 Nina Kaludercic
Nina Kaludercic Fabio Di Lisa
Fabio Di Lisa
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 18 February 2020
Sec. Cardiovascular Metabolism
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00012
This article is part of the Research TopicMitochondrial Dysfunction and Cardiovascular DiseasesView all 10 articles
Diabetic cardiomyopathy is a result of diabetes-induced changes in the structure and function of the heart. Hyperglycemia affects multiple pathways in the diabetic heart, but excessive reactive oxygen species (ROS) generation and oxidative stress represent common denominators associated with adverse tissue remodeling. Indeed, key processes underlying cardiac remodeling in diabetes are redox sensitive, including inflammation, organelle dysfunction, alteration in ion homeostasis, cardiomyocyte hypertrophy, apoptosis, fibrosis, and contractile dysfunction. Extensive experimental evidence supports the involvement of mitochondrial ROS formation in the alterations characterizing the diabetic heart. In this review we will outline the central role of mitochondrial ROS and alterations in the redox status contributing to the development of diabetic cardiomyopathy. We will discuss the role of different sources of ROS involved in this process, with a specific emphasis on mitochondrial ROS producing enzymes within cardiomyocytes. Finally, the therapeutic potential of pharmacological inhibitors of ROS sources within the mitochondria will be discussed.
Chronic hyperglycemia, the major characteristic of type 1 diabetes (T1D), is a life-threatening risk factor that results in organ and tissue damage in the long term. One of the acute metabolic complications associated with mortality includes diabetic ketoacidosis occurring mainly in T1D (1). Instead, type 2 diabetes (T2D) and obesity are characterized by insulin resistance, hyperlipidemia and hyperinsulinemia that might occur before the onset of hyperglycemia. The heart is an insulin-dependent tissue, since insulin promotes glucose utilization and suppresses fatty acid utilization thereby conferring a certain level of metabolic flexibility, i.e., the ability to adapt substrate oxidation rates to substrate availability, in support of cardiac function (2). This metabolic flexibility is largely impaired in diabetic hearts, resulting in minimal glucose utilization, shift to free fatty acid utilization and energetic inefficiency (3). Vascular complications occurring in diabetes account for increased morbidity and mortality associated with this disease. In the long term, diabetes may cause microvascular disease, resulting from the damage of small blood vessels, and/or macrovascular disease, resulting from the damage of the arteries (4). The latter includes coronary artery disease, peripheral arterial disease, and stroke, while microvascular complications result in retinopathy, nephropathy and neuropathy. Diabetic cardiomyopathy (DCM) is a pathology associated with alterations in the myocardial structure and function without the coexistence of other cardiac risk factors such as coronary artery disease, hypertension, valvular disease (5). DCM is one of the deadliest complications associated with diabetes (1). The incidence of heart failure is increased in diabetic patients compared with age-matched individuals, independently of obesity, hypertension, dyslipidemia, and coronary artery disease (6). In addition, diabetes has also been associated with increased rates of cancer, physical and cognitive disability, tuberculosis and depression (7–12).
Reactive oxygen species (ROS) and oxidative stress have been linked both to the onset of diabetes and development of complications associated with this disease (13). Here, we will review the pathophysiological features of DCM, the evidence related to the contribution of ROS to DCM and the role of different sources of ROS involved in this process. The present review will focus on mitochondrial sources of ROS in cardiac myocytes (rather than other cell types in the heart) and will briefly discuss the advantages and disadvantages of targeting mitochondrial enzymes to prevent oxidant damage and postpone or prevent the development of cardiac complications in diabetes.
DCM is a result of diabetes-induced changes in the structure and function of the heart and is diagnosed only if there is cardiac dysfunction not associated with coronary artery disease (14). The clinical outcomes associated with ischemic heart disease, hypertension or heart failure are worse for patients with diabetes and indeed, cardiovascular complications are the leading cause of mortality and morbidity in diabetic patients (5, 15, 16). Thus, a better understanding of DCM-associated pathophysiology and underlying mechanisms is necessary in order to develop tools for early diagnosis and treatment strategies.
As an early complication of diabetes, DCM is characterized by a long latent phase during which the disease progresses, but is completely asymptomatic. This subclinical period includes an increase in the left ventricle (LV) mass, fibrosis, abnormalities in cell signaling and diastolic dysfunction (3, 5). Studies using magnetic resonance imaging demonstrated that hyperglycemia and insulin resistance are associated with an increase in LV mass (3, 17). The increase in cardiac stiffness and fibrosis detected in diabetic patients frequently evolves to heart failure with preserved ejection fraction (HFpEF) (18, 19). In some patients, diastolic dysfunction may progress to pump failure and impairment in systolic function resulting in heart failure with reduced ejection fraction (20, 21). Nevertheless, not all cardiac anomalies observed in T2D are recapitulated in T1D (22, 23). While T2D is characterized by both morphological and functional cardiac abnormalities in patients (i.e., LV hypertrophy, diastolic, and systolic dysfunction), T1D patients show intact systolic function and impairment in diastolic function (23). Moreover, not all studies conducted in T1D patients evidenced an impairment in diastolic function. This may be explained by the fact that T1D patients are normally treated with insulin that normalizes insulin-dependent metabolic processes and therefore likely renders T1D-induced alterations in the heart less evident (23). Regardless of these differences, clinical trials showed that the prevalence of heart failure in diabetic patients ranges from 19 to 26% (24–27), while the mortality rate is 15–20% in diabetic patients with systolic dysfunction (21).
Although the exact mechanism of diabetes-associated LV dysfunction is not known, it appears that hyperglycemia, hyperinsulinemia, and/or lipotoxicity initiate a series of adaptive and maladaptive processes contributing to the development of heart failure. Factors underlying pathological changes in the diabetic heart are multiple. Metabolic alterations such as hyperglycemia, insulin resistance and increased free fatty acid levels, result in the oxidative stress, organelle dysfunction, inflammation, advanced glycation end products (AGEs) formation, activation of protein kinase C (PKC), abnormalities in ion homeostasis, alterations in structural proteins, apoptosis and fibrosis, changes that eventually result in diabetes-induced cardiac dysfunction (Figure 1) (28, 29). Despite a myriad of factors has been shown to collectively contribute to the development and progression of DCM, causal relationships and the exact sequence of events among these cellular and molecular mechanisms are still not entirely clear. Moreover, these factors frequently interact with each other, making DCM a very complex disease to treat.
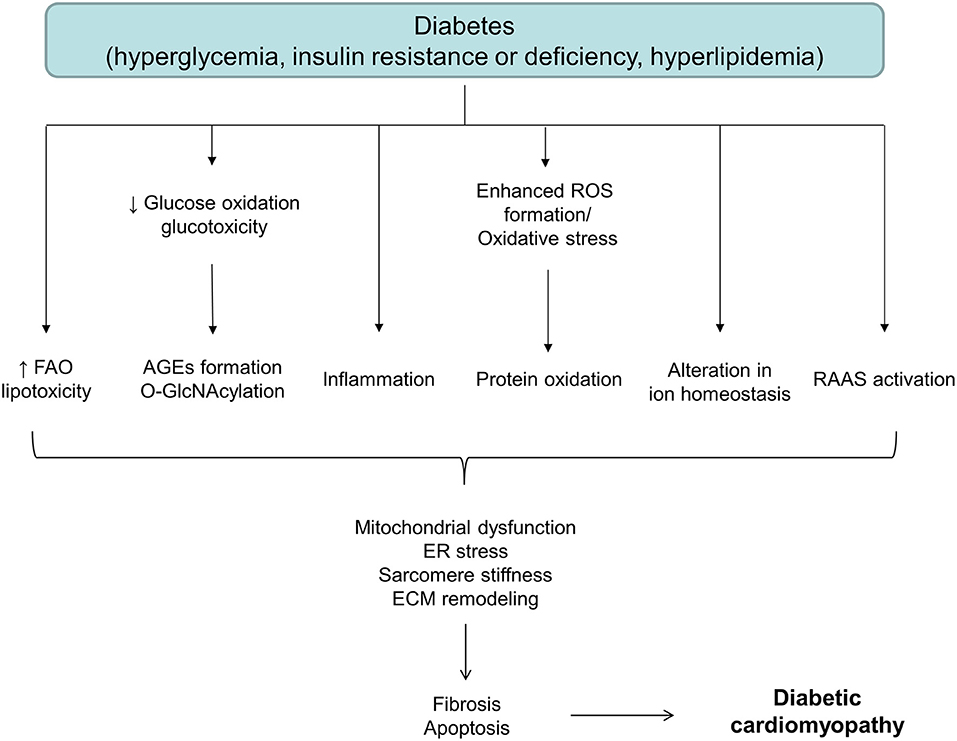
Figure 1. A schematic diagram depicting different factors involved in the onset and development of diabetic cardiomyopathy. AGEs, advanced glycation end products; ECM, extracellular matrix; ER, endoplasmic reticulum; FAO, fatty acid oxidation; RAAS, renin-angiotensin-aldosterone system; ROS, reactive oxygen species.
