects of acute and chronic sunitinib treatment on cardiac function and calcium/calmodulin dependent protein kinase II. Br J Pharmacol. (2015) 172:4342–54. doi: 10.1111/bph.13213
23. Lawan A, Al-Harthi S, Cadalbert L, McCluskey AG, Grant A, Boyd M, et al. Genetic deletion of the DUSP-4 gene reveals an essential non-redundant role for MKP-2 in cell survival. J Biol Chem. (2011) 286:12933–43. doi: 10.1074/jbc.M110.181370
24. Kitagawa D, Yokota K, Gouda M, Narumi Y, Ohmoto H, Nishiwaki E, et al. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells. (2013) 18:110–22. doi: 10.1111/gtc.12022
25. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. (2012) 9:671–5. doi: 10.1038/nmeth.2089
26. Currie S, Martin TP, Salleh H, McCluskey C, Bushell TJ. IP3R contribution to ventricular cardiac fibroblast function in healthy and hypertrophied mouse hearts. FASEB J. (2016) 30:S1–1178.2.
27. Martin TP, Lawan A, Robinson E, Grieve DJ, Plevin RJ, Paul A, et al. Adult cardiac fibroblast proliferation is modulated by calcium/calmodulin dependent protein kinase II in normal and hypertrophied hearts. Pflugers Arch. (2014) 466:319–30. doi: 10.1007/s00424-013-1326-9
28. Martin TP, McCluskey C, Cunningham MR, Beattie J, Paul A, Currie S. Calcium/calmodulin dependent protein kinase II delta interacts with IKKβ and modulates NFκB signalling in mouse heart. Cell Signall. (2018) 51:166–75. doi: 10.1016/j.cellsig.2018.07.008
29. Will Y, Dykens JA, Nadanaciva S, Hirakawa B, Jamieson J, Marroquin LD, et al. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci. (2008) 106:153–61. doi: 10.1093/toxsci/kfn157
30. Wolf A, Couttet P, Dong M, Grenet O, Heron M, Junker U, et al. Imatinib does not induce cardiotoxicity at clinically relevant concentrations in preclinical studies. Leuk Res. (2010) 34:1180–8. doi: 10.1016/j.leukres.2010.01.004
31. Mellor HR, Bell AR, Valentin JP, Roberts RRA. Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicol Sci. (2011) 120:14–32. doi: 10.1093/toxsci/kfq378
32. Bers DM. Calcium cycling and signalling in cardiac myocytes. Ann Rev Physiol. (2008) 70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455
33. Beckendorf J, van den Hoogenhof MMG, Backs J. Physiological and unappreciated roles of CaMKII in the heart. Bas Res Cardiol. (2018) 113:29–41. doi: 10.1007/s00395-018-0688-8
34. Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. (2011) 51:468–73. doi: 10.1016/j.yjmcc.2011.01.012
35. Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodelling after pressure overload. Proc Natl Acad Sci USA. (2009) 106:2342–7. doi: 10.1073/pnas.0813013106
36. Lankheet NA, Knapen LM, Schellens JH, Beijnen JH, Steeghs N, Huitema AD. Plasma concentrations of tyrosine kinase inhibitors imatinib, erlotinib and suntinib in routine clinical outpatient cancer care. Ther Drug Monit. (2014) 36:326–34. doi: 10.1097/FTD.0000000000000004
37. Li GY, Fan B, Zheng YC. Calcium overload is a critical step in programmed necrosis of ARPE-19 cells induced by high concentrations of H2O2. Biomed Envtl Sci. (2010) 23:371–7. doi: 10.1016/S0895-3988(10)60078-5
38. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis. ER-mitochondrial calcium transfer in the control of apoptosis. Oncogene. (2008) 27:6407–18. doi: 10.1038/onc.2008.308
39. Mollova MY, Katus HA, Backs J. Regulation of CaMKII signalling in cardiovascular disease. Front Pharmacol. (2015) 6:178. doi: 10.3389/fphar.2015.00178
40. Erickson JR, Joiner MA, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. (2008) 133:462–74. doi: 10.1016/j.cell.2008.02.048
41. Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV. Angiotensin II induced oxidative stress resets the Ca2+ dependence of CaMKII and promotes a death pathway conserved across different species. Circ Res. (2009) 105:1204–12. doi: 10.1161/CIRCRESAHA.109.204172
42. Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia and oxidative stress-induced myocardial necroptosis. Nat Med. (2016) 22:175–82. doi: 10.1038/nm.4017
43. Giordano FJ. Oxygen, oxidative stress, hypoxia and heart failure. J Clin Inv. (2015) 115:500–8 doi: 10.1172/JCI200524408
44. Mercurio V, Cuomo A, Dessalvi CC, Deidda M, Di Lisi D, Novo G, et al. Redox imbalances in ageing and metabolic alterations: implications in cancer and cardiac diseases. An overview from the working group of cardiotoxicity and cardioprotection of the Italian society of cardiology (SIC). Antioxidants. (2020) 9:641. doi: 10.3390/antiox9070641
45. Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res. 113:690–708. doi: 10.1161/CIRCRESAHA.113.301651
Keywords: sunitinib, cardiac fibroblast, CaMKII, cardiotoxicity, imatinib
Citation: McMullen CJ, Chalmers S, Wood R, Cunningham MR and Currie S (2021) Sunitinib and Imatinib Display Differential Cardiotoxicity in Adult Rat Cardiac Fibroblasts That Involves a Role for Calcium/Calmodulin Dependent Protein Kinase II. Front. Cardiovasc. Med. 7:630480. doi: 10.3389/fcvm.2020.630480
Received: 17 November 2020; Accepted: 29 December 2020;
Published: 01 February 2021.
Edited by:
Carlo Gabriele Tocchetti, University of Naples Federico II, ItalyReviewed by:
Edoardo Bertero, University Hospital Würzburg, GermanyCopyright © 2021 McMullen, Chalmers, Wood, Cunningham and Currie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Currie, c3VzYW4uY3VycmllQHN0cmF0aC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.