 Verena Schwach
Verena Schwach Rolf H. Slaats
Rolf H. Slaats Robert Passier
Robert Passier- 1Applied Stem Cell Technologies, TechMed Centre, University of Twente, Enschede, Netherlands
- 2Department of Anatomy and Embryology, Leiden University Medical Centre, Leiden, Netherlands
Cardiotoxicity is a major cause of high attrition rates among newly developed drugs. Moreover, anti-cancer treatment-induced cardiotoxicity is one of the leading reasons of mortality in cancer survivors. Cardiotoxicity screening in vitro may improve predictivity of cardiotoxicity by novel drugs, using human pluripotent stem cell (hPSC)-derived-cardiomyocytes. Anthracyclines, including Doxorubicin, are widely used and highly effective chemotherapeutic agents for the treatment of different forms of malignancies. Unfortunately, anthracyclines cause many cardiac complications early or late after therapy. Anthracyclines exhibit their potent anti-cancer effect primarily via induction of DNA damage during the DNA replication phase in proliferative cells. In contrast, studies in animals and hPSC-cardiomyocytes have revealed that cardiotoxic effects particularly arise from (1) the generation of oxidative stress inducing mitochondrial dysfunction, (2) disruption of calcium homeostasis, and (3) changes in transcriptome and proteome, triggering apoptotic cell death. To increase the therapeutic index of chemotherapeutic Doxorubicin therapy several protective strategies have been developed or are under development, such as (1) reducing toxicity through modification of Doxorubicin (analogs), (2) targeted delivery of anthracyclines specifically to the tumor tissue or (3) cardioprotective agents that can be used in combination with Doxorubicin. Despite continuous progress in the field of cardio-oncology, cardiotoxicity is still one of the major complications of anti-cancer therapy. In this review, we focus on current hPSC-cardiomyocyte models for assessing anthracycline-induced cardiotoxicity and strategies for cardioprotection. In addition, we discuss latest developments toward personalized advanced pre-clinical models that are more closely recapitulating the human heart, which are necessary to support in vitro screening platforms with higher predictivity. These advanced models have the potential to reduce the time from bench-to-bedside of novel antineoplastic drugs with reduced cardiotoxicity.
Introduction
Cardiovascular diseases and cancer are the two leading causes of death in industrialized countries. Although in recent years new anti-cancer therapies improved long-term survival rates, this is unfortunately also accompanied by an increased risk of cardiovascular complications because of adverse side effects of these anti-cancer drugs. Anthracyclines are a group of widely used anti-cancer drugs that are known to cause cardiac complications, either early or late after start of treatment (1–7). The history of anthracyclines originates in the 1950s when Daunorubicin was isolated from the bacteria Streptomyces peucetius. Ten years later Doxorubicin, a more effective derivative of Daunorubicin, was identified (8). However, Doxorubicin received the ominous nickname “Red Devil” because of the detrimental side effects in combination with its red color. In approximately 11% of patients, Doxorubicin-induced cardiotoxicity leads to an acute response within 2–3 days of administration (early onset of cardiotoxicity), manifested by chest pain resulting from different forms of arrhythmia (3). Chronic Doxorubicin-induced cardiomyopathy has a lower incidence (ranging from 4 to 36% dependent on the dose) and can occur as late as 10 years after the last dose (late onset of chronic cardiotoxicity), with only a 50% 1-year survival prognosis when cardiomyopathy further develops into congestive heart failure (3, 7). Importantly, anthracycline-induced cardiotoxicity endangers cancer patients since the early 70s (9, 10). At present, almost half a century later, success in preventing or counteracting anthracycline-induced cardiotoxicity has been very limited. Moreover, although novel anti-cancer therapeutic compounds to specific target molecules, such as tyrosine kinase inhibitors, have been developed, drug-induced cardiotoxicity remains a persistent problem. One major reason for this is that current in vitro and animal models fail to show sufficient predictive power and limit extrapolation to patients, which consequently leads to high attrition rates during the process of drug development.
Development of preclinical human cell-based models for drug discovery that mimic human physiology and pathophysiology, has the potential to improve the predictability of adverse drug events (11). In particular, human pluripotent stem cells (hPSCs) are excellent candidates for developing these models since they replicate indefinitely and have the capacity to differentiate to any cell type of the human body, including functional cardiomyocytes. This is especially embraced in the cardiac field, since isolated primary human cardiomyocytes are extremely difficult to obtain and maintain in culture. Advances in differentiation and purification of hPSC-derived cardiomyocytes (hPSC-cardiomyocytes) and their unlimited availability has promoted strategies to use these cells for cardiotoxicity assessment of new drugs (12). Previously, the Food and Drug Administration (FDA), the supreme body worldwide that checks the quality and safety of medical products, initiated the so-called “Comprehensive in vitro Proarrhythmia assay (CIPA),” which represents a paradigm shift and encompasses evaluation of life-threatening proarrhythmic risk for all new compounds in a preclinical assay using hPSC-cardiomyocytes (13). This CIPA initiative signifies the enormous potential of hPSC-derived cardiomyocytes for preclinical drug screening. Here, we describe underlying mechanisms of anthracycline-induced cardiotoxicity, as well as therapeutic approaches for cardioprotection. Furthermore, we will discuss the potential to use hPSC-derived cardiac models for improved safety assessment of anti-cancer drugs and strategies to overcome current limitations for developing in vitro drug testing platforms with a higher predictivity.
Mechanism of Anthracycline-Induced Cardiotoxicity in hPSC-Cardiomyocytes
The potent therapeutic anti-cancer effect of Doxorubicin is mediated primarily via inhibition of topoisomerase IIa, an enzyme responsible for unwinding DNA before replication or transcription. This inhibition leads to DNA damage and consequently death of highly proliferating cancer cells, but also affects healthy proliferating cells, such as hematopoietic precursors, epithelial lining of the intestine and hair follicle cells. Fully mature cardiomyocytes are typically quiescent and do not express topoisomerase IIα, however studies performed in hPSC-cardiomyocytes demonstrated that Doxorubicin may bind to the enriched topoisomerase IIβ in cardiomyocytes, and knock-out of this isotype improved cell viability significantly when exposed to Doxorubicin (14, 15). A comprehensive review on damage mechanisms of anthracycline-induced cardiotoxicity has been published elsewhere (16). The underlying mechanisms of Doxorubicin-induced cardiotoxicity is not yet completely understood. However, several studies in hPSC-cardiomyocytes have shown that mitochondrial dysfunction, disruption of calcium homeostasis, as well as altered gene and protein expression levels triggering apoptotic cell death, play important roles in anthracycline-induced cardiotoxicity (Figure 1). Increased oxidative stress by elevated production of reactive oxygen (ROS) and nitrogen species (NOS) in the mitochondria of cardiomyocytes are major factors of Doxorubicin-induced cardiotoxicity, since cardiomyocytes are more sensitive to these molecules because they possess lower levels of antioxidant enzymes than other cell types (1, 3, 4, 17–20). As a consequence, higher stress levels in cardiomyocytes under oxidative stress circumstances can lead to cardiotoxicity (20). An increase in intracellular levels of ROS and NOS cause mitochondrial dysregulation, lipid peroxidation, DNA damage, and protein carbonylation. Cardiac mitochondria are the major site of Doxorubicin-induced ROS/NOS levels due to the localization of the major redox cycling enzymes such as NAD(P)H. Additionally, Doxorubicin becomes nearly irreversibly bound to cardiolipin. Cardiolipin is an essential phospholipid, almost exclusively expressed on the inner mitochondrial membrane. It has been shown that pathological changes in cardiolipin trigger ROS production and impair mitochondrial function (21). In addition, Doxorubicin also increases mitochondrial iron accumulation which further increases ROS production in the mitochondria. Indeed, cellular and mitochondrial (as measured by cellular H2O2 production and intra-mitochondrial O2− levels) ROS production in hPSC-cardiomyocytes increases already 24 h after exposure to Doxorubicin at concentrations as low as 0.01 μM (patient serum levels of Doxorubicin vary in the range of 5–10 μM after single injection). Moreover, short term exposure to 5 μM Doxorubicin induces dramatic reduction of the mitochondrial transmembrane potential indicating mitochondrial dysfunction (14, 22). To meet the high energy demand required to sustain contractile function, cardiomyocytes rely on efficient mitochondrial oxidative metabolism for energy production instead of anaerobic glycolysis. Thus, a disturbed energy metabolism is a high risk for cardiomyocyte survival. Repeated Doxorubicin exposure reduces ATP levels which cannot be restored after Doxorubicin removal suggesting a prolonged effect of Doxorubicin on energy generation in hPSC-cardiomyocytes. Together these effects pinpoint to the fact that Doxorubicin induces mitochondrial dysfunction in hPSC-cardiomyocytes (22, 23). Importantly, structural and functional disturbances are more pronounced after repeated dosing of Doxorubicin (mimicking chronic exposure) (23–25).
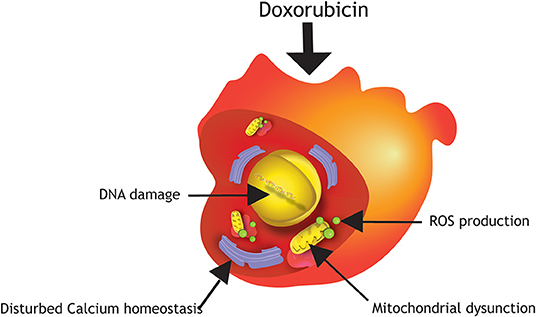
Figure 1. Damage mechanisms of Doxorubicin in hPSC-cardiomyocytes. Major mechanisms of anthracycline-induced cardiotoxicity in hPSC-cardiomyocytes are DNA damage, production of reactive oxygen species (ROS), mitochondrial dysfunction and disturbed calcium homeostasis.
