 Ahmed A. Y. Ragab
Ahmed A. Y. Ragab Gustaf D. S. Sitorus1
Gustaf D. S. Sitorus1 Bianca B. J. J. M. Brundel
Bianca B. J. J. M. Brundel
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 14 February 2020
Sec. Cardiovascular Genetics and Systems Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00014
This article is part of the Research Topic Innovative Approaches to Tackle Atrial Fibrillation: From Bench to Bedside View all 8 articles
Atrial fibrillation (AF) is the most common clinical tachyarrhythmia. In Europe, AF is expected to reach a prevalence of 18 million by 2060. This estimate will increase hospitalization for AF to 4 million and 120 million outpatient visits. Besides being an independent risk factor for mortality, AF is also associated with an increased risk of morbidities. Although there are many well-defined risk factors for developing AF, no identifiable risk factors or cardiac pathology is seen in up to 30% of the cases. The heritability of AF has been investigated in depth since the first report of familial atrial fibrillation (FAF) in 1936. Despite the limited value of animal models, the advances in molecular genetics enabled identification of many common and rare variants related to FAF. The importance of AF heritability originates from the high prevalence of lone AF and the lack of clear understanding of the underlying pathophysiology. A better understanding of FAF will facilitate early identification of people at high risk of developing FAF and subsequent development of more effective management options. In this review, we reviewed FAF epidemiological studies, identified common and rare variants, and discussed their clinical implications and contributions to developing new personalized therapeutic strategies.
Atrial fibrillation (AF) is the most common clinical arrhythmia with a rapidly increasing prevalence (1). By 2050, the prevalence of AF is expected to rise to 5.6–15.6 million in the USA (2, 3). AF is associated with an increased risk of complications such as stroke and heart failure (4). Many risk factors are related to the incidence of AF such as age, sex, valvular heart diseases, obesity, alcohol consumption, and hypertension. However, up to 30% of AF cases have no known cardiac pathology or known risk factors (Lone AF) (1). Inherited AF was first reported in the thirties of the last century (5). Recently, the heritability of AF has been recognized and investigated in depth (6–8).
The importance of studying the genetic contribution to AF comes from the high percentage of lone AF cases and the prevalence differences according to gender and among certain ethnic groups. Understanding the heritable component of AF will also facilitate early identification of people at high risk of developing AF later in their lives. For a long time, the limited value of AF animal models especially murine ones obfuscated the investigation of inherited AF. However, after emerging advances in the molecular genetics, many studies identified both rare and common genetic variants related to AF.
In this review, we highlight the findings of familial AF epidemiological studies, the role of both rare and common genetic variants as well as their clinical and therapeutic implications.
In the Framingham offspring study, those who had one parent with a history of AF had a 1.8-fold increase in the risk of developing AF. Interestingly, the risk was 3-fold higher in subjects <75 years (9). In the Mayo clinic AF registry, 5% of all patients and 15% of lone AF patients had a family history of AF (10). Among 5,000 Icelanders, the first degree relatives of AF patients were 1.77 times more at risk of developing AF than the general population (7). This relative risk reached 4.67 in patients <60 years. In the Danish twins' study, recurrence risk of AF was 12% for monozygotic twins and 22% for dizygotic twins (8). In another Danish cohort, the incidence rate ratio for lone AF was 3.48 in subjects who had affected first degree relatives and 1.64 in those whose second degree relatives were affected (11).
In the past two decades, many researchers tried to elucidate the genetic base of AF by using different types of studies such as linkage analysis, candidate gene analysis, and whole-genome next-generation sequencing. In 1997, Brugada and his coworkers reported the first genetic locus (10q22-q24) related to AF using the linkage analysis approach (12). A few years later, similar studies reported more genetic loci related to AF, namely 6q14-16, 5p13, 10p11-q21, 20q12-13, and 5p15 (13–16).
In 2003, Chen et al. reported the first gain of function mutation (KCNQ1) in the potassium voltage-gated channel in affected Chinese family. However, the candidate gene analysis was costly, time-consuming and restricted to a small number of scanned genes. Also, the causality effect theory of these variants was not clear as more than 30 different variants have been discovered in potassium channels genes.
Since 2003, many studies reported gain of function mutations in genes coding potassium channels (Table 1). Most of the reported variants were gain of function mutations though, loss of function mutation was also reported. The gain of function mutations result in shortening of the effective refractory period thereby increasing AF vulnerability. Other gain of function mutations have also been identified in KCNE1, KCNE2, KCNE, KCNE5, KCNQ1, and KCNJ2 genes (17, 20, 21, 23, 24, 57–59).
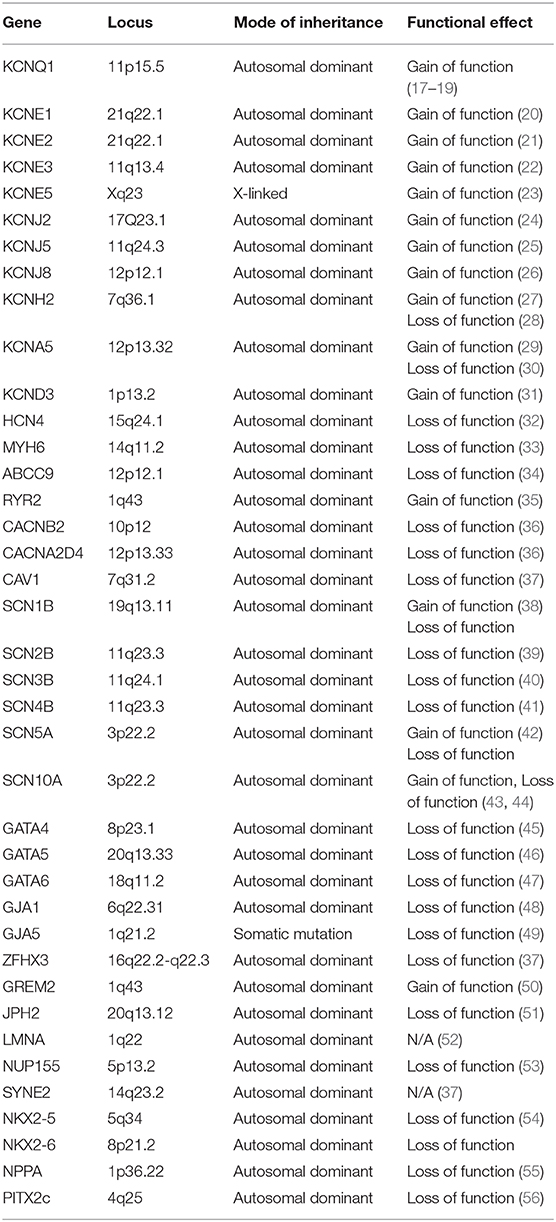
Table 1. Summary of gene loci associated with familial atrial fibrillation.
