 Francesca Bartoli-Leonard
Francesca Bartoli-Leonard Fiona L. Wilkinson
Fiona L. Wilkinson Alex W. W. Langford-Smith
Alex W. W. Langford-Smith M. Y. Alexander
M. Y. Alexander Ria Weston
Ria Weston
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 18 December 2018
Sec. Atherosclerosis and Vascular Medicine
Volume 5 - 2018 | https://doi.org/10.3389/fcvm.2018.00183
This article is part of the Research Topic Vascular Calcification View all 12 articles
Vascular calcification is a major health risk and is highly correlated with atherosclerosis, diabetes, and chronic kidney disease. The development of vascular calcification is an active and complex process linked with a multitude of signaling pathways, which regulate promoters and inhibitors of osteogenesis, the balance of which become deregulated in disease conditions. SIRT1, a protein deacetylase, known to be protective in inhibiting oxidative stress and inflammation within the vessel wall, has been shown as a possible key player in modulating the cell-fate determining canonical Wnt signaling pathways. Suppression of SIRT1 has been reported in patients suffering with cardiovascular pathologies, suggesting that the sustained acetylation of osteogenic factors could contribute to their activation and in turn, lead to the progression of calcification. There is clear evidence of the synergy between β-Catenin and elevated Runx2, and with Wnt signaling being β-Catenin dependent, further understanding is needed as to how these molecular pathways converge and interact, in order to provide novel insight into the mechanism by which smooth muscle cells switch to an osteogenic differentiation programme. Therefore, this review will describe the current concepts of pathological soft tissue mineralization, with a focus on the contribution of SIRT1 as a regulator of Wnt signaling and its targets, discussing SIRT1 as a potential target for manipulation and therapy.
Vascular calcification is a pathology highly correlated with cardiovascular mortality, and although initially described as Monckeberg's sclerosis (1), with calcium being deposited in the medial layer of arteries, it is now known to be an active process, similar to bone development (2–4). Whilst development of calcification occurs naturally in vessels as they age (5), increased calcification occurs in those with diabetes and chronic kidney disease (CKD), in which constant high plasma glucose and an augmented lipid profile; (comprising of low HDL cholesterol, elevated triglycerides, high LDL cholesterol and high total cholesterol) (1) increases their risk of accelerating calcification development. In healthy tissues, vascular smooth muscle cells (vSMCs) exist within the medial layer of the vessel wall in a quiescent, contractile state, expressing a range of contractile proteins, including smooth muscle α-actin, smooth muscle myosin heavy chain, calponin, and smoothelin. However, in response to these local cues they lose expression of these proteins and gain the capability to transdifferentiate from vSMCs to a more synthetic, osteoblastic phenotype, stiffening, and narrowing the vessel wall (6, 7) (Figure 1). Whilst the synthetic phenotype is thought to possess a protective role, contributing to the deposition of a fibrous cap and thus stabilize an atherosclerotic plaque, the intimal SMCs are believed to be detrimental as they acquire foam cell properties, leading to an inflammatory phenotype (8). Many signaling pathway and transcription factors have been shown to govern the contractile, osteogenic or synthetic features of the vasculature (2, 4, 9–12), and with more understanding of the influence of epigenetics on SMC regulation, their role in the pathogenesis of human vascular disease will only expand (13). In vitro models use glucose, calcium and inorganic phosphate as inducers of calcification within vSMCs, with deposition of calcium on the extracellular matrix, and an upregulation of osteogenic markers including alkaline phosphatase (ALP), Runt-relative transcription factor (Runx2), and osteocalcin (14). Calcifying vascular cells (CVCs) are a sub-population of vSMCs susceptible to calcification, which differentiate from stem cell progenitor lineages within the vasculature (15–18). CVCs are characterized as a highly proliferating cell with considerable phenotypic plasticity, where the cells respond to local signals which are activated in disease conditions, including bone morphogenetic proteins (BMPs) and Wnts, and are capable of downregulating contractile proteins and remodeling the extracellular matrix to facilitate migration and differentiation.