Another mechanism by which anthracyclines fight cancer is the induction of apoptosis. Doxorubicin effectively induces apoptotic cell death via activation of so-called death receptors (DRs), such as TNF receptor 1 (TNFR1), Fas receptor, DR4 and DR5, in many cancer types. Activation of these DRs induces the assembly of death inducing signaling complex (DISC), which starts the caspase cascade to mediate cleavage of cellular proteins and ultimately apoptosis of the cell. This DR-mediated apoptosis machinery has been shown to be conserved in human cardiomyocytes. Indeed, a Doxorubicin concentration–dependent increase of caspase 3 and 7 and Annexin V, a marker of early apoptosis, and 7-aminoactinomycin D (7–AAD) or propidium iodide as markers of late apoptosis or necrosis, suggest a strong contribution of programmed cell death to reduced cell survival after exposure to Doxorubicin. Interestingly, in hPSC-cardiomyocytes Doxorubicin induces the expression of all four DRs in a dose-dependent fashion with p53 being the key upstream activator. This suggests a p53-regulated DR-mediated apoptotic pathway a key mechanism involved in early Doxorubicin-induced cardiotoxicity. In accordance with these findings, Doxorubicin-induced apoptosis was effectively blocked by pretreating hPSC-cardiomyocytes with a DR5 neutralizing antibody. This cardiotoxic effect was reversible since expression of DR4 and 5 proteins decreased after recovery for 7 days following washout of Doxorubicin (14, 15, 22, 26, 27). In contrast to these studies with acute exposure to Doxorubicin, Li et al. showed that p53 can protect against chronic Doxorubicin exposure by counteracting mitochondrial DNA depletion after chronic exposure of hPSC-cardiomyocytes (and mice) to low doses of Doxorubicin (28).
Disturbed calcium homeostasis in cardiomyocytes hampers proper cardiac contraction. Doxorubicin has been shown to induce accumulation of intracellular calcium release from the sarcoplasmic reticulum (SR) causing calcium overload with sarcomeric disarray and myofibril deterioration in hPSC-cardiomyocytes. Moreover, Doxorubicin exposure led to downregulation of several ion channel genes, including genes that encode for calcium channels CACNs (14, 22, 26).
In summary this data shows that mitochondrial dysfunction, apoptosis, as well as disturbed calcium homeostasis are the main mechanisms of anthracycline-induced cardiotoxicity. However, in 2018, a new mechanism of Doxorubicin-induced cardiotoxicity via downregulation of the RNA-binding-protein Quaking has been identified (29). Downregulation of Quaking regulates a set of circular RNAs involved in Doxorubicin-induced apoptosis suggesting that other mechanisms cannot be excluded.
Protective Mechanisms to Prevent or Reduce Anthracycline-Induced Cardiotoxicity
To increase the dose window of effective treatment without severe adverse side effects (therapeutic index) of conventional Doxorubicin therapy, several protective mechanisms have been developed, including modifications/analogs of Doxorubicin, targeted delivery or protective agents (Figure 2).
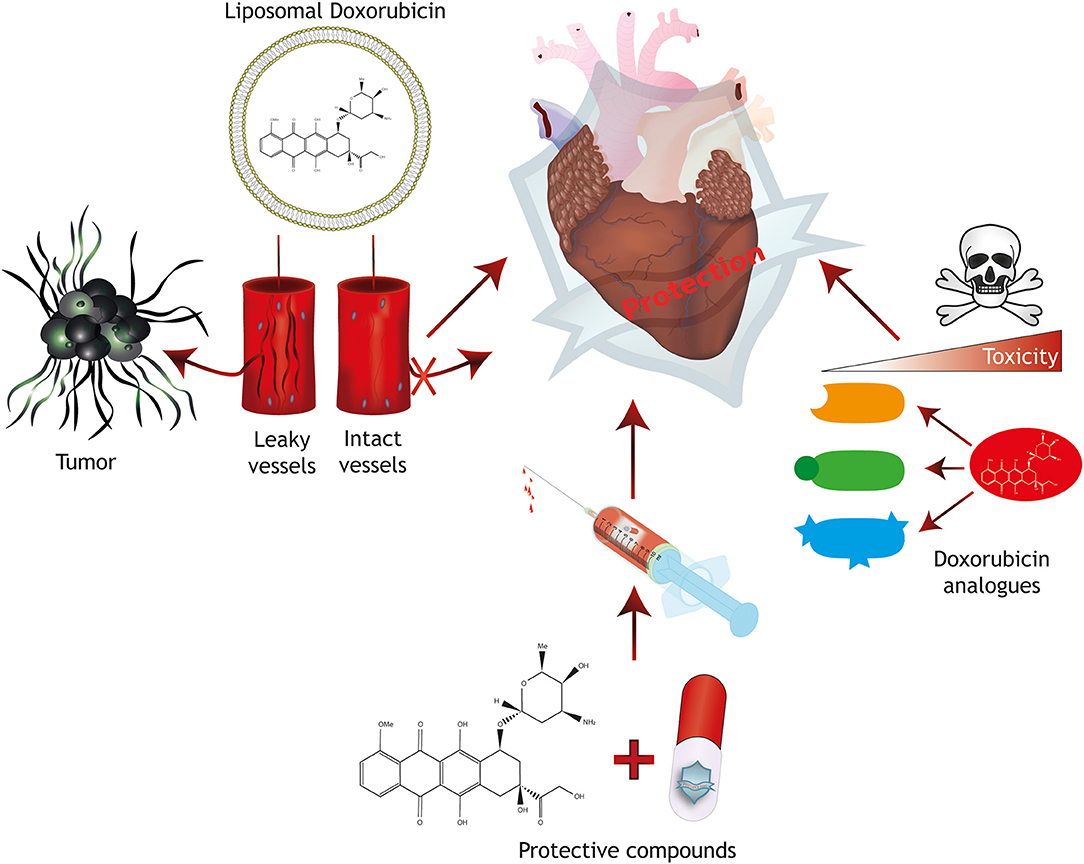
Figure 2. Strategies for cardioprotection. Protective mechanisms for anthracycline-induced cardiotoxicity include the generation of Doxorubicin analogs with reduced toxicity, the combined treatment with protective compounds and targeted therapy approaches including liposomal packaging of Doxorubicin. Liposomal packaging prevents Doxorubicin from exiting the vasculature in tissues with tight endothelial barriers such as the heart, but allows targeting of a tumor with leaky capillaries.
Analogs of Doxorubicin
The cardiotoxic nature of Doxorubicin pushed the development of Doxorubicin analogs into overdrive in the 1970s and 1980s, with over a 1000 analogs manufactured or discovered since (30). However, most of them failed to reach anti-tumor efficacy comparable to Doxorubicin and others were considered too toxic based on trials in rodents (30). Only a handful of analogs reached clinical trial phase, such as epirubicin, pirarubicin (also known as THP), idarubicin, mitoxanthrone, and others (31–42). The success of these analogs varies per clinical setting and malignancy, and a reduction in cardiotoxicity was not always achieved. In patients, cardiotoxic events are classically detected through loss of the left ventricular ejection fraction (LVEF), irregularities in ECG readings (in particular ST-T changes), changes in systolic time intervals (STI, indicating heart muscle failure) and in more recent work, the release of biomarkers in blood, such as cardiac Troponin-T and brain natriuretic peptide (BNP) (31, 33, 35–38, 41–45). Due to the year of discovery, early Doxorubicin analogs have not been tested in vitro using hPSC-cardiomyocytes to evaluate their cardiotoxicity, since these models were not available at the time. Whereas, biomarker release can also be assessed in vitro using hPSC-cardiomyocytes, other clinical read-outs are more difficult to evaluate in vitro. Nevertheless, measurement of changes in action potential, contractility, and cell survival in vitro may be associated with ECG/STI changes and impaired LVEF and therefore it will be of interest to test Doxorubicin analogs in hPSC-cardiomyocytes for cardiac safety.
Targeted Delivery Strategies
An alternative to reducing cardiotoxic properties of anthracyclines is specifically delivering anthracyclines at the location where cancer cells reside. This can be achieved by carriers that encapsulate the compound and release it at the desired location. For example, liposomal encapsulation of anthracycline inhibits transfer from the circulation to tissues with a tight endothelial barrier (e.g., the heart), but allow transfer to tissues with leaky or fenestrated vasculature, such as in tumors, but also in organs such as lung, bone marrow, lymph nodes and liver (39, 46, 47). In the last two decades, numerous trials have been conducted to assess the safety and benefit of liposome-encapsulated Doxorubicin. Success of liposomal encapsulated anthracyclines is inconsistent. Liposomal encapsulation could abolish cardiotoxicity in some clinical trials, allowing a higher cumulative dose for treatment (39, 48–50). However, in some clinical settings, liposomal Doxorubicin did not reduce cardiotoxicity compared to free anthracyclines (51), suggesting that liposomal encapsulation only has limited benefit. Unmasked liposomes such as described thus far are relatively quickly cleared from the circulation by the reticulo-endothelial system (RES), which limits the therapeutic index of so-called non-PEGylated liposomal (NPL) (39).
