
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 25 January 2021
Sec. Cardiac Rhythmology
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.623922
 Tore Kristian Danielsen1,2
Tore Kristian Danielsen1,2 Mani Sadredini1,2
Mani Sadredini1,2 Ravinea Manotheepan1,2
Ravinea Manotheepan1,2 Jan Magnus Aronsen1,3
Jan Magnus Aronsen1,3 Michael Frisk1,2
Michael Frisk1,2 Marie Haugsten Hansen1,2Kjetil Wessel Andressen4Karina Hougen1Finn Olav Levy4
Marie Haugsten Hansen1,2Kjetil Wessel Andressen4Karina Hougen1Finn Olav Levy4 William E. Louch1,2
William E. Louch1,2 Ole Mathias Sejersted1,2Ivar Sjaastad1,2
Ole Mathias Sejersted1,2Ivar Sjaastad1,2 Mathis Korseberg Stokke1,2*
Mathis Korseberg Stokke1,2*Aim: Dysfunction of the cardiac ryanodine receptor (RyR2) is an almost ubiquitous finding in animal models of heart failure (HF) and results in abnormal Ca2+ release in cardiomyocytes that contributes to contractile impairment and arrhythmias. We tested whether exercise training (ET), as recommended by current guidelines, had the potential to stabilize RyR2-dependent Ca2+ release in rats with post-myocardial infarction HF.
Materials and Methods: We subjected male Wistar rats to left coronary artery ligation or sham operations. After 1 week, animals were characterized by echocardiography and randomized to high-intensity interval ET on treadmills or to sedentary behavior (SED). Running speed was adjusted based on a weekly VO2max test. We repeated echocardiography after 5 weeks of ET and harvested left ventricular cardiomyocytes for analysis of RyR2-dependent systolic and spontaneous Ca2+ release. Phosphoproteins were analyzed by Western blotting, and beta-adrenoceptor density was quantified by radioligand binding.
Results: ET increased VO2max in HF-ET rats to 127% of HF-SED (P < 0.05). This coincided with attenuated spontaneous SR Ca2+ release in left ventricular cardiomyocytes from HF-ET but also reduced Ca2+ transient amplitude and slowed Ca2+ reuptake during adrenoceptor activation. However, ventricular diameter and fractional shortening were unaffected by ET. Analysis of Ca2+ homeostasis and major proteins involved in the regulation of SR Ca2+ release and reuptake could not explain the attenuated spontaneous SR Ca2+ release or reduced Ca2+ transient amplitude. Importantly, measurements of beta-adrenoceptors showed a normalization of beta1-adrenoceptor density and beta1:beta2-adrenoceptor ratio in HF-ET.
Conclusion: ET increased aerobic capacity in post-myocardial infarction HF rats and stabilized RyR2-dependent Ca2+ release. Our data show that these effects of ET can be gained without major alterations in SR Ca2+ regulatory proteins and indicate that future studies should include upstream parts of the sympathetic signaling pathway.
Current guidelines recommend exercise training (ET) as part of rehabilitation programs after myocardial infarction (MI) and for patients with heart failure (HF) (1, 2). ET has a range of beneficial cardiovascular effects (3) including improved aerobic capacity and cardiac contractile function (4–6) and decreases mortality after MI and in patients with HF (7, 8). However, approximately half of all patients with HF still die of ventricular arrhythmias (9). To exploit fully the therapeutic potential of ET in HF, we need a better understanding of its effects on the mechanisms for arrhythmias in HF.
The mechanism that underlies increased risk of arrhythmias in post-MI HF is multifactorial and includes fibrosis, altered expression or function of ion channels, and perturbed Ca2+ homeostasis (10). An almost ubiquitous finding in animal models of post-MI HF is a dysfunction of the cardiac ryanodine receptor (RyR2), i.e., the Ca2+ release channel of the sarcoplasmic reticulum (SR) (11, 12). This channel is essential in normal cellular Ca2+ handling and excitation–contraction coupling (13). RyR2 dysfunction can lead to diastolic Ca2+ leak from the SR, which contributes to contractile impairment and arrhythmias (14). Inhibition of SR Ca2+ leak has been found to prevent arrhythmias in animal models of HF, as well as arrhythmogenic events in cardiac tissue taken from patients with HF, and is a potential future therapeutic strategy (15, 16). However, no drugs that specifically target RyR2 are clinically available. Based on results from mouse models that show RyR2 dysfunction, ET might have the potential to stabilize RyR2 function and prevent arrhythmias (17, 18). However, there is a scarcity of data that support such an effect of ET in post-MI HF, and therefore, there is a need for rigorous experimental data from clinically relevant models.
This investigation was approved by the Norwegian National Committee for Animal Welfare under the Norwegian Animal Welfare Act (FOTS ID: 4173 and 6577). It conformed to the National Institutes of Health guidelines (NIH Publication No. 85-23, revised 1996, US).
A total of 52 male Wistar rats were included in the study. They either underwent sham operations or were subjected to MI by left coronary artery ligation. Left coronary artery ligation was performed through a thoracotomy under general anesthesia, which was achieved by inhalation of 65% N2O, 32% O2, and 2.5% isoflurane through an endotracheal tube, as previously described (19). Through a 2-cm incision in the skin over the sternum, the cutis on the left thorax was loosened from the underlying layer, and a left thoracotomy was performed in the fourth intercostal space. The pericardial sac was opened, and the left coronary artery was ligated ~1 mm beneath the left atrium. In the control (sham) rats, the same operating procedure was employed, but the left coronary artery ligation was not performed. The skin was closed by sutures. Buprenorphine 0.2 mg/kg was administered subcutaneously for analgesia.
Rats with MI were evaluated after 1 week by 2D M-mode echocardiography. Based on previously established criteria, rats that exhibited a left atrial diameter of >5.0 mm were included in the HF group (15, 17). Rats within the same group (sham or HF) were paired according to weight and were randomly assigned either to a 5-week high-intensity ET program or to a sedentary, i.e. non-exercising, control group. The rats were housed together in cages under a 12:12 h light:dark cycle, with free access to water and food.
ET was initiated 1 week after coronary artery ligation. Separate treadmill and metabolic chambers were used for exercise training and for weekly measurements of maximal oxygen uptake (VO2max) (Columbus Instruments, OH, USA), respectively. Three days before the first VO2max-test, rats were habituated to the treadmill with daily 15-min exercise bouts at walking pace.
High-intensity exercise training has been shown to increase aerobic capacity in rats more than moderate exercise training (6). Pilot data, as well as previous publications, showed that the effect on VO2max reached a plateau at 5 weeks (6, 20). Therefore, all ET rats completed a 5-week high-intensity ET protocol of treadmill running. Each training session lasted for 1 h. It consisted of a 10-min warm-up period and five 8-min intervals at 80–90% of the running speed that had been found to produce VO2max, interspersed by 2-min rest periods at 60% running speed. The treadmill was set at an inclination of 25° at all times. The running speed was individualized for all rats according to a weekly VO2max-test to keep the intensity constant throughout the ET program. SED rats ran on the treadmill at 60% of the speed that was found to produce VO2max for 15 min, 2 days per week. All rats in the ET groups continued daily exercise until 1 day before euthanasia.
Echocardiographic parameters were recorded with a Vevo2100 System (Fujifilm VisualSonics Inc., Canada) in rats that were anesthetized by a mixture of 65% N2O, 33% O2, and 2% isoflurane by mask ventilation. Echocardiography was performed 1 week after coronary ligation to enable stratification, and it was repeated at completion of the ET program (21).
Ventricular cardiomyocytes were isolated using constant-pressure perfusion of the coronary arteries with collagenase-containing solution (solution containing in mM 130 NaCl, 25 HEPES, 22 D-glucose, 5.4 KCl, 0.5 MgCl2, 0.4 NaH2PO4, adjusted to pH 7.4 with NaOH), as previously described (22). The hearts were mounted on a Langendorff setup and were perfused retrogradely through the aorta with a 37°C solution that contained 0.08 mM Ca2+ and 200 U/ml collagenase type 2 (Worthington Biochemical Corporation, Lakewood, NJ, USA). The hearts were perfused for 20 min before the left ventricular tissue was excised rapidly, and the infarcted area was removed. The ventricular tissue was then cut into small pieces and was gently mixed with a cutoff Pasteur pipette for about 1 min in a buffer that contained 1% bovine serum albumin (BSA) (Sigma Aldrich) and 0.02 U/ml deoxyribonuclease 1 (Worthington Biochemical Corporation, Lakewood, NJ, USA). The solution with the ventricular tissue was then filtered through a nylon mesh (pore diameter, 200 μm). After sedimentation, the cells were resuspended three times in 1% BSA solutions with increasing Ca2+ concentration (0.1, 0.2, and 0.5 mM, respectively). The cells were used for experiments within 10 h.
