 Warisara Parichatikanond
Warisara Parichatikanond Theerut Luangmonkong1
Theerut Luangmonkong1 Supachoke Mangmool
Supachoke Mangmool Hitoshi Kurose
Hitoshi Kurose
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 10 March 2020
Sec. Cardio-Oncology
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00034
This article is part of the Research Topic Emerging Challenges of Cardiovascular and Metabolic Dysfunctions in Cardio-oncology: From Bench to Bedside View all 11 articles
Transforming growth factor-β (TGF-β) is a common mediator of cancer progression and fibrosis. Fibrosis can be a significant pathology in multiple organs, including the heart. In this review, we explain how inhibitors of TGF-β signaling can work as antifibrotic therapy. After cardiac injury, profibrotic mediators such as TGF-β, angiotensin II, and endothelin-1 simultaneously activate cardiac fibroblasts, resulting in fibroblast proliferation and migration, deposition of extracellular matrix proteins, and myofibroblast differentiation, which ultimately lead to the development of cardiac fibrosis. The consequences of fibrosis include a wide range of cardiac disorders, including contractile dysfunction, distortion of the cardiac structure, cardiac remodeling, and heart failure. Among various molecular contributors, TGF-β and its signaling pathways which play a major role in carcinogenesis are considered master fibrotic mediators. In fact, recently the inhibition of TGF-β signaling pathways using small molecule inhibitors, antibodies, and gene deletion has shown that the progression of several cancer types was suppressed. Therefore, inhibitors of TGF-β signaling are promising targets for the treatment of tissue fibrosis and cancers. In this review, we discuss the molecular mechanisms of TGF-β in the pathogenesis of cardiac fibrosis and cancer. We will review recent in vitro and in vivo evidence regarding antifibrotic and anticancer actions of TGF-β inhibitors. In addition, we also present available clinical data on therapy based on inhibiting TGF-β signaling for the treatment of cancers and cardiac fibrosis.
Transforming growth factor-β (TGF-β) is a crucial member of the TGF-β superfamily and its sophisticated signaling pathways have pleiotropic effects that regulate several systems throughout the body such as cell growth, cell differentiation, apoptosis, motility and invasion, tissue remodeling, angiogenesis, and the immune response (1–6). TGF-β signaling dysfunctions are frequently found in tumors and these dysfunctions play critical roles in tumor progression (e.g., development and metastasis) (7–9). In addition, TGF-β is a major profibrotic mediator that plays an important role in the development of fibrosis (10). Due to the significant implication of TGF-β signaling in cancer as well as in fibrosis (Figure 1), drug research into treatments for cancer and fibrosis has aimed to develop various approaches to inhibit TGF-β signaling. Thus, the number of lead compounds used either in animal models or in clinical studies related to cancer and fibrosis is currently growing. Targeting TGF-β signaling pathways could be a novel therapeutic strategy to treat a variety of fibrotic disorders and cancers.
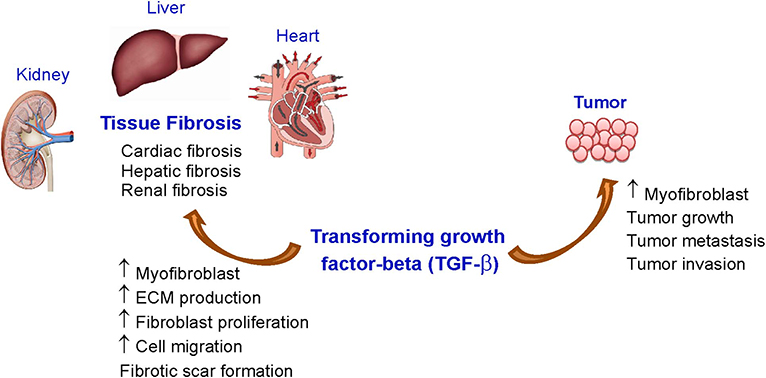
Figure 1. Effects of TGF-β on tissue fibrosis and cancer. ECM, extracellular matrix; TGF-β, transforming growth factor-beta-β.
The synthesis and secretion of TGF-β, including its activity, is markedly increased in experimental models of fibrosis and in patients with tissue fibrosis (e.g., liver, lung, kidney, and heart). Fibrosis is an important pathophysiological phenomenon in many tissues. It is characterized by fibroblast activation and accumulation, an imbalance of extracellular matrix (ECM) production and degradation, and myofibroblast differentiation, which results in the accumulation of fibrotic scar and tissue stiffness, leading to distortions of organ architecture and function [Reviewed in (11, 12)].
Among fibrotic conditions in various organs, cardiac fibrosis is a major pathologic disorder associated with a great number of cardiovascular diseases resulting from an excessive ECM protein deposition in the heart [Reviewed in (11, 12)]. The etiologies of cardiac fibrosis and myocardial stiffness are multifactorially developed in response to multiple risk factors (13, 14) include myocardial infarction (MI), hypertension (15), diabetes (16, 17), aging (16), and excessive alcohol consumptions (18, 19) leading to the excessive deposition of ECM. After cardiac injury, alterations in ECM homeostasis, the upregulation and release of growth factors and cytokines, and differentiation of fibroblasts into myofibroblasts dynamically modulate cardiac fibroblast characteristics and functions, leading to myocardial fibrosis. Myocardial fibrosis is associated with fibrotic scar formation, myocardial stiffness, and the progression of heart failure (HF) (20–23). Treatment of HF and cardiac fibrosis still has limited efficacy and currently there is no drug approved for the treatment of cardiac fibrosis. The main reason is that the underlying mechanism of fibrosis is still unclear. However, cardiovascular diseases remain the leading global cause of death (22, 23) and understanding the pathogenesis of fibrotic myocardial remodeling is crucial to identifying innovative treatment strategies for patients with cardiac fibrosis.
In the heart, activation of cardiac fibroblasts mainly by TGF-β leads to alterations in cardiac ECM and cardiac remodeling that play a major role in the development and progression of heart diseases (10, 22). A significant number of preclinical and clinical studies have reported that inhibition of TGF-β signaling pathways by various strategies exhibited potential effectiveness for the treatment of cardiac fibrosis. Cancers and fibrotic diseases share the most common pathologies associated with the activity of TGF-β (1, 2). Here, we review the molecular mechanisms and signaling pathways of TGF-β and their effect on cancer and cardiac fibrosis, and we also summarize the role of inhibition of TGF-β for anticancer and antifibrotic therapies.
Cancer is defined as a collection of diseases relating to atypical cell growth. In physiological process, new cells can grow, divide, and replace senescent or damaged cells. However, this systemically process fails when cancer develops as aged or injured cells remain survive, together with a proliferation of unneeded new cells. These unnecessary cells can divide, spread, and invade nearby tissues without stopping. Also, the harm cells can possibly travel through the blood or lymph system to invade remote tissues. This atypical cell growth and spreading is known as carcinogenesis (24). Widespread and recognized theory of carcinogenesis is the DNA mutations that disrupt the normal balance between proliferation and cell death. Variants of inherited genes and environmental factors might play a pivotal role in DNA mutations. In addition, viruses containing oncogenes are recently known as a trigger of cancer cell growth (24).
Treatment of cancers can be achieved using several strategies such as surgery, radiation, and especially drugs. Chemotherapy is a conventional treatment by using toxic drugs to kill cancer cells. Beyond fast-growing cancer cells, traditional anticancer drugs using for chemotherapy damage healthy cells that rapidly grow and divide, leading to multiple adverse effects (25). Newer drugs for the treatment of cancers were subsequently developed for a preferable safety issue and prevailing therapeutic efficacy (25). Hormonal therapy is another strategy to cease the growth of cancer which required certain hormones. Due to the blockade, undesired effects of anti-hormone drugs can be seen depending on types of interfered hormone (26, 27). Targeted therapy is a type of cancer treatment using drugs targeting particular molecules required for the pathogenesis of individual cancer. Nevertheless, treated cancer cells can gradually resist to targeted therapy, and conventional chemotherapy might be needed to be co-administered in the regimen for a better outcome (28). Immunotherapy is a novel treatment method by enhancing immune system for eradicating cancer cells. Despite solely activated self-immune cells, overactive immunity against cancer also influences healthy cells and tissues resulting in various adverse effects (29). Described anticancer drug classes and representative drugs among each class are demonstrated in Table 1. However, in-depth review regarding mechanism of drug action, clinical effectiveness, and safety profile of these anticancer drugs are beyond our scope. Furthermore, it should be noted that although anticancer drugs appears to be diverse and abundant, we still need distinct agents to deal with innumerable types of advanced cancers in clinical practice, especially multi-drug resistant cancers (30). Therefore, in this review, we focus on the role of TGF-β and its signaling on the treatment of cancer.
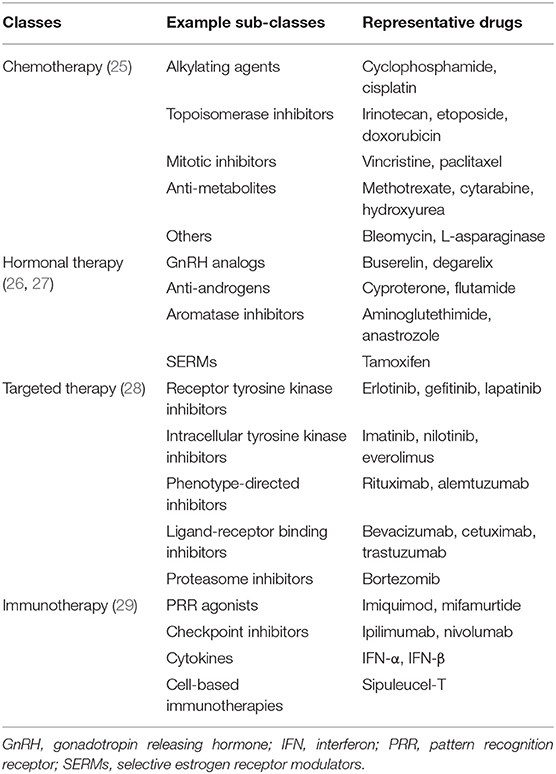
Table 1. Available anticancer drug classes and representative drugs among each class.
Cardiac fibrosis is a pathological remodeling process following cardiac injury, MI, and other heart diseases. Cardiac fibrosis disrupts the communication and function of myocytes and non-myocyte cells in the heart, leading to contractile dysfunction and arrhythmia. Fibrosis also accelerates the remodeling processes that exhibit detrimental effects on the heart (23, 31).
The imbalance between production and degradation of interstitial ECM proteins leads to progressively increased cardiac stiffness and diastolic dysfunction (23). Lines of existed evidence demonstrates that the pathogenesis of diastolic dysfunction caused by cardiac fibrosis (32, 33). In the fibrotic heart, collagens mainly from activated myofibroblasts undergoes cross-linking process contributing to the progression of diastolic dysfunction and the restricted cardiac chamber compliance (34, 35). In addition, ECM overproduction and deposition between the layers of cardiac myocytes results in the disruption of myocardial electrophysiological functions, which leads to contractile dysfunction and an increased risk of cardiac arrhythmia (36, 37). In fact, TGF-β induced cardiac fibrosis is seriously involved in the pathogenesis of arrhythmia by disturbing electrical signal conduction, leading to the generation of re-entry circuits (10).
In the heart, cardiac fibroblasts can be transdifferentiated into myofibroblasts with contractile, migratory, and secretory properties (Figure 2). Myofibroblast is a key regulator that accelerates the fibrotic response in many conditions associated with HF. Regardless of the etiology of cardiac fibrosis, myofibroblast transdifferentiation is a hallmark of the fibrotic response in the heart [Reviewed in (20, 23)].
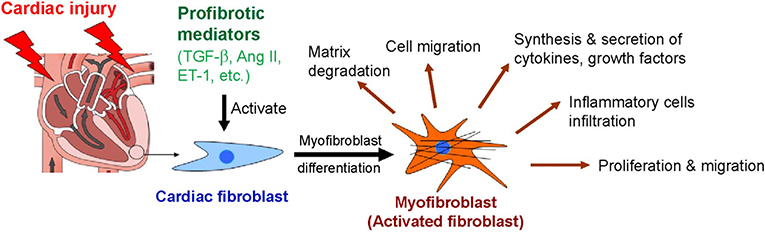
Figure 2. Myofibroblast differentiation and functions of myofibroblasts after cardiac injury. Ang II, angiotensin II; ET-1, endothelin-1; TGF-β, transforming growth factor-β.
Myofibroblasts are the activated form of fibroblasts. They overexpress α-smooth muscle actin (α-SMA) and contain contractile bundles of actin filaments resembling the myofibrils of smooth muscle cells and associated proteins organized into prominent stress fibers (38). The incorporation of α-SMA into contractile bundles is a major characteristic of differentiated myofibroblasts and significantly increases contractile function. Thus, α-SMA has been suggested to be the most significant marker of myofibroblasts (39). Although α-SMA is found in human myocardial scars, the other structural ECM proteins such as collagens, vimentin, and desmin are also present in fibrotic scars (40). Fibroblast differentiation into myofibroblast is controlled by a variety of growth factors and cytokines. Among them, TGF-β is a strong inducer that stimulates myofibroblast formation (Figure 2).
