 Amanda Z. Zucoloto
Amanda Z. Zucoloto Craig N. Jenne
Craig N. Jenne
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 20 June 2019
Sec. Atherosclerosis and Vascular Medicine
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00085
This article is part of the Research Topic Established and Novel Roles of Platelets in Health and Disease View all 20 articles
Well established for their central role in hemostasis, platelets have increasingly been appreciated as immune cells in recent years. This emerging role should not come as a surprise as the central immune cells of invertebrates, hemocytes, are able to phagocytose, secrete soluble mediators and promote coagulation of hemolymph, blurring the line between immunity and hemostasis. The undeniable evolutionary link between coagulation and immunity becomes even clearer as the role of platelets in inflammation is better understood. Platelets exert a range of immune-related functions, many of which involve an intimate interplay with leukocytes. Platelets promote leukocyte recruitment via endothelial activation and can serve as “landing pads” for leukocytes, facilitating cellular adhesion in vascular beds devoid of classic adhesion molecules. Moreover, platelets enhance leukocyte function both through direct interactions and through release of soluble mediators. Among neutrophil-platelets interactions, the modulation of neutrophil extracellular traps (NETs) is of great interest. Platelets have been shown to induce NET formation; and, in turn, NET components further regulate platelet and neutrophil function. While NETs have been shown to ensnare and kill pathogens, they also initiate coagulation via thrombin activation. In fact, increased NET formation has been associated with hypercoagulability in septic patients as well as in chronic vascular disorders. This review will delve into current knowledge of platelet-neutrophil interactions, with a focus on NET-driven coagulation, in the context of infectious diseases. A better understanding of these mechanisms will shed a light on the therapeutic potential of uncoupling immunity and coagulation through targeting of NETs.
Poised at the interface of immunity and coagulation, platelets express a plethora of surface molecules and receptors and carry granules packed with hundreds of biologically active products. Platelets arise from megakaryocytes, which in turn differentiate from pluripotent hematopoietic cells restricted to the bone-proximal osteoblastic niche in the bone marrow (1, 2). Proplatelets are released from this specialized site into the circulation, continue to mature, ultimately releasing mature platelets (3).
Platelets are undoubtedly critical for hemostasis, in a large part by supporting blood coagulation. Upon activation, platelets expose negatively-charged phospholipids on the outer leaflet of their plasma membrane, providing an ideal surface for the assembly of coagulation factors complexes, such as the VIIIa-IXa complex and the Xa-Va complex. Moreover, factor XI and thrombin are brought into close proximity through interactions with the glycoprotein (GP) Ib/V/IX complex on the surface of platelets, facilitating factor XI cleavage by thrombin. This not only sustains the coagulation cascade, but also overcomes coagulation arrest in the presence of TFPI (inhibitor of the extrinsic pathway) (4).
Platelets are equipped with numerous immune receptors. For instance, signaling through TLRs 2/6 and 1/2 triggers platelet activation, marked by increased expression of surface CD62P (P-selectin), degranulation and aggregation (5–7). TLR4 engagement was shown to induce platelet aggregation and interaction with leukocytes as well as affect CD62P expression in a ligand-dependent manner (8–10). Moreover, human platelets have been shown to secrete antimicrobial peptides targeting both bacteria and fungi in response to thrombin, a key enzyme in the coagulation cascade (11). Platelets have also been increasingly recognized for their role in cellular recruitment. In that sense, platelets have been shown to serve as a “landing pad” in endothelial beds devoid of adhesion molecules, such as the brain (12, 13). In a model of hepatitis, CD8+ T cells were also shown to preferentially dock onto platelets adherent within liver sinusoids rather than adhering to endothelial cells themselves, suggesting a role for platelets in adaptive immunity (14).
The active role of platelets in both coagulation and immunity hints at an evolutionary link to the central immune cells of invertebrates, hemocytes. Hemocytes not only provide host defense through secretion of microbicidal peptides and phagocytosis, but also through coagulation of the hemolymph. In these invertebrate organisms, clot formation is a potent host defense mechanism as it isolates and contains the infectious agent (15–17). This ability to sequester and contain pathogens undeniably resembles the role of neutrophil extracellular traps (NETs), an immune effector mechanism of higher vertebrates. The complex interplay between platelets and neutrophils, which is most likely a long-evolving relationship, will be discussed in this review, with a focus on NET-driven coagulation.
The interactions between platelets and neutrophils are orchestrated by both their surface and secreted molecules, with the former allowing for physical interactions between these cells. Activated platelets express CD62P, which binds to P-selectin glycoprotein ligand-1 (PSGL-1) on the surface of neutrophils (Figure 1A) (21, 22). Alternatively, both GPIb or the integrin αIIβ3 on platelets interact with the integrin αMβ2 on leukocytes either directly, or through fibrinogen as a bridging molecule (23–25). Secreted molecules, such as cathepsin G produced by activated neutrophils, can disrupt these interactions through cleavage of GPIb and PSGL-1 (26). Moreover, there is increasing evidence of platelet-derived products modulating neutrophil recruitment, activation and function. For instance, CD40L secreted by platelets has been shown to upregulate integrin expression on neutrophils (27). Serotonin and CXCL4 have also been implicated in platelet-dependent neutrophil recruitment in models of abdominal inflammation and acute pancreatitis (28, 29).