PEGylated liposomal Doxorubicin (PLD) features an anchored polyethylene glycol (PEG) group on the exterior lipid surface that masks them from the RES, thus reducing uptake from the circulation (52). Caelyx (Doxil), a commercialized PLD, showed significant reduction in cardiotoxicity compared to free Doxorubicin in several studies, even when used for patients who have increased risk of cardiac complications (53–55). Unfortunately, PLDs are limited in their use because of increased incidence and severity of non-cardiac side effects such as palmar-plantar erythrodysesthesia (PPE), mucositis and haematogenic disorders (54, 56). Liposomes (PEGylated or not) cannot prevent severe adverse effects of Doxorubicin entering off-target tissues. The mechanism of specific delivery of classical liposomes depends greatly on the difference in endothelial barrier permeability, which is equally high in tumors and several healthy tissues alike, and not all malignancies cohere to the “tumor with a leaky vasculature” phenotype. In an attempt to increase specific targeting of cancer cells, liposomes were conjugated with antibodies that are directed against the human epidermal growth factor receptor 2 (HER2), which is highly expressed in breast cancer cells. In vitro, these liposomes were effective in killing HER2-positive cancer cells, but did not affect hPSC-cardiomyocytes, highlighting their specificity (47). The HERMIONE phase II trial is now being conducted to test the safety and efficacy in patients (57). Another recent development is the encapsulation of anthracyclines in nanocages, constructed of organic (virus-like particles (VLPs), protein, DNA or carbon-based particles) or inorganic particles (supramolecular nanosystems, hybrid metal-organic, gold, or silica-based systems). Advantages of this encapsulation method are: (1) a large carrier storage capacity, (2) a targeted release of the drug at the site of interest, (3) combined with a porous structure and (4) a low immunogenic surface (58). Nanocages have not been tested in clinical trials yet, however in vitro data suggests improved drug targeting of cancer cells and killing efficacy, and a lower cardiotoxicity, when administered in rodents (59–63). Unfortunately, nanocages have not been specifically tested in vitro on hPSC-cardiomyocytes to test their cardiotoxicity.
In addition to Doxorubicin encapsulation, latest modifications to Doxorubicin are being tested on cell cultures prior to animal testing to assess their safety for the heart, using currently available hPSC-cardiomyocyte in vitro models. These modifications of Doxorubicin are aimed at preventing the anthracycline from exerting its toxicity on cardiomyocytes by making the drug delivery more specific. For example, DTS-201, a Doxorubicin prodrug that is injected in a stable, cell-impermeable state, is cleaved by endopeptidases that are released by tumors, enabling entry into nearby cells and specific anti-tumor activity. In a phase I trial, DTS-201 could be administered at a cumulative dose equivalent to three times the recommended dose of free Doxorubicin, without triggering a significant drop in LVEF (64). Another promising technique is the development of Doxorubicin-dendrimer conjugates with acetylgalactosamine attached to the dendrimer surface. These conjugates are specifically taken up by hepatic cancer cells, increasing therapeutic index and reducing cardiac exposure and thus toxicity in mice. In addition, in vitro, these conjugates did not affect hPSC-cardiomyocyte viability and electrophysiology, or induce apoptosis, demonstrating the specificity for drug uptake by the target cell type (65).
Protective Agents to Alleviate Cardiotoxicity
Cardioprotection during anthracycline-based therapies can also be offered by combining the anthracycline with a protective compound. As the cardiotoxic mechanism of anthracyclines involves inducing oxidative stress and mitochondrial dysfunction, protective agents that are able to lower the production of ROS or lower the workload of the heart during anthracycline treatment may be beneficial to reduce anthracycline-induced cardiotoxicity. Factors that may reduce oxidative stress in the heart, such as statins and natural antioxidants, and compounds that reduce the workload of the heart, such as angiotensin-converting-enzyme (ACE) inhibitors and beta-blockers, have been combined with anthracyclines in clinical trials, with mixed success (66–77). As far as known, these compounds have never been tested in hPSC-cardiomyocytes for assessment of their direct cardioprotective potential, even though reducing oxidative stress in particular can be readily modeled in vitro. Since both ischemic heart disease and anthracycline-mediated cardiotoxicity share common pathological pathways, therapies for one disease could also be beneficial for the other. Trials with more original approaches, such as remote ischemic conditioning or physical exercise and caloric restriction, strategies that were beneficial in the setting of ischemic heart disease, have been started with the purpose to reduce cardiotoxicity caused by chemotherapy (78, 79). Results of these trials are not yet available.
In hPSC-cardiomyocytes it has been shown that Doxorubicin causes arrhythmic contractions by inducing the accumulation of calcium in the cell and thereby disturbing calcium homeostasis (14, 22). This would suggest that calcium antagonists can help to reduce Doxorubicin-induced cardiotoxicity. Interestingly, a calcium antagonistic compound called prenylamine, was able to reduce cardiotoxic effects when co-administered with Doxorubicin, before it was withdrawn from the market 1 year later (80). Reason for withdrawal was the pro-arrhythmic properties of prenylamine, inducing long QT syndrome (81). Other calcium agonists are currently primarily used at lower doses to reduce Doxorubicin resistance of cancer cells, by inhibiting their capacity to pump out drugs via the P-glycoprotein ATP-dependent efflux pumps (82). Low doses of these calcium antagonists could be repurposed to reduce Doxorubicin-induced cardiotoxicity.
Neuregulin-1β, an ErbB receptor (HER2) family ligand, has been proven effective against Doxorubicin-induced cardiotoxicity, but is also pro-neoplastic in many cancers via formation of ErbB2/3 interactions (83–86). Even if neuregulin, as key mediator of endothelial–cardiomyocyte crosstalk, is able to protect ventricular cardiomyocytes from anthracycline-induced apoptosis, its use might thus be controversial in the clinic. Nevertheless, phase I, II and III clinical trials have been completed or are ongoing (NCT01251406, NCT1214096, and NCT01541202) (84–86). Patients with symptomatic heart failure (HF) and left ventricular dysfunction showed improved cardiac function with increased LVEF after treatment with recombinant human neuregulin 1. In contrast to native neuregulin-1β, engineered bivalent neuregulin-1β has reduced pro-neoplastic potential because of a shift toward ErbB3 homotypic interactions. Bivalent neuregulin-1β is anti-neoplastic or cytostatic in cancer cells. Importantly, bivalent neuregulin-1β showed similar cardioprotective properties as neuregulin in hPSC-cardiomyocytes and in mice with chronic cardiomyopathy (87). Especially the reduced pro-neoplastic potential of bivalent neuregulin-1β offers translational potential for cardioprotection after anthracycline therapy, however to date, bivalent neuregulin-1β has not been taken into clinical trial.
Dexrazoxane, a catalytic topoisomerase inhibitor and iron chelator, is the only marketed cardioprotective agent that is used specifically to counteract the adverse effects of anthracyclines (88). In children with acute lymphoblastic leukemia, leukemia or lymphoma, the combined treatment with Doxorubicin and Dexrazoxane significantly reduces release of troponin (as biomarker of cardiotoxicity) and Doxorubicin-induced cardiac remodeling. Importantly, the combination with Dexrazoxane improves left ventricular function compared to Doxorubicin alone (89, 90). Dexrazoxane is also effective in relapsed patients with leukemia when administered prior or after a high cumulative dose (91). In breast cancer patients with increased risk of cardiotoxicity because of an enhanced cumulative dose of anthracycline, Dexrazoxane also robustly reduced the incidence of cardiac events, such as decreased ejection fraction, and incidence and severity of CHF (92, 93). Even in a retrospective 2–20 year follow-up, Dexrazoxane demonstrated late-clinical and subclinical cardioprotective effect (94).
Treatment of cardiomyocytes with Dexrazoxane is therefore expected to prevent cardiotoxicity. However, pre– and co–treatment of hPSC-cardiomyocytes with Dexrazoxane could not alleviate Doxorubicin-induced cardiotoxicity and increased cardiotoxicity at concentrations as low as 0.1 μM (22, 95). This discrepancy between in vivo and in vitro may be related to the relative immature character of hPSC-cardiomyocytes. Early stage hPSC-cardiomyocytes may be more sensitive to Doxorubicin because of higher expression levels of topoisomerase IIα and are thus more prone to severe DNA damage (as mentioned earlier: Doxorubicin primarily affects topoisomerase IIα), whereas in mature cardiomyocytes topoisomerase IIα switches to the IIβ isoform. Importantly, Dexrazoxane might exert its protective effects via depletion of topoisomerase IIβ, the major topoisomerase isoform in more mature cardiomyocytes (95, 96). This suggests that more advanced models are needed to assess cardiotoxicity in vitro.
HPSC-Cardiomyocyte Models in Drug Screening of Tyrosine Kinase Inhibitors (TKis)
Not only anthracyclines, but also safety assessment of other classes of anti-cancer drugs may benefit from hPSC-based in vitro models. For example, malignancies caused by hyperactive receptor tyrosine kinases (RTKs), which drive proliferation and survival, are treated with FDA-approved tyrosine-kinase inhibitors (TKis). However, a number of TKis have been associated with adverse cardiac side effects following treatment of cancer patients, such as reduced LVEF, myocardial infarction, arrhythmias and heart failure (97). While animal models have often failed to predict cardiotoxic effects during safety assessment (98), TKis have shown to inhibit viability, contractility, electrophysiology, calcium handling and cardiac oxidative phosphorylation of hPSC-cardiomyocytes (99–101). Different TKis show cell type specific cytotoxicity, either in hPSC-derived cardiomyocytes, endothelial cells and fibroblasts or hPSCs, suggesting that TKis differently affect cardiovascular and non-cardiovascular cell types (99).