Whole-cell Ca2+ imaging was performed with a Zeiss Axiovert 200M microscope (Carl Zeiss Microscopy, LCC, NY, USA). Left-ventricular cardiomyocytes were field stimulated at 1, 2, and 4 Hz by a 3-ms symmetrical bipolar pulse, which was ~20% above the voltage threshold for contraction of the individual cardiomyocytes. The experimental superfusate was based on modified HEPES–Tyrode's solution, and contained (in mM) HEPES 5, NaCl 140, KCl 5.4, MgCl2 0.5, glucose 5.5, NaH2PO4 0.4, and CaCl2 1. pH was adjusted to 7.4 with NaOH. Cytosolic Ca2+ was visualized the by use of 5 μM fluo-4 acetomethyl (AM) ester (Molecular Probes, Eugene, Oregon, USA), with 10-min loading before experiments. All experiments were performed at 37°C. For beta-adrenoceptor activation, 10 nM isoprenaline sulfate (ISO) (NAF, Norway) was added to the modified HEPES–Tyrode's solution. After addition of ISO, the Ca2+ transient amplitude reached steady state after ~1 min. The cardiomyocytes were stimulated for another 30 s before measurements were made of the Ca2+ transients. Ca2+ waves were measured in a 10-s pause after the stimulation period. SR Ca2+ removal rate was estimated as the average rate of decay (k = 1/tau) that was taken from the last three Ca2+ transients before a pause. SR Ca2+ content was measured as peak fluorescence after rapid application of 10 mM caffeine at 1 Hz in the presence and absence of ISO. SERCA2-dependent Ca2+ removal was measured as the difference between the decay rate of Ca2+ transients and the decay rate of SR Ca2+ release after exposure to 10 mM caffeine. Ca2+ removal rate by the sodium–calcium exchanger (NCX) and plasma membrane Ca2+ ATPase (PMCA) was measured as the decay rate of the caffeine-induced Ca2+ release.
The relative increase in diastolic Ca2+ during pacing was measured as the difference in mean F0 between 1, 2, and 4 Hz for three Ca2+ transients after 30 s stimulation.
Fractional release was measured as mean F of 3 Ca2+ transients after 30 s stimulation divided with F for caffeine release.
Confocal microscopy line-scan imaging was performed using a Zeiss LSM 7 Live confocal microscope (Carl Zeiss Microscopy, LCC, NY, USA). Cardiomyocytes were stimulated at 1 and 4 Hz in a protocol that was similar to the one described for whole-cell Ca2+ imaging measurements of Ca2+ waves, but with a 6-s post-stimulation rest period in which Ca2+ sparks were recorded.
To assess beta-adrenoceptor density, a radioligand binding assay was performed on left ventricles snap frozen in liquid nitrogen. Crude cell membrane fractions were prepared as described by Krobert et al. (23). Radioligand binding was performed as described by Ramberg et al. (24), where membranes were incubated with [125I]-(–)iodocyanopincolol (0.066 nM) and the indicated concentration of either CGP20712A or ICI118551 for 2 h at 23°C. Data were fitted to a two-site binding model, and high and low binding affinities (pKi) were calculated in GraphPad Prism 8.0.1 using a Kd of 0.04 nM (affinity of [125I]-(–)iodocyanopincolol was determined in the left ventricular membranes). Beta1-adrenoceptor density was determined as an average of high-affinity CGP20712A and low-affinity ICI118551 binding in the same heart. Similarly, beta2-adrenoceptor density was determined as an average of high-affinity ICI118551 and low-affinity CGP20712A binding in the same heart.
Western blots were performed on total protein homogenates from the left ventricles that had been stored at −70°C. Homogenates were denatured at 100°C for 5 min or at 37°C for 10 min in a sample buffer that contained 50% sucrose, 7.5% sodium dodecyl sulfate (SDS), 62.5 mM Tris–HCl, 2 mM ethylenediaminetetraacetic acid (EDTA), 0.2 M dithiothreitol (DTT), and 0.01% bromophenol blue. Proteins were then fractionated according to size on 4–15% Criterion TGX gels (26 wells, 15 μl, Cat no. 567-1085, Bio-Rad Laboratories, Oslo, Norway) and blotted on 0.45 μM polyvinylidene fluoride (PVDF) membranes (GE Healthcare, Oslo, Norway). The examination of phospholamban (Plb) was performed through use of 18% Criterion TGX gels (26 wells, 15 μl, Cat no. 567-1075, Bio-Rad Laboratories, Oslo, Norway). Membranes were blocked in 5% non-fat milk in Tris-buffered saline with 0.1% polysorbate 20 for 1 h at room temperature. Then, they were incubated with primary antibody overnight at 4°C and with secondary antibody for 1 h at room temperature. Blots were developed by application of enhanced chemiluminescence (ECL prime, GE Healthcare, Oslo, Norway), and signals were quantified using ImageQuant software (GE Healthcare, Oslo, Norway).
The primary antibodies for protein detection were anti-RYR2 phosphoserine-2814 (A010-31, Badrilla, Leeds, UK), anti-RYR2 phosphoserine-2808 (A010-30, Badrilla, Leeds, UK), ryanodine receptor (MA3-916, Thermo Fisher Scientific Inc., Rockford, IL, USA), anti-CaMKII (phospho-threonine286) (ab32678, Abcam PLC, Cambridge, UK), CaMKIIδ (22), anti-phospholamban phosphoserine-16, (A010-12, Badrilla, Leeds, UK), anti-phospholamban phospho-threonine-17, (A010-13, Badrilla, Leeds, UK), and anti-SERCA2a, Cat no. MA3-919 (Thermo Fisher Scientific Inc., Rockford, IL, USA), beta1 adrenergic receptor antibody (PA1-049, Thermo Fisher), and monoclonal antivinculin antibody produced in mouse (V9131, Sigma-Aldrich). Secondary antibodies were antirabbit or antimouse immunoglobulin G (IgG) horseradish peroxidase (HRP)-linked whole antibodies, Cat no. NA934/NA931 (GE Healthcare, Oslo, Norway), diluted in the ratio 1:5,000. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (v-18, sc-20357, Santa Cruz Biotechnology Inc., CA, USA) was used as the loading control for all Western blots, except for phosholamban, and for beta1 adrenergic receptor, for which vinculin was used. The membrane was reprobed with the specified antibodies with stripping of the membrane between each antibody when GAPDH was used as the loading control. HeLa Whole-cell lysate (sc-2200, Santa-Cruz) was used as a positive control for Western blots of beta1-adrenoceptor.
All experiments and analyses were performed by investigators who were blinded to the phenotype and group identity of each cell and animal. Statistical tests were selected based on advice from an external statistical advisor. A paired Student's t-test was used to compare the effect of exercise training on VO2max at baseline and after 5 weeks within each group. Unpaired nested ANOVA was used to compare ET and SED for all results from whole-cell Ca2+ imaging. In addition, two-way ANOVA was used to compare difference between ET and SED in the presence or absence of ISO at all frequencies for Ca2+ waves, Ca2+ transient amplitude, and Ca2+ decay rate. A unpaird Student's t-test was used to compare differences in Ca2+ waves frequency across frequencies and in absense and presence of ISO for HF cardiomyocytes. Ca2+ spark frequency was analyzed by application of a Poisson test to adjust for skewed distribution. One-way ANOVA was used when comparing beta-adrenoceptors. Bonferroni, Holm–Sidak, or Tukey's correction was performed when appropriate. Normal distribution was assessed with Shapiro–Wilk test. All statistics were performed by using IBM SPSS statistics 27, Sigmaplot 12.5 or R software (version 3.0.2, The R Foundation for Statistical Computing). P < 0.05 was considered statistically significant. Results are reported as the mean ± standard error of mean (SEM).
We performed left coronary artery ligation on 23 male Wistar rats to induce large left ventricular myocardial infarctions. Sham operations were performed on 29 rats. One week after surgery, all rats were examined by echocardiography before randomization to ET or standard sedentary conditions (SED) (Figure 1A).