Mutations of KCNE1 and KCNQ1 affect Iks potassium channels by a gain of function effect which accelerates repolarization and hence, shortens the refractory period. However, mutations of KCNA5 affect Ikur potassium channels but with a loss of function mutation (30, 60). This mutation introduced an alternative mechanism for AF including delayed repolarization and prolongation of the effective refractory period.
In 2005, Olson et al. was the first to report an SCN5A mutation related to AF (61). These reported mutations are encoding α-subunit in Na 1.5 sodium channel. α-subunit gene mutations including genes encoding the four regulatory β-subunits (SCN1B, SCN2B, SCN3B, and SCN4B) are all related to AF (Table 1) (42, 61, 62). Uncovering the underlying mechanisms of these mutations has multiple challenges such as the mixed phenotypes reported, how both loss and gain of function mutations could cause these different phenotypes and the lack of an animal model with pure AF phenotype. Mutations in the SCN10A gene are related to AF. This gene encodes the NA 1.8 sodium channels which is believed to be responsible for late sodium currents and can be modulated by SCN5A level of expression (63).
Increased diastolic Ca2+ leak is one of the pathophysiological pathways to AF. Phosphorylation of RyR at PKA or CAMKII sites would lead to increased RyR opening probability and increased Ca2+ leak from sarcoplasmic reticulum (SR). Recently, a study showed that AF patients have less miRNA-106b-25 cluster with consequent increase in RyR expression and Ca2+ leak (64).
Gollob et al. described the first three somatic mutations in GJA5 gene related to AF; these mutations are responsible for impaired cell to cell coupling (49). This impairment is caused by depletion of atrial specific connexin 40. Moreover, Christophersen et al. described a germline mutation in the same gene. Mutations in gene encoding atrial natriuretic peptide (ANP) have been reported to be related to AF. It is believed that this mutation in ANP protein would shorten the action potential. In 2008, a mutation in the NUP155 gene encoding nucleoporin of the nuclear envelope was discovered. This mutation leads to alteration in nuclear envelope permeability. Many mutations in transcription factors genes have been reported to be related to AF such as NKX2-5, PITX2, ZFHX3, GATA4, GATA5, and GATA6 genes. GATA and PITX2 genes affect the development of the pulmonary venous myocardium which is involved in the initiation of AF (Table 1). Several studies reported an increased risk of AF with polymorphism of RAAS system genes encoding angiotensin converting enzyme inhibitor, angiotensin gene promotor, and angiotensinogen (24, 65).
in vitro methodologies for functionally characterizing the role of ion channels variants have drawbacks. For instance, AF cell lines continuously proliferate and are affected by rapid maturation, increased number of cells, and disorganized three-dimensional structure. In addition, not all areas within cell lines have the same metabolic activity. The evolving induced pluripotential stem cells is one step closer to the optimal in vivo conditions such as conduction properties, contraction and relaxation velocity, action potential duration, and repolarization fraction. Repolarization fraction is a parameter to distinguish between atrial and ventricular like human induced pluripotent stem cells (hiPSCs) and it is calculated based on the following equation: (APD90–APD50)/APD90), APD90; is action potential duration at 90% repolarization and APD 50 is action potential duration at 50% repolarization. However, these type of cells are electrophysiologically different from adult atrial cardiomyocytes in respect to Ca2+ handling and the predominance of ventricular like cells; ventricular contribution to the cell population can be minimized to <10% by using timed retinoic acid exposure.
In recent decades, murine models have drawn the attention of many investigators attempting to decode electrophysiological mechanism underlying AF. Murine models were considered a good candidate because of the conservation of development and signaling pathways between homo sapiens and mice, the ease of genetic manipulation, and rapid maturation.
Potassium channels mutation models have been studied such as the knockout models for KCNE1and SK2 channels (66–69). Moreover, sodium channel genes have been a target for transgenic models. ΔKPQ-SCN5A models showed more susceptibility to atrial arrhythmia (70–74). SCN3B subunit knockout models also showed conduction disturbances (75). Non-ion channels models also showed promising results such as connexin 40 and 43 models (76–78), Ankyrin B (79), and PITX2 (80). Knock out mice of spinophilin-1 leads to increased RyR phosphorylation and increases Ca2+ leak (81). The same results were also shown in junctophilin and FKBP-12.6 knock out models (51, 82).
Despite the value of these murine models, they have several limitations. One of the main limitations of these models is that AF was always induced in a non-physiological way. Other factors involved in clinical AF such as environmental factors, diet, and abuse of toxic substances were omitted. Although there is similarity in signaling pathways between mice and humans, there are important differences in heart rate, ion currents, calcium handling, and predominant myosin isoform.
