 Fuyi Xu1†
Fuyi Xu1† Jun Gao1†
Jun Gao1† Undral Munkhsaikhan2,3
Undral Munkhsaikhan2,3 Ning Li2,3,4
Ning Li2,3,4 Qingqing Gu1,5
Qingqing Gu1,5 Joseph F. Pierre2,3
Joseph F. Pierre2,3 Athena Starlard-Davenport1Jeffrey A. Towbin2,3,6
Athena Starlard-Davenport1Jeffrey A. Towbin2,3,6 Yan Cui1Enkhsaikhan Purevjav2,3*
Yan Cui1Enkhsaikhan Purevjav2,3* Lu Lu1*
Lu Lu1*- 1Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN, United States
- 2Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN, United States
- 3Children's Foundation Research Institute, Le Bonheur Children's Hospital, Memphis, TN, United States
- 4Department of Cardiology, Second Affiliated Hospital, Harbin Medical University, Harbin, China
- 5Department of Cardiology, The Affiliated Hospital of Nantong University, Nantong, China
- 6Pediatric Cardiology, St. Jude Children's Research Hospital, Memphis, TN, United States
Background: A high inflammatory and cytokine burden that induces vascular inflammation, myocarditis, cardiac arrhythmias, and myocardial injury is associated with a lethal outcome in COVID-19. The SARS-CoV-2 virus utilizes the ACE2 receptor for cell entry in a similar way to SARS-CoV. This study investigates the regulation, gene network, and associated pathways of ACE2 that may be involved in inflammatory and cardiovascular complications of COVID-19.
Methods: Cardiovascular traits were determined in the one of the largest mouse genetic reference populations: BXD recombinant inbred strains using blood pressure, electrocardiography, and echocardiography measurements. Expression quantitative trait locus (eQTL) mapping, genetic correlation, and functional enrichment analysis were used to identify Ace2 regulation, gene pathway, and co-expression networks.
Results: A wide range of variation was found in expression of Ace2 among the BXD strains. Levels of Ace2 expression are negatively correlated with cardiovascular traits, including systolic and diastolic blood pressure and P wave duration and amplitude. Ace2 co-expressed genes are significantly involved in cardiac- and inflammatory-related pathways. The eQTL mapping revealed that Cyld is a candidate upstream regulator for Ace2. Moreover, the protein–protein interaction (PPI) network analysis inferred several potential key regulators (Cul3, Atf2, Vcp, Jun, Ppp1cc, Npm1, Mapk8, Set, Dlg1, Mapk14, and Hspa1b) for Ace2 co-expressed genes in the heart.
Conclusions: Ace2 is associated with blood pressure, atrial morphology, and sinoatrial conduction in BXD mice. Ace2 co-varies with Atf2, Cyld, Jun, Mapk8, and Mapk14 and is enriched in the RAS, TGFβ, TNFα, and p38α signaling pathways, involved in inflammation and cardiac damage. We suggest that all these novel Ace2-associated genes and pathways may be targeted for preventive, diagnostic, and therapeutic purposes in cardiovascular damage in patients with systemic inflammation, including COVID-19 patients.
Introduction
Angiotensin (Ang) converting enzyme 2 (ACE2) is a protein found as both a membrane-associated and secreted enzyme in cardiac, vascular, pulmonary, neuronal, and reproductive organs (1). In the renin-angiotensin-aldosterone system (RAS or RAAS), ACE2 catalyzes the conversion of AngII to Ang1–7, which acts as a vasodilator (2) and plays a critical role in the control of cardiovascular and renal functions by maintaining the physiological homeostasis of blood pressure (BP) and electrolyte balance (3). ACE2, in turn, contributes to the cardioprotective effects of Ang1–7 after myocardial infarction (4), ischemic cardiomyopathy (5), and myocardial ischemia/reperfusion (6). Moreover, ACE2 counterbalances with ACE and functions as a negative regulator of the RAS (7), playing a critical protective role against heart failure with reduced and preserved ejection fraction, myocardial infarction, and hypertension as well as against lung disease and diabetes (8). Knockout of ACE2 in mice resulted in severe cardiac contractile deficiency due to increased AngII levels and upregulation of hypoxia-induced genes in the heart, suggesting that ACE2 is an essential regulator of cardiac function (9). However, the full spectrum of underlying genetic pathways and networks of ACE2 in the heart remains to be understood.
Coronavirus disease (COVID-19), caused by a novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a recent global pandemic infectious disease with almost unprecedented speed of spread from an initial outbreak in November or December 2019 (10). SARS-CoV-2 causes primary pneumonia while also causing acute myocardial injury and chronic damage to the cardiovascular system (11). In addition, COVID-19 is associated with a high inflammatory burden that can induce vascular inflammation, myocarditis, and cardiac arrhythmias (12). In particular, inflammation and cytokine surge may be a potential mechanism for myocardial injury (13). Similar to SARS-CoV, SARS-CoV-2 uses ACE2 as the receptor to enter the host cell facilitating the viral endocytosis (14, 15). Therefore, the ACE2 expression pattern in cardiac tissue may play a critical role in the cardiovascular damage from the SARS-CoV-2 infection, and elucidation of its regulatory mechanisms would be beneficial for preventive, diagnostic, and therapeutic purposes of cardiovascular damage in COVID-19.
BXD recombinant inbred (RI) strains, one of the largest and best-characterized mouse genetic reference populations (GRPs) currently, have been constructed for systems genetics study in unraveling the genetic architecture of polygenic traits. Currently, ~152 BXD progeny strains derived from crosses between C57BL/6J (B6) and DBA/2J (D2) parents have been generated and maintained in our lab (16). Both parental strains and BXD progeny strains have been sequenced, which showed that they segregate more than 6 million variants, similar to humans with a mean minor allele frequency close to 0.5 (17). Each BXD strain with the same genetic background can be replicated in large numbers per genome as a murine GRP for systems genetics analysis, enabling uncovering candidate genes and mechanisms related to complex traits. It facilitates more precise phenotypic estimates for mapping complex traits with low-to-moderate heritability as well (16). We have successfully used our BXD cohort for identification of candidate genes in hypertension, cardiac and renal damage, and cardiac fibrosis (18–21).
In this study, we aimed to assess the expression variation of Ace2 in the heart tissue of BXD mice and the association with cardiovascular traits. Through the systems genetics approach, we sought to identify the Ace2-correlated genes, potential pathways, and candidate upstream or downstream regulators that may contribute to cardiovascular damage upon systemic inflammatory background.
Materials and Methods
Heart Expression Data Set
The gene expression data, EPFL/LISP BXD CD Heart Affy Mouse Gene 2.0 ST (Jan14) RMA, used in this study was generated through collaborative efforts and can be accessed at our GeneNetwork (GN) website (http://genenetwork.org/) with the GN485 accession number (22).
Mice
Male mice of 40 BXDs and their two founder strains B6 and D2, representing murine GRP, were used in this study. All mice were fed a chow diet (6% kcal/fat, 20% protein, 74% carbohydrate, Harlan, 2918) throughout life after weaning until sacrifice at around 29 weeks of age. For cardiac tissue collection, animals were sacrificed under isoflurane anesthesia after an overnight fast. All animal procedures were approved by the Swiss cantonal veterinary authorities of Vaud under licenses 2257.0 and 2257.1.
RNA Isolation and Microarray
Hearts were later shattered in liquid nitrogen, usually broken into 2–4 pieces, and around half of the sample (at random) was taken for preparation. Total RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA). RNA samples from ~5 mice of the same strain were pooled equally (by microgram of RNA) into a single RNA sample. The pooled RNA samples were then purified using RNEasy (Qiagen, Hilden, Germany). Samples that passed quality control (RIN > 8.0) were run on Affymetrix Mouse Gene 2.0 ST in a single batch at the University of Tennessee Health Science Center (UTHSC).