TKis can be subdivided into small molecules, including crizotinib, sunitinib, or sorafenib, and monoclonal antibodies. Importantly, different TKis have distinct toxicity profiles and not all TKIs induce cardiotoxicity (97). The broad-range TKi sunitinib, for example, decreased hPSC-cardiomyocyte viability, as well as AMP-activated protein kinase (AMPK), increased lipid accumulation and induced arrhythmic events in sunitinib-treated hPSC-cardiomyocytes. Crizotinib, an ALK/MET inhibitor, increased ROS production and caspase activation, induced cholesterol accumulation and an irregular beat pattern. Similarly, Nilotinib, a second generation Bcr-Abl inhibitor, increased ROS generation and caspase activation and induced arrhythmic beating. Interestingly, compared to the TKis sunitinib, crizotinib and nilotinib, the relatively cardiac-safe TKi erlotinib only displayed minor effects on hPSC-cardiomyocyte health in the same study (98) which suggests that hPSC-cardiomyocytes may represent a reliable in vitro model for predicting cardiotoxic side effects.
RTK activity may also be reduced by blocking the human epidermal growth factor receptor 2 (HER2), which activation is a common feature of a subset of malignant diseases, particularly breast cancer (102). While trastuzumab (TZM), a TKi monoclonal antibody blocking HER2, greatly improves treatment against HER2 positive malignancies, inhibition of HER2 signaling was found to be detrimental for cardiac function (102–104). In a subset of breast cancer patients, treatment with TZM led to mild to severe decrease in LVEF without apparent myocardial tissue damage or cell loss. Using patient-derived hPSC-cardiomyocyte 2D monolayer cultures it was shown that TZM affected metabolic and mitochondrial processes, of which the severity depended on genetic variation. Moreover, this disease phenotype could be rescued using metabolism-stimulating agents, such as AMPK (102). Additionally, HER2 activation triggered by exogenous neuregulin-1 could reduce hPSC-cardiomyocyte damage and cell loss during Doxorubicin exposure, an effect that was lost upon inhibition of the HER2 receptor by TZM (103, 104). These findings indicate that hPSC-cardiomyocyte 2D models may serve as a valuable tool to identify cardioprotective compounds in a high throughput system, which can be further validated in more advanced human-based 3D cardiac models.
Generation of Advanced hPSC-Derived in vitro Models for Accurate Assessment of Cardiac Safety
Although cardiotoxic effects could be observed in hPSC-cardiomyocyte based models, not all aspects of in vivo cardiotoxicity are recapitulated in current human in vitro models. One of the important shortcomings of hPSC-cardiomyocyte models is their relative immature phenotype. Reviews on the topic of the immaturity of hPSC-cardiomyocytes, including strategies to increase the level of maturity have already been described elsewhere (105–107). Figure 3 shows a description of current in vitro models utilizing hPSC-cardiomyocytes, including their advantages and limitations. It is therefore crucial to develop innovative advanced human models that approximate the adult human heart. In order to achieve this, defined multicellular models, consisting of the main cell-types of the heart, such as atrial or ventricular cardiomyocytes, endothelial cells, smooth muscle cells and fibroblasts, need to be constructed.
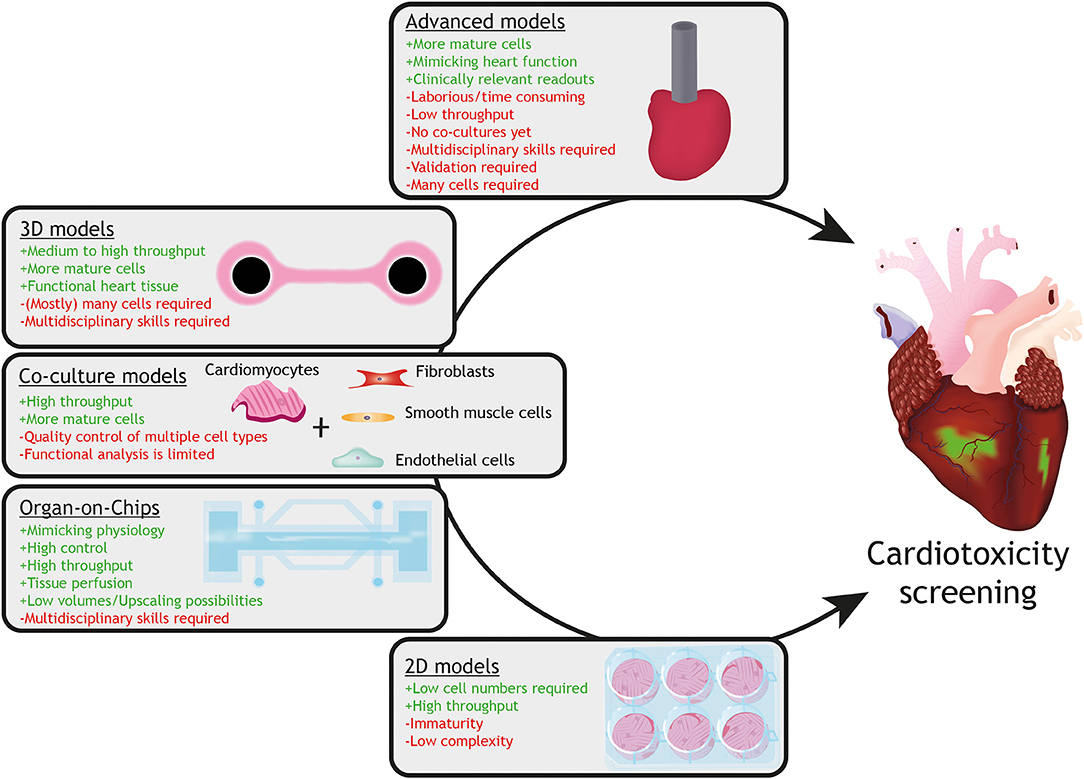
Figure 3. Current in vitro models with hPSC-cardiomyocytes and their advantages (+ green) and limitations (– red). Models with increasing complexity (from 2D monolayers to 3D advanced models) are available for assessment of drug-induced cardiotoxicity utilizing hPSC-cardiomyocytes. Whereas 2D monolayer cardiomyocyte cultures can be used for high throughput-screening for specific cellular responses, they lack the physiology, multicellular interactions and tissue organization, which can be provided by organ-on-chip, co-culture or 3D tissue models (for instance engineered heart tissues). Advanced cardiac models, such as an engineered mini heart tube with the capacity to pump fluid, mimic clinically-relevant function(s) of the heart.
Cardiac tissue formation in 3D tissues, such as cardiac microtissues, more closely mimics native heart tissue which might allow studying more physiologically relevant dosing profiles and deciphering acute vs. chronic cardiotoxicity (25). For generating other 3D advanced cardiac models, so-called engineered heart tissues (EHTs), hPSC-cardiomyocytes are mixed with fibroblasts (or other cardiac cells) in a hydrogel and poured into a casting mold around silicon posts, leading to beating structures within the first week under continuous mechanical strain. Previously, it has been shown that 3D EHTs display a higher degree of maturation based on sarcomeric organization, formation of T-tubules and functional characterization (e.g., contraction force, electrophysiology) (108–110). Cardiotoxicity of anti-cancer agents, other than anthracyclines, such as the TKi sunitinib, has already been assessed in EHTs, which resulted in triggered activation of apoptosis and loss of contractile force, spontaneous beating and mitochondrial membrane potential (111).
Until now studies did not consider subtype-specific cardiomyocytes for cardiotoxicity testing. This would be of high importance, as different cardiac subtypes may exhibit different susceptibility to Doxorubicin. Especially, because of the well-established arrhythmogenic potential of Doxorubicin (31, 112, 113), it is highly relevant to assess arrhythmogenic effects of Doxorubicin on hPSC-derived atrial and ventricular cardiomyocytes. Moreover, in a trial comparing Doxorubicin to Epirubicin in treatment of breast cancer, a quarter of all cardiac side effects of Doxorubicin was identified as sinus tachycardia, for Epirubicin this was half of the cardiotoxic responses in the trial population (31). In addition, a recent study showed that genetic loci are associated with both increased sensitivity to Doxorubicin-induced hPSC-pacemaker cell death and a higher risk of arrhythmia in patients (114), which justifies to perform cardiotoxicity testing on pacemaker cells. Previously, we and others have shown efficient production of atrial, ventricular and pacemaker cardiomyocytes from hPSCs (115–118).
In the cardiac microenvironment in vivo, each cardiomyocyte is surrounded by 3–4 capillaries with a distance between each cardiomyocyte and an endothelial cell of about 1 μm (119). This delicate build-up allows for defined cardiomyocyte-endothelium crosstalk via paracrine signaling and cell-cell contact (119–122). This data suggests that the crosstalk of human cardiomyocytes with highly metabolic active cardiac endothelial cells is very important. Recent advances in micro-engineering and stem cell technologies enabled development of microfluidic devices in which living cells (mostly several cell-types) are cultured in channels or small compartments and perfused in a controlled manner, mimicking the microenvironment and responses of tissues or organs (123–125). These so-called organ-on-chips can be combined with integrated sensors (for example for biochemical and electrical readouts) and are compatible with live imaging (for example using stem cell-based fluorescent reporter lines) (126) and human cell and tissue sampling. Very recently, Weng et al. (127) generated a multiple chambered tissue chip, a so-called organ-on-a-chip, which allowed simultaneous culturing of tumor tissue and hPSC-cardiomyocytes while being connected via a microfluidic channel, lined with endothelial cells. This design enables simultaneous testing of potential anti-tumor effects and cardiotoxicity of drugs administered via the endothelial layer. Another important aspect of drug efficacy and safety assessment is to predict pharmacokinetic parameters, determined by absorption, distribution, metabolism and excretion (ADME) of drugs, which are difficult to measure or model in vitro. As mentioned before, organ-on-chips mimic organ-like function and responses in perfusable mircophysiological systems and may offer an opportunity to overcome current limitations related to pharmacokinetic modeling. Multiple organ-on-chip devices with organ-level functionality could be connected to each other with vascularized, endothelium-lined channels mimicking blood circulation and recapitulating tissue-tissue interfaces, thus modeling organ crosstalk and metabolism of compounds in a single system (128, 129). For predictive physiologically based pharmacokinetic (PBPK) modeling, it is of paramount importance to combine defined hPSC culture methods and 3D tissue engineering or organoid formation with technical advances in the field of organ-on-chip technology, ultimately leading to interconnected multiple human organ-on-chip platforms (for example, combining heart-, liver- and gut-on-chip models).