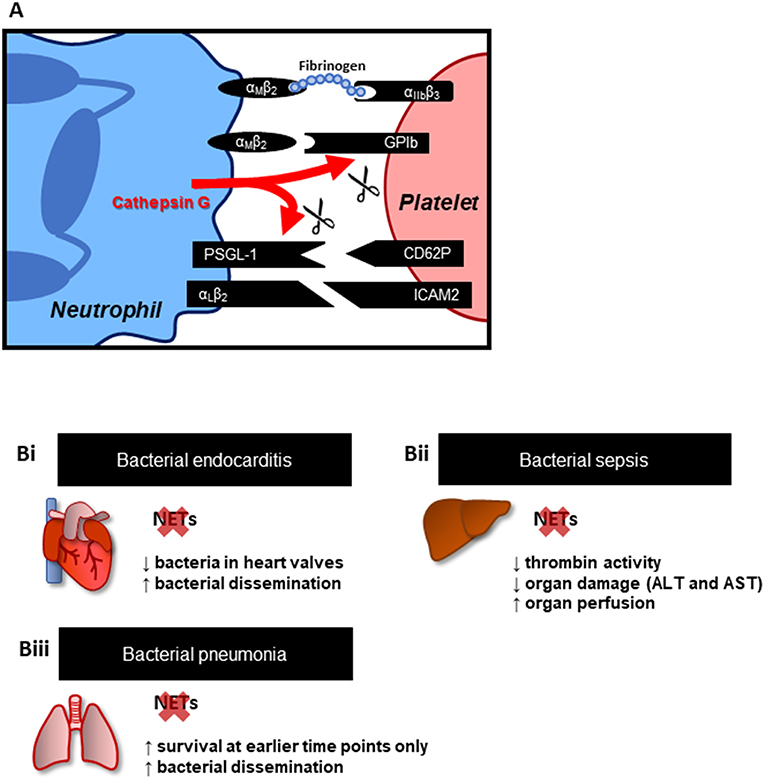
Figure 1. Platelets-neutrophil interactions and NETs. (A) Key adhesion molecules involved in platelet-neutrophil interactions. These interactions not only provide mechanisms of cell attachment but may also trigger intracellular signaling, promoting cell activation, resulting in the upregulation of additional adhesion and effector molecules. Effector molecules from both cells, such as cathepsin G from neutrophils, in turn modulate neutrophil-platelet physical interactions through cleavage of PSGL-1 and GPIb. (B) Effect of NET inhibition (PAD4−/− mice) or disruption (DNase treatment) in animal models of (Bi) endocarditis (18), (Bii) bacterial sepsis (19), and (Biii) bacterial pneumonia (20). Overall, targeting NETs is associated with reduced inflammation and organ damage; however, this effect has been shown to favor bacterial dissemination.
Platelets mediate leukocyte recruitment via two main mechanisms: (a) by serving as a docking site for immune cells along the endothelium surrounding the inflammatory focus and (b) through secretion of chemoattractants. The extent to which platelets promote cellular recruitment appears to be tissue- and model-dependent. For instance, neutrophil infiltration into the peritoneal cavity, skin and brain in response to LPS was shown to be platelet-dependent whereas in the lung, a compensatory mechanism characterized by the upregulation of CXCL1 and CCL5 was able to overcome platelet depletion (30). In a model of Pseudomonas aeruginosa pulmonary infection, however, platelet depletion was shown to reduce neutrophil infiltration in the lung (31). Similarly, platelet-driven neutrophil recruitment to the colon and kidney has been demonstrated in models of dextran sodium sulfate (DSS)-induced acute colitis and cecal ligation and puncture (CLP) (32, 33). In these studies, platelet depletion was shown to improve clinical and histopathological scores, whereas in the aforementioned model of P. aeruginosa pulmonary infection, platelet depletion led to increased bacterial dissemination and mortality (31–33). These differences further emphasize the complexity of the interplay between platelets and other immune cells, such as neutrophils.
Indeed, platelets are not limited to providing a port-of-entry to neutrophils into sites of tissue insult. Platelets have been shown to directly stimulate the production of NETs through the process of NETosis (18, 34–37). In turn, NETs amplify platelet activation, aggregation and thrombin activation, and all three act in synergy to promote intravascular coagulation in sepsis (19, 38, 39). Evidence supporting the deleterious effects of NET-induced coagulation in infectious diseases will be discussed in the next section.