ROS formation has gained significant experimental and clinical evaluation amongst the various mechanisms proposed (13, 20, 30). Notably, the aforementioned pathogenic factors and changes either induce or result from oxidative stress. ROS can be dangerous for biological systems for their capacity to interact with numerous macromolecules, such as proteins, lipids and DNA. ROS-induced modification of DNA can be mutagenic, especially if DNA damage cannot be repaired (31). ROS may lead to DNA strand breakage and formation of 8-hydroxydeoxyguanosine, a prominent feature in diabetic hearts (32, 33). While protein oxidation can be reversible and serve for signaling purposes, oxidative stress may lead to protein carbonylation that cannot be reversed and results in toxic aggregate accumulation if carbonylated molecules are not promptly degraded (34, 35). Membrane lipids are rich in polyunsaturated fatty acids that can easily be oxidized by ROS, a process that is also involved in the generation of atherosclerotic plaques (36). Lipid oxidation results in excess formation of carbonyl compounds, such as prostanoids and aldehydes, toxic metabolites that can promote numerous pathologies (37). In addition to direct macromolecule targeting, high ROS formation can also decrease the antioxidant capacity of the diabetic myocardium, contributing thereby to oxidative stress and resulting myocardial damage. This concept is further supported by studies demonstrating that overexpression of antioxidant defense proteins, such as metallothionein or catalase, could prevent oxidative stress and maladaptive remodeling of the diabetic hearts (22, 38–41). Mitochondrial ROS production underlies several hyperglycemia-induced pathogenic mechanisms, such as GAPDH inhibition, activation of polyol pathway, formation of AGEs, activation of PKC, glucose auto-oxidation, and activation of the 12/15-lipoxygenases pathway (13, 30, 42, 43). Activation of these pathways can, in turn, exacerbate oxidative stress. For instance, the polyol pathway utilizes NAPDH which is required for GSH regeneration, while binding of AGEs to their receptor results in ROS formation (44). Inhibition of AGE formation or AGE receptor gene knock-down attenuates the development of DCM (45). Moreover, activation of p53 signaling in T1D and T2D mouse models by an initial oxidative trigger leads to the upregulation of cytochrome c oxidase assembly protein, mitochondrial respiration, fatty acid oxidation, and mitochondrial ROS generation (46). However, hyperglycemia is not the only factor responsible for cardiac complications in diabetes, as mentioned before. Lipotoxicity and increased oxidation of free fatty acids also lead to oxidative stress, mitochondrial and ER stress, and activation of pro-inflammatory signals (47–51). Damage to mitochondria results in enhanced ROS generation and activation of the NLRP3 inflammasome (52) which, in turn, may promote or exacerbate cardiac fibrosis. Moreover, high glucose and inflammation provide a synergistic effect and further enhance ROS formation and all the downstream events leading to cell dysfunction (53, 54). Inflammation, angiogenesis, cardiomyocyte hypertrophy and apoptosis, fibrosis and contractile dysfunction, are processes susceptible to ROS-dependent modulation in the diabetic heart (55). Diastolic abnormalities observed in HFpEF are largely due to increased collagen and cardiomyocyte stiffness (56). ROS are well-known to target sarcomere proteins thereby inducing oxidative changes that may impact on sarcomere and cardiomyocyte stiffness (57, 58). While oxidation of the proteins forming the thick and thin filaments is mostly associated with impaired contractility, post-translational modifications of the elastic filament protein titin are tightly related to changes in LV stiffness (59). The passive stiffness of cardiac muscle was shown to be redox-dependent through titin oxidation and disulfide bridge formation that lead to increased cardiac stiffness (60). In addition to direct mechanisms, ROS can modulate sarcomere function also by affecting key protein kinases or phosphatases to induce post-translational modifications (57). In that regard, reduced titin phosphorylation is an important determinant of diastolic stiffness in HFpEF (59, 61, 62). This is particularly relevant in diabetic and obese patients in which oxidative stress impairs NO/cGMP/PKG signaling and leads to titin hypophosphorylation (59, 63) and increased cardiomyocyte stiffness along with collagen and AGEs deposition (59). Thus, enhanced ROS formation and alteration in the redox status are deeply intertwined with numerous alterations observed in diabetic hearts, suggesting that targeting ROS formation/elimination may represent an attractive therapeutic strategy for the treatment of DCM. Several cellular and subcellular sources that may account for enhanced ROS production were described in diabetic cardiovascular system and other tissues. Enzymes involved in deleterious ROS generation associated with diabetic complications include nicotinamide adenine dinucleotide phosphate oxidases (NOXs) (64–66), xanthine oxidase/oxidoreductase (XO) (67, 68), arachidonic acid cascade and microsomal enzymes, uncoupled nitric oxide synthase (NOS) (69), and mitochondria (13, 70–72).
NOX is a family of membrane-bound enzyme complexes composed of plasma membrane spanning and cytosolic components (73, 74). The active NOX complex allows for the transfer of electrons to molecular oxygen to generate superoxide (75). NOXs are considered to be one of the major cellular ROS sources and prominent players in several pathological conditions (74, 76, 77). NOX2, located in the cell membrane, and NOX4, localized in perinuclear ER and/or mitochondria, are expressed in the heart (78, 79). Increased cardiac NOX2 expression/activity has been described both in T1D and T2D, and contributes to hyperglycemia-induced ROS production (64–66, 80). NOX4 expression and NOX4-derived ROS are increased ~14 days after the induction of T1D in rats and contribute to the development of cardiomyopathy (81). Importantly, reducing either NOX2 or NOX4 activity in streptozotocin-induced diabetic hearts blunts myocardial oxidative stress, remodeling and improves cardiac function (81–83). ROS formation through NOX following high glucose administration has been associated with pathways involving sodium/glucose co-transporter 1 (SGLT1), PKCβ, and calcium/calmodulin dependent kinase II (CaMKII) (84). SGLT1-mediated glucose transport is responsible for NOX2 activation, since its inhibition efficiently abolished ROS production induced by exposure to high glucose (85). Importantly, PKCβ activation by RhoA/Rho kinase pathway activates Rac1 that, in turn, determines p47phox translocation to the membrane, event required for NOX2 activation (86). Indeed, PKCβ inhibition by ruboxistaurin prevented NOX2 activation and subsequent ROS formation in cardiomyocytes treated with high glucose (87). An additional mechanism responsible for NOX activation in hyperglycemic conditions involves CaMKII activation. High glucose causes an increase in intracellular levels of Ca2+ that leads to CaMKII hyperphosphorylation and activation (32). Activated CaMKII is likely responsible for activation of PKCβ and downstream cascade of events (86). In that regard, inhibition of CaMKII prevented both the upregulation of p47phox and p67phox as well as oxidative stress in streptozotocin-induced model of T1D (32), suggesting that CaMKII may indeed play a major role in NOX-induced ROS formation.
XO is a cytoplasmic enzyme that catalyzes the oxidation of hypoxanthine to xanthine and further converts xanthine to uric acid (88). It uses oxygen as electron acceptor and produces superoxide and hydrogen peroxide (H2O2). In addition to their role in cardiac damage induced by ischemia/reperfusion injury or pacing-induced heart failure in dogs (89), hypoxanthine and XO activity are also increased in diabetic subjects (90). The role of XO in hyperglycemia-induced oxidative stress is documented by increased ROS formation in the muscle and development of fibrosis of hyperglycemic streptozotocin-induced diabetic mice (68, 91, 92). Some investigators reported evidence for beneficial vascular effects of XO inhibitors in hypercholesterolemic and diabetic patients (72, 93). Indeed, in T1D patients XO inhibition reduced the degree of oxidative stress, whereas in T2D patients it led to significant improvements in peripheral endothelium-dependent vasorelaxation (67, 90, 93).
NOS uncoupling results in superoxide formation, oxidative stress and decreased NO bioavailability that may have important vascular effects in diabetic subjects (94). Indeed, a decrease in the dimer to monomer ratio, indicative of the enzyme uncoupling, has been reported within the myocardium of diabetic animals (95). Consequently, inhibition of NOS activity and uncoupling by L-NAME, insulin-like growth factor, sepiapterin, ascorbic acid or N-acetyl-cysteine improved LV function in the diabetic heart (66, 96–100). In addition to uncoupling, NOS expression may also be increased in the diabetic hearts (33, 64, 101) and this is associated with an increase in lipid peroxidation and peroxynitrite generation (72). Peroxynitrite in turn may also lead to NOS uncoupling (102). Taken together, these studies suggest that the increased production of superoxide and peroxynitrite through NOS uncoupling is a major contributor to suppressed contractile performance in diabetes (72, 99, 100).
For detailed discussion related to XO, NOX or uncoupled NOS involvement in DCM, readers are referred to other excellent reviews (67, 72, 74, 84).
The role of mitochondrial ROS formation and dysfunction in the pathogenesis of diabetes and its complications is well-established (13, 20, 28). Indeed, cardiac mitochondria from diabetic patients are dysfunctional, displaying increased mitochondrial H2O2 emission, impaired mitochondrial respiratory capacity and increased levels of oxidized or hydroxynonenal-modified proteins (103–105). Several mechanisms are likely responsible for mitochondrial dysfunction in diabetic hearts, including fatty acid-induced mitochondrial uncoupling, changes in mitochondrial morphology, increased ROS formation, mitochondrial proteome remodeling, impaired mitochondrial calcium handling and altered mitochondrial turnover (20, 28, 106–108). All these events might lead to compromised cardiac ATP generation and ultimately to cardiac dysfunction. Impairment in the activity of ATP synthase also affects mitochondrial function in the diabetic heart. A recent study very elegantly showed that hyperglycemia-induced calpain-1 upregulation in the mitochondria cleaves the ATP synthase α subunit, resulting in the reduction in the ATP synthase activity and increased mitochondrial ROS formation (109) that eventually contribute to the development of DCM. In addition, excessive mitochondrial ROS formation results in the increased propensity to permeability transition pore (PTP) opening that eventually leads to cell death (110). A tight relationship also exists between alterations in mitochondrial morphology and ROS formation that may reciprocally modulate each other. Cardiomyocytes from animal models of T1D, T2D, and from diabetic patients show increased levels of ROS and altered mitochondrial morphology, including mitochondrial fragmentation, cristae disruption and swelling (107, 108). Of interest, mitochondrial fragmentation induced by chronic hyperglycemia can be reversed with antioxidants, suggesting that ROS are causally related to this pro-fission phenotype and that controlling mitochondrial morphology and dynamics might represent a therapeutic strategy for the treatment of DCM (107, 111). Altered mitochondrial function may inhibit insulin signaling by interfering with oxidation of fatty acyl-CoA, accumulation of intracellular lipid and diacylglycerol, PKC activation and through generation of ROS (112). Both processes lead to insulin receptor substrate 1 phosphorylation and interference with insulin signal transduction. Reduction in mitochondrial ROS formation obtained either through cardiac-specific Mn-SOD overexpression or following stimulation of AMPK activity, prevented mitochondrial damage and many fatty acid- or hyperglycemia-induced events, both in vitro and in vivo (113–116).
Given the tight relationship between mitochondrial ROS formation, structure/function, and diabetes-induced complications, it is crucial to dissect and identify sites responsible for ROS formation in mitochondria exposed to diabetic milieu. Electron transport chain (ETC), p66Shc, and monoamine oxidase (MAO) are the major sources of ROS formation in mitochondria (Figure 2).
