
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 25 January 2021
Sec. Cardiovascular Genetics and Systems Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.588347
This article is part of the Research TopicEpigenetic Regulation in Cardiovascular DiseasesView all 16 articles
 Somayeh Saadat1Mahdi Noureddini1Maryam Mahjoubin-Tehran2Sina Nazemi3Layla Shojaie4Michael Aschner5Behnaz Maleki1Mohammad Abbasi-kolli6Hasan Rajabi Moghadam7*Behrang Alani8*
Somayeh Saadat1Mahdi Noureddini1Maryam Mahjoubin-Tehran2Sina Nazemi3Layla Shojaie4Michael Aschner5Behnaz Maleki1Mohammad Abbasi-kolli6Hasan Rajabi Moghadam7*Behrang Alani8* Hamed Mirzaei9*
Hamed Mirzaei9*Unintended cardiac fibroblast proliferation in many pathophysiological heart conditions, known as cardiac fibrosis, results in pooling of extracellular matrix (ECM) proteins in the heart muscle. Transforming growth factor β (TGF-β) as a pivotal cytokine/growth factor stimulates fibroblasts and hastens ECM production in injured tissues. The TGF-β receptor is a heterodimeric receptor complex on the plasma membrane, made up from TGF-β type I, as well as type II receptors, giving rise to Smad2 and Smad3 transcription factors phosphorylation upon canonical signaling. Phosphorylated Smad2, Smad3, and cytoplasmic Smad4 intercommunicate to transfer the signal to the nucleus, culminating in provoked gene transcription. Additionally, TGF-β receptor complex activation starts up non-canonical signaling that lead to the mitogen-stimulated protein kinase cascade activation, inducing p38, JNK1/2 (c-Jun NH2-terminal kinase 1/2), and ERK1/2 (extracellular signal–regulated kinase 1/2) signaling. TGF-β not only activates fibroblasts and stimulates them to differentiate into myofibroblasts, which produce ECM proteins, but also promotes fibroblast proliferation. Non-coding RNAs (ncRNAs) are important regulators of numerous pathways along with cellular procedures. MicroRNAs and circular long ncRNAs, combined with long ncRNAs, are capable of affecting TGF-β/Smad signaling, leading to cardiac fibrosis. More comprehensive knowledge based on these processes may bring about new diagnostic and therapeutic approaches for different cardiac disorders.
Excessive aggregation of ECM, which is mainly produced by myofibroblasts, results in fibrosis (1). In addition, α-smooth muscle actin (α-SMA), a highly contractile protein, is expressed by myofibroblasts. ECM deposition is shown to be reversible, and improved cardiac function and coronary flow result in a minor collagen volume fraction regression (20% relative change and 1% absolute change) (2), an important indicator of ECM content. Additionally, patients with heart failure (HF) are commonly treated with renin–angiotensin–aldosterone system modulators, which lessen cardiac fibrosis (3, 4). Cardiac fibrosis induces pathological processes, which lead to chamber dilatation, muscular hypertrophy, and apoptosis, eventually developing into congestive HF (5). Cardiac fibrosis pathogenesis is complex with no efficient treatment options (6).
Transforming growth factor β1 (TGF-β1) is the principal isoform of TGF-β in cardiac tissue, which can cause Smad2/Smad3 (its downstream mediator) phosphorylation, which in turn can stimulate cardiac fibrosis development. It has been shown in mice that cardiac fibrosis related to pressure overload can be diminished by specific deletion of TGF-β1 or Smad3 gene in the triggered cardiac fibroblasts (CFs) (7). Non-coding RNAs (ncRNAs) include small microRNAs (miRNAs or miRs; > ~22 nucleotides) and long non-coding RNAs (lncRNAs; > ~200 nucleotides), as well as circular RNAs (circRNAs; > ~200 circular nucleotides) (1), all of which are involved in regulating several signaling pathways, including TGF-β and Smad, for the control of cytokine release, along with ECM production (8–10). Evidence corroborates the existence of cross-regulation between the two ncRNAs mediated fibrosis-stimulating pathways and its role in cardiac fibrosis pathophysiology. Recognizing mechanisms associated with such cross-regulation provides possibilities for the development of new therapeutic approaches to reverse cardiac fibrosis (10–12). The present review examines TGF-β, as well as Smad signaling, followed by their contribution in the cardiac fibrosis pathogenesis. In addition, evidence regarding TGF-β and Smad signaling involvement in vascular and cardiac remodeling across fibrotic events is detailed. Finally, ncRNAs (consisting of miRNAs, lncRNAs, and circRNAs) roles in TGF-β and Smad signaling in the heart are discussed. Specifically, the review will focus on the role of TGF-β/Smad signaling in ECM overproduction, cardiac fibrotic event, and myofibroblast alterations, which is the aim of this study. We point out the impacts of miRNAs and lncRNAs, as well as circular lncRNAs, on cardiac fibrosis via interaction with the signaling pathways of TGF-β/Smad.
Cardiac fibrosis, namely, the accumulation of scar tissue in the heart, is a product of mismatch between production and degradation of ECM and is strongly associated with cardiac and endocrine disorders (13). Upon stimulation, circulation and myocardial fibrosis–promoting growth factors as well as cytokines levels will increase and initiate a fibrotic response (14). Attachment of the fibrotic-promoting growth factors and cytokines takes place in the corresponding receptors in fibroblasts, namely, cytokine receptors, integrins, syndecans, and CD44 (15), after which signaling pathways and transcriptional factors, such as Smad, mitogen-stimulated protein kinases (MAPKs), nuclear factor κB, and protein kinase B (also called AKT), are activated. These activations induce CFs to transform into myofibroblasts, capable of expressing the strongly contractile protein α-SMA and producing certain tissue inhibitor of metalloproteinases (TIMPs), as well as matrix metalloproteinases (MMPs) for the modulation of ECM homeostasis (14). Additionally, synthesis and release of fibrotic-promoting growth factors and cytokines in CFs are controlled by these transcriptional factors (16). The growth factors and cytokines secreted via CFs or other cells, such as cardiomyocytes, and endothelial cells affect CFs or cardiomyocytes and create a positive feedback with final enhancement of the fibrotic responses (16).
In addition to various cell types (such as inflammatory, epithelial, endothelial, and other cells) that contribute to fibrogenesis, three cellular signaling transduction pathways contribute significantly during fibrosis: MAPKs, TGF-β, and integrins. The first pathway, which includes c-Jun NH2-terminal kinase (JNK), p38 MAPK, and extracellular signal–modulated kinase in mammals, has mediating effects on signaling, initiated by extracellular stimulation, such as growth factors and cytokines, or stimulation within the cells (17). The second pathway contributes significantly to the regulation of cellular functions, such as proliferation, differentiation, apoptosis, and survival. Integrins include subunits of α and β, which surface receptors on every cell type with the exception of red blood cells (18). Alongside extracellular receptors, signals transducing pathways engaged in fibrogenesis are triggered by integrins working in coordination with integrin-associated kinases within the cells (18–21).
TGF-β can be described as a cytokine with multifunctionality, whose expression takes place by various kinds of cells (22). The superfamily of TGF-β included the TGF-β isoforms (TGF-β1, TGF-β2, and TGF-β3) and activins, as well as inhibins, growth-differentiating factors, bone morphogenetic proteins (BMPs), together with anti-müllerian hormones (AMH) as suborders (23, 24). TGF-β plays a role in different diseases such as cardiac abnormality, cardiac fibrosis, failure of the heart, and remodeling of chamber, as well as cardiac hypertrophy (22) (Figure 1). TGF-β isoforms function with activins toward stimulating signals within the cells through Smad2/3 transcribing factors (25). TGF-β ligand complex has seven different type I receptors (which are sometimes called activin-like kinase or ALK receptors) or five type II receptors (ActRIIA, ActRIIB, TGFBRII, BMPRII, and AMHRII) (26, 27).
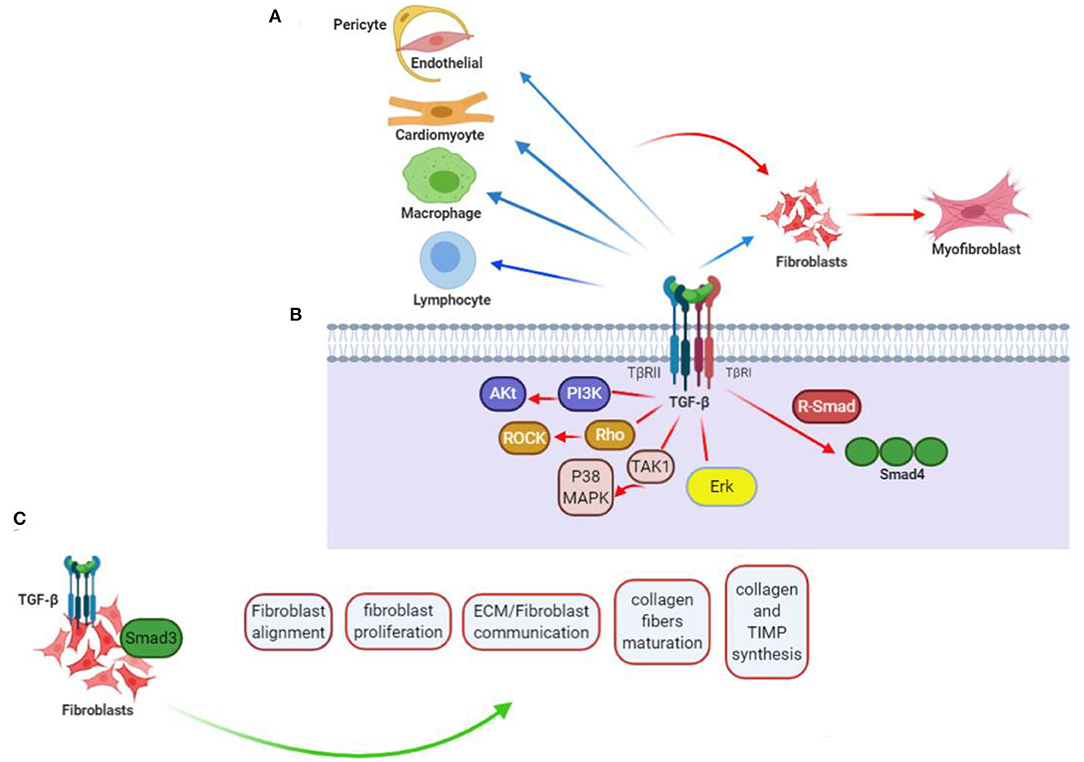
Figure 1. TGF-β contribution to cardiac fibrotic events. (A) TGF-β affects phenotype along with operation in every cell somehow engaged in myocardial fibrotic event. Straightforward effects on conversing fibroblast (F) to myofibroblast (MF) and activating myofibroblast can be probably of more importance, but fibrogenesis caused by TGF-βs can further relate with its impacts on the phenotype of macrophage (Ma), differentiation and function of lymphocyte (L), and cardiomyocyte (CM) viability, as well as gene expression. Apart from that, TGF-β can encourage pericyte (P) to fibroblast transformation and endothelial-to-mesenchymal transdifferentiation, when induced by vascular cells to express fibrosis-associated genes. (B) TGF-βs regulate phenotypes of the cells through activation of Smad-related together with non-Smad signaling pathways. (C) TGF-β/Smad3 signaling effect on CFs. Current researches taking advantage of loss-of-function procedures associated with specific cells drew conclusion that activating Smad3 contributes significantly to the formation of organized myofibroblast arrays after myocardial infarction. Lack of Smad3 in fibroblasts deranges infarcted heart reparations, resulting in higher risks of late cardiac rupture and undesirable chamber dilation. The evidence points out the reparative operation of fibroblasts activated in the infarcted myocardium. Mediation of the Smad3 effects is being carried out by the integrin–ROS axis arousal. This figure was adapted from Frangogiannis (15).