Unsurprisingly, platelet and neutrophil interactions are greatly increased during inflammatory responses. These interactions are, for the most part, initiated by soluble mediators, which directly activate these cells (Table 1). Co-incubation of healthy platelets and neutrophils with plasma from septic patients has been shown to promote platelet adhesion to neutrophils in a TLR4-dependent manner, a result similar to what is observed following co-incubation of platelets, neutrophils and LPS (34, 36). Moreover, LPS-induced intravascular NETosis and trapping of Escherichia coli in NETs has been shown to be augmented in the presence of platelets (34). Platelet-driven NETosis has been observed in the presence of all classic platelet agonists (i.e., thrombin, ADP, collagen, arachidonic acid) as well as several TLR ligands; however, in these models, NET formation does not occur in the absence of platelet activation (35–37). Hence, it is consensus that, for platelet-induced NETosis in vitro, platelets must be first activated. CD62P is also required for platelet-induced NETosis as CD62P−/− platelets have been shown to fail to promote the release of NETs whereas overexpression of platelet CD62P enhanced phorbol 12-myristate 13-acetate (PMA)- and ionomycin-induced NETosis (35). The role of other surface molecules involved in platelet-neutrophil interactions, GPIb/IIa, CD11b (integrin α-chain L), and CD18 (integrin β-chain 2), remains debatable as some studies have shown these molecules to be dispensable (37) while other models have indicated a clear need for integrin-mediated platelet adhesion in the induction of NETosis (69). Although there is strong evidence that platelet-neutrophil adhesion plays a central role in platelet-induced NET formation, physical interactions between platelets and neutrophils may not be absolutely required as activated platelets are known to shed CD62P. Indeed, neutrophils, in the presence of Streptococcus mutans and soluble CD62P (sCD62P) have been shown to produce NETs, thus indicating that direct interactions are dispensable in some situations, at least in vitro (18).
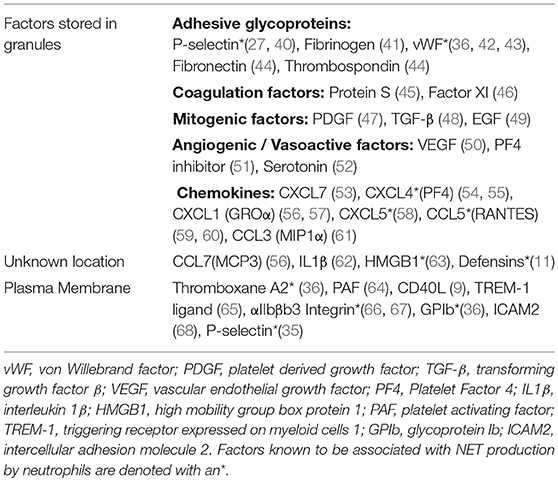
Table 1. Platelet molecules that modulate neutrophil activation.
In addition, to CD62P/PSGL-1 signaling, other mediators have been shown to be involved in triggering NET release. For instance, antibody-mediated blockade of platelet-derived high-mobility box group 1 protein (HMBG1) has been shown to inhibit NET formation in vitro (37). In fact, NET release was inhibited in the presence of anti-HMBG1 to a greater extent than in the presence of anti-CD62P. Moreover, HMGB1 was shown to be required for activation of autophagy pathways, which are required for NETosis in a RAGE-dependent manner, a key receptor for HMGB1 (37). A role for thromboxane A2 (TXA2) has also been demonstrated in platelet-driven NET release. In a study by Caudrillier et al. (66), activated, but not resting, platelets were shown to induce NET formation in vitro. This process was shown to be dependent on TXA2 receptor-mediated signaling, which activates the MAPK pathway (66). Moreover, platelet-derived β-defensin 1 was shown to induce NET formation in a ROS-dependent manner (70). Human platelets store β-defensin 1, an antimicrobial peptide primarily expressed by epithelial cells, in extragranular cytoplasmic compartments. Thus, β-defensin is not released during classic platelet degranulation stimulated by agonists such as thrombin or platelet activating factor (PAF). Instead, β-defensin 1 was shown to be released when platelets were stimulated in the presence of Staphylococcus aureus-derived α-toxin but not LPS (15). This α-toxin-platelet-β-defensin 1 axis could represent a novel mechanism by which platelets directly induce NETs in response to Gram-positive infections. The generation of NETs, however, was only demonstrated in the presence of purified β-defensin 1 (70). Co-incubation of α-toxin, platelets and neutrophils could provide more conclusive evidence of this putative interplay.
The downstream effects of platelet-induced NET release have also been studied. Activated, but not resting, platelets and neutrophils were shown to result in NET release and increased monolayer permeability in LPS-stimulated endothelial cells in vitro (66). Importantly, NETs also affect platelet function and direct evidence demonstrates that the platelet-NET axis is by no means a one-way road. In a report by Elaskalani et al. (38), cell-free NETs (collected from PMA-stimulated neutrophils) were incubated with human platelets where the presence of NETs alone (with no other platelet agonists added) was shown to promote platelet aggregation, secretion of ATP and ADP, and increased expression of CD62L and phosphatidylserine (PS) on the surface of the platelets (38). An increase in protein phosphorylation at tyrosine residues pointed to NET-mediated activation of intracellular signaling in platelets. Accordingly, Fuchs et al. (39) have demonstrated a role for DNA as well as histones H3 and H4 in NET-induced platelet aggregation in vitro (39). These results strongly hint at a positive feedback loop between platelets and NETs. NET-dependent platelet aggregation, however, was shown to be unaffected by DNase or heparin, suggesting mechanisms independent of the DNA scaffold and thrombin. Inhibition of cathepsin G, a NET component, reduced surface expression of CD62P and PS exposure in platelets whereas blockade of GPIIb/IIIa significantly inhibited platelet aggregation without affecting CD62P expression (38). These observations suggest multiple pathways are likely involved in NET-induced platelet activation and aggregation.