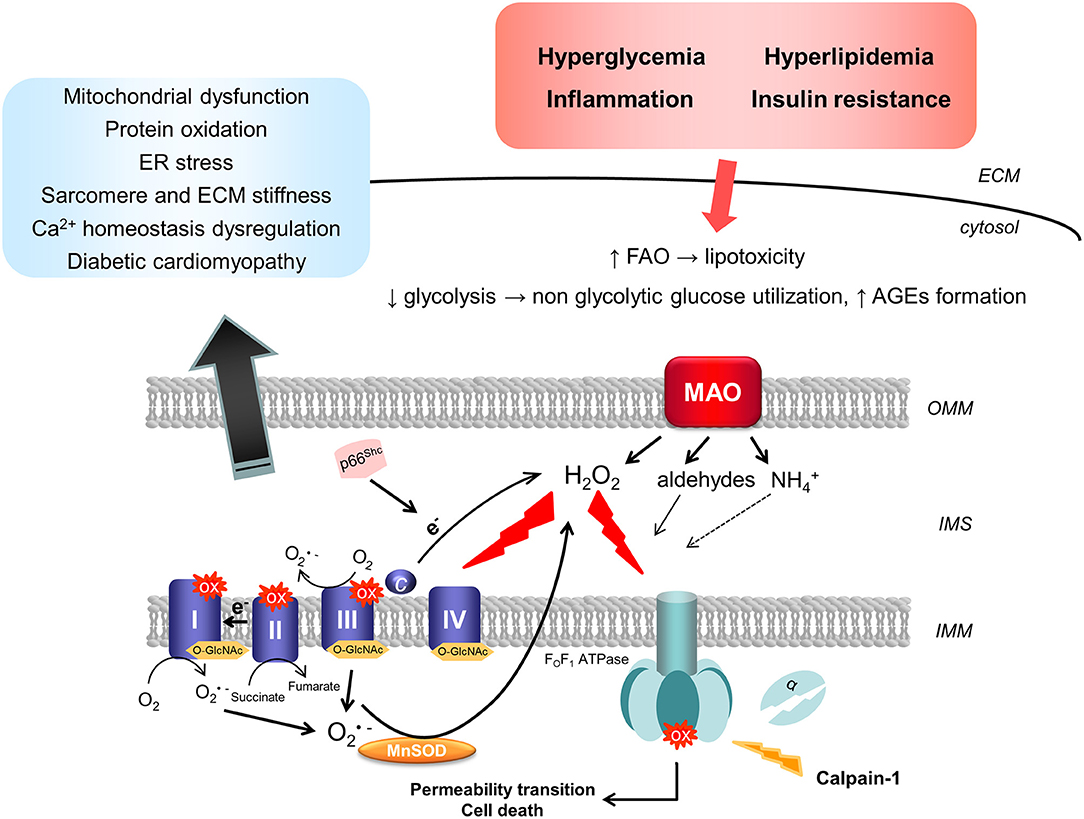
Figure 2. Mitochondrial sources of ROS in diabetic cardiomyopathy. Diabetic milieu, characterized by hyperglycemia, hyperlipidemia, and inflammation, results in the up-regulation in the activity of mitochondrial ROS-producing enzymes. Superoxide can be produced by the respiratory chain through forward or reverse electron transport. In addition, calpain-1 translocates to the mitochondrial matrix in the diabetic heart and cleaves the α subunit of the ATP synthase, leading therefore to the reduction in its activity and mitochondrial dysfunction. On the other hand, in situations of stress, p66Shc is phosphorylated and translocates to the IMS where it catalyzes the electron transfer from cytochrome c to oxygen (O2) leading to the formation of hydrogen peroxide (H2O2). Finally, up-regulation of MAO activity upon exposure to high glucose and pro-inflammatory stimuli results in enhanced formation of H2O2 that can directly increase the susceptibility of mitochondria to undergo permeability transition. Post-translational modifications, such as oxidation or O-GlcNAcylation of respiratory chain complexes, can impair mitochondrial bioenergetics and function. All these events are implicated in the pathogenesis of diabetic cardiomyopathy by promoting mitochondrial and ER stress, leading to protein oxidation and Ca2+ homeostasis impairment, as well as sarcomere and ECM stiffness. AGEs, advanced glycation end products; ECM, extracellular matrix; ER, endoplasmic reticulum; FAO, fatty acid oxidation; IMM, inner mitochondrial membrane; IMS, intermembrane space; MAO, monoamine oxidase; MnSOD, manganese superoxide dismutase; OMM, outer mitochondrial membrane; O-GlcNAc, β-linked N-acetylglucosamine; ox, oxidation.
ETC is by far the major site of ATP production in mitochondria inside any given cell, and especially in cardiomyocytes (more than 90%). At the inner mitochondrial membrane (IMM), electrons from NADH and FADH2 are transferred across the respiratory chain to oxygen, which is reduced to water at the level of complex IV (117). This process powers the movement of protons into the intermembrane space and generates a proton gradient that drives the synthesis of ATP by the ATP synthase. A small amount of electrons (about 0.1%) can leak from the ETC and cause superoxide formation due to the partial reduction of oxygen (118). Superoxide generation may occur under conditions that decrease the flow of electrons, particularly at the level of the first three complexes where flavins or quinones are able to act as single electron donors (117, 119, 120). Notably, ROS formation can also result from the reverse electron flow through complex I (121). A recent study supported this pathophysiological concept demonstrating that succinate accumulates during cardiac ischemia in vivo (121, 122). Upon reperfusion, accumulated succinate is oxidized by complex II leading to dramatic ROS formation that is likely attributable to the reverse electron flow through complex I (122).
Seminal discoveries implicating ETC superoxide production as the central event in hyperglycemia-induced pathogenic mechanisms were provided by Brownlee's group back in 2000 using endothelial cells (123, 124). High intracellular glucose levels and glucose-derived pyruvate promote mitochondrial respiration by increasing the availability of reducing equivalents for the ETC and resulting in mitochondrial membrane hyperpolarization and superoxide production (123, 125). Furthermore, hyperglycemia-induced ROS formation is prevented by several interventions, such as via inhibition of ETC complex II activity, uncoupling of oxidative phosphorylation, by overexpression of uncoupling protein-1 and/or Mn-SOD (123). Normalizing levels of mitochondrial ROS with each of these agents prevents glucose-induced activation of PKC, hexosamine pathway, formation of AGEs, sorbitol accumulation, and NFκB activation. A further confirmation that ETC superoxide production is responsible for these events comes from experiments performed in Rho zero (ρ0) endothelial cells lacking mitochondrial ETC function (30). When exposed to high glucose, ρ0 cells do not display an increase in ROS production. Similar mechanism has also been shown to be at play in cardiomyocytes exposed to high glucose. Indeed, ROS formation is reduced in cardiomyocytes isolated from diabetic animals in which complex I and II activity is inhibited or which overexpress catalase, further denoting the crucial role of ETC in ROS generation in diabetes (41, 86, 126). Interestingly, the protective effect afforded by complex I or II inhibition suggests that ETC superoxide production upon high glucose exposure likely occurs through the reverse electron transport. It remains to be elucidated whether succinate accumulation occurs at some point during the development of cardiovascular complications in diabetes. Cardiac lipotoxicity is also mediated by mitochondrial ROS formation. Indeed, exposure to palmitate enhances mitochondrial ROS generation and leads to increased mitochondrial fission by modulating DRP1 phosphorylation levels and proteolytic processing of OPA1 (47).
An initial ROS trigger produced by ETC can promote activation of processes that eventually amplify the signal and lead to oxidative stress. Such processes involve the occurrence of post-translational modifications, such as (but not limited to) diabetes–induced defects caused by oxidation, increased methylglyoxal adduct formation and increased O-GlcNAcylation, that contribute to the impairment in mitochondrial and systolic function (127–129). Hyperglycemia alters the function of respiratory chain in mitochondria via dysregulation of O-GlcNAcylation (130, 131). O-GlcNAc transferase (OGT) enzyme is located in the IMM and interacts with complex IV of the respiratory chain in normal conditions. In streptozotocin-treated rats this enzyme is improperly localized to the mitochondrial matrix and the impairment in the OGT-complex IV interaction results in the loss of complex IV activity and reduced mitochondrial membrane potential (130). O-GlcNAcylation of proteins involved in mitochondrial dynamics, such as DRP-1 and OPA1, also contributes to mitochondrial fragmentation that further exacerbates organelle dysfunction (132, 133). On the other hand, methylglyoxal-induced modifications affect Ca2+ homeostasis and indirectly affect mitochondrial function. Indeed, in the diabetic heart methylglyoxal preferentially forms adducts with proteins involved in the intracellular calcium handling such as ryanodine receptor 2 and SERCA2a (134, 135). Ryanodine receptor glycation is associated with impaired Ca2+ cycling, increased mitochondrial Ca2+ levels and mitochondrial dysfunction (136). Collectively, these studies underline the importance of ETC-derived superoxide in diabetic conditions and mitochondria as their source and target.
p66Shc is another important source of ROS in mitochondria. p66Shc is a cytosolic adaptor protein and, along with p46Shc and p52Shc, is encoded by the ShcA gene (137, 138). p46Shc and p52Shc isoforms are formed through alternative translation start sites (137, 139). While p46Shc and p52Shc isoforms are ubiquitously expressed, p66Shc promoter may bear epigenetic modifications resulting in cell type- or specific condition-restricted expression (140). Under stress conditions, PKCβ phosphorylates p66Shc at Ser-36, event required for its translocation to mitochondria (141). Once in the intermembrane space, p66Shc catalyzes the electron transfer from cytochrome c to oxygen resulting in the formation of H2O2 (142). In addition to this mechanism, p66Shc can promote oxidative stress by activating membrane-bound NOX or through down-regulation of antioxidant enzymes synthesis (143). Accordingly, cells and mice lacking p66Shc show reduction in markers of oxidative stress (139, 144).
A number of studies characterized the pathophysiological role of p66Shc in cardiovascular diseases, such as maladaptive hypertrophy, heart failure and ischemia/reperfusion injury (137–139, 145). Importantly, excessive ROS generation is a major contributing factor to those cardiac pathologies (146). Since PKC activation plays a major role in the intracellular signaling leading to oxidative stress, cell dysfunction and tissue damage in hyperglycemia, and is required for p66Shc translocation to mitochondria in response to stress (70), it is tempting to hypothesize that p66Shc may play a role in cardiovascular complications induced by hyperglycemia acting as a downstream target following high glucose-induced PKCβ activation. Indeed, p66Shc−/− mice have an increased resistance to ROS (70), less atherosclerosis and preserved aortic endothelium-dependent vasorelaxation following high-fat diet and in a model of streptozotocin-induced T1D (147, 148). Moreover, lack of p66Shc prevented oxidative damage in cardiac progenitor cells and cardiomyocytes in streptozotocin-induced DCM (149). Unlike diabetic wild type animals characterized by cardiomyocyte loss, diabetic p66Shc−/− hearts displayed preserved cardiac progenitor cell replication and turnover, along with unaltered wall thickness, chamber volume, LV end-diastolic pressure and diastolic wall stress (149).