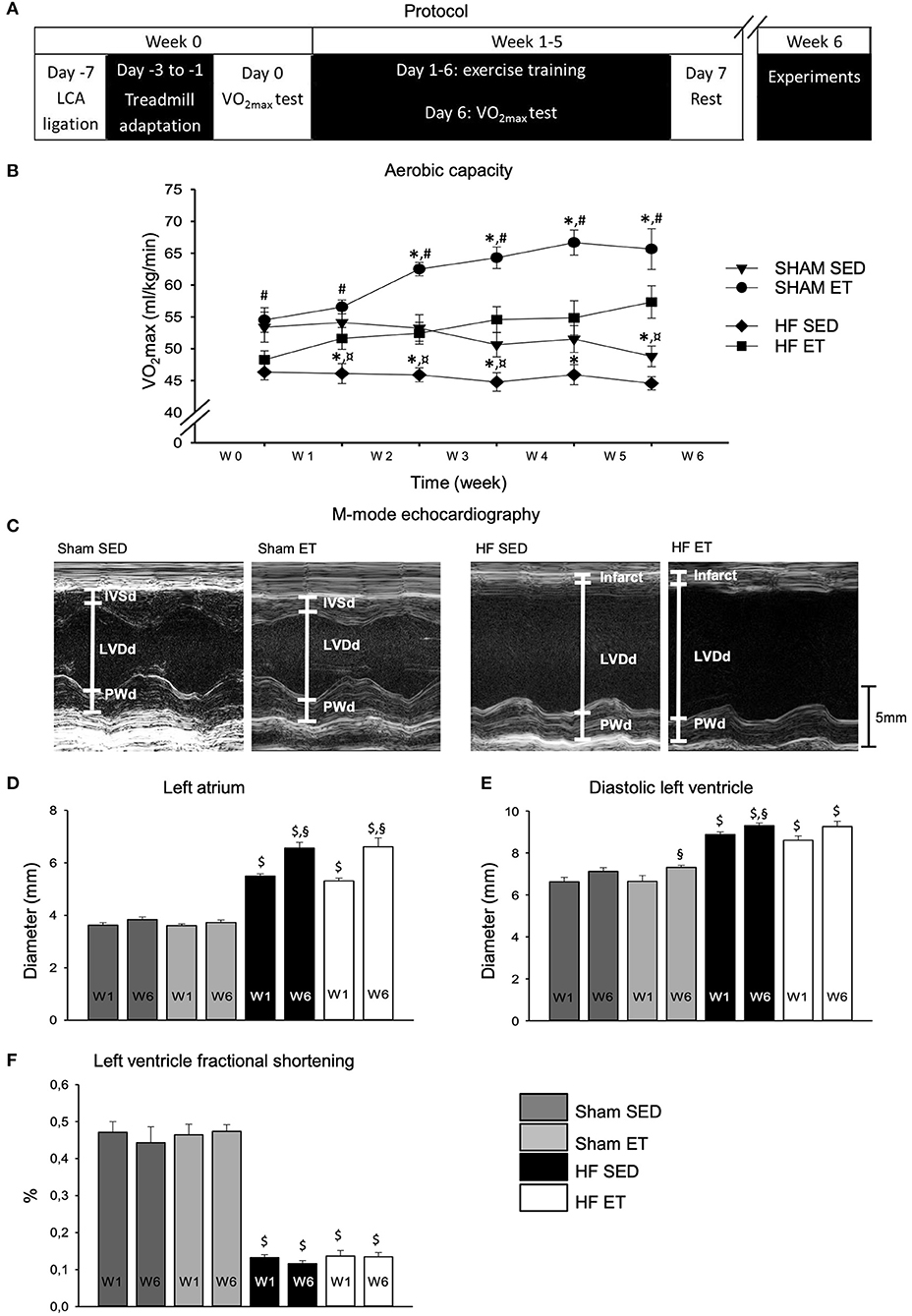
Figure 1. Aerobic capacity and echocardiographic parameters. High-intensity exercise training improved aerobic capacity in sham-ET and HF-ET rats. (A) Exercise training protocol. (B) Mean VO2max measurements from sham and heart failure (HF) rats. Number of rats in each group (SED/ET): sham 15/14, HF 13/10. Echocardiographic parameters were used for stratification of rats to the sham and HF groups. Representative M-mode images from the left ventricle in the parasternal long axis are shown for sham-SED (C) (left, n = 9), sham-ET (middle left, n = 8), HF-SED (middle right, n = 12), and HF-ET rats (left, n = 8). Bar graphs of the (D) left atrium diameter, (E) left ventricular diameter in diastole, and (F) left ventricle fractional shortening represent mean ± SEM. P < 0.05: $sham vs. HF; *SED vs. ET; #HF-ET vs. sham-ET; §week 1 vs. week 6; ¤HF SED vs. sham SED (Paired Student's t-test).
One week after surgery, the aerobic capacity of HF-ET rats was 86% of that of the sham-ET rats (P < 0.05) but did not differ from that of the HF-SED rats (Figure 1B). During the ET protocol, VO2max increased in both ET groups, as expected. After 5 weeks of ET, VO2max in the HF-ET group was 127% of that of the HF-SED group (P < 0.05), while VO2max in the sham-ET group was 119% of that of the sham-SED group (P < 0.05, Figure 1B). However, VO2max in the HF-ET group was only 88% of that of the sham-ET group (Figure 1B, P < 0.05).
Echocardiography was performed before randomization and repeated after completion of the ET protocol (Figure 1C). As per definition, the left atrial diameter was larger in the HF groups than in the sham groups at the time of randomization 1 week after surgery and had increased further in both HF groups when examined at week 6 (P < 0.05, Figure 1D). One week after surgery, HF rats also showed a clear phenotype with increased left ventricular diameter and reduced fractional shortening (P < 0.05, Figures 1E,F). During 5 weeks of ET, the left ventricular diastolic diameter (Figure 1E) and fractional shortening remained unchanged in the HF-ET group (Figure 1F), and ET did not affect any of the echocardiographic parameters in sham-operated rats (Figures 1D–F).
We isolated cardiomyocytes from the left ventricles of the HF-ET and HF-SED rats at week 6 for the analysis of RyR2-dependent spontaneous Ca2+ release. We first recorded cardiomyocyte-wide propagated Ca2+ release events, i.e. Ca2+ waves, after a train of electrical stimuli at increasing frequencies (Figures 2A,B). We performed this protocol in the absence and presence of ISO to simulate sympathetic activation with beta-adrenoceptor stimulation. Ca2+ wave frequency increased with increasing pacing frequency and during exposure to ISO (Figure 2B). Importantly, Ca2+ wave frequency during ISO was lower in HF-ET cardiomyocytes than in those of HF-SED rats at 1 Hz (Figure 2B) and when considered across all frequencies with two-way ANOVA (Figure 2C). This finding indicated that ET stabilized Ca2+ handling in HF.
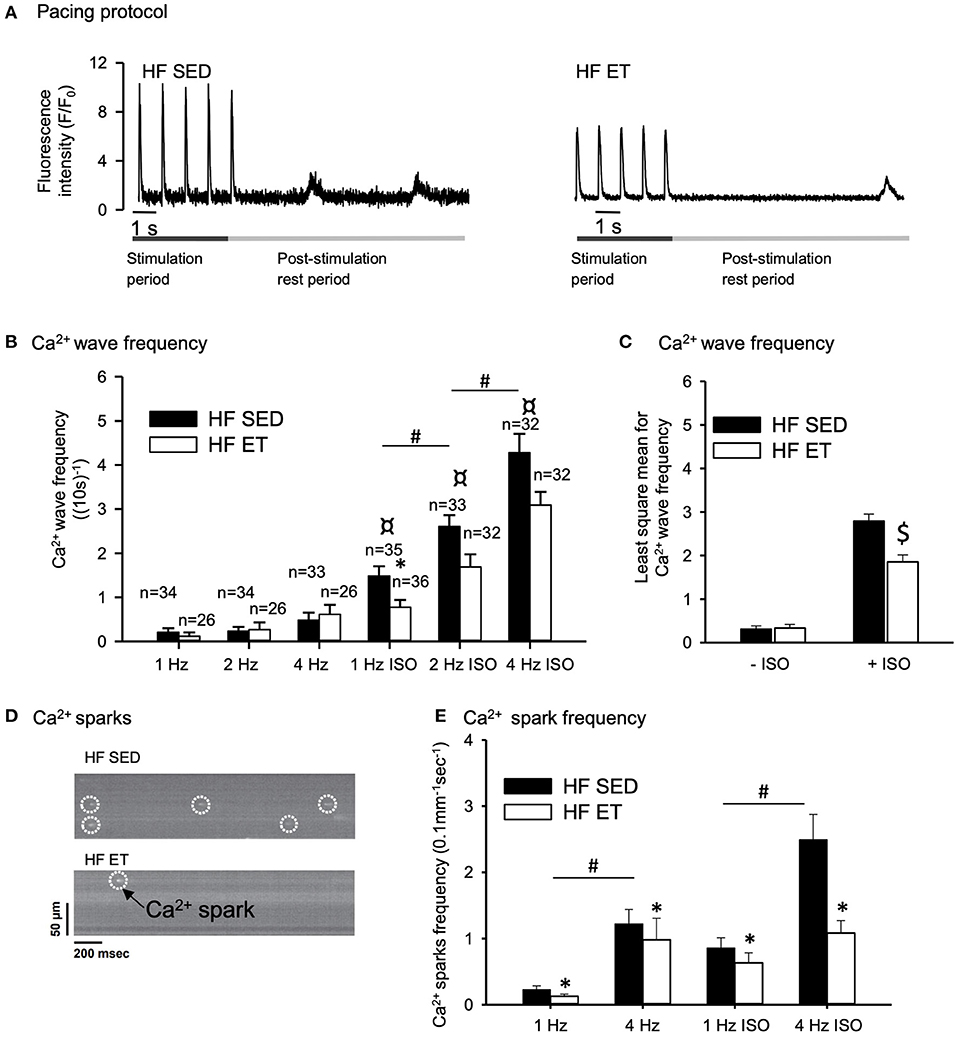
Figure 2. Effect of exercise training on spontaneous Ca2+ release in heart failure (HF) cardiomyocytes. (A) Representative Ca2+ wave recordings and (D) confocal images of Ca2+ sparks are shown after field stimulation of HF-SED and HF-ET cardiomyocytes that were exposed to isoprenaline sulfate (ISO). Pacing at 1, 2, and 4 Hz was provided, and ISO was added to the experimental solution. Bar graphs of (B) Ca2+ wave frequency, (C) Ca2+ wave frequency across all pacing frequencies, and (E) Ca2+ spark frequency represent mean ± SEM. Number of rats in each group (SED/ET): Ca2+ wave frequency: n = 6/5, Ca2+ spark frequency: n = 10/8. Number of cells used for measurements of Ca2+ spark frequency (SED/ET): without ISO 101/81, with ISO 88/75. P < 0.05: *HF-SED vs. HF-ET (unpaired nested ANOVA); *(Poisson test for Ca2+ spark frequency, Bonferroni); #1 Hz vs. 2 and 4 Hz (Unpaired Student's t-test); ¤ +ISO vs. –ISO at same frequency (Unpaired Student's t-test), $least square mean for HF-SED vs. HF-ET (two-way ANOVA, Holm-Sidak).