It has been previously demonstrated that TGF-β-stimulated clone 22 (TSC-22) could facilitate TGF-β signaling by antagonizing Smad7 activity secondary to enhanced receptor stability. TSC-22 increases TGF-β-induced transcriptional responsiveness and phosphorylation of Smad2/3 (28). Furthermore, the stimulatory effect of TSC-22 is Smad7-dependent, and silencing the expression of Smad7 abolishes TSC-22's effect. TSC-22 can interact with TβRI (TGF-β type I receptor) and Smad7 and prevent the Smad7/Smurfs and TβRI association and receptor degradation. TSC-22 also promotes cardiac myofibroblast differentiation by increasing fibrotic gene expression for α-SMA, fibronectin, plasminogen activator inhibitor 1 (PAI-1), and collagen I, consistent with TSC-22 upregulation and phospho-Smad2/3 in myocardial fibrotic hearts. Therefore, it has been suggested that TSC-22 could regulate TGF-β signaling through a positive-feedback mechanism and may lead to myocardial fibrosis (28).
Binding of type II receptor TGFBRII with TGF-β1 ligands leads to phosphorylation of the type I receptor ALK-5. Various ligands may bind to cell surface TGF-β receptors, which lead to activation of signaling effectors and the Sma- and Mad-related proteins (Smads), as well as interacting with deoxyribonucleic acid (29). TGF-β, myostatin, or activin activates both Smad2 and Smad3, whereas activation of Smad1, Smad5, and Smad8 is performed with BMPs, leading to interactions with Smad4, bringing forth modulating the target gene expression (24, 30, 31). It is noteworthy that TGF-β pathway activation will lead to upregulation of Smad6 and Smad7 expression as well, in turn deactivating the pathways (29). Several ncRNAs and their substrates play a role in the TGF-β signal transduction pathway regulation (21).
Smad2/3 activation affects various profibrotic gene expression, consisting of collagens [COL1A1, COL3A1, COL5A2, COL6A1, COL6A3, COL7A1, (32)], PAI-1 (33, 34), various proteoglycans (35–37), integrins (38), connective tissue growth factor (CTGF) (39), and MMPs (27, 40).
Considerable increase in the levels of TGF-β was observed in individuals experiencing ischemic cardiomyopathy (ICM) and dilated cardiomyopathy (DCM), showing that TGF-β levels correlate with phosphorylated Smad2, along with collagen types I and III, triggering further myocardial fibrotic events in ICM and DCM secondary to activation of TGF-β (41). Fibulin 2 is an essential ECM protein for TGF-β/Smad signaling. Moreover, phosphorylation of Smad2 is achieved only in the presence of fibulin-2 (42). Peroxisome proliferator–activated receptor γ (PPARγ) activation was thought to moderate cardiac fibrosis. A study showed that TGF-β1 directly suppresses PPARγ expression by increasing binding of Smad2/3, Smad4, histone deacetylase 1 (HDAC1), and decreasing binding of HDAC3 to the PPARγ promoter in CFs (43). Another study has shown that reactive oxygen species (ROS) derived from NADPH oxidase 4 (Nox4) enhanced myocardial fibroblasts reaction against TGF-β1 through TGF-β Smad signaling pathways (44). Wnt/β-catenin pathway in inflammatory DCM has been shown to be activated by secretion of Wnt proteins in response to TGF-β signaling, mediated by Smad-independent TGF-β-activated kinase 1 (TAK1) (45, 46). Wnt inactivation or Wnt secretion hindrance impeded TGF-β-mediated CF transformation into pathogenic myofibroblasts, making Wnt protein secretion a neoteric downstream process of TGF-β-modulated cardiac fibrotic development (46). It has been demonstrated that CTGF, also known as CCN2, may play roles in the hypertension-induced myocardial fibrosis through regulation of TGF-β expression (22, 47).
ncRNAs are short RNAs that act as epigenetic regulators (48). The regulation of these molecules is related to modulation of several physiological properties such as apoptosis, cell proliferation, metabolism, and differentiation. Deregulation of these molecules shows associations with the onset and progress of various diseases, such as cardiovascular diseases, diabetes, cancer, and inflammatory disorders (49). According to existing evidence, ncRNAs can be categorized into two main groups: (i) short ncRNAs possessing fewer than 200 small nucleotides in their length (i.e., snoRNAs, siRNA, piwi-RNA, and miRNAs), (ii) lncRNAs possessing more than 200 nucleotides in their length including lncRNAs and circRNAs (16, 50). Cardiac fibrosis is a common feature in many types of heart diseases. ncRNA deregulation has been posited to be associated with cardiac fibrosis development and occurrence (49). Table 1 summarizes the role of different ncRNAs contributing to cardiac fibrosis pathogenesis.
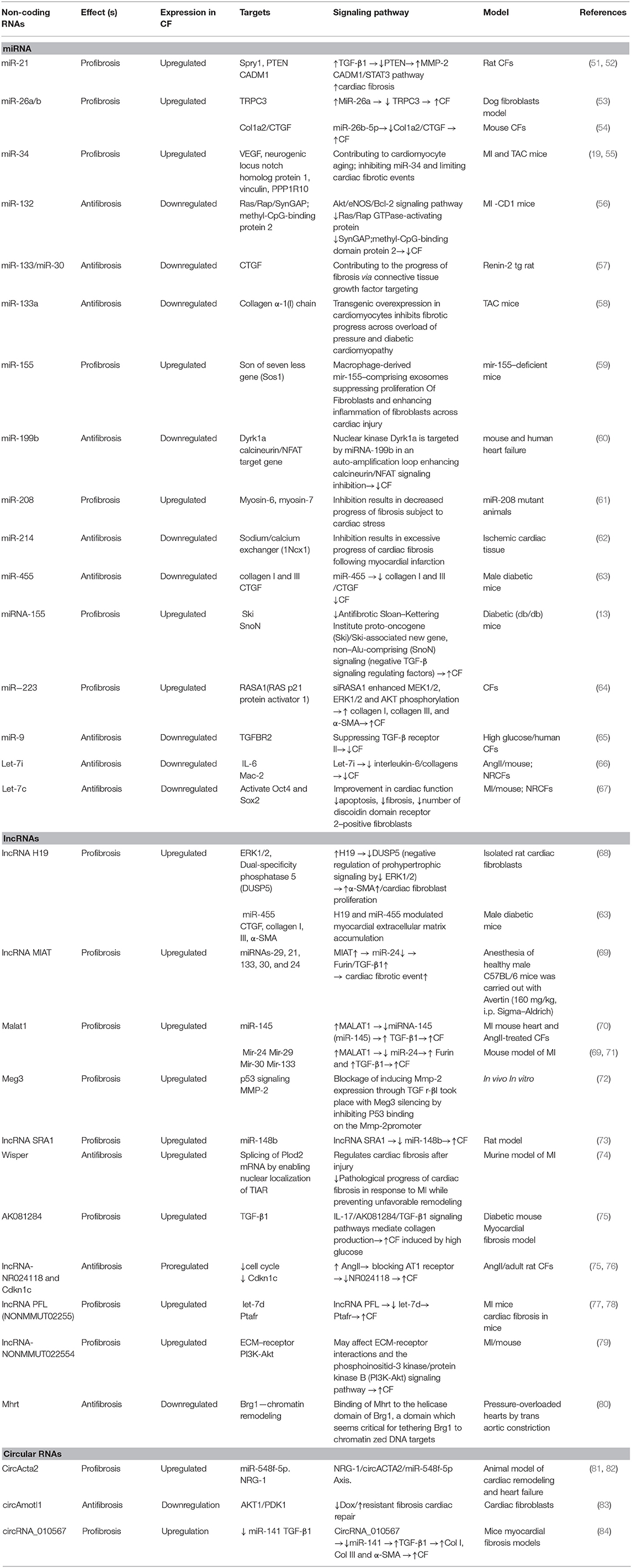
Table 1. ncRNAs contributing to cardiac fibrosis.
As mentioned previously, miRNAs can be defined as short ncRNAs with a length of 18 to 24 nucleotides (85, 86). miRNAs are capable of regulating the function of proteins by binding to target messenger RNA. This may result in the induction of mRNA degradation and/or suppression of protein translation. It has been shown that these molecules modulate myocardial fibrosis pathogenesis (Table 1) (87). Cardiac fibrosis is a complicated process involving the concerted interaction of multiple miRNAs. In this respect, different miRNAs are related to same pathologically fibrotic process. For instance, miR-24, miR-21, miR-34a, miR-29, and miR-433 contribute to fibrosis following infarction, and miR-26a, miR-21, and miR-125b are associated with pressure-overload fibrosis, which is caused by transverse aortic constriction (88–91). In addition, various miRNAs could be classified into antifibrotic (e.g., miR-15 family, miR-101a, miR-145, miR-378, miR-122, miR-142-3p) or profibrotic miRNAs (e.g., miR-29, miR-21, miR-34, miR-208, miR-155, miR-223) (88–91). miRNAs exert their regulatory effects on cardiac fibrosis, although affecting a sequence of cellular and molecular pathways, such as TGF-β/Smad system, MRTF/SRF axis RhoA/ROCK cascade, Wnt signaling, AngII/MAPK signaling, and the cationic channels that regulate calcium responses (92). Callis et al. evaluated miR-208a role in cardiac fibrosis induction. They indicated that miR-208a plays its role via targeting THRAP-1 and myostatin in myocardial hypertrophy (93). Furthermore, they showed miR-208a can induce cardiac fibrosis through increased endogen expression (93). Other study demonstrated that the upregulation of miR-208b is related to myocardial function enhancement and could inhibit type I collagen and alias α-SMA. In agreement, miR-208b exerts protection against post-infarction myocardial fibrosis by targeting GATA4 (94).
TGF-β1 can be associated with collagen secretion and activation in myocardial fibroblasts, which play a role in cardiac fibrosis development with other risk factors (95). Furin can modulate TGF-β activation by targeting AngII (96). Bearing that in mind, furin can exert its functions by TGF-β activation (97). Chen et al. showed that miR-24 downregulation is associated with cardiac infarction. Their findings confirmed that miR-24 exerts its effects by inhibiting TGF-β1 with having impact on furin. TGF-β1 and furin levels were elevated, indicating a critical role of miR-24 deregulation in myocardial fibrotic events following myocardial infarction (98).
