 Clément Mercier
Clément Mercier Marina Rousseau
Marina Rousseau Pedro Geraldes
Pedro Geraldes- Department of Medicine, Division of Endocrinology, Research Center of the Centre Hospitalier Universitaire de Sherbrooke, Université de Sherbrooke, Sherbrooke, QC, Canada
Peripheral artery disease is caused by atherosclerosis of lower extremity arteries leading to the loss of blood perfusion and subsequent critical ischemia. The presence of diabetes mellitus is an important risk factor that greatly increases the incidence, the progression and the severity of the disease. In addition to accelerated disease progression, diabetic patients are also more susceptible to develop serious impairment of their walking abilities through an increased risk of lower limb amputation. Hyperglycemia is known to alter the physiological development of collateral arteries in response to ischemia. Deregulation in the production of several critical pro-angiogenic factors has been reported in diabetes along with vascular cell unresponsiveness in initiating angiogenic processes. Among the multiple molecular mechanisms involved in the angiogenic response, protein tyrosine phosphatases are potent regulators by dephosphorylating pro-angiogenic tyrosine kinase receptors. However, evidence has indicated that diabetes-induced deregulation of phosphatases contributes to the progression of several micro and macrovascular complications. This review provides an overview of growth factor alterations in the context of diabetes and peripheral artery disease, as well as a description of the role of phosphatases in the regulation of angiogenic pathways followed by an analysis of the effects of hyperglycemia on the modulation of protein tyrosine phosphatase expression and activity. Knowledge of the role of phosphatases in diabetic peripheral artery disease will help the development of future therapeutics to locally regulate phosphatases and improve angiogenesis.
Methods of Bibliographic Search
The bibliographic search was performed in the PubMed and Google Scholar databases. All papers published before June 2020 were included in the review. A large set of keywords was used including growth factor, tyrosine kinase receptor, diabetic PAD, angiogenesis, blood flow reperfusion, endothelial dysfunction, endothelial cells, perivascular cells, hindlimb ischemia, phosphatase, and hyperglycemia.
Introduction
Current data reveals that more than 400 million people are suffering from diabetes mellitus (DM) and projections estimate that numbers of this world-wide issue will almost triple by 2030 (1). DM is a major risk factor of vascular complications altering both micro- and macrovascular beds. Peripheral artery disease (PAD) belongs to the macrovascular complications of diabetes with an estimated prevalence of 20% in diabetic patients over 40 years of age reaching 29% in patients over 50 years of age (2). The severity and the duration of DM are important predictors of both the incidence and the extent of PAD which appears more frequently, more severe, and at a younger age compared to non-diabetic PAD (3). Diabetic patients with PAD are also less responsive to revascularization procedures leading to a dramatic 5 to 15 times higher risk of major amputation (4). The pathophysiology of PAD is characterized by obstructive atherosclerosis of the lower limb arteries gradually reducing blood flow to the tissues and ultimately leading to critical ischemia (5). Despite both major angiogenic inducers, hypoxia and oxidative stress, being elevated in diabetic PAD, collateral vessel development is insufficient to support the loss of blood flow through occluded arteries. The deregulation of growth factors represents a hallmark of diabetic vascular complications occurring differentially in various tissues (6). Although excessive activity and overproduction of pro-angiogenic growth factors contribute to the progression of diabetic nephropathy or retinopathy, the angiogenic paradox suggests that diabetic PAD and myocardial infarction (MI) are rather characterized by defects in the production and/or responsiveness to pro-angiogenic growth factors (7). Thus, identifying the underlying molecular mechanisms that are involved in the deregulation of pro-angiogenic factors has generated increased interest in the context of diabetic PAD. Protein tyrosine phosphatases (PTP) are critical regulators of pro-angiogenic tyrosine kinase receptor (RTK) signaling (8). Not only do PTP shut down RTK, they also finely modulate tyrosine phosphorylation of these receptors to specifically regulate several cellular angiogenic properties. Thus, any slight dysregulation in PTP expression or activity will trigger pathological angiogenesis. Evidence has emerged concerning the role of PTP in the development of diabetes and the progression of diabetic vascular complications including nephropathy (9) and retinopathy (10). Therefore, this review aims to summarize and discuss the deregulation of the main growth factors in diabetic PAD with a translational view from clinical studies to mechanistic preclinical models. We next sought to discuss the role of PTP in the regulation of angiogenic pathways and highlight how PTP expression and activity are regulated in the hypoxic environment of diabetic PAD.
Deregulation of Pro-Angiogenic Factors in Diabetic PAD
Angiogenesis is a complex, dynamic, and well-coordinated physiological process leading to the formation of new vessels from pre-existing vasculature in response to ischemia. This process involves temporal interactions between multiple growth factors and vascular cells, which have been extensively summarized in excellent reviews (11, 12). Briefly, vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), angiopoietins (Ang-1 and Ang-2), and stromal cell-derived factor 1 (SDF-1) are the main actors in angiogenesis and the most studied in diabetic PAD (Table 1). Overall, SDF-1 is required for the recruitment of undifferentiated endothelial progenitor cells (EPC) to the hypoxic environment, while VEGF initializes the proliferation and sprouting of endothelial cells (EC) to form immature neo-vessels. In a final step, PDGF and Ang-1 drive the recruitment of pericytes and vascular smooth muscle cells (VSMC) which wrap around and stabilize neo-vessels to develop mature capillaries. The next section provides an overview of the current understanding of main growth factor deregulation in diabetic PAD (Figure 1). Clinical studies will be briefly summarized while preclinical models (in vitro and in vivo) will be reported to highlight the underlying molecular mechanisms leading to the alteration of the angiogenic process following hindlimb ischemia in diabetes. Findings based on other diabetic macrovascular complications sharing a similar pathophysiology as PAD will be reported where preclinical data was not available in the context of diabetic PAD.
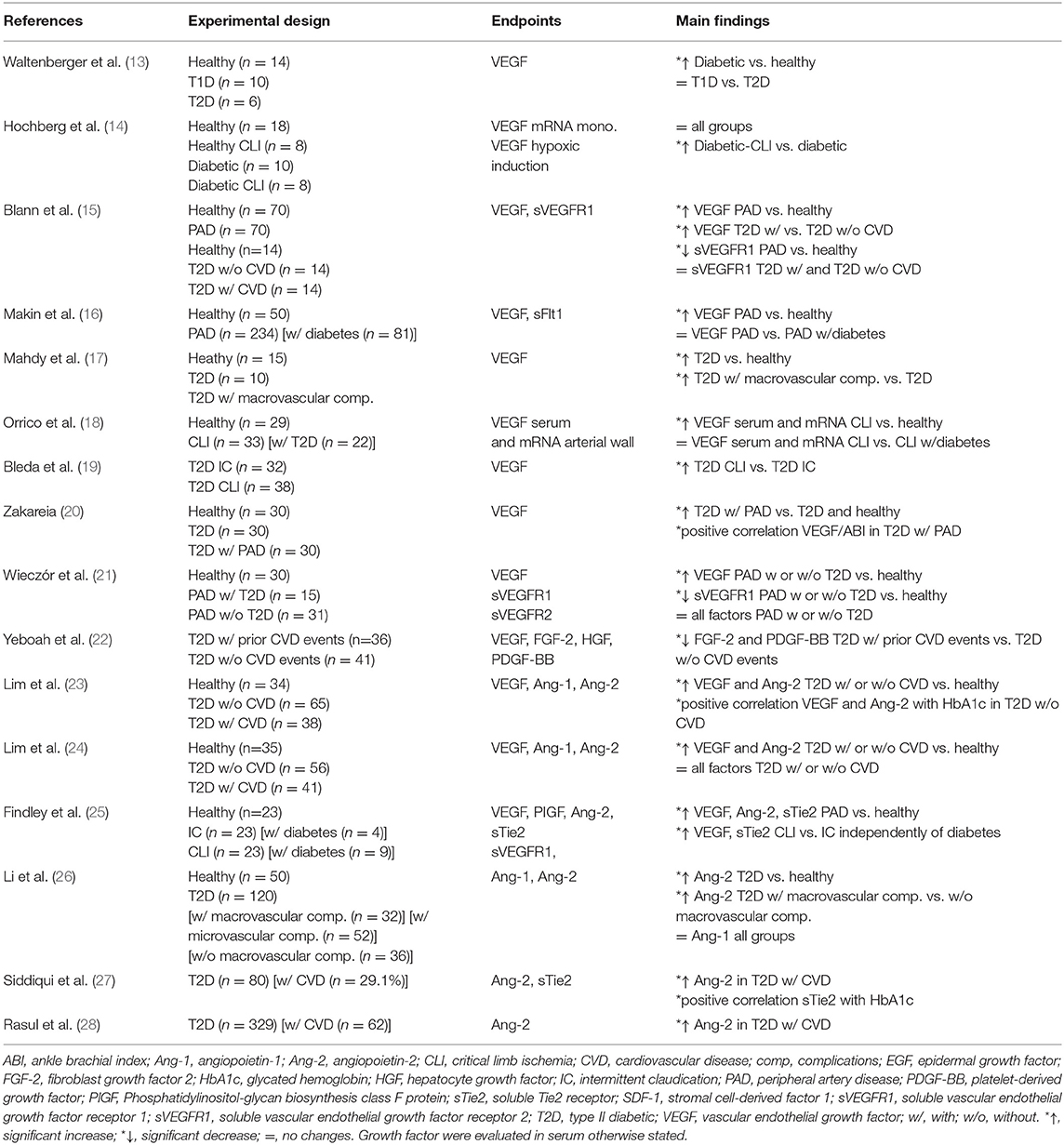
Table 1. Clinical investigations of growth factors in diabetic PAD.
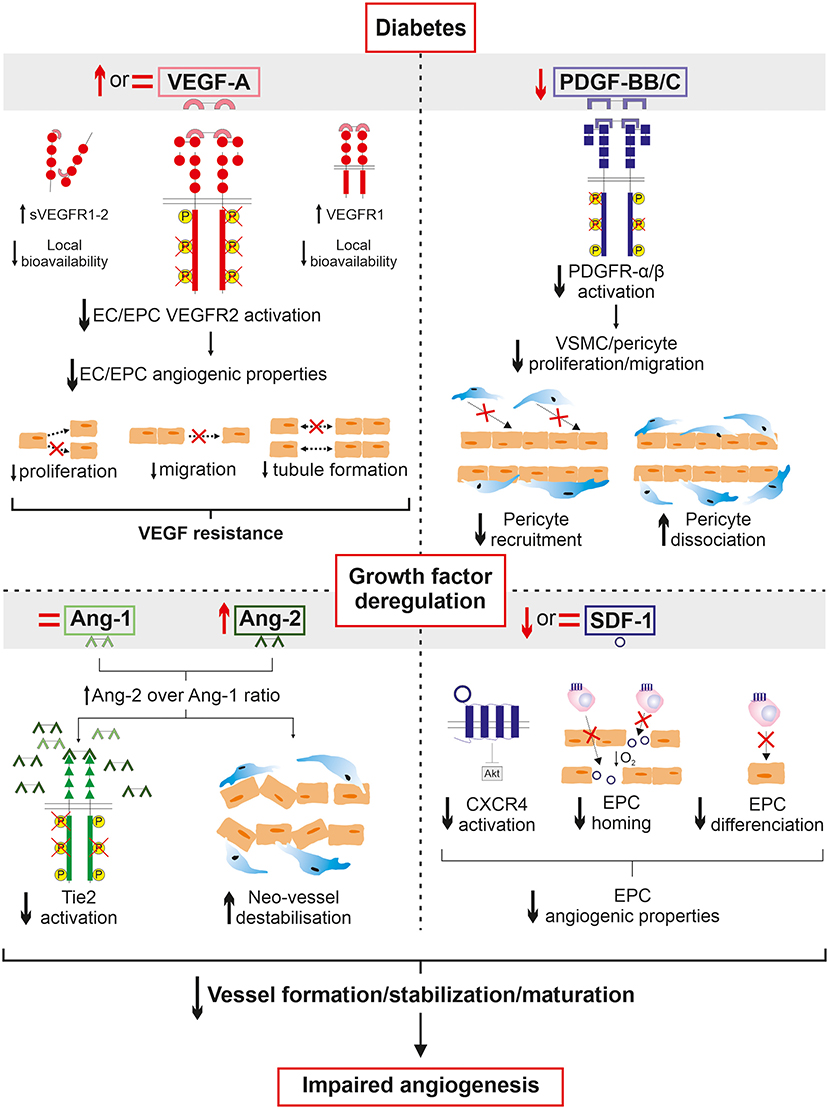
Figure 1. Overview of growth factor deregulation in diabetic PAD. A history of diabetes deeply altered circulating growth factor expression in response to ischemia in PAD. Despite an increased production of VEGF, decreased cell responses to VEGF are observed in diabetes. The VEGF resistance paradox could be partially attributed to a lack of local bioavailability due to increased expression of both the soluble forms of VEGF receptor and VEGFR1 acting together as decoys to sequestrate VEGF. In addition, VEGF signaling is impaired leading to the alteration of endothelial cell angiogenic properties which fail to initiate the angiogenic process upon VEGF stimulation. Concurrently, PDGF production and PDGFR signaling are decreased therefore contributing to reduced pericyte/vascular smooth muscle cell coverage of neo-vessels in addition to promoting their dissociation. Angiopoietin balance is also deregulated to promote Ang-2 over Ang-1 expression. This implies decreased activation of Tie2 receptor subsequently leading to Ang-2-induced destabilization and regression of neo-vessels. Finally, SDF-1 production and CXCR4 activation are altered resulting in endothelial progenitor cell dysfunctions and consequently failing to efficiently migrate to the ischemic environment and trigger endothelial differentiation. Taken together, the deregulation of growth factors in diabetes negatively affects all fundamental steps of neo-vessel generation to ultimately alter angiogenesis in the ischemic lower limb of patients suffering from PAD.