Fibroblasts are abundant in normal hearts and can differentiate into myofibroblasts via profibrotic mediators such as TGF-β (41, 42). This process suggests that the activation of resident fibroblasts represents a major source of myofibroblasts in hearts with fibrosis. In addition, proliferating myofibroblasts are commonly found in high numbers in the infracted area of the heart (41, 42).
Following cardiac fibroblast activation, inflammatory cells (e.g., macrophages, monocytes, and mast cells) infiltrate the site of remodeling myocardium and secrete various types of profibrotic mediators, including growth factors and cytokines [Reviewed in (43)]. These mediators have been found to promote myofibroblast formation, but the most significant and common inducer is TGF-β (44). TGF-β accelerates the differentiation of resident fibroblasts, epithelial cells, and endothelial cells into myofibroblasts (44). Thus, agents that inhibit myofibroblast differentiation might provide a tool to prevent the maladaptive myocardial remodeling that occurs in response to profibrotic stimuli and for fibrosis prevention.
Alterations in ECM homeostasis, especially in terms of ECM overproduction, lead to cardiac dysfunction. Several mediators, including angiotensin II (Ang II), and TGF-β, regulate ECM production by cardiac fibroblasts (45). In response to cardiac injury, myocardial fibrosis results from an imbalance of both ECM synthesis and degradation, leading to an accumulation of collagen type I and III in the heart (20, 23). Deposition of ECM proteins is significantly increased in the hearts of patients with cardiac diseases (46). In addition, the levels of cardiac fibrosis are associated with cardiac dysfunction (46). Moreover, ECM deposition and fibroblast activation contribute to the impairment of ventricular compliance and filling due to increased ventricular stiffness (20, 23). Furthermore, overproduction of ECM interrupts the electrophysiological functions in the heart, leading to arrhythmias (10).
According to cardiac fibrosis is associated with cardiac remodeling and is involved in the pathogenesis of HF, the prevention and reversal of cardiac fibrosis is an important therapeutic target for the treatment of HF. Numerous signaling pathways, through a variety of profibrotic mediators (e.g., Ang II, endothelin-1 [ET-1], and TGF-β), have been implicated in the activation of cardiac fibroblasts and the development of cardiac fibrosis. Modulation of these signaling pathways using inhibitors is of great interest for the treatment and prevention of cardiac fibrosis. Below, we summarize the update and important roles of several agents that act against cardiac fibrosis (Table 2). Although, both angiotensin converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs) have already demonstrated significant efficacy in reducing cardiac fibrosis in human and animal models of HF, neither ACEIs nor ARBs have been approved for the treatment of cardiac fibrosis. Further studies are required to establish the molecular mechanisms of ACEIs and ARBs not only for treatment but also for reversal of fibrotic remodeling in HF.

Table 2. Therapeutic targets/strategies for treatment of cardiac fibrosis.
TGF-β is a member of the TGF-β superfamily, which is comprised of TGF-β, bone morphogenetic proteins (BMPs), growth differentiation factors (GDFs), activin and inhibin (65). Members of this diversify superfamily are the pleiotropic multifunctional polypeptides that play a role in a wide range of physiological cellular activities such as growth, proliferation, differentiation, and apoptosis (65). Among these polypeptides, TGF-β has been proven to be one of the major factors driving the fibrotic response in most organs (2). In mammals, there are 3 isoforms of TGF-β: TGF-β1, TGF-β2, and TGF-β3. These highly homologous polypeptides, encoded by various genes, are synthesized, processed and regulated in a similar fashion. However, these 3 isoforms are secreted by various types of cells and signals through the same receptors, but they exhibit distinct patterns of distribution in different tissues (3, 66). Even though any isoform can be found in fibrotic tissues, the progression of organ fibrosis, in particular cardiac fibrosis, is predominantly attributed to TGF-β1 (67). To date, information on isoform-specific activities of various isoforms of TGF-β in a specific pathology is lacking and needs further investigation. Next, the signaling of TGF-β, excluding conclusions regarding specific isoforms, is discussed in detail.
The synthesis, release, and activation of TGF-β is a complex process (Figure 3). Following intracellular biosynthesis, a dimer of TGF-β is secreted as an inactive protein complex (latent TGF-β), which is retained in the ECM. Active TGF-β1 can be liberated from ECM by multiple activators such as reactive oxygen species (ROS), plasmin, thrombospondin-1, and αvβ6 integrin (68). Once active TGF-β is released from ECM, it binds to transmembrane TGF-β receptor type II (TβRII) of a target cell. This receptor-ligand interaction induces serine/threonine kinase activity of TβRII for autophosphorylation (69). The canonical pathway of TGF-β signaling is initiated after phosphorylated TβRII forms a stable heteromeric complex with TGF-β receptor type I (TβRI), also known as activin receptor-like kinase 5 (ALK5), for the transphosphorylation of residual phosphate to TβRI (70). This receptor binding complex, which is a heterotetrameric combination between two molecules of TβRII and another two of TβRI, recruits and phosphorylates the downstream signaling proteins Smad2 or Smad3, which are called receptor-activated Smads. After phosphorylation, Smad2 or Smad3 is released and forms an intracellular complex with Smad4, the mediator Smad. This intracellular complex between Smad2/4 or Smad3/4 moves from the cytoplasm into the nucleus, where it binds to promoter regions of the genes involved in physiological process of induction of specific gene expression (71). For an example of fibrogenesis, gene encoding α-SMA, collagens, and fibronectin are significantly upregulated via the Smad3-dependent pathway (72). The expression of these fibrosis-related genes plays a pivotal role in the cellular transdifferentiation that generates myofibroblasts and the production/deposition of ECM by myofibroblasts in fibrotic tissue (72). In addition to fibrogenesis, the Smad-mediated signaling pathway is also a significant intracellular process activated by TGF-β that increases genes associated with carcinogenesis (73). Furthermore, the activation of TGF-β signaling results in the expression of Smad7, an inhibitory SMAD, which acts as a negative regulator by interacting with Smad2 or Smad3, thereby mitigating signaling through receptor-activated Smads and further decreasing TGF-β actions (74).
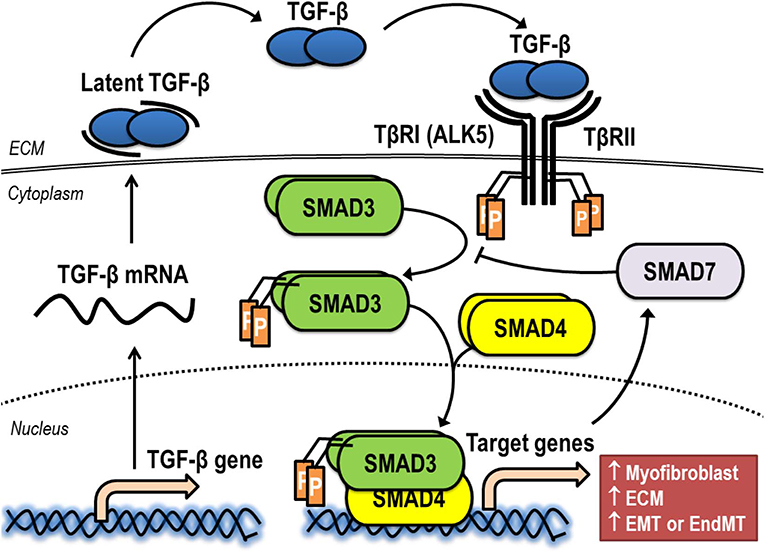
Figure 3. Synthesis, release, and activation of TGF-β signaling via the canonical pathway. ALK5, activin receptor-like kinase 5; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; EndMT, endothelial-to-mesenchymal transition; TβRI, TGF-β receptor type I; TβRII, TGF-β receptor type II.
Beyond canonical pathways or Smad-mediated signaling, TGF-β might mediate signaling directly by activating kinase enzymes via non-Smad signaling pathways, which are also known as non-canonical pathways (Figure 4). The non-Smad signaling pathways are initially propagated by either or both phosphorylated TβRI and TβRII for modulating downstream cellular responses. It has been reported that crosstalk between canonical and non-canonical pathways appeared to occur in most TGF-β-mediated effects (75). Epithelial-to-mesenchymal transition (EMT) plays a significant role in the pathogenesis of cancer. In part, this process requires an activation of ERK by TGF-β to upregulate the genes involving in remodeling of cell-matrix adhesion, thereby promoting the motility of the transformed cells (76). Also, EMT might be induced by TGF-β via both TβRI and TβRII through the activation of TNF receptor-associated factor 6 (TRAF6). TRAF6 is capable of recruiting TGF-β-activated kinase 1 (TAK1) to subsequently allow the activation of c-Jun amino terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38-MAPK) (77). In addition, the TRAF6-TAK1-JNK/p38 pathway is believed to be an essential pathway for TGF-β-induced apoptosis (78). Similar to the ERK and JNK/p38-MAPK pathway, the Ras homolog gene family member A (RhoA) is also a signaling mediator of EMT. TGF-β-induce RhoA degradation by phosphorylating partitioning-defective 6 (Par6), which subsequently recruits Smad-specific E3 ubiquitin protein ligase (Smurf1) to loosen tight junctions and rearrange the actin cytoskeleton, a prerequisite step for EMT (79). Another non-Smad signaling pathway contributing to TGF-β-promoted EMT is the phosphoinositide 3-kinase (PI3K)/Akt (protein kinase B) pathway, which subsequently activates the mammalian target of rapamycin (mTOR) and phosphorylation of S6 kinase (S6K) (80, 81). In addition, TGF-β1 signaling can be regulated at the post-transcriptional level via the expression of microRNAs (miRNAs), and the expression of miRNAs might play a role in TGF-β1-mediated EMT also (82).
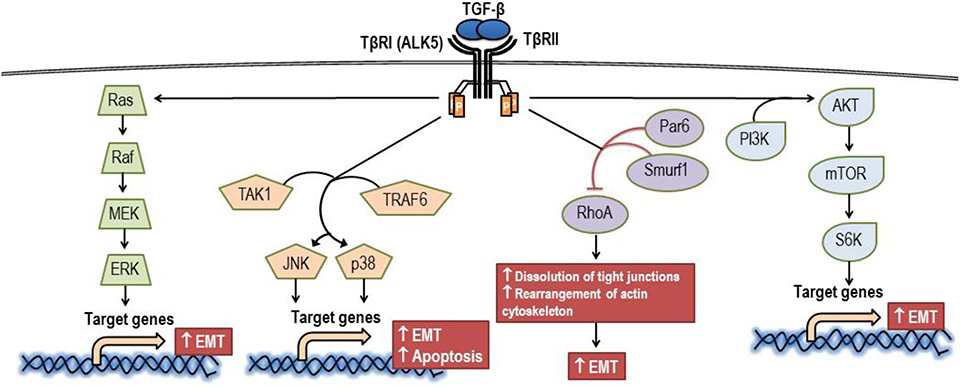
Figure 4. Signaling via the non-canonical pathway of TGF-β. AKT, protein kinase B; ALK5, activin receptor-like kinase 5; EMT, epithelial to mesenchymal transition; ERK, extracellular signal-regulated kinase; JNK, c-Jun amino terminal kinase; MEK, mitogen-activated protein kinase kinase; mTOR, mammalian target of rapamycin; Par6, partitioning-defective 6; PI3K, phosphoinositide 3-kinase; p38, p38 mitogen-activated protein kinase; Raf, Raf proto-oncogene serine/threonine-protein kinase; Ras, Ras GTPase; RhoA, Ras homolog gene family member A; Smurf1, SMAD specific E3 ubiquitin protein ligase; S6K, phosphorylation of S6 kinase; TAK1, TGF-β-activated kinase 1; TRAF6, tumor necrosis factor receptor-associated factor 6; TβRI, TGF-β receptor type I; TβRII, TGF-β receptor type II.
For the ultimate outcome of TGF-β-mediated responses in any pathological condition, it is apparent that a combination of canonical and non-canonical pathways are coordinated (1). Cancers and fibrotic diseases are the most common pathologies associated with the activity of TGF-β. Currently, most putative drugs affecting TGF-β for the treatment of cardiac fibrosis were initially developed for the management of cancer; therefore, we next discuss the signaling of TGF-β in carcinogenesis.