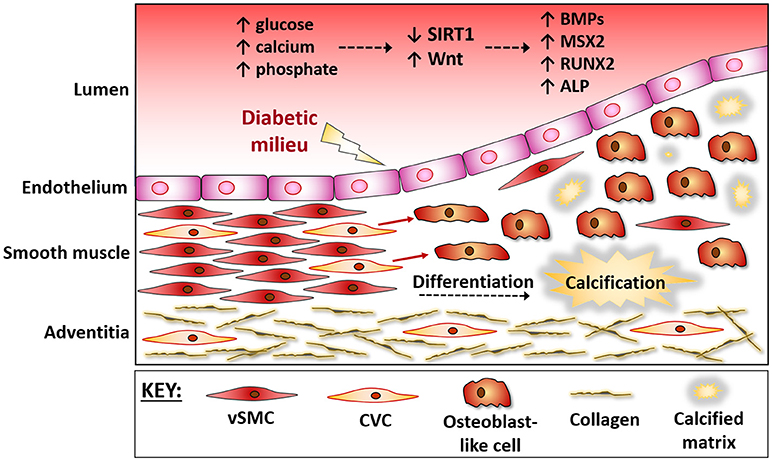
Figure 1. The vessel wall during osteogenic differentiation. The vessel wall responds to the micro-environment within the circulation. During diabetes hyperglycaemia and mineral ion imbalances lead to endothelial damage. The medial layer then responds via triggering a repair response, which often gets masked and further damage ensues. Progenitor cells within the media, often referred to as calcifying vascular cells (CVCs), are believed to up-regulate osteogenic factors and differentiate into bone-forming osteoblasts that contribute to vessel stiffening. These vascular progenitor cells directly sense extracellular signals, including a down regulation of SIRT1 and activation of Wnt signaling, and the protective mechanisms are over-ridden, causing a differentiation of CVCs into bone-forming osteoblasts. BMPs, Bone Morphogenic Proteins; MSX2, msh homeobox 2; RUNX2, Runt related transcription factor 2; OCN, Osteocalcin; CVC, Calcifying Vascular Cells; vSMC, vascular Smooth Muscle Cells.
Sirtuin 1, (SIRT1) has been identified as a highly conserved nicotinamide adenine dinucleotide-dependent deacetylase, interacting with a range of protein targets involved in Wnt signaling, glucose homeostasis, insulin regulation, and calcium signaling (19), making SIRT1 an attractive candidate for control of calcification. Smooth muscle specific acetylation sites have been identified which allow repression or access to the cellular transcriptional machinery and are regulated via a range of stimuli including transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF) and oxidized phospholipids, which execute their actions by modulating SMC chromatin structure (20). Wnt signaling and its downstream mediators affect a range of biological processes, first identified in embryonic development (21). The Wnt family is a highly conserved group of 19 genes encoding cysteine-rich-secreted glycoproteins, first identified in Drosophilia melanogaster as a mutant wingless gene (22). Subsequent studies demonstrated sequence homology with the Int-1 gene present in vertebrae and thus the nomenclature Wnt was coined in 1991 (23). Being highly conserved and well-studied in eukaryotes, Wnt signaling became recognized as one of the cornerstones for embryonic development, regulating cellular proliferation, polarity, and apoptosis and subsequently becoming suppressed in adults (23). Recent studies have shown a reactivation of Wnt signaling in a variety of cardiovascular pathologies (24, 25), acting as a cell fate determination switch, allowing cellular differentiation to occur, where aberrant Wnt signaling is diverted toward disease progression. This review will discuss the role of SIRT1 in vascular calcification, as well as an overview on Wnt signaling and a summary of potential therapeutic interventions that could modulate osteogenic differentiation, thus linking both SIRT1 and Wnt signaling to vascular calcification.
The development of smooth muscle calcification occurs in the presence of hyperphosphatemia, often coupled with hyperglycaemia, in patients with diabetes and CKD. The histone deacetylase SIRT1, known to ameliorate calcification (26), is shown to be decreased in diabetic models (27, 28). The suppression of SIRT1 within blood or tissue allows a build-up of sodium-dependent phosphate co-transporters (29), increasing the concentration of phosphate systemically and within vessels, which is recognized as a key trigger in the development of calcification. Furthermore, diabetic SIRT1 +/– mice exhibited a greater propensity to undergo calcification within the aorta (30). Elevated phosphate within the circulation increases expression of systemic osteogenic and inflammatory factors, activating Wnt signaling and osteogenic transcription factors Msx2 and Runx2 (31). Subsequently, levels of osteocalcin, RANKL, Sclerostin, Osterix, BMPs, and ALP (32, 33) activity are increased. Elevated BMPs form a positive feedback loop, activating the SMAD pathway, sustaining Wnt activation and its downstream targets, Msx2 and Runx2. Runx2 has also been linked to vascular fibrosis, in the absence of overt calcification, highlighting the important role of SIRT1 regulated signals in the multistep processes that control osteogenic programming and calcium deposition (34).