In 2007, the first GWAS study on AF was published. By using a p-value of < 5 × 10−8 to minimize false positives, variant frequencies were compared between affected and non-affected subjects. The first detected locus was on chromosome 4q25 (83). However, this locus is in a non-coding area; studies revealed its role in regulating the closest gene (PITX2). This gene is essential for cardiac development and suppression of a sinus node development in pulmonary vein myocardium (left-right asymmetry). PITX2 knockout mice model showed a decrease in sodium and potassium channels expression and caused a conduction block at the atrioventricular node (84). Herraiz-Martínez et al. recently investigated whether chr4q25 risk variants alter the intracellular calcium homoeostasis. Patients carrying the rs13143308T risk variant show increased SERCA2a expression, SR calcium load, and RyR2 phosphorylation. These changes lead to excessive calcium release and a higher risk for AF (85).
In 2009, a novel locus on chromosome 16q22 was described in a cohort of European descent (86). The closest single nucleotide polymorphism (SNP) to this locus was intron to the zinc finger homeobox 3 (ZFHX3). This motif binding factor is required for regulation of the Pituitary-specific positive transcription factor 1 (POU1F1) which interacts with the PITX2 gene. In 2010, Ellinor et al. described a novel locus on chromosome 1q21. This SNP is located near KCNN3 gene which encodes voltage-independent calcium-activated potassium channel protein. These SK3 channels are essential for the repolarization phase of the cardiac action potential (87). These SK3 channels are also located in the inner mitochondrial membrane and opening of these channels using agonists have a protective effect against oxidative stress-induced injury resulting from Ca2+ overload (88).
Chromosome 15q24 also contained a locus related to AF and sinus node dysfunction. The closest gene to this SNP was HCN4 which encodes channels proteins regulating funny current of the sinoatrial node in the left atrium (89). Recently, two SNPs were discovered in the Japanese population on chromosome 12q24. The first SNP is located near NEURL gene. Knocking out this gene in zebrafish lead to action potential prolongation. The other SNP was intronic to CUX2 gene, however, the mechanism leading to AF is not clear yet.
An AF GWAS risk SNP on chr14q23 in the SYNE2 encodes nesprin-2 which is part of nuclear outer membrane and sarcomere (90, 91). Another non ion channel gene showed AF related SNP on chromosome 7q31, this locus is intronic to CAVI (caveolin-1) which has a role in repolarization phase of action potential and also has a structural role by regulating TGF-β-1 and fibrosis (92).
Recently, many studies investigated the role of cytoskeletal proteins in the pathogenesis of FAF. Two Islandic cohorts reported two novel SNPs in MYH6 and MYL4 genes (93, 94). MYH6 encodes the alpha myosin heavy chain subunit. Mutations in this subunit have been reported to affect cardiac contractility and muscles fibers integrity (95, 96). MYL4 encodes the essential myosin light chain subunit which is known as atrial light chain1. In vitro experiments on zebrafish with mutant MYL4 revealed loss of cardiac contractility and absence of sarcomere structure (97, 98). Another study supported the role of myocardial structure in FAF by the discovery of a missense variant in the PLEC gene (99). This gene encodes a cross-linking protein (plectin) which has a role in keeping the integrity of cardiac muscles. These studies suggest a strong role of cytoskeletal proteins in the pathogenesis of AF. A recent large GWAS meta-analysis showed that AF is associated with variants in 18 structural genes and also variants in 13 genes with a cardiac fetal developmental role such as ARNT2 and EPHA3 (100). This could explain the pathophysiology of AF as a result of atrial cardiomyopathy via cardiac structural remodeling either during fetal development or during adult life.
Another large GWAS study identified 134 AF associated loci among 93,000 AF cases and more than 1 million referents (101). This study showed that TBX3, TBX5, and NKX2-5 genes encode transcriptional factors that regulate development of the cardiac conduction system. This study also highlights the overlap between AF and other atrial arrhythmias and the pleiotropy of genes which are responsible for cardiac morphology and function. Nielsen et al. showed the relationship between AF and cardiac development and suggested that AF variants play a role in the developing heart or in reactivating fetal genes or pathways during adulthood as a response to stress and remodeling (100).
Despite the revolutionary output of GWAS studies, this approach of investigating heritability of FAF has several limitations. A large number of detected loci has only explained a small fraction of the missing heritability. This fact limits the clinical usage of outcomes of GWAS studies and urges the need for studies investigating gene-gene and gene-environment interactions. Another challenge is that approximately 80% of the discovered SNPs are in non-coding regions of the genome and this requires additional research to explore the exact causal variant by deploying techniques such as fine mapping, functional analyses, and evolutionary genetics.
There is no doubt that FAF is part of the uprising field of personalized medicine. Technological advances in genetics and a large amount of newly available data have encouraged many researchers to investigate the possible clinical value of this data to develop more efficient prediction models and personalized management strategies. The ORBIT-AF registry showed that FAF patients experienced more symptoms than non-FAF patients. However, there was no difference between the two groups regarding AF recurrences, hospitalization rate, complications, and all-cause mortality (102, 103). On the other hand, risk stratification based on genotype showed promising results. Husser et al. and Shoemaker et al. showed that patients with 4q25 SNP rs2200733 had an increased risk of developing recurrent AF after ablation (104, 105). Another study showed that AF patients with the same 4q25 SNP also had higher risks of developing AF recurrences after direct current cardioversion (HR:2.1, 95% CI: 1.21–3.3; P = 0.008) (106). The main limitations of these results are the small sample sizes and using the time to the first symptomatic episode which is a poor quantitative metrics for AF. Time to the first symptomatic AF episode does not take into account the frequency and length of AF episodes. Advances in continuous rhythm monitoring devices and AF detection algorithms will facilitate using AF burden as a more realistic, accurate and quantitative parameter for AF and also as a surrogate outcome after treatment. The effect of genotype on the success of ablative therapy was tested; likewise, response to antiarrhythmic drugs. Parvez et al. showed that the SNP rs10033464 at 4q25 is an independent predictor for success in rhythm control in both discovery and validation cohorts. Furthermore, they showed this same SNP is a predictor for AF recurrence in the same cohorts (107). Another study showed that flecainide potency is increased in AF patients with β1AR Arg389Arg genotype (108). Also, AF patients with the same genotype have a better response to rate control therapy and required lower doses of these drugs (109). One of the main limitations of these studies is the lack of randomization. Data were analyzed retrospectively and drug response was evaluated a priori without knowledge of the genotype.