We also measured local Ca2+ release events, i.e., Ca2+ sparks, by confocal microscopy after a train of electrical stimuli (Figure 2D). The frequency of Ca2+ sparks increased with stimulation frequency (from 1 to 4 Hz), both in the presence and absence of ISO (P < 0.05), and was clearly lower in the HF-ET than in the HF-SED cardiomyocytes (Figure 2E). This further indicated that ET stabilized SR Ca2+ handling in HF.
If ET is to be used as an antiarrhythmic intervention, negative effects on systolic Ca2+ release responsible for contraction should not outweigh the positive effects of ET on RyR2-dependent spontaneous Ca2+ release associated with arrhythmias. Surprisingly, however, when we analyzed Ca2+ transients across different pacing frequencies, the amplitude was lower in HF-ET than HF-SED both in the absence and presence of ISO (Figures 3A–D). Ca2+ transient decay rate was also lower in HF-ET than HF-SED, but only during exposure to ISO (Figures 3E,F). To explain these findings, SR Ca2+ content and Ca2+ removal were analyzed during caffeine exposure (Figure 3G). However, no difference in SR Ca2+ content was observed between HF-ET and HF-SED (Figure 3H) nor could the slowed removal be attributed to SERCA2-dependent or sarcolemmal Ca2+ removal alone (Figures 3I,J). We also analyzed the increase in diastolic Ca2+ that is expected in response to increased pacing frequencies but found no difference between HF-ET and HF-SED (Figure 3K). Lastly, we calculated fractional SR Ca2+ release but only observed a small increase in HF-ET compared HF-SED during ISO, which cannot explain stabilized RyR2-dependent SR Ca2+ release (Figure 3L) (25).
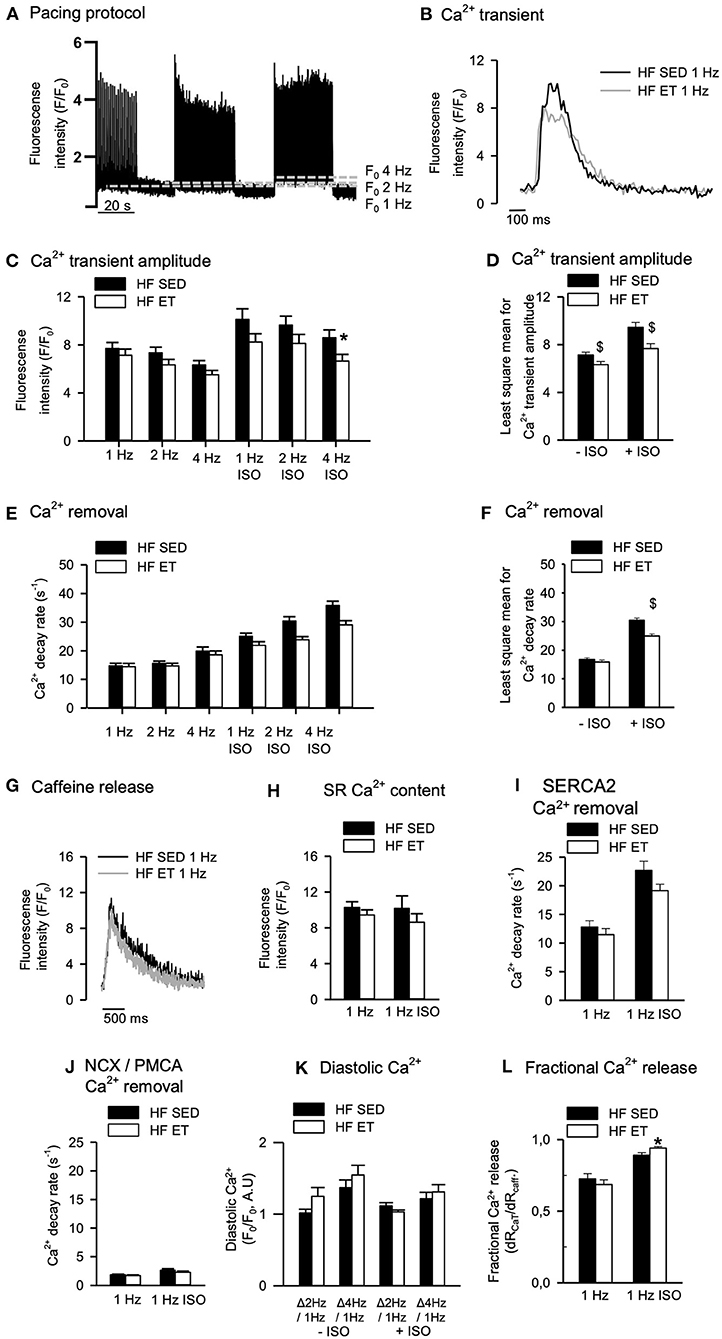
Figure 3. Effects of exercise training on Ca2+ handling in heart failure (HF) cardiomyocytes. (A) Representative pacing protocol and (B) Ca2+ transients for HF-SED and HF-ET cardiomyocytes during 1 Hz stimulation and exposure to isoprenaline sulfate (ISO). (C) Ca2+ transient amplitude was reduced after exercise training at high stimulation frequency and ISO exposure and (D) across all frequencies with and without ISO exposure. (E,F) Ca2+ decay rate was lower across all frequencies with ISO exposure. (G) Representative tracings of SR Ca2+ content, measured by peak F/F0 in response to a rapid application of 10 mM caffeine. Exercise training (H) did not affect SR Ca2+ content and (I) did not alter Ca2+ removal by SERCA2a or sodium–calcium exchanger (NCX) and plasma membrane Ca2+ ATPase (PMCA), as (J) measured by the decay rate of the caffeine-induced Ca2+ release. (K) Exercise training did not alter diastolic Ca2+; (L) however, fractional release was higher in HF-ET compared to HF-SED when exposed to ISO. Number of rats (SED/ET): 6/5. Number of cells in C–F, K (SED/ET): –ISO 34/26, +ISO 35/36. Number of cells in H–J, L (ET/SED): HF 26/22, ISO 22/28. Bar graphs represent mean ± SEM. P < 0.05: *HF-SED vs. HF-ET (unpaired nested ANOVA), $least square mean for HF-SED vs. HF-ET (two-way ANOVA, Holm-Sidak).
To explore other potential explanations for the stabilization of RyR2-dependent spontaneous Ca2+ release that was observed after ET, phosphoproteins levels were quantified in HF-ET and HF-SED (Figure 4). We focused on RyR2 and the SERCA/Plb system as the proteins that are mainly responsible for SR Ca2+ release and reuptake. However, no differences in these proteins or their major regulatory phosphorylation sites were observed (Figures 4A–I). The Plb/SERCA2a ratio was also unaltered in HF-ET compared with that in HF-SED (P = 0.68).