This influx of pro-osteogenic activators coincides with the loss of endogenous contractile vSMC genes such as SM22α and smooth muscle-actin, with a concordant up-regulation of calcification inhibitor proteins including osteopontin in an attempt to counteract the effect. However, under disease conditions the inhibitory effects are masked by the more dominant stimuli and this generally leads to a change in cellular morphology and an increase in extracellular matrix deposition (35). Whilst CVCs have historically been reported to be differentiated vSMCs (36), staining of cells within atherosclerotic plaques have identified both macrophage and stem cell markers, suggesting that CVCs may be sourced from a range of progenitor-like cells within the media or adventitia (18, 37, 38). Recently, SIRT1 has been shown to translocate to the nucleus during neuronal differentiation and repress the Notch3 target Hes1 (39), suggesting a role for SIRT1 during cellular differentiation. Notch3 and Hes1 have also been associated with osteogenic differentiation of vSMCs in vitro (9) and mutations in Notch have been shown to lead to aortic valve calcification (40). Therefore, the loss of SIRT1 in diabetes may allow activation of Notch3 and Hes1, leading to osteogenic differentiation of vSMCs and subsequent deposition of a calcified matrix. Whether Wnt, SIRT1, and Notch signaling pathways directly interact, in terms of vSMCs osteogenic differentiation merits further study.
Wnt signaling is controlled by the Wnt ligand; a 350 residue hydrophobic protein with a post-translational fatty acid O-acylation modification (41), which is essential for secretion and signal propagation. Post translational modification of Wnt occurs in the endoplasmic reticulum (42) before shutting to the golgi and exportation to the extracellular space (43). Secretion of Wnt is tightly regulated via the wntless transmembrane protein, allowing both paracrine and autocrine activation of the Wnt pathways (44). Activation of Wnt triggers one of three different pathways, all dependent on the Wnt protein firstly becoming palmitoylated on conserved serine residues, then glycosylated and binding to a frizzled cell membrane receptor, propagating signaling within the cell via Disheveled (45–47) (Figure 2). The canonical pathway, involving β-Catenin, has the strongest links to cardiovascular disease and vascular dysfunction (48). Classically, Wnts have been characterized into pro and anti-osteogenic factors, where Wnts such as Wnt5a (49) and Wnt3a are osteogenic, whereas Wnt1 is anti-osteogenic, enforcing a contractile phenotype (46). However, recent work has demonstrated a more complex pathway, in which not only does the active Wnt ligand dictate cell fate, but its interaction with the co-factors and cell surface receptors dictate the emerging osteogenic profile.
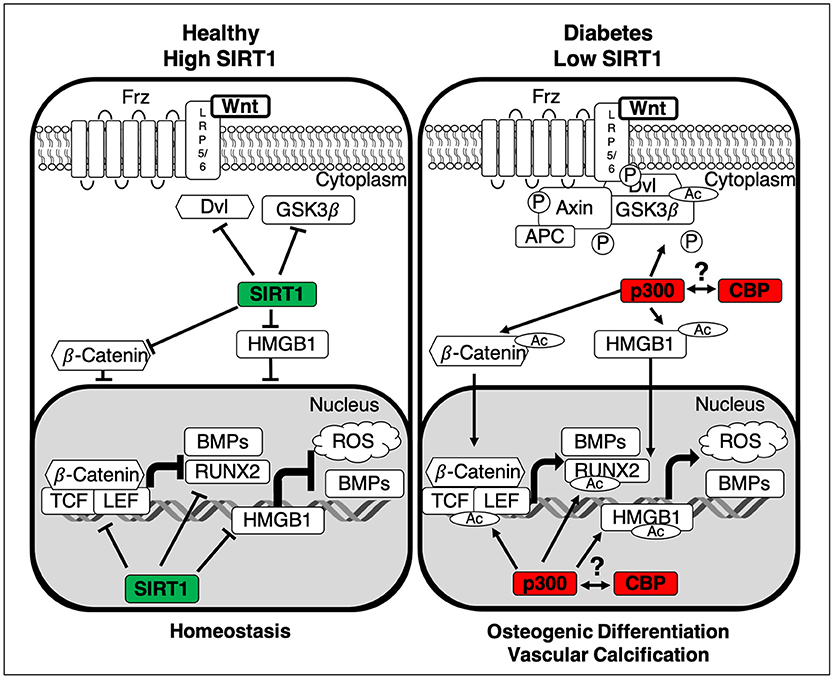