Few studies tried to implement genotype into prediction models of de novo AF. In 2013, AF-genetic risk score (AF-GRS) was introduced. This score consisted of 12 risk alleles in nine loci. They investigated the predictive value of this score in 20000 females without cardiovascular disease at baseline. Adding this score to the main prediction model increased the area under the curve to (0.74) (110). In 2014, Tada and his colleagues showed that multiple single nucleotide polymorphisms can improve the prediction to develop AF and ischemic stroke. GRS score showed a potential value as an indicator for anticoagulant therapy (111). The main limitation of these studies is their lack of external validity to other ethnic groups such as Africans or Asians.
For postoperative AF, few studies have tried to replicate this approach but results are still controversial to improve prediction models performance as these studies lacked large sample sizes and did not use continuous ECG monitoring to identify AF episodes (112–114). In 2016, Lin and his colleagues investigated if gene-gene interaction would affect AF susceptibility. However, this study could not find any significant association and a larger cohort containing participants from other ethnic groups is indeed justified (115).
Translating the advances achieved in genetic technology into clinical practice still has many limitations with respect to genetic based prediction models and personalized therapeutic strategies.
Firstly, prediction models still have insufficient discriminative ability between low and high-risk individuals for several reasons such as testing small number of variants, potential gene-gene interactions, and gene-environment interactions. Moreover, the cost and logistic aspects have to be considered while moving this prediction model into clinical use. Secondly, applications of pharmacogenetics guided therapy are limited.
Another limitation is that pathophysiological pathways underlying AF genetic variants are not clear which delays attempts to target certain pathways caused by specific genetic variants. The multifactorial complex nature of AF could also limit the efficacy of any new drug development. In addition, involvement of multiple genetic variants in a patient is more challenging for a personalized efficient treatment strategy.
Despite the advances in our understanding of FAF, there are still many challenges and questions to be addressed. Firstly, large cohorts are needed to study the effect of gene-gene and gene-environment interactions on AF. These cohorts should consider larger sample sizes, participation of non-European ancestry and analyzing interactions between more than two variants. Secondly, randomized controlled trials are needed to validate the effect of genotype guided treatment strategies. Advances in rhythm monitoring devices and rhythm detection algorithms are needed in addition to using AF burden as a reliable parameter to quantify AF.
Larger cohorts are needed to investigate the effect of genotype guided prediction models of AF incidence, AF complications and mortality. Last but not least, large and effective screening studies for families with FAF is advised to uncover part of the missing heritability of FAF. For instance exome sequencing and whole genome sequencing projects would discover more missing rare and structural variants which GWAS studies cannot identify.
Genetic basis and heritability of AF is part of the complexity of this arrhythmia and a lot of progress has been achieved in many aspects such as risk stratification for AF, identification of novel therapeutic targets, and genome-based prediction models. There is no doubt that better understanding of AF heritability will not only improve AF prediction models but will also be the next step toward more efficient personalized treatment strategies.
AR and GS: drafting article and interpretation. BB and NG: critical revision and approval of article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. (2014) 129:837–47. doi: 10.1161/CIRCULATIONAHA.113.005119
2. Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. (2001) 285:2370–5. doi: 10.1001/jama.285.18.2370
3. Miyasaka Y, Barnes ME, Gersh BJ, Cha SS, Bailey KR, Abhayaratna WP, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. (2006) 114:119–25. doi: 10.1161/CIRCULATIONAHA.105.595140
4. Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. (2014) 114:1453–68. doi: 10.1161/CIRCRESAHA.114.303211
5. Orgain ES, Wolff L, White PD. Uncomplicated auricular fibrillation and auricular flutter: Frequent occurrence and good prognosis in patients without other evidence of cardiac disease. Arch Internal Med. (1936) 57:493–513. doi: 10.1001/archinte.1936.00170070018002
6. Lubitz SA, Yin X, Fontes JD, Magnani JW, Rienstra M, Pai M, et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA. (2010) 304:2263–9. doi: 10.1001/jama.2010.1690
7. Arnar DO, Thorvaldsson S, Manolio TA, Thorgeirsson G, Kristjansson K, Hakonarson H, et al. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. (2006) 27:708–12. doi: 10.1093/eurheartj/ehi727
8. Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. (2009) 2:378–83. doi: 10.1161/CIRCEP.108.786665
9. Fox CS, Parise H, D'Agostino RB Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. (2004) 291:2851–5. doi: 10.1001/jama.291.23.2851
10. Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, et al. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol. (2003) 41:2185–92. doi: 10.1016/S0735-1097(03)00465-0
11. Øyen N, Ranthe MF, Carstensen L, Boyd HA, Olesen MS, Olesen S-P, et al. Familial aggregation of lone atrial fibrillation in young persons. J Am Coll Cardiol. (2012) 60:917–21. doi: 10.1016/j.jacc.2012.03.046
12. Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, Mont L, et al. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med. (1997) 336:905–11. doi: 10.1056/NEJM199703273361302
13. Ellinor PT, Shin JT, Moore RK, Yoerger DM, MacRae CA. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation. (2003) 107:2880–3. doi: 10.1161/01.CIR.0000077910.80718.49
14. Oberti C, Wang L, Li L, Dong J, Rao S, Du W, et al. Genome-wide linkage scan identifies a novel genetic locus on chromosome 5p13 for neonatal atrial fibrillation associated with sudden death and variable cardiomyopathy. Circulation. (2004) 110:3753–9. doi: 10.1161/01.CIR.0000150333.87176.C7
15. Volders PG, Zhu Q, Timmermans C, Eurlings PM, Su X, Arens YH, et al. Mapping a novel locus for familial atrial fibrillation on chromosome 10p11-q21. Heart Rhythm. (2007) 4:469–75. doi: 10.1016/j.hrthm.2006.12.023
16. Darbar D, Hardy A, Haines JL, Roden DM. Prolonged signal-averaged P-wave duration as an intermediate phenotype for familial atrial fibrillation. J Am Coll Cardiol. (2008) 51:1083–9. doi: 10.1016/j.jacc.2007.11.058
17. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. (2003) 299:251–4. doi: 10.1126/science.1077771
18. Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. (2005) 68:433–40. doi: 10.1016/j.cardiores.2005.06.023
19. Das S, Makino S, Melman YF, Shea MA, Goyal SB, Rosenzweig A, et al. Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart Rhythm. (2009) 6:1146–53. doi: 10.1016/j.hrthm.2009.04.015
20. Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, et al. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet. (2012) 13:24. doi: 10.1186/1471-2350-13-24
21. Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. (2004) 75:899–905. doi: 10.1086/425342
22. Lundby A, Ravn LS, Svendsen JH, Hauns S, Olesen SP, Schmitt N. KCNE3 mutation V17M identified in a patient with lone atrial fibrillation. Cell Physiol Biochem. (2008) 21:47–54. doi: 10.1159/000113746
23. Ravn LS, Aizawa Y, Pollevick GD, Hofman-Bang J, Cordeiro JM, Dixen U, et al. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm. (2008) 5:427–35. doi: 10.1016/j.hrthm.2007.12.019
24. Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res. (2012) 93:666–73. doi: 10.1093/cvr/cvr329
25. Noriaki Y, Yoshihiro A, Masashi F, Satoru Y, Atsushi I, Norio M, et al. Mutant KCNJ3 and KCNJ5 potassium channels as novel molecular targets in bradyarrhythmias and atrial fibrillation. Circulation. (2019) 139:2157–69. doi: 10.1161/CIRCULATIONAHA.118.036761
26. Delaney JT, Muhammad R, Blair MA, Kor K, Fish FA, Roden DM, et al. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace. (2012) 14:1428–32. doi: 10.1093/europace/eus150
27. Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. (2005) 16:394–6. doi: 10.1046/j.1540-8167.2005.40621.x
28. Sinner MF, Pfeufer A, Akyol M, Beckmann BM, Hinterseer M, Wacker A, et al. The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: results from a systematic candidate gene-based analysis of KCNH2 (HERG). Eur Heart J. (2008) 29:907–14. doi: 10.1093/eurheartj/ehm619
29. Christophersen IE, Olesen MS, Liang B, Andersen MN, Larsen AP, Nielsen JB, et al. Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur Heart J. (2013) 34:1517–25. doi: 10.1093/eurheartj/ehs442
30. Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. (2006) 15:2185–91. doi: 10.1093/hmg/ddl143
31. Olesen MS, Refsgaard L, Holst AG, Larsen AP, Grubb S, Haunso S, et al. A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc Res. (2013) 98:488–95. doi: 10.1093/cvr/cvt028
32. Macri V, Mahida SN, Zhang ML, Sinner MF, Dolmatova EV, Tucker NR, et al. A novel trafficking-defective HCN4 mutation is associated with early-onset atrial fibrillation. Heart Rhythm. (2014) 11:1055–62. doi: 10.1016/j.hrthm.2014.03.002
33. Orr N, Arnaout R, Gula LJ, Spears DA, Leong-Sit P, Li Q, et al. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun. (2016) 7:11303. doi: 10.1038/ncomms11303
34. Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, et al. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. (2007) 4:110–6. doi: 10.1038/ncpcardio0792
35. Zhabyeyev P, Hiess F, Wang R, Liu Y, Wayne Chen SR, Oudit GY. S4153R is a gain-of-function mutation in the cardiac Ca(2+) release channel ryanodine receptor associated with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. Can J Cardiol. (2013) 29:993–6. doi: 10.1016/j.cjca.2012.12.019
36. Zhang Q, Chen J, Qin Y, Wang J, Zhou L. Mutations in voltage-gated L-type calcium channel: implications in cardiac arrhythmia. Channels. (2018) 12:201–18. doi: 10.1080/19336950.2018.1499368
37. Tsai CT, Hsieh CS, Chang SN, Chuang EY, Juang JM, Lin LY, et al. Next-generation sequencing of nine atrial fibrillation candidate genes identified novel de novo mutations in patients with extreme trait of atrial fibrillation. J Med Genet. (2015) 52:28–36. doi: 10.1136/jmedgenet-2014-102618