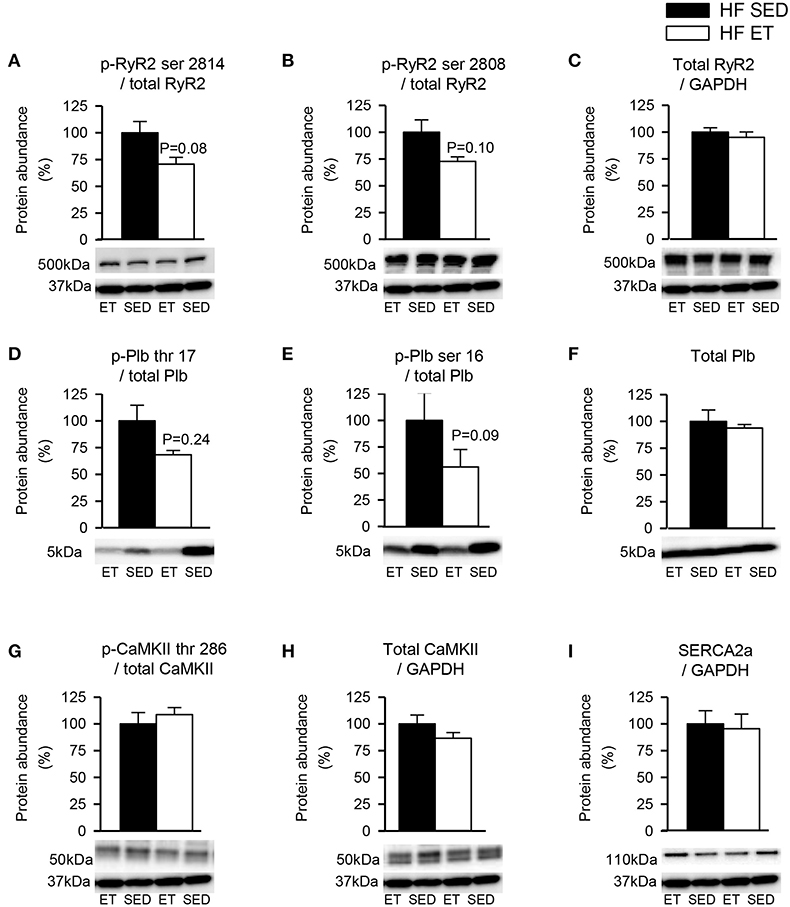
Figure 4. Protein phosphorylation levels in heart failure (HF) left ventricles. Exercise did not decrease phosphorylation at sites that are known to be involved in destabilization of RyR2. Phosphoprotein levels were normalized to total protein levels. RyR2 phosphorylated at (A) serine 2814 (p-RyR2 ser 2814) and (B) serine 2808 (p-RyR2 ser 2808) were normalized to (C) total RyR2. Plb phosphorylated at (D) threonine 17 (p-Plb thr 17) and (E) serine 16 (p-Plb ser 16) were normalized to (F) total Plb and CaMKII phosphorylated at (G) threonine 286 (p-CaMKII thr 286) was normalized to (H) total CaMKII. (I) SERCA2a level was unaltered by exercise training. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as loading control, except for Plb, and the membrane was reprobed with the specified antibodies with stripping of the membrane between each antibody, shown at 37 kDa. (C,F,H,I) Total protein levels were normalized to GAPDH levels. Number of rats (SED/ET): 6/5.
To control for untoward effects of ET, we performed most experiments in parallel in sham-ET and sham-SED rats (Figures 5, 6). The ET protocol lowered Ca2+ wave frequency even in sham-ET compared to sham-SED both in the presence and absence of ISO when compared across all frequencies with two-way ANOVA (Figures 5A–C). However, Ca2+ spark frequency was similar in sham-ET and sham-SED (Figures 5D,E). ET had no effect on Ca2+ transients amplitude (Figures 6A–C), Ca2+ removal rate, diastolic Ca2+ (Figures 6D–F), SR Ca2+ content (Figures 6G,H), or RyR2 phosphorylation (Figures 6I,J) in sham-operated animals.
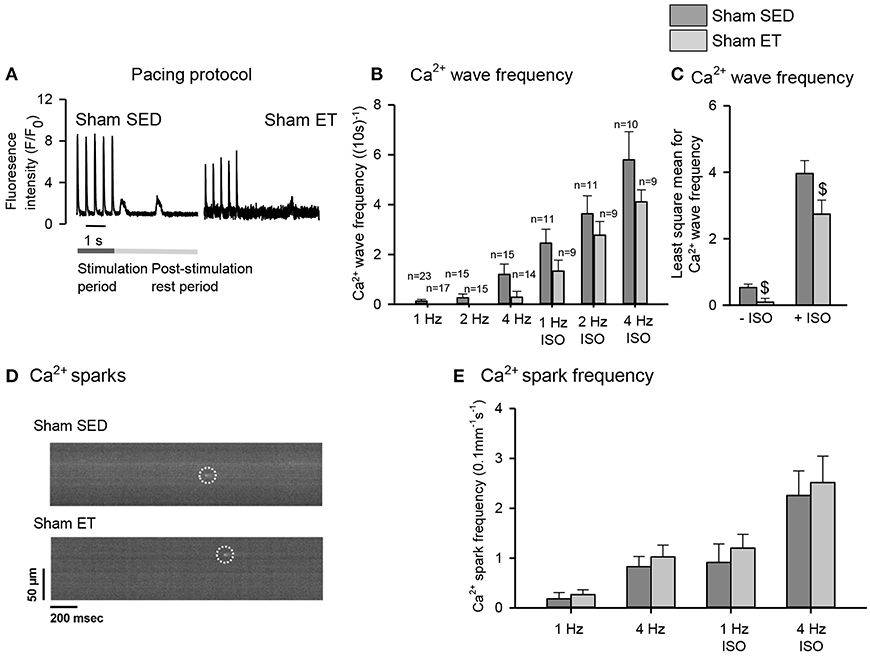
Figure 5. Effect of exercise training on spontaneous Ca2+ release in sham cardiomyocytes. (A) Representative Ca2+ wave recordings, bar graphs of (B) Ca2+ wave frequencies at different pacing frequencies in the absence and presence of isoprenaline sulfate (ISO) and (C) two-way ANOVA comparing HF-ET with HF-SED. (D) Confocal images of Ca2+ sparks, and (E) bar graphs of Ca2+ spark frequency after different pacing frequencies. Number of rats (SED/ET): Ca2+ waves 4/3; Ca2+ sparks 7/6; number of cells (SED/ET): Ca2+ sparks: –ISO (60/53), +ISO (46/48); Ca2+ handling: –ISO (23/17), +ISO (10/9). P < 0.05: $least square mean for sham-SED vs. sham-ET (two-way ANOVA, Holm-Sidak).
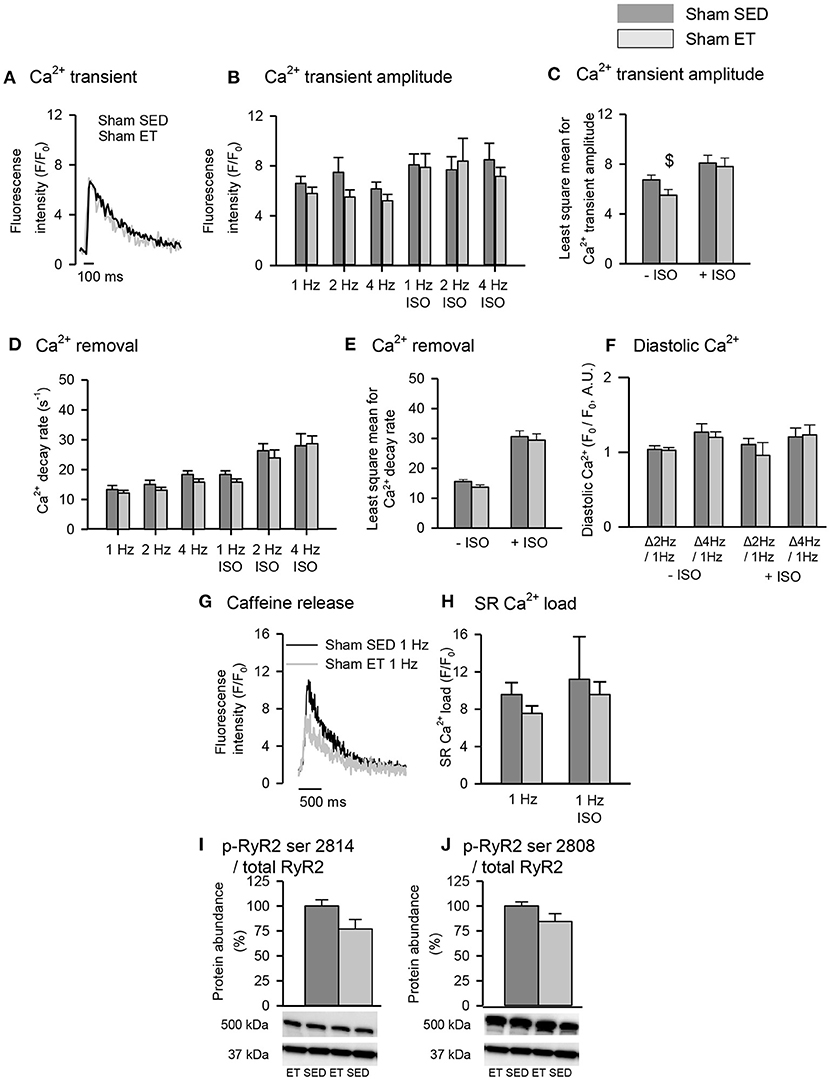
Figure 6. Effect of exercise training on Ca2+ handling in sham cardiomyocytes. (A) Representative Ca2+ transient recordings is shown after field-stimulation of sham-SED and sham-ET cardiomyocytes. Stimulation of 1, 2, and 4 Hz was provided, and isoprenaline sulfate (ISO) was added to the experimental solution. Bar graphs of (B,C) Ca2+ transient amplitude, (D,E) Ca2+ removal and (F) diastolic Ca2+ represent mean ± SEM. (G) Representative tracings of SR Ca2+ content, measured by peak F/F0 in response to a rapid application of 10 mM caffeine at 1 Hz. (H) Bar graphs of SR Ca2+ load, represents mean ± SEM. Phosphoprotein levels were normalized to total protein levels. RyR2 phosphorylated at (I) serine 2814 (p-RyR2 ser 2814) and (J) serine 2808 (p-RyR2 ser 2808). (I,J) Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as loading control, and the membrane was reprobed with the specified antibodies with stripping of the membrane between each antibody, shown at 37 kDa. Number of rats (SED/ET): Ca2+ handling (4/3); proteins (5/5). Number of cells (SED/ET): Ca2+ handling: –ISO (23/17), +ISO (10/9). P < 0.05: $least square mean for sham-SED vs. sham-ET (two-way ANOVA, Holm-Sidak).