Future Outlook
Because of the previously mentioned shortcomings, a full recapitulation of clinical cardiotoxic manifestations is beyond the capacity of current in vitro models. Interestingly, recent developments have shown that it is feasible to build human in vitro models that are more closely resembling functional human hearts for assessment of clinically relevant parameters, which may greatly enhance the predictability of these in vitro models for safety pharmacology and disease phenotypes. Recently, Li et al. created for the first time a human ventricular-like cardiac organoid chamber (hvCOC) with hPSC-cardiomyocytes and dermal fibroblasts (130). Similarly, Macqueen and colleagues generated a one cell layer thick ventricular-like tube from hPSC-cardiomyocytes (131). Both HvCOCs and ventricular tubes were able to pump fluid, which allowed measuring ejection fraction and pressure-volume loops as readouts for cardiac function. Although these developments are promising, combination with other cardiac cells, such as endothelial and smooth muscle cells and fibroblasts, may lead to further maturation and improvement of function. Future studies will be required in order to evaluate the predictive values of these human advanced 3D cardiac models in preclinical drug testing, their potential to reduce time for bringing new drugs from bench-to-bedside and repurposing of drugs.
Moreover, state-of-the-art multiple organ-on-chip platforms will also advance predictability of efficacy and toxicity of combinatorial drug (cardioprotective) treatment and specific target delivery.
It is evident that interpatient variability further complicates development of predictive in vitro models for drug screening. This is also true for anthracycline-induced cardiotoxicity, which varies tremendously from patient to patient. These variations may be caused due to environmental factors, life-style or genetic variance. Patient-derived (or genetically modified) hPSCs provide the possibility to include disease-associated genetic risk factors in these in vitro models, which can be directly compared to isogenic control hPSC lines. Interestingly, studies with hPSC-cardiomyocytes have shown that cardiotoxic effects of Doxorubicin were dependent on the genetic background of patient-derived hPSC-cardiomyocytes (22). Similarly, characterization of hPSC-cardiomyocytes from 45 individuals showed inter-individual variation in transcriptional response after 24 h exposure to different concentrations of Doxorubicin, which were predictive of in vitro cell damage as measured by cardiac troponin release of cardiomyocytes (132). These findings highlight the importance of personalized medicine and risk stratification, which will allow us to predict for which patients new therapies will be effective and safe and for which not. This will reduce the attrition rate, costs and time of drug development and facilitate repurposing of drugs that have shown a lack of efficacy or risk of toxicity. Consequently, successful implementation of these advanced hPSC-based models will lead to better and safer drugs.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by ERA-CVD 2016T092 and the Dutch Heart Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Christian B. Schwach for illustrations.
References
1. Šimunek T, Štěrba M, Popelová O, Adamcová M, Hrdina R, Gerši V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. (2009) 61:154–71. doi: 10.1016/S1734-1140(09)70018-0
2. Albini A, Pennesi G, Donatelli F, Cammarota R, De Flora S, Noonan DM. Cardiotoxicity of anticancer drugs: The need for cardio-oncology and cardio-oncological prevention. J Natl Cancer Inst. (2010) 102:14–25. doi: 10.1093/jnci/djp440
3. Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. (2010) 115:155–62. doi: 10.1159/000265166
4. Štěrba M, Popelová O, Vávrová A, Jirkovský E, Kovaríková P, Geršl V, et al. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal. (2013) 18:899–929. doi: 10.1089/ars.2012.4795
5. Franco VI, Lipshultz SE. Cardiac complications in childhood cancer survivors treated with anthracyclines. Cardiol Young. (2015) 25:107–16. doi: 10.1017/S1047951115000906
6. Kankeu C, Clarke K, Passante E, Huber HJ. Doxorubicin-induced chronic dilated cardiomyopathy—the apoptosis hypothesis revisited. J Mol Med. (2017) 95:239–48. doi: 10.1007/s00109-016-1494-0
7. Dong J, Chen H. Cardiotoxicity of anticancer therapeutics. Front Cardiovasc Med. (2018) 5:9. doi: 10.3389/fcvm.2018.00009
8. Volkova M, Russell R. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. (2012) 7:214–20. doi: 10.2174/157340311799960645
9. Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. (1973) 32:302–14. doi: 10.1002/1097-0142(197308)32:2<302::AID-CNCR2820320205>3.0.CO;2-2
10. Rinehart JJ, Lewis RP, Balcerzak SP, Riehart JJ, Lewis RP, Balcerzak SP. Adriamycin cardiotoxicity in man. Ann Intern Med. (1974) 81:475–8. doi: 10.7326/0003-4819-81-4-475
11. Moreno L, Pearson ADJ. How can attrition rates be reduced in cancer drug discovery? Expert Opin Drug Discov. (2013) 8:363–8. doi: 10.1517/17460441.2013.768984
12. Devalla HD, Passier R. Cardiac differentiation of pluripotent stem cells and implications for modeling the heart in health and disease. Sci Transl Med. (2018) 10:1–14. doi: 10.1126/scitranslmed.aah5457
13. Colatsky T, Fermini B, Gintant G, Pierson JB, Sager P, Sekino Y, et al. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative — Update on progress. J Pharmacol Toxicol Methods. (2016) 81:15–20. doi: 10.1016/j.vascn.2016.06.002
14. Maillet A, Tan K, Chai X, Sadananda SN, Mehta A, Ooi J, et al. Modeling doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci Rep. (2016) 6:25333. doi: 10.1038/srep25333
15. Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep. (2017) 7:44735. doi: 10.1038/srep44735
16. Nebigil CG, Désaubry L. Updates in anthracycline-mediated cardiotoxicity. Front Pharmacol. (2018) 9:1–13. doi: 10.3389/fphar.2018.01262
17. Arena E, D'Alessandro N, Dusonchet L, Geraci M, Rausa L, Sanguedolce R. Repair kinetics of DNA, RNA and proteins in the tissues of mice treated with doxorubicin. Arzneimittelforschung. (1979) 29:901−2.
18. Abou El Hassan MAI, Verheul HMW, Jorna AS, Schalkwijk C, Van Bezu J, Van Der Vijgh WJF, et al. The new cardioprotector Monohydroxyethylrutoside protects against doxorubicin-induced inflammatory effects in vitro. Br J Cancer. (2003) 89:357–62. doi: 10.1038/sj.bjc.6601022
19. Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
20. Cappetta D, De Angelis A, Sapio L, Prezioso L, Illiano M, Quaini F, et al. Oxidative stress and cellular response to doxorubicin: a common factor in the complex milieu of anthracycline cardiotoxicity. Oxid Med Cell Longev. (2017) 2017:1–13. doi: 10.1155/2017/1521020
21. Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol. (2017) 5:90. doi: 10.3389/fcell.2017.00090
22. Burridge PW, Li YF, Matsa E, Wu H, Ong S-G, Sharma A, et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. (2016) 22:547–56. doi: 10.1038/nm.4087
23. Chaudhari U, Ellis JK, Wagh V, Nemade H, Hescheler J, Keun HC, et al. Metabolite signatures of doxorubicin induced toxicity in human induced pluripotent stem cell-derived cardiomyocytes. Amino Acids. (2017) 49:1955–63. doi: 10.1007/s00726-017-2419-0
24. Louisse J, Wüst RCI, Pistollato F, Palosaari T, Barilari M, Macko P, et al. Assessment of acute and chronic toxicity of doxorubicin in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Vitr. (2017) 42:182–90. doi: 10.1016/j.tiv.2017.04.023
25. Verheijen M, Schrooders Y, Gmuender H, Nudischer R, Clayton O, Hynes J, et al. Bringing in vitro analysis closer to in vivo: Studying doxorubicin toxicity and associated mechanisms in 3D human microtissues with PBPK-based dose modelling. Toxicol Lett. (2018) 294:184–92. doi: 10.1016/j.toxlet.2018.05.029
26. Holmgren G, Sartipy P, Andersson CX, Lindahl A, Synnergren J. Expression profiling of human pluripotent stem cell-derived cardiomyocytes exposed to doxorubicin-Integration and visualization of multi-omics data. Toxicol Sci. (2018) 163:182–95. doi: 10.1093/toxsci/kfy012
27. McSweeney KM, Bozza WP, Alterovitz WL, Zhang B. Transcriptomic profiling reveals p53 as a key regulator of doxorubicin-induced cardiotoxicity. Cell Death Discov. (2019) 5:1–11. doi: 10.1038/s41420-019-0182-6
28. Li J, Wang PY, Long NA, Zhuang J, Springer DA, Zou J, et al. P53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc Natl Acad Sci USA. (2019) 116:19626–34. doi: 10.1073/pnas.1904979116
29. Gupta SK, Garg A, Bär C, Chatterjee S, Foinquinos A, Milting H, et al. Quaking inhibits doxorubicin-mediated cardiotoxicity through regulation of cardiac circular RNA expression short communication. Circ Res. (2018) 122:246–54. doi: 10.1161/CIRCRESAHA.117.311335
30. Weiss RB, Sarosy G, Clagett-Carr K, Russo M, Leyland-Jones B. Anthracycline analogs: the past, present, and future. Cancer Chemother Pharmacol. (1986) 18:185–97. doi: 10.1007/bf00273384
31. Ambrosini G, Balli M, Garusi G, Demicheli R, Jirillo A, Bonciarelli G, et al. Phase III randomized study of fluorouracil, epirubicin, and cyclophosphamide v fluorouracil, doxorubicin, and cyclophosphamide in advanced breast cancer: an Italian multicentre trial. Italian Multicentre Breast Study with Epirubicin. J Clin Oncol. (1988) 6:976–82. doi: 10.1200/JCO.1988.6.6.976
32. Dittrich C, Baur M, Mader R, Schlappack O, Dudczak R, Leitha T, et al. Phase I-II study on weekly administration of pirarubicin in patients with metastatic breast cancer. Am J Clin Oncol Cancer Clin Trials. (1990) 13:S29–39. doi: 10.1097/00000421-199012001-00008
33. Drings P, Günther IU, Gatzemeier U, Berdel W, Stahl M, Salewski E, et al. Pirarubicin in advanced non-small cell lung cancer: a trial of the phase i/ii study group of the association for medical oncology of the german cancer society. Oncol Res Treat. (1990) 13:180–4. doi: 10.1159/000216754
34. Dhingra K, Frye D, Newman RA, Walters R, Theriault R, Fraschini G, et al. Phase II clinical and pharmacological study of pirarubicin in combination with 5-fluorouracil and cyclophosphamide in metastatic breast cancer. Clin Cancer Res. (1995) 1:691–7.