Data Preprocessing
Raw microarray data were first normalized using the robust multichip array (RMA) method (23), and then the data were logged and Z normalized (24). Instead of leaving the mean at 0 and the standard deviation of 1 unit, we shift up to a mean of 8 units and increase the spread by having a standard deviation of 2 units (what we call 2Z + 8 normalized data). This removes negative values from the tables.
Cardiac Traits Measurement
The cardiovascular system–related phenotype was collected in 3- to 4-month-old mice. This included 94 mice (50 strains) for collection of 18 electrocardiogram (ECG) traits, 425 mice (45 strains) for 16 echocardiogram traits, and 424 mice (24 strains) for 5 BP traits. The CODA non-invasive tail-cuff BP system with a volume pressure recording (VPR) sensor (Kent Scientific Corporation) measured systolic and diastolic BP. Before measuring BP, mice were acclimated to the restraint holder placed on a heating pad for 3 days. For BP recording, the VPR cuff was placed at the base of the tail, and sessions consisted of 15 inflation and deflation cycles with the first five being acclimation cycles. At least 5 cycles per mouse were used in the analysis. Transthoracic echocardiography was used to evaluate heart function by short and long cross-sectional, two-dimensional, color Doppler using a Vevo2100 Micro-Imaging System (VisualSonics Inc., Toronto, Canada) with a 30-MmHz transducer. Single-lead ECG tracings were recorded for 5 min at a sampling rate of 200 Hz using BIOPAC (Goleta, CA USA) with AcqKnowledge 3.9.2 software. Normal and arrhythmic heart rhythms were distinguished relying on the minimum and maximum RR intervals using LabChart 7 software.
eQTL Mapping
Two methods were used for eQTL mapping for Ace2 expression on GN, the fast linear mapping (25) method and the genome-wide efficient mixed model association (GEMMA) method (26). Fast linear mapping used the likelihood ratio statistics (LRS) to measure the linkages between the investigated phenotype (clinical or intermediate traits) and genotype markers. The genome-wide significant and suggestive QTLs were determined with 2,000~10,000 permutation tests (suggestive LRS = 11.3 and significant LRS = 18.4). For GEMMA, a linear mixed model method was used to effectively correct the kinship among samples. In addition, it incorporates the leave one chromosome out (LOCO) method to ensure that the correction for kinship does not remove useful genetic variance near each marker. The suggestive and significant threshold for a genome-wide scan is –log (p) of 2.5 and 4.0. The threshold was based on one unit of –log(p) being roughly equivalent to 1 unit of logarithm of the odds (LOD) value, where LOD = LRS/4.61. Both methods used a total of 7,321 informative SNP genotype markers for the analysis. The BXD genotype file can be accessed and reviewed on GN.
Correlation Analysis
Genetic correlation analysis was performed with the Pearson correlation coefficient to identify gene–gene and gene–phenotype relationships. The literature correlation quantifies genes that are described by similar terminology in published papers extracted from MEDLINE/PubMed abstracts. Similarity values, known as literature correlations, are computed for a matrix of about 20,000 genes using latent semantic indexing (27, 28). These values are always positive and range from 0 to 1. Both analyses were done on GN.
To evaluate the false discovery rate (FDR) for gene–gene, we computed the adjusted p-value with Westfall and Young's multiple testing procedure (29). Briefly, we randomly permuted the Ace2 expression data 1,000 times. For each permutation, we computed the p-value of Pearson's correlation between the randomized Ace2 and the other cardiac transcripts (41,355 probe sets). The adjusted P-value was determined by ranking the correlation coefficient. For gene–phenotype relationships, the same analysis was done between the randomized Ace2 and phenotypes as well as between the randomized Cyld and phenotypes in each cardiac trait categories (BP, ECG, and echocardiogram).
Gene Enrichment Analysis
Genes with significant correlations with Ace2 (FDR <0.05 and r > 0.3) were used for gene set over representation analysis for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Mammalian Phenotype Ontology (MPO) on the Webgestalt website (http://bioinfo.vanderbilt.edu/webgestalt/) (30). Mouse protein-coding genes were used as a reference gene set with a minimum of 5 genes for each category.
Genetic Variations
Genetic variations between the founder strains B6 and D2 were obtained from whole genome sequencing analysis in our previous publication (31). Here, we focused on the variants located in the coding sequence region, such as stop gain, stop loss, frameshift, and missense variants.
Exploration of Gene Function
In order to identify genes with functions related to inflammation and the cardiovascular system. We retrieved lists of genetically engineered alleles, transgenes, or QTL variants from The Phenotypes/Alleles project (http://www.informatics.jax.org/allele) in Mouse Genome Informatics (MGI) (32) with the key words “inflammation” or “cardiovascular.” This portal enables access to spontaneous, induced, and genetically engineered mutations and their strain-specific phenotypes.
Candidate Gene Selection
For the Ace2 modulating eQTL, all positional candidates in the confidence interval were selected for further analysis based on the following criteria: (1) mRNA correlation with Ace2, including genetic correlation (FDR <0.05) and literature correlation (r > 0.3); (2) presence of cis regulation, defined as LRS > 12 for the peak SNP within the 5-Mb interval of its gene's physical position; (3) presence of coding sequence variation between B6 and D2; (4) mRNA correlation with the cardiovascular traits; and (5) gene function related to inflammation or cardiovascular system.
PPI Network
In order to further discover the key regulators in Ace2 co-expressed genes, we created and evaluated the PPI network with the NetworkAnalyst (www.networkanalyst.ca) (33), in which the International Molecular Exchange (IMEx) Interactome database was used (34). The IMEx consortium is a publicly available database of curated and non-redundant set of protein interactions.
Results
Levels of Ace2 mRNA in the Heart Are Significantly Correlated With Cardiovascular Traits
Ace2 has been reported to be associated with several cardiovascular system–related phenotypes under various mouse genome backgrounds (9, 35–37). To define an association between Ace2 and cardiovascular traits in the BXD mice, we performed Pearson correlation analysis of Ace2 mRNA level against cardiovascular traits collected by BP measurements, ECG tracings, and transthoracic echocardiography. Results show that Ace2 expression is significantly and negatively correlated with both systolic (r = −0.797, FDR = 0.004, Figure 1A) and diastolic (r = 0.722, FDR = 0.010, Figure 1B) BP, indicating that higher Ace2 levels were observed in BXD strains with lower systolic and diastolic pressure. Further, Ace2 has a negative association with the P duration (r = −0.509, p = 0.011, FDR = 0.026, Figure 1C) and P amplitude (r = −0.479, FDR = 0.035, Figure 1D), indicating that BXD strains showing narrower and lower P waves on ECG tracings have higher Ace2 expression. P-wave amplitude and duration changes together have important diagnostic values in hemodynamic and morphological changes of the left atrium (LA) and sinoatrial conduction in patients with hypertension, left ventricular (LV) hypertrophy, and diastolic dysfunction (38). Thus, we calculated a Pearson correlation and found no association between BP and P-wave amplitude and duration in BXDs (p = 0.61 and 0.65 for systolic/diastolic BP vs. P-wave amplitude; p = 0.30 and 0.31 for systolic/diastolic BP vs. P-wave duration). Further, we found no association between Ace2 expression and echocardiographic parameters of LV function, including EF%, FS%, and LV volumes or wall thickness in BXD strains. Collectively, phenotype–genotype correlation predicted a strong association of Ace2 with the genes regulating BP as well as atrial morphology and sinoatrial conduction while no association was predicted between Ace2 and LV dysfunction.