38. Olesen MS, Holst AG, Svendsen JH, Haunso S, Tfelt-Hansen J. SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm. (2012) 9:770–3. doi: 10.1016/j.hrthm.2011.12.005
39. Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. (2009) 2:268–75. doi: 10.1161/CIRCEP.108.779181
40. Olesen MS, Jespersen T, Nielsen JB, Liang B, Moller DV, Hedley P, et al. Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc Res. (2011) 89:786–93. doi: 10.1093/cvr/cvq348
41. Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu X, et al. Mutations of the SCN4B-encoded sodium channel beta4 subunit in familial atrial fibrillation. Int J Mol Med. (2013) 32:144–50. doi: 10.3892/ijmm.2013.1355
42. Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. (2008) 117:1927–35. doi: 10.1161/CIRCULATIONAHA.107.757955
43. Jabbari J, Olesen MS, Yuan L, Nielsen JB, Liang B, Macri V, et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ Cardiovasc Genet. (2015) 8:64–73. doi: 10.1161/CIRCGENETICS.113.000442
44. Savio-Galimberti E, Weeke P, Muhammad R, Blair M, Ansari S, Short L, et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc Res. (2014) 104:355–63. doi: 10.1093/cvr/cvu170
45. Yang YQ, Wang MY, Zhang XL, Tan HW, Shi HF, Jiang WF, et al. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin Chim Acta. (2011) 412:1825–30. doi: 10.1016/j.cca.2011.06.017
46. Wang XH, Huang CX, Wang Q, Li RG, Xu YJ, Liu X, et al. A novel GATA5 loss-of-function mutation underlies lone atrial fibrillation. Int J Mol Med. (2013) 31:43–50. doi: 10.3892/ijmm.2012.1189
47. Li J, Liu WD, Yang ZL, Yang YQ. Novel GATA6 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. (2012) 30:783–90. doi: 10.3892/ijmm.2012.1068
48. Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, et al. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation. (2010) 122:236–44. doi: 10.1161/CIRCULATIONAHA.110.961227
49. Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. (2006) 354:2677–88. doi: 10.1056/NEJMoa052800
50. Müller II, Melville DB, Tanwar V, Rybski WM, Mukherjee A, Shoemaker MB, et al. Functional modeling in zebrafish demonstrates that the atrial-fibrillation-associated gene < em>GREM2 < /em> regulates cardiac laterality, cardiomyocyte differentiation and atrial rhythm. Dis Models Mech. (2013) 6:332–41. doi: 10.1242/dmm.010488
51. Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. (2013) 62:2010–9. doi: 10.1016/j.jacc.2013.06.052
52. Pan H, Richards AA, Zhu X, Joglar JA, Yin HL, Garg V. A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm. (2009) 6:707–10. doi: 10.1016/j.hrthm.2009.01.037
53. Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. (2008) 135:1017–27. doi: 10.1016/j.cell.2008.10.022
54. Yuan F, Qiu XB, Li RG, Qu XK, Wang J, Xu YJ, et al. A novel NKX2–5 loss-of-function mutation predisposes to familial dilated cardiomyopathy and arrhythmias. Int J Mol Med. (2015) 35:478–86. doi: 10.3892/ijmm.2014.2029
55. Ren X, Xu C, Zhan C, Yang Y, Shi L, Wang F, et al. Identification of NPPA variants associated with atrial fibrillation in a Chinese GeneID population. Clin Chim Acta. (2010) 411:481–5. doi: 10.1016/j.cca.2009.12.019
56. Wang J, Zhang D-F, Sun Y-M, Yang Y-Q. A novel PITX2c loss-of-function mutation associated with familial atrial fibrillation. Eur J Med Genet. (2014) 57:25–31. doi: 10.1016/j.ejmg.2013.11.004
57. Otway R, Vandenberg JI, Guo G, Varghese A, Castro ML, Liu J, et al. Stretch-sensitive KCNQ1 mutation A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J Am Coll Cardiol. (2007) 49:578–86. doi: 10.1016/j.jacc.2006.09.044
58. Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR, Ackerman MJ. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm. (2008) 5:704–9. doi: 10.1016/j.hrthm.2008.02.007
59. Kharche S, Garratt CJ, Boyett MR, Inada S, Holden AV, Hancox JC, et al. Atrial proarrhythmia due to increased inward rectifier current (I(K1)) arising from KCNJ2 mutation–a simulation study. Prog Biophys Mol Biol. (2008) 98:186–97. doi: 10.1016/j.pbiomolbio.2008.10.010
60. Yang Y, Li J, Lin X, Yang Y, Hong K, Wang L, et al. Novel KCNA5 loss-of-function mutations responsible for atrial fibrillation. J Hum Genet. (2009) 54:277–83. doi: 10.1038/jhg.2009.26
61. Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. (2005) 293:447–54. doi: 10.1001/jama.293.4.447
62. Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, et al. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol. (2008) 52:1326–34. doi: 10.1016/j.jacc.2008.07.013
63. Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. (2012) 111:322–32. doi: 10.1161/CIRCRESAHA.112.265173
64. Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, et al. Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ Arrhythm Electrophysiol. (2014) 7:1214–22. doi: 10.1161/CIRCEP.114.001973
65. Zhao LQ, Wen ZJ, Wei Y, Xu J, Chen Z, Qi BZ, et al. Polymorphisms of renin-angiotensin-aldosterone system gene in chinese han patients with nonfamilial atrial fibrillation. PLoS ONE. (2015) 10:e0117489. doi: 10.1371/journal.pone.0117489
66. Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, et al. Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci. (2004) 24:5301–6. doi: 10.1523/JNEUROSCI.0182-04.2004
67. Li N, Timofeyev V, Tuteja D, Xu D, Lu L, Zhang Q, et al. Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol. (2009) 587(Pt 5):1087–100. doi: 10.1113/jphysiol.2008.167718