Furthermore, NETs have been implicated in directly inducing thrombin generation through both platelet-dependent and independent mechanisms. NETs released from PMA-stimulated neutrophils have been shown to increase thrombin generation in platelet-poor plasma. This activation required coagulation factors XII and XI, pointing to the involvement of the intrinsic coagulation pathway (71). Importantly, this mechanism was DNA-dependent, as thrombin generation was abrogated when DNase was added to the system (71, 72), although the exact role of DNA scaffold in this context is unknown. Given that coagulation factors optimally assemble on negatively-charged surfaces (such as the PS-rich plasma membrane of activated platelets and microparticles), and that DNA is a negatively-charged molecule, perhaps the DNA backbone of NETs serves as a somewhat ideal surface for the formation of coagulation factor complexes. Importantly, some studies have demonstrated a failure of purified NETs to induce thrombin generation. Whereas, the purified DNA backbone from NETs was able to induce thrombin generation in platelet-poor plasma, intact NETs, failed to generate active thrombin pointing out clear differences between in vivo and in vitro assays and suggesting qualitative differences in NETs themselves exist (73). In the presence of platelets, NETs were also shown to contribute to thrombin generation. The effect was dependent on platelet expression of TLR-2 and TLR-4, suggesting that NET components may act as receptor agonists, inducing platelet activation. Interestingly, DNase potentiated platelet- and NET-driven thrombin generation, likely because dismantling of NETs led to enhanced release of NET components, making them more readily available to act as platelet agonists (71).
In addition to NET production, PMA-stimulated neutrophils have also been shown to release microparticles, which attach themselves to NETs via PS residues. Blocking this interaction was implicated in reduced NET-dependent thrombin generation pointing to a role for neutrophil-derived microparticles in addition to DNA and other NET components (72). While these reports were largely done in vitro, the direct effect of NETs on coagulation in vivo has been also been investigated (19, 72). Using a model of sepsis following CLP, increased cell-free DNA in plasma was shown to be largely derived from neutrophils. Plasma levels of DNA-histone complexes were significantly inhibited in neutrophil-depleted animals, strongly suggesting that neutrophils undergo NETosis in the vasculature during sepsis. Thrombin-antithrombin (TAT) complex levels, a marker of thrombin generation, were increased 24 h after CLP. Moreover, thrombin generation ex vivo was decreased in these animals due to consumption of coagulation factors, a clinical manifestation of disseminated intravascular coagulation (DIC) in septic patients. In animals treated with DNase prior to CLP, thrombin generation ex vivo was restored, placing NETs as critical factor for the depletion of coagulation factors during sepsis (72). Additionally, blockade of NETs has been shown to reduce thrombin activity in the liver and lung microvasculature and inhibition of thrombin prevents NET-induced liver damage in an in vivo model of E. coli-induced sepsis (19), providing a direct link between NETs and coagulation. Interestingly, NETs have been shown to contribute to the occlusion of vessels in the lung microvasculature independently of thrombin generation in animals deficient for DNase1 and 3 during sepsis (74). These data indicate that NETs engage multiple mechanisms leading to microvascular obstruction, impairing organ perfusion and driving tissue damage.
Taken together, these studies have contributed greatly to mapping of the molecular mechanisms underlying platelet-NET interplay. While these mechanisms remain incompletely known, crucial molecules, such as CD62P and HMGB1 in platelets, PSGL1 in neutrophils and cathepsin G, histones and DNA in NETs, have been identified as potential targets for uncoupling immunity and coagulation. Targeting of these interactions, however, may have profound impact on host defense, underscoring the need for studying platelet-neutrophil crosstalk in in vivo models of infection and inflammatory disease.
In support of experimental evidence of NET-induced coagulation, a correlation between NETs and thrombosis has been previously demonstrated in clinical studies (75–77). Various components of NETs (cell-free DNA, citrullinated H3 and nucleosome) were shown to be significantly increased following acute ischemic stroke. Levels of citrullinated H3 were also associated with mortality at the 1-year follow up assessment of the study, which included over 200 patients with acute ischemic stroke (75). Of note, H3 citrullination weakens histone binding to negatively charged DNA, which in turn favors chromatin decondensation, a critical step for NETosis (78). Furthermore, a descriptive study of the composition of thrombi retrieved from ischemic stroke patients has revealed the presence of activated neutrophils (CD66b+ and neutrophil elastase+ granulocytes) and as well as NETs (extracellular DNA with citrullinated H3) (76). In the context of infectious diseases, a study by Yang et al. (77) has compared the ability of neutrophils from septic patients to undergo NETosis and its implications in thrombin and fibrin generation (77). Neutrophils from septic patients were able to promote thrombin and fibrin generation in the presence of control plasma in a DNA-dependent manner. Thus, NET release alone was associated with risk of thromboembolism. However, it is still unclear if NETs are causative of thrombosis or if NETosis is a consequence of thrombus formation.