Figure 2. Summary of the interactions between SIRT1 and the canonical Wnt pathway. In healthy homeostatic conditions SIRT1 binds to p300, diminishing its ability to acetylate the β-Catenin complex, comprised of Dvl, GSK3β and β-Catenin. Additionally, SIRT1 deacetylates both β-Catenin and HMGB1 thus inhibiting their translocation to the nucleus, inactivating Wnt signaling and stopping the development of vascular calcification. In the absence of SIRT1 the β-Catenin complex is activated by p300-mediated acetylation and GSK3β-catalyzed phosphorylation, inhibiting its ability to degrade β-Catenin. Additionally, HMGB1 and β-Catenin are also acetylated via p300, facilitating their translocation to the nucleus. Subsequently β-Catenin binds cofactors TCF/LEF and following their acetylation transcription of osteogenic factors Runx2 and BMPs is induced. Whilst the epigenetic roles of CBP and p300 are distinct, the role of CBP still remains an area for further investigation. LRP5/6, Low-density lipoprotein Receptor-related Protein 5/6; APC, Adenomatous Polyposis Coli; GSK3β, Glycogen Synthase Kinase-3 beta; Dvl, Dishelved Protein; TCF/LEF, T-Cell Factor/Lymphoid enhancer factor; Runx2, Runt-Related Transcription Factor; BMPs, Bone Morphogenic Proteins; HMGB1, High Mobility Group Box 1; ROS, Reactive Oxygen Species; Ac, Acetylation; P, Phosphorylation.
Activation of the canonical pathway is characterized by the accumulation of β-Catenin in the cytoplasm and subsequent translocation to the nucleus. When inactive, β-Catenin is constantly degraded via the β-Catenin complex, firstly phosphorylated (50, 51) then ubiquitinated and degraded (52). Conversely when Wnt activates the canonical pathway by binding the transmembrane Frz protein complex (53) it disrupts the β-Catenin complex (54) allowing its translocation to the nucleus (53–55). Without ubiquitination β-Catenin becomes acetylated (56), potentially facilitated by the loss of SIRT1, and binds to target gene promotor elements, triggering increased transcription of a range of osteogenic genes (48). Whilst the role of β-Catenin in diabetes is still illusive, there is clear evidence that hyperglycaemia increases nuclear accumulation of β-Catenin, creating an osteogenic environment. Chronic hyperglycaemic conditions are also shown to displace and inactivate SIRT1 from within the nucleus, allowing acetylation of β-Catenin via p300 (57), promoting the glucose-dependent amplification of Wnt-dependent transcription of osteogenic factors. However, the p300 related acetyltransferase CBP is also engaged in SIRT1 regulatory circuits, including the HIF pathway, which is central in bone formation, Wnt signaling, and vascular calcification. It is increasingly clear that the epigenetic roles of p300 and CBP are distinct, and the role of SIRT1 and CBP in vascular Wnt signaling remains to be elucidated (58–60).
Whilst Wnt signaling is a recognized orchestrator of vascular development within embryos, increasing neovascularisation and proliferation (61) it has been shown to be cell type dependent, with SIRT1 demonstrating varied responses under normal and disease conditions (47). SIRT1 knock-out (62) and knock-in mice (63) clearly demonstrate that a reduction of SIRT1 activates Wnt/β-Catenin signaling (64, 65) and loss of SIRT7 enhances osteogenesis of mesenchymal stem cell differentiation via Wnt signaling (66, 67), both of which contribute to osteogenic differentiation. Therefore, it seems reasonable to suggest that Wnt may be involved in cell fate determination of vSMCs, stimulating their switch to an osteoblastic-like phenotype, and increasing calcified matrix deposition.