Clinical Investigations
VEGF
From the extensive list of pro-angiogenic factors, VEGF is by far the most clinically investigated. As a result of blunted cellular response to hypoxia, hypoxia-inducible factor-1 alpha (HIF-1α) itself and most of its target genes, including VEGF, are reduced in a diabetic state (29). Thus, a slightly lower non-significant expression of VEGF-A was observed in the serum of type 2 diabetic (T2D) PAD patients compared with non-diabetic PAD individuals (21). On the other hand, PAD is characterized by a tremendously favorable ischemic environment triggering VEGF production and an ample number of investigations have suggested that patients with PAD and coexisting diabetes display higher or at least unchanged serum levels of VEGF-A (Table 1) (13–20, 22–25). Therefore, the altered collateral vessel formation in diabetic PAD might be attributed to defects in downstream signal transduction rather than in VEGF production in response to ischemia (13, 14). This hypothesis is further supported by increased levels of both soluble vascular endothelial growth factor receptor 1 (sVEGFR1) and 2 (sVEGFR2) in the serum of T2D patients with PAD compared with nondiabetic PAD individuals (21) that could act as decoys to sequestrate VEGF-A (15). Collectively, the lack of significant improvement of the PAD following exogenous administration of VEGF could be attributed to decreased local bioavailability and unresponsiveness of VEGF in diabetic individuals.
PDGF
Unlike VEGF, the impact of hyperglycemia on PDGF expression and activation in the context of PAD has been poorly clinically investigated. To date, only one pilot study has reported lower serum levels of PDGF-BB in T2D patients with a previous cardiovascular event compared to those that have not (22) (Table 1). Although this investigation did not provide definitive evidence of additive effects of diabetes, it suggested that PDGF-BB could be a predictive marker of cardiovascular diseases (CVD) in T2D.
Angiopoietins
Two consecutive cross-sectional studies investigated the serum levels of Ang-1 and Ang-2 in control and T2D patients, with or without CVD, including PAD (23, 24) (Table 1). Only Ang-2 serum levels were significantly raised in T2D patients compared to controls, which correlated with an increased index of endothelial dysfunction (24). Interestingly, Ang-2, but not Ang-1, serum levels were increased in T2D patients compared to healthy controls and positively correlated with circulating glycated hemoglobin (HbA1c) (23). In addition, several clinical investigations showed that Ang-2 levels were increased in T2D subjects with macrovascular complications compared to those without (26–28). Increased Ang-2 levels were also reported in the serum of PAD patients along with the soluble Tie2 (sTie2) receptor. Interestingly, sTie2 levels were correlated with an increased severity of PAD but were not associated with several cardiovascular risk factors including diabetes (26). Similarly, Ang-2 but not sTie2 levels were significantly associated with the presence of macrovascular complications including PAD (28). Adding to hyperglycemia-induced increase Ang-2 serum levels, these two last studies raised the hypothesis that sTie2 could be a marker of PAD severity regardless of diabetes.
SDF-1
The role of SDF-1 in diabetic vascular complications has gained interest since an early study found a significant decrease in the number of circulating EPC in T1D patients compared to controls (30). The reduction in circulating EPC was further diminished in diabetic subjects with PAD compared to non-diabetic individuals with PAD, demonstrating a noxious additive effect of hyperglycemia (31). Interestingly, Egan et al. reported lower numbers of CXCR4-positive circulating cells, SDF-1's main receptor in T2D patients which was further decreased in T2D individuals suffering from peripheral atherosclerosis (32).
In vivo Studies
Animal studies are based on models of hindlimb ischemia (HLI) that mimic the pathophysiology of PAD and allow to assess ischemia-induced angiogenesis/arteriogenesis as well as subsequent blood flow reperfusion. Several different surgical approaches are used to induce ischemia in the lower limb of small animals (33, 34). The easier approach is a one-point distal ligation of the femoral artery while the most reported is a distal and proximal ligation followed by the excision between these two ligation points. These methods will result in a mild (ligation) and acute (ligation and excision) ischemia that ultimately influence both the temporality and the efficiency of blood flow reperfusion in the ischemic limb (33, 34). Although these variations might be responsible on the variability observed in local therapeutic studies, no conflicting results were found in the angiogenic outcomes reported in the context of this review.
VEGF
Early studies reported decreased or unchanged expression of VEGF in the ischemic muscle of non-obese T1D and db/db T2D mice, which was illustrated by a significant reduction in capillary density and blood flow reperfusion 35 days post HLI-surgery (35, 36). In the same time frame, blood flow reperfusion was also dampened in high-fat diet T2D mice despite ischemia-induced upregulation of VEGF production (37). Interestingly, sVEGFR1 and VEGFR1 expression were gradually increased and correlated with significantly higher VEGF binding in the diabetic ischemic muscle compared to that of non-diabetic mice sustaining the hypothesis of a locally limited VEGF bioavailability suggested by clinical studies (13, 14). In addition to an alteration in VEGF production, a lower expression and/or activation of its receptor VEGFR2 were reported in the 28-day ischemic muscle of db/db T2D mice (38) as well as in streptozotocin (STZ)-induced T1D mice 3, 14 and 28 days post HLI-surgery, respectively (39–42). This was illustrated by reduced capillary density and blood reperfusion suggesting angiogenic defects in the diabetic lower limb. The reduced phosphorylation of VEGFR2 was notably dependent on the activation of protein kinase C delta (PKCδ) and angiotensin II receptor type 2 (ATR2) in a diabetic state (41, 42). In line with the impaired activation of VEGFR2, a decreased phosphorylation of several downstream effectors of the VEGF pathway including protein kinase B (Akt) (30, 34–36), endothelial nitric oxide synthase (eNOS) (40, 43, 44) and extracellular signal-regulated kinase (ERK) (41) was reported in the ischemic muscle of both T1D and T2D mice.
PDGF
In the HLI mouse model, a decreased expression of PDGF-BB was reported in the ischemic muscle of both STZ-induced T1D and ob/ob T2D mice compared to wild-type counterparts 7 days post-surgery which was correlated with an increased activation of PKC-α (45, 46). The impaired blood flow reperfusion in the ischemic diabetic limb was associated with an apparent dissociation of pericytes from the endothelial capillary tube (45). Interestingly, a reduction of PDGF expression along with an alteration of PDGF receptor beta (PDGFR-β) phosphorylation was found in the ischemic muscle of STZ-induced T1D mice compared to controls 28 days post-ligation of the femoral artery reflecting persisting defects in PDGF signaling (40).
Angiopoietins
Unfortunately, no HLI-based animal studies have yet been conducted to elucidate the role of angiopoietins in the revascularization of diabetic lower limbs. Although, interesting findings were observed in MI, a well-known diabetic macrovascular complication in the same category as PAD. Hence, the expression of Ang-2 was significantly upregulated in the ischemic heart of both T2D and T1D mice compared to ischemic controls while Tie2 expression and signaling were downregulated and Ang-1 was unaffected (47, 48). Hyperglycemia-induced disruption of Tie2 signaling by favoring Ang-2 expression over Ang-1 promoted EC apoptosis that subsequently aggravated the size of myocardial infarction (47). Importantly, Ang-1-induced recruitment of VSMC and EC coverage were impaired in the diabetic ischemic heart suggesting alterations in the maturation process of capillaries. This was illustrated by a significant reduction of myocardial capillary density, arteriole formation and vessel outgrowth (48).
SDF-1
In a HLI model, the recruitment of EPC was completely abolished in T2D rats due to a decrease in SDF-1 serum concentrations 3 days post-surgery that was illustrated by an impaired blood flow recovery in the diabetic limb (31). Interestingly, bone marrow progenitor cells from T1D mice were unable to differentiate into an endothelial phenotype following SDF-1 stimulation thus showing that hyperglycemia also deregulates the CXCR4 signaling pathway (49). An elegant study recently found a significant reduction of SDF-1 levels in both serum and ischemic muscle of STZ-induced diabetic mice 3 days post femoral artery ligation. This was accompanied with blunted activation of Akt leading to decreased recruitment of EPC to the ischemic sites triggering insufficient blood reperfusion (50).
Cell-Based Mechanistical Studies
Endothelial cells/Endothelial Progenitor Cells
At the cellular level, a recent study showed increased production of VEGF-A165 but decreased mRNA expression and blunted phosphorylation of extracellular signal-regulated kinases (ERK) 1/2 in HUVEC simultaneously exposed to hyperglycemia and CoCl2-induced hypoxia compared to controls leading to reduced migration (51). This could be attributed to a downregulation of VEGFR2 expression under concurrent hypoxic and hyperglycemic environments (39). Recently, exposure of HUVEC to methylglyoxal (MGO), a glucose metabolite, reduced the expression and production of PDGF-BB while raising VEGF-A expression, which ultimately resulted in pericyte recruitment inhibition (52). In time course experiments on EC exposed to high glucose, both Akt and eNOS activation were decreased and delayed overtime as the expression of Ang-2 increased compared to normal glucose concentrations (48). Despite unaltered expression of Ang-1, Ang-1-stimulated Tie2 activation was decreased in HUVEC exposed to high glucose levels leading to reduced Akt phosphorylation (53). However, the angiogenic outcomes related to diabetes-induced angiopoietin imbalance remain to be identified.
Several alterations in growth factor expression have also been reported in the EPC niche in hyperglycemia. Exposure of EPC to high glucose blunted the activation of Akt and eNOS, which contributed to the inhibition of VEGF-induced EPC proliferation, migration, or tubule formation and ultimately triggered abnormal neovasculogenesis (54, 55). Interestingly, mRNA levels of Ang-1 were decreased in the EPC from healthy volunteers. Furthermore, exposing these cells to hyperglycemia and exogenous Ang-1 improved EPC angiogenic properties (56). Similarly, EPC extracted from diabetic mice exhibited low expression of Ang-1 compared to non-diabetic counterparts (57). Finally, EPC exposed both to hypoxia and high glucose levels exhibited decreased protein levels of SDF-1 and Akt phosphorylation, which was associated with an impairment of SDF-1-induced tubule formation (50).
Vascular Smooth Muscle Cells/Pericytes
In contrast to the endothelial lineage, only few studies have investigated the molecular mechanisms underlying growth factor deregulation in hyperglycemic perivascular cells. The expression of PDGFR-α and β were decreased in VSMC exposed to high glucose levels due to the upregulation of PKC-α. On the other hand, PDGF-C was only decreased in VSMC suggesting that perivascular cells might be more responsible for decreased expression of PDGF-C previously reported in the whole diabetic muscle (58, 59). Similarly, non-toxic MGO concentrations altered PDGF-BB-induced PDGFR-β and ERK1/2-activation as well as the subsequent proliferation of VSMC (60).
Monocytes
Monocytes extracted from diabetic patients with CLI do not exhibit impairment in the ability to produce VEGF in hypoxic conditions (14). Nevertheless, monocytes extracted from diabetic subjects presented severe impairment in the chemotaxis response to VEGF stimulation despite a significantly higher concentration of VEGF in the serum of diabetic patients (13). However, functional angiogenic outcomes related to monocyte dysfunction in diabetes still needs to be evaluated.
Emerging Role of Phosphatases in the Regulation of Angiogenesis
As described above, the expression and activation of growth factors are deeply impaired in diabetes leading to blunted pro-angiogenic signaling pathways and poor collateral vessel formation in response to a PAD ischemic environment. Pro-angiogenic RTK, including VEGFR, PDGFR and Tie2, are mainly regulated by a fine balance between tyrosine phosphorylation and dephosphorylation. Any slight alteration in the equilibrium between protein tyrosine kinase and protein tyrosine phosphatase (PTP) activity will trigger abnormalities in cellular processes including proliferation, migration, and differentiation, thereby resulting in a global deregulation of angiogenesis (Table 2). Additionally, phosphatases can directly act on several downstream RTK effectors to modulate specific pathways and cell functions (87). Over the years, the influence of PTP in both micro and macrovascular complications of diabetes has been progressively discovered. Hyperglycemia-induced modification of PTP expression or activity has been shown to deregulate the insulin (88), nephrin (89), and PDGFR-β (10) pathways contributing to insulin resistance, nephropathy, and retinopathy, respectively. Early evidence concerning the potential role of PTP in the regulation of collateral vessel angiogenesis emerged when administration of broad-spectrum PTP inhibitors improved blood flow reperfusion following HLI in mice (Figure 2) (90, 91). Given that several PTP have been identified in the vascular lineage since (92), the following section summarizes and discusses evidence of PTP contribution in the regulation of pro-angiogenic RTK as well as their emerging role in the pathophysiology of PAD (Table 3). In addition, a focus was made to highlight current knowledge concerning the effects of hyperglycemia in the modulation of PTP and related angiogenic outcomes at the mechanistical cellular level (Figure 3) and in the context of HLI models reflecting the microenvironment of PAD (Table 4).