In the pathogenesis of cancer, TGF-β acts as a tumor suppressor in early stages of the disease. However, in later stages, TGF-β turns into a tumor promoter. This paradoxical role of TGF-β is due to a bypass of the cytostatic effect of TGF-β in tumor cells (4). The tumor suppressive effect of TGF-β is derived from various cellular effects. TGF-β stabilizes the cell cycle of epithelial cells by upregulating multiple cyclin-dependent kinases: p15, p21, and p27, via the canonical pathway (83). Also, via the Smad-dependent pathway, TGF-β downregulates genes associated with cell proliferation, such as c-Myc (84). In addition, the canonical pathway contributes to the tumor suppressive effects of TGF-β by inducing gene encoding B-cell lymphoma 2 (BCL2) and subsequently activating BIM for apoptotic processes in human B cells (85). Conversely, non-canonical pathways might mediate the apoptotic effect of TGF-β by inducing caspase-8 expression and activating BID in human gastric carcinoma cells (86). The difference in signaling of TGF-β-mediated apoptosis indicates that the cellular context is essential for controlling the main pathway in the tumor suppressive effects of TGF-β. The tumor promoting effects of TGF-β such as EMT, invasion, metastasis, and angiogenesis emerge when cancer progresses to a later stage (5, 87). The upregulation of miR-106b-25 cluster targets Smad7 to ameliorate the TGF-β signaling that is not generally found in normal tissues is an excellent example of this phenomenon. In human breast cancer, increased miR-106b-25 leads to the inhibition of tumor suppressive protein p21 and BIM, thereby allowing tumor cells to grow via the activation of TGF-β (88). Interestingly, TGF-β also regulates the functions of various immune cells, including the modulation of cytokines released from these cells. Impairment of TGF-β signaling pathways leads to immune dysregulation, fibrosis, and cancer [Reviewed in (7)]. TGF-β is produced as a complex with latency associated peptide (LAP). This complex associates with ECM by binding to latent TGF-β binding protein (LTBP) or glycoprotein A repetitions predominant (GARP) expressed on T cells, especially on Tregs, or platelets. Integrins bind to the complex and stimulate the release of TGF-β from the complex. The release of active TGF-β promotes oncogenesis and immune tolerance in breast cancer (89). Inhibition of αvβ8 integrins potentiates cytotoxic T cell responses and recruitment of immune cells to tumor centers. Cancer cells can evade host immunity by mobilizing active TGF-β1 through αvβ8 integrins (90). Thus, TGF-β acts as a significant suppressor of immune responses during tumor progression.
In general, tissue fibrosis is considered a main step in triggering cancer development. An apparent example is hepatocellular carcinoma, the most common form of liver cancer. Cirrhosis, which is known as the end-stage of liver fibrosis, occurs in most patients who ultimately develop hepatocellular carcinoma (91). Interestingly, the progression of fibrosis to cancer in the heart is rare. The low incidence of cardiac cancer might be due to the fact that cardiac cells, in particular cardiomyocytes, are fully differentiated cells. Moreover, the regenerative capacity of cardiomyocytes is considered to be negligibly low. Thus, cardiomyocytes appear to resist further transformation and proliferation processes such as EMT in the development of cancer (92). Accordingly, signaling of TGF-β in fibrogenesis of the heart might not be identical to that occurring in other organs where progressive fibrosis ultimately develops cancers.
During tissue injury, TGF-β expression is increased to play a role in the tissue repair process and scar formation. In the heart tissue following MI, TGF-β signaling plays an important role in reparative, angiogenetic, and fibrotic responses by modulating inflammation (93). Studies on mice and dogs have revealed that TGF-β1 and TGF-β2 were upregulated in the early phase after MI, and then TGF-β3 was increased in a later stage post-infarction myocardium (94). Among various cells that release TGF-β, a significant amount of TGF-β might be released from infiltrated macrophages that migrate to the injured area to engulf the damaged cardiomyocytes, as shown in a mouse model (95). On the other hand, a study using a porcine model of chronic coronary constriction revealed that cardiomyocytes were a significant source of TGF-β (96). Another study suggested that TGF-β was found in the extracellular fluid of ischemic canine myocardium tissue (97). Multiple pathways involving integrins and thrombospondin-1 were found to be associated with the release of TGF-β from the cardiac ECM-bound TGF-β (98, 99). Following the release of active TGF-β, TGF-β binds to the receptors, as described earlier, to activate intracellular responses in the infarcted tissue. The TGF-β-mediated effects can be classified into 4 actions in the following order: cardiomyocyte survival, immune cell-related action, formation of myofibroblasts, and production/deposition of ECM, all of which modulate the effects on myocardial endothelial cells.
TGF-β-mediated effects on cardiomyocyte survival in MI appear to be dependent on the time period after MI. In the early phase, exogenous TGF-β administered before or immediately after ischemic injury to an isolated perfused heart showed cardioprotective effects by reducing the amount of superoxide anions, maintaining coronary relaxation, and reducing injurious responses of exogenous TNF-α (100). Similarly, a study has shown that the infarct size of intact rat hearts receiving TGF-β during early reperfusion was reduced, and this reduction was due to activation of MAPK (101). However, the mechanism underlying cardioprotection remains poorly understood. Conversely, a proapoptotic effect of TGF-β via interplay with Ang II was demonstrated in a study using rat cardiomyocytes (102). The findings showed that the actions of exogenous TGF-β are likely dependent on the timing of administration.
Immune cells play a pivotal role in fibrogenesis, and TGF-β regulates both the phenotype and function of the immune cells. It is worth noting that TGF-β can be either a pro- or anti-inflammatory mediator of the immune response in in vitro studies [Reviewed in (93)]. Factors that determine the effects of TGF-β include the types of cytokines and the origin of the tissue (103). In an in vivo study, TGF-β suppressed T cell-mediated inflammation in genetically modified mice with T cell-specific loss of TβRII. Thus, the results from this in vivo study implicate an immunosuppressive effect of TGF-β (104). Nevertheless, the specific TGF-β-mediated effects on the phenotype of immune cells, together with its signaling and significance in the regulation of fibrosis, in the infarcted tissue remain unknown in the infarcted tissue.
TGF-β-mediated effects on the formation of myofibroblasts and on the induction of transformed myofibroblasts to further produce/deposit ECM are currently recognized central to the role of TGF-β in the pathogenesis of fibrosis. In cardiac fibrosis, Smad3-deficient mice that underwent reperfused MI showed significantly less fibroblast proliferation and ECM when compared to those of wild-type mice (105, 106). Even though the origin of the cells that underwent transformation has been debated (107), a recent study using fibroblast-specific, TGF-β signaling pathway knockout mice demonstrated that myofibroblasts in cardiac fibrosis are derived from resident fibroblasts, which activated via the TGF-β-Smad2/3 signaling pathway (72). These results suggest that the canonical pathway of TGF-β is principally involved in the pathogenesis of cardiac fibrosis. Interestingly, it was found that the Smad3-dependent pathway is essential for the upregulation of connective tissue growth factor (CTGF), which in turn acts as a mediator to stimulate fibroblast differentiation and collagen synthesis (108). Beyond the formation of myofibroblasts, genes encoding collagen type I and III were upregulated in cardiac fibroblasts isolated from rabbit hearts following treatment with TGF-β (109). The TAK1/p38-MAPK pathway in the cardiomyocytes of non-infarcted myocardium was found to be activated in rats after acute MI, suggesting a role for this non-canonical pathway in ventricular hypertrophy and remodeling (110). Nevertheless, the significance of Smad-independent pathways in the transformation of cardiac fibroblasts appears to be less proven than that of renal and pulmonary fibrosis (111, 112). Finally, a study on TGF-β-overexpressed mice showed increase expression of tissue inhibitors of matrix metalloproteinases (TIMPs), which regulate the remodeling of ECM in the cardiac tissue. However, the signaling of TGF-β was not evaluated in this study (113).
In addition to cardiomyocytes, immune cells, and transformed myofibroblasts, vascular endothelial cells might also play an important role in cardiac fibrosis. It has been found that endothelial cells served as a source of chemokines and played a role in recruiting neutrophils and monocytes to the heart after MI (114). Interestingly, although TGF-β plays a role in angiogenesis in cancers (8), information on the effects of TGF-β on angiogenesis in infarcted myocardium is limited at present. Moreover, although most cardiac myofibroblasts originate from resident fibroblasts, a study has shown that endothelial cells might be activated by the TGF-β via Smad3-dependent pathway and transform into myofibroblasts, thereby inducing cardiac fibrosis (115).
TGF-β suppresses cell proliferation leading to apoptosis in the early phase of tumor development, whereas it aggravates tumor invasion and metastasis via boosting immune escape, angiogenesis, and EMT of tumors at an advanced stage (116). The paradoxical impact of TGF-β signaling in various tumors raises concerns that anti-TGF-β signaling might lead to a poor prognosis due to its tumor suppressor role. This concern has delayed progression in the development of TGF-β inhibitors as therapeutic agents. In addition, some experimental models have revealed that TβRI inhibitors aggravated the potential for cardiotoxicity (117).
However, several potential approaches to interfering with TGF-β signaling to prevent TGF-β production and block its signaling pathway have emerged. Next, we summarize the results of TGF-β inhibitors that have been studied in preclinical or clinical trials on carcinogenesis. The studies can be mainly categorized into 3 levels: (1) The ligand level: Direct blockage of TGF-β ligand synthesis by antisense molecules; (2) The ligand-receptor level: Inhibition of TGF-β ligand-receptor interaction using monoclonal antibodies or soluble TGF-β decoy receptors (traps); and (3) The intracellular level: Suppression of the TGF-β signaling pathway by tyrosine kinase inhibitors that disturb the downstream signaling of TGF-β related proteins (9, 118). The examples of current therapeutic agents in preclinical and clinical development in oncology are summarized in Tables 3, 4.
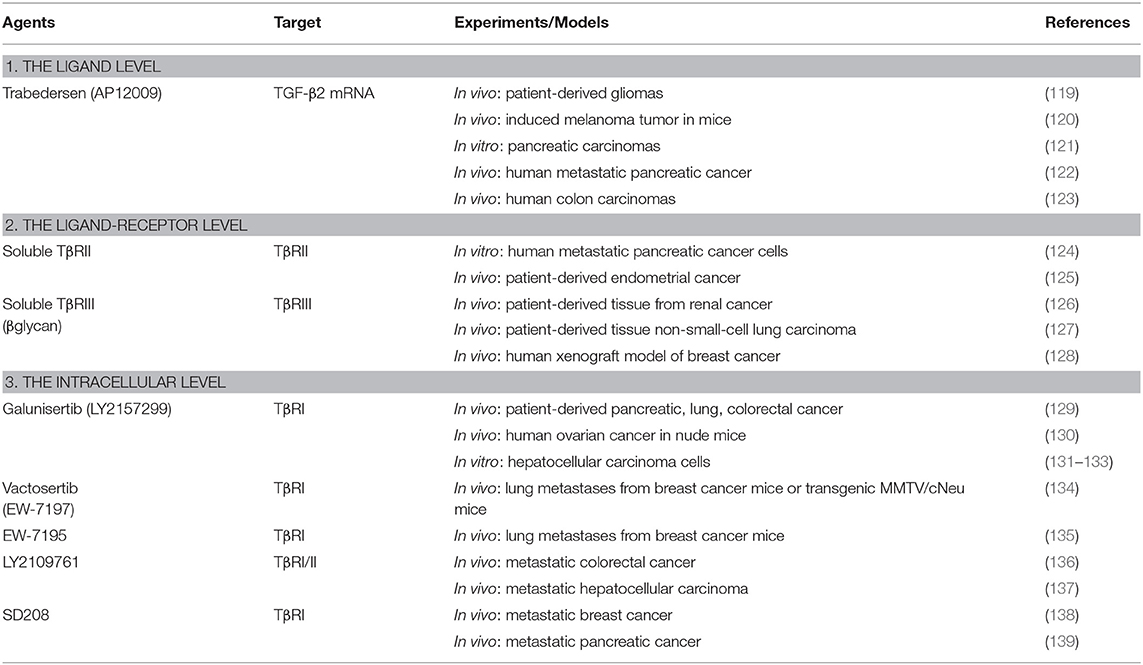
Table 3. Preclinical studies of TGF-β inhibitors for cancer treatment.