BMPs belong to the TGF-β superfamily and are responsible for orchestrating much of the tissue architecture throughout the body (68). Whilst BMPs are key regulators of bone ossification (69) the relationship between BMPs, SIRT1, Wnt signaling, and vascular calcification remains unclear regarding which acts as the initial trigger, or whether they act in concert (70, 71). BMPs are secreted throughout the vessel wall, predominately by pericytes and endothelial cells in a paracrine manner (46) and by vSMCs in an autocrine loop (72). Of the twenty BMPs discovered, BMP2 (73), BMP4 (74), and BMP9 (75) are the most widely reported inducers of bone ossification and vascular calcification in both in-vitro and in-vivo models. The BMP signaling pathway is activated when BMP binds serine/threonine receptor kinases BMPRI and BMPRII, causing them to heterodimerise and phosphorylate one another (76). Once bound, BMPRI/II phosphorylate receptor-regulated SMADs (R-SMADs) (77, 78), which subsequently bind with co-SMAD4, allowing gene specific promotor binding and osteogenic gene expression (79).
Whilst elevated BMP2 independently induces calcification (80), when combined with β-Catenin activation, the effect is synergistic, increasing mineralized matrix deposition (71, 81). BMP2 is upregulated by hyperglycaemia (82), activating the ligand high mobility group box 1 (HMGB1), which translocates to the nucleus, binding to a cAMP response element (CRE) region of the BMP2 promoter, inducing its expression (83). In contrast, deacetylation of HMGB1 via SIRT1, prevents upregulation of BMP2 (84) (Figure 2), reducing induction of inflammatory markers, including TNFα and reactive oxygen species (85), thus limiting an inflammatory environment conducive to calcification progression. Hyperphosphatemia stimulates BMP2 production, which in turn increases Pit-1 co-transporter expression throughout the cell membrane, allowing an influx of phosphate ions (86). Increased phosphate can downregulate SIRT1 production (28), allowing sustained acetylation of downstream proteins and facilitating hyperacetylation of β-Catenin (87) and Runx2 (88) via p300, thus accelerating calcification.
Although activation of BMP in adults is associated with increased cardiovascular risk and the development of calcification, BMPs have been shown to mediate increased vascular development during embryogenesis, via β-Catenin activation (89). Wnt also controls the development and stability of newly formed bones (90), and as only a proportion of vSMCs calcify within the vessel, it may be suggested that this process is in overdrive by the reactivation of the CVCs, causing bone-like development to form along vessel walls. Downstream of BMPs, phosphorylation of Smad proteins dictates the duration and efficacy of osteogenic gene expression (91). Degradation of β-Catenin, coupled with the inhibition of Smad signaling (79), and prevention of the subsequent signaling pathways impedes the progression of osteogenic differentiation of vSMCs. However, activation and binding of BMPs to their cognate receptor, allows Smad phosphorylation and acetylation via p300 (91), inhibiting Smad ubiquitination (79) and thus propagating the activation of downstream osteogenic genes (92). The suppression of SIRT1 in CVD patients, leads to a reduction in competitive inhibition of p300, thereby increasing BMP/SMAD/p300 signaling and subsequent osteogenic gene transcription.
Human T Cell/Lymphoid Enhancing-Factor (TCF/LEF) (93) regulates cell fate markers, including Wnt, by binding β-catenin alongside co-factors within the nucleus at the TCF response element, to govern gene expression (92), thus causing the progression of vascular calcification. Multiple TCF/LEF response binding elements are present throughout the promotor region of the BMP2 gene (94) which, when bound by a β-Catenin/TCF complex, increases BMP2 transcript production. Inhibition of this activation is achieved via endogenous inhibitor Noggin, suggesting that a positive feedback loop exists between the activation and inhibition of β-Catenin and TCF binding onto the BMP2 promotor (72). Modulation of these proteins within individuals may be the missing link to distinguish between patients more susceptible to calcification and those who appear to be protected.
Runx2; a key osteoblastic transcription factor (95), is essential for chondrocyte maturation and osteogenic differentiation. Originally thought to be expressed solely during bone development, it is now known to be activated in both the intimal and medial layers of the vasculature during calcification development. The activation of Runx2 triggers expression of a range of downstream osteogenic effectors, leading to the differentiation of vSMCs from a contractile to an osteogenic phenotype in a diabetic environment. Hyperphosphatemia downregulates the expression of secreted frizzled-related proteins (SFRPs) (96), which act as a decoy for Wnt signaling, inhibiting internalization of phosphate transporters (97), thereby facilitating the constant entry of phosphate into the cells and propagating the development of calcification. SIRT1 has been shown to regulate SFRPs via deacetylation, directly contributing to their aberrant epigenetic silencing within histone 3 and 4 (47).