68. Kupershmidt S, Yang T, Anderson ME, Wessels A, Niswender KD, Magnuson MA, et al. Replacement by homologous recombination of the minK gene with lacZ reveals restriction of minK expression to the mouse cardiac conduction system. Circ Res. (1999) 84:146–52. doi: 10.1161/01.RES.84.2.146
69. Temple J, Frias P, Rottman J, Yang T, Wu Y, Verheijck EE, et al. Atrial fibrillation in KCNE1-null mice. Circ Res. (2005) 97:62–9. doi: 10.1161/01.RES.0000173047.42236.88
70. Dautova Y, Zhang Y, Sabir I, Grace AA, Huang CL. Atrial arrhythmogenesis in wild-type and Scn5a+/delta murine hearts modelling LQT3 syndrome. Pflugers Arch. (2009) 458:443–57. doi: 10.1007/s00424-008-0633-z
71. Blana A, Kaese S, Fortmuller L, Laakmann S, Damke D, van Bragt K, et al. Knock-in gain-of-function sodium channel mutation prolongs atrial action potentials and alters atrial vulnerability. Heart Rhythm. (2010) 7:1862–9. doi: 10.1016/j.hrthm.2010.08.016
72. Guzadhur L, Pearcey SM, Duehmke RM, Jeevaratnam K, Hohmann AF, Zhang Y, et al. Atrial arrhythmogenicity in aged Scn5a+/DeltaKPQ mice modeling long QT type 3 syndrome and its relationship to Na+ channel expression and cardiac conduction. Pflugers Arch. (2010) 460:593–601. doi: 10.1007/s00424-010-0851-z
73. Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, et al. Paced electrogram fractionation analysis of arrhythmogenic tendency in DeltaKPQ Scn5a mice. J Cardiovasc Electrophysiol. (2005) 16:1329–40. doi: 10.1111/j.1540-8167.2005.00200.x
74. Wu J, Zhang Y, Zhang X, Cheng L, Lammers WJ, Grace AA, et al. Altered sinoatrial node function and intra-atrial conduction in murine gain-of-function Scn5a+/DeltaKPQ hearts suggest an overlap syndrome. Am J Physiol Heart Circ Physiol. (2012) 302:H1510–23. doi: 10.1152/ajpheart.00357.2011
75. Hakim P, Brice N, Thresher R, Lawrence J, Zhang Y, Jackson AP, et al. Scn3b knockout mice exhibit abnormal sino-atrial and cardiac conduction properties. Acta Physiol. (2010) 198:47–59. doi: 10.1111/j.1748-1716.2009.02048.x
76. Hagendorff A, Schumacher B, Kirchhoff S, Luderitz B, Willecke K. Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation. (1999) 99:1508–15. doi: 10.1161/01.CIR.99.11.1508
77. Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, et al. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol. (2007) 292:H1561–7. doi: 10.1152/ajpheart.00285.2006
78. Kirchhof P, Marijon E, Fabritz L, Li N, Wang W, Wang T, et al. Overexpression of cAMP-response element modulator causes abnormal growth and development of the atrial myocardium resulting in a substrate for sustained atrial fibrillation in mice. Int J Cardiol. (2013) 166:366–74. doi: 10.1016/j.ijcard.2011.10.057
79. Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. (2011) 124:1212–22. doi: 10.1161/CIRCULATIONAHA.111.023986
80. Wang J, Klysik E, Sood S, Johnson RL, Wehrens XH, Martin JF. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc Natl Acad Sci USA. (2010) 107:9753–8. doi: 10.1073/pnas.0912585107
81. Chiang DY, Li N, Wang Q, Alsina KM, Quick AP, Reynolds JO, et al. Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovasc Res. (2014) 103:178–87. doi: 10.1093/cvr/cvu123
82. Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, et al. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. (2008) 5:1047–54. doi: 10.1016/j.hrthm.2008.03.030
83. Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. (2007) 448:353–7. doi: 10.1038/nature06007
84. Tessari A, Pietrobon M, Notte A, Cifelli G, Gage PJ, Schneider MD, et al. Myocardial Pitx2 differentially regulates the left atrial identity and ventricular asymmetric remodeling programs. Circ Res. (2008) 102:813–22. doi: 10.1161/CIRCRESAHA.107.163188
85. Herraiz-Martínez A, Llach A, Tarifa C, Gandía J, Jiménez-Sabado V, Lozano-Velasco E, et al. The 4q25 variant rs13143308T links risk of atrial fibrillation to defective calcium homoeostasis. Cardiovasc Res. (2018) 115:578–89. doi: 10.1093/cvr/cvy215
86. Gudbjartsson DF, Holm H, Gretarsdottir S, Thorleifsson G, Walters GB, Thorgeirsson G, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. (2009) 41:876–8. doi: 10.1038/ng.417
87. Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. (2010) 42:240–4. doi: 10.1038/ng.537
88. Yang M, Camara AKS, Aldakkak M, Kwok W-M, Stowe DF. Identity and function of a cardiac mitochondrial small conductance Ca2+-activated K+ channel splice variant. Biochim Biophys Acta Bioenerg. (2017) 1858:442–58. doi: 10.1016/j.bbabio.2017.03.005
89. Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. (2012) 44:670–5. doi: 10.1038/ng.2261
90. Homer RJ, Herzog EL. Recent advances in pulmonary fibrosis: implications for scleroderma. Curr Opin Rheumatol. (2010) 22:683–9. doi: 10.1097/BOR.0b013e32833ddcc9
91. He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. (2009) 20:765–76. doi: 10.1681/ASN.2008060566
92. Volonte D, McTiernan CF, Drab M, Kasper M, Galbiati F. Caveolin-1 and caveolin-3 form heterooligomeric complexes in atrial cardiac myocytes that are required for doxorubicin-induced apoptosis. Am J Physiol Heart Circ Physiol. (2008) 294:H392–401. doi: 10.1152/ajpheart.01039.2007
93. Holm H, Gudbjartsson DF, Sulem P, Masson G, Helgadottir HT, Zanon C, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. (2011) 43:316–20. doi: 10.1038/ng.781