Intra-action of the cell death and inflammation to myocardial fibrosis is crucial (99). Pyroptosis, namely, cell death triggered by inflammatory reactions, is described by apoptosis and necrosis (100). Nod-like receptor protein 3 (NLRP3) inflammasome expression in cardiac fibrosis is activated by inflammation; subsequently, it activates the cleaved caspase (101). Recent studies have corroborated the contribution of pyroptosis in myocardial fibrosis pathogenesis (102). Nonetheless, the initiating mechanisms for cardiac fibrosis and fibroblast-derived pyroptosis have yet to be determined. Thus, identification of the pathological mechanisms along with efficient treatment targets of myocardial fibrosis is essential. Growth arrest–specific 5 (GAS5), a lncRNA, whose encoding takes place by the GAS5 gene, has been introduced as a tumor suppressor in variety of cancer types (103). GAS5 contributes critically to cell apoptosis and pyroptosis (104). She et al. (105) identified lncRNA-GAS5 as the initiator of pyroptosis in CFs and cardiac fibrotic events. Upon lipopolysaccharide (LPS) stimulation, they detected ISO-induced CF pyroptosis and myocardial fibrosis. Proteins associated with pyroptosis include caspase 1, NLRP3, and DNMT1, higher in cardiac fibrotic tissues, with reduced GAS5 expression. Furthermore, lncRNA GAS5 overexpression enhances and prevents CF pyroptosis and also decreases the expression of caspase 1 and NLRP3 in CF. Other research showed that treating with DNMT inhibitors, 5-aza-2-deoxycytidine, or downregulating DNMT1 caused an increase in expression of GAS5 by reversing promoter hypermethylation in CF. Notably, it has been shown that DNMT1 methylation of lncRNA GAS5 results in CF pyroptosis when NLRP3 axis is affected, suggesting a novel regulatory mechanism regarding CF pyroptosis subject to LPS stress (105).
RNA component of mitochondrial RNA processing endoribonuclease (RMRP) is known as a lncRNA (106). RMRP forms a distinct ribonucleoprotein complex by interaction with the telomerase reverse transcriptase catalytic subunit, which exhibits the activity of RNA-dependent RNA polymerase and makes double-stranded RNAs, which with getting processed can turn into small interfering RNA (siRNA) (106). Prior work has examined the contribution of RMRP to various cancers, such as in lung cancer, gastric cancer, and glioma (107–109). Additionally, Wang et al. (110) reported that the level of RMRP expression in nucleus pulposus tissues correlates with grade of disc degeneration. Another investigation gas demonstrated that overexpression of RMRP could induce nucleus pulposus cell growth and regulate the ECM expression with targeting miR-206. In a recent study, Greco et al. profiled 83-lncRNA expression in biopsies taken from left ventricle of patients suffering HF and corroborated remarkable upregulation of RMRP in these patients (111). Steinbusch et al. (112) found associations of RMRP with chondrocyte hypertrophy and determined chondrogenic differentiation, proposing the contribution of RMRP to the modulation of the dynamic balance of ECM degradation and synthesis. Zhang et al. (113) explored the biological role and mechanisms behind CF induction by the lncRNA, RNA component of RMRP. The findings showed that RMRP expression in an abdominal aortic banding–treated rat model was upregulated in the presence of myocardial fibrosis. Treatment with AngII enhanced RMRP expression in CFs, whereas RMRP knockdown by small-interfering RNA prevented CF proliferation and differentiation as well as collagen accumulation. Based on these findings, RMRP might regulate miR-613 negatively in CFs. Moreover, it was showed that miR-613 mediates the positive effect of RMRP on activation of CF. Based on the present study, RMRP increased CF activation with serving as a competing endogenous RNA for miR-613. Thus, RMRP may represent as a novel target to prevent or treat cardiac fibrosis (113).
miR-125b induces fibrotic process and upregulation in CFs, indicating numerous binding sites of miR-125b for circ_LAS1L, with inverse association of their expression in those with acute myocardial infarction (AMI) and CFs. RNA immunoprecipitation (RIP), pull-down, and dual-luciferase reporter gene assay supported direct binding of miR-125b bound to circ_LAS1L (114). Overexpressed Circ_LAS1L led to promotion of the downstream target gene secreted frizzled-associated protein 5 (SFRP5) expressions, while reducing α-SMA, collagen I, and collagen III expression; hindering CF proliferation and migration; and increasing apoptosis. Cotransfection with miR-125b mimics and circ_LAS1L overexpression vector did not show considerable changes. However, cotransfection of SFRP5 siRNA and circ_LAS1L overexpression vector resulted downregulation of SFRP5 expression and upregulation of collagen I, collagen III, and α-SMA, as well as enhancement in proliferation and migration of CFs. Accordingly, circ_LAS1L reduces miR-125b activities through its adsorption, consequently increasing SFRP5 and subsequent regulation of the CFs biological properties. Such results can be regarded as a significant experimental basis for regulating myocardial fibrosis following myocardial infarction. CircRNAs contribute critically to the cardiovascular diseases; however, little research has been done on their effect on the myocardial fibrosis. Sun et al. investigated that circ_LAS1L in those suffering AMI and CFs was downregulated and was capable of direct binding to miR-125b, consequently enhancing the downstream target gene secreted frizzled-related protein 5 (SFRP5) expression, finally repressing CF activating, proliferating, and migrating, along with inducing apoptosis. Thus, it is has been posited that the circ_LAS1L/miR-125b/SFRP5 pathway is capable of modulating the biological characteristics of CF and can contribute vitally to the process of cardiac fibrosis, therefore offering a significant theoretical basis to regulate cardiac fibrotic event following myocardial infarction (114).
Gu et al. (115) explored circRNA expression profile and identified circRNA contributions to myocardial fibrosis. Utilization of TGF-β1 aimed at establishing an in vitro cardiac fibrotic model in CFs. CircRNA sequencing unveiled that an overall number of 283 circRNAs was expressed abnormally in fibrotic CFs, of which 79 were experiencing upregulation and 204 receiving downregulation. Alterations in randomly selected circRNA expression could be verified with the use of real-time polymerase chain reaction. Establishment of a circRNA-based competing endogenous RNA network 1,755 nodes and 30,394 edges was followed by module analyses performed with implementation of the plug-in MCODE. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses targeted mRNAs, engaging in the top three enriched modules. It was found that these mRNAs were enriched in myocardial fibrosis–associated signaling pathways, namely, the AMPK signaling pathway, TGF-β signaling pathway, MAPK signaling pathway, and PI3K-Akt signaling pathway. The predicted ceRNAs and bioinformatics analysis unveiled the possible contribution of circRNAs in myocardial fibrotic event, providing novel knowledge on the mechanisms and searching for efficient preventive, as well as therapeutic targets for myocardial fibrosis (115).
Based on existing evidence, expression of abnormal circRNA takes place in the cardiac fibrotic process. During promotion of CF activated by TGF-β1 or AngII, marked suppression in circRNA circ_BMP2K and miR-455-3p expression has been observed, along with induction of SUMO1 expression (116). RIP, pull-down assay, and dual-luciferase reporter gene assay, demonstrating direct binding of miR-455-3p to circ_BMP2K and their induction of each other's expression. SUMO1 served as a target gene for miR-455-3p, and circ_BMP2K boosted the miR-455-3p inhibiting on the expression of the SUMO1. According to several studies, both circ_BMP2K and miR-455-3p suppressed expressing α-SMA and types I and III collagen, but SUMO1 increased their expression, and the regulatory impacts of circ_BMP2K and miR-455-3p were reversed by miR-455-3p inhibitors or SUMO1 overexpression. Circ_BMP2K and miR-455-3p reduced CF proliferation and migration, concomitantly inducing their apoptosis; however, SUMO1 effect was the opposite; circ_BMP2K and miR-455-3 upregulation on biological characteristics was reversed by miR-455-3p inhibitors or overexpression of SUMO1. Therefore, circ_BMP2K induces expression of miR-455-3p with subsequent downregulation of SUMO1 expression and ultimately prevents CF activation, growth, and migration (116).
Various ligands have the ability of binding to TGF-β receptors on the surfaces of cells, permitting regulatory messages transfer to the cells through activation of the signaling effectors, as well as the Sma- and Mad-associated proteins (Smads) and finally, showing interactions with deoxyribonucleic acid (29). Activation of Smad2 and Smad3 are carried out with TGF-β, myostatin, or activin, whereas Smad1, Smad5, and Smad8 are activated by BMPs; activating such proteins leads to interactions with Smad4, resulting in target gene expression modulation (117). Notably, the TGF-β pathway activation additionally leads to upregulation of Smad6 and Smad7 expression, which may end in the pathway deactivation (24, 29). Smad2 and Smad7 lessen fibrosis, but Smad3 results in the promotion of fibrosis (118) (Figure 2). Several miRNAs and their substrates contribute to regulating TGF-β signal transduction pathways (Table 2, Figure 3) (21).
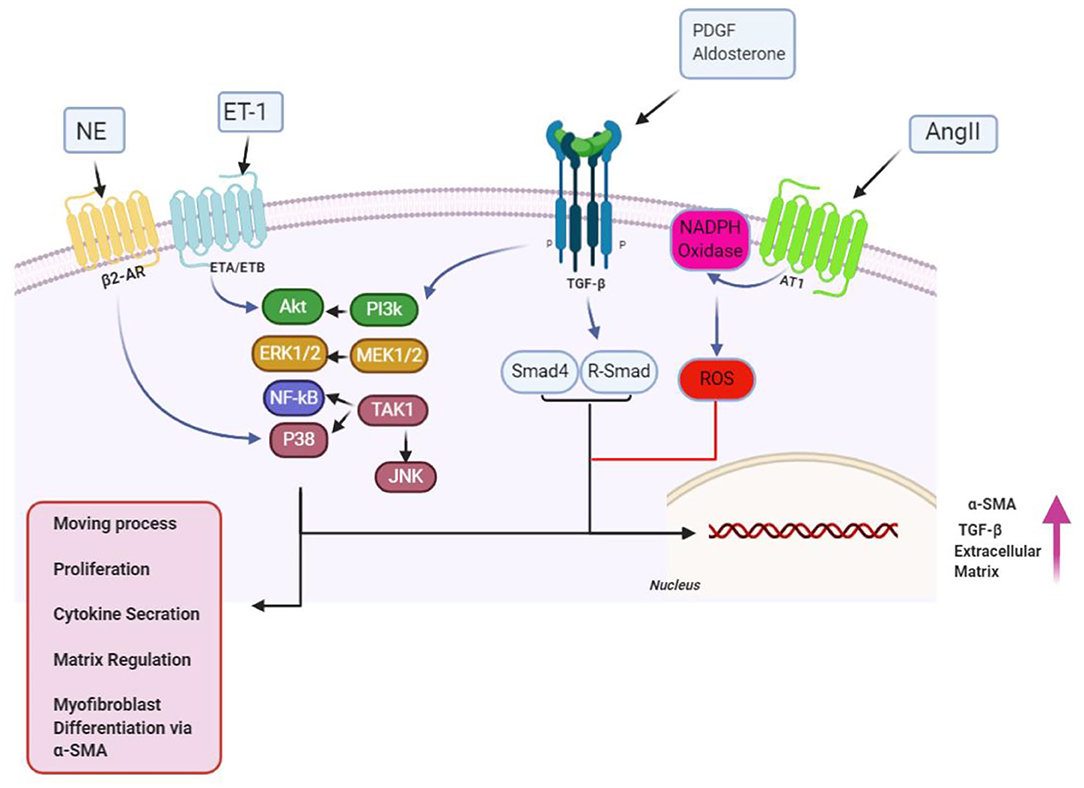
Figure 2. Over the proliferation phase of infarct remedial, fibrogenic growth, as well as neurohumoral mediating factors, stimulates myofibroblast proliferating and migrating, along with activating. A broad scope of fibrogenic mediators, engaged in this phase of cardiac, has been considered in activation of myofibroblast. Neurohumoral mediating factors, including angiotensin II (AngII), aldosterone, and norepinephrine (NE), and growth factors [transforming growth factor (TGF-β), fibroblast growth factor, and platelet-derived growth factor (PDGFs)], together with special matrix proteins, including ED-A fibronectin and matricellular proteins, have cooperation in activating intracellular signaling pathways, which enhance conversion, as well as proliferation of myofibroblast, while modulating ECM protein expression, and of genes related to matrix metabolism. Design of the cartoon took place according to Servier Medical Art (https://smart.servier.com). AR1/4, adrenergicreceptor; ET1/4, endothelin; MMP1/4, matrix metalloproteinase; NF1/4, nuclear factor; ROS1/4, reactive oxygen species 1/4; SMA1/4, smooth muscle actin; TIMP1/4, tissue inhibitor of metalloproteinase. This figure was adapted from Humeres and Frangogiannis (119).