Sclerostin, a selective inhibitor of Low-density lipoprotein Receptor-related Protein (LRPs), a member of the Frz membrane complex downstream of Wnt, is increased during CVC differentiation (98). Increased activation of Sclerostin decreases Wnt signal propagation via competitive inhibition, temporarily halting calcification until phosphate and calcium build up within the serum, reactivating the Wnt pathway and further calcifying the vessel. β-Catenin nuclear translocation induces Runx2 expression via SMAD activation in vSMCs (99), binds to the proximal region of the SOST promotor, activating osteogenic transcription, similar to that which occurs in bone (100). Runx2 together with Osterix (Osx) may actively limit the expression of Sclerostin, with polymorphic variations of Runx2 transcriptionally regulating Sclerostin expression in a negative feedback loop (101). Additionally, SIRT1 may decrease SOST gene expression in bone via the deacetylation of histone 3 at lys9 within the promotor region, inhibiting the mechanical loading and subsequent transcription (102). With the development of vascular calcification considered the paradox of bone development, the reduction of SIRT1 in the CVD patient may lead to a decrease in SOST, perpetuating Wnt signaling and vascular calcification.
Runx2 expression is controlled by Wnt signaling via the direct binding of TCF/LCF co-transcription factors. Direct β-Catenin binding to TCF/LEF at the Runx2 promotor site is enhanced by p300 acetylation (23), propagating the Runx2 signaling pathway. Additionally, indirect DNA binding of SMADs and TCF via protein-protein interaction enhances Runx2 expression (57) alongside production of downstream osteogenic proteins. β-Catenin binding to TCF1 at TB1 and TBE2 sites within the Runx2 promotor region has been shown to increase endogenous Runx2 expression 10–20-fold, with damage to these sites attenuating selective Runx2 expression (99). It is clear that vascular calcification involves a network of signaling pathways, not restricted to SIRT1, Runx2, and Wnt and that further studies involving analysis of vSMCs harvested from relevant patient groups could provide insight into the profile of vSMCs during early and late osteogenic differentiation, offering stratified treatment plans to combat calcification development.
Activation of Sirtuins, in particular SIRT1, has been shown to decrease Wnt signaling and reduce the risk of cardiovascular disease in both animal and human models (103–105), and administration of resveratrol has been shown to reduce arterial calcification, in both non-human primates and in uremic rats (106, 107) suggesting SIRT1 may be a good candidate molecule for sustained control of vascular calcification. SIRT1 regulates metabolic pathways including 5'AMP and canonical Wnt signaling, both of which underpin key biological events, such as proliferation and differentiation. Developmental studies have demonstrated the crucial role of β-Catenin signaling in bone development, with Wnt-floxed mice developing spontaneous fractures and an inability to develop mature osteoblasts (108). Additionally, the fate of resident stem cells appears to be regulated via the upregulation of the SIRT1/Wnt/β-Catenin pathway, in which MSCs undergo an osteogenic differentiation programme (109, 110). Taken together, it can be suggested that CVCs are part of a resident stem cell population not dissimilar to bone, responding to the absence of SIRT1 and the upregulation of Wnt, causing bone to develop within the vasculature. Bone deposition in the vessel wall is thought to occur in conjunction with bone loss, where calcium and mineral is released from bone, and taken up in the vessel wall, inducing aberrant signaling pathways that result in an osteogenic differentiation of progenitor cells residing in the vessel wall. With the close links between SIRT1 and Wnt signaling, and the activation of Wnt signaling in driving an osteogenic differentiation program by vSMCs (99), understanding Wnt signaling-related interactions with SIRT1 will add insight into the pathogenesis of vascular calcification and enable the development of anti-calcification strategies. Thus, in a pathology where vSMCs are thought to undergo an aberrant differentiation programme, further understanding of SIRT1 signaling in this context, could allow the development of SIRT1 modulators for prevention of this debilitating process (111–113).