Clinical evidence has supported the role of NETs in driving coagulation and ultimately, vascular dysfunction in both primarily cardiovascular disorders (i.e., ischemic stroke) as well as conditions initiated by infections, such as sepsis. These reports, however, did not delve into the implications of disrupting NET-induced coagulation for disease outcome. Perhaps the main concern when considering this strategy is the impairment of the innate immune response. To address this issue, animal models of systemic or highly invasive infections have been very insightful (Figure 1B). Infective endocarditis is a condition involving thrombi formation following an immune response to bacterial colonization in the heart. Vegetations, a pathological feature of endocarditis, are composed of bacteria embedded in a mass of platelets, fibrin, and immune cells, such as neutrophils. In a rat model of Streptococcus mutans-induced endocarditis, NETs were identified in vegetations on damaged heart valves (18). Accordingly, pre-treatment with DNase I, which dismantles the NET web-like structure, led to a decrease in vegetation weight and bacterial load. However, the effect was also accompanied by increased bacterial dissemination. These results suggest a protective role of thrombus and NET formation on heart valves, at least in restricting pathogen invasion and dissemination throughout the body. Indeed, in vitro, in the presence of neutrophils, platelets were shown to form aggregates around bacteria. The effect was abrogated in the presence of DNase I, supporting the involvement of NETs in pathogen trapping (18).
Furthermore, in models of bacterial sepsis, NET formation, platelet aggregation and thrombin activity were associated with impaired perfusion of the liver microvasculature and organ damage. PAD4−/− mice (unable to release NETs from neutrophils) or pre-treatment with DNase I markedly reduced thrombin activity, suggesting that NETs directly contributed to thrombin activation, likely through platelet-dependent mechanisms. Moreover, organ perfusion and function were improved in the absence of NETs, suggesting that direct targeting of NETs may be beneficial in the context of disseminated infections characterized by overt inflammation, such as sepsis (19). Importantly, this beneficial effect of NET prevention/dismantlement is very much context dependent. In a study by Lefrançais et al. (20), NETs, as expected, were associated with increased inflammation, lung damage and early mortality in a model of bacteria-induced lung injury (20). However, NET-deficient mice or removal of NETs with DNAse I, was associated with increased bacterial loads, demonstrating a clear role for NETs in restricting pathogen dissemination. Critically, the survival rate of animals in this model was only improved if NETs were targeted at earlier time points post-infection. At later time points (>40 h from the i.t., administration of S. aureus), blockade of NETs was no longer protective. These results suggest that although NETs play a role in immunopathology early in infection, their role in trapping and sequestering microorganisms is also critical to limit dissemination of infection and as such, complete abrogation of NET production may be just as deleterious to the host as NET-induced pathology.
Platelet-neutrophil interactions are undoubtedly a two-way relationship due, in large part, to the role of NETs in modulating both platelet and neutrophil function. Moreover, clinical and experimental studies have supported that NET-mediated coagulation and immunity are critical to disease outcome. Perhaps not surprisingly, NETs seem to play a dual role in models of infectious diseases: they orchestrate both immunopathology and infection clearance. Interestingly, the direct participation of NETs in amplifying coagulation through platelet activation, thrombin generation and microparticle release, seems to ultimately underlie NET-induced immunopathology. Importantly, while blocking or dismantling NETs ameliorates coagulation dysfunction, it may also impair pathogen clearance. Although separating immunity and coagulopathy is the ultimate goal, the question still remains… is this a matter of simply fine tuning of platelet-neutrophil interactions, or, is targeting the procoagulant components of NETs the key to this therapeutic avenue? In the end, a more thorough understanding of the molecular mechanisms underlying NET-driven coagulation will be needed if we are to uncouple immunity and coagulation in the setting of infectious disease.