As an alternative explanation for our observations, we examined if beta-adrenoceptor density was affected by exercise. For this, we used radioligand binding on membranes from left ventricular cardiomyocytes. The beta1-adrenoceptor selective ligand CGP20712A displayed a pKi−high of 9.21 ± 0.02 and a pKi−low of 5.7 ± 0.03 (Figure 7A), while the beta2-adrenoceptor selective ligand ICI118551 displayed a pKi−high of 9.32 ± 0.04 and a pKi−low of 6.9 ± 0.04 (Figure 7B), similar to previously reported values (26). The total beta-adrenoceptor density did not differ between the four groups (HF-ET, HF-SED, sham-ET, sham-SED, Figure 7C). Importantly, however, the beta1-adrenoceptor density was significantly reduced in the HF-SED compared to sham-SED but increased back to normal levels in HF-ET (Figures 7D,E). The beta2-adrenoceptor density, on the other hand, was increased in HF-SED compared to sham-SED and was not affected by ET (Figure 7F). In contrast to the effects of ET on membrane beta1-adrenoceptor density, total abundance of the receptor measured by Western blotting was not altered by ET (Supplementary Figure 1).
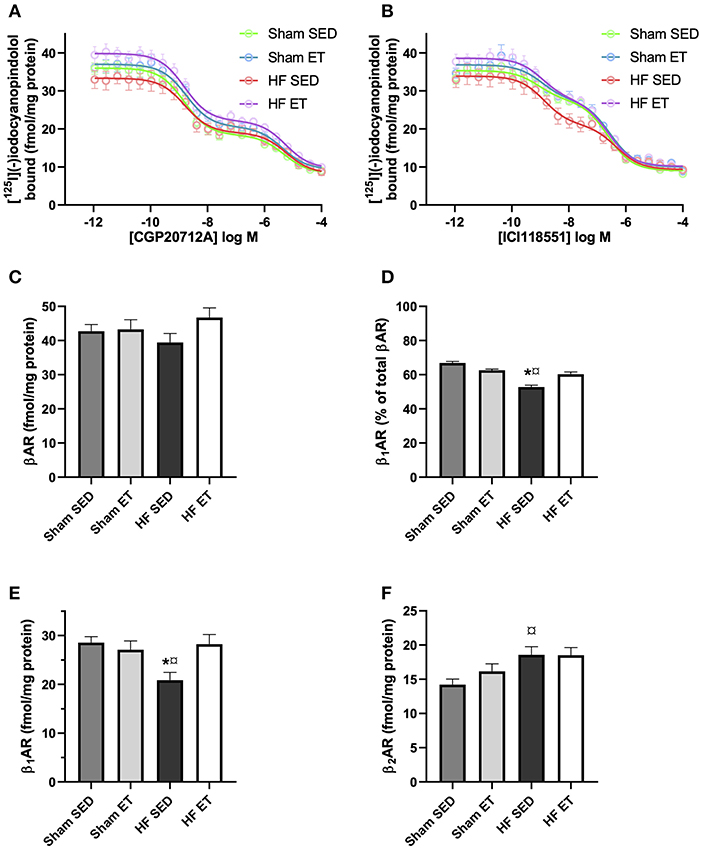
Figure 7. Beta-adrenoceptor density in heart failure (HF) and sham left ventricles. Competitive binding assays between [125I]-(–)iodocyanopindolol and the (A) beta1-adrenoceptor selective ligand CGP20712A or the (B) beta2-adrenoceptor selective ligand ICI118551 in membranes from the left ventricle from animals with the indicated treatment. The (C) total beta-adrenoceptor density and (D) percentage of beta1-adrenoceptor was determined by a two-site binding model, where the average of high-affinity CGP20712A and low-affinity of ICI118551 is presented. (E) Beta1-adrenoceptor density was determined as the average of high affinity of CGP20712ACFP20712A displacement and low affinity of ICI118551 displacement. (F) Beta2-adrenoceptor density was determined as the average of high affinity of ICI118551 displacement and low affinity of CGP20712ACFP20712A displacement. The data shown are mean ± SEM of five (sham SED), four (sham ET), six (HF SED), and six (HF ET) animals. P < 0.05: *SED vs. ET, ¤HF SED vs. sham SED (one-way ANOVA, Tukey's correction).
We tested the hypothesis that ET could stabilize RyR2-dependent SR Ca2+ release associated with arrhythmias in post-MI HF rats. We subjected rats with HF to a 5-week ET protocol that was initiated 1 week after the induction of MI by ligation of the left coronary artery. This protocol increased the aerobic capacity of HF-ET rats to 127% of that of HF-SED rats but did not prevent a further increase in atrial diameter and had no impact on left ventricular diameter or on contractile function measured as fractional shortening. ET reduced the frequency of spontaneous Ca2+ release events in left ventricular cardiomyocytes, as indicated by a reduced frequency of Ca2+ waves and Ca2+ sparks in HF-ET cells compared with the HF-SED cells. In situations associated with increased adrenergic stress and beta-adrenoceptor stimulation, the HF-ET group exhibited lower Ca2+ transient amplitudes and decay rates than the HF-SED group, but no significant changes in RyR2, SERCA2 or their major regulatory proteins or phosphorylation sites. Sham-operated ET rats also exhibited a 135% increase in aerobic capacity compared to sham-SED and lower Ca2+ wave frequency when measured across all frequencies. However, we observed no changes in echocardiographic parameters or changes in Ca2+ measurements or SR Ca2+ regulatory proteins that can explain this. Since the Ca2+ regulatory proteins could not explain our main observation, we tested an alternative hypothesis and found that beta1-adrenoceptors were downregulated in HF-SED but normalized with ET.
The pathophysiological changes that occur in post-MI HF are highly complex and progress over time (27). After a large MI, structural and mechanical alterations occur at the organ level, as well as in cardiomyocytes and the extracellular matrix (28). The progression of these alterations leads to different mechanisms being responsible for increased risk of arrhythmias at different time points after an MI (29). Altered Ca2+ handling due to perturbed expression and function of Ca2+ handling proteins is involved in arrhythmogenesis at all stages after MI, mainly because these alterations increase the risk of triggered activity (30, 31). Dysfunctional RyR2 is key in this arrhythmia mechanism and has been observed in several animal models of established post-MI HF (32–34). Our model is in line with other models of HF, with reduced aerobic capacity, severely dilated left ventricular diameters, and increased left-atrial diameters as a sign of long-standing increase in end-diastolic pressure, as well as reduced density of beta1-adrenoceptors (35, 36). Many models of HF with a similar phenotype show post-translational modifications of RyR2, especially increased phosphorylation (12). Thus, dysfunction and modification of RyR2 have received much attention as potential targets for prevention of arrhythmias in HF (37). Our model is therefore highly relevant to test the hypothesis that ET can stabilize RyR2.