Sirtuin activators have generally been described for SIRT1, and resveratrol, a natural compound found as a constituent of grapes and red wine (114), is the most commonly used activator of SIRT1. Resveratrol has been shown to cause resistance to oxidative stress and inflammation and is used widely in the diabetic and age-related decline in heart function and neuronal loss (115, 116). However, because of its modest bioavailability, resveratrol has been reformulated with more efficient small molecules being designed commonly known as synthetic sirtuin activating compounds (STACs) (105, 117), (resVida, Longevinex®, SRT501) (106, 107). Furthermore, molecules that are structurally unrelated to resveratrol (SRT1720 and SRT2379) have been also developed with increased potency, although most have not yet reached the clinic (118, 119). SRT2104 was used recently in a small group of diabetic patients (NCT01031108). It was well-tolerated, but had little beneficial effect on a range of measures of cardiovascular health. However, the study was small, not focussed on human vascular calcification and further investigations will be required to confirm any enhanced metabolic effects (120).
With an increasingly aging population throughout the Western world, vascular calcification has become a major health concern, correlating with cardiovascular disease development and mortality. Although, the molecular mechanism underpinning vascular calcification remains closely linked to bone formation, the association between loss of SIRT1, activation of Wnt signaling and the upregulation of major osteogenic factors add to the growing armament of deranged osteogenic signaling pathways occurring under pathological conditions in the vessel wall. Additionally, SIRT1, a longevity factor and deacetylase, may act at an epigenetic level to control these converging pathways and cardiovascular risk factors. Furthermore, SIRT7 and SIRT1 have been shown to co-ordinately enhance Sp7/Osx activity and support orthotropic bone formation, and thus further studies could add to the understanding of the role of osteotropic signals during mineralization in skeletal and vascular environments. Indeed, it may be, that other members of the sirtuin family, may contribute to cardiovascular Wnt signaling (121) and either osteoporosis or vascular calcification.
Although the hyperglycaemia and hyperphosphatemia present in many CVD patients is known to suppress SIRT1 expression, there is a growing need for a comprehensive single-cell differentiation pathway for vSMC phenotypic switching, to identify the temporal activity of relevant signaling pathways, and the importance of their early inhibition and reactivation at later time-points. Only then, will we be able to extend our current knowledge of osteogenic vSMC differentiation, which potentially could have implications for future research and clinical application in this field.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. McCullough P A, Chinnaiyan K M, Agrawal V, Danielewicz E, Abela GS. Amplification of atherosclerotic calcification and Monckeberg's sclerosis: a spectrum of the same disease process. Adv Chronic Kidney Dis. (2008) 15:396–412. doi: 10.1053/j.ackd.2008.07.009
2. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
3. Andrews J, Psaltis P, Bartolo B, Nicolls S, Puri R. Coronary arterial calcification: a review of mechanisms, promoters and imaging. Trends Cardiovasc Med. (2018) 28:491–501. doi: 10.1016/j.tcm.2018.04.007
4. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. (2014) 34:715–23. doi: 10.1161/ATVBAHA.113.302070
5. Leopold JA. Vascular calcification: an age-old problem of old age. Circulation (2013) 127:2380–2. doi: 10.1161/CIRCULATIONAHA.113.003341
6. Leopold JA. Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. (2015) 25:267–74. doi: 10.1016/j.tcm.2014.10.021
7. Iyemere VP, Proudfoot D, Weissberg PL, Shanahan CM. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med. (2006) 260:192–210. doi: 10.1111/j.1365-2796.2006.01692.x