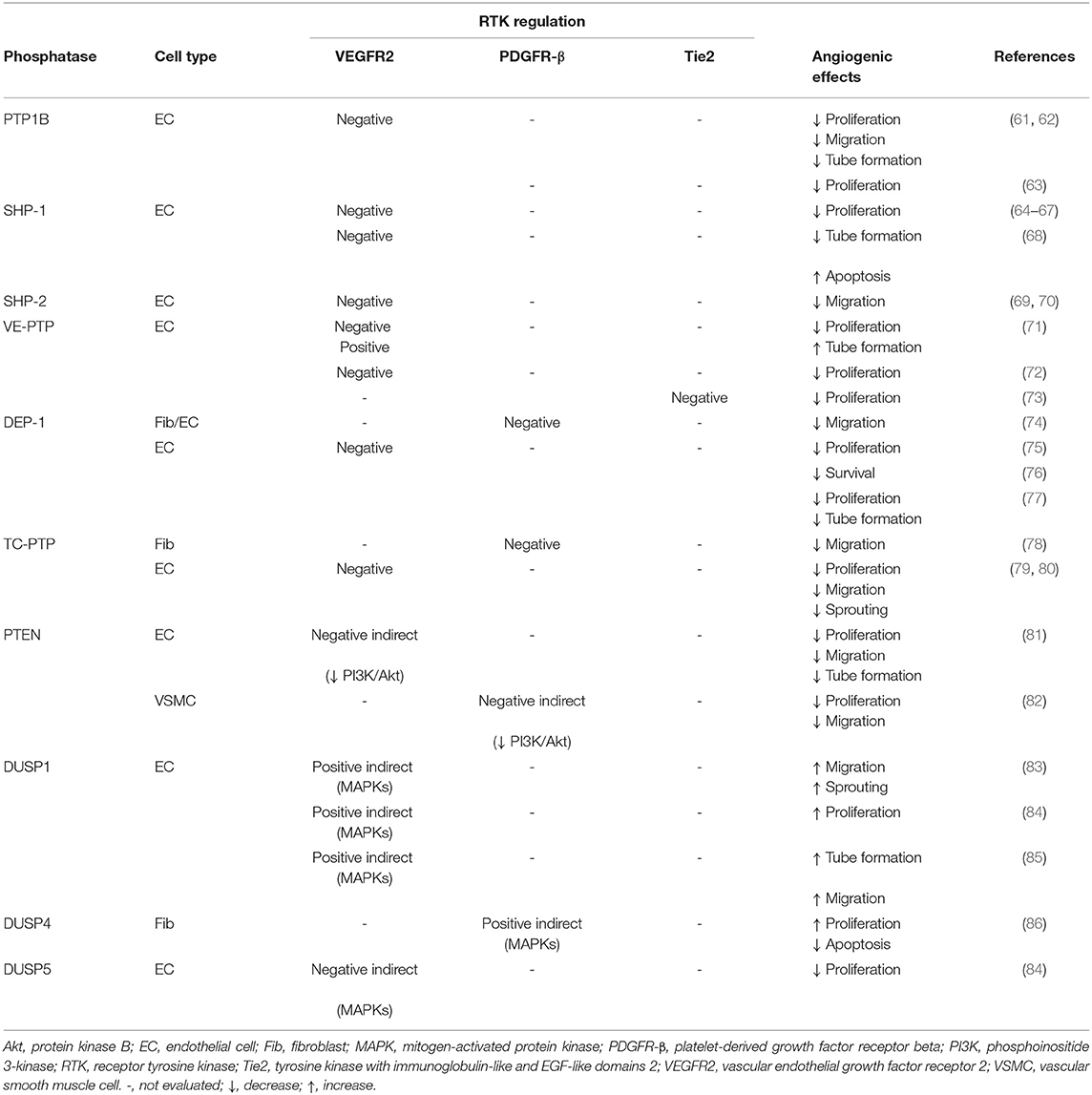
Table 2. Baseline regulation of RTK by phosphatases in angiogenesis.
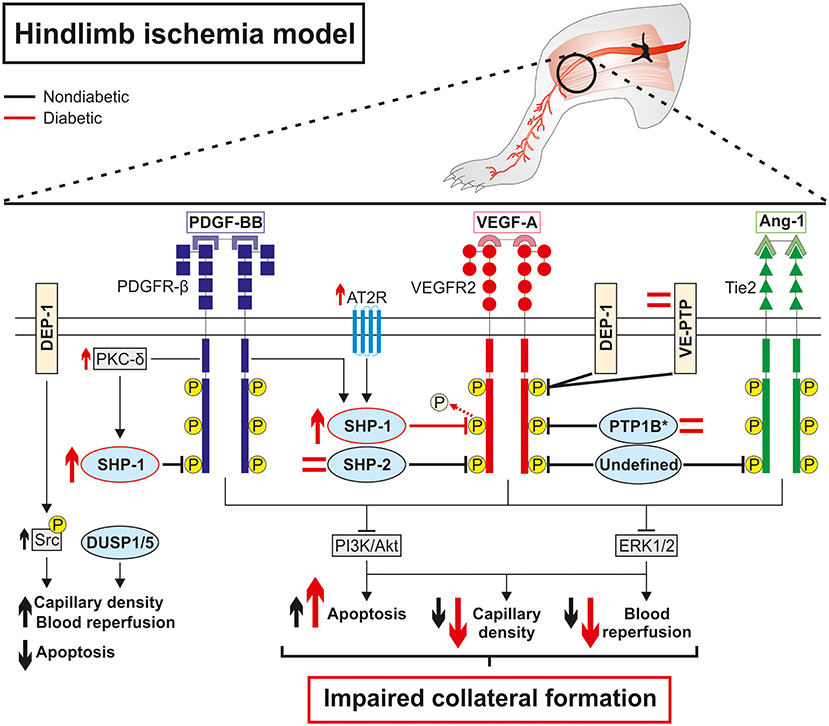
Figure 2. Diabetes impaired collateral vessel formation by deregulating phosphatase expression and activity in a hindlimb ischemia model. Overall, protein tyrosine phosphatases are direct negative regulators of pro-angiogenic receptors tyrosine kinase PDGFR-β, VEGFR2, and Tie2 at baseline, which limits collateral vessel formation in the hindlimb ischemia model. In contrast, DEP-1 exerted indirect positive regulation of VEGFR2 by dephosphorylating inhibitory Y529 on Src while DUSP1/5 promoted capillary formation and blood reperfusion through the reduction of endothelial cell apoptosis in an RTK-independent way. Diabetes activates the PKC-δ isoform and the angiotensin II receptor type 2 that in turn increases the expression of SHP-1 and its phosphatase activity against VEGFR2, ultimately leading to blunted downstream signaling and aggravating impaired collateral vessel formation following ischemia. Conversely, SHP-2, PTP1B, and VE-TPT expressions are unchanged in endothelial cells (*PTP1B) or whole diabetic ischemic muscle compared to non-diabetic counterparts suggesting a lesser role of these phosphatases in diabetic PAD. The regulation and the subsequent influence of DEP-1 and DUSP1/5 in hindlimb angiogenesis still needs to be investigated in the context of diabetic PAD.
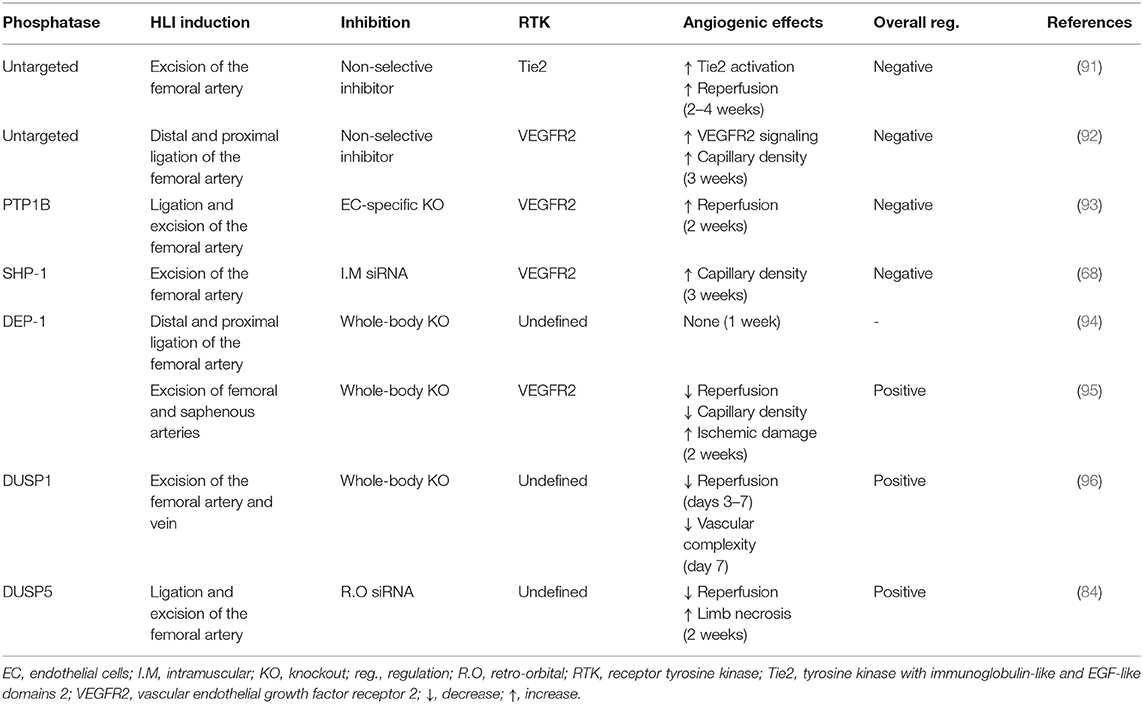
Table 3. Baseline regulation of collateral angiogenesis by PTP in HLI models.
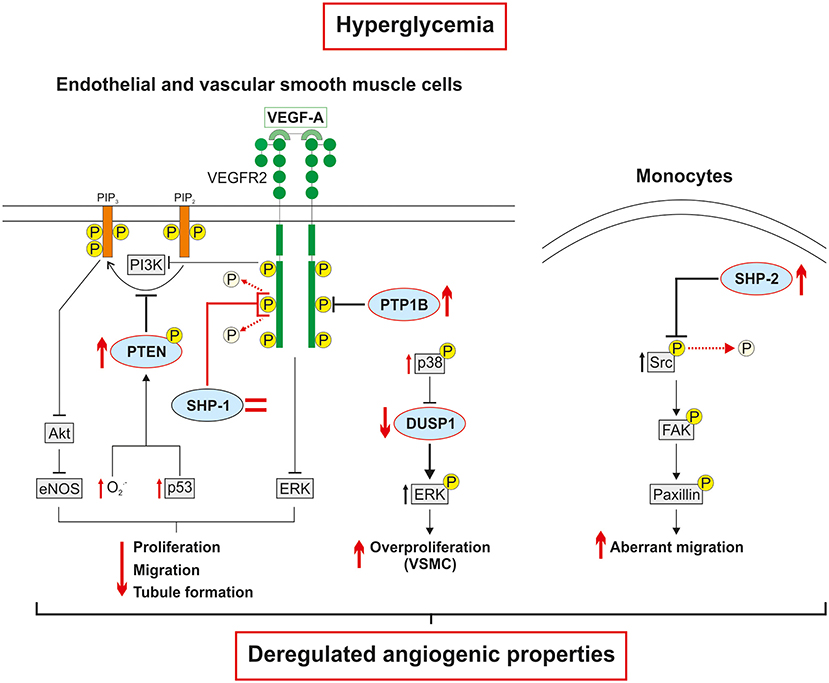
Figure 3. Hyperglycemia-induced phosphatase deregulation is associated with dysfunctions in cellular angiogenic responses. The expression of PTP1B, a negative regulator of VEGFR2, is increased in hyperglycemia. Conversely, the expression of SHP-1 is unchanged in endothelial cells exposed to high glucose. However, SHP-1 phosphatase activity and interaction with VEGFR2 are increased leading to decreased VEGFR2 activation and downstream signaling. The hyperglycemia-induced overproduction of free radicals and upregulation of p53 contribute to increased PTEN expression and phosphorylation which in turn reduced the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3). The direct deactivation of VEGFR2 signaling followed by the indirect decrease in the production of downstream effectors by phosphatases, ultimately impair the pro-angiogenic properties of endothelial cells. In vascular smooth muscle cells, high glucose levels increase p38 expression and activation, which reduces DUSP1 expression, leads to sustained overactivation of ERK and subsequent abnormal proliferation. In monocytes, hyperglycemia increases the expression of SHP-2 and its phosphatase activity on an inhibitory tyrosine residue of Src kinase. The overactivation of Src activates the FAK/paxillin axis resulting in aberrant and ineffective migration of monocytes subjected to VEGF stimulation.