AZ and CJ contributed to manuscript generation and revision and read and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. (2004) 10:64–71. doi: 10.1038/nm973
2. Larson MK, Watson SP. A product of their environment: do megakaryocytes rely on extracellular cues for proplatelet formation? Platelets. (2006) 17:435–40. doi: 10.1080/09537100600772637
3. Italiano JE Jr, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. (1999) 147:1299–312. doi: 10.1083/jcb.147.6.1299
4. Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. (2002) 88:186–93. doi: 10.1055/s-0037-1613209
5. Damien P, Cognasse F, Payrastre B, Spinelli SL, Blumberg N, Arthaud CA, et al. NF-kappaB Links TLR2 and PAR1 to soluble immunomodulator factor secretion in human platelets. Front Immunol. (2017) 8:85. doi: 10.3389/fimmu.2017.00085
6. Assinger A, Kral JB, Yaiw KC, Schrottmaier WC, Kurzejamska E, Wang Y, et al. Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler Thromb Vasc Biol. (2014) 34:801–9. doi: 10.1161/ATVBAHA.114.303287
7. Keane C, Tilley D, Cunningham A, Smolenski A, Kadioglu A, Cox D, et al. Invasive Streptococcus pneumoniae trigger platelet activation via toll-like receptor 2. J Thromb Haemost. (2010) 8:2757–65. doi: 10.1111/j.1538-7836.2010.04093.x
8. Rigg RA, Healy LD, Nowak MS, Mallet J, Thierheimer ML, Pang J, et al. Heat shock protein 70 regulates platelet integrin activation, granule secretion and aggregation. Am J Physiol Cell Physiol. (2016) 310:C568–75. doi: 10.1152/ajpcell.00362.2015
9. Carestia A, Rivadeneyra L, Romaniuk MA, Fondevila C, Negrotto S, Schattner M. Functional responses and molecular mechanisms involved in histone-mediated platelet activation. Thromb Haemost. (2013) 110:1035–45. doi: 10.1160/TH13-02-0174
10. Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional toll-like receptor-4. Blood. (2005) 106:2417–23. doi: 10.1182/blood-2005-03-0916
11. Tang YQ, Yeaman MR, Selsted ME. Antimicrobial peptides from human platelets. Infect Immun. (2002) 70:6524–33. doi: 10.1128/IAI.70.12.6524-6533.2002
12. Langer HF, Choi EY, Zhou H, Schleicher R, Chung KJ, Tang Z, et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ Res. (2012) 110:1202–10. doi: 10.1161/CIRCRESAHA.111.256370
13. Carvalho-Tavares J, Hickey MJ, Hutchison J, Michaud J, Sutcliffe IT, Kubes P. A role for platelets and endothelial selectins in tumor necrosis factor-alpha-induced leukocyte recruitment in the brain microvasculature. Circ Res. (2000) 87:1141–8. doi: 10.1161/01.RES.87.12.1141
14. Iannacone M. Platelet-mediated modulation of adaptive immunity. Semin Immunol. (2016) 28:555–60. doi: 10.1016/j.smim.2016.10.008
15. Salzet M. Vertebrate innate immunity resembles a mosaic of invertebrate immune responses. Trends Immunol. (2001) 22:285–8. doi: 10.1016/S1471-4906(01)01895-6
16. Iwanaga S, Lee BL. Recent advances in the innate immunity of invertebrate animals. J Biochem Mol Biol. (2005) 38:128–50. doi: 10.5483/BMBRep.2005.38.2.128
17. Jenne CN, Kubes P. Platelets in inflammation and infection. Platelets. (2015) 26:286–92. doi: 10.3109/09537104.2015.1010441
18. Jung CJ, Yeh CY, Hsu RB, Lee CM, Shun CT, Chia JS. Endocarditis pathogen promotes vegetation formation by inducing intravascular neutrophil extracellular traps through activated platelets. Circulation. (2015) 131:571–81. doi: 10.1161/CIRCULATIONAHA.114.011432
19. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
20. Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. (2018) 3:98178. doi: 10.1172/jci.insight.98178
21. Yang J, Furie BC, Furie B. The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost. (1999) 81:1–7. doi: 10.1055/s-0037-1614407
22. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. (1999) 93:876–85.
23. Diacovo TG, deFougerolles AR, Bainton DF, Springer TA. A functional integrin ligand on the surface of platelets: intercellular adhesion molecule-2. J Clin Invest. (1994) 94:1243–51. doi: 10.1172/JCI117442
24. Kazzaz NM, Sule G, Knight JS. Intercellular interactions as regulators of NETosis. Front Immunol. (2016) 7:453. doi: 10.3389/fimmu.2016.00453
25. Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. (2000) 192:193–204. doi: 10.1084/jem.192.2.193
26. Gardiner EE, De LM, McNally T, Michelson AD, Andrews RK, Berndt MC. Regulation of P-selectin binding to the neutrophil P-selectin counter-receptor P-selectin glycoprotein ligand-1 by neutrophil elastase and cathepsin G. Blood. (2001) 98:1440–7. doi: 10.1182/blood.V98.5.1440
27. Rahman M, Zhang S, Chew M, Ersson A, Jeppsson B, Thorlacius H. Platelet-derived CD40L (CD154) mediates neutrophil upregulation of Mac-1 and recruitment in septic lung injury. Ann Surg. (2009) 250:783–90. doi: 10.1097/SLA.0b013e3181bd95b7
28. Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. (2013) 121:1008–15. doi: 10.1182/blood-2012-06-437392
29. Wetterholm E, Linders J, Merza M, Regner S, Thorlacius H. Platelet-derived CXCL4 regulates neutrophil infiltration and tissue damage in severe acute pancreatitis. Transl Res. (2016) 176:105–18. doi: 10.1016/j.trsl.2016.04.006
30. Giles JA, Greenhalgh AD, Denes A, Nieswandt B, Coutts G, McColl BW, et al. Neutrophil infiltration to the brain is platelet-dependent, and is reversed by blockade of platelet GPIbalpha. Immunology. (2018) 154:322–8. doi: 10.1111/imm.12892
31. Amison RT, O'Shaughnessy BG, Arnold S, Cleary SJ, Nandi M, Pitchford SC, et al. Platelet depletion impairs host defense to pulmonary infection with pseudomonas aeruginosa in mice. Am J Respir Cell Mol Biol. (2018) 58:331–40. doi: 10.1165/rcmb.2017-0083OC
32. Li XH, Qian YB, Meng XX, Wang RL. Effect of platelet-derived P-selectin on neutrophil recruitment in a mouse model of sepsis-induced acute kidney injury. Chin Med J. (2017) 130:1694–9. doi: 10.4103/0366-6999.209889
33. Yu C, Zhang S, Wang Y, Zhang S, Luo L, Thorlacius H. Platelet-derived CCL5 regulates CXC chemokine formation and neutrophil recruitment in acute experimental colitis. J Cell Physiol. (2016) 231:370–6. doi: 10.1002/jcp.25081
34. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
35. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
36. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol. (2016) 99:153–62. doi: 10.1189/jlb.3A0415-161R
37. Maugeri N, Campana L, Gavina M, Covino C, De MM, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. (2014) 12:2074–88. doi: 10.1111/jth.12710
38. Elaskalani O, bdol Razak NB, Metharom P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun Signal. (2018) 16:24. doi: 10.1186/s12964-018-0235-0
39. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
40. Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, et al. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. (1989) 59:305–12. doi: 10.1016/0092-8674(89)90292-4
41. Wencel-Drake JD, Painter RG, Zimmerman TS, Ginsberg MH. Ultrastructural localization of human platelet thrombospondin, fibrinogen, fibronectin, and von Willebrand factor in frozen thin section. Blood. (1985) 65:929–38.
42. Lenting PJ, Christophe OD, Denis CV. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. (2015) 125:2019–28. doi: 10.1182/blood-2014-06-528406
43. Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. (2017) 15:1285–94. doi: 10.1111/jth.13696
44. Suzuki H, Katagiri Y, Tsukita S, Tanoue K, Yamazaki H. Localization of adhesive proteins in two newly subdivided zones in electron-lucent matrix of human platelet alpha-granules. Histochemistry. (1990) 94:337–44. doi: 10.1007/BF00266440
45. Schwarz HP, Heeb MJ, Wencel-Drake JD, Griffin JH. Identification and quantitation of protein S in human platelets. Blood. (1985) 66:1452–5.
46. Zucker M, Hauschner H, Seligsohn U, Rosenberg N. Platelet factor XI: intracellular localization and mRNA splicing following platelet activation. Blood Cells Mol Dis. (2018) 69:30–7. doi: 10.1016/j.bcmd.2017.04.006
47. Tzeng DY, Deuel TF, Huang JS, Senior RM, Boxer LA, Baehner RL. Platelet-derived growth factor promotes polymorphonuclear leukocyte activation. Blood. (1984) 64:1123–8.
48. Assoian RK, Sporn MB. Type beta transforming growth factor in human platelets: release during platelet degranulation and action on vascular smooth muscle cells. J Cell Biol. (1986) 102:1217–23. doi: 10.1083/jcb.102.4.1217
49. Kiuru J, Viinikka L, Myllyla G, Pesonen K, Perheentupa J. Cytoskeleton-dependent release of human platelet epidermal growth factor. Life Sci. (1991) 49:1997–2003. doi: 10.1016/0024-3205(91)90642-O
50. Gunsilius E, Petzer A, Stockhammer G, Nussbaumer W, Schumacher P, Clausen J, et al. Thrombocytes are the major source for soluble vascular endothelial growth factor in peripheral blood. Oncology. (2000) 58:169–74. doi: 10.1159/000012095
51. Struyf S, Burdick MD, Proost P, Van Damme J, Strieter RM. Platelets release CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and potent inhibitor of angiogenesis. Circ Res. (2004) 95:855–7. doi: 10.1161/01.RES.0000146674.38319.07
52. Mauler M, Herr N, Schoenichen C, Witsch T, Marchini T, Hardtner C, et al. Platelet serotonin aggravates myocardial ischemia/reperfusion injury via neutrophil degranulation. Circulation. (2019) 139:918–31. doi: 10.1161/CIRCULATIONAHA.118.033942
53. Brown AJ, Sepuru KM, Sawant KV, Rajarathnam K. Platelet-derived chemokine CXCL7 dimer preferentially exists in the glycosaminoglycan-bound form: implications for neutrophil-platelet crosstalk. Front Immunol. (2017) 8:1248. doi: 10.3389/fimmu.2017.01248