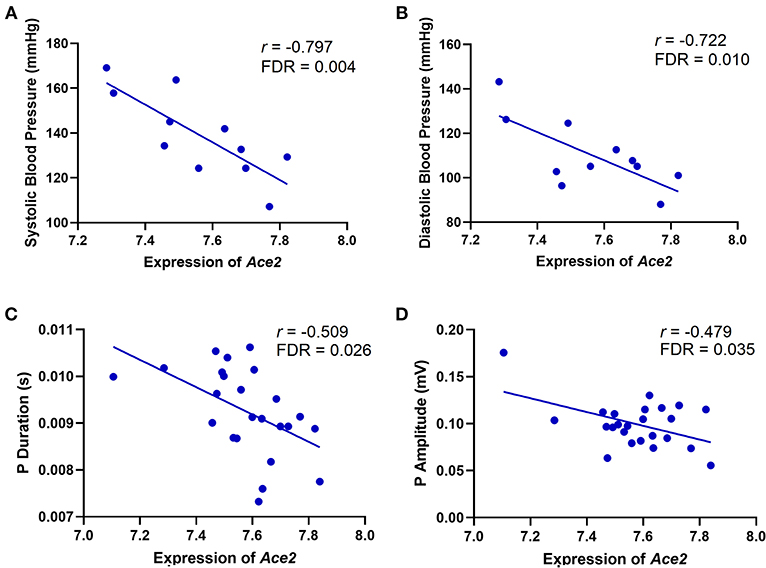
Figure 1. Scatterplots of the correlations of Ace2 expression with systolic BP (A), diastolic BP (B), P duration (C), and P amplitude (D). The Pearson correlation coefficient was used to determine the relationship. Pearson correlation r and FDR are indicated. Gene expression levels are log2 transformed.
eQTL Mapping Identifies an Ace2 Regulating Locus on Chromosome 8
We observed a 1.8-fold change of the Ace2 expression across the 40 BXD strains and their parents (Figure 2A). The average expression of Ace2 across the strains is 7.53 ± 0.20 SD. BXD70 and D2 mice have the lowest and highest expression of 7.1 and 8.0, respectively.
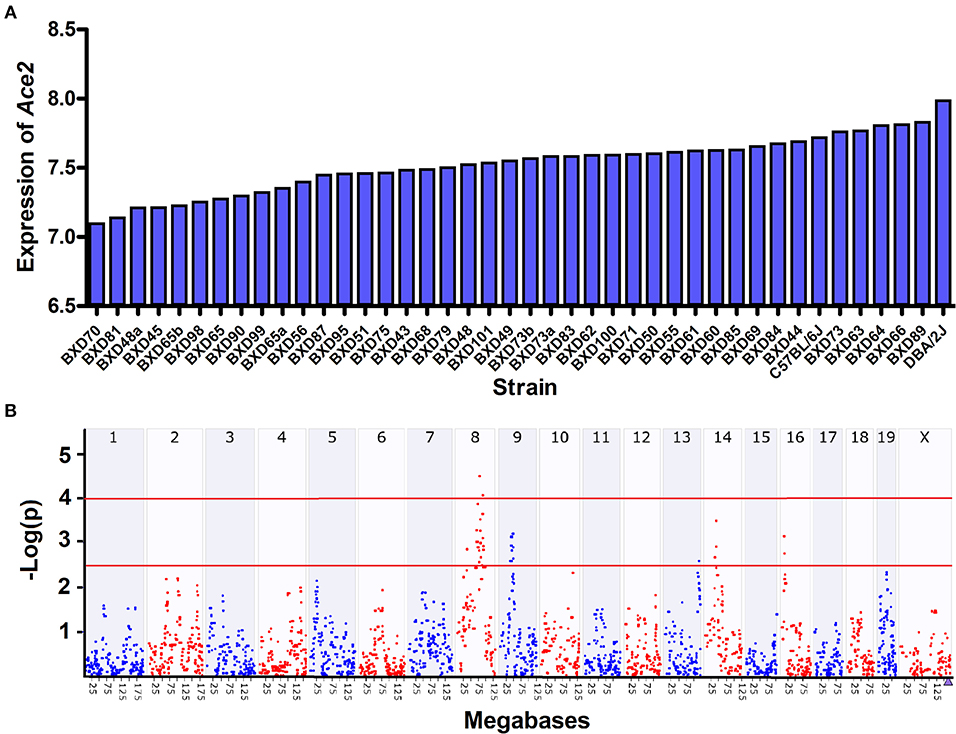
Figure 2. Expression and eQTL mapping of Ace2 in BXD family. (A) Bar plots of the Ace2 expression levels of heart across the BXD mice. The x-axis shows the BXD strains and the two parental strains. The y-axis shows the normalized log2 expression levels of Ace2. (B) Manhattan plot of genome-wide Ace2-regulated genomic loci. The x-axis denotes a position on the mouse genome, in megabases (Mb), while the y-axis gives the –log (p), a measurement of the linkage between Ace2 expression, and genomic region. The purple triangle indicates the genomic position of Ace2. The red line indicates suggestive (–log (p) of 2.5) and significant (–log (p) of 4.0) genome-wide thresholds. eQTL mapping was conducted with GEMMA on GN.
We then looked up the genetic variations in Ace2 in the two founder strains across our whole genome sequencing data. Although we did not find any protein-coding variants, three variants (rs52428988, rs387358736, and rs230145272) were located at upstream of Ace2. Next, we performed eQTL mapping for Ace2 expression. One suggestive eQTL was mapped to Chromosome (Chr) 8 at 88.4 Mb using interval mapping methods (Peak SNP LRS = 15, suggestive LRS = 11.3, and significant LRS = 18.4), and achieved significance with GEMMA after correcting the kinship among samples (Figure 2B). This locus is apart from the Ace2 genomic location (Chr X at 164.14 Mb), suggesting this is a trans-acting eQTL.
Cyld Is a Candidate Upstream Regulator for Ace2
The 1.5-LOD interval for Chr 8 QTL encompasses 33 Mb from 63 to 96 Mb. Among the genes in the QTL region, we identified 17 genes (Table 1) that had significant genetic correlation (FDR <0.05) and literature correlation (r > 0.3) with Ace2. In order to further prioritize the candidates, we first performed eQTL mapping for those 17 genes. This resulted in the identification of 6 genes (Jund, Il15, Nfix, Prdx2, Cyld, and Coq9) that were cis-regulated. Moreover, by comparing the DNA sequence differences between the B6 and D2 mice, 5 genes (Lpl, Clgn, Il27ra, Cc2d1a, and Ces1c) were identified to harbor missense variants. Ten of the genes (Klf2, Ciapin1, Jund, Il15, Coq9, Lpl, Cc2d1a, Dnase2a, Cyld, and Il27ra) have been implicated in the cardiovascular system or inflammation response in various natural, induced, or genetically engineered mice according to the Phenotypes/Alleles project (http://www.informatics.jax.org/allele) in the MGI (32).
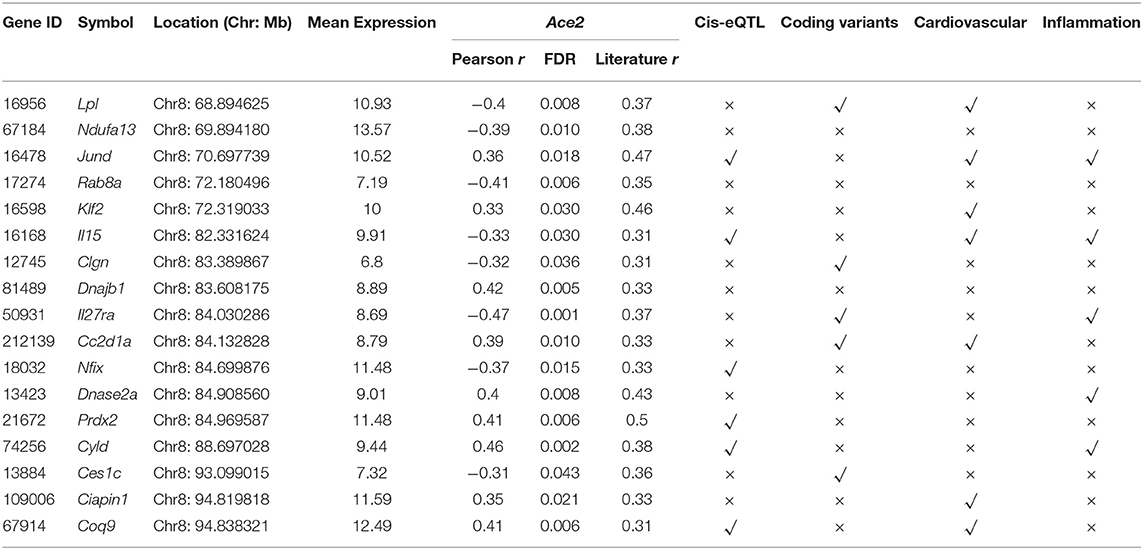
Table 1. Lists of the candidate genes in Chr 8 QTL interval.