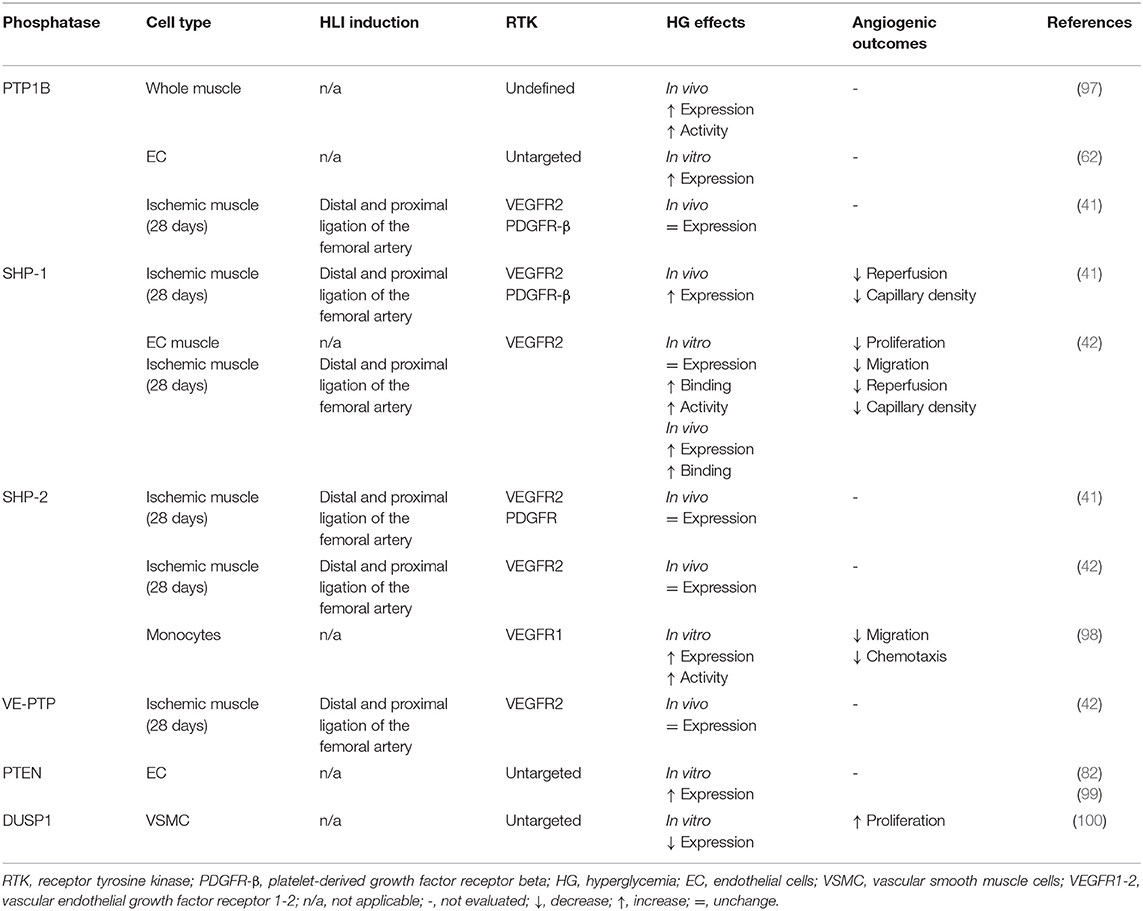
Table 4. Hyperglycaemia-induced PTP modulation.
Protein Tyrosine Phosphatase 1B
Protein tyrosine phosphatase 1B (PTP1B, gene: PTPN1) is an intracellular ubiquitous PTP. The systemic deletion of PTP1B results in viable and fertile mice with enhanced insulin sensitivity and accelerated wound healing (88). PTP1B has been initially described as a critical negative regulator of insulin signaling through its interaction with the insulin receptor (101). Early evidence in the role PTP1B in angiogenesis has emerged when a PTP1B inhibitor was shown to enhance VEGF-mediated VEGFR2 activation, migration, and proliferation of EC (61). The overexpression of PTP1B in EC inhibited VEGF-induced phosphorylation of VEGFR2 and signaling, which lead to altered EC proliferation whereas EC extracted from Ptpn1−/− mice exhibited increased angiogenic properties (63). Interestingly, hyperglycemia was found to increase PTP1B expression in macrovascular EC leading to endothelial dysfunction (62).
In the setting of PAD, PTP1B expression and activity were upregulated by hypoxia in the ischemic limb of mice 7 days post-HLI surgery (63) and endothelial-specific PTP1B deletion was sufficient to significantly improve blood flow reperfusion and capillary density (93). These findings emphasized PTP1B as a negative regulator of ischemia-induced angiogenesis through inhibition of the VEGF pathway in the endothelial lineage. Unfortunately, the regulation and influence of PTP1B remain to be clarified in a diabetic PAD environment. Indeed, PTP1B expression and activity was upregulated in skeletal muscle of Goto-Kakizaki T2D rats (97) while its expression remained unchanged in the ischemic muscle of STZ-induced T1D mice 28 days post-ligation of the femoral artery (41). These data suggest that the type of diabetes, presence and duration of hypoxia could influence PTP1B regulation in the lower limb.
However, PTP1B levels and activity were increased in the wounded skin of STZ-induced T1D (102) and ob/ob T2D (103) mice compared to wild-type counterparts, which was associated with impaired neovascularization and subsequent delayed wound healing, a well-known complication of diabetic PAD (104). Administration of a selective inhibitor significantly accelerated the wound healing process by decreasing PTP1B activity, which promoted a local recovery of VEGFR2 phosphorylation that contributed to the restoration of EC angiogenic properties (102, 103). Hence, diabetic wound-healing studies highlighted that hyperglycemia exacerbated PTP1B-induced downregulation of angiogenesis in macrovascular complications.
Src Homology Region 2 Domain-Containing Phosphatase-1
Src homology region 2 domain-containing phosphatase-1 (SHP-1, gene: PTPN6) is a cytoplasmic tyrosine phosphatase. SHP-1 null mice showed chronic systemic inflammation and died within 3 weeks of birth from interstitial pneumonia (105). Initially, SHP-1 was found to have a role in VEGF signaling due to its association with VEGFR2 following VEGF-A stimulation in EC (106). Further studies confirmed that SHP-1 interacted with VEGFR2 following TNF-α stimulation, reducing the proliferation of primary EC (64, 65) while silencing SHP-1 increased the activation of VEGFR2 pathway resulting in improved VEGF-induced EC proliferation and capillary formation (66, 67). In the meantime, Sugano et al. was the first to report that inhibition of ischemia-induced SHP-1 expression markedly increased VEGFR2 activation and capillary density in the HLI rat model (68).
The influence of SHP-1 in diabetic collateral vessel angiogenesis has been partially brought to light. Using the HLI model, a first study uncovered that hyperglycemia-driven PKC-δ activation increased both mRNA and protein expression of SHP-1 in the muscle of STZ-induced T1D mice 28 days post-ligation of the femoral artery. The upregulation of SHP-1 was accompanied by a decrease in both VEGFR2 and PDGFR-β signaling, leading to reduced capillary density and blood flow reperfusion. Whole-body deletion of PKC-δ re-established the angiogenic pathways in the diabetic ischemic limb by suppressing SHP-1 overexpression (41). In the same model, our group later reported that the deletion of AT2 receptor suppressed diabetes-induced binding of SHP-1 to VEGFR2, therefore restoring its signaling to improve blood flow reperfusion following 28 days of HLI (42). Interestingly, the exposure of BAEC to high glucose had no effect on SHP-1 expression but increased its phosphatase activity and direct interaction with VEGFR2 through the activation of AT2R, resulting in decreased EC migration (42). Finally, Chen et al. evaluated the effect of hyperglycemia on SHP-1 interaction with the Ang-1/Tie2 pathway in MI. SHP-1 was upregulated in the heart of T2D mice which was associated with blunted vessel outgrowth and capillary density. Administration of a broad-spectrum PTP inhibitor suppressed SHP-1 expression and improved Tie2 signaling. Further cell-based assays uncovered that high glucose levels increased SHP-1/Tie2 interaction and reduced Tie2 phosphorylation by decreasing Ang-1-mediated SHP-1/Tie2 dissociation in EC (48).
Src Homology Region 2 Domain-Containing Phosphatase-2
Src homology 2 domain-containing phosphatase 2 (SHP-2, gene: PTPN11) is a ubiquitously expressed non-transmembrane phosphatase sharing a similar structure and phosphatase activity mechanism as SHP-1. Unlike SHP-1, the global deletion of SHP-2 is embryonically lethal (107). SHP-2 have been shown to interact with several pro-angiogenic RTK including PDGFR-β (108), VEGFR2 (106) and Tie2 (109). In contrast to the exclusively negative role of SHP-1, SHP-2 exerts a dual regulatory role on RTK. Overall, SHP-2 has been widely described as a positive regulator of angiogenesis through the activation of the Ras/ERK pathway (87). However, silencing SHP-2 or inhibiting its catalytic domain increased ERK activation following VEGF-A stimulation which contradicts its role as a positive regulator (110). In addition, SHP-2 has been shown to directly bind to the Y1175 residue and dephosphorylate VEGFR2 upon VEGF-A stimulation, therefore reducing aortic EC migration (111). Conversely, overexpressing an inactive form or SHP-2 knockdown restored VEGFR2 activation and angiogenic properties of EC (69, 111). Moreover, SHP-2 is a positive regulator of the PI3K/Akt axis. Hence, inhibition of SHP-2 decreased the activation of Src and downstream Akt/eNOS effectors in response to VEGF-A in HUVEC through hyperactivity of the C-terminal SRC kinase (Csk) (110). SHP-2 was also critically required to fully activate Src/Akt in EC and subsequently induce migration and capillary formation following association with the docking adaptor Gab1 (70). SHP-2 has also been shown to bind, dephosphorylate and ultimately inhibit Tie2 signaling (92).
Despite its dual role, little is known about the regulation of SHP-2 in PAD. Indeed, SHP-2 levels remained unchanged in the ischemic hindlimb muscle of rats 3 weeks post excision of the femoral artery (68). Similarly, no variations in mRNA and protein levels of SHP-2 were observed in the ischemic muscle of T1D mice compared to ischemic controls, suggesting that SHP-2 might have a lesser impact than SHP-1 in collateral vessel formation in diabetic PAD (41, 42). However, a recent study reported that MGO-exposed human monocytes or monocytes collected from T2D patients and db/db T2D mice showed excessive and abnormal migration through enhanced activity of SHP-2, leading to dephosphorylation of Src and consequent paxillin activation (98). This suggests that hyperglycemia-induced upregulation of SHP-2 could have a critical role in monocyte dysfunction by promoting VEGF resistance to ultimately alter arteriogenesis in diabetic PAD patients.
Vascular Endothelial PTP
Vascular endothelial PTP (VE-PTP, gene: PTPRB) belongs to the receptor PTP (PTPR) family anchored at the cell membrane. VE-PTP is exclusively expressed in EC (92). Ptprb−−/− mouse embryos die between E9.5-10 due to severe disorganization of the primary vascular plexus and lack of functional blood vessels (112, 113). Due to its predominant expression in EC, it is not surprising that VEGFR2 is the main target of VE-PTP (114). Indeed, upon VEGF-A stimulation, VE-PTP was shown to bind and quickly dephosphorylate VEGFR2. Silencing VE-PTP with an siRNA further increased the degree of VEGF-A-induced VEGFR2 phosphorylation and signaling. This lead to an increase in EC proliferation but at the expense of tubule formation (114). Interestingly, VE-PTP required the recruitment of Tie2 to form a complex and dephosphorylate VEGFR2 in EC. The deletion of VE-PTP induced hyperproliferation of stalk cells following VEGF-A stimulation and caused an excessive sprouting of non-functional lumen-free neo-vessels (71). Interestingly, Tie2 was previously found to be an independent direct target of VE-PTP in mice EC (115). Ang-1 stimulation increased the binding of VE-PTP with Tie2 in HUVEC. In addition, silencing VE-PTP enhanced Tie2 signaling, ERK activation, and EC over-proliferation, ultimately triggering pathological vessel enlargement in juvenile mice (72). Taken together these studies uncovered VE-PTP as a physiological molecular brake of VEGFR2/Tie2 pro-angiogenic pathways favoring vessel maturation.
VE-PTP has been poorly investigated in PAD and only an unchanged expression was found in the diabetic ischemic muscle of STZ-induced T1D mice 28-days post ligation of the femoral artery (42). Nevertheless, a recent investigation showed that VE-PTP was upregulated in the renal microvasculature, which resulted in reduced Tie2 activation, increased neo-vessel destabilization and ultimately the progression of diabetic nephropathy (73). Similarly, local inhibition of VE-PTP promoted Tie2 activation, reduced vascular leakage, and neovascularization in diabetic macular edema (116). Although VE-PTP was viewed as a potent player in the progression of diabetic microvascular complications through the deregulation of VEGFR/Tie2 angiogenic pathways, its role in lower limb angiogenesis still need to be clarified.