There is widespread interest in HF pathophysiology, RyR2 dysfunction, and the beneficial effects of ET in HF. Therefore, it is a surprise that data on the effects of ET on RyR2 function in post-MI HF are scarce. Bonilla et al. subjected dogs with anterior MI to 10 weeks of ET and showed a highly reduced tendency toward ventricular fibrillation compared with sedentary dogs (38). In line with our study, they showed that the frequency of Ca2+ sparks in ventricular cardiomyocytes was reduced in dogs in the exercise group and that the abundance of RyR2 that was phosphorylated at serine 2814 was also reduced, compared with the sedentary group. However, there was no data that indicated that the dogs had HF. Kemi et al. performed a study of post-MI HF rats that showed many similarities with our study, although they employed a different rat strain (39). They too found a positive effect on Ca2+ wave frequency, but they did not analyze phosphoproteins. In other publications, the same group has provided compelling evidence that ET can have beneficial effects on Ca2+ handling and on T-tubule structure in normal rats (5, 17, 20, 40). Based on these and other data of disease models that show dysfunctional RyR2 (17, 18), we hypothesized that ET could have beneficial effects on RyR2 function. We focused specifically on serine 2808 and 2814 in RyR2, as there is mounting evidence that phosphorylation of these residues contributes to SR Ca2+ leak in HF and that prevention of such phosphorylation can prevent arrhythmogenic Ca2+ release (41, 42). It is therefore interesting that we observed beneficial effects on spontaneous SR Ca2+ release, without significant alterations in the abundance of key SR Ca2+ handling proteins, or CaMKII-or protein kinase A (PKA)-dependent phosphorylation. Although previous studies have clearly shown that ET can exert beneficial effects on SR Ca2+ handling through attenuated phosphorylation by CaMKII especially, our results show that other mechanisms may also contribute to a similar result. Interestingly, we observed that Ca2+ transient amplitude and reuptake of Ca2+ was lower in HF-ET rats during adrenergic stress. It was therefore somewhat surprising that this coincided with increased beta1-adrenoceptor density compared to HF-SED. Our radioligand binding experiments show that the number of beta1-adrenoceptors was reduced in HF rats and increased to normal levels by ET (Figure 7). Our experiments do not provide an explanation for how this is compatible with reduced reuptake or why the increased density was not reflected in the total abundance of the receptors when measured by Western blotting. We can only speculate that ET altered the relative pools of these receptors at the cell surface and receptors associated with the SR. Recent studies have shown that internalized and SR-bound beta1-adrenoceptors exert an important regulatory role in SR Ca2+ release (43). These possible effects of ET should be explored further in future studies.
We investigated Ca2+ handling after MI and ET and focused specifically on SR Ca2+ handling. Our studies lead us toward a role of beta-adrenoceptors in the effects of ET in HF rats, although the focus of the study did not allow us to conclude if this represents an explanation to reduced frequency of spontaneous Ca2+ release events after ET. Still, there is a risk that we missed a difference in phosphoprotein abundance after ET, due to low sample size (type II error) in our analysis, and such effects should therefore not be excluded. The reader should be aware that a one-sided test of CaMKII-dependent phosphorylation of RyR2 would have resulted in a P < 0.05. Based on previous data, the a priori assumption that ET would decrease (not increase) CaMKII activity could be claimed, thus supporting a one-sided test. Furthermore, our analyses of phosphoproteins were only performed in the absence of sympathetic stimulation. Analysis performed after f.ex. exposure to ISO could potentially have clarified some of the non-significant observations.
Another potentially contributing factor that should be included in future studies is altered cell ultrastructure. ET can effect cell size and has been shown to alter t-tubule organization (40). These aspects could be of importance but were not included in our study.
Our study is limited to cellular phenomena and does not include measurements of in vivo arrhythmias. This limits the potential for extrapolation to propensity for arrhythmias in vivo. However, other mechanisms are also important for antiarrhythmic effects of ET, such as alterations in repolarization reserve (38) or reductions in fibrosis levels (44), although the latter is debated after findings in healthy rats (45). As these other mechanisms were not the focus of our study, we would not have been able to ascertain a causal link between the cellular phenomena and arrhythmias regardless of any observed effects on arrhythmias.
Our model of HF is based on induction of large MIs, which leads to clear mechanical and structural alterations in the left ventricle at an early time point after the MI. Any extrapolation to HF that develops after smaller MIs, which involves less remodeling, is unclear. Especially, we did not separate cardiomyocytes according to relative localization or distance from the MI, although the effects of ET could vary between regions (46, 47).
We chose to start high-intensity ET at an early time point after MI. High-intensity ET was chosen based on previous studies in rats (6). Moderate-intensity ET might have been more in line with what patients can perform, although high-intensity interval training is feasible even in patients with severe HF (48). Any extrapolation to correspondingly large MIs in humans is speculative, and no conclusion regarding the time at which ET should be initiated in patients can be drawn from studies in rats. The early initiation that was chosen in our study could mean that remodeling processes were still highly active, which could have affected our results. However, it could be argued that this is a reason to stress early initiation of ET in order to affect beneficially the remodeling process. We did not compare effects of exercise in HF and sham rats for all data, as this was not the focus of our study. The effects of ET could be different in diseased and normal animals, and the therapeutic potential should be tested in relevant disease models.
ET can stabilize RyR2-dependent Ca2+ release in post-MI HF. Our data indicate that the protective mechanism involves regulation of both SR Ca2+ release and reuptake, and that effects of ET at several levels of the sympathetic signaling pathways controlling SR Ca2+ release should be explored in future studies.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Norwegian National Committee for Animal Welfare.
TD, MS, RM, JA, MF, MH, KA, KH, and IS conceived and designed the analysis, collected data, contributed to data analysis, performed the analysis, and wrote the paper. FL, WL, OS, and MS conceived and designed the analysis, contributed to data analysis, and wrote the paper. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2020.623922/full#supplementary-material
1. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the european society of cardiology (ESC) developed with the special contribution of the heart failure association (HFA) of the ESC. Eur Heart J. (2016) 37:2129–200. doi: 10.1093/eurheartj/ehw128
2. Piepoli MF, Hoes AW, Agewall S, Albus C, Brotons C, Catapano AL, et al. 2016 European guidelines on cardiovascular disease prevention in clinical practice: the sixth joint task force of the european society of cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European association for cardiovascular prevention & rehabilitation (EACPR). Eur Heart J. (2016) 37:2315–81. doi: 10.1093/eurheartj/ehw106
3. Shephard RJ, Balady GJ. Exercise as cardiovascular therapy. Circulation. (1999) 99:963–72. doi: 10.1161/01.CIR.99.7.963
4. Kemi OJ, Ellingsen Ø, Ceci M, Grimaldi S, Smith GL, Condorelli G, et al. Aerobic interval training enhances cardiomyocyte contractility and Ca2+ cycling by phosphorylation of CaMKII and Thr-17 of phospholamban. J Mol Cell Cardiol. (2007) 43:354–61. doi: 10.1016/j.yjmcc.2007.06.013
5. Wisløff U, Loennechen JPÑ, Falck G, Beisvag V, Currie S, Smith G, et al. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc Res. (2001) 50:495–508. doi: 10.1016/S0008-6363(01)00210-3
6. Kemi OJ, Haram PM, Loennechen JP, Osnes JBr, Skomedal T, Wisløff U, et al. Moderate vs. high exercise intensity: differential effects on aerobic fitness, cardiomyocyte contractility, and endothelial function. Cardiovasc Res. (2005) 67:161–72. doi: 10.1016/j.cardiores.2005.03.010
7. Cattadori G, Segurini C, Picozzi A, Padeletti L, Anzà C. Exercise and heart failure: an update. ESC Heart Fail. (2018) 5:222–32. doi: 10.1002/ehf2.12225