Based on the above criteria of candidate gene identification, we selected Cyld as the best candidate among the genes in this QTL interval. This gene encodes CYLD lysine 63 deubiquitinase and is located at the QTL peak position (Chr 8 at 88 Mb). Although Cyld harbors no coding sequence variants, more than 30 3′-UTR variants were unveiled between the two founder B6 and D2 strains, and eQTL mapping demonstrated that this gene is cis-regulated (Figure 3A). Besides, Cyld was positively correlated with Ace2 expression (r = 0.46, FDR = 0.002, Figure 3B). Importantly, we found the levels of Cyld mRNA negatively correlated with echocardiography parameters, such pulmonary valve (PV) peak velocity (r = −0.463, FDR = 0.028, Figure 3C) and PV peak pressure (r = −0.441, FDR = 0.036, Figure 3D). These echocardiographic correlations suggest that the BXD strains with faster blood flow through PV and higher pressure on PV have significantly lower Cyld expression, suggesting that decreased Cyld expression is associated with elevated pulmonary artery pressure and pulmonary hypertension (PH). In addition, the levels of Cyld mRNA negatively correlated with heart rate (r = −0.680, FDR = 0.0009, Figure 3E) in BXD mice, showing that the BXD strains with lower Cyld expression have an increased heart rate.
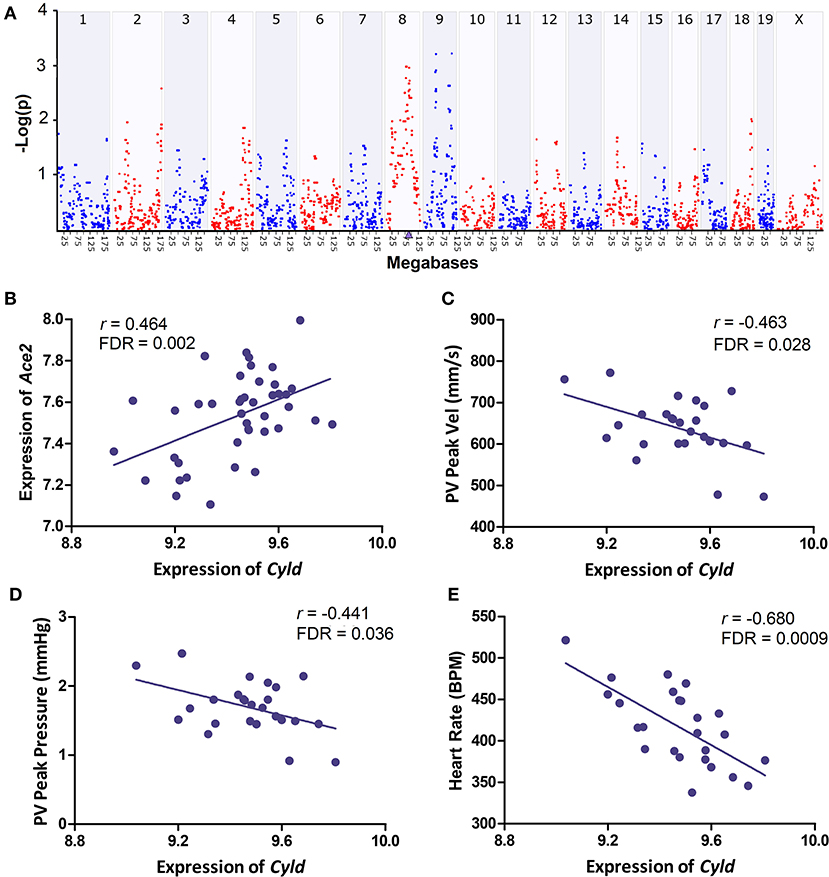
Figure 3. Genetic mapping and correlation analysis of Cyld. (A) Manhattan plot of genome-wide Cyld regulated genomic loci. The x-axis denotes a position on the mouse genome, in megabases (Mb), while the y-axis gives the –log(p), a measurement of the linkage between Cyld expression and genomic region. The purple triangle indicates the genomic position of Cyld. Genome-wide eQTL mapping was conducted with GEMMA on GN. (B–E) Scatterplots of the correlations of Cyld with Ace2, PV peak velocity (Vel), PV peak pressure, and heart rate. Pearson correlation coefficient was used to determine the relationship. Pearson correlation r and FDR are indicated. Gene expression levels are log2 transformed.
Ace2 Co-expressed Genes Are Involved in Cardiac and Inflammatory Functions
To gain insight into the pathways and biological functions of Ace2 involvement in the heart, we performed genetic correlation analysis for Ace2 against the whole heart transcriptome. We identified a total of 2,800 transcripts that are significantly (FDR <0.05) correlated with the expression of Ace2 in the heart, among which 440 genes shown literature correlation r > 0.3 with Ace2. Those genes were further submitted to WebGestalt (http://www.webgestalt.org/) for the KEGG pathway and MPO enrichment analysis. Results clearly demonstrated that the genes were overrepresented in two biological systems: the cardiovascular system as well as inflammation and immune response Figure 4. The KEGG pathways included renin secretion, mitogen-activated protein kinase (MAPK), RAS, tumor necrosis factor (TNF), and transforming growth factor beta (TGFβ) signaling pathways (Figure 4A). The MPO revealed abnormal inflammatory response, abnormal innate immunity, abnormal muscle physiology, and abnormal cardiovascular system physiology (Figure 4B).
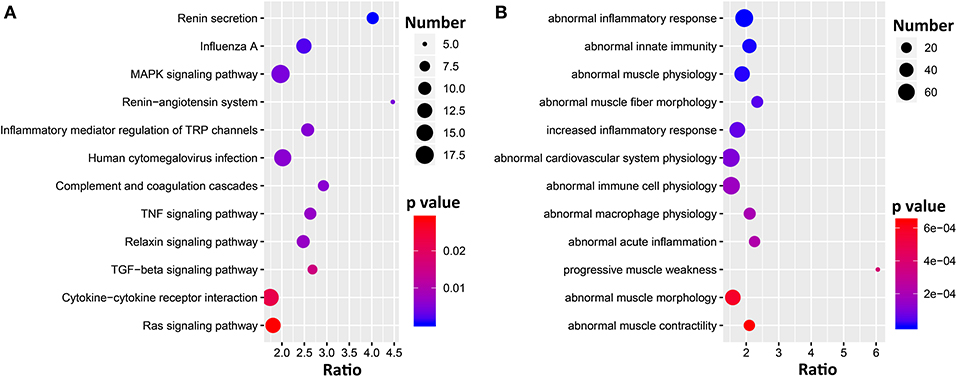
Figure 4. Bubble charts of the KEGG pathways (A) and MPO (B) enriched for Ace2 co-variates. Gene over-representation analysis for KEGG pathway and MPO of the Ace2 correlated genes (FDR <0.05 and r > 0.3) were performed with WebGestalt (http://www.webgestalt.org/). The x-axis represents an enriched ratio, and the y-axis represents enriched pathways/terms. The size of the dots represents the number of genes, and the color indicates the p-values. An enriched ratio is defined as the number of observed divided by the number of expected genes from the annotation category in the gene list.