Monoamine oxidases (MAOs) are flavoenzymes localized at the level of the outer mitochondrial membrane. MAOs exist in two isoforms, A and B, differing in structure, substrate preference, inhibitor specificity and tissue distribution (150–153). The physiological role of MAOs consists in the catalysis of the oxidative deamination of its substrates (i.e., endogenous and exogenous amines, neurotransmitters). MAOs generate H2O2, ammonia and corresponding aldehydes as products of catalysis (154, 155). Over the last decade, several studies have shown that alterations in redox balance cause by enhanced MAO activity play a prominent role in promoting the development of cardiovascular disorders and causing oxidative damage to cardiomyocytes (37, 146, 156–158). Indeed, MAO contributes to ischemia/reperfusion injury, maladaptive hypertrophy, heart failure and vascular dysfunction (37, 139, 159–162). Of note, evidence for MAO involvement in cardiac disease has also been demonstrated in patients. Up-regulation of MAO activity and consequent ROS formation has been identified as a prominent contributor to the impaired myocardial redox balance in patients and a major risk factor and predictor for the postoperative atrial fibrillation (163). In addition, MAO activity was shown to be increased in left and right ventricles from patients with ischemic heart disease (164). With regard to the possible involvement of MAO in diabetes, one study showed an improvement in blood glucose levels and systolic and diastolic pressures in a patient with T1D administered with the MAO inhibitor tranylcypromine (165). Unexpectedly, it has been demonstrated that pioglitazione, used as an antidiabetic drug in T2D patients, is a specific and reversible MAO-B inhibitor (166). These findings support a possible MAO involvement in diabetes-induced complications.
A clear and undeniable evidence for the role of MAO in the pathogenesis and progression of DCM came from animal models of T1D showing that MAO inhibition prevents cardiac dysfunction, death and fibrosis in diabetic mice and rats (71, 167). Data from our laboratory indicates that MAO activity is responsible for diastolic stiffness and dysfunction, some of the earliest signs of DCM in diabetic mice (71). Indeed, administration of MAO inhibitors is able to prevent oxidative changes, diastolic dysfunction and myocardial fibrosis in streptozotocin-treated hearts. In addition, MAO inhibition prevented mast cell degranulation in diabetic hearts, event that can contribute to fibrotic remodeling of the myocardial tissue. This evidence suggests that MAO-generated ROS are at the basis of diabetes-induced cardiovascular complications and, in addition to cardiomyocytes, affect also other cell types present in the heart. Oxidative stress induced by enhanced MAO activity has also been implicated in cardiomyocyte and mesenchymal stromal cell senescence (168–170). It remains to be elucidated whether ROS produced by MAO may also promote cardiac progenitor cell senescence during remodeling induced by diabetes, as is the case with p66Shc.
Up to date, the mechanisms underlying MAO toxicity have mostly been attributed to excessive H2O2 and aldehyde formation that leads to impaired mitochondrial function (146). Our recent work showed that incubation of primary cardiomyocytes with high glucose and pro-inflammatory cytokine IL-1β leads to a MAO-dependent increase in ROS that, in addition to causing PTP opening and mitochondrial dysfunction, also results in the endoplasmic reticulum (ER) stress (71). This evidence indicates that, in addition to mitochondrial ROS being a trigger for inflammasome activation, inflammatory processes can also promote mitochondrial ROS formation by up-regulating MAO activity. MAO inhibition prevented mitochondrial dysfunction and ER stress, factors that eventually contribute to the progression of DCM, suggesting that cardiomyocyte targeting of pro-inflammatory stimuli occurs in a MAO-dependent manner (71). Given that MAO is localized at the outer mitochondrial membrane and faces the cytosol, it is conceivable to imagine that H2O2 produced by MAO can also affect the function of neighboring organelles. Notably, ER and mitochondria are adjacent organelles, connected both at structural and functional level (171). Although our data suggests that MAO-induced mitochondrial dysfunction occurs upstream of ER stress, it is tempting to hypothesize that MAO may directly modulate ER function also through physical interaction with ER-resident proteins (mitochondria associated membrane proteins, for instance). Finally, it cannot be excluded that other products of MAO activity, such as aldehydes, may also contribute to diabetes-induced alterations. MAO-dependent oxidative stress may lead to the inhibition of aldehyde dehydrogenase 2 (ALDH2) resulting in further accumulation of toxic and reactive aldehydes (37). In that regard, it has been demonstrated that stimulation of ALDH2 activity protects from streptozotocin-induced cardiac damage (172), suggesting that accumulation of aldehydes may promote cardiac remodeling in diabetes independently or in concert with high ROS levels (173).
An intense cross-talk between different cellular ROS sources is likely to exist since many papers report that inhibition of a single ROS source prevents the development of cardiac pathology triggered by oxidative stress (146). For instance, hyperglycemia does not induce ROS formation in the ρ0 cells in which the respiratory chain is disrupted, as well as upon NOX or MAO inhibition (30). In addition, mitochondrial superoxide scavenging using mitochondria-targeted antioxidants is able to reduce NOX2 expression and activity in diabetic myocardium (174), while genetic inhibition of NOX2 and consequent reduction in superoxide formation at the mitochondrial level suggest that mitochondrial ROS formation in hyperglycemic hearts might be NOX2-dependent (82–84). Such evidence strongly supports the existence of an “amplification mechanism,” whereby an initial stress (i.e., hyperglycemia and/or inflammation), induces the formation of ROS that, in turn, activates other ROS producing enzymes to start producing free radicals thus amplifying the original oxidative trigger (146). The hypothesis of the feed-forward/amplification mechanism is also supported by the characterization of the so-called ROS-induced ROS release mechanism, whereby an initial ROS trigger induces PTP opening that leads to further ROS formation, instituting thereby a positive feedback loop for the ROS-induced ROS release (175, 176). This is indeed the case in adult cardiomyocytes that, when exposed to high glucose and pro-inflammatory stimuli, display an increase in MAO-dependent ROS formation that causes PTP opening and mitochondrial and ER stress (71). Moreover, other processes may participate in such amplification loop, such as for instance impairment in autophagy. While low/moderate ROS levels are required for autophagy initiation, excessive oxidative damage can impair autophagy resulting in the aberrant clearance of damaged proteins and/or organelles (177–180). For instance, AGE accumulation in an experimental model of diabetes inhibits autophagy, induces ER stress and promotes ROS formation (181). Either autophagy stimulation with rapamycin or inhibition of ER stress due to ER chaperone administration alleviate AGEs-induced deleterious effects on cardiomyocytes, suggesting that these processes are involved in diabetes-induced cardiac remodeling. Impairment in the elimination of damaged and dysfunctional mitochondria in diabetic hearts results in the accumulation of ROS-producing fragmented mitochondria (182, 183). Either inhibition of mitochondrial fragmentation during exposure to high glucose or stimulation of organelle removal through mitophagy in high-fat diet fed animals prevents oxidative stress as well as mitochondrial and cardiac dysfunction (182, 184, 185). However, it appears that autophagy and mitophagy are independently controlled in T2D, since autophagy flux was attenuated following 6 weeks of high-fat diet while mitophagy continued to increase even after 2 months (184). This suggests that mitophagy may occur through a non-canonical, alternative autophagy pathway. In this regard, it has been previously shown that Rab9 is mobilized to the mitochondria in early stages of diabetes where it induces activation of alternative autophagy for mitophagy (186, 187). The mechanisms controlling the activation of canonical vs. non-canonical autophagy remain unknown to date. Mitochondrial ROS are implicated in the activation of the canonical autophagy (178), but on the other hand excessive mitochondrial ROS formation impairs lysosomal biogenesis, function and the autophagy process in the cardiac myocytes (156, 168). Whether alterations in the redox status may represent the switch for autophagy to become maladaptive, and/or for the activation of canonical vs. non-canonical autophagy remains to be defined.
Given the large body of evidence linking aberrant ROS formation and oxidative stress to the development of cardiac diseases, it is quite straightforward to hypothesize that reducing redox burden would protect the heart against deleterious changes induced by diabetes or other pathologies. Nevertheless, large scale clinical trials using antioxidant therapies have not produced the desired results (188, 189). Whether this is a consequence of particular antioxidant molecules used in clinical trials, their limited absorption and/or reduced cardiac availability, or it can be explained by the fact that a certain level of ROS is beneficial and required for signaling and physiological processes, including the response to insulin mediated by p66Shc-dependent ROS (190), remains to be elucidated. Another attractive explanation is that interfering with the complex redox network might result in compensatory changes (191). Currently, there are no efficient therapies to treat HFpEF in patients with diabetes. In that regard, antidiabetic SGLT2 inhibitors (such as empagliflozin) afforded cardioprotective effects in patients with diabetes (192). SGLT2 inhibitors lead to the reduction in plasma volume and reduced preload, events that have a favorable effect on cardiac function and structure (193, 194). Importantly, human and rodent hearts do not express SGLT2 (195–197), suggesting that the direct cardioprotective effects of SGLT2 inhibitors are independent of their action on SGLT2. Indeed, it has been demonstrated that SGLT2 inhibitors can directly affect cardiomyocytes by targeting Na+/H+ exchanger 1, reducing intracellular Na+ and Ca2+ levels, improving mitochondrial function and reducing inflammation and AMPK activity (197, 198). In addition, SGLT2 inhibitors are able to reduce oxidative stress through Nrf2/ARE signaling activation and it is likely that these off-target effects contribute to the cardioprotection observed in clinical trials (198–200). Another strategy to modulate an oxidative stress-related pathway is the use of the soluble guanylate cyclase stimulator vericiguat that targets the cGMP pathway in an ROS/NO-independent manner (201). HFpEF is associated with excessive ROS formation by the coronary microvasculature that limits NO bioavailability, reduces cGMP levels and therefore lowers PKG activity (as discussed in section 3). A recent clinical trial demonstrated an improvement in quality of life in patients with HFpEF receiving vericiguat for 12 weeks, suggesting that it could be a promising therapeutic agent in HFpEF (201). To foster the development of a specific and successful therapy, future studies should aim either at identifying the molecular ROS targets (191), the pathways of redox signaling or the specific sources of ROS that are responsible for deleterious changes in the diseased heart. While the first two options are just beginning to become accessible and are still far from being conclusively elucidated, inhibition of specific ROS sources might prove to be a useful strategy to prevent alterations in the redox status, and myocardial structure and function.