8. O'Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, et al. Efficacy and safety of exercise training in patients with chronic heart failure. JAMA. (2009) 301:1439–50. doi: 10.1001/jama.2009.454
9. Kannel WB, Plehn JF, Cupples LA. Cardiac failure and sudden death in the Framingham Study. Am Heart J. (1988) 115:869–75. doi: 10.1016/0002-8703(88)90891-5
10. Di Diego JM, Antzelevitch C. Ischemic ventricular arrhythmias: experimental models and their clinical relevance. Heart Rhythm. (2011) 8:1963–8. doi: 10.1016/j.hrthm.2011.06.036
11. Kolstad TR, van den Brink J, MacQuaide N, Lunde PK, Frisk M, Aronsen JM, et al. Ryanodine receptor dispersion disrupts Ca(2+) release in failing cardiac myocytes. eLife. (2018) 7:e39427. doi: 10.7554/eLife.39427
12. Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, et al. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. (2011) 90:493–502. doi: 10.1093/cvr/cvr025
13. Bers DM. Cardiac excitation-contraction coupling. Nature. (2002) 415:198–205. doi: 10.1038/415198a
14. Kushnir A, Wajsberg B, Marks AR. Ryanodine receptor dysfunction in human disorders. Biochimica et Biophysica Acta (BBA)Mol Cell Res. (2018) 1865:1687–97. doi: 10.1016/j.bbamcr.2018.07.011
15. Mohamed BA, Hartmann N, Tirilomis P, Sekeres K, Li W, Neef S, et al. Sarcoplasmic reticulum calcium leak contributes to arrhythmia but not to heart failure progression. Sci Transl Med. (2018) 10:eaano724. doi: 10.1126/scitranslmed.aan0724
16. Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. (2011) 4:128–35. doi: 10.1161/CIRCEP.110.959916
17. Stølen TO, Høydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, et al. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res. (2009) 105:527–36. doi: 10.1161/CIRCRESAHA.109.199810
18. Manotheepan R, Danielsen TK, Sadredini M, Anderson ME, Carlson CR, Lehnart SE, et al. Exercise training prevents ventricular tachycardia in CPVT1 due to reduced CaMKII-dependent arrhythmogenic Ca2+ release. Cardiovasc Res. (2016) 111:295–306. doi: 10.1093/cvr/cvw095
19. Lunde IG, Aronsen JM, Kvaløy H, Qvigstad E, Sjaastad I, Tønnessen T, et al. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol Genomics. (2012) 44:162–72. doi: 10.1152/physiolgenomics.00016.2011
20. Wisløff U, Loennechen JP, Currie S, Smith GL, Ellingsen Ø. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc Res. (2002) 54:162–74. doi: 10.1016/S0008-6363(01)00565-X
21. Sjaastad I, Sejersted OM, Ilebekk A, Bjørnerheim R. Echocardiographic criteria for detection of postinfarction congestive heart failure in rats. J Appl Physiol. (2000) 89:1445–54. doi: 10.1152/jappl.2000.89.4.1445
22. Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. (2011) 51:288–98. doi: 10.1016/j.yjmcc.2011.06.012
23. Krobert KA, Bach T, Syversveen T, Kvingedal AM, Levy FO. The cloned human 5-HT7 receptor splice variants: a comparative characterization of their pharmacology, function and distribution. Naunyn-Schmiedeberg's Arch Pharmacol. (2001) 363:620–32. doi: 10.1007/s002100000369
24. Ramberg H, Eide T, Krobert KA, Levy FO, Dizeyi N, Bjartell AS, et al. Hormonal regulation of beta2-adrenergic receptor level in prostate cancer. Prostate. (2008) 68:1133–42. doi: 10.1002/pros.20778
25. Eisner DA, Kashimura T, O'Neill SC, Venetucci LA, Trafford AW. What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J Mol Cell Cardiol. (2009) 46:474–81. doi: 10.1016/j.yjmcc.2008.12.005
26. Levy FO, Zhu X, Kaumann AJ, Birnbaumer L. Efficacy of beta 1-adrenergic receptors is lower than that of beta 2-adrenergic receptors. Proc Natl Acad Sci USA. (1993) 90:10798–802. doi: 10.1073/pnas.90.22.10798
27. Kemp CD, Conte JV. The pathophysiology of heart failure. Cardiovasc Pathol. (2012) 21:365–71. doi: 10.1016/j.carpath.2011.11.007
28. Gómez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation. (2001) 104:688–93. doi: 10.1161/hc3201.092285
29. Kalarus Z, Svendsen JH, Capodanno D, Dan GA, De Maria E, Gorenek B, et al. Cardiac arrhythmias in the emergency settings of acute coronary syndrome and revascularization: an European Heart Rhythm Association (EHRA) consensus document, endorsed by the European Association of Percutaneous Cardiovascular Interventions (EAPCI), and European Acute Cardiovascular Care Association (ACCA). Europace. (2019) 21:1603–4. doi: 10.1093/europace/euz163
30. Antoons G, Oros A, Bito V, Sipido KR, Vos MA. Cellular basis for triggered ventricular arrhythmias that occur in the setting of compensated hypertrophy and heart failure: considerations for diagnosis and treatment. J Electrocardiol. (2007) 40:S8–14. doi: 10.1016/j.jelectrocard.2007.05.022
31. Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure. Circ Res. (2001) 88:1159–67. doi: 10.1161/hh1101.091193
32. Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, et al. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. (2012) 110:569–77. doi: 10.1161/CIRCRESAHA.111.260455
33. Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice. J Clin Invest. (2010) 120:4375–87. doi: 10.1172/JCI37649
34. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/CalmodulinΓÇôDependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. (2005) 97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89
35. Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. (2003) 93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA
36. Johnson DM, Antoons G. Arrhythmogenic mechanisms in heart failure: linking β-adrenergic stimulation, stretch, and calcium. Front Physiol. (2018) 9:1453. doi: 10.3389/fphys.2018.01453
37. McCauley MD, Wehrens XHT. Targeting ryanodine receptors for anti-arrhythmic therapy. Acta Pharmacol Sin. (2011) 32:749–57. doi: 10.1038/aps.2011.44
38. Bonilla IM, Belevych AE, Sridhar A, Nishijima Y, Ho HT, He Q, et al. Endurance exercise training normalizes repolarization and calcium-handling abnormalities, preventing ventricular fibrillation in a model of sudden cardiac death. J Appl Physiol. (2012) 113:1772–83. doi: 10.1152/japplphysiol.00175.2012
39. Kemi OJ, MacQuaide N, Høydal MA, Ellingsen O, Smith GL, Wisloff U. Exercise training corrects control of spontaneous calcium waves in hearts from myocardial infarction heart failure rats. J Cell Physiol. (2012) 227:20–6. doi: 10.1002/jcp.22771
40. Kemi OJ, Hoydal MA, MacQuaide N, Haram PM, Koch LG, Britton SL, et al. The effect of exercise training on transverse tubules in normal, remodeled, and reverse remodeled hearts. J Cell Physiol. (2011) 226:2235–43. doi: 10.1002/jcp.22559
41. van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. (2010) 122:2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298
42. Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, et al. Role of RyR2 phosphorylation at S2814 during heart failure progression/novelty and significance. Circ Res. (2012) 110:1474–83. doi: 10.1161/CIRCRESAHA.112.268094
43. Wang Y, Shi Q, Li M, Zhao M, Raghavender Reddy G, Teoh J-P, et al. Intracellular β1-adrenergic receptors and organic cation transporter 3 mediate phospholamban phosphorylation to enhance cardiac contractility. Circ Res. (2020). doi: 10.1161/CIRCRESAHA.120.317452. [Epub ahead of print].
44. Jia D, Hou L, Lv Y, Xi L, Tian Z. Postinfarction exercise training alleviates cardiac dysfunction and adverse remodeling via mitochondrial biogenesis and SIRT1/PGC-1α/PI3K/Akt signaling. J Cell Physiol. (2019) 234:23705–18. doi: 10.1002/jcp.28939
45. Benito B, Gay-Jordi G, Serrano-Mollar A, Guasch E, Shi Y, Tardif JC, et al. Cardiac arrhythmogenic remodeling in a rat model of long-term intensive exercise training. Circulation. (2011) 123:13–22. doi: 10.1161/CIRCULATIONAHA.110.938282
46. Frisk M, Ruud M, Espe EK, Aronsen JM, Røe Å T, Zhang L, et al. Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc Res. (2016) 112:443–51. doi: 10.1093/cvr/cvw111
47. Dries E, Amoni M, Vandenberk B, Johnson DM, Gilbert G, Nagaraju CK, et al. Altered adrenergic response in myocytes bordering a chronic myocardial infarction underlies in vivo triggered activity and repolarization instability. J Physiol. (2020) 598:2875–95. doi: 10.1113/JP278839
Keywords: exercise training, arrhythmias, cardiac ryanodine receptor, heart failure, myocardial infarction
Citation: Danielsen TK, Sadredini M, Manotheepan R, Aronsen JM, Frisk M, Hansen MH, Andressen KW, Hougen K, Levy FO, Louch WE, Sejersted OM, Sjaastad I and Stokke MK (2021) Exercise Training Stabilizes RyR2-Dependent Ca2+ Release in Post-infarction Heart Failure. Front. Cardiovasc. Med. 7:623922. doi: 10.3389/fcvm.2020.623922
Received: 30 October 2020; Accepted: 17 December 2020;
Published: 25 January 2021.
Edited by:
Antonio Zaza, University of Milano-Bicocca, ItalyReviewed by:
Godfrey Smith, University of Glasgow, United KingdomCopyright © 2021 Danielsen, Sadredini, Manotheepan, Aronsen, Frisk, Hansen, Andressen, Hougen, Levy, Louch, Sejersted, Sjaastad and Stokke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathis Korseberg Stokke, bS5rLnN0b2trZUBtZWRpc2luLnVpby5ubw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.