Density Enhanced Phosphatase 1
Density enhanced phosphatase 1 (DEP-1, gene: PTPRJ) is the second member of the PTPR family and shares mechanistic analogies with VE-PTP. Nevertheless, since DEP-1 expression is not restricted to EC, it can bind a larger range of pro-angiogenic RTK mainly VEGFR2 (117) and PDGFR-β (76). Hence, DEP-1 directly bound and selectively dephosphorylated the latter upon PDGF-BB stimulation, which in turn negatively affected PDGFR signaling in EC and fibroblasts (118) as well as in VSMC (74). Similar findings were made regarding VEGF signaling since DEP-1 bound (117) and dephosphorylated VEGFR2 at multiple sites following VEGF-A stimulation in HUVEC (119) leading to blunted VEGFR2/ERK axis and EC angiogenic properties (75). In contrast, DEP-1 exerted an indirect positive regulation of VEGFR2 by dephosphorylating inhibitory Y529 residue leading to enhanced EC angiogenic properties and permeability (77).
Although molecular mechanisms of DEP-1 are well-establish in vitro, little is known about its role in ischemia-induced hindlimb angiogenesis in PAD. Using the HLI model, a first study failed to detect variations in blood reperfusion or vessel generation in Ptprj−/− mice 7 days post-surgery (94). The deletion of DEP-1 reduced limb reperfusion and capillary density in HLI mice 14 days post-surgery compared to controls, which correlated with a loss of mobility and an increased amount of necrotic fingers (95). These data suggest that, despite a prominent negative regulatory role of pro-angiogenic RTK in vitro, DEP-1 might be critically required in lower limb collateral angiogenesis in vivo through the positive regulation of pro-angiogenic RTK downstream effectors. Nevertheless, the impact of diabetes on the regulation of DEP-1 and further angiogenic outcomes still need to be evaluated in both in vitro and in vivo preclinical models.
T-Cell Protein Tyrosine Phosphatase
T-cell PTP (TC-PTP, gene: PTPN2) is an intracellular ubiquitously expressed non-receptor PTP. TC-PTP was found to be expressed in both EC and VSMC (92) as well as involved in the regulation of VEGFR2 (120), PDGFR-β (108) but also epithelial growth factor receptor (EGFR) signaling (79). Using site-selective antibodies, Persson et al. showed an increase in the auto-phosphorylation of four PDGFR-β tyrosine residues following PDGF-BB stimulation in TC-PTP KO mice fibroblasts compared to control mice. The deletion of TC-PTP raised the phosphorylation of PDGFR-β at Y1021, which led to increase PLCγ activation and subsequent fibroblast migration (121). Besides PDGFR-β, TC-PTP co-precipitated with VEGFR2 upon VEGF-A stimulation in EC and selectively dephosphorylated several residues but not Y1175. Consequently, VEGF-A-driven HUVEC proliferation was modestly improved by silencing TC-PTP whereas migration and sprout formation were increased (120, 122).
The regulation of TC-PTP expression or activity has never been explored in preclinical models of diabetic PAD and the associated angiogenic outcomes are currently unknown. However, a recent study has reported a decrease in TC-PTP expression in the renal cortex of T1D mice, which was associated with increased fibrosis and progression of diabetic nephropathy suggesting a protective role of TC-PTP (78). Unlike promoting excessive angiogenesis in diabetic nephropathy, TC-PTP could be likely upregulated in the context of diabetic PAD leading to VEGF unresponsiveness. Nonetheless, this assumption must be assessed with further experiments using diabetic HLI models and cultured EC exposed to high glucose levels.
Phosphatase and Tensin Homolog
Phosphatase and tensin homolog (PTEN, gene: PTEN) is a ubiquitous lipid and protein phosphatase. PTEN is also the main regulator of the PI3K/Akt pathway by dephosphorylating phosphatidylinositol-3,4,5-trisphosphate (PIP3) into phosphatidylinositol-3,4-bisphosphate (PIP2), therefore indirectly preventing RTK-induced Akt activation (80). A mechanistic study reported that the overexpression of wild-type PTEN dampened VEGF-A-induced Akt activation and subsequent proliferation, migration, and tubule formation in HUVEC. Furthermore, overexpressing a dominant-negative form of PTEN rescued the Akt pathway and EC angiogenic properties (123). In a similar study, overexpressing PTEN inhibited the PDGF-BB-induced FAK/Akt/p70S6K cascade reducing proliferation and migration of VSMC (124). Interestingly, PTEN expression was upregulated by NF-κB in hypoxic EC and normalized following Ang-1 treatment (81).
PTEN was also upregulated in both aortic homogenates from T1D mice and EC exposed to high glucose concentrations which was associated with increased apoptosis. On the other hand, silencing PTEN restored Akt activation and EC viability similar to normoglycemic exposed cells (82). The upregulation of PTEN in HG-exposed EC and aorta of STZ-induced diabetic mice was dependent on endothelial upregulation of p53 (99).
Although the above-mentioned studies demonstrate that the anti-angiogenic role of PTEN could be potentiated by both ischemia and hyperglycemia, no data are currently available in the setting of diabetic PAD. Nevertheless, a clinical investigation revealed significantly increased expression of PTEN along with a decreasing trend in the activation of Akt/eNOS in atrial biopsies of T2D patients compared to healthy individuals. Consequently, this raises the assumption that similar PTEN activity may occur in ischemic diabetic limbs (125).
Dual-Specificity Phosphatases
Dual specificity protein phosphatases (DUSPs) are either cytoplasmic or nuclear phosphatases acting upon tyrosine or serine/threonine residues which are responsible for dephosphorylation and regulation of MAPKs (ERK, p38, and JNK). Therefore, DUSPs indirectly participate in the regulation of RTK-induced angiogenesis. Several studies reported a pro-angiogenetic role of multiple DUSPs. For instance, silencing DUSP1 blunted VEGF-A-induced migration (83, 96), proliferation (85), and tubule formation in HUVEC (96) as well as ex vivo EC sprouting (96). Fibroblasts isolated from DUSP4 KO mice exhibited reduced proliferation and increased apoptosis due to a sustained overactivation of ERK and JNK, respectively (86). Conversely, overexpressing DUSP1 and DUSP5 abolished p38/ERK activation and subsequent HUVEC migration/proliferation suggesting that a proper balance in DUSPs expression is critical for maintaining angiogenic properties of EC (85). High glucose exposure was shown to downregulate DUSP1 expression in VSMC causing increased and sustained activation of p38/ERK which was associated with excessive proliferation of VSMC (100). Recently, DUSP5 expression was found to be increased in hypoxic HUVEC but not in VSMC (84).
In the HLI model, the deletion of DUSP1 led to a significant reduction in blood flow reperfusion at days 3 and 7 post-surgery, gradually recovering to levels similar to controls by day 28. Muscle vessel density was altered in the same time frame highlighting the critical role of DUSP1 in the early response to hindlimb ischemia (96). Knocking-down DUSP5 also significantly reduced perfusion recovery and was correlated with an increase in limb necrosis which persisted 4 weeks post-surgery (84). Unfortunately, no studies have yet been conducted in the setting of diabetic PAD. Nevertheless, since diabetes-induced downregulation of DUSP1 (126) and DUSP4 (89) have been associated with worsened diabetic nephropathy, it is likely that any variations in DUSPs expression or activity could also contribute to the alteration of collateral vessel formation in the context of diabetic PAD.
Concluding Remarks and Future Perspectives
Hyperglycemia is deeply involved in the progression of macrovascular complications by deregulating cellular responses to hypoxia. Diabetic PAD is characterized by insufficient angiogenesis of the lower extremities which is attributed to impaired production of pro-angiogenic factors and defects in signal transduction in both vascular and perivascular cells. Although diabetes-induced alteration of VEGF signaling is extensively proven, there is still a lack of conclusive large-scale clinical studies concerning other growth factors such as PDGF and angiopoietins. Hence, larger clinical studies supported by suitable preclinical mechanistic models are required to guide and optimize the development of new therapeutic targets based on the administration of exogenous growth factors which are currently lacking sustainable effects. The source of vascular complications in diabetes is tremendously multifactorial and PTP have progressively emerged as potent players in vascular cell unresponsiveness to growth factors. Although the role of PTP in the modulation of pro-angiogenic RTK signaling is well-documented at a physiological level, their regulation in the complex hypoxic and hyperglycemic environment in PAD remains partially understood. Because the expression of PTP is not strictly limited to the endothelial lineage but extended to perivascular cells, the use of simultaneous cell and phosphatase-specific knockout models represent the next step in improving knowledge on this subject. In addition, since diabetic PAD is in part characterized by vascular and perivascular cell unresponsiveness to pro-angiogenic growth factors due to the upregulation or downregulation of PTP activity, the development of PTP inhibitors and activators is also an exciting avenue to modulate pro-angiogenic RTK activity (Figure 4). From a pharmacological point of view, the development of such drugs should ideally meet three criteria: PTP selectivity, cell population specificity, and ability to be locally delivered. PTP selectivity is crucial since not all PTP are exclusively negative regulators of pro-angiogenic RTK. Some PTP are critically required for proper angiogenic signal transduction through the dephosphorylation of inhibitory residues in downstream effectors (e.g., Y527 inhibitory on Src). The importance of cell population specificity could be found in the tight balance between angiogenesis and atherogenesis. Hence, the inhibition of PTP (e.g., SHP-1) in EC could promote enhanced VEGFR2 activation to improve EC angiogenic properties and the subsequent formation of neo-vessels. However, simultaneous overactivation of PDGFR-β through the inhibition of SHP-1 in VSMC may lead to uncontrolled proliferation and migration, representing a hallmark of restenosis. Finally, local delivery of PTP drugs exclusively in the ischemic muscle of PAD patients is desirable as pathological events can arise from the modulation of these ubiquitous proteins in other body locations. In a context of vascular complications, the systemic inhibition of PTP could trigger excessive angiogenesis in non-targeted tissues leading to the progression and worsening of diabetic retinopathy (127) or nephropathy (128). PTP drug-induced exacerbated and uncontrolled angiogenesis should also be avoided in the ischemic limb as it can promote atherosclerosis and plaque instability to subsequently increase the risk of PAD (129, 130). Local growth factor inhibitors such as anti-VEGF (131) or anti-Ang-2 (132) antibodies could be used along with PTP drugs to slow down the formation of atherosclerotic lesions and stabilize the neo-vasculature.
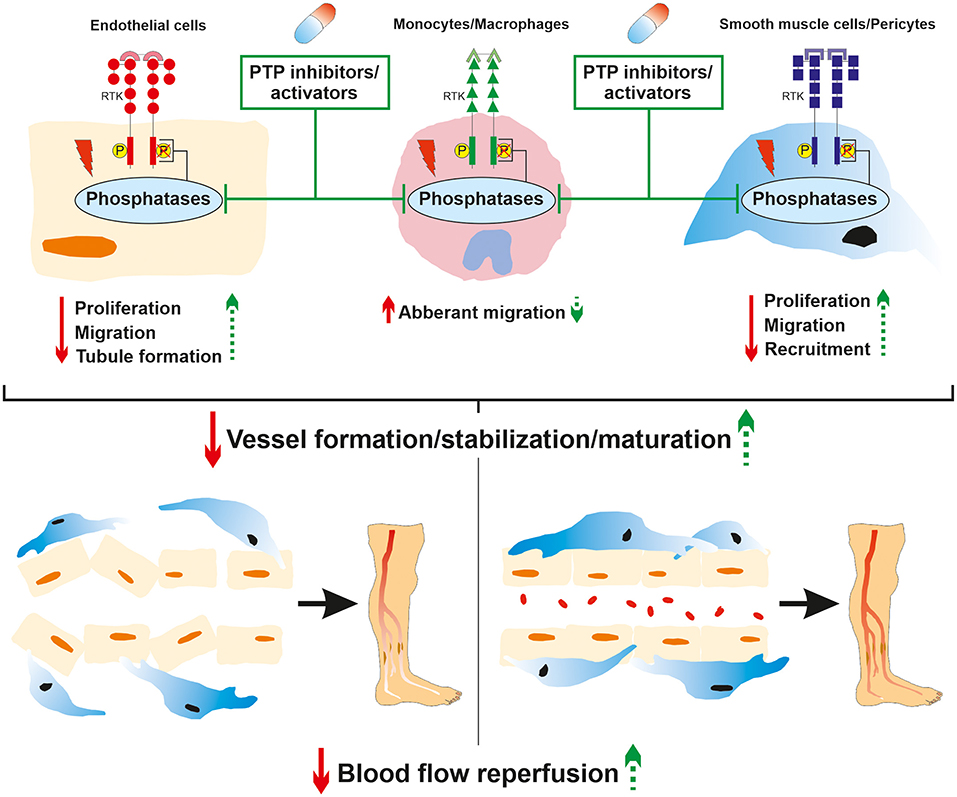
Figure 4. Role of phosphatases in diabetic PAD and therapeutic potential of targeted PTP drugs. Hyperglycemia-induced modulation of phosphatase expression or activity contributes to pro-angiogenic receptor tyrosine kinase signaling deregulation. This led to dysfunctions in endothelial cell, macrophage/monocyte and VSMC/pericyte angiogenic properties that subsequently impaired proper neo-vessel formation resulting in insufficient angiogenesis in the ischemic lower limb. The local administration of both PTP and cell specific inhibitors/activators could partially restore endothelial, immune, and perivascular cell angiogenic properties. This could contribute to improve neo-vessel generation, maturation, and stabilization to ultimately enhance blood flow reperfusion in the ischemic lower limb of diabetic PAD patients.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Author Contributions
CM and PG conceived the idea for the review. CM performed the literature search and wrote the manuscript. PG and MR critically revised the work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Canadian Institute of Health Research [PTJ159627 to PG]. This work was performed at the CHUS research center, funded by the Fonds de Recherche du Québec–Santé' (FRQ-S). PG is currently the holder of the Canada Research Chair in Vascular Complications of Diabetes.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Akt, protein kinase B; CLI, critical limb ischemia; CVD, cardiovascular disease; DUSP, dual-specificity phosphatase; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; EPC, endothelial progenitor cell; ERK, extracellular signal-regulated kinase; PAD, peripheral artery disease; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; PTP, protein tyrosin phosphatase; RTK, receptor tyrosine kinase; SDF-1, stromal cell-derived factor 1; STZ, streptozotocin; sVEGFR, soluble vascular endothelial growth factor receptor; T1D, Type I diabetes; T2D, Type II diabetes; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; VSMC, vascular smooth muscle cells; PTP1B, protein tyrosine phosphatase 1B; VE-PTP, Vascular endothelial PTP; PTEN, Phosphatase and tensin homolog; HbA1c, glycated hemoglobin; HLI, Hindlimb ischemia; PKC, protein kinase C; AT2R, angiotensin type 2 receptor; Ang-1/2, angiopoietin 1/2; SHP-1/2, Src homology region 2 domain-containing phosphatase-1/2.