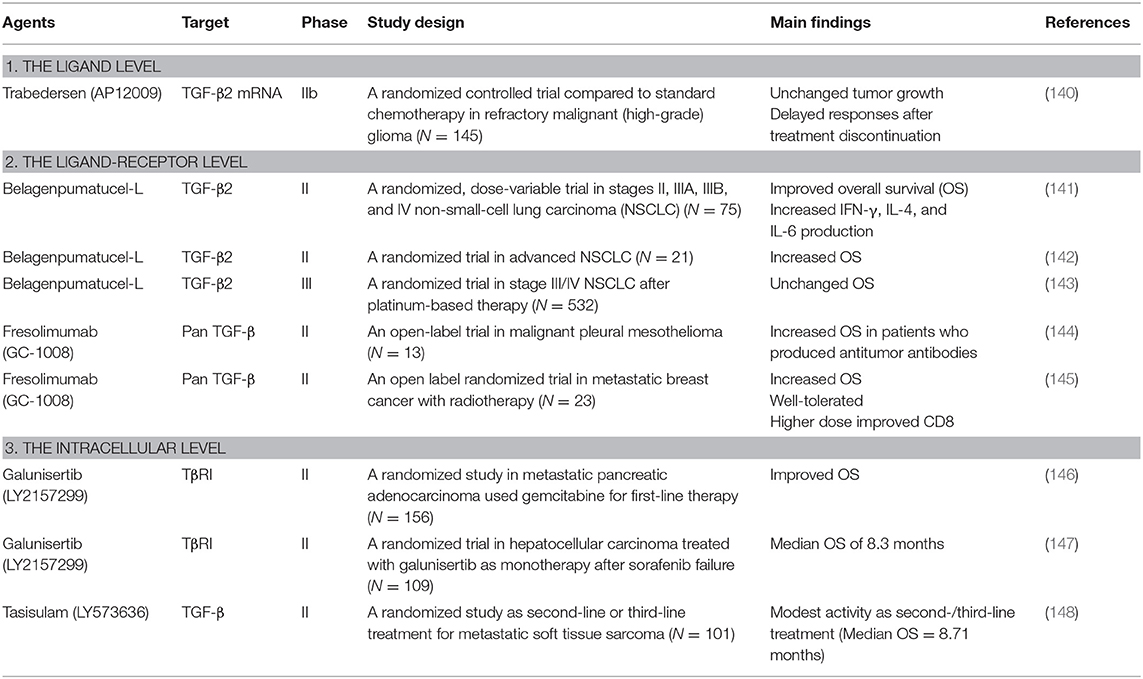
Table 4. Clinical studies of TGF-β inhibitors for cancer treatment.
Trabedersen (AP12009, Antisense Pharma) is a synthetic, 18-oligomer phosphorothioate antisense oligonucleotide (ASO). It was developed as an ASO specifically targeting human TGF-β2 mRNA, which leads to a reduction in TGF-β2 expression, cellular proliferation, and cellular migration in various types of tumors in vitro and in vivo, including gliomas (119), melanoma (120), pancreatic carcinomas (121, 122), and colorectal cancer (123). Trabedersen has been shown to reduce cell proliferation, tumor growth, cell migration or metastasis, and vascularization in human pancreatic cancer cells and in mouse model of human metastatic pancreatic cancer (122).
After several preclinical studies provided evidence of potential clinical efficacy, trabedersen was moved to phase I/II trials in patients with recurrent high-grade gliomas (119, 140, 149). Trabedersen was initially assessed for its safety and efficacy in phase I/II dose escalation studies in patients with high-grade gliomas and found a significant increase of median survival time after recurrence, exceeding that of standard chemotherapy (149). Similarly, prolonged survival and high response rates after treatment with trabedersen were observed in phase I/II studies in patients with recurrent or refractory malignant glioma, WHO grade III or IV (119). However, trabedersen was further compared with standard chemotherapy (temozolomide or procarbazine/lomustine/vincristine) in patients with recurrent or refractory malignant glioma (WHO grade III or IV) in a phase IIb trial. The results revealed that trabedersen did not control tumor growth, but delayed responses were observed after discontinuation of treatment (140).
The principle of anti-TGF-β cancer vaccines is to deliver antisense molecules of TGF-β into cancer cells and overturn the effects of immunosuppression in host cells, as well as to enhance antitumor immunity (9). Belagenpneumatucel–L (Lucanix, NovaRx) is a TGF-β2, antisense, gene-modified non-viral based allogenic tumor cell vaccine. It was developed from non-small cell lung cancer (NSCLC) and modified to express ASO, which leads to suppression of the immunosuppressive activity implicit in TGF-β2 overexpressing cancer cells (141).
Currently, an anti-TGF-β cancer vaccine, belagenpumatucel-L, has entered a phase III study to determine whether it improves overall survival (OS) and might be useful for stimulating immune reactions. A dose-related survival difference was achieved in patients who received belagenpumatucel-L at least 2.5 × 107 cells/injection in a phase II trial involving patients with stages II, III, and IV NSCLC. Moreover, immune function measurements revealed an increase in cytokine production, including IFN-γ, IL-6, and IL-4, among clinical responders, who also displayed an elevated antibody-mediated response to the vaccine human leukocyte antigens (HLAs) (141). Likewise, a further study to evaluate its safety and response at the previously defined optimal dose found the median survival of patients with fewer than 2 circulating tumor cells (CTCs) at baseline was longer than patients with 2 or more CTCs. Thus, plasma levels of CTCs are associated with the OS of patients with stage IV NSCLC (142). Nevertheless, in a phase III trial with 532 patients with stage III/IV NSCLC who did not progress after platinum-based induction chemotherapy with or without irradiation, belagenpumatucel-L did not increase survival compared with placebo (143).
Fresolimumab (GC1008, Genzyme/Sanofi) is a fully human monoclonal antibody blocking pan-TGF-β (TGF-β1, TGF-β2, and TGF-β3) [Reviewed in (150)]. Fresolimumab demonstrated acceptable safety and preliminary evidence of antitumor activity in a phase I trial on patients with previously treated malignant melanoma or renal cell carcinoma (151). In a phase II trial on 13 patients with malignant pleural mesothelioma, 3 patients showed stable disease for at least 3 months, and those who produced antitumor antibodies had an increased median OS. However, treatment with fresolimumab had no effect on the expression of NK, CD4+, or CD8+ T cell activating and inhibitory markers, other than a decrease in the expression of CD244 (also known as 2B4) and CD266 (best known as DNAM1) on NK cells (144). A phase II trial on 23 patients with metastatic breast cancer undergoing radiotherapy has reported that fresolimumab in combination with focal radiotherapy significantly increased OS and was well-tolerated in a dose-dependent manner. Higher doses of fresolimumab correlated with an improved CD8+ pool, leading to a favorable systemic immune response and longer median OS (145).
Galunisertib monohydrate (LY2157299, Eli Lilly) is a small-molecule inhibitor of TβRI that robustly downregulate the phosphorylation of Smad2 in pancreatic, lung, colorectal (129), and ovarian cancer (130). Galunisertib effectively demonstrated potent inhibition of both canonical and non-canonical pathways in a variety of in vitro hepatocellular carcinoma cells regardless of TGF-β pathway protein expression (131, 132). Nevertheless, the antiproliferative activity of TGF-β pathway inhibitors is quite limited. It has been reported that TGF-β inhibited cell proliferation while inducing apoptosis in cell lines with low endogenous levels of TGF-β and Smad7 and strong transcriptional Smad3 activity (PLC/PRF/5, HepG2, Hep3B, HuH7). However, cancer cells were sensitive to TGF-β-dependent growth inhibition and displayed limited sensitivity to galunisertib in another group of cell lines expressing high quantities of TGF-β and Smad7 and showing significantly reduced Smad3 signaling (SK-HEP1, SK-Suni, SK-Sora, JHH6, HLE, HLF, and FLC-4) (132, 133). Despite limited antiproliferative activity in vitro, galunisertib exhibited antiproliferative effects in ex vivo models, indicating that inhibition of TGF-β can exert anticancer properties (131, 133). Nevertheless, from the reports on several preclinical studies, treatment with TGF-β inhibitors as monotherapy might display limited efficacy. However, the immunological effects of galunisertib are strongly augmented in combination with other checkpoint inhibitors (152, 153).
Among small molecule inhibitors, galunisertib is one of the most advanced. It has shown promising results in clinical trials due to its safety profile, with no cardiac potential toxicity in humans, which was a primary concern with first-generation TGF-β inhibitors (154). A phase I study on 28 patients with Grade IV glioma showed galunisertib was well-tolerated. The dose limiting toxicities included pulmonary embolism and thrombocytopenia, but no cardiotoxicities were observed (155). In addition, the safety of galunisertib was confirmed by a first-in-human dose study with 79 cancer patients with glioma and solid tumors treated with galunisertib as monotherapy or in combination with lomustine. No medically relevant cardiac toxicity or signs of cardiovascular injury were found, including increased blood pressure, troponin I, BNP, or hs-CRP or reductions in cystatin C levels (156). Likewise, no safety concerns or dose limiting toxicities was observed after treatment with galunisertib in patients with glioblastoma based on a pharmacokinetic/pharmacodynamic (PK/PD) model (157). Galunisertib as monotherapy and as second-line therapy after sorafenib failure in a subset of 109 patients with hepatocellular carcinoma yielded a median OS of 8.3 months in a phase II trial (147). Interestingly, patients who had decreased expression levels of specified blood biomarkers [e.g., alpha-fetoprotein (AFP), TGF-β1, and CDH1] had improved clinical outcomes, indicating that the effects of galunisertib might be more pronounced in patients with a poor prognosis due to elevated AFP at baseline (147). Similarly, galunisertib in combination with gemcitabine improved OS with minimal added toxicity in a phase II study on patients with locally advanced or metastatic pancreatic adenocarcinoma who were considered candidates for first-line chemotherapy with gemcitabine (146).
Vactosertib (EW-7197 or TEW-7197), a novel small molecule inhibitor of ALK5, has been recently developed as a more potent and specific antitumoral compound than galunisertib. Vactosertib and EW-7195 expressed potent antimetastatic activity in vivo via an inhibition of TGF-β1-induced Smad/TGFβ signaling, cell migration, invasion, EMT, and breast tumor metastasis to the lung in xenografted nude mice and transgenic MMTV/cNeu mice (134, 135). In addition, vactosertib expressed the potential to boost cytotoxic T lymphocyte function in 4T1 orthotopic-grafted mice and prolonged the lifespan of 4T1 breast tumor-bearing mice (134).
Vactosertib is currently being tested in phase I/II clinical trials for several cancer types in combination with chemotherapy or antibodies against immune checkpoints. A phase I study is evaluating the safety and tolerability of the drug in combination with paclitaxel in 12 metastatic gastric cancer patients (NCT03698825). The phase Ib/IIa trials include a study of vactosertib in combination with durvalumab in patients with advanced NSCLC who progressed following platinum-based chemotherapy (N = 63) (NCT03732274). A combination with pembrolizumab is being employed for metastatic or locally advanced colorectal or gastric/gastroesophageal junction adenocarcinoma (N = 67) (NCT03724851), and a combination with imatinib is being employed for patients with advanced desmoid tumors (N = 24) (NCT03802084). The latest phase II trial aims to determine whether administration of vactosertib with durvalumab will provide meaningful increases in the overall response rate in patients with urothelial cancers that fail to achieve a CR with anti-PD-1/PD-L1 based regimens (N = 48) (NCT04064190).
Remarkably, given TGF-β signaling plays a crucial role in fibrotic states, vactosertib has recently been investigated as an antifibrotic agent to delay the development of fibrosis in primary organs including the liver, kidney, and lung. Vactosertib was found to suppress fibrosis-induced accumulation of ROS and ECM proteins (collagen, α-SMA, fibronectin, and integrins) in the liver, lungs, and kidneys of mice due to its antifibrotic mechanism via inhibition of both TGF-β1/Smad2/3 and ROS signaling (158). A study on a rat model of Peyronie's disease showed that vactosertib suppressed phospho-Smad2 expression and recruitment of inflammatory cells, leading to a decline in fibrotic plaques (159). Thus, vactosertib and EW-7195 could be a promising antifibrotic compound for the treatment of fibrotic diseases.
Tasisulam has completed many trials in various oncologic diseases, including phase I studies on patients with essential thrombocythemia and acute myeloid leukemia (NCT00718159) and solid tumors (NCT01214668) and phase II trials on patients with ovarian cancer (NCT00428610), metastatic breast cancer (NCT00992225), NSCL cancer (NCT00363766), and malignant melanoma (NCT00383292). A phase II study on tasisulam as second- or third-line treatment for 101 patients with unresectable or metastatic soft tissue sarcoma reported that tasisulam demonstrated modest activity with a median OS of 8.71 months (148). Consequently, the synergistic and additive effects of tasisulam combined with other anticancer agents are currently of interest. Currently there is an ongoing phase I trial of tasisulam in combination with sunitinib, a multiple tyrosine kinase, in renal cancer patients (NCT01258348), and with pemetrexed, an inhibitor of purine synthesis, in patients with solid tumors (NCT01215916).
Interestingly, recent preclinical study has been reported that M7824 (MSB0011359C) which is a dual inhibitor of programmed death ligand 1 (PD-L1) and TGF-β inhibited tumor growth and metastasis more effectively than treatment with TGF-β inhibitor alone. Thus, M7824 (an inhibitor of PD-L1 and TGF-β) exhibits potent and superior antitumor effects compared to that of TGF-β inhibitor monotherapy and is likely to help minimize potential side effects (160).