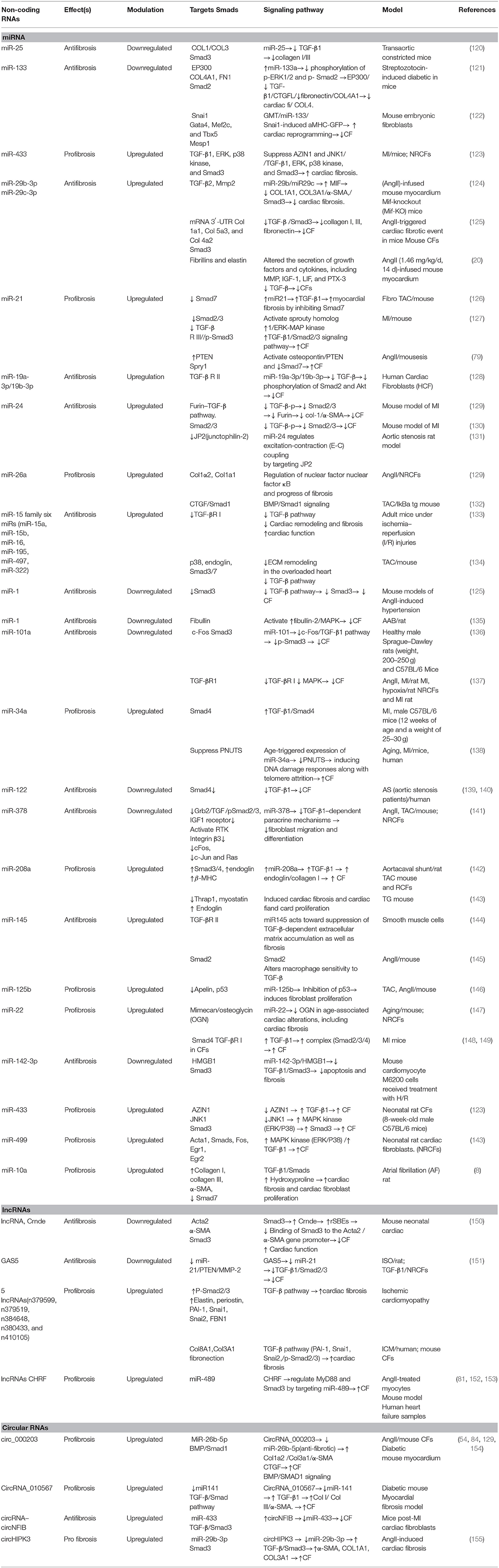
Table 2. ncRNAs and TGF-β/Smad signaling in cardiac fibrosis.
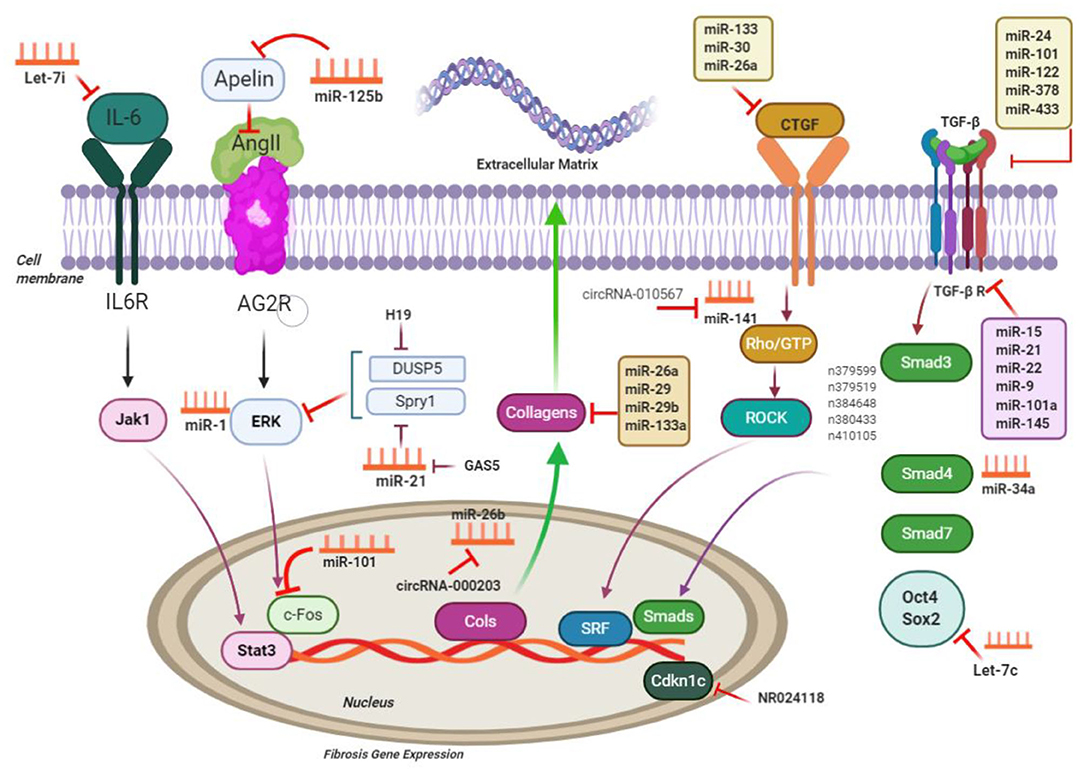
Figure 3. ncRNAs engaged in the pathways of cardiac fibrosis; ncRNAs regulate processes related to cardiac fibrosis via targeting the main molecules mediating ECM gene transcription and performing TGF-β signaling; CTGF, connective tissue growth factor; Rho-GTP, Rho-GTPase-stimulating protein; ROCK, Rho related coiled-coil comprising protein kinase; SRF, serum response factor; MMP, matrix metalloproteinases; IL6, interleukin-6; Jak1, Janus kinase 1; Stat3, signal transducers and activators of transcription 3; c-Fos, FBJ murine osteosarcoma viral oncogene homolog; Spry1, sprouty homolog 1; ERK extracellular signal–regulated kinases; DUSP5, dual-specificity phosphatase 5. This figure was adapted from Chen et al. (49).
miRNA-associated TGF-β pathways in cardiac fibrotic event exert their effects when they target the common ECM protein CTGF. Moreover, it was demonstrated that miR-101 inhibited interstitial fibrosis and then, by inhibition of a c-Fos/TGF-β1 axis, may promote myocardial infarction (136). Downregulation of miR-101 was evident in infarcted myocardium in mice and in angiotensin-cultured CFs. Interestingly, miR-101 overexpression inhibited proliferating and producing COL through suppression of its target c-Fos and the downstream protein TGF-β1 (136). Transfection of miR-101 mimic significantly suppressed the expression of TGF-β RI and p-Smad3, CF differentiation, and collagen content (137). According to He et al., miR-21 may reinforce the TGF-β1/Smad signaling pathway in atrial fibrosis stimulated by AF, through Smad7 downregulation (126). A reciprocal loop was ascertained between miR-21 and its target TGF receptor III, causing ECM remodeling and fibrotic process. Upregulation of cardiac miR-21 occurred in infarcted myocardium as a result of TGF-β1/Smad2/3 signaling pathway activation, whereas downregulation of its target gene (TGF receptor III) was evident. Nevertheless, lower expression of the TGF receptor III reinforced TGF-β1/Smad2/3 signaling pathway (126, 156).
Thum et al. (127) showed promotion of myocardial fibrosis by miR-21 by targeting extracellular modulated kinase inhibitor sprouty homolog 1 (Spry1) while activating MAPK signaling in cardiac fibroblasts. In a myocardial ischemia–reperfusion model, miR-21 was found to target Pten, subsequently leading to an increase in matrix metalloprotease 2 (Mmp2). Consistently, miR-21 antagonism leads to increased Pten in cardiac fibroblasts (157). miR-24 overexpression reduced secretion of the TGF-β and phosphorylation of the Smad2/3 in CFs (130). miR-24 showed protective features against myocardial fibrosis following myocardial infarction, which was dependent on the inhibitive effects on its target gene FURIN, suppressing the TGF-β signaling pathways (98, 130). Wang et al. (130) demonstrated interference of miR-24 with TGF signaling by targeting the pro-protein convertase, furin, and then downregulation of TGF level in cardiac fibroblasts with targeting CTGF. miR-18a and miR-19b downregulated the expression of the collagen (COL) 1A1 as well as COL3A1, reducing cardiac fibrosis in age-related cardiac failure triggered through activation of TGF-β (19, 124, 128). Functional examinations are consistent with prevention of HCF autophagy by miR-19a-3p/19b-3p with targeting TGF-β R II mRNA. Furthermore, autophagy development releases suppressive effects of miR-19a3p/19b-3p on Smad2 and Akt phosphorylation via TGF-βRII signaling (128).
In addition, many other miRNAs were also recognized to target collagens and TGF signaling to contribute to the fiwere also. For example, Let-7i and miR-26a reduce collagen deposition and impose their effects by targeting Col12 and Col11, correspondingly (66, 132, 158). miR-29b upregulation because of TGF/Smad3 inactivation downregulated profibrotic genes, such as ECM genes elastin (159), fibrillin 1 (Fbn1), collagen type I, 1 and 2 (Col11, Col12), and collagen type III, 1 (Col31) (160) and enhanced cardioprotective impacts of carvedilol vs. myocardial fibrosis triggered by AMI (79, 125). It was shown that insulin-like growth factor 1 and leukemia inhibitory factor, which are targeted by miR-29b, play roles in activating CF and proliferating ECM (20, 124).