35. Zinzani PL, Martelli M, Storti S, Musso M, Cantonetti M, Leone G, et al. Phase III comparative trial using CHOP vs CIOP in the treatment of advanced intermediate-grade non-Hodgkin's lymphoma. Leuk Lymphoma. (1995) 19:329–35. doi: 10.3109/10428199509107906
36. Llombart-Cussac A, Pivot X, Rhor-Alvarado A, Le Cesne A, Le Chevalier T, Tursz T, et al. First-line vinorelbine-mitoxantrone combination in metastatic breast cancer patients relapsing after an adjuvant anthracycline regimen: results of a phase II study. Oncology. (1998) 55:384–90. doi: 10.1159/000011883
37. Nielsen OS, Dombernowsky P, Mouridsen H, Crowther D, Verweij J, Buesa J, et al. High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer. (1998) 78:1634–9. doi: 10.1038/bjc.1998.735
38. Srokowski TP, Liebmann JE, Modiano MR, Cohen GI, Pro B, Romaguera JE, et al. Pixantrone dimaleate in combination with fludarabine, dexamethasone, and rituximab in patients with relapsed or refractory indolent non-Hodgkin lymphoma. Cancer. (2011) 117:5067–73. doi: 10.1002/cncr.26121
39. Rivankar S. An overview of doxorubicin formulations in cancer therapy. J Cancer Res Ther. (2014) 10:853–8. doi: 10.4103/0973-1482.139267
40. Xue K, Gu JJ, Zhang Q, Liu X, Wang J, Li XQ, et al. Cardiotoxicity as indicated by LVEF and troponin T sensitivity following two anthracycline-based regimens in lymphoma: Results from a randomized prospective clinical trial. Oncotarget. (2016) 7:32519–31. doi: 10.18632/oncotarget.8685
41. Hara T, Yoshikawa T, Goto H, Sawada M, Yamada T, Fukuno K, et al. R-THP-COP versus R-CHOP in patients younger than 70 years with untreated diffuse large B cell lymphoma: a randomized, open-label, noninferiority phase 3 trial. Hematol Oncol. (2018) 36:638–44. doi: 10.1002/hon.2524
42. Xu PP, Fu D, Li J-Y, Hu J-D, Wang X, Zhou J-F, et al. Anthracycline dose optimisation in patients with diffuse large B-cell lymphoma: a multicentre, phase 3, randomised, controlled trial. Lancet Haematol. (2019) 6:e328–37. doi: 10.1016/S2352-3026(19)30051-1
43. Kang YK, Ryoo BY, Kim TY, Im YH, Kim BS, Park YH, et al. A phase II trial of DA-125, a novel anthracycline, in advanced non- small-cell lung cancer. Cancer Chemother Pharmacol. (1999) 44:518–21. doi: 10.1007/s002800051127
44. Obasaju C, Manola J, Hudes GR, Khandekar JD, Citrin DL, Carbone P, et al. Phase II evaluation of menogaril in advanced prostate cancer: Eastern Cooperative Oncology Group EST P-A885. Am J Clin Oncol. (2001) 24:150–4. doi: 10.1097/00000421-200104000-00011
45. Hofheinz RD, Porta C, Hartung G, Santoro A, Hanauske AR, Kutz K, et al. BBR 3438, a novel 9-aza-anthrapyrazole, in patients with advanced gastric cancer: A phase II study group trial of the central European Society of Anticancer-Drug Research (CESAR). Invest New Drugs. (2005) 23:363–8. doi: 10.1007/s10637-005-1445-z
46. Tardi PG, Boman NL, Cullis PR. Liposomal doxorubicin. J Drug Target. (1996) 4:129–40. doi: 10.3109/10611869609015970
47. Reynolds JG, Geretti E, Hendriks BS, Lee H, Leonard SC, Klinz SG, et al. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol Appl Pharmacol. (2012) 262:1–10. doi: 10.1016/j.taap.2012.04.008
48. Giotta F, Lorusso V, Maiello E, Filippelli G, Valerio MR, Caruso M, et al. Liposomal-encapsulated doxorubicin plus cyclophosphamide as first-line therapy in metastatic breast cancer: a phase II multicentric study. Ann Oncol. (2007) 18(Suppl. 6):vi66–9. doi: 10.1093/annonc/mdm228
49. Xing M, Yan F, Yu S, Shen P. Efficacy and cardiotoxicity of liposomal doxorubicin-based chemotherapy in advanced breast cancer: a meta-analysis of ten randomized controlled trials. PLoS ONE. (2015) 10:e0133569. doi: 10.1371/journal.pone.0133569
50. Fridrik MA, Jaeger U, Petzer A, Willenbacher W, Keil F, Lang A, et al. Cardiotoxicity with rituximab, cyclophosphamide, non-pegylated liposomal doxorubicin, vincristine and prednisolone compared to rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone in frontline treatment of patients with diffuse large B-. Eur J Cancer. (2016) 58:112–21. doi: 10.1016/J.EJCA.2016.02.004
51. Chan S, Davidson N, Juozaityte E, Erdkamp F, Pluzanska A, Azarnia N, et al. Phase III trial of liposomal doxorubicin and cyclophosphamide compared with epirubicin and cyclophosphamide as first-line therapy for metastatic breast cancer. Ann Oncol. (2004) 15:1527–34. doi: 10.1093/annonc/mdh393
52. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. (2003) 42:419–36. doi: 10.2165/00003088-200342050-00002
53. Lyass O, Uziely B, Ben-Yosef R, Tzemach D, Heshing NI, Lotem M, et al. Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma. Cancer. (2000) 89:1037–47. doi: 10.1002/1097-0142(20000901)89:5<1037::aid-cncr13>3.0.co;2-z
54. O'Brien MER. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYXTM/Doxil") versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. (2004) 15:440–9. doi: 10.1093/annonc/mdh097
55. Gil-Gil MJ, Bellet M, Morales S, Ojeda B, Manso L, Mesia C, et al. Pegylated liposomal doxorubicin plus cyclophosphamide followed by paclitaxel as primary chemotherapy in elderly or cardiotoxicity-prone patients with high-risk breast cancer: results of the phase II CAPRICE study. Breast Cancer Res Treat. (2015) 151:597–606. doi: 10.1007/s10549-015-3415-2
56. Ansari L, Shiehzadeh F, Taherzadeh Z, Nikoofal-Sahlabadi S, Momtazi-borojeni AA, Sahebkar A, et al. The most prevalent side effects of pegylated liposomal doxorubicin monotherapy in women with metastatic breast cancer: a systematic review of clinical trials. Cancer Gene Ther. (2017) 24:189–93. doi: 10.1038/cgt.2017.9
57. Miller K, Cortes J, Hurvitz SA, Krop IE, Tripathy D, Verma S, et al. HERMIONE: a randomized Phase 2 trial of MM-302 plus trastuzumab versus chemotherapy of physician's choice plus trastuzumab in patients with previously treated, anthracycline-naïve, HER2-positive, locally advanced/metastatic breast cancer. BMC Cancer. (2016) 16:352. doi: 10.1186/s12885-016-2385-z
58. Karimi M, Zangabad PS, Mehdizadeh F, Malekzad H, Ghasemi A, Bahrami S, et al. Nanocaged platforms: modification, drug delivery and nanotoxicity. Opening synthetic cages to release the tiger. Nanoscale. (2017) 9:1356–92. doi: 10.1039/c6nr07315h
59. Zhen Z, Tang W, Chen H, Lin X, Todd T, Wang G, et al. RGD modified apoferritin nanoparticles for efficient drug delivery to tumors NIH Public Access. ACS Nano. (2013) 7:4830–7. doi: 10.1021/nn305791q
60. Du B, Jia S, Wang Q, Ding X, Liu Y, Yao H, et al. A self-targeting, Dual ROS/pH-responsive apoferritin nanocage for spatiotemporally controlled drug delivery to breast cancer. Biomacromolecules. (2018) 19:1026–36. doi: 10.1021/acs.biomac.8b00012
61. Han X, Jiang Y, Li S, Zhang Y, Ma X, Wu Z, et al. Multivalent aptamer-modified tetrahedral DNA nanocage demonstrates high selectivity and safety for anti-tumor therapy. Nanoscale. (2018) 11:339–47. doi: 10.1039/c8nr05546g
62. Ji M, Qiu X, Hou L, Huang S, Li Y, Liu Y, et al. Construction and application of a liver cancer-targeting drug delivery system based on core-shell gold nanocages. Int J Nanomed. (2018) 13:1773–89. doi: 10.2147/IJN.S151043