The renin-angiotensin system (RAS) inhibitors are currently used as standard therapy for HF and have been shown to inhibit activation of fibroblast and differentiation into myofibroblast. However, cardiac fibrosis persists in patients with HF even when treated with these conventional RAS inhibitors, indicating a need to develop novel and effective antifibrotic therapies for heart disease (161). Currently, due to its established role in cardiac fibrosis, there is great interest in inhibiting the TGF-β signaling pathway (6, 161). TGF-β is considered a mediator of cancer and fibrosis. Thus, blockades of TGF-β signaling activity using receptor antagonists, inhibition via antibody or antisense oligonucleotide, or even using gene deletion of TGF-β signaling molecules are potential therapeutic strategies.
Anti-TGF-β1 neutralizing antibodies have also been under investigation as potential antifibrotic agents by interfering with TGF-β signaling. Administration with anti-TGF-β1 antibody attenuated cardiac fibrosis and diastolic abnormalities in a rat model of pressure overload (47) (Table 2). Although these antibodies attenuated fibroblast activation and collagen synthesis, no improvements in overall cardiac functions were found in pressure-overloaded rats (47). Furthermore, anti-TGF-β neutralizing antibody inhibited ECM proteins synthesis and reduced cardiac fibrosis in a rat model induced by a chronic blockade of nitric oxide synthesis (162). However, in a mouse model of MI, a neutralizing anti-TGF antibody administered before or after coronary artery ligation resulted in increased mortality rates and left ventricular (LV) dilation after MI (163).
Alternative approaches have included inhibition of the expression of TGF-β using antisense oligonucleotides (164), and the use of a soluble TβRII, which either acts by adsorbing TGF-β or acting as a dominant negative receptor (165). Inhibitors of ALK5 (TβRI) are under investigation for antifibrotic effects in the heart. Inhibitor of ALK5 which decrease TGF-β activity can rescue cardiac dysfunction and ameliorate cardiac remodeling in post-MI hearts (50). Moreover, ALK5 inhibitors can also suppress the collagen synthesis and attenuate the progression of fibrosis in animal model of pressure overload induced by transverse aortic constriction, and inhibit TGF-β-mediated collagen synthesis in cardiac fibroblasts (51) (Table 2).
In addition to the canonical Smad-mediated signaling pathway, TGF-β also stimulates the non-canonical MAPK signaling pathways such as JNK-dependent and p38-MAPK-dependent pathways (166–168). These MAPK signaling pathways are involved in TGF-β-mediated activation of TAK1 which is thought to play a role in cardiac fibrosis and remodeling. Cardiac specific overexpression of the active form of TAK1 induced myocardial hypertrophy and HF (166–168), suggesting that TAK1 is a major effector of TGF-β signaling. Blockade of TAK1 activity attenuated TGF-β-mediated ECM protein overproduction in cardiac fibroblasts (48) (Table 2). In addition to inhibition of TAK1, inhibition of p38-MAPK is being investigated for its efficacy in the treatment of cardiac fibrosis. Inhibitors of p38-MAPK suppress myofibroblast activation and expression of ECM proteins and α-SMA induced by TGF-β, while overexpression of p38-MAPK induces myofibroblast differentiation in cardiac fibroblasts (49).
Two promising antifibrotic agents include tranilast and pirfenidone, which inhibit the actions of TGF-β as well as other pathogenic growth factors by unclear mechanisms (169). Current agents and therapeutic targets in preclinical and clinical development for the treatment of cardiac fibrosis and heart-related diseases are summarized in Tables 5, 6.

Table 5. Preclinical studies of TGF-β inhibitors for treatment of cardiac fibrosis.
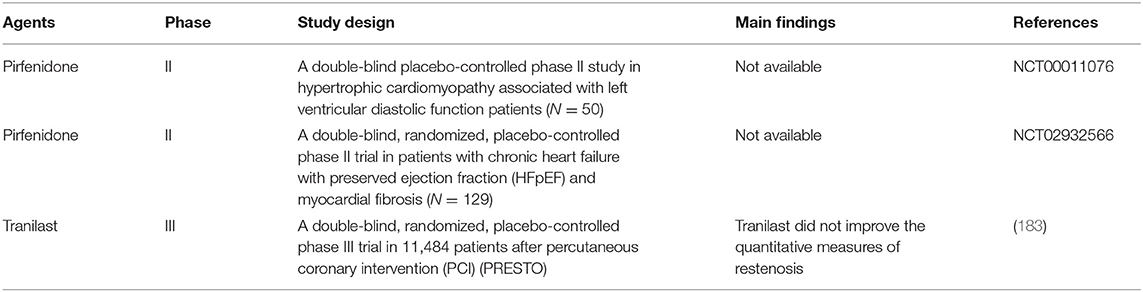
Table 6. Clinical studies of TGF-β inhibitors for treatment of cardiac fibrosis.
GW788388 is a potent inhibitor of both ALK5 and TGβRII with an improved pharmacokinetic profile (184) and minimal toxic effects (185). Several studies have been demonstrated that GW788388 pre-clinically reduces cardiac fibrosis in various models. GW788388 inhibited the development of cardiac fibrosis by suppression of collagen I and fibronectin synthesis, increased survival, and improved cardiac function in an experimental murine model of Chagas heart disease (170). Deletion of SCN5A, a gene encoding the main cardiac sodium channel NaV1.5, has been associated with inherited progressive cardiac conduction disease. GW788388 chronically inhibited TGF-β receptors and prevented fibrosis in a Scn5a heterozygous knockout (Scn5a+/−) mouse model of progressive cardiac conduction disease (171). Furthermore, treatment with GW788388 attenuated systolic dysfunction and delayed LV remodeling by reducing the phosphorylated Smad2, α-SMA, and collagen I in a rat model of HF following MI (50). Taken together, GW788388 appears to be a promising antifibrotic agent, although further studies are warranted.
Pirfenidone is an oral antifibrotic drug initially approved for the treatment of idiopathic pulmonary fibrosis (186). Pirfenidone inhibited TGF-β expression and also inhibited the profibrotic effects of TGF-β signaling (187). Thus, pirfenidone might be a promising agent for the treatment of cardiac fibrosis. A reduction in ventricular hypertrophy without lowering systolic blood pressure has been detected in the deoxycorticosterone acetate (DOCA)-salt hypertensive rats after pirfenidone treatment (172). Moreover, pirfenidone decreased total and non-scar myocardial fibrosis, which has been associated with decreased infarct scarring, improved LV function, and decreased ventricular tachycardia in rat MI model (173). Administration of pirfenidone reversed cardiac fibrosis, including renal fibrosis, and attenuated myocardial stiffness in streptozotocin (STZ)-diabetic rats (176).
Given pirfenidone has significant antifibrotic and anti-inflammatory properties, the anti-inflammatory effects of pirfenidone have been investigated. Pirfenidone inhibited NLRP3 expression and formation, contributing to a reduction in IL-1β synthesis, and attenuation of IL-1β-induced inflammatory and profibrotic responses in a mouse model with transverse aortic constriction (TAC)-induced LV remodeling (174). Similar effects were observed in murine pressure-overload injury; pirfenidone increased survival and attenuated fibrosis through suppression of myocardial fibrosis and vascular permeability in pressure-overloaded hearts (175). Therefore, pirfenidone might be a potential treatment for cardiac fibrosis.
Although pirfenidone has shown efficacy in the treatment of idiopathic pulmonary fibrosis in humans (186), clinical trials for the treatment of cardiac fibrosis are ongoing and the results have not yet been published. A phase II study of pirfenidone in patients with hypertrophic cardiomyopathy associated with LV diastolic function aims to examine the effectiveness of pirfenidone in improving heart function and reducing of myocardial fibrosis. The study was completed with unpublished data (NCT00011076). Another phase II trial is ongoing and will finish in Jan 2020. This trial is exploring the antifibrotic effects of pirfenidone on patients with chronic heart failure with preserved ejection fraction (HFpEF) and cardiac fibrosis by determining changes in myocardial ECM volume and investigating the relationship between myocardial fibrosis and myocardial energetics (PIROUETTE study, NCT02932566) (188).
Tranilast has been used to treat allergic disorders (e.g., allergic rhinitis, asthma, and atopic dermatitis); however, tranilast might also be useful for other medical conditions due to its ability to suppress TGF-β expression and activity. The molecular mechanisms underlying its antifibrotic actions are not completely understood, but tranilast might inhibit several profibrotic growth factors such as TGF-β and platelet-derived growth factor (PDGF) (22). The effects of tranilast on inhibition of cardiac fibrosis have also been supported by multiple animal models of cardiomyopathy. In STZ-induced (mRen-2)27 diabetic rats, tranilast treatment attenuated cardiac matrix deposition in association with reductions in phospho-Smad2 of the heart (177). In a similar model, administration of tranilast attenuated cardiac dysfunction and structural abnormalities in diabetic cardiomyopathy with improved LV systolic and diastolic function, while tranilast did not affect Smad phosphorylation but it significantly attenuated TGF-β-induced p44/42 MAPK phosphorylation (178).
The underlying mechanisms of the antifibrotic effects of tranilast have been attributed to its regulation of TGF-β signaling and to suppression of the infiltration of inflammatory cells, including monocytes and macrophages. The mRNA levels of TGF-β1, plasminogen activator inhibitor 1 (PAI-1), monocyte chemotactic protein-1 (MCP-1), IL-6, procollagens were attenuated, and myocardial fibrosis and collagen accumulation were suppressed in DOCA/salt hypertensive rats receiving tranilast (179). Similar findings were observed in other animal models of renovascular hypertensive rats (180) and hypertensive (mRen-2)27 rats (182). Interestingly, tranilast-mediated inhibition of cardiac fibrosis is independent of changes in blood pressure in these studies, suggesting that tranilast directly targeted cardiac fibrosis and might be beneficial for HF treatment in addition to current therapeutic strategies (181).
Restenosis after percutaneous coronary intervention (PCI) is a major adverse outcome following stent placement. In limited trials, administration of tranilast reduced the frequency of angiographic restenosis after PCI (189). Accordingly, the Prevention of Restenosis With Tranilast and Its Outcomes (PRESTO) trial was designed as a phase III trial with a large group of patients after PCI to investigate major adverse cardiovascular events of tranilast. It was found that tranilast did not improve restenosis or its clinical sequelae in patients receiving successful PCI (183). However, the number of events of MI was significantly reduced with tranilast treatment. The most commonly reported adverse events were laboratory test abnormalities consisting of hyperbilirubinemia, elevations in hepatic enzymes, and increased serum creatinine (183).
TGF-β is a multifunctional cytokine regulator acting through transmembrane serine/threonine kinase receptors and intracellular Smad transcriptional regulators. Once TGF-β is activated, it regulates ECM remodeling and promotes a fibroblast to myofibroblast transition, which is essential for fibrotic processes. Given TGF-β plays a major role in various stages of cancer progression and in the development of cardiac fibrosis, TGF-β and its signaling pathway offer opportunities for novel treatment strategies in patients with cancer and cardiac fibrosis. Research on the underlying mechanisms and the therapeutic targets of TGF-β inhibitors for cancer and cardiac fibrosis has advanced significantly in recent decades. The inhibitors of TGF-β signaling for cancer and fibrosis have been extensively studied in animal models and clinical studies; however, translation of these findings into human pathologic conditions has been limited due to the broad range of responses to TGF-β and its role in tissue homeostasis. Currently, various types of TGF-β inhibitors are challenged and tested their efficacies in patients with cancers. A few of TGF-β inhibitors are subjected into the clinical studies for treatment of cardiac fibrosis. The development of more specific agents targeting TGF-β signaling pathways such as M7824, a bifunctional fusion protein composed of TGF-β trap, and a monoclonal antibody against programmed death ligand 1 (PD-L1) are likely to help minimize potential side effects and enhances efficacy for treatment of cancers. Furthermore, the combination of anti-TGF-β therapies with various mechanisms of action might have greater efficacy against cancer and cardiac fibrosis.
WP, TL, and SM wrote the manuscript. HK reviewed and edited. All authors agree to submit the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by grants from JSPS KAKENHI Grant Number JP17H01525 and the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIP) (2017K1A1A2004511) (to HK), and Thailand Research Fund [Grant RSA6080061] (to SM).
1. Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor-β in human disease. N Engl J Med. (2000) 342:1350–8. doi: 10.1056/NEJM200005043421807
2. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. (2016) 12:325–8. doi: 10.1038/nrneph.2016.48
3. Roberts AB. Molecular and cell biology of TGF-beta. Miner Electrolyte Metab. (1998) 24:111–9. doi: 10.1159/000057358
4. Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-β pathway for cancer therapy. Clin Cancer Res. (2012) 18:4514–21. doi: 10.1158/1078-0432.CCR-11-3224
5. Xie F, Ling L, van Dam H, Zhou F, Zhang L. TGF-β signaling in cancer metastasis. Acta Biochim Biophys Sin. (2018) 50:121–32. doi: 10.1093/abbs/gmx123
6. Walton KL, Johnson KE, Harrison CA. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol. (2017) 8:461. doi: 10.3389/fphar.2017.00461
7. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
8. Pardali E, ten Dijke P. Transforming growth factor-β signaling and tumor angiogenesis. Front Biosci. (2009) 14:4848–61. doi: 10.2741/3573
9. Haque S, Morris JC. Transforming growth factor-β: a therapeutic target for cancer. Hum Vaccin Immunother. (2017) 13:1741–50. doi: 10.1080/21645515.2017.1327107
10. Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor-β in cardiomyopathy, valvular disease and arrhythmia. Immunology. (2006) 118:10–24. doi: 10.1111/j.1365-2567.2006.02336.x
11. Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Fact. (2011) 29:196–202. doi: 10.3109/08977194.2011.595714
12. Pohlers D, Brenmoehl J, Löffler I, Müller CK, Leipner C, Schultze-Mosgau S, et al. TGF-beta and fibrosis in different organs- molecular pathway imprints. Biochim Biophys Acta. (2009) 1792:746–56. doi: 10.1016/j.bbadis.2009.06.004
13. Najafi F, Jamrozik K, Dobson AJ. Understanding the 'epidemic of heart failure: a systematic review of trends in determinants of heart failure. Eur J Heart Fail. (2009) 11:472–9. doi: 10.1093/eurjhf/hfp029
14. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling- concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. (2000) 35:569–82. doi: 10.1016/S0735-1097(99)00630-0
15. Norton GR, Tsotetsi J, Trifunovic B, Hartford C, Candy GP, Woodiwiss AJ. Myocardial stiffness is attributed to alterations in cross-linked collagen rather than total collagen or phenotypes in spontaneously hypertensive rats. Circulation. (1997) 96:1991–8. doi: 10.1161/01.CIR.96.6.1991
16. Liu J, Masurekar MR, Vatner DE, Jyothirmayi GN, Regan TJ, Vatner SF, et al. Glycation end-product cross-link breaker reduces collagen and improves cardiac function in aging diabetic heart. Am J Physiol Heart Circ Physiol. (2003) 285:H2587–91. doi: 10.1152/ajpheart.00516.2003
17. Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor-β (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract. (2017) 133:124–30. doi: 10.1016/j.diabres.2017.08.018
18. Voskoboinik A, Costello BT, La Gerche A, Prabhu S, Wong G, Flannery MD, et al. Relation of alcohol consumption to left ventricular fibrosis using cardiac magnetic resonance imaging. Am J Cardiol. (2019) 123:460–5. doi: 10.1016/j.amjcard.2018.10.026
19. El Hajj EC, El Hajj MC, Voloshenyuk TG, Mouton AJ, Khoutorova E, Molina PE, et al. Alcohol modulation of cardiac matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs favors collagen accumulation. Alcohol Clin Exp Res. (2014) 38:448–56. doi: 10.1111/acer.12239
20. Ma ZG, Yuan YP, Wu HM, Zhang X, Tang QZ. Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci. (2018) 14:1645–57. doi: 10.7150/ijbs.28103
21. Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol. (2016) 90:84–93. doi: 10.1016/j.yjmcc.2015.12.011
22. Edgley AJ, Krum H, Kelly DJ. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor-β. Cardiovasc Ther. (2012) 30:e30–40. doi: 10.1111/j.1755-5922.2010.00228.x
23. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. (2014) 71:549–74. doi: 10.1007/s00018-013-1349-6
24. DeVita VT Jr, Rosenberg SA. Two hundred years of cancer research. N Engl J Med. (2012) 366:2207–14. doi: 10.1056/NEJMra1204479
25. Shewach DS, Kuchta RD. Introduction to cancer chemotherapeutics. Chem Rev. (2009) 109:2859–61. doi: 10.1021/cr900208x
26. Lumachi F, Luisetto G, Basso SM, Basso U, Brunello A, Camozzi V. Endocrine therapy of breast cancer. Curr Med Chem. (2011) 18:513–22. doi: 10.2174/092986711794480177
27. Tammela TA. Endocrine treatment of prostate cancer. J Steroid Biochem Mol Biol. (2004) 92:287–95. doi: 10.1016/j.jsbmb.2004.10.005
28. Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol. (2015) 12:3–20. doi: 10.2174/1570163812666150602144310
29. Zhang H, Chen J. Current status and future directions of cancer immunotherapy. J Cancer. (2018) 9:1773–81. doi: 10.7150/jca.24577
30. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. (2017) 7:339–48. doi: 10.15171/apb.2017.041
31. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5:1841–75. doi: 10.1002/cphy.c150006
32. Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. (2004) 94:1533–42. doi: 10.1161/01.RES.0000129254.25507.d6
33. Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. (2002) 27:92–8. doi: 10.1007/s00059-002-2354-y
34. López B, Querejeta R, González A, Larman M, Díez J. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension. (2012) 60:677–83. doi: 10.1161/HYPERTENSIONAHA.112.196113
35. Woodiwiss AJ, Tsotetsi OJ, Sprott S, Lancaster EJ, Mela T, Chung ES, et al. Reduction in myocardial collagen cross-linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation. (2001) 103:155–60. doi: 10.1161/01.CIR.103.1.155
36. Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin Electrophysiol. (1997) 20:397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x
37. de Bakker JM, van Capelle FJ, Janse MJ, Tasseron S, Vermeulen JT, de Jonge N, et al. Fractionated electrograms in dilated cardiomyopathy: origin and relation to abnormal conduction. J Am Coll Cardiol. (1996) 27:1071–8. doi: 10.1016/0735-1097(95)00612-5
38. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. (2002) 3:349–63. doi: 10.1038/nrm809
39. Sappino AP, Schürch W, Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Lab Invest. (1990) 63:144–61.
40. Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. (1994) 145:868–75.
41. van Putten S, Shafieyan Y, Hinz B. Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol. (2016) 93:133–42. doi: 10.1016/j.yjmcc.2015.11.025
42. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. (2016) 118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565
43. Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. (2019) 65:70–99. doi: 10.1016/j.mam.2018.07.001
44. Kurose H, Mangmool S. Myofibroblasts and inflammatory cells as players of cardiac fibrosis. Arch Pharm Res. (2016) 39:1100–13. doi: 10.1007/s12272-016-0809-6
45. Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. (2006) 291:H1015–26. doi: 10.1152/ajpheart.00023.2006
46. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogene Tissue Repair. (2012) 5:15. doi: 10.1186/1755-1536-5-15
47. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, et al. Transforming growth factor-β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. (2002) 106:130–5. doi: 10.1161/01.CIR.0000020689.12472.E0
48. Ono K, Ohtomo T, Ninomiya-Tsuji J, Tsuchiya M. A dominant negative TAK1 inhibits cellular fibrotic responses induced by TGF-β. Biochem Biophys Res Commun. (2003) 307:332–7. doi: 10.1016/S0006-291X(03)01207-5
49. See F, Thomas W, Way K, Tzanidis A, Kompa A, Lewis D, et al. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J Am Coll Cardiol. (2004) 44:1679–89. doi: 10.1016/j.jacc.2004.07.038
50. Tan SM, Zhang Y, Connelly KA, Gilbert RE, Kelly DJ. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. (2010) 298:H1415–25. doi: 10.1152/ajpheart.01048.2009
51. Engebretsen KV, Skårdal K, Bjørnstad S, Marstein HS, Skrbic B, Sjaastad I, et al. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J Mol Cell Cardiol. (2014) 76:148–57. doi: 10.1016/j.yjmcc.2014.08.008
52. Lucas JA, Zhang Y, Li P, Gong K, Miller AP, Hassan E, et al. Inhibition of transforming growth factor-β signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am J Physiol Heart Circ Physiol. (2010) 298:H424–32. doi: 10.1152/ajpheart.00529.2009
53. Xavier S, Piek E, Fujii M, Javelaud D, Mauviel A, Flanders KC, et al. Amelioration of radiation-induced fibrosis: inhibition of transforming growth factor-β signaling by halofuginone. J Biol Chem. (2004) 279:15167–76. doi: 10.1074/jbc.M309798200
54. Shimada YJ, Passeri JJ, Baggish AL, O'Callaghan C, Lowry PA, Yannekis G, et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. (2013) 1:480–7. doi: 10.1016/j.jchf.2013.09.001
55. Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. (2000) 102:1388–93. doi: 10.1161/01.CIR.102.12.1388
56. Galie PA, Russell MW, Westfall MV, Stegemann JP. Interstitial fluid flow and cyclic strain differentially regulate cardiac fibroblast activation via AT1R and TGF-β1. Exp Cell Res. (2012) 318:75–84. doi: 10.1016/j.yexcr.2011.10.008
57. Singh AD, Amit S, Kumar OS, Rajan M, Mukesh N. Cardioprotective effects of bosentan, a mixed endothelin type A and B receptor antagonist, during myocardial ischaemia and reperfusion in rats. Basic Clin Pharmacol Toxicol. (2006) 98:604–10. doi: 10.1111/j.1742-7843.2006.pto_405.x
58. Ammarguellat F, Larouche I, Schiffrin EL. Myocardial fibrosis in DOCA-salt hypertensive rats: effect of endothelin ETA receptor antagonism. Circulation. (2001) 103:319–24. doi: 10.1161/01.CIR.103.2.319
59. Wakeno M, Minamino T, Seguchi O, Okazaki H, Tsukamoto O, Okada K, et al. Long-term stimulation of adenosine A2B receptors begun after myocardial infarction prevents cardiac remodeling in rats. Circulation. (2006) 114:1923–32. doi: 10.1161/CIRCULATIONAHA.106.630087
60. Phosri S, Arieyawong A, Bunrukchai K, Parichatikanond W, Nishimura A, Nishida M, et al. Stimulation of adenosine A2B receptor inhibits endothelin-1-induced cardiac fibroblast proliferation and α-smooth muscle actin synthesis through the cAMP/Epac/PI3K/Akt-signaling pathway. Front Pharmacol. (2017) 8:428. doi: 10.3389/fphar.2017.00428
61. Phosri S, Bunrukchai K, Parichatikanond W, Sato VH, Mangmool S. Epac is required for exogenous and endogenous stimulation of adenosine A2B receptor for inhibition of angiotensin II-induced collagen synthesis and myofibroblast differentiation. Purinergic Signal. (2018) 14:141–56. doi: 10.1007/s11302-017-9600-5
62. Le Elizabeth D, Pascotto M, Leong-Poi H, Sari I, Micari A, Kaul S. Anti-inflammatory and pro-angiogenic effects of beta blockers in a canine model of chronic ischemic cardiomyopathy: comparison between carvedilol and metoprolol. Basic Res Cardiol. (2013) 108:384. doi: 10.1007/s00395-013-0384-7
63. Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. (2008) 103:413–22. doi: 10.1161/CIRCRESAHA.107.168336
64. Nuamnaichati N, Sato VH, Moongkarndi P, Parichatikanond W, Mangmool S. Sustained β-AR stimulation induces synthesis and secretion of growth factors in cardiac myocytes that affect on cardiac fibroblast activation. Life Sci. (2018) 193:257–69. doi: 10.1016/j.lfs.2017.10.034
65. Heldin CH, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. (1997) 390:465–71. doi: 10.1038/37284
66. Roth DA, Gold LI, Han VK, McCarthy JG, Sung JJ, Wisoff JH, et al. Immunolocalization of transforming growth factor-β1, -β2, and -β3 and insulin-like growth factor I in premature cranial suture fusion. Plast Reconstr Surg. (1997) 99:300–9. doi: 10.1097/00006534-199702000-00002
67. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. (2011) 51:600–6. doi: 10.1016/j.yjmcc.2010.10.033
68. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-β structure and activation. Nature. (2011) 474:343–9. doi: 10.1038/nature10152
69. Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-β receptor. Nature. (1994) 370:341–7. doi: 10.1038/370341a0
70. Loomans HA, Andl CD. Activin receptor-like kinases: a diverse family playing an important role in cancer. Am J Cancer Res. (2016) 6:2431–47.