Tao et al. investigated that miR-433 was related to cardiac fibrosis and is a potential target to mitigate cardiac fibrosis. Their study has found that cardiac fibrosis induces miR-433, subsequently decreasing the expression of AZIN1 and JNK1. Downregulated AZIN1 induces TGF-β1 pathway, whereas decreased JNK1 results in ERK and p38 kinase activation, causing Smad3 activation and eventually leading to cardiac fibrosis (123). In another study in that same year, Ooi et al. (161) suggested that AZIN1 expression reduction induces TGF-β/Smad3 signaling activation in CFs; (III) reduced JNK level would enhance ERK, P38 kinase, and Smad3 phosphorylation, and that is in turn associated with proliferation and differentiation of fibroblast into myofibroblasts.
miR-133a contribution to cardiac fibrosis and electrical repolarization in adult hearts with pressure overload can potentially indicate its regulatory impacts on Col11 A1, Serca2a, and calcineurin expression (58, 162). Based on existing evidence, miR-133a overexpression has prevented myocardial fibrotic event in both AngII-related hypertension and diabetes, even though the effector proteins were different in diabetes (fibronectin and COL4A1) and AngII-related hypertension (COL1A1) (21, 121, 162). Moreover, overexpression of the cardiac miR-133a inhibited ERK1/2 and Smad2 phosphorylation. Accordingly, it is posited that miR-133a may show efficacy in treating myocardial events triggered by diabetes (90, 121).
miR-15 family members are also regarded as having antifibrotic characteristics, through functions against TGF-β-mediated actions (163). miR-15 family members (miR-15a, miR-15b, miR-16, miR-195, miR-497, miR-322) can be observed in a variety of cardiac cell types, and with cardiac stress, they are expressed at higher levels (134, 163, 164). miR-15, in fibroblasts, targets some of TGF-β signaling cascade members, such as TGF-β1, p38, endoglin, Smad3, and Smad7, and as a result, leads to negative regulation of ECM production. Correspondingly, in vivo miR-15 suppression with LNA-based anti-miRs in mice resulted in higher levels of fibrosis following transverse aortic constriction (163). However, the miR-15 family inhibition in a mouse model of reperfusion injury led to smaller infarct sizes and lesser cardiac remodeling (134). Table 2 lists various non-coding RNAs in the CF via activation/inhibition of Smad/TGF signaling pathway.
Several lncRNAs contribute to the TGF-β pathways affecting the ECM gene expression along with myofibroblast differentiation (165). According to Huang et al., regulation of lncRNAs expression took place in ICM dynamically, in which several lncRNAs further attend in the TGF-pathways provoking gene expression associated with accumulating collagen along with ECM protein encoding genes (e.g., COL14A1, COL16A1, COL12A1, COL8A1) and myofibroblast differentiation. Huang et al. reported altered lncRNA expression in ICM and demonstrated that CF-enriched lncRNAs such as n379599, n379519, n384648, n380433, and n410105 in mouse modulate the fimouse-associated gene expression by targeting TGF-β signaling (165). TGF-β expression targets PAI-1, Snai1, and Snai2 in CF, and several lncRNA overexpression indicated induction of these target gene expression by lncRNAs. It was also demonstrated that lncRNAs induced phosphorylated Smad2/3 and not Smad2/3 protein (165, 166).
Tao et al. recently studied the lncRNA growth arrest–specific 5 (GAS5) role and function in cardiac fibrosis and concluded that GAS5 via negative miR-21 regulation plays its suppressive role in cardiac fibrosis. Moreover, they demonstrated that the modulation of miR-21 regulated MMP-2 expression via a phosphatase as well as tensin homolog (PTEN) pathway in CFs (151). miR-21 down regulation decreased secretion of TGF-β and phosphorylation of Smad2/3 in CFs (126).
lncRNAs and cardiac fibrosis CHRF (cardiac hypertrophy–related factor) upregulation was noted in myocytes treated with AngII and in the heart of a mouse model with transverse aortic constriction and human HF sample (152). CHRF knockdown increased miR-489 level but decreased Myd88 level in myocytes. CHRT overexpression reduced miR-489 level and upregulated Myd88 level and resulted in hypertrophic responses. Cardiac fibrosis was decreased in Myd88-knockout mice. CHRF regulates MyD88 and Smad3 by targeting miR-489. This study proposed CHRF as a role player in cardiac fibrosis by miR-489 and Myd88 adjustment (81, 152). lncRNA Crnde, by means of Smad3-Crnde negative feedback in diabetic cardiomyopathy, alleviates cardiac fibrosis. Crnde overexpression markedly prevents α-SMA promoter activity induced by TGF-β. Crnde stops Smad3 transcriptional activity via rSBEs (RNA SBEs) (49, 150, 165, 166).
Zhou et al. (84) showed that circRNA-010567 boosts myocardial fibrosis through suppression of miR-141 suppression along with targeting TGF-β1 in a mice model with diabetes. In another recent article, it was shown that upregulation of CircRNA_000203 took place in diabetic mice cardiac muscle and in AngII-triggered fibroblasts in the animal's heart (54). CircRNA_000203 characterizes as a miR-26-5p sponge and interacts with miR-26-5p and fibrosis-related genes Col1a2, Col3a1, and α-SMA and CTGF in fibroblasts in mouse heart (54, 167).
Zhu et al. suggested that the circNFIB–miR-433 axis can potentially provide new therapeutic target to treat fibrotic diseases. circNFIB overexpression decreased pro-proliferative impacts stimulated by means of the miR-433 mimic, while inhibiting circNFIB led to contrary results. circNFIB was recognized as a miR-433 endogenous sponge. circNFIB upregulation also lessened the activation of p38, ERK kinases, and the Smad3 signaling pathways were indicated through reduced ratios of p-p38/p38, p-ERK/ERK, and p-Smad3/Smad3 (168).
CircHIPK3 expression led to a significant increase in CFs and heart tissues following AngII treatments. CircHIPK3 silencing decreased CFs proliferating as well as migrating and the α-SMA expression level upregulation triggered by AngII in vitro. circHIPK3 served as a miR-29b-3p sponge, and circHIPK3 overexpression reversed miR-29b-3p–triggered inhibition of CF proliferation and migration, while altering miR-29b-3p targeting genes (α-SMA, COL1A1, COL3A1) expression levels in vitro. circHIPK3 silencing and miR-29b-3p overexpression conjointly exerted more severe effects on suppression of cardiac fibrotic event in vivo compared to either of them alone. In addition, the expression of circHIPK3 was also markedly increased after TGF-β1 treatment (155). Their data suggested that circHIPK3 functions as a miR-29b-3p sponge in the adjustment of CF proliferating, migrating, and promoting cardiac fibrotic event, introducing possible novel targets to be explored in preventing cardiac fibrosis triggered by AngII (155).
The uncompromising progress of fibrosis represents a pathological finding inherent to multiple cardiac diseases. Gaining insight into these fibrotic processes in terms of the functional characteristics and molecular profiling could make it possible to prevent and treat fibrotic lesions in the heart. An enlarging body of evidence addresses the cross-talk between the TGF-β and Smad signaling pathways and its contribution to cardiac fibrosis pathogenesis. Despite the fact that the TGF-β and Smad pathways have been extensively studied, their contributions to profibrotic pathways in cardiac diseases are yet to be known. ncRNAs have been identified as possible role players in strategies for mitigating CVDs, as discussed before. Current research on ncRNAs described herein focuses on the role of ncRNAs in regulating cell signaling pathways, particularly TGF-β and Smad signaling. The identified signaling pathways discussed herein, which have roles in the involvement of ncRNAs in cardiac fibrosis, may offer novel putative targets for therapeutic approaches for cardiac fibrosis. More studies are required to better understand the mechanisms by which the ncRNA network induces cardiac fibrotic events via TGF-β/Smad signaling. In addition, the potential clinical significance of the TGF-β/Smad-associated ncRNAs, including miRNAs implemented as therapeutic instruments and circRNAs employed as diagnostic/prognostic biomarkers for cardiac fibrotic cases, needs testing in additional animal models as well as clinical conditions.
HM involved in conception, design, statistical analysis, and drafting of the manuscript. LS, SN, MAs, MM-T, SS, BA, MN, BM, MAb, and HR contributed in data collection and manuscript drafting. All authors approved the final version for submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Yousefi F, Shabaninejad Z, Vakili S, Derakhshan M, Movahedpour A, Dabiri H, et al. TGF-β and WNT signaling pathways in cardiac fibrosis: non-coding RNAs come into focus. Cell Commun Signal. (2020) 18:87. doi: 10.1186/s12964-020-00555-4
2. Schelbert EB, Fonarow GC, Bonow RO, Butler J, Gheorghiade M. Therapeutic targets in heart failure: refocusing on the myocardial interstitium. J Am Coll Cardiol. (2014) 63:2188–98. doi: 10.1016/j.jacc.2014.01.068
3. Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Invest Circ. (2000) 102:2700–6. doi: 10.1161/01.CIR.102.22.2700
4. Izawa H, Murohara T, Nagata K, Isobe S, Asano H, Amano T, et al. Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy: a pilot study. Circulation. (2005) 112:2940–45. doi: 10.1161/CIRCULATIONAHA.105.571653
5. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. (2016) 118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565
6. Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG (2010). The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 48, 504–511. doi: 10.1016/j.yjmcc.2009.07.015
7. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. (2017) 127:3770–83. doi: 10.1172/JCI94753
8. Li P-F, He R-H, Shi S-B, Li R, Wang Q-T, Rao G-T, et al. Modulation of miR-10a-mediated TGF-β1/Smads signaling affects atrial fibrillation-induced cardiac fibrosis and cardiac fibroblast proliferation. Biosci Rep. (2019) 39:BSR20181931. doi: 10.1042/BSR20181931
9. Wang L, Jiang P, He Y, Hu H, Guo Y, Liu X, et al. A novel mechanism of Smads/miR-675/TGFβR1 axis modulating the proliferation and remodeling of mouse cardiac fibroblasts. J Cell Physicol. (2019) 234:20275–85. doi: 10.1002/jcp.28628
10. Wei Y, Wu Y, Feng K, Zhao Y, Tao R, Xu H, et al. Astragaloside IV inhibits cardiac fibrosis via miR-135a-TRPM7-TGF-β/Smads pathway. J Ethnopharmacol. (2020) 249:112404. doi: 10.1016/j.jep.2019.112404
11. Carthy JM, Garmaroudi FS, Luo Z, McManus BM. Wnt3a induces myofibroblast differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS ONE. (2011) 6:e19809. doi: 10.1371/journal.pone.0019809
12. Tran BH, Yu Y, Chang L, Tan B. A novel liposomal s-propargyl-cysteine: a sustained release of hydrogen sulfide reducing myocardial fibrosis via TGF-β1/Smad Pathway. Int J Nanomed. (2019) 14:10061–77. doi: 10.2147/IJN.S216667
13. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. (1991) 83:1849–65. doi: 10.1161/01.CIR.83.6.1849
14. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. (2014) 71:549–74. doi: 10.1007/s00018-013-1349-6
15. Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. (2019) 65:70–99. doi: 10.1016/j.mam.2018.07.001
16. Ma Z-G, Yuan Y-P, Wu H-M, Zhang X, Tang Q-Z. Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci. (2018) 14:1645. doi: 10.7150/ijbs.28103
17. Kim EK, Choi E-J. Compromised MAPK signaling in human diseases: an update. Arch Toxicol. (2015) 89:867–82. doi: 10.1007/s00204-015-1472-2
18. Beauvais S, Drevelle O, Jann J, Lauzon M-A, Foruzanmehr M, Grenier G, et al. Interactions between bone cells and biomaterials: an update. Front Biosci (Sch Ed). (2016) 8:227–63. doi: 10.2741/s460
19. van Almen GC, Verhesen W, van Leeuwen RE, van de Vrie M, Eurlings C, Schellings MW, et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. (2011) 10:769–79. doi: 10.1111/j.1474-9726.2011.00714.x
20. Abonnenc M, Nabeebaccus AA, Mayr U, Barallobre-Barreiro J, Dong X, Cuello F, et al. Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c. Circ Res. (2013) 113:1138–47. doi: 10.1161/CIRCRESAHA.113.302400
21. Yang C, Zheng SD, Wu HJ, Chen SJ. Regulatory mechanisms of the molecular pathways in fibrosis induced by microRNAs. Chin Med J (Engl). (2016) 129:2365–72. doi: 10.4103/0366-6999.190677
22. An Z, Yang G, Zheng H, Nie W, Liu G. Biomarkers in patients with myocardial fibrosis. Open Life Sci. (2017) 12:337–44. doi: 10.1515/biol-2017-0039
23. Bai J, Xi Q. Crosstalk between TGF-β signaling and epigenome. Acta biochimica et biophysica Sinica. (2018) 50:60–7. doi: 10.1093/abbs/gmx122
24. Hanna A, Frangogiannis NG. The role of the TGF-beta superfamily in myocardial infarction. Front Cardiovasc Med. (2019) 6:140. doi: 10.3389/fcvm.2019.00140
25. Hata A, Chen Y-G. TGF-β signaling from receptors to Smads. Cold Spring Harb Perspect Biol. (2016) 8:a022061. doi: 10.1101/cshperspect.a022061
26. Aykul S, Martinez-Hackert E. Transforming growth factor-β family ligands can function as antagonists by competing for type II receptor binding. J Biol Chem. (2016) 291:10792–804. doi: 10.1074/jbc.M115.713487
27. Walton KL, Johnson KE, Harrison CA. Targeting TGF-beta mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol. (2017) 8:461. doi: 10.3389/fphar.2017.00461
28. Yan X, Zhang J, Pan L, Wang P, Xue H, Zhang L, et al. TSC-22 promotes transforming growth factor β-mediated cardiac myofibroblast differentiation by antagonizing Smad7 activity. Mol Cell Biol. (2011) 31:3700–9. doi: 10.1128/MCB.05448-11
29. MacDonald EM, Cohn RD. TGFβ signaling: its role in fibrosis formation and myopathies. Curr Opin Rheumatol. (2012) 24:628–34. doi: 10.1097/BOR.0b013e328358df34
30. Rebbapragada A, Benchabane H, Wrana J, Celeste A, Attisano L. Myostatin signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Mol Cell Biol. (2003) 23:7230–42. doi: 10.1128/MCB.23.20.7230-7242.2003
31. Hudnall AM, Arthur JW, Lowery JW. Clinical relevance and mechanisms of antagonism between the BMP and activin/TGF-β signaling pathways. J Am Osteopath Assoc. (2016) 116:452. doi: 10.7556/jaoa.2016.089
32. Verrecchia F, Chu M-L, Mauviel A. Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. (2001) 276:17058–62. doi: 10.1074/jbc.M100754200
33. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. (1998) 17:3091–100. doi: 10.1093/emboj/17.11.3091
34. Hua X, Liu X, Ansari DO, Lodish HF. Synergistic cooperation of TFE3 and smad proteins in TGF-β-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. (1998) 12:3084–95. doi: 10.1101/gad.12.19.3084
35. Schönherr E, Järveläinen H, Sandell L, Wight T. Effects of platelet-derived growth factor and transforming growth factor-beta 1 on the synthesis of a large versican-like chondroitin sulfate proteoglycan by arterial smooth muscle cells. J Biol Chem. (1991) 266:17640–7.