References
1. Rowley WR, Bezold C, Arikan Y, Byrne E, Krohe S. Diabetes 2030: insights from yesterday, today, and future trends. Popul Health Manag. (2017) 20:6–12. doi: 10.1089/pop.2015.0181
2. Jude EB, Eleftheriadou I, Tentolouris N. Peripheral arterial disease in diabetes-a review: peripheral arterial disease in diabetes. Diabet Med. (2010) 27:4–14. doi: 10.1111/j.1464-5491.2009.02866.x
3. Jude EB, Oyibo SO, Chalmers N, Boulton AJM. Peripheral arterial disease in diabetic and nondiabetic patients: a comparison of severity and outcome. Diabetes Care. (2001) 24:1433–7. doi: 10.2337/diacare.24.8.1433
4. Faglia E, Clerici G, Clerissi J, Gabrielli L, Losa S, Mantero M, et al. Early and five-year amputation and survival rate of diabetic patients with critical limb ischemia: data of a cohort study of 564 patients. Eur J Vasc Endovasc Surg. (2006) 32:484–90. doi: 10.1016/j.ejvs.2006.03.006
5. McDermott MM. Lower extremity manifestations of peripheral artery disease: the pathophysiologic and functional implications of leg ischemia. Circ Res. (2015) 116:1540–50. doi: 10.1161/CIRCRESAHA.114.303517
6. Fadini GP, Albiero M, Bonora BM, Avogaro A. Angiogenic abnormalities in diabetes mellitus: mechanistic and clinical aspects. J Clin Endocrinol Metab. (2019) 104:5431–44. doi: 10.1210/jc.2019-00980
7. Waltenberger J. VEGF resistance as a molecular basis to explain the angiogenesis paradox in diabetes mellitus. Biochem Soc Trans. (2009) 37:1167–70. doi: 10.1042/BST0371167
8. Östman A. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. (2001) 11:258–66. doi: 10.1016/S0962-8924(01)01990-0
9. Denhez B, Geraldes P. Regulation of nephrin phosphorylation in diabetes chronic kidney injury. In: ed. Atassi MZ, Protein Reviews Advances In Experimental Medicine Biology. Singapore: Springer Singapore, 149–61. doi: 10.1007/5584_2017_62
10. Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, et al. Activation of PKC-δ and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. (2009) 15:1298–306. doi: 10.1038/nm.2052
11. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. (2000) 6:389–95. doi: 10.1038/74651
12. Otrock Z, Mahfouz R, Makarem J, Shamseddine A. Understanding the biology of angiogenesis: review of the most important molecular mechanisms. Blood Cells Mol Dis. (2007) 39:212–20. doi: 10.1016/j.bcmd.2007.04.001
13. Waltenberger J, Lange J, Kranz A. Vascular endothelial growth factor-a-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: a potential predictor for the individual capacity to develop collaterals. Circulation. (2000) 102:185–90. doi: 10.1161/01.CIR.102.2.185
14. Hochberg I, Hoffman A, Levy AP. Regulation of VEGF in diabetic patients with critical limb ischemia. Ann Vasc Surg. (2001) 15:388–92. doi: 10.1007/s100160010089
15. Blann AD, Belgore FM, McCollum CN, Silverman S, Lip PL, Lip GYH. Vascular endothelial growth factor and its receptor, Flt-1, in the plasma of patients with coronary or peripheral atherosclerosis, or Type II diabetes. Clin Sci. (2002) 102:187–94. doi: 10.1042/cs1020187
16. Makin AJ, Chung NAY, Silverman SH, Lip GYH. Vascular endothelial growth factor and tissue factor in patients with established peripheral artery disease: a link between angiogenesis and thrombogenesis? (2003) 104:397–404. doi: 10.1042/CS20020182
17. Mahdy RA, Nada WM, Hadhoud KM, El-Tarhony SA. The role of vascular endothelial growth factor in the progression of diabetic vascular complications. Eye. (2010) 24:1576–84. doi: 10.1038/eye.2010.86
18. Orrico C, Pasquinelli G, Foroni L, Muscarà D, Tazzari PL, Ricci F, et al. Dysfunctional vasa vasorum in diabetic peripheral artery obstructive disease with critical lower limb ischaemia. Eur J Vasc Endovasc Surg. (2010) 40:365–74. doi: 10.1016/j.ejvs.2010.04.011
19. Bleda S, De Haro J, Varela C, Esparza L, Ferruelo A, Acin F. Vascular endothelial growth factor polymorphisms are involved in the late vascular complications in Type II diabetic patients. Diab Vasc Dis Res. (2012) 9:68–74. doi: 10.1177/1479164111426162
20. Zakareia FA. Correlation of peripheral arterial blood flow with plasma chemerin and VEGF in diabetic peripheral vascular disease. Biomark Med. (2012) 6:81–7. doi: 10.2217/bmm.11.85
21. Wieczór R, Gadomska G, Ruszkowska-Ciastek B, Stankowska K, Budzyński J, Fabisiak J, et al. Impact of type 2 diabetes on the plasma levels of vascular endothelial growth factor and its soluble receptors type 1 and type 2 in patients with peripheral arterial disease. J Zhejiang Univ Sci B. (2015) 16:948–56. doi: 10.1631/jzus.B1500076
22. Yeboah K, Agyekum JA, Baafour EK, Antwi DA, Adjei AB, Boima V, et al. Circulating angiogenic growth factors in diabetes patients with peripheral arterial disease and exertional leg pain in ghana. Int J Vasc Med. (2017) 2017:1–7. doi: 10.1155/2017/2390174
23. Lim HS, Blann AD, Chong AY, Freestone B, Lip GYH. Plasma vascular endothelial growth factor, angiopoietin-1, and angiopoietin-2 in diabetes: implications for cardiovascular risk and effects of multifactorial intervention. Diabetes Care. (2004) 27:2918–24. doi: 10.2337/diacare.27.12.2918
24. Lim HS, Lip GY, Blann AD. Angiopoietin-1 and angiopoietin-2 in diabetes mellitus: relationship to VEGF, glycaemic control, endothelial damage/dysfunction and atherosclerosis. Atherosclerosis. (2005) 180:113–8. doi: 10.1016/j.atherosclerosis.2004.11.004
25. Findley CM, Mitchell RG, Duscha BD, Annex BH, Kontos CD. Plasma levels of soluble Tie2 and vascular endothelial growth factor distinguish critical limb ischemia from intermittent claudication in patients with peripheral arterial disease. J Am Coll Cardiol. (2008) 52:387–93. doi: 10.1016/j.jacc.2008.02.045
26. Li L, Qian L, Yu ZQ. Serum angiopoietin-2 is associated with angiopathy in type 2 diabetes mellitus. J Diabetes Compl. (2015) 29:568–71. doi: 10.1016/j.jdiacomp.2015.02.006
27. Siddiqui K, Joy SS, Nawaz SS. Serum Angiopoietin-2 levels as a marker in type 2 diabetes mellitus complications. Int J Diabetes Dev Ctries. (2019) 39:387–93. doi: 10.1007/s13410-018-0679-1
28. Rasul S, Reiter MH, Ilhan A, Lampichler K, Wagner L, Kautzky-Willer A. Circulating angiopoietin-2 and soluble Tie-2 in type 2 diabetes mellitus: a cross-sectional study. Cardiovasc Diabetol. (2011) 10:55. doi: 10.1186/1475-2840-10-55
29. Bento CF, Pereira P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia. (2011) 54:1946–56. doi: 10.1007/s00125-011-2191-8
30. Loomans CJM, de Koning EJP, Staal FJT, Rookmaaker MB, Verseyden C, de Boer HC, et al. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. (2004) 53:195–9. doi: 10.2337/diabetes.53.1.195
31. Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. (2005) 45:1449–57. doi: 10.1016/j.jacc.2004.11.067
32. Egan CG, Lavery R, Caporali F, Fondelli C, Laghi-Pasini F, Dotta F, et al. Generalised reduction of putative endothelial progenitors and CXCR4-positive peripheral blood cells in type 2 diabetes. Diabetologia. (2008) 51:1296–305. doi: 10.1007/s00125-008-0939-6
33. Krishna SM, Omer SM, Golledge J. Evaluation of the clinical relevance and limitations of current pre-clinical models of peripheral artery disease. Clin Sci. (2016) 130:127–50. doi: 10.1042/CS20150435
34. Hellingman AA, Bastiaansen AJNM, de Vries MR, Seghers L, Lijkwan MA, Löwik CW, et al. Variations in surgical procedures for hind limb ischaemia mouse models result in differences in collateral formation. Eur J Vasc Endovasc Surg. (2010) 40:796–803. doi: 10.1016/j.ejvs.2010.07.009
35. Rivard A, Silver M, Chen D, Kearney M, Magner M, Annex B, et al. Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am J Pathol. (1999) 154:355–63. doi: 10.1016/S0002-9440(10)65282-0
36. Schiekofer S, Galasso G, Sato K, Kraus BJ, Walsh K. Impaired revascularization in a mouse model of type 2 diabetes is associated with dysregulation of a complex angiogenic-regulatory network. Arterioscler Thromb Vasc Biol. (2005) 25:1603–9. doi: 10.1161/01.ATV.0000171994.89106.ca
37. Hazarika S, Dokun AO, Li Y, Popel AS, Kontos CD, Annex BH. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res. (2007) 101:948–56. doi: 10.1161/CIRCRESAHA.107.160630
38. Amin AH, Abd Elmageed ZY, Nair D, Partyka MI, Kadowitz PJ, Belmadani S, et al. Modified multipotent stromal cells with epidermal growth factor restore vasculogenesis and blood flow in ischemic hind-limb of type II diabetic mice. Lab Invest. (2010) 90:985–96. doi: 10.1038/labinvest.2010.86
39. Dokun AO, Chen L, Lanjewar SS, Lye RJ, Annex BH. Glycaemic control improves perfusion recovery and VEGFR2 protein expression in diabetic mice following experimental PAD. Cardiovasc Res. (2014) 101:364–72. doi: 10.1093/cvr/cvt342
40. Robillard S, Mercier C, Breton V, Paquin-Veillette J, Guay A, Lizotte F, et al. Ablation of angiotensin type 2 receptor prevents endothelial nitric oxide synthase glutathionylation and nitration in ischaemic abductor muscle of diabetic mice. Diab Vasc Dis Res. (2020) 17:147916411988397. doi: 10.1177/1479164119883978
41. Lizotte F, Paré M, Denhez B, Leitges M, Guay A, Geraldes P. PKCd impaired vessel formation and angiogenic factor expression in diabetic ischemic limbs. Diabetes. (2013) 62:2948–57. doi: 10.2337/db12-1432
42. Paquin-Veillette J, Lizotte F, Robillard S, Béland R, Breton M-A, Guay A, et al. Deletion of AT2 receptor prevents SHP-1-induced VEGF inhibition and improves blood flow reperfusion in diabetic ischemic hindlimb. Arterioscler Thromb Vasc Biol. (2017) 37:2291–300. doi: 10.1161/ATVBAHA.117.309977
43. Emanueli C. Type-2 diabetic leprdb/db mice show a defective microvascular phenotype under basal conditions and an impaired response to angiogenesis gene therapy in the setting of limb ischemia. Front Biosci. (2007) 12:2003. doi: 10.2741/2205
44. Yan J, Tie G, Park B, Yan Y, Nowicki PT, Messina LM. Recovery from hind limb ischemia is less effective in type 2 than in type 1 diabetic mice: roles of endothelial nitric oxide synthase and endothelial progenitor cells. J Vasc Surg. (2009) 50:1412–22. doi: 10.1016/j.jvs.2009.08.007
45. Tanii M, Yonemitsu Y, Fujii T, Shikada Y, Kohno R, Onimaru M, et al. Diabetic microangiopathy in ischemic limb is a disease of disturbance of the platelet-derived growth factor-BB/protein kinase C axis but not of impaired expression of angiogenic factors. Circ Res. (2006) 98:55–62. doi: 10.1161/01.RES.0000197842.38758.45
46. Fujii T, Onimaru M, Yonemitsu Y, Kuwano H, Sueishi K. Statins restore ischemic limb blood flow in diabetic microangiopathy via eNOS/NO upregulation but not via PDGF-BB expression. Am J Physiol Heart Circ Physiol. (2008) 294:H2785–H91. doi: 10.1152/ajpheart.00149.2008