Next, we focused on exploring the Ace2 co-expressed genes in the two enriched biological systems, inflammation and cardiovascular, using the Phenotypes/Alleles project (http://www.informatics.jax.org/allele). This resulted in 90 and 133 genes with spontaneous, induced, or genetically engineered mutations that are associated with inflammation- or cardiovascular-related traits (Figure 5). It is worth noting that 48 genes (Figure 5) met the above two criteria, including genes Dsg, Prkag2, Sgca, Syne1, and Ren1.
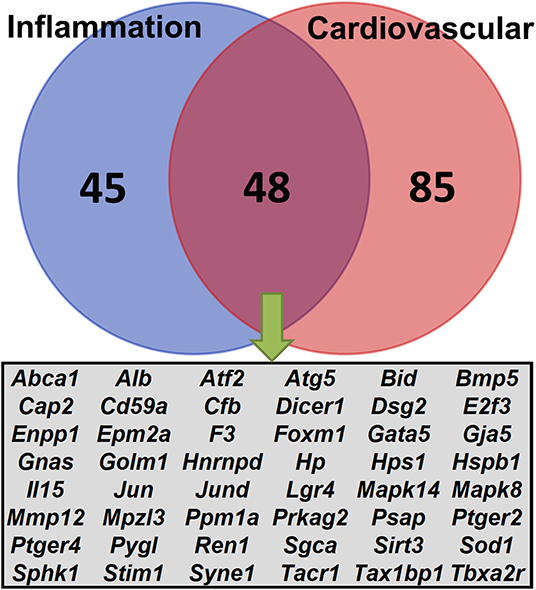
Figure 5. Venn diagrams of the Ace2 correlated genes. A total of 440 genes were correlated (FDR <0.05 and r > 0.3) with Ace2. Lists of genetically engineered alleles, transgenes, or QTL variants were retrieved from the Phenotypes/Alleles project (http://www.informatics.jax.org/allele) in the MGI (32) with key words “inflammation” or “cardiovascular.” The Venn diagram shows the number of overlapping genes between the two categories.
PPI Network Analysis Infers Key Regulators for Ace2 Co-expressed Genes in the Heart
Finally, in order to investigate the key regulators among the Ace2 co-expressed genes, we submitted the 440 transcripts that are significantly correlated with Ace2 (FDR <0.05 and r > 0.3) to the NetworkAnalyst (www.networkanalyst.ca) (33) and evaluated the PPI network with the curated and non-redundant set of protein interactions in the IMEx consortium database (34). The PPI network suggests that 12 genes (Cul3, Atf2, Vcp, Jun, Ppp1cc, Cyld, Npm1, Mapk8, Set, Dlg1, Mapk14, and Hspa1b) are at the central node of the network and have the most connections to other genes (Figure 6). Notably, most of these genes are involved in the regulation of cardiovascular and inflammation-related traits (Table 2).
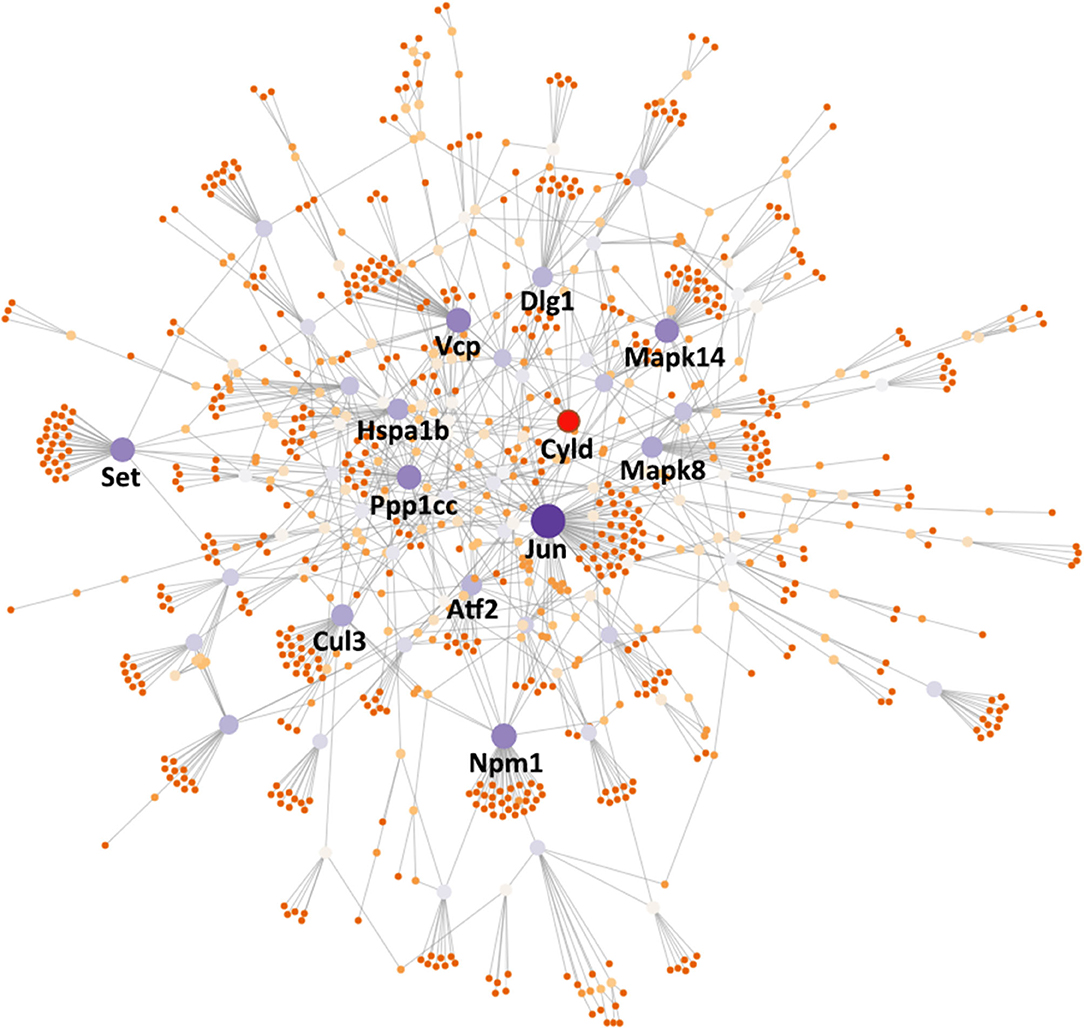
Figure 6. PPI network analysis inferred key regulators of Ace2. The PPI network was constructed using NetworkAnalyst (https://www.networkanalyst.ca/) with the input of Ace2 co-expressed genes. The nodes in the network represent genes. Key node genes were indicated with gene symbols. Generic PPI was constructed using the IMEx Interactome database.
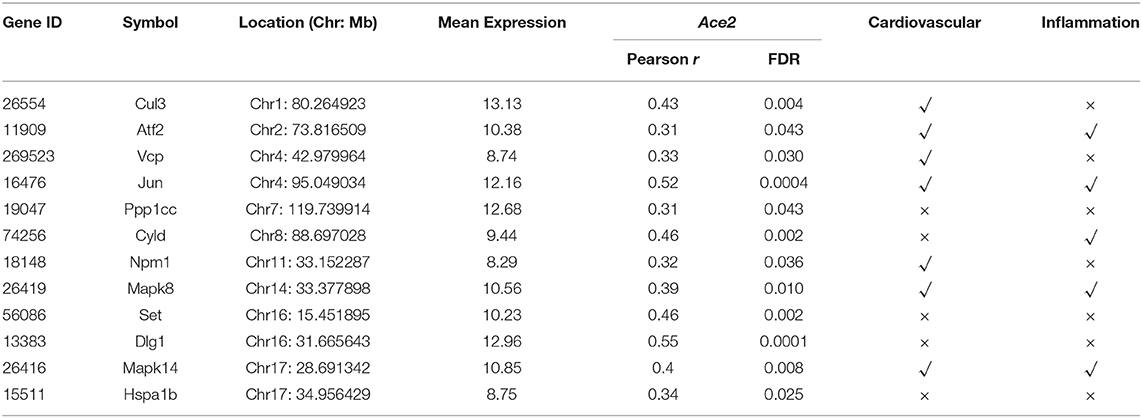
Table 2. List of 13 prioritized genes associated with the inflammation and cardiovascular traits.