In this regard, inhibitors for some of the ROS sources outlined in this review are being developed and/or tested in the clinic. Data obtained in experimental models of diabetes identified NOX4 as a therapeutic target (81). Indeed, NOX4 inhibitors are currently being tested for various cardiovascular indications (76, 202). For instance, GKT-831 is a NOX1/4 dual inhibitor and the only NOX inhibitor that has reached the clinical trial stage; in fact, it is currently being tested in clinical trial phase II for diabetic nephropathy. It remains to be established whether NOX inhibitors would be effective in limiting cardiovascular complications in diabetic patients.
In line with the concept of mitochondria as major ROS producers, employment of mitochondria-targeted antioxidants such as MitoTEMPO proved to be cardioprotective in experimental models of DCM (203). On the other hand, MitoQ was never tested in such setting and neither of the compounds was ever tested in clinical trials. The paucity of studies concerning the use of mitochondrial antioxidants in DCM urges for studies adopting strategies that target specific mitochondrial ROS sources or their downstream targets (204). In that regard, it is not possible to inhibit the respiratory chain in humans in the long term without jeopardizing a wide array of vital functions. Although genetic inhibition of p66Shc has proven protective in many cardiovascular pathologies, pharmacological inhibitors of p66Shc are not yet available. On the contrary, MAO inhibitors are clinically available and employed for the treatment of depression and neurodegenerative diseases (76, 164, 205–207). As mentioned before, administration of a non-selective MAO inhibitor to a patient with T1D led to several improvements, including those at the cardiovascular level (165). Side-effects associated with the “old” irreversible MAO-A inhibitors have been eliminated since reversible MAO-A inhibitors or selective MAO-B inhibitors have been developed (207). Taking into consideration recent findings obtained in experimental models of DCM, it is worth assessing whether molecules such as moclobemide or safinamide could be repurposed for the treatment of patients with DCM.
Current consensus is that exacerbated ROS generation due to hyperglycemia and/or fatty acid oxidation causes oxidative stress, that in turn promotes the development and progression of diabetes and its complications. In addition to the cytosolic sources of ROS, it is now well-documented that mitochondrial sources represent the major ROS burden in multiple tissues in both animal and human diabetic subjects. Pharmacological targeting of specific ROS sources may prove as a successful therapeutic strategy for the treatment of DCM. Alternatively, identification of processes and targets downstream of mitochondrial ROS may hold more promise in correcting cellular structural and functional derangements in diabetic individuals.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work has been supported by the Leducq Foundation Transatlantic Network of Excellence (grant no. 16CVD04).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. (2013) 93:137–88. doi: 10.1152/physrev.00045.2011
2. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. (1963) 1:785–9. doi: 10.1016/S0140-6736(63)91500-9
3. Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, et al. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the translational research committee of the heart failure association-european society of cardiology. Eur Heart J. (2018) 39:4243–54. doi: 10.1093/eurheartj/ehy596
4. Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. (2008) 26:77–82. doi: 10.2337/diaclin.26.2.77
5. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
6. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. (1974) 34:29–34. doi: 10.1016/0002-9149(74)90089-7
7. Carstensen B, Jorgensen ME, Friis S. The epidemiology of diabetes and cancer. Curr Diab Rep. (2014) 14:535. doi: 10.1007/s11892-014-0535-8
8. Lu FP, Lin KP, Kuo HK. Diabetes and the risk of multi-system aging phenotypes: a systematic review and meta-analysis. PLoS ONE. (2009) 4:e4144. doi: 10.1371/journal.pone.0004144
9. Wong E, Backholer K, Gearon E, Harding J, Freak-Poli R, Stevenson C, et al. Diabetes and risk of physical disability in adults: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. (2013) 1:106–14. doi: 10.1016/S2213-8587(13)70046-9
10. Jeon CY, Murray MB. Diabetes mellitus increases the risk of active tuberculosis: a systematic review of 13 observational studies. PLoS Med. (2008) 5:e152. doi: 10.1371/journal.pmed.0050152
11. Riza AL, Pearson F, Ugarte-Gil C, Alisjahbana B, van de Vijver S, Panduru NM, et al. Clinical management of concurrent diabetes and tuberculosis and the implications for patient services. Lancet Diabetes Endocrinol. (2014) 2:740–53. doi: 10.1016/S2213-8587(14)70110-X
12. Roy T, Lloyd CE. Epidemiology of depression and diabetes: a systematic review. J Affect Disord. (2012) 142:S8–21. doi: 10.1016/S0165-0327(12)70004-6
13. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. (2010) 107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545
14. Bugger H, Bode C. The vulnerable myocardium. Diabetic cardiomyopathy. Hamostaseologie. (2015) 35:17–24. doi: 10.5482/HAMO-14-09-0038
15. Solang L, Malmberg K, Ryden L. Diabetes mellitus and congestive heart failure. Further knowledge needed. Eur Heart J. (1999) 20:789–95. doi: 10.1053/euhj.1998.1472
16. Bauters C, Lamblin N, McFadden EP, Van Belle E, Millaire A, de Groote P. Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc Diabetol. (2003) 2:1. doi: 10.1186/1475-2840-2-1
17. Yoneyama K, Venkatesh BA, Wu CO, Mewton N, Gjesdal O, Kishi S, et al. Diabetes mellitus and insulin resistance associate with left ventricular shape and torsion by cardiovascular magnetic resonance imaging in asymptomatic individuals from the multi-ethnic study of atherosclerosis. J Cardiovasc Magn Reson. (2018) 20:53. doi: 10.1186/s12968-018-0472-9
18. Boyer JK, Thanigaraj S, Schechtman KB, Perez JE. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am J Cardiol. (2004) 93:870–5. doi: 10.1016/j.amjcard.2003.12.026
19. van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. (2008) 117:43–51. doi: 10.1161/CIRCULATIONAHA.107.728550
20. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. (2014) 57:660–71. doi: 10.1007/s00125-014-3171-6
21. Teupe C, Rosak C. Diabetic cardiomyopathy and diastolic heart failure – difficulties with relaxation. Diabetes Res Clin Pract. (2012) 97:185–94. doi: 10.1016/j.diabres.2012.03.008
22. Wold LE, Ceylan-Isik AF, Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol Sin. (2005) 26:908–17. doi: 10.1111/j.1745-7254.2005.00146.x
23. Holscher ME, Bode C, Bugger H. Diabetic cardiomyopathy: does the type of diabetes matter? Int J Mol Sci. (2016) 17:2136. doi: 10.3390/ijms17122136
24. Ryden L, Armstrong PW, Cleland JG, Horowitz JD, Massie BM, Packer M, et al. Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk, including those with diabetes mellitus. Results from the ATLAS trial. Eur Heart J. (2000) 21:1967–78. doi: 10.1053/euhj.2000.2311
25. Thrainsdottir IS, Aspelund T, Thorgeirsson G, Gudnason V, Hardarson T, Malmberg K, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care. (2005) 28:612–6. doi: 10.2337/diacare.28.3.612
26. Shindler DM, Kostis JB, Yusuf S, Quinones MA, Pitt B, Stewart D, et al. Diabetes mellitus, a predictor of morbidity and mortality in the Studies of Left Ventricular Dysfunction (SOLVD) trials and registry. Am J Cardiol. (1996) 77:1017–20. doi: 10.1016/S0002-9149(97)89163-1
27. Aronow WS, Ahn C. Incidence of heart failure in 2,737 older persons with and without diabetes mellitus. Chest. (1999) 115:867–8. doi: 10.1378/chest.115.3.867
28. Bugger H, Abel ED. Mitochondria in the diabetic heart. CardiovascRes. (2010) 88:229–40. doi: 10.1093/cvr/cvq239
29. Neel S, Singla DK. Induced pluripotent stem (iPS) cells inhibit apoptosis and fibrosis in streptozotocin-induced diabetic rats. Mol Pharm. (2011) 8:2350–7. doi: 10.1021/mp2004675
30. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. (2005) 54:1615–25. doi: 10.2337/diabetes.54.6.1615
31. Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. (2000) 21:361–70. doi: 10.1093/carcin/21.3.361
32. Nishio S, Teshima Y, Takahashi N, Thuc LC, Saito S, Fukui A, et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J Mol Cell Cardiol. (2012) 52:1103–11. doi: 10.1016/j.yjmcc.2012.02.006
33. Farhangkhoee H, Khan ZA, Mukherjee S, Cukiernik M, Barbin YP, Karmazyn M, et al. Heme oxygenase in diabetes-induced oxidative stress in the heart. J Mol Cell Cardiol. (2003) 35:1439–48. doi: 10.1016/j.yjmcc.2003.09.007
34. Pacifici RE, Davies KJ. Protein, lipid and DNA repair systems in oxidative stress: the free-radical theory of aging revisited. Gerontology. (1991) 37:166–80. doi: 10.1159/000213257
35. Sitte N, Huber M, Grune T, Ladhoff A, Doecke WD, Von Zglinicki T, et al. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB J. (2000) 14:1490–8. doi: 10.1096/fj.99-0843com
36. Halliwell B. The role of oxygen radicals in human disease, with particular reference to the vascular system. Haemostasis. (1993) 23(Suppl. 1):118–26. doi: 10.1159/000216921
37. Kaludercic N, Carpi A, Nagayama T, Sivakumaran V, Zhu G, Lai EW, et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid Redox Signal. (2014) 20:267–80. doi: 10.1089/ars.2012.4616
38. Wold LE, Ceylan-Isik AF, Fang CX, Yang X, Li SY, Sreejayan N, et al. Metallothionein alleviates cardiac dysfunction in streptozotocin-induced diabetes: role of Ca2+ cycling proteins, NADPH oxidase, poly(ADP-Ribose) polymerase and myosin heavy chain isozyme. Free Radic Biol Med. (2006) 40:1419–29. doi: 10.1016/j.freeradbiomed.2005.12.009
39. Dong F, Li Q, Sreejayan N, Nunn JM, Ren J. Metallothionein prevents high-fat diet induced cardiac contractile dysfunction: role of peroxisome proliferator activated receptor γ coactivator 1α and mitochondrial biogenesis. Diabetes. (2007) 56:2201–12. doi: 10.2337/db06-1596