36. ROMARiS M, Bassols A, David G. Effect of transforming growth factor-β1 and basic fibroblast growth factor on the expression of cell surface proteoglycans in human lung fibroblasts. Enhanced glycanation and fibronectin-binding of CD44 proteoglycan, and down-regulation of glypican. Biochem J. (1995) 310:73–81. doi: 10.1042/bj3100073
37. Dadlani H, Ballinger ML, Osman N, Getachew R, Little PJ. Smad and p38 MAP kinase-mediated signaling of proteoglycan synthesis in vascular smooth muscle. J Biol Chem. (2008) 283:7844–52. doi: 10.1074/jbc.M703125200
38. Margadant C, Sonnenberg A. Integrin–TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. (2010) 11:97–105. doi: 10.1038/embor.2009.276
39. Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A. CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int. (2002) 62:1149–59. doi: 10.1111/j.1523-1755.2002.kid567.x
40. Yuan W, Varga J. Transforming growth factor-β repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J Biol Chem. (2001) 276:38502–10. doi: 10.1074/jbc.M107081200
41. Khan S, Joyce J, Margulies KB, Tsuda T. Enhanced bioactive myocardial transforming growth factor-β in advanced human heart failure. Circ J. (2014) 78:2711–8. doi: 10.1253/circj.CJ-14-0511
42. Khan SA, Dong H, Joyce J, Sasaki T, Chu M-L, Tsuda T. Fibulin-2 is essential for angiotensin II-induced myocardial fibrosis mediated by transforming growth factor (TGF)-β. Lab Invest. (2016) 96:773. doi: 10.1038/labinvest.2016.52
43. Gong K, Chen Y-F, Li P, Lucas JA, Hage FG, Yang Q, et al. Transforming growth factor-β inhibits myocardial PPARγ expression in pressure overload-induced cardiac fibrosis and remodeling in mice. J Hypertens. (2011) 29:1810. doi: 10.1097/HJH.0b013e32834a4d03
44. Yu Q-G, Zhang Y. Transforming growth factor-β1 mediates NADPH oxidase 4: a significant contributor to the pathogenesis of myocardial fibrosis. Int J Cardiol. (2017) 227:53–4. doi: 10.1016/j.ijcard.2016.10.118
45. Kumawat K, Menzen MH, Slegtenhorst RM, Halayko AJ, Schmidt M, Gosens R. TGF-β-activated kinase 1 (TAK1) signaling regulates TGF-β-induced WNT-5A expression in airway smooth muscle cells via Sp1 and β-catenin. PLoS ONE. (2014) 9:e94801. doi: 10.1371/journal.pone.0094801
46. Blyszczuk P, Müller-Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J. (2017) 38:1413–25. doi: 10.1093/eurheartj/ehw116
47. Zhang Y, Yan H, Guang G-c, Deng Z-r. Overexpressed connective tissue growth factor in cardiomyocytes attenuates left ventricular remodeling induced by angiotensin II perfusion. Clin Exp Hyperten. (2017) 39:168–74. doi: 10.1080/10641963.2016.1226893
48. Hashemian SM, Pourhanifeh MH, Fadaei S, Velayati AA, Mirzaei H, Hamblin MR. Non-coding RNAs and exosomes: their role in the pathogenesis of sepsis. Mol Ther Nucleic Acids. (2020) 21:51–74. doi: 10.1016/j.omtn.2020.05.012
49. Chen Z, Li C, Lin K, Cai H, Ruan W, Han J, et al. Non-coding RNAs in cardiac fibrosis: emerging biomarkers and therapeutic targets. Cardiol J. (2018) 25:732–41. doi: 10.5603/CJ.a2017.0153
50. Tao H, Yang J-J, Shi K-H. Non-coding RNAs as direct and indirect modulators of epigenetic mechanism regulation of cardiac fibrosis. Expert Opin Ther Targets. (2015) 19:707–16. doi: 10.1517/14728222.2014.1001740
51. Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park D-H, Thum T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler Thromb Vasc Biol. (2012) 32:361–9. doi: 10.1161/ATVBAHA.111.234286
52. Cao W, Shi P, Ge JJ. miR-21 enhances cardiac fibrotic remodeling and fibroblast proliferation via CADM1/STAT3 pathway. BMC Cardiovasc Disord. (2017) 17:88. doi: 10.1186/s12872-017-0520-7
53. Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. (2012) 126:2051–64. doi: 10.1161/CIRCULATIONAHA.112.121830
54. Tang CM, Zhang M, Huang L, Hu ZQ, Zhu JN, Xiao Z, et al. CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Sci Rep. (2017) 7:40342. doi: 10.1038/srep40342
55. Bernardo BC, Gao X-M, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci. (2012) 109:17615–20. doi: 10.1073/pnas.1206432109
56. Katare R, Riu F, Mitchell K, Gubernator M, Campagnolo P, Cui Y, et al. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132. Circ Res. (2011) 109:894–906. doi: 10.1161/CIRCRESAHA.111.251546
57. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. (2009) 104:170–8, 176p following 178. doi: 10.1161/CIRCRESAHA.108.182535
58. Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. (2010) 106:166–75. doi: 10.1161/CIRCRESAHA.109.202176
59. Wang C, Zhang C, Liu L, Xi A, Chen B, Li Y, et al. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther. (2017) 25:192–204. doi: 10.1016/j.ymthe.2016.09.001
60. da Costa Martins PA, Salic K, Gladka MM, Armand A-S, Leptidis S, El Azzouzi H, et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol. (2010) 12:1220. doi: 10.1038/ncb2126
61. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. (2007) 316:575–9. doi: 10.1126/science.1139089
62. Aurora AB, Mahmoud AI, Luo X, Johnson BA, Van Rooij E, Matsuzaki S, et al. (2012). MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca 2+ overload and cell death. J Clin Invest. 122:1222–32. doi: 10.1172/JCI59327
63. Huang ZW, Tian LH, Yang B, Guo RM. Long noncoding RNA H19 acts as a competing endogenous RNA to mediate CTGF expression by sponging miR-455 in cardiac fibrosis. DNA Cell Biol. (2017) 36:759–66. doi: 10.1089/dna.2017.3799
64. Liu X, Xu Y, Deng Y, Li H. MicroRNA-223 regulates cardiac fibrosis after myocardial infarction by targeting RASA1. Cell Physiol Biochem. (2018) 46:1439–54. doi: 10.1159/000489185
65. Li J, Dai Y, Su Z, Wei G. MicroRNA-9 inhibits high glucose-induced proliferation, differentiation and collagen accumulation of cardiac fibroblasts by down-regulation of TGFBR2. Biosci Rep. (2016) 36:e00417. doi: 10.1042/BSR20160346
66. Wang X, Wang H-X, Li Y-L, Zhang C-C, Zhou C-Y, Wang L, et al. MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension. (2015) 66:776–85. doi: 10.1161/HYPERTENSIONAHA.115.05548
67. Tolonen AM, Magga J, Szabo Z, Viitala P, Gao E, Moilanen AM, et al. Inhibition of Let-7 micro RNA attenuates myocardial remodeling and improves cardiac function postinfarction in mice. Pharmacol Res Perspect. (2014) 2:e00056. doi: 10.1002/prp2.56
68. Tao H, Cao W, Yang JJ, Shi KH, Zhou X, Liu LP, et al. Long noncoding RNA H19 controls DUSP5/ERK1/2 axis in cardiac fibroblast proliferation and fibrosis. Cardiovasc Pathol. (2016) 25:381–9. doi: 10.1016/j.carpath.2016.05.005
69. Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S, et al. MIAT Is a pro-fibrotic long non-coding RNA governing cardiac fibrosis in post-infarct myocardium. Sci Rep. (2017) 7:42657. doi: 10.1038/srep42657
70. Huang S, Zhang L, Song J, Wang Z, Huang X, Guo Z, et al. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J Cell Physiol. (2019) 234:2997–3006. doi: 10.1002/jcp.27117
71. Hobuss L, Bar C, Thum T. Long non-coding RNAs: at the heart of cardiac dysfunction? Front Physiol. (2019) 10:30. doi: 10.3389/fphys.2019.00030
72. Piccoli M-T, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, et al. Inhibition of the cardiac fibroblast–enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res. (2017) 121:575–83. doi: 10.1161/CIRCRESAHA.117.310624
73. Guo M, Liu T, Zhang S, Yang L. RASSF1-AS1, an antisense lncRNA of RASSF1A, inhibits the translation of RASSF1A to exacerbate cardiac fibrosis in mice. Cell Biol Int. (2018) 43:1163–73. doi: 10.1002/cbin.11085
74. Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting C-C, Alexanian M, et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med. (2017) 9:eaai9118. doi: 10.1126/scitranslmed.aai9118
75. Zhang Y, Zhang YY, Li TT, Wang J, Jiang Y, Zhao Y, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol. (2018) 115:64–72. doi: 10.1016/j.yjmcc.2018.01.001