Discussion
Accumulated evidence indicates that ACE2, a gatekeeper of RAS that coronaviruses use as a host functional receptor, is a potential therapeutic target due to its degrading effects on AngII in hypertension, LV hypertrophy, fibrosis, and diastolic dysfunction, including cardiovascular complication in COVID-19 (9, 39). The current pandemic COVID-19 infection has been associated with multiple cardiovascular complications, and at least 8.0% of COVID-19 patients suffer acute cardiac injury (40). Moreover, the high inflammatory burden of SARS-CoV-2 has been shown to induce vascular inflammation, myocarditis, and cardiac arrhythmias (13). Since a complicated picture has emerged on the association of ACE2 effects on cardiovascular physiology in the COVID-19 pandemic, we hypothesized that dissection of the cardiac genetic regulatory network of ACE2 using a systems genetics approach would uncover the underlying genetic determinants in ACE2-driven cardiovascular function, the results of which would benefit in the diagnosis and care of patients with cardiovascular disease, including cardiovascular complications in COVID-19. In the present study, we collected cardiovascular traits in murine GRP of BXD strains and correlated those traits with their cardiac gene expression. Then, we identified co-expressed genes of Ace2, upstream and downstream regulators, networks, and pathways to uncover the underlying genetic determinants in ACE2-driven cardiovascular function and inflammation as well as immune response.
The important cardiovascular traits, systolic and diastolic BPs, are found to be significantly negatively correlated with Ace2 expression in BXDs, demonstrating that the lowest Ace2 levels are associated with hypertension. Supporting our results, Ace2 deficiency resulted in BP increase (9) while Ace2 ablation also exacerbates AngII-mediated inflammation and myocardial injury resulting in cardiac dysfunction in Ace2 knockout mice (41). In the Yamamoto et al. (36) study, Ace2−/y mice received transverse aortic constriction developed cardiac hypertrophy and dilatation, and this event was associated with significantly increased concentration in cardiac AngII and MAPK activity. In our study, Ace2 expression is significantly and negatively correlated with the P-wave duration and amplitude in ECG tracings of BXD mice. These changes in P-wave were not associated with BP while Ace2 expression had no association with echocardiography parameters of LV function. These results strongly suggest an underlying association of Ace2 with the genes regulating BP as well as atrial function (diastolic dysfunction and atrial dilation) and sinoatrial conduction abnormalities.
To uncover the genetic correlates and pathways of Ace2, gene set enrichment analysis was performed, which found several Ace2 correlated genes involved in two main KEGG pathway categories, such as RAS system-related (renin secretion, RAS, and RAS signaling pathway) and inflammation-related (inflammatory mediator regulation of TRP channels, MAPK, TNF, TGFβ, and cytokine–cytokine receptor interaction) signaling pathways (Figure 4A). The Ren1 encoding renin, an RAS initiative enzyme (42), was found to be significantly associated with ACE2, emerging in two signals: renin secretion and the RAS KEGG pathway. In mice, some strains (e.g., C57BL/6) have only Ren1, whereas others (e.g., DBA/2, J129) have two, Ren1 and Ren2, genes (43). An increase in Ren1, in particular, causes a striking overexpression of renin in kidney tubules in vivo, partially explaining the association between Ace2 and BP in BXDs (44). For the inflammation pathway, TNFα is a master cytokine that mediates inflammation and innate immune responses (45). MAPKs not only regulate TNFα expression by several mechanisms (45), but also involved in different facets of cardiac hypertrophy, cardioprotection vs. myocardial cell death or cardiac remodeling (46). Through the PPI network analysis, we elucidated the key regulators of the Ace2 co-expressed genes on a protein level involved in heart function, such as Atf2, Cul3, Cyld, Dlg1, Hspa1b, Jun, Mapk8, Mapk14, Npm1, Ppp1cc, Set, and Vcp. Among those genes, Mapk8 (also known as Jnk1) and Mapk14 (also known as p38α) belong to the MAPK family. Jnk1 encodes four different isoforms of c-Jun N-terminal kinases (JNK) (47) involved in inflammation (48), and it also has been implicated in cardiac fibrosis and cardiomyocyte apoptosis (49). In addition, JNK alters the activity of many proteins that reside in mitochondria or act on the nucleus through phosphorylation, including activating transcription factor 2 (ATF2), one of the node genes found in our PPI network. Mapk14 is a critical regulator of vascular smooth muscle inflammation, proliferation, and migration (50), and Mapk14 deficiency in mice blocked cardiac fibroblast differentiation into myofibroblasts (51).
A key node gene found in our PPI network of the Ace2 co-expressed genes is deubiquitinating enzyme cylindromatosis (Cyld), which is also identified as a candidate upstream regulator for Ace2 by eQTL mapping. Although the regulatory relationship between Cyld and Ace2 predicted in our study needs to be further validated, accumulating evidence suggests that Cyld is a crucial regulator of diverse cellular processes, such as immune responses, inflammation, death, and proliferation (52, 53). Cyld has a C-terminal catalytic domain characteristic of deubiquitinating enzymes, which is essential for Cyld to remove ubiquitin from certain proteins that positively mediate signaling through nuclear factor-kappa B (NF-kB) and JNK pathways (54); thus, Cyld acts a negative regulator of JNK and NF-kB signaling (55). Cyld is expressed in the intimal layer of the vessels as well as in cardiomyocytes. In the cardiomyocytes of hypertrophic and failing human and murine hearts, Cyld is highly upregulated, suggesting that Cyld may mediate cardiac injury and dysfunction during inflammation and vascular damage (55). Conversely, knockout of Cyld improved a survival rate and alleviated cardiac hypertrophy, fibrosis, apoptosis, oxidative stress, and contractile dysfunction in mice with sustained pressure overload after transverse aortic constriction (56). In this study, expression of Cyld was significantly negatively correlated with increased heart rate in BXD strains, further supporting the importance of Cyld in cardiac function.
Related to respiratory damages, our systems genetics approach found that levels of Cyld, but not Ace2, is correlated with increased circulation in pulmonary artery and related PH, evidenced by the fact that the BXD strains with lower Cyld expression have increased PV peak volume and PV peak pressure on echocardiograms. Echocardiographic signs of acute cor pulmonale, including RV dilation and dysfunction (RVD), paradoxical septal motion, and PH were the common symptoms in COVID-19 patients with acute respiratory distress (57). Moreover, PH, but not RVD, was associated with clinical signs of more severe COVID-19 and with worse in-hospital clinical outcome (58). Combining murine and human results, we suggest that CYLD may represent a novel target gene for PH and cardiopulmonary diseases. Interestingly, treatment with recombinant TNFα significantly increased CYLD expression in endothelial and vascular smooth muscle cells (59).