54. Giddings JC, Brookes LR, Piovella F, Bloom AL. Immunohistological comparison of platelet factor 4 (PF4), fibronectin (Fn) and factor VIII related antigen (VIIIR:Ag) in human platelet granules. Br J Haematol. (1982) 52:79–88. doi: 10.1111/j.1365-2141.1982.tb03863.x
55. Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun. (2019) 10:1322. doi: 10.1038/s41467-019-09160-7
56. Boehlen F, Clemetson KJ. Platelet chemokines and their receptors: what is their relevance to platelet storage and transfusion practice? Transfus Med. (2001) 11:403–17. doi: 10.1046/j.1365-3148.2001.00340.x
57. Jin L, Batra S, Jeyaseelan S. Diminished neutrophil extracellular trap (NET) formation is a novel innate immune deficiency induced by acute ethanol exposure in polymicrobial sepsis, which can be rescued by CXCL1. PLoS Pathog. (2017) 13:e1006637. doi: 10.1371/journal.ppat.1006637
58. Power CA, Furness RB, Brawand C, Wells TN. Cloning of a full-length cDNA encoding the neutrophil-activating peptide ENA-78 from human platelets. Gene. (1994) 151:333–4. doi: 10.1016/0378-1119(94)90682-3
59. Appay V, Rowland-Jones SL. RANTES: a versatile and controversial chemokine. Trends Immunol. (2001) 22:83–7. doi: 10.1016/S1471-4906(00)01812-3
60. Rossaint J, Herter JM, Van Aken H, Napirei M, Doring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood. (2014) 123:2573–84. doi: 10.1182/blood-2013-07-516484
61. Klinger MH, Wilhelm D, Bubel S, Sticherling M, Schroder JM, Kuhnel W. Immunocytochemical localization of the chemokines RANTES and MIP-1 alpha within human platelets and their release during storage. Int Arch Allergy Immunol. (1995) 107:541–6. doi: 10.1159/000237097
62. Brown GT, Narayanan P, Li W, Silverstein RL, McIntyre TM. Lipopolysaccharide stimulates platelets through an IL-1beta autocrine loop. J Immunol. (2013) 191:5196–203. doi: 10.4049/jimmunol.1300354
63. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep. (2018) 8:2068. doi: 10.1038/s41598-018-20479-x
64. Gill P, Jindal NL, Jagdis A, Vadas P. Platelets in the immune response: revisiting platelet-activating factor in anaphylaxis. J Allergy Clin Immunol. (2015) 135:1424–32. doi: 10.1016/j.jaci.2015.04.019
65. Jolly L, Lemarie J, Carrasco K, Popovic B, Derive M, Boufenzer A, et al. Triggering receptor expressed on myeloid cells-1: a new player in platelet aggregation. Thromb Haemost. (2017) 117:1772–81. doi: 10.1160/TH17-03-0156
66. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. (2012) 122:2661–71. doi: 10.1172/JCI61303
67. Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. (2013) 13:169–80. doi: 10.1016/j.chom.2013.01.005
68. Kuijper PH, Gallardo Tores HI, Lammers JW, Sixma JJ, Koenderman L, Zwaginga JJ. Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb Haemost. (1998) 80:443–8. doi: 10.1055/s-0037-1615227
69. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. (2012) 12:324–33. doi: 10.1016/j.chom.2012.06.011
70. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. (2011) 7:e1002355. doi: 10.1371/journal.ppat.1002355
71. Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. (2014) 34:1977–84. doi: 10.1161/ATVBAHA.114.304114
72. Wang Y, Luo L, Braun OO, Westman J, Madhi R, Herwald H, et al. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci Rep. (2018) 8:4020. doi: 10.1038/s41598-018-22156-5
73. Noubouossie DF, Whelihan MF, Yu YB, Sparkenbaugh E, Pawlinski R, Monroe DM, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood. (2017) 129:1021–9. doi: 10.1182/blood-2016-06-722298
74. Jimenez-Alcazar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. (2017) 358:1202–6. doi: 10.1126/science.aam8897
75. Valles J, Lago A, Santos MT, Latorre AM, Tembl JI, Salom JB, et al. Neutrophil extracellular traps are increased in patients with acute ischemic stroke: prognostic significance. Thromb Haemost. (2017) 117:1919–29. doi: 10.1160/TH17-02-0130
76. Laridan E, Denorme F, Desender L, Francois O, Andersson T, Deckmyn H, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. (2017) 82:223–32. doi: 10.1002/ana.24993
77. Yang S, Qi H, Kan K, Chen J, Xie H, Guo X, et al. Neutrophil extracellular traps promote hypercoagulability in patients with sepsis. Shock. (2017) 47:132–9. doi: 10.1097/SHK.0000000000000741
Keywords: platelets, neutrophils, neutrophil extracellular traps, NETs, coagulation, inflammation
Citation: Zucoloto AZ and Jenne CN (2019) Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 6:85. doi: 10.3389/fcvm.2019.00085
Received: 26 March 2019; Accepted: 06 June 2019;
Published: 20 June 2019.
Edited by:
Marie Lordkipanidzé, Université de Montréal, CanadaReviewed by:
Nigel S. Key, University of North Carolina at Chapel Hill, United StatesCopyright © 2019 Zucoloto and Jenne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig N. Jenne, Y25qZW5uZUB1Y2FsZ2FyeS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.