76. Kirabo A, Ryzhov S, Gupte M, Sengsayadeth S, Gumina RJ, Sawyer DB, et al. Neuregulin-1beta induces proliferation, survival and paracrine signaling in normal human cardiac ventricular fibroblasts. J Mol Cell Cardiol. (2017) 105:59–69. doi: 10.1016/j.yjmcc.2017.03.001
77. Leisegang MS. LET's sponge: how the lncRNA PFL promotes cardiac fibrosis. Theranostics. (2018) 8:874–7. doi: 10.7150/thno.23364
78. Liang H, Pan Z, Zhao X, Liu L, Sun J, Su X, et al. LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics. (2018) 8:1180–94. doi: 10.7150/thno.20846
79. Zhu J-N, Chen R, Fu Y-H, Lin Q-X, Huang S, Guo L-L, et al. Smad3 inactivation and MiR-29b upregulation mediate the effect of carvedilol on attenuating the acute myocardium infarction-induced myocardial fibrosis in rat. PLoS ONE. (2013) 8:e75557. doi: 10.1371/journal.pone.0075557
80. Han P, Li W, Lin C-H, Yang J, Shang C, Nurnberg ST, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. (2014) 514:102. doi: 10.1038/nature13596
81. Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. (2014) 114:1377–88. doi: 10.1161/CIRCRESAHA.114.302476
82. Sun Y, Yang Z, Zheng B, Zhang XH, Zhang ML, Zhao XS, et al. A novel regulatory mechanism of smooth muscle alpha-actin expression by NRG-1/circACTA2/miR-548f-5p axis. Circ Res. (2017) 121:628–35. doi: 10.1161/CIRCRESAHA.117.311441
83. Lin J-F, Chen P-C, Thomas I, Hwang S. Autophagy modulation by dysregulated micrornas in human bladder cancer. Urological Sci. (2019) 30:46. doi: 10.4103/UROS.UROS_97_18
84. Zhou B, Yu JW. A novel identified circular RNA, circRNA_010567, promotes myocardial fibrosis via suppressing miR-141 by targeting TGF-beta1. Biochem Biophys Res Commun. (2017) 487:769–75. doi: 10.1016/j.bbrc.2017.04.044
85. Pourhanifeh MH, Mahjoubin-Tehran M, Karimzadeh MR, Mirzaei HR, Razavi ZS, Sahebkar A, et al. Autophagy in cancers including brain tumors: role of MicroRNAs. Cell Commun Signal. (2020) 18:88. doi: 10.1186/s12964-020-00587-w
86. Pourhanifeh MH, Vosough M, Mahjoubin-Tehran M, Hashemipour M, Nejati M, Abbasi-Kolli M, et al. Autophagy-related microRNAs: possible regulatory roles and therapeutic potential in and gastrointestinal cancers. Pharmacol Res. (2020) 161:105133. doi: 10.1016/j.phrs.2020.105133
87. Thum T. Noncoding RNAs and myocardial fibrosis. Nat Rev Cardiol. (2014) 11:655–63. doi: 10.1038/nrcardio.2014.125
88. Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. (2009) 6:418. doi: 10.1038/nrcardio.2009.56
89. Dong D-l, Yang B-f. Role of microRNAs in cardiac hypertrophy, myocardial fibrosis and heart failure. Acta Pharmaceutica Sinica B. (2011) 1:1–7. doi: 10.1016/j.apsb.2011.04.010
90. Wang J, Liew OW, Richards AM, Chen YT. Overview of MicroRNAs in cardiac hypertrophy, fibrosis, and apoptosis. Int J Mol Sci. (2016) 17:749. doi: 10.3390/ijms17050749
91. Colpaert RM, Calore M. microRNAs in cardiac diseases. Cells. (2019) 8:737. doi: 10.3390/cells8070737
92. Piccoli MT, Bar C, Thum T. Non-coding RNAs as modulators of the cardiac fibroblast phenotype. J Mol Cell Cardiol. (2016) 92:75–81. doi: 10.1016/j.yjmcc.2015.12.023
93. Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. (2009) 119:2772–86. doi: 10.1172/JCI36154
94. Zhou C, Cui Q, Su G, Guo X, Liu X, Zhang J. MicroRNA-208b alleviates post-infarction myocardial fibrosis in a rat model by inhibiting GATA4. Med Sci Monit. (2016) 22:1808–16. doi: 10.12659/MSM.896428
95. Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. (2015) 6:82. doi: 10.3389/fphys.2015.00082
96. Stawowy P, Margeta C, Kallisch H, Seidah NG, Chretien M, Fleck E, et al. Regulation of matrix metalloproteinase MT1-MMP/MMP-2 in cardiac fibroblasts by TGF-beta1 involves furin-convertase. Cardiovasc Res. (2004) 63:87–97. doi: 10.1016/j.cardiores.2004.03.010
97. Dogar AM, Towbin H, Hall J. Suppression of latent transforming growth factor (TGF)-beta1 restores growth inhibitory TGF-beta signaling through microRNAs. J Biol Chem. (2011) 286:16447–58. doi: 10.1074/jbc.M110.208652
98. Chen C, Ponnusamy M, Liu C, Gao J, Wang K, Li P. MicroRNA as a therapeutic target in cardiac remodeling. Biomed Res Int. (2017) 2017:1278436. doi: 10.1155/2017/1278436
99. Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest. (2017) 127:383–401. doi: 10.1172/JCI83822
100. Del Re DP, Amgalan D. Fundamental mechanisms of regulated cell death and implications for heart disease. Physicol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
101. Cáceres FT, Gaspari TA, Samuel CS, Pinar AA. Serelaxin inhibits the profibrotic TGF-β1/IL-1β axis by targeting TLR-4 and the NLRP3 inflammasome in cardiac myofibroblasts. Faseb J. (2019) 33:14717–33. doi: 10.1096/fj.201901079RR
102. Tschöpe C, Müller I, Xia Y, Savvatis K, Pappritz K, Pinkert S, et al. NOD2 (Nucleotide-Binding Oligomerization Domain 2) is a major pathogenic mediator of coxsackievirus B3-induced myocarditis. Circ Heart Fail. (2017) 10:e003870. doi: 10.1161/CIRCHEARTFAILURE.117.003870
103. Xu C, Zhang Y, Wang Q, Xu Z, Jiang J, Gao Y, et al. Long non-coding RNA GAS5 controls human embryonic stem cell self-renewal by maintaining NODAL signalling. Nat Commun. (2016) 7:13287. doi: 10.1038/ncomms13287
104. Wu N, Zhang X, Bao Y, Yu H, Jia D, Ma C. Down-regulation of GAS5 ameliorates myocardial ischaemia/reperfusion injury via the miR-335/ROCK1/AKT/GSK-3β axis. J Cell Mol Med. (2019) 23:8420–31. doi: 10.1111/jcmm.14724
105. She Q, Shi P, Xu SS, Xuan HY, Tao H, Shi KH. DNMT1 methylation of LncRNA GAS5 leads to cardiac fibroblast pyroptosis via affecting NLRP3 axis. Inflamation. (2020) 43:1065–76. doi: 10.1007/s10753-020-01191-3
106. Rosenbluh J, Nijhawan D, Chen Z, Wong KK, Masutomi K, Hahn WC. RMRP is a non-coding RNA essential for early murine development. PLoS ONE. (2011) 6:e26270. doi: 10.1371/journal.pone.0026270
107. Meng Q, Ren M, Li Y, Song X. LncRNA-RMRP acts as an oncogene in lung cancer. PLoS ONE. (2016) 11:e0164845. doi: 10.1371/journal.pone.0164845
108. Shao Y, Ye M, Li Q, Sun W, Ye G, Zhang X, et al. LncRNA-RMRP promotes carcinogenesis by acting as a miR-206 sponge and is used as a novel biomarker for gastric cancer. Oncotarget. (2016) 7:37812–24. doi: 10.18632/oncotarget.9336
109. Feng W, Li L, Xu X, Jiao Y, Du W. Up-regulation of the long non-coding RNA RMRP contributes to glioma progression and promotes glioma cell proliferation and invasion. Arch Med Sci. (2017) 13:1315–21. doi: 10.5114/aoms.2017.66747
110. Wang X, Peng L, Gong X, Zhang X, Sun R, Du J. LncRNA-RMRP promotes nucleus pulposus cell proliferation through regulating miR-206 expression. J Cell Mol Med. (2018) 22:5468–76. doi: 10.1111/jcmm.13817
111. Greco S, Zaccagnini G, Perfetti A, Fuschi P, Valaperta R, Voellenkle C, et al. Long noncoding RNA dysregulation in ischemic heart failure. J Transl Med. (2016) 14:183. doi: 10.1186/s12967-016-0926-5
112. Steinbusch MMF, Caron MMJ, Surtel DAM, Friedrich F, Lausch E, Pruijn GJM, et al. Expression of RMRP RNA is regulated in chondrocyte hypertrophy and determines chondrogenic differentiation. Sci Rep. (2017) 7:6440. doi: 10.1038/s41598-017-06809-5
113. Zhang SY, Huang SH, Gao SX, Wang YB, Jin P, Lu FJ. Upregulation of lncRNA RMRP promotes the activation of cardiac fibroblasts by regulating miR-613. Mol Med Rep. (2019) 20:3849–57. doi: 10.3892/mmr.2019.10634
114. Sun LY, Zhao JC, Ge XM, Zhang H, Wang CM, Bie ZD. Circ_LAS1L regulates cardiac fibroblast activation, growth, and migration through miR-125b/SFRP5 pathway. Cell Biochem Funct. (2020) 38:443–50. doi: 10.1002/cbf.3486
115. Gui X, Li Y, Zhang X, Su K, Cao W. Circ_LDLR promoted the development of papillary thyroid carcinoma via regulating miR-195-5p/LIPH axis. Cancer Cell Int. (2020) 20:241. doi: 10.1186/s12935-020-01327-3
116. Ma W, Zhao P, Zang L, Zhang K, Liao H, Hu Z. CircTP53 promotes the proliferation of thyroid cancer via targeting miR-1233-3p/MDM2 axis. J Endocrinol Invest. (2020). doi: 10.1007/s40618-020-01317-2. [Epub ahead of print].