40. Ye G, Metreveli NS, Ren J, Epstein PN. Metallothionein prevents diabetes-induced deficits in cardiomyocytes by inhibiting reactive oxygen species production. Diabetes. (2003) 52:777–83. doi: 10.2337/diabetes.52.3.777
41. Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, et al. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. (2004) 53:1336–43. doi: 10.2337/diabetes.53.5.1336
42. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. (2003) 112:1049–57. doi: 10.1172/JCI18127
43. Nascimento NR, Lessa LM, Kerntopf MR, Sousa CM, Alves RS, Queiroz MG, et al. Inositols prevent and reverse endothelial dysfunction in diabetic rat and rabbit vasculature metabolically and by scavenging superoxide. Proc Natl Acad Sci USA. (2006) 103:218–23. doi: 10.1073/pnas.0509779103
44. Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM, et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. (2006) 113:1235–43. doi: 10.1161/CIRCULATIONAHA.105.581397
45. Ma H, Li SY, Xu P, Babcock SA, Dolence EK, Brownlee M, et al. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med. (2009) 13:1751–64. doi: 10.1111/j.1582-4934.2008.00547.x
46. Nakamura H, Matoba S, Iwai-Kanai E, Kimata M, Hoshino A, Nakaoka M, et al. p53 promotes cardiac dysfunction in diabetic mellitus caused by excessive mitochondrial respiration-mediated reactive oxygen species generation and lipid accumulation. Circ Heart Fail. (2012) 5:106–15. doi: 10.1161/CIRCHEARTFAILURE.111.961565
47. Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res. (2018) 122:58–73. doi: 10.1161/CIRCRESAHA.117.311307
48. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. (2007) 56:2457–66. doi: 10.2337/db07-0481
49. Li S, Zhang L, Ni R, Cao T, Zheng D, Xiong S, et al. Disruption of calpain reduces lipotoxicity-induced cardiac injury by preventing endoplasmic reticulum stress. Biochim Biophys Acta. (2016) 1862:2023–33. doi: 10.1016/j.bbadis.2016.08.005
50. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. (2016) 57:2099–114. doi: 10.1194/jlr.R066514
51. Wende AR, Symons JD, Abel ED. Mechanisms of lipotoxicity in the cardiovascular system. Curr Hypertens Rep. (2012) 14:517–31. doi: 10.1007/s11906-012-0307-2
52. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
53. Lafuente N, Matesanz N, Azcutia V, Romacho T, Nevado J, Rodriguez-Manas L, et al. The deleterious effect of high concentrations of D-glucose requires pro-inflammatory preconditioning. J Hypertens. (2008) 26:478–85. doi: 10.1097/HJH.0b013e3282f331fb
54. Peiro C, Romacho T, Azcutia V, Villalobos L, Fernandez E, Bolanos JP, et al. Inflammation, glucose, and vascular cell damage: the role of the pentose phosphate pathway. Cardiovasc Diabetol. (2016) 15:82. doi: 10.1186/s12933-016-0397-2
55. Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. (2018) 104:293–9. doi: 10.1136/heartjnl-2017-311448
56. Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. (2004) 94:1533–42. doi: 10.1161/01.RES.0000129254.25507.d6
57. Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. (2011) 50:777–93. doi: 10.1016/j.freeradbiomed.2011.01.003
58. Steinberg SF. Oxidative stress and sarcomeric proteins. Circ Res. (2013) 112:393–405. doi: 10.1161/CIRCRESAHA.111.300496
59. Breitkreuz M, Hamdani N. A change of heart: oxidative stress in governing muscle function? Biophys Rev. (2015) 7:321–41. doi: 10.1007/s12551-015-0175-5
60. Grutzner A, Garcia-Manyes S, Kotter S, Badilla CL, Fernandez JM, Linke WA. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys J. (2009) 97:825–34. doi: 10.1016/j.bpj.2009.05.037
61. Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. (2009) 104:87–94. doi: 10.1161/CIRCRESAHA.108.184408
62. Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, et al. PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. (2009) 105:631–8. doi: 10.1161/CIRCRESAHA.109.198465
63. Beckendorf L, Linke WA. Emerging importance of oxidative stress in regulating striated muscle elasticity. J Muscle Res Cell Motil. (2015) 36:25–36. doi: 10.1007/s10974-014-9392-y
64. Rajesh M, Mukhopadhyay P, Batkai S, Patel V, Saito K, Matsumoto S, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am College Cardiol. (2010) 56:2115–25. doi: 10.1016/j.jacc.2010.07.033
65. Rajesh M, Batkai S, Kechrid M, Mukhopadhyay P, Lee WS, Horvath B, et al. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes. (2012) 61:716–27. doi: 10.2337/db11-0477
66. Roe ND, Thomas DP, Ren J. Inhibition of NADPH oxidase alleviates experimental diabetes-induced myocardial contractile dysfunction. Diabetes Obes Metab. (2011) 13:465–73. doi: 10.1111/j.1463-1326.2011.01369.x
67. Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. (2006) 58:87–114. doi: 10.1124/pr.58.1.6
68. Rajesh M, Mukhopadhyay P, Batkai S, Mukhopadhyay B, Patel V, Hasko G, et al. Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J Cell Mol Med. (2009) 13:2330–41. doi: 10.1111/j.1582-4934.2008.00564.x
69. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. (2007) 87:315–424. doi: 10.1152/physrev.00029.2006
70. Francia P, Cosentino F, Schiavoni M, Huang Y, Perna E, Camici GG, et al. p66(Shc) protein, oxidative stress, and cardiovascular complications of diabetes: the missing link. J Mol Med. (2009) 87:885–91. doi: 10.1007/s00109-009-0499-3
71. Deshwal S, Forkink M, Hu CH, Buonincontri G, Antonucci S, Di Sante M, et al. Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes. Cell Death Differ. (2018) 25:1671–85. doi: 10.1038/s41418-018-0071-1
72. Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P, Pacher P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta. (2015) 1852:232–42. doi: 10.1016/j.bbadis.2014.06.030
73. Braunersreuther V, Montecucco F, Asrih M, Pelli G, Galan K, Frias M, et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. (2013) 64:99–107. doi: 10.1016/j.yjmcc.2013.09.007
74. Casas AI, Dao VT, Daiber A, Maghzal GJ, Di Lisa F, Kaludercic N, et al. Reactive oxygen-related diseases: therapeutic targets and emerging clinical indications. Antioxid Redox Signal. (2015) 23:1171–85. doi: 10.1089/ars.2015.6433
75. Sirker A, Zhang M, Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol. (2011) 106:735–47. doi: 10.1007/s00395-011-0190-z
76. Dao VT, Casas AI, Maghzal GJ, Seredenina T, Kaludercic N, Robledinos-Anton N, et al. Pharmacology and clinical drug candidates in redox medicine. Antioxid Redox Signal. (2015) 23:1113–29. doi: 10.1089/ars.2015.6430
77. Santos CX, Raza S, Shah AM. Redox signaling in the cardiomyocyte: from physiology to failure. Int J Biochem Cell Biol. (2016) 74:145–51. doi: 10.1016/j.biocel.2016.03.002
78. Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. (2010) 107:15565–70. doi: 10.1073/pnas.1002178107
79. Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA. (2010) 107:18121–6. doi: 10.1073/pnas.1009700107
80. Varga ZV, Kupai K, Szucs G, Gaspar R, Paloczi J, Farago N, et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol. (2013) 62:111–21. doi: 10.1016/j.yjmcc.2013.05.009
81. Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, et al. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am J Physiol Cell Physiol. (2012) 302:C597–604. doi: 10.1152/ajpcell.00331.2011
82. Shen E, Li Y, Li Y, Shan L, Zhu H, Feng Q, et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. (2009) 58:2386–95. doi: 10.2337/db08-0617
83. Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes. (2010) 59:2033–42. doi: 10.2337/db09-1800
84. Hansen SS, Aasum E, Hafstad AD. The role of NADPH oxidases in diabetic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1908–13. doi: 10.1016/j.bbadis.2017.07.025
85. Balteau M, Tajeddine N, de Meester C, Ginion A, Des Rosiers C, Brady NR, et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res. (2011) 92:237–46. doi: 10.1093/cvr/cvr230
86. Roul D, Recchia FA. Metabolic alterations induce oxidative stress in diabetic and failing hearts: different pathways, same outcome. Antioxid Redox Signal. (2015) 22:1502–14. doi: 10.1089/ars.2015.6311
87. Wang Z, Zhang Y, Guo J, Jin K, Li J, Guo X, et al. Inhibition of protein kinase C βII isoform rescues glucose toxicity-induced cardiomyocyte contractile dysfunction: role of mitochondria. Life Sci. (2013) 93:116–24. doi: 10.1016/j.lfs.2013.06.002
88. Saliaris AP, Amado LC, Minhas KM, Schuleri KH, Lehrke S, St John M, et al. Chronic allopurinol administration ameliorates maladaptive alterations in Ca2+ cycling proteins and β-adrenergic hyporesponsiveness in heart failure. Am J Physiol Heart Circ Physiol. (2007) 292:H1328–35. doi: 10.1152/ajpheart.00461.2006
89. Amado LC, Saliaris AP, Raju SV, Lehrke S, St John M, Xie J, et al. Xanthine oxidase inhibition ameliorates cardiovascular dysfunction in dogs with pacing-induced heart failure. J Mol Cell Cardiol. (2005) 39:531–6. doi: 10.1016/j.yjmcc.2005.04.008
90. Desco MC, Asensi M, Marquez R, Martinez-Valls J, Vento M, Pallardo FV, et al. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. (2002) 51:1118–24. doi: 10.2337/diabetes.51.4.1118
91. Bravard A, Bonnard C, Durand A, Chauvin MA, Favier R, Vidal H, et al. Inhibition of xanthine oxidase reduces hyperglycemia-induced oxidative stress and improves mitochondrial alterations in skeletal muscle of diabetic mice. Am J Physiol Endocrinol Metab. (2011) 300:E581–91. doi: 10.1152/ajpendo.00455.2010
92. Gao X, Xu Y, Xu B, Liu Y, Cai J, Liu HM, et al. Allopurinol attenuates left ventricular dysfunction in rats with early stages of streptozotocin-induced diabetes. Diabetes Metab Res Rev. (2012) 28:409–17. doi: 10.1002/dmrr.2295
93. Szwejkowski BR, Gandy SJ, Rekhraj S, Houston JG, Lang CC, Morris AD, et al. Allopurinol reduces left ventricular mass in patients with type 2 diabetes and left ventricular hypertrophy. J Am College Cardiol. (2013) 62:2284–93. doi: 10.1016/j.jacc.2013.07.074
94. Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Curr Pharm Des. (2014) 20:3579–94. doi: 10.2174/13816128113196660748
95. Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. (2002) 109:817–26. doi: 10.1172/JCI0214442
96. Jo H, Otani H, Jo F, Shimazu T, Okazaki T, Yoshioka K, et al. Inhibition of nitric oxide synthase uncoupling by sepiapterin improves left ventricular function in streptozotocin-induced diabetic mice. Clin Exp Pharmacol Physiol. (2011) 38:485–93. doi: 10.1111/j.1440-1681.2011.05535.x
97. Okazaki T, Otani H, Shimazu T, Yoshioka K, Fujita M, Iwasaka T. Ascorbic acid and N-acetyl cysteine prevent uncoupling of nitric oxide synthase and increase tolerance to ischemia/reperfusion injury in diabetic rat heart. Free Radic Res. (2011) 45:1173–83. doi: 10.3109/10715762.2011.605361
98. Ren J, Duan J, Thomas DP, Yang X, Sreejayan N, Sowers JR, et al. IGF-I alleviates diabetes-induced RhoA activation, eNOS uncoupling, and myocardial dysfunction. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R793–802. doi: 10.1152/ajpregu.00713.2007
99. Smith JM, Paulson DJ, Romano FD. Inhibition of nitric oxide synthase by L-NAME improves ventricular performance in streptozotocin-diabetic rats. J Mol Cell Cardiol. (1997) 29:2393–402. doi: 10.1006/jmcc.1997.0474
100. Esberg LB, Ren J. Role of nitric oxide, tetrahydrobiopterin and peroxynitrite in glucose toxicity-associated contractile dysfunction in ventricular myocytes. Diabetologia. (2003) 46:1419–27. doi: 10.1007/s00125-003-1183-8
101. Stockklauser-Farber K, Ballhausen T, Laufer A, Rosen P. Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochim Biophys Acta. (2000) 1535:10–20. doi: 10.1016/S0925-4439(00)00078-8
102. Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, Kamath V, et al. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes. (2014) 63:1381–93. doi: 10.2337/db13-0577
103. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am College Cardiol. (2009) 54:1891–8. doi: 10.1016/j.jacc.2009.07.031
104. Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol. (2011) 300:H118–24. doi: 10.1152/ajpheart.00932.2010
105. Katunga LA, Gudimella P, Efird JT, Abernathy S, Mattox TA, Beatty C, et al. Obesity in a model of gpx4 haploinsufficiency uncovers a causal role for lipid-derived aldehydes in human metabolic disease and cardiomyopathy. Mol Metab. (2015) 4:493–506. doi: 10.1016/j.molmet.2015.04.001
106. Guo W, Kan JT, Cheng ZY, Chen JF, Shen YQ, Xu J, et al. Hydrogen sulfide as an endogenous modulator in mitochondria and mitochondria dysfunction. Oxid Med Cell Longev. (2012) 2012:878052. doi: 10.1155/2012/878052
107. Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal. (2015) 22:1545–62. doi: 10.1089/ars.2015.6293
108. Jarosz J, Ghosh S, Delbridge LM, Petzer A, Hickey AJ, Crampin EJ, et al. Changes in mitochondrial morphology and organization can enhance energy supply from mitochondrial oxidative phosphorylation in diabetic cardiomyopathy. Am J Physiol Cell Physiol. (2017) 312:C190–7. doi: 10.1152/ajpcell.00298.2016
109. Ni R, Zheng D, Xiong S, Hill DJ, Sun T, Gardiner RB, et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes. (2016) 65:255–68. doi: 10.2337/db15-0963
110. Bernardi P, Di Lisa F, Fogolari F, Lippe G. From ATP to PTP and back: a dual function for the mitochondrial ATP Synthase. Circ Res. (2015) 116:1850–62. doi: 10.1161/CIRCRESAHA.115.306557
111. Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. (2010) 53:1783–94. doi: 10.1007/s00125-010-1770-4
112. Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. (2010) 12:537–77. doi: 10.1089/ars.2009.2531
113. Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. (2006) 55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039
114. Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. (2006) 55:120–7. doi: 10.2337/diabetes.55.01.06.db05-0943
115. Viglino C, Foglia B, Montessuit C. Chronic AICAR treatment prevents metabolic changes in cardiomyocytes exposed to free fatty acids. Pflugers Arch. (2019) 471:1219–34. doi: 10.1007/s00424-019-02285-0
116. Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, et al. Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation. (2019) 139:1913–36. doi: 10.1161/CIRCULATIONAHA.118.033552
117. Ramsay RR. Electron carriers and energy conservation in mitochondrial respiration. ChemTexts. (2019) 5:9. doi: 10.1007/s40828-019-0085-4
118. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
119. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. (2003) 552:335–44. doi: 10.1113/jphysiol.2003.049478
120. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. (2002) 277:44784–90. doi: 10.1074/jbc.M207217200
121. Scialo F, Fernandez-Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol. (2017) 8:428. doi: 10.3389/fphys.2017.00428
122. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
123. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. (2000) 404:787–90. doi: 10.1038/35008121
124. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci USA. (2000) 97:12222–6. doi: 10.1073/pnas.97.22.12222
125. Teshima Y, Takahashi N, Nishio S, Saito S, Kondo H, Fukui A, et al. Production of reactive oxygen species in the diabetic heart. Roles of mitochondria and NADPH oxidase. Circ J. (2014) 78:300–6. doi: 10.1253/circj.CJ-13-1187
126. Sedlic F, Muravyeva MY, Sepac A, Sedlic M, Williams AM, Yang M, et al. Targeted modification of mitochondrial ROS production converts high glucose-induced cytotoxicity to cytoprotection: effects on anesthetic preconditioning. J Cell Physiol. (2017) 232:216–24. doi: 10.1002/jcp.25413
127. Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY, Murad F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J Biol Chem. (2003) 278:33972–7. doi: 10.1074/jbc.M303734200
128. Lashin OM, Szweda PA, Szweda LI, Romani AM. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic Biol Med. (2006) 40:886–96. doi: 10.1016/j.freeradbiomed.2005.10.040
129. Baseler WA, Dabkowski ER, Williamson CL, Croston TL, Thapa D, Powell MJ, et al. Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction. Am J Physiol Regul Integr Comp Physiol. (2011) 300:R186–200. doi: 10.1152/ajpregu.00423.2010
130. Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci USA. (2015) 112:6050–5. doi: 10.1073/pnas.1424017112
131. Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, et al. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem. (2009) 284:547–55. doi: 10.1074/jbc.M808518200
132. Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, et al. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Compar Physiol. (2011) 300:R1296–302. doi: 10.1152/ajpregu.00437.2010
133. Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. (2012) 287:30024–34. doi: 10.1074/jbc.M112.390682
134. Tian C, Alomar F, Moore CJ, Shao CH, Kutty S, Singh J, et al. Reactive carbonyl species and their roles in sarcoplasmic reticulum Ca2+ cycling defect in the diabetic heart. Heart Fail Rev. (2014) 19:101–12. doi: 10.1007/s10741-013-9384-9
135. Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. (2016) 118:1808–29. doi: 10.1161/CIRCRESAHA.116.306923
136. Ruiz-Meana M, Minguet M, Bou-Teen D, Miro-Casas E, Castans C, Castellano J, et al. Ryanodine receptor glycation favors mitochondrial damage in the senescent heart. Circulation. (2019) 139:949–64. doi: 10.1161/CIRCULATIONAHA.118.035869
137. Di Lisa F, Giorgio M, Ferdinandy P, Schulz R. New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. Br J Pharmacol. (2017) 174:1690–703. doi: 10.1111/bph.13478
138. Di Lisa F, Kaludercic N, Carpi A, Menabo R, Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol. (2009) 104:131–9. doi: 10.1007/s00395-009-0008-4
139. Carpi A, Menabo R, Kaludercic N, Pelicci P, Di Lisa F, Giorgio M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta. (2009) 1787:774–80. doi: 10.1016/j.bbabio.2009.04.001
140. Messina E, Giacomello A. Diabetic cardiomyopathy: a “cardiac stem cell disease” involving p66Shc, an attractive novel molecular target for heart failure therapy. Circ Res. (2006) 99:1–2. doi: 10.1161/01.RES.0000233141.65522.3e
141. Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, et al. Protein kinase C β and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. (2007) 315:659–63. doi: 10.1126/science.1135380
142. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. (2005) 122:221–33. doi: 10.1016/j.cell.2005.05.011
143. Galimov ER. The role of p66shc in oxidative stress and apoptosis. Acta Naturae. (2010) 2:44–51. doi: 10.32607/20758251-2010-2-4-44-51
144. Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. (2002) 21:3872–8. doi: 10.1038/sj.onc.1205513
145. Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. (2001) 89:279–86. doi: 10.1161/hh1501.094115
146. Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol. (2014) 73:34–42. doi: 10.1016/j.yjmcc.2013.12.032
147. Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, et al. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci USA. (2007) 104:5217–22. doi: 10.1073/pnas.0609656104
148. Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, et al. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci USA. (2003) 100:2112–6. doi: 10.1073/pnas.0336359100
149. Rota M, LeCapitaine N, Hosoda T, Boni A, De AA, Padin-Iruegas ME, et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. CircRes. (2006) 99:42–52. doi: 10.1161/01.RES.0000231289.63468.08
150. Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol. (2002) 9:22–6. doi: 10.1038/nsb732
151. De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci USA. (2005) 102:12684–9. doi: 10.1073/pnas.0505975102
152. Youdim MB, Finberg JP. New directions in monoamine oxidase A and B selective inhibitors and substrates. BiochemPharmacol. (1991) 41:155–62. doi: 10.1016/0006-2952(91)90471-G
153. Finberg JP, Youdim MB. Selective MAO A and B inhibitors: their mechanism of action and pharmacology. Neuropharmacology. (1983) 22:441–6. doi: 10.1016/0028-3908(83)90194-6
154. Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F. Structure and mechanism of monoamine oxidase. CurrMedChem. (2004) 11:1983–93. doi: 10.2174/0929867043364784
155. Ramsay RR. Monoamine oxidases: the biochemistry of the proteins as targets in medicinal chemistry and drug discovery. Curr Top Med Chem. (2012) 12:2189–209. doi: 10.2174/156802612805219978
156. Santin Y, Sicard P, Vigneron F, Guilbeau-Frugier C, Dutaur M, Lairez O, et al. Oxidative stress by monoamine oxidase-a impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid Redox Signal. (2016) 25:10–27. doi: 10.1089/ars.2015.6522
157. Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, et al. p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal. (2013) 18:5–18. doi: 10.1089/ars.2011.4373
158. Kaludercic N, Carpi A, Menabo R, Di Lisa F, Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta. (2011) 1813:1323–32. doi: 10.1016/j.bbamcr.2010.09.010
159. Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. (2010) 106:193–202. doi: 10.1161/CIRCRESAHA.109.198366
160. Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation. (2005) 112:3297–305. doi: 10.1161/CIRCULATIONAHA.104.528133
161. Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, Leducq N, et al. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ Res. (2007) 100:41–9. doi: 10.1161/01.RES.0000253900.66640.34
162. Sturza A, Leisegang MS, Babelova A, Schroder K, Benkhoff S, Loot AE, et al. Monoamine oxidases are mediators of endothelial dysfunction in the mouse aorta. Hypertension. (2013) 62:140–6. doi: 10.1161/HYPERTENSIONAHA.113.01314
163. Anderson EJ, Efird JT, Davies SW, O'Neal WT, Darden TM, Thayne KA, et al. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. J Am Heart Assoc. (2014) 3:e000713. doi: 10.1161/JAHA.113.000713
164. Manni ME, Rigacci S, Borchi E, Bargelli V, Miceli C, Giordano C, et al. Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: an intriguing therapeutic target. Oxid Med Cell Longev. (2016) 2016:4375418. doi: 10.1155/2016/4375418
165. Emory H, Mizrahi N. Glycaemic control by monoamine oxidase inhibition in a patient with type 1 diabetes. Diab Vasc Dis Res. (2016) 14:163–5. doi: 10.1177/1479164116675492
166. Binda C, Aldeco M, Geldenhuys WJ, Tortorici M, Mattevi A, Edmondson DE. Molecular insights into human monoamine oxidase B inhibition by the glitazone anti-diabetes drugs. ACS Med Chem Lett. (2011) 3:39–42. doi: 10.1021/ml200196p
167. Umbarkar P, Singh S, Arkat S, Bodhankar SL, Lohidasan S, Sitasawad SL. Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radic Biol Med. (2015) 87:263–73. doi: 10.1016/j.freeradbiomed.2015.06.025
168. Manzella N, Santin Y, Maggiorani D, Martini H, Douin-Echinard V, Passos JF, et al. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell. (2018) 17:e12811. doi: 10.1111/acel.12811
169. Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. (2019) 38:e100492. doi: 10.15252/embj.2018100492
170. Martini H, Iacovoni JS, Maggiorani D, Dutaur M, Marsal DJ, Roncalli J, et al. Aging induces cardiac mesenchymal stromal cell senescence and promotes endothelial cell fate of the CD90 + subset. Aging Cell. (2019) 18:e13015. doi: 10.1111/acel.13015
171. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008) 456:605–10. doi: 10.1038/nature07534
172. Zhang Y, Babcock SA, Hu N, Maris JR, Wang H, Ren J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: role of GSK3β and mitochondrial function. BMC Med. (2012) 10:40. doi: 10.1186/1741-7015-10-40
173. Pan G, Munukutla S, Kar A, Gardinier J, Thandavarayan RA, Palaniyandi SS. Type-2 diabetic aldehyde dehydrogenase 2 mutant mice (ALDH 2*2) exhibiting heart failure with preserved ejection fraction phenotype can be determined by exercise stress echocardiography. PLoS ONE. (2018) 13:e0195796. doi: 10.1371/journal.pone.0195796
174. Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, et al. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med. (2016) 90:12–23. doi: 10.1016/j.freeradbiomed.2015.11.013
175. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. (2000) 192:1001–14. doi: 10.1084/jem.192.7.1001
176. Antonucci S, Mulvey JF, Burger N, Di Sante M, Hall AR, Hinchy EC, et al. Selective mitochondrial superoxide generation in vivo is cardioprotective through hormesis. Free Radic Biol Med. (2019) 134:678–87. doi: 10.1016/j.freeradbiomed.2019.01.034
177. Liang L, Shou XL, Zhao HK, Ren GQ, Wang JB, Wang XH, et al. Antioxidant catalase rescues against high fat diet-induced cardiac dysfunction via an IKKβ-AMPK-dependent regulation of autophagy. Biochim Biophys Acta. (2015) 1852:343–52. doi: 10.1016/j.bbadis.2014.06.027
178. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. (2007) 26:1749–60. doi: 10.1038/sj.emboj.7601623
179. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. (2015) 22:377–88. doi: 10.1038/cdd.2014.150
180. Pajares M, Cuadrado A, Engedal N, Jirsova Z, Cahova M. The role of free radicals in autophagy regulation: implications for ageing. Oxid Med Cell Longev. (2018) 2018:2450748. doi: 10.1155/2018/2450748
181. Pei Z, Deng Q, Babcock SA, He EY, Ren J, Zhang Y. Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: Role of autophagy and ER stress. Toxicol Lett. (2018) 284:10–20. doi: 10.1016/j.toxlet.2017.11.018
182. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. (2006) 103:2653–8. doi: 10.1073/pnas.0511154103
183. Kaludercic N, Maiuri MC, Kaushik S, Fernandez AF, de Bruijn J, Castoldi F, et al. Comprehensive autophagy evaluation in cardiac diseases models. Cardiovasc Res. (2019). doi: 10.1093/cvr/cvz233. [Epub ahead of print].
184. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. (2019) 124:1360–71. doi: 10.1161/CIRCRESAHA.118.314607
185. Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. (2012) 125:1134–46. doi: 10.1161/CIRCULATIONAHA.111.078212
186. Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. (2013) 288:18077–92. doi: 10.1074/jbc.M113.474650
187. Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. (2019) 129:802–19. doi: 10.1172/JCI122035
188. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, et al. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the american heart association. Circ Res. (2016) 119:e39–75. doi: 10.1161/RES.0000000000000110
189. Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. (2005) 4:5. doi: 10.1186/1475-2840-4-5
190. Berniakovich I, Trinei M, Stendardo M, Migliaccio E, Minucci S, Bernardi P, et al. p66Shc-generated oxidative signal promotes fat accumulation. J Biol Chem. (2008) 283:34283–93. doi: 10.1074/jbc.M804362200
191. Brandes RP, Rezende F, Schroder K. Redox Regulation Beyond ROS: why ROS should not be measured as often. Circ Res. (2018) 123:326–8. doi: 10.1161/CIRCRESAHA.118.313146
192. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
193. Lytvyn Y, Bjornstad P, Udell JA, Lovshin JA, Cherney DZI. Sodium glucose cotransporter-2 inhibition in heart failure: potential mechanisms, clinical applications, and summary of clinical trials. Circulation. (2017) 136:1643–58. doi: 10.1161/CIRCULATIONAHA.117.030012
194. Matsumura K, Sugiura T. Effect of sodium glucose cotransporter 2 inhibitors on cardiac function and cardiovascular outcome: a systematic review. Cardiovasc Ultrasound. (2019) 17:26. doi: 10.1186/s12947-019-0177-8
195. Di Franco A, Cantini G, Tani A, Coppini R, Zecchi-Orlandini S, Raimondi L, et al. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: a new potential pharmacological target. Int J Cardiol. (2017) 243:86–90. doi: 10.1016/j.ijcard.2017.05.032
196. Mustroph J, Wagemann O, Lucht CM, Trum M, Hammer KP, Sag CM, et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. (2018) 5:642–8. doi: 10.1002/ehf2.12336
197. Uthman L, Baartscheer A, Schumacher CA, Fiolet JWT, Kuschma MC, Hollmann MW, et al. Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Front Physiol. (2018) 9:1575. doi: 10.3389/fphys.2018.01575
198. Li C, Zhang J, Xue M, Li X, Han F, Liu X, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. (2019) 18:15. doi: 10.1186/s12933-019-0816-2
199. Tanaka H, Hirata KI. Potential impact of SGLT2 inhibitors on left ventricular diastolic function in patients with diabetes mellitus. Heart Fail Rev. (2018) 23:439–44. doi: 10.1007/s10741-018-9668-1
200. Lam CSP, Chandramouli C, Ahooja V, Verma S. SGLT-2 Inhibitors in heart failure: current management, unmet needs, and therapeutic prospects. J Am Heart Assoc. (2019) 8:e013389. doi: 10.1161/JAHA.119.013389
201. Pieske B, Maggioni AP, Lam CSP, Pieske-Kraigher E, Filippatos G, Butler J, et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur Heart J. (2017) 38:1119–27. doi: 10.1093/eurheartj/ehw593
202. Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, et al. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. (2012) 69:2327–43. doi: 10.1007/s00018-012-1010-9
203. Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. (2013) 123:1262–74. doi: 10.1172/JCI65268
204. Schilling JD. The mitochondria in diabetic heart failure: from pathogenesis to therapeutic promise. Antioxid Redox Signal. (2015) 22:1515–26. doi: 10.1089/ars.2015.6294
205. Youdim MB, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. (2006) 7:295–309. doi: 10.1038/nrn1883
206. Riederer P, Lachenmayer L, Laux G. Clinical applications of MAO-inhibitors. Curr Med Chem. (2004) 11:2033–43. doi: 10.2174/0929867043364775
Keywords: diabetic cardiomyopathy, reactive oxygen species, mitochondria, oxidative stress, diabetic complication
Citation: Kaludercic N and Di Lisa F (2020) Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 7:12. doi: 10.3389/fcvm.2020.00012
Received: 31 October 2019; Accepted: 28 January 2020;
Published: 18 February 2020.
Edited by:
Junichi Sadoshima, University of Medicine and Dentistry of New Jersey, United StatesReviewed by:
Susumu Minamisawa, Jikei University School of Medicine, JapanCopyright © 2020 Kaludercic and Di Lisa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Kaludercic, bmluYS5rYWx1ZGVyY2ljQHVuaXBkLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.