71. Euler-Taimor G, Heger J. The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res. (2006) 69:15–25. doi: 10.1016/j.cardiores.2005.07.007
72. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. (2017) 127:3770–83. doi: 10.1172/JCI94753
73. Zhao M, Mishra L, Deng CX. The role of TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. (2018) 14:111–23. doi: 10.7150/ijbs.23230
74. Yan X, Liao H, Cheng M, Shi X, Lin X, Feng XH, et al. Smad7 protein interacts with receptor-regulated Smads (R-Smads) to inhibit transforming growth factor-β (TGF-β)/Smad signaling. J Biol Chem. (2016) 291:382–92. doi: 10.1074/jbc.M115.694281
75. Zhang YE. Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb Perspect Biol. (2017) 9:a022129. doi: 10.1101/cshperspect.a022129
76. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-β1-induced EMT in vitro. Neoplasia. (2004) 6:603–10. doi: 10.1593/neo.04241
77. Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. (2002) 115:3193–206. Retrived from: https://jcs.biologists.org/content/115/15/3193
78. Yu L, Hébert MC, Zhang YE. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. (2002) 21:3749–59. doi: 10.1093/emboj/cdf366
79. Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science. (2005) 307:1603–9. doi: 10.1126/science.1105718
80. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor-β-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. (2000) 275:36803–10. doi: 10.1074/jbc.M005912200
81. Lamouille S, Derynck R. Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. (2007) 178:437–51. doi: 10.1083/jcb.200611146
82. Suzuki HI. MicroRNA control of TGF-β signaling. Int J Mol Sci. (2018) 19:1901. doi: 10.3390/ijms19071901
83. Robson CN, Gnanapragasam V, Byrne RL, Collins AT, Neal DE. Transforming growth factor-β1 up-regulates p15, p21 and p27 and blocks cell cycling in G1 in human prostate epithelium. J Endocrinol. (1999) 160:257–66. doi: 10.1677/joe.0.1600257
84. Yagi K, Furuhashi M, Aoki H, Goto D, Kuwano H, Sugamura K, et al. C-myc is a downstream target of the smad pathway. J Biol Chem. (2002) 277:854–61. doi: 10.1074/jbc.M104170200
85. Spender LC, O'Brien DI, Simpson D, Dutt D, Gregory CD, Allday MJ, et al. TGF-β induces apoptosis in human B cells by transcriptional regulation of BIK and BCL-XL. Cell Death Differ. (2009) 16:593–602. doi: 10.1038/cdd.2008.183
86. Kim SG, Jong HS, Kim TY, Lee JW, Kim NK, Hong SH, et al. Transforming growth factor-β1 induces apoptosis through Fas ligand-independent activation of the Fas death pathway in human gastric SNU-620 carcinoma cells. Mol Biol Cell. (2004) 15:420–34. doi: 10.1091/mbc.e03-04-0201
87. Zonneville J, Safina A, Truskinovsky AM, Arteaga CL, Bakin AV. TGF-β signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer. (2018) 18:670. doi: 10.1186/s12885-018-4587-z
88. Smith AL, Iwanaga R, Drasin DJ, Micalizzi DS, Vartuli RL, Tan AC, et al. The miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and induces EMT and tumor initiating cell characteristics downstream of six1 in human breast cancer. Oncogene. (2012) 31:5162–71. doi: 10.1038/onc.2012.11
89. Metelli A, Wu BX, Fugle CW, Rachidi S, Sun S, Zhang Y, et al. Surface expression of TGFβ docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res. (2016) 76:7106–17. doi: 10.1158/0008-5472.CAN-16-1456
90. Takasaka N, Seed RI, Cormier A, Bondesson AJ, Lou J, Elattma A, et al. Integrin αvβ8-expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI Insight. (2018) 3:e122591. doi: 10.1172/jci.insight.122591
91. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. (2004) 127:S35–50. doi: 10.1053/j.gastro.2004.09.014
92. Taguchi S. Comprehensive review of the epidemiology and treatments for malignant adult cardiac tumors. Gen Thorac Cardiovasc Surg. (2018) 66:257–62. doi: 10.1007/s11748-018-0912-3
93. Frangogiannis NG. The role of transforming growth factor (TGF)-β in the infarcted myocardium. J Thorac Dis. (2017) 9(Suppl. 1):S52–63. doi: 10.21037/jtd.2016.11.19
94. Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. (2004) 164:665–77. doi: 10.1016/S0002-9440(10)63154-9
95. van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. (2007) 170:818–29. doi: 10.2353/ajpath.2007.060547
96. Wünsch M, Sharma HS, Markert T, Bernotat-Danielowski S, Schott RJ, Kremer P, et al. In situ localization of transforming growth factor-β1 in porcine heart: enhanced expression after chronic coronary artery constriction. J Mol Cell Cardiol. (1991) 23:1051–62. doi: 10.1016/0022-2828(91)91640-D
97. Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, et al. Complement C5a, TGF-β1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. (1997) 95:684–92. doi: 10.1161/01.CIR.95.3.684
98. Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H, et al. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc Res. (2014) 102:407–17. doi: 10.1093/cvr/cvu053
99. Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, et al. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. (2005) 111:2935–42. doi: 10.1161/CIRCULATIONAHA.104.510354
100. Lefer AM, Tsao P, Aoki N, Palladino MA Jr. Mediation of cardioprotection by transforming growth factor-β. Science. (1990) 249:61–4. doi: 10.1126/science.2164258
101. Baxter GF, Mocanu MM, Brar BK, Latchman DS, Yellon DM. Cardioprotective effects of transforming growth factor-β1 during early reoxygenation or reperfusion are mediated by p42/p44 MAPK. J Cardiovasc Pharmacol. (2001) 38:930–9. doi: 10.1097/00005344-200112000-00015
102. Schröder D, Heger J, Piper HM, Euler G. Angiotensin II stimulates apoptosis via TGF-β1 signaling in ventricular cardiomyocytes of rat. J Mol Med. (2006) 84:975–83. doi: 10.1007/s00109-006-0090-0
103. Celada A, Maki RA. Transforming growth factor-β enhances the M-CSF and GM-CSF-stimulated proliferation of macrophages. J Immunol. (1992) 148:1102–5.
104. Gorelik L, Flavell RA. Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. (2000) 12:171–81. doi: 10.1016/S1074-7613(00)80170-3
105. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. (2010) 107:418–28. doi: 10.1161/CIRCRESAHA.109.216101
106. Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. (2007) 116:2127–38. doi: 10.1161/CIRCULATIONAHA.107.704197
107. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. (2010) 225:631–7. doi: 10.1002/jcp.22322
108. Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, et al. Connective tissue growth factor mediates transforming growth factor-β-induced collagen synthesis: down-regulation by cAMP. FASEB J. (1999) 13:1774–86. doi: 10.1096/fasebj.13.13.1774
109. Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B. Differential effects of transforming growth factor-β1 and phorbol myristate acetate on cardiac fibroblasts. regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res. (1991) 69:483–90. doi: 10.1161/01.RES.69.2.483
110. Matsumoto-Ida M, Takimoto Y, Aoyama T, Akao M, Takeda T, Kita T. Activation of TGF-β1-TAK1-p38 MAPK pathway in spared cardiomyocytes is involved in left ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. (2006) 290:H709–15. doi: 10.1152/ajpheart.00186.2005
111. Wang S, Wilkes MC, Leof EB, Hirschberg R. Imatinib mesylate blocks a non-Smad TGF-β pathway and reduces renal fibrogenesis in vivo. FASEB J. (2005) 19:1–11. doi: 10.1096/fj.04-2370com
112. Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-β and prevents bleomycin-mediated lung fibrosis. J Clin Invest. (2004) 114:1308–16. doi: 10.1172/JCI200419603
113. Seeland U, Haeuseler C, Hinrichs R, Rosenkranz S, Pfitzner T, Scharffetter-Kochanek K, et al. Myocardial fibrosis in transforming growth factor-β1 (TGF-β1) transgenic mice is associated with inhibition of interstitial collagenase. Eur J Clin Invest. (2002) 32:295–303. doi: 10.1046/j.1365-2362.2002.00985.x
114. Frangogiannis NG. The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities. J Cardiovasc Pharmacol. (2014) 63:185–95. doi: 10.1097/FJC.0000000000000003
115. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. (2007) 13:952–61. doi: 10.1038/nm1613
116. Chen Y, Di C, Zhang X, Wang J, Wang F, Yan JF, et al. Transforming growth factor β signaling pathway: a promising therapeutic target for cancer. J Cell Physiol. (2020) 253:1903–14. doi: 10.1002/jcp.29108
117. Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. (2011) 39:916–24. doi: 10.1177/0192623311416259
118. Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. (2015) 147:22–31. doi: 10.1016/j.pharmthera.2014.11.001
119. Hau P, Jachimczak P, Schlingensiepen R, Schulmeyer F, Jauch T, Steinbrecher A, et al. Inhibition of TGF-β2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. (2007) 17:201–12. doi: 10.1089/oli.2006.0053
120. Zhang C, Zhang F, Tsan R, Fidler IJ. Transforming growth factor-β2 is a molecular determinant for site-specific melanoma metastasis in the brain. Cancer Res. (2009) 69:828–35. doi: 10.1158/0008-5472.CAN-08-2588
121. Jonson T, Albrechtsson E, Axelson J, Heidenblad M, Gorunova L, Johansson B, et al. Altered expression of TGFβ receptors and mitogenic effects of TGFβ in pancreatic carcinomas. Int J Oncol. (2001) 19:71–81. doi: 10.3892/ijo.19.1.71
122. Schlingensiepen KH, Jaschinski F, Lang SA, Moser C, Geissler EK, Schlitt HJ, et al. Transforming growth factor-β2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. (2011) 102:1193–200. doi: 10.1111/j.1349-7006.2011.01917.x
123. Bellone G, Carbone A, Tibaudi D, Mauri F, Ferrero I, Smirne C, et al. Differential expression of transforming growth factors-β1, -β2 and -β3 in human colon carcinoma. Eur J Cancer. (2001) 37:224–33. doi: 10.1016/S0959-8049(00)00391-9
124. Rowland-Goldsmith MA, Maruyama H, Kusama T, Ralli S, Korc M. Soluble type II transforming growth factor-beta (TGF-β) receptor inhibits TGF-β signaling in COLO-357 pancreatic cancer cells in vitro and attenuates tumor formation. Clin Cancer Res. (2001) 7:2931–40. Retrived from: https://clincancerres.aacrjournals.org/content/7/9/2931.article-info
125. Sakaguchi J, Kyo S, Kanaya T, Maida Y, Hashimoto M, Nakamura M, et al. Aberrant expression and mutations of TGF-β receptor type II gene in endometrial cancer. Gynecol Oncol. (2005) 98:427–33. doi: 10.1016/j.ygyno.2005.04.031
126. Copland JA, Luxon BA, Ajani L, Maity T, Campagnaro E, Guo H, et al. Genomic profiling identifies alterations in TGFβ signaling through loss of TGFβ receptor expression in human renal cell carcinogenesis and progression. Oncogene. (2003) 22:8053–62. doi: 10.1038/sj.onc.1206835
127. Finger EC, Turley RS, Dong M, How T, Fields TA, Blobe GC. TβRIII suppresses non-small cell lung cancer invasiveness and tumorigenicity. Carcinogenesis. (2008) 29:528–35. doi: 10.1093/carcin/bgm289
128. Bandyopadhyay A, López-Casillas F, Malik SN, Montiel JL, Mendoza V, Yang J, et al. Antitumor activity of a recombinant soluble betaglycan in human breast cancer xenograft. Cancer Res. (2002) 62:4690–5. Retrived from: https://cancerres.aacrjournals.org/content/62/16/4690
129. Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. (2015) 47:320–9. doi: 10.1038/ng.3225
130. Alsina-Sanchis E, Figueras A, Lahiguera Á, Vidal A, Casanovas O, Graupera M, et al. The TGFβ pathway stimulates ovarian cancer cell proliferation by increasing IGF1R levels. Int J Cancer. (2016) 139:1894–903. doi: 10.1002/ijc.30233
131. Dituri F, Mazzocca A, Fernando J, Peidrò FJ, Papappicco P, Fabregat I, et al. Differential inhibition of the TGF-β signaling pathway in HCC cells using the small molecule inhibitor LY2157299 and the D10 monoclonal antibody against TGF-β receptor type II. PLoS ONE. (2013) 8:e67109. doi: 10.1371/journal.pone.0067109
132. Dzieran J, Fabian J, Feng T, Coulouarn C, Ilkavets I, Kyselova A, et al. Comparative analysis of TGF-β/Smad signaling dependent cytostasis in human hepatocellular carcinoma cell lines. PLoS ONE. (2013) 8:e72252. doi: 10.1371/journal.pone.0072252
133. Serova M, Tijeras-Raballand A, Dos Santos C, Albuquerque M, Paradis V, Neuzillet C, et al. Effects of TGF-β signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget. (2015) 6:21614–27. doi: 10.18632/oncotarget.4308
134. Son JY, Park SY, Kim SJ, Lee SJ, Park SA, Kim MJ, et al. EW-7197, a novel ALK-5 kinase inhibitor, potently inhibits breast to lung metastasis. Mol Cancer Ther. (2014) 13:1704–16. doi: 10.1158/1535-7163.MCT-13-0903