117. Miyazawa K, Miyazono K. Regulation of TGF-β family signaling by inhibitory Smads. Cold Spring Harb Perspect Biol. (2017) 9:a022095. doi: 10.1101/cshperspect.a022095
118. Lan HY, Chung AC-K. TGF-β/Smad signaling in kidney disease. Semin Nephrol. (2012) 32:236–43. doi: 10.1016/j.semnephrol.2012.04.002
119. Humeres C, Frangogiannis NG. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl Sci. (2019) 4:449–67. doi: 10.1016/j.jacbts.2019.02.006
120. Divakaran V, Adrogue J, Ishiyama M, Entman ML, Haudek S, Sivasubramanian N, et al. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ Heart Fail. (2009) 2:633–42. doi: 10.1161/CIRCHEARTFAILURE.108.823070
121. Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW II, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med. (2014) 18:415–21. doi: 10.1111/jcmm.12218
122. Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. (2014) 33:1565–81. doi: 10.15252/embj.201387605
123. Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H, et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. (2016) 6:2068–83. doi: 10.7150/thno.15007
124. Liang JN, Zou X, Fang XH, Xu JD, Xiao Z, Zhu JN, et al. The Smad3-miR-29b/miR-29c axis mediates the protective effect of macrophage migration inhibitory factor against cardiac fibrosis. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2441–50. doi: 10.1016/j.bbadis.2019.06.004
125. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol Ther. (2014) 22:974–85. doi: 10.1038/mt.2014.25
126. He X, Zhang K, Gao X, Li L, Tan H, Chen J, et al. Rapid atrial pacing induces myocardial fibrosis by down-regulating Smad7 via microRNA-21 in rabbit. Heart Vessels. (2016) 31:1696–708. doi: 10.1007/s00380-016-0808-z
127. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. (2008) 456:980. doi: 10.1038/nature07511
128. Zou M, Wang F, Gao R, Wu J, Ou Y, Chen X, et al. Autophagy inhibition of hsa-miR-19a-3p/19b-3p by targeting TGF-beta R II during TGF-beta1-induced fibrogenesis in human cardiac fibroblasts. Sci Rep. (2016) 6:24747. doi: 10.1038/srep24747
129. Wang H, Cai J. The role of microRNAs in heart failure. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:2019–30. doi: 10.1016/j.bbadis.2016.11.034
130. Wang J, Huang W, Xu R, Nie Y, Cao X, Meng J, et al. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med. (2012) 16:2150–60. doi: 10.1111/j.1582-4934.2012.01523.x
131. Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, et al. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res. (2012) 111:837–41. doi: 10.1161/CIRCRESAHA.112.277418
132. Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G, et al. NF-κB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol. (2013) 228:1433–42. doi: 10.1002/jcp.24296
133. Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci USA. (2013) 110:187–92. doi: 10.1073/pnas.1208863110
134. Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. (2012) 110:71–81. doi: 10.1161/CIRCRESAHA.111.244442
135. Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, et al. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. (2013) 2:e000078. doi: 10.1161/JAHA.113.000078
136. Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway. Circulation. (2012) 126:840–50. doi: 10.1161/CIRCULATIONAHA.112.094524
137. Zhao X, Wang K, Liao Y, Zeng Q, Li Y, Hu F, et al. MicroRNA-101a inhibits cardiac fibrosis induced by hypoxia via targeting TGFbetaRI on cardiac fibroblasts. Cell Physiol Biochem. (2015) 35:213–26. doi: 10.1159/000369689
138. Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. (2013) 495:107. doi: 10.1038/nature11919
139. Beaumont J, Lopez B, Hermida N, Schroen B, San José G, Heymans S, et al. microRNA-122 down-regulation may play a role in severe myocardial fibrosis in human aortic stenosis through TGF-β1 up-regulation. Clin Sci. (2014) 126:497–506. doi: 10.1042/CS20130538
140. Sun Y, Wang H, Li Y, Liu S, Chen J, Ying H. miR-24 and miR-122 negatively regulate the transforming growth factor-beta/smad signaling pathway in skeletal muscle fibrosis. Mol Ther Nucleic Acids. (2018) 11:528–37. doi: 10.1016/j.omtn.2018.04.005
141. Nagalingam RS, Sundaresan NR, Noor M, Gupta MP, Solaro RJ, Gupta M. Deficiency of cardiomyocyte-specific microRNA-378 contributes to the development of cardiac fibrosis involving a transforming growth factor beta (TGFbeta1)-dependent paracrine mechanism. J Biol Chem. (2014) 289:27199–214. doi: 10.1074/jbc.M114.580977
142. Tao H, Shi KH, Yang JJ, Huang C, Liu LP, Li J. Epigenetic regulation of cardiac fibrosis. Cell Signal. (2013) 25:1932–8. doi: 10.1016/j.cellsig.2013.03.024
143. Huang Y, Li J. MicroRNA208 family in cardiovascular diseases: therapeutic implication and potential biomarker. J Physiol Biochem. (2015) 71:479–86. doi: 10.1007/s13105-015-0409-9
144. Zhao N, Koenig SN, Trask AJ, Lin CH, Hans CP, Garg V, et al. MicroRNA miR145 regulates TGFBR2 expression and matrix synthesis in vascular smooth muscle cells. Circ Res. (2015) 116:23–34. doi: 10.1161/CIRCRESAHA.115.303970
145. Louafi F, Martinez-Nunez RT, Sanchez-Elsner T. MicroRNA-155 targets SMAD2 and modulates the response of macrophages to transforming growth factor-{beta}. J Biol Chem. (2010) 285:41328–36. doi: 10.1074/jbc.M110.146852
146. Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, et al. MiR-125b Is Critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation. (2016) 133:291–301. doi: 10.1161/CIRCULATIONAHA.115.018174
147. Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A, et al. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age. (2013) 35:747–62. doi: 10.1007/s11357-012-9407-9
148. Hong Y, Cao H, Wang Q, Ye J, Sui L, Feng J, et al. MiR-22 may suppress fibrogenesis by targeting TGFbetaR I in cardiac fibroblasts. Cell Physiol Biochem. (2016) 40:1345–53. doi: 10.1159/000453187
149. Chen Z, Lu S, Xu M, Liu P, Ren R, Ma W. Role of miR-24, furin, and transforming growth factor-β1 signal pathway in fibrosis after cardiac infarction. Med Sci Monit Int Med J Exp Clin Res. (2017) 23:65. doi: 10.12659/MSM.898641
150. Zheng D, Zhang Y, Hu Y, Guan J, Xu L, Xiao W, et al. Long noncoding RNA Crnde attenuates cardiac fibrosis via Smad3-Crnde negative feedback in diabetic cardiomyopathy. FEBS J. (2019) 286:1645–55. doi: 10.1111/febs.14780
151. Tao H, Zhang JG, Qin RH, Dai C, Shi P, Yang JJ, et al. LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology. (2017) 386:11–8. doi: 10.1016/j.tox.2017.05.007
152. Wu Q, Han L, Yan W, Ji X, Han R, Yang J, et al. miR-489 inhibits silica-induced pulmonary fibrosis by targeting MyD88 and Smad3 and is negatively regulated by lncRNA CHRF. Sci Rep. (2016) 6:30921. doi: 10.1038/srep30921
153. Chen L, Yan K-P, Liu X-C, Wang W, Li C, Li M, et al. Valsartan regulates TGF-β/Smads and TGF-β/p38 pathways through lncRNA CHRF to improve doxorubicin-induced heart failure. Arch Pharm Res. (2018) 41:101–9. doi: 10.1007/s12272-017-0980-4
154. Bayoumi AS, Aonuma T, Teoh JP, Tang YL, Kim IM. Circular noncoding RNAs as potential therapies and circulating biomarkers for cardiovascular diseases. Acta Pharmacol Sin. (2018) 39:1100–9. doi: 10.1038/aps.2017.196
155. Hurtado MD, Vella A. What is type 2 diabetes? Medicine. (2019) 47:10–5. doi: 10.1016/j.mpmed.2018.10.010
156. Liang H, Zhang C, Ban T, Liu Y, Mei L, Piao X, et al. A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int J Biochem Cell Biol. (2012) 44:2152–60. doi: 10.1016/j.biocel.2012.08.019
157. Roy S, Khanna S, Hussain S-RA, Biswas S, Azad A, Rink C, et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. (2009) 82:21–9. doi: 10.1093/cvr/cvp015
158. He W, Huang H, Xie Q, Wang Z, Fan Y, Kong B, et al. MiR-155 knockout in fibroblasts improves cardiac remodeling by targeting tumor protein p53-inducible nuclear protein 1. J Cardiovasc Pharmacol Ther. (2016) 21:423–35. doi: 10.1177/1074248415616188
159. Manzoni GM, Castelnuovo G, Proietti R. Assessment of psychosocial risk factors is missing in the 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. J Am Coll Cardiol. (2011) 57:1569–70. doi: 10.1016/j.jacc.2010.12.015
160. Van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci. (2008) 105:13027–32. doi: 10.1073/pnas.0805038105
161. Ooi JY, Bernardo BC, McMullen JR. Therapeutic potential of targeting microRNAs to regulate cardiac fibrosis: miR-433 a new fibrotic player. Ann Transl Med. (2016) 4:548. doi: 10.21037/atm.2016.12.01
162. Castoldi G, Di Gioia CR, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, et al. MiR-133a regulates collagen 1A1: potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J Cell Physiol. (2012) 227:850–6. doi: 10.1002/jcp.22939
163. Tijsen AJ, van der Made IM, van den Hoogenhof M, Wijnen WJ, van Deel ED, de Groot NE, et al. The microRNA-15 family inhibits the TGFbeta-pathway in the heart. Cardiovasc Res. (2014) 104:61–71. doi: 10.1093/cvr/cvu184
164. Van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci. (2006) 103:18255–60. doi: 10.1073/pnas.0608791103
165. Huang ZP, Ding Y, Chen J, Wu G, Kataoka M, Hu Y, et al. Long non-coding RNAs link extracellular matrix gene expression to ischemic cardiomyopathy. Cardiovasc Res. (2016) 112:543–54. doi: 10.1093/cvr/cvw201
166. Tang PM, Zhang YY, Lan HY. LncRNAs in TGF-beta-driven tissue fibrosis. Noncoding RNA. (2018) 4:26. doi: 10.3390/ncrna4040026
167. Wang X, Ding Z, Wu J, Wang S, Zou Y. Circular RNA in cardiovascular disease. Noncoding RNA Invest. (2017) 1:5. doi: 10.21037/ncri.2017.08.04
Keywords: cardiac fibrosis, non-coding RNAs, Smad, TGF—transforming growth factor, microRNA
Citation: Saadat S, Noureddini M, Mahjoubin-Tehran M, Nazemi S, Shojaie L, Aschner M, Maleki B, Abbasi-kolli M, Rajabi Moghadam H, Alani B and Mirzaei H (2021) Pivotal Role of TGF-β/Smad Signaling in Cardiac Fibrosis: Non-coding RNAs as Effectual Players. Front. Cardiovasc. Med. 7:588347. doi: 10.3389/fcvm.2020.588347
Received: 28 July 2020; Accepted: 15 October 2020;
Published: 25 January 2021.
Edited by:
Suowen Xu, University of Science and Technology of China, ChinaReviewed by:
Osvaldo Contreras, Victor Chang Cardiac Research Institute, AustraliaCopyright © 2021 Saadat, Noureddini, Mahjoubin-Tehran, Nazemi, Shojaie, Aschner, Maleki, Abbasi-kolli, Rajabi Moghadam, Alani and Mirzaei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hasan Rajabi Moghadam, aHJtY2FyZGlvQGdtYWlsLmNvbQ==; Behrang Alani, YmVocmFuZ2FAeWFob28uY29t; Hamed Mirzaei, bWlyemFlaS1oQGthdW1zLmFjLmly; aC5taXJ6YWVpMjAwMkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.