94. Gudbjartsson DF, Holm H, Sulem P, Masson G, Oddsson A, Magnusson OT, et al. A frameshift deletion in the sarcomere gene MYL4 causes early-onset familial atrial fibrillation. Eur Heart J. (2016) 38:27–34. doi: 10.1093/eurheartj/ehw379
95. Elisa C, Matthew RGT, Gianfranco S, Andrea Di L, Lisa K, Pamela RF, et al. α-myosin heavy chain. Circulation. (2005) 112:54–9. doi: 10.1161/CIRCULATIONAHA.104.507699
96. Granados-Riveron JT, Ghosh TK, Pope M, Bu'Lock F, Thornborough C, Eason J, et al. α-Cardiac myosin heavy chain (MYH6) mutations affecting myofibril formation are associated with congenital heart defects. Human Mol Genet. (2010) 19:4007–16. doi: 10.1093/hmg/ddq315
97. Meder B, Laufer C, Hassel D, Just S, Marquart S, Vogel B, et al. A single serine in the carboxyl terminus of cardiac essential myosin light chain-1 controls cardiomyocyte contractility in vivo. Circ Res. (2009) 104:650–9. doi: 10.1161/CIRCRESAHA.108.186676
98. Chen Z, Huang W, Dahme T, Rottbauer W, Ackerman MJ, Xu X. Depletion of zebrafish essential and regulatory myosin light chains reduces cardiac function through distinct mechanisms. Cardiovasc Res. (2008) 79:97–108. doi: 10.1093/cvr/cvn073
99. Thorolfsdottir RB, Sveinbjornsson G, Sulem P, Helgadottir A, Gretarsdottir S, Benonisdottir S, et al. A missense variant in PLEC increases risk of atrial fibrillation. J Am Coll Cardiol. (2017) 70:2157–68. doi: 10.1016/j.jacc.2017.09.005
100. Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. (2018) 50:1234–9. doi: 10.1038/s41588-018-0171-3
101. Roselli C, Chaffin MD, Weng LC, Aeschbacher S, Ahlberg G, Albert CM, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. (2018) 50:1225–33. doi: 10.1038/s41588-018-0133-9
102. Gundlund A, Fosbol EL, Kim S, Fonarow GC, Gersh BJ, Kowey PR, et al. Family history of atrial fibrillation is associated with earlier-onset and more symptomatic atrial fibrillation: Results from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF) registry. Am Heart J. (2016) 175:28–35. doi: 10.1016/j.ahj.2016.01.020
103. Gundlund A, Olesen JB, Staerk L, Lee C, Piccini JP, Peterson ED, et al. Outcomes associated with familial versus nonfamilial atrial fibrillation: a matched nationwide cohort study. J Am Heart Assoc. (2016) 5:e003836. doi: 10.1161/JAHA.116.003836
104. Shoemaker MB, Andreas B, Steven AL, Laura U, Harsimran S, Jay M, et al. Common genetic variants and response to atrial fibrillation ablation. Circ Arrhythm Electrophysiol. (2015) 8:296–302. doi: 10.1161/CIRCEP.114.001909
105. Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol. (2010) 55:747–53. doi: 10.1016/j.jacc.2009.11.041
106. Parvez B, Shoemaker MB, Muhammad R, Richardson R, Jiang L, Blair MA, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm. (2013) 10:849–55. doi: 10.1016/j.hrthm.2013.02.018
107. Parvez B, Vaglio J, Rowan S, Muhammad R, Kucera G, Stubblefield T, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol. (2012) 60:539–45. doi: 10.1016/j.jacc.2012.01.070
108. Nia AM, Caglayan E, Gassanov N, Zimmermann T, Aslan O, Hellmich M, et al. Beta1-adrenoceptor polymorphism predicts flecainide action in patients with atrial fibrillation. PLoS ONE. (2010) 5:e11421. doi: 10.1371/journal.pone.0011421
109. Parvez B, Chopra N, Rowan S, Vaglio JC, Muhammad R, Roden DM, et al. A common beta1-adrenergic receptor polymorphism predicts favorable response to rate-control therapy in atrial fibrillation. J Am Coll Cardiol. (2012) 59:49–56. doi: 10.1016/j.jacc.2011.08.061
110. Everett BM, Cook NR, Conen D, Chasman DI, Ridker PM, Albert CM. Novel genetic markers improve measures of atrial fibrillation risk prediction. Eur Heart J. (2013) 34:2243–51. doi: 10.1093/eurheartj/eht033
111. Tada H, Shiffman D, Smith JG, Sjogren M, Lubitz SA, Ellinor PT, et al. Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke. (2014) 45:2856–62. doi: 10.1161/STROKEAHA.114.006072
112. Kolek MJ, Muehlschlegel JD, Bush WS, Parvez B, Murray KT, Stein CM, et al. Genetic and clinical risk prediction model for postoperative atrial fibrillation. Circ Arrhythm Electrophysiol. (2015) 8:25–31. doi: 10.1161/CIRCEP.114.002300
113. Virani SS, Brautbar A, Lee VV, Elayda M, Sami S, Nambi V, et al. Usefulness of single nucleotide polymorphism in chromosome 4q25 to predict in-hospital and long-term development of atrial fibrillation and survival in patients undergoing coronary artery bypass grafting. Am J Cardiol. (2011) 107:1504–9. doi: 10.1016/j.amjcard.2011.01.026
114. Body SC, Collard CD, Shernan SK, Fox AA, Liu KY, Ritchie MD, et al. Variation in the 4q25 chromosomal locus predicts atrial fibrillation after coronary artery bypass graft surgery. Circ Cardiovasc Genet. (2009) 2:499–506. doi: 10.1161/CIRCGENETICS.109.849075
Keywords: familial atrial fibrillation, genetics, electrophysiology, arrhythmia, personalized medicine
Citation: Ragab AAY, Sitorus GDS, Brundel BBJJM and Groot NMSd (2020) The Genetic Puzzle of Familial Atrial Fibrillation. Front. Cardiovasc. Med. 7:14. doi: 10.3389/fcvm.2020.00014
Received: 08 October 2019; Accepted: 28 January 2020;
Published: 14 February 2020.
Edited by:
Georges Nemer, American University of Beirut, LebanonReviewed by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, United StatesCopyright © 2020 Ragab, Sitorus, Brundel and Groot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natasja M. S. de Groot, bi5tLnMuZGVncm9vdEBlcmFzbXVzbWMubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.