63. Xu Q, Wan J, Bie N, Song X, Yang X, Yong T, et al. A biomimetic gold nanocages-based nanoplatform for efficient tumor ablation and reduced inflammation. Theranostics. (2018) 8:5362–78. doi: 10.7150/thno.27631
64. Schöffski P, Delord JPP, Brain E, Robert J, Dumez H, Gasmi J, et al. First-in-man phase I study assessing the safety and pharmacokinetics of a 1-hour intravenous infusion of the doxorubicin prodrug DTS-201 every 3 weeks in patients with advanced or metastatic solid tumours. Eur J Cancer. (2017) 86:240–7. doi: 10.1016/j.ejca.2017.09.009
65. Kuruvilla SP, Tiruchinapally G, Crouch AC, ElSayed MEH, Greve JM. Dendrimer-doxorubicin conjugates exhibit improved anticancer activity and reduce doxorubicin-induced cardiotoxicity in a murine hepatocellular carcinoma model. PLoS ONE. (2017) 12:1–24. doi: 10.1371/journal.pone.0181944
66. Georgakopoulos P, Roussou P, Matsakas E, Karavidas A, Anagnostopoulos N, Marinakis T, et al. Cardioprotective effect of metoprolol and enalapril in doxorubicin-treated lymphoma patients: a prospective, parallel-group, randomized, controlled study with 36-month follow-up. Am J Hematol. (2010) 85:894–6. doi: 10.1002/ajh.21840
67. Zhang H, Shen WS, Gao CH, Deng LC, Shen D. Protective effects of salidroside on epirubicin-induced early left ventricular regional systolic dysfunction in patients with breast cancer. Drugs R D. (2012) 12:101–6. doi: 10.2165/11632530-000000000-00000
68. Seicean S, Seicean A, Plana JC, Budd GT, Marwick TH. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. J Am Coll Cardiol. (2012) 60:2384–90. doi: 10.1016/j.jacc.2012.07.067
69. Chotenimitkhun R, D'Agostino R, Lawrence JA, Hamilton CA, Jordan JH, Vasu S, et al. Chronic statin administration may attenuate early anthracycline-associated declines in left ventricular ejection function. Can J Cardiol. (2015) 31:302–7. doi: 10.1016/j.cjca.2014.11.020
70. Tashakori Beheshti A, Mostafavi Toroghi H, Hosseini G, Zarifian A, Homaei Shandiz F, Fazlinezhad A. Carvedilol administration can prevent doxorubicin-induced cardiotoxicity: a double-blind randomized trial. Cardiology. (2016) 134:47–53. doi: 10.1159/000442722
71. Hao W, Liu S, Qin Y, Sun C, Chen L, Wu C, et al. Cardioprotective effect of Platycodon grandiflorum in patients with early breast cancer receiving anthracycline-based chemotherapy: study protocol for a randomized controlled trial. Trials. (2017) 18:386. doi: 10.1186/s13063-017-2140-z
72. Janbabai G, Nabati M, Faghihinia M, Azizi S, Borhani S, Yazdani J. Effect of enalapril on preventing anthracycline-induced cardiomyopathy. Cardiovasc Toxicol. (2017) 17:130–9. doi: 10.1007/s12012-016-9365-z
73. Nabati M, Janbabai G, Baghyari S, Esmaili K, Yazdani J. Cardioprotective effects of carvedilol in inhibiting doxorubicin-induced cardiotoxicity. J Cardiovasc Pharmacol. (2017) 69:279–85. doi: 10.1097/FJC.0000000000000470
74. Skrypnyk I, Maslova G, Lymanets T, Gusachenko I. L-arginine is an effective medication for prevention of endothelial dysfunction, a predictor of anthracycline cardiotoxicity in patients with acute leukemia. Exp. Oncol. (2017) 39:308–11.
75. Abuosa AM, Elshiekh AH, Qureshi K, Abrar MB, Kholeif MA, Kinsara AJ, et al. Prophylactic use of carvedilol to prevent ventricular dysfunction in patients with cancer treated with doxorubicin. Indian Heart J. (2018) 70:S96–S100. doi: 10.1016/j.ihj.2018.06.011
76. Cardinale D, Ciceri F, Latini R, Franzosi MG, Sandri MT, Civelli M, et al. Anthracycline-induced cardiotoxicity: a multicenter randomised trial comparing two strategies for guiding prevention with enalapril: The International CardioOncology Society-one trial. Eur J Cancer. (2018) 94:126–37. doi: 10.1016/j.ejca.2018.02.005
77. Gupta V, Kumar Singh S, Agrawal V, Bali Singh T. Role of ACE inhibitors in anthracycline-induced cardiotoxicity: A randomized, double-blind, placebo-controlled trial. Pediatr Blood Cancer. (2018) 65:e27308. doi: 10.1002/pbc.27308
78. Chung R, Maulik A, Hamarneh A, Hochhauser D, Hausenloy DJ, Walker JM, et al. Effect of remote ischaemic conditioning in oncology patients undergoing chemotherapy: rationale and design of the ERIC-ONC Study - a single-center, blinded, randomized controlled trial. Clin Cardiol. (2016) 39:72–82. doi: 10.1002/clc.22507
79. Kirkham AA, Paterson DI, Prado CM, Mackey JM, Courneya KS, Pituskin E, et al. Rationale and design of the Caloric Restriction and Exercise protection from Anthracycline Toxic Effects (CREATE) study: A 3-arm parallel group phase II randomized controlled trial in early breast cancer. BMC Cancer. (2018) 18:864. doi: 10.1186/s12885-018-4778-7
80. Milei J, Marantz A, Alé J, Vazquez A, Buceta JE. Prevention of adriamycin-induced cardiotoxicity by prenylamine: a pilot double blind study. Cancer Drug Deliv. (1987) 4:129–36. doi: 10.1089/cdd.1987.4.129
81. Shah RR, Stonier PD. Withdrawal of prenylamine: perspectives on pharmacological, clinical and regulatory outcomes following the first QT-related casualty. Ther Adv Drug Saf. (2018) 9:475–93. doi: 10.1177/2042098618780854
82. Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. (2006) 7:861–79. doi: 10.2174/138945006777709593
83. Stove C, Bracke M. Roles for neuregulins in human cancer. Clin Exp Metastasis. (2005) 21:665–84. doi: 10.1007/s10585-004-6917-6
84. Gao R, Zhang J, Cheng L, Wu X, Dong W, Yang X, et al. A phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol. (2010) 55:1907–14. doi: 10.1016/j.jacc.2009.12.044
85. Galindo CL, Ryzhov S, Sawyer DB. Neuregulin as a heart failure therapy and mediator of reverse remodeling. Curr Heart Fail Rep. (2014) 11:40–9. doi: 10.1007/s11897-013-0176-2
86. Lenihan DJ, Koren M, Eisen A, Caggiano A. Sustained improvement of left ventricular ejection fraction with intravenous cimaglermin in patients with symptomatic systolic dysfunction. J Am Coll Cardiol. (2018) 71:A667. doi: 10.1016/s0735-1097(18)31208-7
87. Jay SM, Murthy AC, Hawkins JF, Wortzel JR, Steinhauser ML, Alvarez LM, et al. An engineered bivalent neuregulin protects against doxorubicin-induced cardiotoxicity with reduced proneoplastic potential. Circulation. (2013) 128:152–61. doi: 10.1161/CIRCULATIONAHA.113.002203
88. Zhang J, Cui X, Yan Y, Li M, Yang Y, Wang J, et al. Research progress of cardioprotective agents for prevention of anthracycline cardiotoxicity. Am J Transl Res. (2016) 8:2862–75.
89. Lipshultz SE, Rifai N, Dalton VM, Levy DE, Silverman LB, Lipsitz SR, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. (2004) 351:145–53. doi: 10.1056/NEJMoa035153
90. Asselin BL, Devidas M, Chen L, Franco VI, Pullen J, Borowitz MJ, et al. Cardioprotection and safety of dexrazoxane in patients treated for newly diagnosed t-cell acute lymphoblastic leukemia or advanced-stage lymphoblastic non-hodgkin lymphoma: a Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol. (2016) 34:854–62. doi: 10.1200/JCO.2015.60.8851
91. Lemez P, Maresová J. Efficacy of dexrazoxane as a cardioprotective agent in patients receiving mitoxantrone- and daunorubicin-based chemotherapy. Semin Oncol. (1998) 25:61–5.
92. Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, Spicer D, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol. (1997) 15:1318–32. doi: 10.1200/JCO.1997.15.4.1318
93. Marty M, Espié M, Llombart A, Monnier A, Rapoport BL, Stahalova V. Multicenter randomized phase III study of the cardioprotective effect of dexrazoxane (Cardioxane®) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann Oncol. (2006) 17:614–22. doi: 10.1093/annonc/mdj134
94. Elbl L, Hrstkova H, Tomaskova I, Michalek J. Late anthracycline cardiotoxicity protection by dexrazoxane (ICRF-187) in pediatric patients: echocardiographic follow-up. Support Care Cancer. (2006) 14:128–36. doi: 10.1007/s00520-005-0858-8
95. Cui N, Wu F, Lu WJ, Bai R, Ke B, Liu T, et al. Doxorubicin-induced cardiotoxicity is maturation dependent due to the shift from topoisomerase IIα to IIβ in human stem cell derived cardiomyocytes. J Cell Mol Med. (2019) 23:4627–39. doi: 10.1111/jcmm.14346
96. Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Gödtel-Armbrust U, et al. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both Topoisomerase II isoforms. BMC Cancer. (2014) 14:1–11. doi: 10.1186/1471-2407-14-842
97. Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. (2007) 7:332–44. doi: 10.1038/nrc2106
98. Doherty KR, Wappel RL, Talbert DR, Trusk PB, Moran DM, Kramer JW, et al. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol Appl Pharmacol. (2013) 272:245–55. doi: 10.1016/j.taap.2013.04.027
99. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. (2017) 9:1–13. doi: 10.1126/scitranslmed.aaf2584
100. Li C, Zou R, Zhang H, Wang Y, Qiu B, Qiu S, et al. Upregulation of phosphoinositide 3-kinase prevents sunitinib-induced cardiotoxicity in vitro and in vivo. Arch Toxicol. (2019) 93:1697–712. doi: 10.1007/s00204-019-02448-z
101. Wang H, Sheehan RP, Palmer AC, Everley RA, Boswell SA, Ron-Harel N, et al. Adaptation of human iPSC-derived cardiomyocytes to tyrosine kinase inhibitors reduces acute cardiotoxicity via metabolic reprogramming. Cell Syst. (2019) 8:412–26.e7. doi: 10.1016/j.cels.2019.03.009
102. Kitani T, Ong SG, Lam CK, Rhee JW, Zhang JZ, Oikonomopoulos A, et al. Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation. (2019) 139:2451–65. doi: 10.1161/CIRCULATIONAHA.118.037357
103. Eldridge S, Guo L, Mussio J, Furniss M, Hamre J, Davis M. Examining the protective role of ErbB2 modulation in human-induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci. (2014) 141:547–59. doi: 10.1093/toxsci/kfu150
104. Kurokawa YK, Shang MR, Yin RT, George SC. Modeling trastuzumab-related cardiotoxicity in vitro using human stem cell-derived cardiomyocytes. Toxicol Lett. (2018) 285:74–80. doi: 10.1016/j.toxlet.2018.01.001
105. Veerman CC, Kosmidis G, Mummery CL, Casini S, Verkerk AO, Bellin M. Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev. (2015) 24:1035–52. doi: 10.1089/scd.2014.0533
106. Denning C, Borgdorff V, Crutchley J, Firth KSA, George V, Kalra S, et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim Biophys Acta. (2016) 1863:1728–48. doi: 10.1016/j.bbamcr.2015.10.014
107. Schwach V, Passier R. Native cardiac environment and its impact on engineering cardiac tissue. Biomater Sci. (2019) 7:3566–80. doi: 10.1039/c8bm01348a
108. Hansen A, Eder A, Bönstrup M, Flato M, Mewe M, Schaaf S, et al. Development of a drug screening platform based on engineered heart tissue. Circ Res. (2010) 107:35–44. doi: 10.1161/CIRCRESAHA.109.211458
109. Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Börnchen C, Müller C, et al. Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol. (2014) 74:151–61. doi: 10.1016/j.yjmcc.2014.05.009
110. Wijnker PJM, Friedrich FW, Dutsch A, Reischmann S, Eder A, Mannhardt I, et al. Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue. J Mol Cell Cardiol. (2016) 97:82–92. doi: 10.1016/j.yjmcc.2016.03.003
111. Truitt R, Mu A, Corbin EA, Vite A, Brandimarto J, Ky B, et al. Increased Afterload Augments Sunitinib-Induced Cardiotoxicity in an engineered cardiac microtissue model. JACC Basic to Transl Sci. (2018) 3:265–76. doi: 10.1016/j.jacbts.2017.12.007
112. Kilickap S, Barista I, Akgul E, Aytemir K, Aksoy S, Tekuzman G. Early and late arrhythmogenic effects of doxorubicin. South Med J. (2007) 100:262–5. doi: 10.1097/01.smj.0000257382.89910.fe
113. Buza V, Rajagopalan B, Curtis AB. Cancer treatment-induced arrhythmias: focus on chemotherapy and targeted therapies. Circ Arrhythmia Electrophysiol. (2017) 10:1–12. doi: 10.1161/CIRCEP.117.005443
114. Chen S. Pharmacogenetics and drug discovery for anthracycline-induced cardiotoxicity enabled by sinoatrial node-like cells derived from human pluripotent stem cells. Circ Res. (2019) doi: 10.1161/res.125.suppl_1.476. [Epub ahead of print].
115. Birket MJ, Ribeiro MC, Verkerk AO, Ward D, Leitoguinho AR, Den Hartogh SC, et al. Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nat Biotechnol. (2015) 33:970–9. doi: 10.1038/nbt.3271
116. Devalla HD, Schwach V, Ford JW, Milnes JT, El-Haou S, Jackson C, et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol Med. (2015) 7:394–410. doi: 10.15252/emmm.201404757
117. Protze SI, Liu J, Nussinovitch U, Ohana L, Backx PH, Gepstein L, et al. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat Biotechnol. (2016) 351:35–56. doi: 10.1038/nbt.3745
118. Schwach V, Passier R. Generation and purification of human stem cell-derived cardiomyocytes. Differentiation. (2016) 91:126–38. doi: 10.1016/j.diff.2016.01.001
119. Strijdom H, Lochner A. Cardiac endothelium: more than just a barrier. SA Hear. (2017) 2:174–85. doi: 10.24170/6-3-1987
120. Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK, et al. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol. (2011) 51:803–11. doi: 10.1016/j.yjmcc.2011.06.026
121. Talman V, Kivelä R. Cardiomyocyte—endothelial cell interactions in cardiac remodeling and regeneration. Front Cardiovasc Med. (2018) 5:101. doi: 10.3389/fcvm.2018.00101
122. Colliva A, Braga L, Giacca M, Zacchigna S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J Physiol. (2019). doi: 10.1113/JP276758. [Epub ahead of print].
123. Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. (2010) 328:1662–8. doi: 10.1126/science.1188302
124. Passier R, Orlova V, Mummery C. Complex tissue and disease modeling using hiPSCs. Cell Stem Cell. (2016) 18:309–21. doi: 10.1016/j.stem.2016.02.011
125. Van Den Berg A, Mummery CL, Passier R, Van der Meer AD. Personalised organs-on-chips: functional testing for precision medicine. Lab Chip. (2019) 19:198–205. doi: 10.1039/c8lc00827b
126. Den Hartogh SC, Passier R. Concise review: fluorescent reporters in human pluripotent stem cells: contributions to cardiac differentiation and their applications in Cardiac Disease and Toxicity. Stem Cells. (2016) 34:13–26. doi: 10.1002/stem.2196
127. Weng C, Kurokawa YK, Hajek BS, Paladin JA, Shirure VS, George SC. Human iPS-cardiac-endothelial-tumor-on-a-chip to assess anti-cancer efficacy and cardiotoxicity. Mary Ann Liebert. (2019) 26:1–40. doi: 10.1089/ten.TEC.2019.0248
128. Skardal A, Shupe T, Atala A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov Today. (2016) 21:1399–411. doi: 10.1016/j.drudis.2016.07.003
129. Prantil-Baun R, Novak R, Das D, Somayaji MR, Przekwas A, Ingber DE. Physiologically based pharmacokinetic and pharmacodynamic analysis enabled by microfluidically linked organs-on-chips. Annu Rev Pharmacol Toxicol. (2018) 58:37–64. doi: 10.1146/annurev-pharmtox-010716-104748
130. Li RA, Keung W, Cashman TJ, Backeris PC, Johnson BV, Bardot ES, et al. Bioengineering an electro-mechanically functional miniature ventricular heart chamber from human pluripotent stem cells. Biomaterials. (2018) 163:116–27. doi: 10.1016/j.biomaterials.2018.02.024
131. MacQueen LA, Sheehy SP, Chantre CO, Zimmerman JF, Pasqualini FS, Liu X, et al. A tissue-engineered scale model of the heart ventricle. Nat Biomed Eng. (2018) 2:930–41. doi: 10.1038/s41551-018-0271-5
Keywords: human pluripotent stem cells, cardiomyocytes, cardio-oncology, organ-on-chip, cardiotoxicity
Citation: Schwach V, Slaats RH and Passier R (2020) Human Pluripotent Stem Cell-Derived Cardiomyocytes for Assessment of Anticancer Drug-Induced Cardiotoxicity. Front. Cardiovasc. Med. 7:50. doi: 10.3389/fcvm.2020.00050
Received: 28 December 2019; Accepted: 16 March 2020;
Published: 08 April 2020.
Edited by:
Canan G. Nebigil, École Supérieure de Biotechnologie Strasbourg, FranceReviewed by:
Fabrice Jaffré, Cornell University, United StatesAlessandra Ghigo, University of Turin, Italy
Copyright © 2020 Schwach, Slaats and Passier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Passier, cm9iZXJ0LnBhc3NpZXJAdXR3ZW50ZS5ubA==