47. Tuo Q, Zeng H, Stinnett A, Yu H, Aschner JL, Liao DF, et al. Critical role of angiopoietins/Tie-2 in hyperglycemic exacerbation of myocardial infarction and impaired angiogenesis. Am J Physiol Heart Circ Physiol. (2008) 294:H2547–57. doi: 10.1152/ajpheart.01250.2007
48. Chen JX, Stinnett A. Disruption of Ang-1/Tie-2 signaling contributes to the impaired myocardial vascular maturation and angiogenesis in type II diabetic mice. Arterioscler Thromb Vasc Biol. (2008) 28:1606–13. doi: 10.1161/ATVBAHA.108.169235
49. De Falco E, Avitabile D, Totta P, Straino S, Spallotta F, Cencioni C, et al. Altered SDF-1-mediated differentiation of bone marrow-derived endothelial progenitor cells in diabetes mellitus. J Cell Mol Med. (2010) 13:3405–14. doi: 10.1111/j.1582-4934.2008.00655.x
50. Wang K, Dai X, He J, Yan X, Yang C, Fan X, et al. Endothelial overexpression of metallothionein prevents diabetes mellitus-induced impairment in ischemia angiogenesis via preservation of HIF-1α/SDF-1/VEGF signaling in endothelial progenitor cells. Diabetes. (2020) 69:1779–92. doi: 10.2337/db20-605-P
51. Bakhashab S, Ahmed F, Schulten HJ, Ahmed F, Glanville M, Al-Qahtani M, et al. Proangiogenic effect of metformin in endothelial cells is via upregulation of VEGFR1/2 and their signaling under hyperglycemia-hypoxia. Int J Mol Sci. (2018) 19:293. doi: 10.3390/ijms19010293
52. Nakano T, Kumiko T, Mizumasa T, Kuroki Y, Tsuruya K, Kitazono T. The glucose degradation product methylglyoxal induces immature angiogenesis in patients undergoing peritoneal dialysis. Biochem Biophys Res Commun. (2020) 525:767–72. doi: 10.1016/j.bbrc.2020.02.048
53. Singh H, Brindle NPJ, Zammit VA. High glucose and elevated fatty acids suppress signaling by the endothelium protective ligand angiopoietin-1. Microvasc Res. (2010) 79:121–7. doi: 10.1016/j.mvr.2010.01.005
54. Chen YH, Lin SJ, Lin FY, Wu TC, Tsao CR, Huang PH, et al. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes. (2007) 56:1559–68. doi: 10.2337/db06-1103
55. Nakamura N, Naruse K, Kobayashi Y, Matsuki T, Hamada Y, Nakashima E, et al. High glucose impairs the proliferation and increases the apoptosis of endothelial progenitor cells by suppression of Akt: high glucose impairs EPC functions. J Diabetes Investig. (2011) 2:262–70. doi: 10.1111/j.2040-1124.2010.00093.x
56. Jiraritthamrong C, Kheolamai P, U-Pratya Y, Chayosumrit M, Supokawej A, Manochantr S, et al. In vitro vessel-forming capacity of endothelial progenitor cells in high glucose conditions. Ann Hematol. (2012) 91:311–20. doi: 10.1007/s00277-011-1300-6
57. You J, Sun J, Ma T, Yang Z, Wang X, Zhang Z, et al. Curcumin induces therapeutic angiogenesis in a diabetic mouse hindlimb ischemia model via modulating the function of endothelial progenitor cells. Stem Cell Res Ther. (2017) 8:182. doi: 10.1186/s13287-017-0636-9
58. Moriya J. Platelet-derived growth factor C promotes revascularization in ischemic limbs of diabetic mice. J Vasc Surg. (2014) 59:1402–09. doi: 10.1016/j.jvs.2013.04.053
59. Moriya J, Ferrara N. Inhibition of protein kinase C enhances angiogenesis induced by platelet-derived growth factor C in hyperglycemic endothelial cells. Cardiovasc Diabetol. (2015) 14:19. doi: 10.1186/s12933-015-0180-9
60. Cantero A, Portero-Otín M, Ayala V, Auge N, Sanson M, Elbaz M, et al. Methylglyoxal induces advanced glycation end product (AGEs) formation and dysfunction of PDGF receptor-β: implications for diabetic atherosclerosis. FASEB J. (2007) 21:3096–106. doi: 10.1096/fj.06-7536com
61. Soeda S, Shimada T, Koyanagi S, Yokomatsu T, Murano T, Shibuya S, et al. An attempt to promote neo-vascularization by employing a newly synthesized inhibitor of protein tyrosine phosphatase. FEBS Lett. (2002) 524:54–8. doi: 10.1016/S0014-5793(02)03002-8
62. Herre DJ, Norman JB, Anderson R, Tremblay ML, Huby A-C, Belin de Chantemèle EJ. Deletion of protein tyrosine phosphatase 1B (PTP1B) enhances endothelial cyclooxygenase 2 expression and protects mice from type 1 diabetes-induced endothelial dysfunction. PLoS ONE. (2015) 10:e0126866. doi: 10.1371/journal.pone.0126866
63. Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, et al. Role of protein tyrosine phosphatase 1b in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res. (2008) 102:1182–91. doi: 10.1161/CIRCRESAHA.107.167080
64. Guo D-Q, Wu L-W, Dunbar JD, Ozes ON, Mayo LD, Kessler KM, et al. Tumor necrosis factor employs a protein-tyrosine phosphatase to inhibit activation of kdr and vascular endothelial cell growth factor-induced endothelial cell proliferation. J Biol Chem. (2000) 275:11216–21. doi: 10.1074/jbc.275.15.11216
65. Nakagami H, Cui TX, Iwai M, Shiuchi T, Takeda-Matsubara Y, Wu L, et al. Tumor necrosis factor-α inhibits growth factor-mediated cell proliferation through SHP-1 activation in endothelial cells. Arterioscler Thromb Vasc Biol. (2002) 22:238–42. doi: 10.1161/hq0202.104001
66. Bhattacharya R, Kwon J, Wang E, Mukherjee P, Mukhopadhyay D. Src homology 2 (SH2) domain containing protein tyrosine phosphatase-1 (SHP-1) dephosphorylates VEGF receptor-2 and attenuates endothelial DNA synthesis, but not migration. J Mol Signal. (2008) 3:8. doi: 10.1186/1750-2187-3-8
67. Cho S-W, Hartle L, Son SM, Yang F, Goldberg M, Xu Q, et al. Delivery of small interfering RNA for inhibition of endothelial cell apoptosis by hypoxia and serum deprivation. Biochem Biophys Res Commun. (2008) 376:158–63. doi: 10.1016/j.bbrc.2008.08.123
68. Sugano M, Tsuchida K, Maeda T, Makino N. SiRNA targeting SHP-1 accelerates angiogenesis in a rat model of hindlimb ischemia. Atherosclerosis. (2007) 191:33–9. doi: 10.1016/j.atherosclerosis.2006.04.021
69. Sinha S, Vohra PK, Bhattacharya R, Dutta S, Sinha S, Mukhopadhyay D. Dopamine regulates phosphorylation of VEGF receptor 2 by engaging Src-homology-2-domain-containing protein tyrosine phosphatase 2. J Cell Sci. (2009) 122:3385–92. doi: 10.1242/jcs.053124
70. Laramée M, Chabot C, Cloutier M, Stenne R, Holgado-Madruga M, Wong AJ, et al. The scaffolding adapter gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J Biol Chem. (2007) 282:7758–69. doi: 10.1074/jbc.M611327200
71. Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun. (2013) 4:1672. doi: 10.1038/ncomms2683
72. Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, et al. VE-PTP controls blood vessel development by balancing tie-2 activity. J Cell Biol. (2009) 185:657–71. doi: 10.1083/jcb.200811159
73. Carota IA, Kenig-Kozlovsky Y, Onay T, Scott R, Thomson BR, Souma T, et al. Targeting VE-PTP phosphatase protects the kidney from diabetic injury. J Exp Med. (2019) 216:936–49. doi: 10.1084/jem.20180009
74. Kappert K, Paulsson J, Sparwel J, Leppänen O, Hellberg C, Östman A, et al. Dynamic changes in the expression of DEP-1 and other PDGF receptor-antagonizing PTPs during onset and termination of neointima formation. FASEB J. (2007) 21:523–34. doi: 10.1096/fj.06-6219com
75. Ortuso F, Paduano F, Carotenuto A, Gomez-Monterrey I, Bilotta A, Gaudio E, et al. Discovery of PTPRJ Agonist peptides that effectively inhibit in vitro cancer cell proliferation and tube formation. ACS Chem Biol. (2013) 8:1497–506. doi: 10.1021/cb3007192
76. Kovalenko M, Denner K, Sandström J, Persson C, Groβ S, Jandt E, et al. Site-selective dephosphorylation of the platelet-derived growth factor β-receptor by the receptor-like protein-tyrosine phosphatase DEP-1. J Biol Chem. (2000) 275:16219–26. doi: 10.1074/jbc.275.21.16219
77. Spring K, Chabot C, Langlois S, Lapointe L, Trinh NTN, Caron C, et al. Tyrosine phosphorylation of DEP-1/CD148 as a mechanism controlling Src kinase activation, endothelial cell permeability, invasion, and capillary formation. Blood. (2012) 120:2745–56. doi: 10.1182/blood-2011-12-398040
78. Li Y, Zhou H, Li Y, Han L, Song M, Chen F, et al. PTPN2 improved renal injury and fibrosis by suppressing STAT-induced inflammation in early diabetic nephropathy. J Cell Mol Med. (2019) 23:4179–95. doi: 10.1111/jcmm.14304
79. Tiganis T, Bennett AM, Ravichandran KS, Tonks NK. Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase. Mol Cell Biol. (1998) 18:1622–34. doi: 10.1128/MCB.18.3.1622
80. Georgescu M-M. PTEN tumor suppressor network in PI3K-akt pathway control. Genes Cancer. (2010) 1:1170–7. doi: 10.1177/1947601911407325
81. Lee SW, Youn SW, Kim TY, Suh JW, Koh GY, Kwon YW, et al. Angiopoietin-1 protects endothelial cells from hypoxia-induced apoptosis via inhibition of phosphatase and tensin homologue deleted from chromosome ten. Korean Circ J. (2009) 39:57. doi: 10.4070/kcj.2009.39.2.57
82. Song P, Wu Y, Xu J, Xie Z, Dong Y, Zhang M, et al. Reactive nitrogen species induced by hyperglycemia suppresses akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation. (2007) 116:1585–95. doi: 10.1161/CIRCULATIONAHA.107.716498
83. Kinney CM, Chandrasekharan UM, Mavrakis L, DiCorleto PE. VEGF and thrombin induce MKP-1 through distinct signaling pathways: role for MKP-1 in endothelial cell migration. Am J Physiol Cell Physiol. (2008) 294:C241–C50. doi: 10.1152/ajpcell.00187.2007
84. Alleboina S, Ayalew D, Peravali R, Chen L, Wong T, Dokun AO. Dual specificity phosphatase 5 regulates perfusion recovery in experimental peripheral artery disease. Vasc Med. (2019) 24:395–404. doi: 10.1177/1358863X19866254
85. Bellou S, Hink MA, Bagli E, Panopoulou E, Bastiaens PIH, Murphy C, et al. VEGF autoregulates its proliferative and migratory ERK1/2 and p38 cascades by enhancing the expression of DUSP1 and DUSP5 phosphatases in endothelial cells. Am J Physiol Cell Physiol. (2009) 297:C1477–C89. doi: 10.1152/ajpcell.00058.2009
86. Lawan A, Al-Harthi S, Cadalbert L, McCluskey AG, Shweash M, Grassia G, et al. Deletion of the dual specific phosphatase-4 (DUSP-4) gene reveals an essential non-redundant role for MAP kinase phosphatase-2 (MKP-2) in proliferation and cell survival. J Biol Chem. (2011) 286:12933–43. doi: 10.1074/jbc.M110.181370
87. Corti F, Simons M. Modulation of VEGF receptor 2 signaling by protein phosphatases. Pharmacol Res. (2017) 115:107–23. doi: 10.1016/j.phrs.2016.11.022
88. Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. (1999) 283:1544–8. doi: 10.1126/science.283.5407.1544
89. Denhez B, Rousseau M, Dancosst D-A, Lizotte F, Guay A, Auger-Messier M, et al. Diabetes-Induced DUSP4 reduction promotes podocyte dysfunction and progression of diabetic nephropathy. Diabetes. (2019) 68:1026–39. doi: 10.2337/db18-0837