The protective effects of ACE2 and its product Ang1-7 against alveolar epithelial cell death by reducing JNK phosphorylation have been shown (60). The main cellular substrate activated by JNK-mediated phosphorylation is C-Jun encoded by Jun (61). Interestingly, our PPI network shows that C-Jun has the most connections with other genes. Ablation of Jun resulted in progressive myocardial fibrosis, cardiomyocyte apoptosis, and sarcomeric disorganization in vivo. These alterations were exacerbated in response to mechanical pressure overload resulting in premature heart failure (62), potentially explaining the genetic basis of cardiopulmonary complications in COVID-19 patients. Recently, supporting our murine results, the Human Coronavirus (HCoV)-host interactome network study has reported that JUN and NPM1 (both are found in our PPI network) are the important hub proteins selected out of 119 CoV-associated host proteins assembled using four known human (SARS-CoV, MERS-CoV, HCoV-229E, and HCoV-NL63), one mouse (MHV), and one avian (IBV) coronaviruses. These host proteins are either the direct targets of HCoV proteins or are involved in crucial pathways of HCoV infection (63). Although there are still many unknowns needed to validate in the future, we believe that our systems genetics approach based on Ace2 and cardiovascular traits associations found in BXD mice provide important regulatory relationship between Ace2 and the node genes, opening surplus avenues in hunting novel targets in cardiovascular and cardiopulmonary diseases, including cardiovascular complications in COVID-19 infection.
Conclusion
In summary, our study aimed to understand the underlying genetic basis of cardiovascular complications through the dissection of Ace2 genetic correlates in murine GRP of BXD mice. We have found that Ace2 expression is associated with systolic and diastolic BP, P-wave duration, and P-wave amplitude. Ace2 co-varies with many genes that are enriched in pathways related to inflammation and cardiac damage, suggesting that all these novel genes (Cyld, Jun, Mapk8, and Mapk14) and pathways (RAS, TGFβ, TNFα, and p38α) can be targeted for preventive, diagnostic, and therapeutic purposes in patients with cardiac damage, including COVID-19 patients.
Data Availability Statement
The gene expression data EPFL/LISP BXD CD Heart Affy Mouse Gene 2.0 ST (Jan14) RMA used in this study was generated through collaborative efforts and can be accessed at our GeneNetwork (GN) website (http://genenetwork.org/) with the GN485 accession number (http://gn1.genenetwork.org/webqtl/main.py?FormID=sharinginfo&GN_AccessionId=485) (22).
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Tennessee Health Science Center (UTHSC).
Author Contributions
LL and EP conceived the study. UM and NL conducted the experiments. FX and JG performed data analysis. FX, JG, LL, and EP wrote the manuscript, prepared the figures, and tables. UM, QG, JP, AS-D, JT, and YC edited the manuscript. All authors read and approved the final version of the manuscript for publication.
Funding
This work was partly supported by National Institutes of Health grants R01HL128350 (LL) and R01HL151438 (LL, EP, and JT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. (2000) 87:E1–9. doi: 10.1161/01.RES.87.5.e1
2. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. (2004) 383:45–51. doi: 10.1042/BJ20040634
3. Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. (2008) 93:543–8. doi: 10.1113/expphysiol.2007.040048
4. Loot AE, Roks AJ, Henning RH, Tio RA, Suurmeijer AJ, Boomsma F, et al. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation. (2002) 105:1548–50. doi: 10.1161/01.CIR.0000013847.07035.B9
5. Averill DB, Ishiyama Y, Chappell MC, Ferrario CM. Cardiac angiotensin-(1-7) in ischemic cardiomyopathy. Circulation. (2003) 108:2141–6. doi: 10.1161/01.CIR.0000092888.63239.54
6. Ferreira AJ, Santos RA, Almeida AP. Angiotensin-(1-7): cardioprotective effect in myocardial ischemia/reperfusion. Hypertension. (2001) 38:665–8. doi: 10.1161/01.HYP.38.3.665
7. Imai Y, Kuba K, Penninger JM. The renin–angiotensin system in acute respiratory distress syndrome. Drug Discov Today Dis Mech. (2006) 3:225–9. doi: 10.1016/j.ddmec.2006.06.012
8. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system. Circ Res. (2020) 126:1456–74. doi: 10.1161/CIRCRESAHA.120.317015
9. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. (2002) 417:822–8. doi: 10.1038/nature00786
10. Wu P, Hao X, Lau EHY, Wong JY, Leung KSM, Wu JT, et al. Real-time tentative assessment of the epidemiological characteristics of novel coronavirus infections in Wuhan, China, as at 22 January 2020. Euro Surveill. (2020) 25:2000044. doi: 10.2807/1560-7917.ES.2020.25.3.2000044
11. Zheng Y-Y, Ma Y-T, Zhang J-Y, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. (2020) 17:259–60. doi: 10.1038/s41569-020-0360-5
12. Kochi AN, Tagliari AP, Forleo GB, Fassini GM, Tondo C. Cardiac and arrhythmic complications in patients with COVID-19. J Cardiovasc Electrophysiol. (2020) 31:1003–8. doi: 10.1111/jce.14479
13. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:811–8. doi: 10.1001/jamacardio.2020.1017
14. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. (2003) 426:450–4. doi: 10.1038/nature02145
15. Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
16. Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. (2004) 5:7. doi: 10.1186/1471-2156-5-7
17. Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG, et al. The expanded BXD family of mice: a cohort for experimental systems genetics and precision medicine. bioRxiv [Preprint]. (2019). doi: 10.1101/672097
18. Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart: role of oxidative stress. Am J Pathol. (2002) 161:1773–81. doi: 10.1016/S0002-9440(10)64454-9
19. Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Reactive oxygen species promote angiogenesis in the infarcted rat heart. Int J Exp Pathol. (2009) 90:621–9. doi: 10.1111/j.1365-2613.2009.00682.x
20. Liu X, Chen Y, Mccoy CW, Zhao T, Quarles DL, Pi M, et al. Differential regulatory role of soluble klothos on cardiac fibrogenesis in hypertension. Am J Hypertens. (2016) 29:1140–7. doi: 10.1093/ajh/hpw062
21. Zhao W, Zhao T, Chen Y, Bhattacharya SK, Lu L, Sun Y. Differential expression of hypertensive phenotypes in BXD mouse strains in response to angiotensin II. Am J Hypertens. (2018) 31:108–14. doi: 10.1093/ajh/hpx144
22. Gu Q, Mendsaikhan U, Khuchua Z, Jones BC, Lu L, Towbin JA, et al. Dissection of Z-disc myopalladin gene network involved in the development of restrictive cardiomyopathy using system genetics approach. World J Cardiol. (2017) 9:320–31. doi: 10.4330/wjc.v9.i4.320
23. Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. (2003) 19:185–93. doi: 10.1093/bioinformatics/19.2.185
24. Chesler EJ, Lu L, Shou S, Qu Y, Gu J, Wang J, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet. (2005) 37:233. doi: 10.1038/ng1518
25. Haley CS, Knott SA. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity. (1992) 69:315–24. doi: 10.1038/hdy.1992.131
26. Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. (2012) 44:821–4. doi: 10.1038/ng.2310
27. Homayouni R, Heinrich K, Wei L, Berry MW. Gene clustering by latent semantic indexing of MEDLINE abstracts. Bioinformatics. (2005) 21:104–15. doi: 10.1093/bioinformatics/bth464
28. Xu L, Furlotte N, Lin Y, Heinrich K, Berry MW, George EO, et al. Functional cohesion of gene sets determined by latent semantic indexing of PubMed abstracts. PLoS ONE. (2011) 6:e18851. doi: 10.1371/journal.pone.0018851
29. Westfall PH, Young SS. Resampling-Based Multiple Testing: Examples and Methods for P-Value Adjustment. New York, NY: John Wiley & Sons (1993).
30. Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. (2019) 47:W199–205. doi: 10.1093/nar/gkz401
31. Wang X, Pandey AK, Mulligan MK, Williams EG, Mozhui K, Li Z, et al. Joint mouse–human phenome-wide association to test gene function and disease risk. Nat Commun. (2016) 7:10464. doi: 10.1038/ncomms10464
32. Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE. Mouse genome database (MGD) 2019. Nucleic Acids Res. (2019) 47:D801–6. doi: 10.1093/nar/gky1056
33. Zhou G, Soufan O, Ewald J, Hancock RE, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. (2019) 47:W234–41. doi: 10.1093/nar/gkz240
34. Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, et al. Protein interaction data curation: the international molecular exchange (IMEx) consortium. Nat Methods. (2012) 9:345–50. doi: 10.1038/nmeth.1931
35. Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, et al. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest. (2006) 116:2218–25. doi: 10.1172/JCI16980
36. Yamamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M, et al. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension. (2006) 47:718–26. doi: 10.1161/01.HYP.0000205833.89478.5b
37. Oudit GY, Kassiri Z, Patel MP, Chappell M, Butany J, Backx PH, et al. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc Res. (2007) 75:29–39. doi: 10.1016/j.cardiores.2007.04.007
38. Dagli N, Karaca I, Yavuzkir M, Balin M, Arslan N. Are maximum P wave duration and P wave dispersion a marker of target organ damage in the hypertensive population? Clin Res Cardiol. (2008) 97:98–104. doi: 10.1007/s00392-007-0587-8
39. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. (2020) 46:586–90. doi: 10.1007/s00134-020-05985-9
40. Li B, Yang J, Zhao F, Zhi L, Wang X, Liu L, et al. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin Res Cardiol. (2020) 109:531–8. doi: 10.1007/s00392-020-01626-9
41. Song B, Zhang ZZ, Zhong JC, Yu XY, Oudit GY, Jin HY, et al. Loss of angiotensin-converting enzyme 2 exacerbates myocardial injury via activation of the CTGF-fractalkine signaling pathway. Circ J. (2013) 77:2997–3006. doi: 10.1253/circj.CJ-13-0805
42. Wong MK. Renin. In: Takei Y, Ando H, Tsutsui K, editors. Handbook of Hormones. Cambridge: Elsevier (2016). p. 255–e229A–252. doi: 10.1016/B978-0-12-801028-0.00176-8
43. Li YC. Vitamin D regulation of the renin–angiotensin system. J Cell Biochem. (2003) 88:327–31. doi: 10.1002/jcb.10343
44. Pleasant L, Ma Q, Devarajan M, Parameswaran P, Drake K, Siroky B, et al. Increased susceptibility to structural acute kidney injury in a mouse model of presymptomatic cardiomyopathy. Am J Physiol Renal Physiol. (2017) 313:F699–705. doi: 10.1152/ajprenal.00505.2016
45. Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. (2014) 26:237–45. doi: 10.1016/j.smim.2014.02.009
46. Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. (2010) 90:1507–46. doi: 10.1152/physrev.00054.2009
47. Waetzig V, Herdegen T. Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends Pharmacol Sci. (2005) 26:455–61. doi: 10.1016/j.tips.2005.07.006
48. Oltmanns U, Issa R, Sukkar MB, John M, Chung KF. Role of c-jun N-terminal kinase in the induced release of GM-CSF, RANTES and IL-8 from human airway smooth muscle cells. Br J Pharmacol. (2003) 139:1228–34. doi: 10.1038/sj.bjp.0705345
49. Tachibana H, Perrino C, Takaoka H, Davis RJ, Prasad SVN, Rockman HA. JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem Biophys Res Commun. (2006) 343:1060–6. doi: 10.1016/j.bbrc.2006.03.065
50. Wu W, Zhang W, Choi M, Zhao J, Gao P, Xue M, et al. Vascular smooth muscle-MAPK14 is required for neointimal hyperplasia by suppressing VSMC differentiation and inducing proliferation and inflammation. Redox Biol. (2019) 22:101137. doi: 10.1016/j.redox.2019.101137
51. Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. (2017) 136:549–61. doi: 10.1161/CIRCULATIONAHA.116.026238
52. Lim JH, Jono H, Koga T, Woo C-H, Ishinaga H, Bourne P, et al. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS ONE. (2007) 2:e1032. doi: 10.1371/journal.pone.0001032
53. Lim JH, Ha UH, Woo CH, Xu H, Li JD. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cell Microbiol. (2008) 10:2247–56. doi: 10.1111/j.1462-5822.2008.01204.x
54. Glittenberg M, Ligoxygakis P. CYLD: a multifunctional deubiquitinase. Fly. (2007) 1:330–2. doi: 10.4161/fly.5399
55. Mathis BJ, Lai Y, Qu C, Janicki JS, Cui T. CYLD-mediated signaling and diseases. Curr Drug Targets. (2015) 16:284–94. doi: 10.2174/1389450115666141024152421
56. Wang H, Lai Y, Mathis BJ, Wang W, Li S, Qu C, et al. Deubiquitinating enzyme CYLD mediates pressure overload-induced cardiac maladaptive remodeling and dysfunction via downregulating Nrf2. J Mol Cell Cardiol. (2015) 84:143–53. doi: 10.1016/j.yjmcc.2015.04.012
57. Beneduce A, Esposito A, Ingallina G, Palumbo D, Palmisano A, Ancona F, et al. Heart and lung multimodality imaging in COVID-19. Cardiovasc Imaging. (2020) 13:1792–808. doi: 10.1016/j.jcmg.2020.05.017
58. Pagnesi M, Baldetti L, Beneduce A, Calvo F, Gramegna M, Pazzanese V, et al. Pulmonary hypertension and right ventricular involvement in hospitalised patients with COVID-19. Heart. (2020) 106:1324–31. doi: 10.1136/heartjnl-2020-317355
59. Takami Y, Nakagami H, Morishita R, Katsuya T, Hayashi H, Mori M, et al. Potential role of CYLD (Cylindromatosis) as a deubiquitinating enzyme in vascular cells. Am J Pathol. (2008) 172:818–29. doi: 10.2353/ajpath.2008.070312
60. Uhal BD, Dang M, Dang V, Llatos R, Cano E, Abdul-Hafez A, et al. Cell cycle dependence of ACE-2 explains downregulation in idiopathic pulmonary fibrosis. Eur Respir J. (2013) 42:198–210. doi: 10.1183/09031936.00015612
61. Yarza R, Vela S, Solas M, Ramirez MJ. c-Jun N-terminal kinase (JNK) signaling as a therapeutic target for Alzheimer's disease. Front Pharmacol. (2016) 6:321. doi: 10.3389/fphar.2015.00321
62. Windak R, Müller J, Felley A, Akhmedov A, Wagner EF, Pedrazzini T, et al. The AP-1 transcription factor c-Jun prevents stress-imposed maladaptive remodeling of the heart. PLoS ONE. (2013) 8:e73294. doi: 10.1371/journal.pone.0073294
Keywords: BXD mice, cardiovascular, ACE2, gene regulaiton, COVID-19
Citation: Xu F, Gao J, Munkhsaikhan U, Li N, Gu Q, Pierre JF, Starlard-Davenport A, Towbin JA, Cui Y, Purevjav E and Lu L (2020) The Genetic Dissection of Ace2 Expression Variation in the Heart of Murine Genetic Reference Population. Front. Cardiovasc. Med. 7:582949. doi: 10.3389/fcvm.2020.582949
Received: 13 July 2020; Accepted: 02 September 2020;
Published: 20 November 2020.
Edited by:
Christian F. Deschepper, Institute of Clinical Research de Montreal (IRCM), CanadaReviewed by:
Jessica Wang, University of California, Los Angeles, United StatesChristoph D. Rau, University of North Carolina at Chapel Hill, United States
Copyright © 2020 Xu, Gao, Munkhsaikhan, Li, Gu, Pierre, Starlard-Davenport, Towbin, Cui, Purevjav and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lu Lu, bHVsdUB1dGhzYy5lZHU=; Enkhsaikhan Purevjav, ZXB1cmV2amFAdXRoc2MuZWR1
†These authors have contributed equally to this work