135. Park CY, Son JY, Jin CH, Nam JS, Kim DK, Sheen YY. EW-7195, a novel inhibitor of ALK5 kinase inhibits EMT and breast cancer metastasis to lung. Eur J Cancer. (2011) 47:2642–53. doi: 10.1016/j.ejca.2011.07.007
136. Zhang B, Halder SK, Zhang S, Datta PK. Targeting transforming growth factor-β signaling in liver metastasis of colon cancer. Cancer Lett. (2009) 277:114–20. doi: 10.1016/j.canlet.2008.11.035
137. Mazzocca A, Fransvea E, Lavezzari G, Antonaci S, Giannelli G. Inhibition of transforming growth factor-β receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology. (2009) 50:1140–51. doi: 10.1002/hep.23118
138. Ge R, Rajeev V, Ray P, Lattime E, Rittling S, Medicherla S, et al. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-β type I receptor kinase in vivo. Clin Cancer Res. (2006) 12:4315–30. doi: 10.1158/1078-0432.CCR-06-0162
139. Gaspar NJ, Li L, Kapoun AM, Medicherla S, Reddy M, Li G, et al. Inhibition of transforming growth factor-β signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol Pharmacol. (2007) 72:152–61. doi: 10.1124/mol.106.029025
140. Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. (2011) 13:132–42. doi: 10.1093/neuonc/noq142
141. Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. (2006) 24:4721–30. doi: 10.1200/JCO.2005.05.5335
142. Nemunaitis J, Nemunaitis M, Senzer N, Snitz P, Bedell C, Kumar P, et al. Phase II trial of Belagenpumatucel-L, a TGF-β2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. (2009) 16:620–4. doi: 10.1038/cgt.2009.15
143. Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhász E, Ramlau R, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. (2015) 51:2321–9. doi: 10.1016/j.ejca.2015.07.035
144. Stevenson JP, Kindler HL, Papasavvas E, Sun J, Jacobs-Small M, Hull J, et al. Immunological effects of the TGFβ-blocking antibody GC1008 in malignant pleural mesothelioma patients. Oncoimmunology. (2013) 2:e26218. doi: 10.4161/onci.26218
145. Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal irradiation and systemic TGFβ blockade in metastatic breast cancer. Clin Cancer Res. (2018) 24:2493–504. doi: 10.1158/1078-0432.CCR-17-3322
146. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. (2018) 119:1208–14. doi: 10.1038/s41416-018-0246-z
147. Faivre SJ, Santoro A, Kelley RK, Merle P, Gane E, Douillard JY, et al. A phase 2 study of a novel transforming growth factor-β (TGF-β) receptor I kinase inhibitor, LY2157299 monohydrate (LY), in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol. (2014) 32:173. doi: 10.1200/jco.2014.32.3_suppl.lba173
148. Ryan CW, Matias C, Agulnik M, Lopez-Pousa A, Williams C, de Alwis DP, et al. A phase II study of tasisulam sodium (LY573636 sodium) as second-line or third-line treatment for patients with unresectable or metastatic soft tissue sarcoma. Invest New Drugs. (2013) 31:145–51. doi: 10.1007/s10637-012-9819-5
149. Schlingensiepen KH, Schlingensiepen R, Steinbrecher A, Hau P, Bogdahn U, Fischer-Blass B, et al. Targeted tumor therapy with the TGF-β2 antisense compound AP 12009. Cytokine Growth Factor Rev. (2006) 17:129–39. doi: 10.1016/j.cytogfr.2005.09.002
150. David JM, Dominguez C, Palena C. Pharmacological and immunological targeting of tumor mesenchymalization. Pharmacol Ther. (2017) 170:212–25. doi: 10.1016/j.pharmthera.2016.11.011
151. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-β (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE. (2014) 9:e90353. doi: 10.1371/journal.pone.0090353
152. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer. (2018) 6:47. doi: 10.1186/s40425-018-0356-4
153. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
154. de Gramont A, Faivre S, Raymond E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology. (2017) 6:e1257453. doi: 10.1080/2162402X.2016.1257453
155. Rodon J, Carducci MA, Sepulveda-Sánchez JM, Azaro A, Calvo E, Seoane J, et al. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res. (2015) 21:553–60. doi: 10.1158/1078-0432.CCR-14-1380
156. Kovacs RJ, Maldonado G, Azaro A, Fernández MS, Romero FL, Sepulveda-Sánchez JM, et al. Cardiac safety of TGF-β receptor I kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc Toxicol. (2015) 15:309–23. doi: 10.1007/s12012-014-9297-4
157. Gueorguieva I, Cleverly AL, Stauber A, Sada Pillay N, Rodon JA, Miles CP, et al. Defining a therapeutic window for the novel TGF-β inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br J Clin Pharmacol. (2014) 77:796–807. doi: 10.1111/bcp.12256
158. Park SA, Kim MJ, Park SY, Kim JS, Lee SJ, Woo HA, et al. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell Mol Life Sci. (2015) 72:2023–39. doi: 10.1007/s00018-014-1798-6
159. Song KM, Chung DY, Choi MJ, Ghatak K, Nguyen NM, Limanjaya A, et al. Vactosertib, a novel, orally bioavailable activin receptor-like kinase 5 inhibitor, promotes regression of fibrotic plaques in a rat model of Peyronie's disease. World J Mens Health. (2019). doi: 10.5534/wjmh.190071. [Epub ahead of print].
160. Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med. (2018) 10:eaan5488. doi: 10.1126/scitranslmed.aan5488
161. Fang L, Murphy AJ, Dart AM. A clinical perspective of anti-fibrotic therapies for cardiovascular disease. Front Pharmacol. (2017) 8:186. doi: 10.3389/fphar.2017.00186
162. Tomita H, Egashira K, Ohara Y, Takemoto M, Koyanagi M, Katoh M, et al. Early induction of transforming growth factor-β via angiotensin II type 1 receptors contributes to cardiac fibrosis induced by long-term blockade of nitric oxide synthesis in rats. Hypertension. (1998) 32:273–9. doi: 10.1161/01.HYP.32.2.273
163. Frantz S, Hu K, Adamek A, Wolf J, Sallam A, Maier SK, et al. Transforming growth factor-β inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. (2008) 103:485–92. doi: 10.1007/s00395-008-0739-7
164. Ihn H, Yamane K, Kubo M, Tamaki K. Blockade of endogenous transforming growth factor-β signaling prevents up-regulated collagen synthesis in scleroderma fibroblasts: association with increased expression of transforming growth factor-β receptors. Arthritis Rheum. (2001) 44:474–80. doi: 10.1002/1529-0131(200102)44:2<474::AID-ANR67>3.0.CO;2-#
165. Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, et al. Postinfarction gene therapy against transforming growth factor-β signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. (2005) 111:2430–7. doi: 10.1161/01.CIR.0000165066.71481.8E
166. Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci USA. (2001) 98:12283–8. doi: 10.1073/pnas.211086598
167. Yan W, Wang P, Zhao CX, Tang J, Xiao X, Wang DW. Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor-β/Smad and p38 mitogen- activated protein kinase signaling pathways. Hum Gene Ther. (2009) 20:1190–200. doi: 10.1089/hum.2008.204
168. Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, et al. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. (2000) 6:556–63. doi: 10.1038/75037
169. Park S, Nguyen NB, Pezhouman A, Ardehali R. Cardiac fibrosis: potential therapeutic targets. Transl Res. (2019) 209:121–37. doi: 10.1016/j.trsl.2019.03.001
170. de Oliveira FL, Araújo-Jorge TC, de Souza EM, de Oliveira GM, Degrave WM, Feige JJ, et al. Oral administration of GW788388, an inhibitor of transforming growth factor-β signaling, prevents heart fibrosis in Chagas disease. PLoS Negl Trop Dis. (2012) 6:e1696. doi: 10.1371/journal.pntd.0001696
171. Derangeon M, Montnach J, Cerpa CO, Jagu B, Patin J, Toumaniantz G, et al. Transforming growth factor-β receptor inhibition prevents ventricular fibrosis in a mouse model of progressive cardiac conduction disease. Cardiovasc Res. (2017) 113:464–74. doi: 10.1093/cvr/cvx026
172. Mirkovic S, Seymour AM, Fenning A, Strachan A, Margolin SB, Taylor SM, et al. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol. (2002) 135:961–8. doi: 10.1038/sj.bjp.0704539
173. Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. (2010) 7:1438–45. doi: 10.1016/j.hrthm.2010.04.030
174. Wang Y, Wu Y, Chen J, Zhao S, Li H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology. (2013) 126:1–11. doi: 10.1159/000351179
175. Yamagami K, Oka T, Wang Q, Ishizu T, Lee JK, Miwa K, et al. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am J Physiol Heart Circ Physiol. (2015) 309:H512–22. doi: 10.1152/ajpheart.00137.2015
176. Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol. (2001) 133:687–94. doi: 10.1038/sj.bjp.0704131
177. Martin J, Kelly DJ, Mifsud SA, Zhang Y, Cox AJ, See F, et al. Tranilast attenuates cardiac matrix deposition in experimental diabetes: role of transforming growth factor-β. Cardiovasc Res. (2005) 65:694–701. doi: 10.1016/j.cardiores.2004.10.041
178. Kelly DJ, Zhang Y, Connelly K, Cox AJ, Martin J, Krum H, et al. Tranilast attenuates diastolic dysfunction and structural injury in experimental diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2007) 293:H2860–9. doi: 10.1152/ajpheart.01167.2006
179. Kagitani S, Ueno H, Hirade S, Takahashi T, Takata M, Inoue H. Tranilast attenuates myocardial fibrosis in association with suppression of monocyte/macrophage infiltration in DOCA/salt hypertensive rats. J Hypertens. (2004) 22:1007–15. doi: 10.1097/00004872-200405000-00024
180. Hocher B, Godes M, Olivier J, Weil J, Eschenhagen T, Slowinski T, et al. Inhibition of left ventricular fibrosis by tranilast in rats with renovascular hypertension. J Hypertens. (2002) 20:745–51. doi: 10.1097/00004872-200204000-00034
181. See F, Watanabe M, Kompa AR, Wang BH, Boyle AJ, Kelly DJ, et al. Early and delayed tranilast treatment reduces pathological fibrosis following myocardial infarction. Heart Lung Circ. (2013) 22:122–32. doi: 10.1016/j.hlc.2012.08.054
182. Pinto YM, Pinto-Sietsma SJ, Philipp T, Engler S, Kossamehl P, Hocher B, et al. Reduction in left ventricular messenger RNA for transforming growth factor-β1 attenuates left ventricular fibrosis and improves survival without lowering blood pressure in the hypertensive TGR(mRen2)27 rat. Hypertension. (2000) 36:747–54. doi: 10.1161/01.HYP.36.5.747
183. Holmes DJ, Savage M, LaBlanche JM, Grip L, Serruys PW, Fitzgerald P, et al. Results of prevention of REStenosis with tranilast and its outcomes (PRESTO) trial. Circulation. (2002) 106:1243–50. doi: 10.1161/01.CIR.0000028335.31300.DA
184. Gellibert F, de Gouville AC, Woolven J, Mathews N, Nguyen VL, Bertho-Ruault C, et al. Discovery of 4-{4-[3-(pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl}-N-(tetrahydro-2H- pyran-4-yl)benzamide (GW788388): a potent, selective, and orally active transforming growth factor-β type I receptor inhibitor. J Med Chem. (2006) 49:2210–21. doi: 10.1021/jm0509905
185. Petersen M, Thorikay M, Deckers M, van Dinther M, Grygielko ET, Gellibert F, et al. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. (2008) 73:705–15. doi: 10.1038/sj.ki.5002717
186. King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2083–92. doi: 10.1056/NEJMoa1402582
187. Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on transforming growth factor-β gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. (1999) 291:367–73.
188. Lewis GA, Schelbert EB, Naish JH, Bedson E, Dodd S, Eccleson H, et al. Pirfenidone in heart failure with preserved ejection fraction-rationale and design of the PIROUETTE trial. Cardiovasc Drugs Ther. (2019) 33:461–70. doi: 10.1007/s10557-019-06876-y
Keywords: anticancer, antifibrotic, cancer, cardiac fibrosis, inhibitors of TGF-β signaling, transforming growth factor-β (TGF-β)
Citation: Parichatikanond W, Luangmonkong T, Mangmool S and Kurose H (2020) Therapeutic Targets for the Treatment of Cardiac Fibrosis and Cancer: Focusing on TGF-β Signaling. Front. Cardiovasc. Med. 7:34. doi: 10.3389/fcvm.2020.00034
Received: 30 November 2019; Accepted: 24 February 2020;
Published: 10 March 2020.
Edited by:
Tienush Rassaf, University Hospital Essen, GermanyReviewed by:
Alessandra Cuomo, Federico II University Hospital, ItalyCopyright © 2020 Parichatikanond, Luangmonkong, Mangmool and Kurose. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hitoshi Kurose, aGlrdXJvc2VAcGhhci5reXVzaHUtdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.