90. Carr AN, Davis MG, Eby-Wilkens E, Howard BW, Towne BA, Dufresne TE, et al. Tyrosine phosphatase inhibition augments collateral blood flow in a rat model of peripheral vascular disease. Am J Physiol-Heart Circ Physiol. (2004) 287:H268–H76. doi: 10.1152/ajpheart.00007.2004
91. Sugano M, Tsuchida K, Makino N. A protein tyrosine phosphatase inhibitor accelerates angiogenesis in a rat model of hindlimb ischemia. J Cardiovasc Pharmacol. (2004) 44:460–5. doi: 10.1097/01.fjc.0000143275.45289.0a
92. Kappert K, Peters K, Bohmer F. Tyrosine phosphatases in vessel wall signaling. Cardiovasc Res. (2005) 65:587–98. doi: 10.1016/j.cardiores.2004.08.016
93. Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, et al. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation. (2014) 130:902–9. doi: 10.1161/CIRCULATIONAHA.114.009683
94. Hackbusch D, Dülsner A, Gatzke N, Krüger J, Hillmeister P, Nagorka S, et al. Knockout of density-enhanced phosphatase-1 impairs cerebrovascular reserve capacity in an arteriogenesis model in mice. BioMed Res Int. (2013) 2013:1–9. doi: 10.1155/2013/802149
95. Fournier P, Dussault S, Fusco A, Rivard A, Royal I. Tyrosine phosphatase PTPRJ/DEP-1 is an essential promoter of vascular permeability, angiogenesis, and tumor progression. Cancer Res. (2016) 76:5080–91. doi: 10.1158/0008-5472.CAN-16-1071
96. Boerckel JD, Chandrasekharan UM, Waitkus MS, Tillmaand EG, Bartlett R, DiCorleto PE. Mitogen-activated protein kinase phosphatase-1 promotes neovascularization and angiogenic gene expression. Arterioscler Thromb Vasc Biol. (2014) 34:1020–31. doi: 10.1161/ATVBAHA.114.303403
97. Dadke SS, Li HC, Kusari AB, Begum N, Kusari J. Elevated expression and activity of protein-tyrosine phosphatase 1B in skeletal muscle of insulin-resistant type II diabetic goto-kakizaki rats. Biochem Biophys Res Commun. (2000) 274:583–9. doi: 10.1006/bbrc.2000.3188
98. Dorenkamp M, Müller JP, Shanmuganathan KS, Schulten H, Müller N, Löffler I, et al. Hyperglycaemia-induced methylglyoxal accumulation potentiates VEGF resistance of diabetic monocytes through the aberrant activation of tyrosine phosphatase SHP-2/SRC kinase signalling axis. Sci Rep. (2018) 8:14684. doi: 10.1038/s41598-018-33014-9
99. Yokoyama M, Shimizu I, Nagasawa A, Yoshida Y, Katsuumi G, Wakasugi T, et al. p53 plays a crucial role in endothelial dysfunction associated with hyperglycemia and ischemia. J Mol Cell Cardiol. (2019) 129:105–17. doi: 10.1016/j.yjmcc.2019.02.010
100. Begum N, Ragolia L. High glucose and insulin inhibit VSMC MKP-1 expression by blocking iNOS via p38 MAPK activation. Am J Physiol Cell Physiol. (2000) 278:C81–C91. doi: 10.1152/ajpcell.2000.278.1.C81
101. Seely BL, Staubs PA, Reichart DR, Berhanu P, Milarski KL, Saltiel AR, et al. Protein tyrosine phosphatase 1b interacts with the activated insulin receptor. Diabetes. (1996) 45:1379–85. doi: 10.2337/diabetes.45.10.1379
102. Zhang Y, Li Q, Youn JY, Cai H. Protein phosphotyrosine phosphatase 1B (PTP1B) in calpain-dependent feedback regulation of vascular endothelial growth factor receptor (VEGFR2) in endothelial cells: implications in vegf-dependent angiogenesis and diabetic wound healing. J Biol Chem. (2017) 292:407–16. doi: 10.1074/jbc.M116.766832
103. Zhang J, Li L, Li J, Liu Y, Zhang CY, Zhang Y, et al. Protein tyrosine phosphatase 1B impairs diabetic wound healing through vascular endothelial growth factor receptor 2 dephosphorylation. Arterioscler Thromb Vasc Biol. (2015) 35:163–74. doi: 10.1161/ATVBAHA.114.304705
104. Brownrigg JRW, Apelqvist J, Bakker K, Schaper NC, Hinchliffe RJ. Evidence-based management of PAD & the diabetic foot. Eur J Vasc Endovasc Surg. (2013) 45:673–81. doi: 10.1016/j.ejvs.2013.02.014
105. Wo Tsui H, Siminovitch KA, de Souza L, Tsui FWL. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. (1993) 4:124–9. doi: 10.1038/ng0693-124
106. Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem. (1997) 272:32521–27. doi: 10.1074/jbc.272.51.32521
107. Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. (1997) 16:2352–64. doi: 10.1093/emboj/16.9.2352
108. Markova B, Herrlich P, Rönnstrand L, Böhmer F-D. Identification of protein tyrosine phosphatases associating with the PDGF receptor †. Biochemistry. (2003) 42:2691–9. doi: 10.1021/bi0265574
109. Huang L, Turck CW, Rao P, Peters KG. GRB2 and SH-PTP2: potentially important endothelial signaling molecules downstream of the TEK/TIE2 receptor tyrosine kinase. Oncogene. (1995) 11:2097–103.
110. Ha CH, Bennett AM, Jin ZG. A novel role of vascular endothelial cadherin in modulating c-Src activation and downstream signaling of vascular endothelial growth factor. J Biol Chem. (2008) 283:7261–70. doi: 10.1074/jbc.M702881200
111. Mitola S, Brenchio B, Piccinini M, Tertoolen L, Zammataro L, Breier G, et al. Type I collagen limits VEGFR-2 signaling by a SHP2 protein-tyrosine phosphatase-dependent mechanism 1. Circ Res. (2006) 98:45–54. doi: 10.1161/01.RES.0000199355.32422.7b
112. Bäumer S, Keller L, Holtmann A, Funke R, August B, Gamp A, et al. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood. (2006) 107:4754–62. doi: 10.1182/blood-2006-01-0141
113. Dominguez MG, Hughes VC, Pan L, Simmons M, Daly C, Anderson K, et al. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci USA. (2007) 104:3243–8. doi: 10.1073/pnas.0611510104
114. Mellberg S, Dimberg A, Bahram F, Hayashi M, Rennel E, Ameur A, et al. Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J. (2009) 23:1490–502. doi: 10.1096/fj.08-123810
115. Fachinger G, Deutsch U, Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor tie-2. Oncogene. (1999) 18:5948–53. doi: 10.1038/sj.onc.1202992
116. Campochiaro PA, Sophie R, Tolentino M, Miller DM, Browning D, Boyer DS, et al. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates tie2. Ophthalmology. (2015) 122:545–54. doi: 10.1016/j.ophtha.2014.09.023
117. Chabot C, Spring K, Gratton JP, Elchebly M, Royal I. New role for the protein tyrosine phosphatase DEP-1 in akt activation and endothelial cell survival. Mol Cell Biol. (2009) 29:241–53. doi: 10.1128/MCB.01374-08
118. Jandt E, Denner K, Kovalenko M, Östman A, Böhmer F-D. The protein-tyrosine phosphatase DEP-1 modulates growth factor-stimulated cell migration and cell-matrix adhesion. Oncogene. (2003) 22:4175–85. doi: 10.1038/sj.onc.1206652
119. Lampugnani MG, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, et al. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, β-catenin, and the phosphatase DEP-1/CD148. J Cell Biol. (2003) 161:793–804. doi: 10.1083/jcb.200209019
120. Mattila E, Auvinen K, Salmi M, Ivaska J. The protein tyrosine phosphatase TCPTP controls VEGFR2 signalling. J Cell Sci. (2008) 121:3570–80. doi: 10.1242/jcs.031898
121. Persson C, Savenhed C, Bourdeau A, Tremblay ML, Markova B, Bohmer FD, et al. Site-Selective regulation of platelet-derived growth factor? receptor tyrosine phosphorylation by t-cell protein tyrosine phosphatase. Mol Cell Biol. (2004) 24:12. doi: 10.1128/MCB.24.5.2190-2201.2004
122. Mattila E, Marttila H, Sahlberg N, Kohonen P, Tähtinen S, Halonen P, et al. Inhibition of receptor tyrosine kinase signalling by small molecule agonist of T-cell protein tyrosine phosphatase. BMC Cancer. (2010) 10:7. doi: 10.1186/1471-2407-10-7
123. Huang J, Kontos CD. PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. J Biol Chem. (2002) 277:10760–66. doi: 10.1074/jbc.M110219200
124. Huang J, Kontos CD. Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arterioscler Thromb Vasc Biol. (2002) 22:745–51. doi: 10.1161/01.ATV.0000016358.05294.8D
125. Wang B, Raedschelders K, Shravah J, Hui Y, Safaei HG, Chen DDY, et al. Differences in myocardial PTEN expression and akt signalling in type 2 diabetic and nondiabetic patients undergoing coronary bypass surgery: myocardial PTEN and akt-related signalling in patients with diabetes. Clin Endocrinol. (2011) 74:705–13. doi: 10.1111/j.1365-2265.2011.03979.x
126. Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J, et al. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J Cell Physiol. (2019) 234:3043–57. doi: 10.1002/jcp.27124
127. Crawford T, Alfaro D III, Kerrison J, Jablon E. Diabetic retinopathy and angiogenesis. Curr Diabetes Rev. (2009) 5:8–13. doi: 10.2174/157339909787314149
128. Nakagawa T, Kosugi T, Haneda M, Rivard CJ, Long DA. Abnormal angiogenesis in diabetic nephropathy. Diabetes. (2009) 58:1471–8. doi: 10.2337/db09-0119
129. Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R. Angiogenesis in the atherosclerotic plaque. Redox Biol. (2017) 12:18–34. doi: 10.1016/j.redox.2017.01.007
130. Slevin M, Krupinski J, Badimon L. Controlling the angiogenic switch in developing atherosclerotic plaques: possible targets for therapeutic intervention. J Angiogenesis Res. (2009) 1:4. doi: 10.1186/2040-2384-1-4
131. de Vries MR, Parma L, Peters HAB, Schepers A, Hamming JF, Jukema JW, et al. Blockade of vascular endothelial growth factor receptor 2 inhibits intraplaque haemorrhage by normalization of plaque neovessels. J Intern Med. (2019) 285:59–74. doi: 10.1111/joim.12821
132. Theelen TL, Lappalainen JP, Sluimer JC, Gurzeler E, Cleutjens JP, Gijbels MJ, et al. Angiopoietin-2 blocking antibodies reduce early atherosclerotic plaque development in mice. Atherosclerosis. (2015) 241:297–304. doi: 10.1016/j.atherosclerosis.2015.05.018
133. Li Y, Hazarika S, Xie D, Pippen AM, Kontos CD, Annex BH. In mice with type 2 diabetes, a vascular endothelial growth factor (VEGF)-activating transcription factor modulates VEGF signaling and induces therapeutic angiogenesis after hindlimb ischemia. Diabetes. (2007) 56:656–65. doi: 10.2337/db06-0999
Keywords: peripheral arterial disease, diabetes, growth factor, phosphatase, endothelia cell dysfunction
Citation: Mercier C, Rousseau M and Geraldes P (2021) Growth Factor Deregulation and Emerging Role of Phosphatases in Diabetic Peripheral Artery Disease. Front. Cardiovasc. Med. 7:619612. doi: 10.3389/fcvm.2020.619612
Received: 20 October 2020; Accepted: 10 December 2020;
Published: 07 January 2021.
Edited by:
Paul H. A. Quax, Leiden University, NetherlandsReviewed by:
Bernard Van Den Berg, Leiden University Medical Center, NetherlandsMª Carmen Duran-Ruiz, University of Cádiz, Spain
Janice Tsui, University College London, United Kingdom
Copyright © 2021 Mercier, Rousseau and Geraldes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Geraldes, Pedro.Geraldes@USherbrooke.ca