8. Allahverdian S, Chaabane C, Boukais K, Francis G A, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. (2018) 114:540–50. doi: 10.1093/cvr/cvy022
9. Liu Y, Wang T, Yan J, Jiagbogu N, Heideman DA, Canfield AE, et al. HGF/c-met signalling promotes Notch3 activation and human vascular smooth muscle cell osteogenic differentiation in vitro. Atherosclerosis (2011) 219:440–7. doi: 10.1016/j.atherosclerosis.2011.08.033
10. Yan J, Stringer SE, Hamilton A, Charlton-Menys V, Gotting C, Muller B, et al. Decorin GAG synthesis and TGF-beta signaling mediate Ox-LDL-induced mineralization of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2011) 31:608–15. doi: 10.1161/ATVBAHA.110.220749
11. Zhu D, Mackenzie NC, Shanahan CM, Shroff RC, Farquharson C, MacRae VE. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. J Cell Mol Med. (2015) 19:165–74. doi: 10.1111/jcmm.12373
12. Liu Y, Shanahan CM. Signalling pathways and vascular calcification. Front Biosci. (2011) 16:1302–14. doi: 10.2741/3790
13. Liu R, Leslie KL, Martin KA. Epigenetic regulation of smooth muscle cell plasticity. Biochim Biophys Acta (2015) 1849:448–53. doi: 10.1016/j.bbagrm.2014.06.004
14. Giachelli CM. The emerging role of phosphate in vascular calcification. Kidney Int. (2009) 75:890–7. doi: 10.1038/ki.2008.644
15. Hirschi KK, Majesky MW. Smooth muscle stem cells. Anat Rec A Discov Mol Cell Evol Biol. (2004) 276:22–33. doi: 10.1002/ar.a.10128
16. Leszczynska A, Murphy JM. Vascular calcification: is it rather a stem/progenitor cells driven phenomenon? Front Bioeng Biotechnol. (2018) 6:10. doi: 10.3389/fbioe.2018.00010
17. Hortells L, Sur S, St Hilaire C. Cell phenotype transitions in cardiovascular calcification. Front Cardiovasc Med. (2018) 5:27. doi: 10.3389/fcvm.2018.00027
18. Tintut Y, Alfonso Z, Saini T, Radcliff K, Watson K, Bostrom K, et al. Multilineage potential of cells from the artery wall. Circulation (2003) 108:2505–10. doi: 10.1161/01.CIR.0000096485.64373.C5
19. Kwon EJ, Park EJ, Yu H, Huh JS, Kim J, Cho M. SIRT-1 regulates TGF-beta-induced dermal fibroblast migration via modulation of Cyr61 expression. Connect Tissue Res. (2018) 59:245–54. doi: 10.1080/03008207.2017.1360293
20. Spin JM, Maegdefessel L, Tsao PS. Vascular smooth muscle cell phenotypic plasticity: focus on chromatin remodelling. Cardiovasc Res. (2012) 95:147–55. doi: 10.1093/cvr/cvs098
21. MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
23. Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene (2004) 341:19–39. doi: 10.1016/j.gene.2004.06.044
24. Albanese I, Khan K, Barratt B, Al Kindi H, Schwertani A. Atherosclerotic calcification: Wnt is the hint. J Am Heart Assoc. (2018) 7:e007356. doi: 10.1161/JAHA.117.007356
25. Jin X, Rong S, Yuan W, Gu L, Jia J, Wang L, et al. High mobility group box 1 promotes aortic calcification in chronic kidney disease via the wnt/beta-catenin pathway. Front Physiol. (2018) 9:665. doi: 10.3389/fphys.2018.00665
26. Zhang P, Li Y, Du Y, Li G, Wang L, Zhou F. Resveratrol ameliorated vascular calcification by regulating Sirt-1 and Nrf2. Transplant Proc. (2016) 48:3378–86. doi: 10.1016/j.transproceed.2016.10.023
27. Peng Y, Zhang G, Tang H, Dong L, Gao C, Yang X, et al. Influence of SIRT1 polymorphisms for diabetic foot susceptibility and severity. Medicine (2018) 97:e11455. doi: 10.1097/MD.0000000000011455
28. Takemura A, Iijima K, Ota H, Son B K, Ito Y, Ogawa S, et al. Sirtuin 1 retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2011) 31:2054–62. doi: 10.1161/ATVBAHA.110.216739
29. Miyagawa A, Tatsumi S, Takahama W, Fujii O, Nagamoto K, Kinoshita E, et al. The sodium phosphate cotransporter family and nicotinamide phosphoribosyltransferase contribute to the daily oscillation of plasma inorganic phosphate concentration. Kidney Int. (2018) 93:1073–85. doi: 10.1016/j.kint.2017.11.022
30. Akiyoshi T, Ota H, Iijima K, Son BK, Kahyo T, Setou M, et al. A novel organ culture model of aorta for vascular calcification. Atherosclerosis (2016) 244:51–8. doi: 10.1016/j.atherosclerosis.2015.11.005
31. Matsubara T, Kida K, Yamaguchi A, Hata K, Ichida F, Meguro H, et al. BMP2 regulates osterix through Msx2 and Runx2 during osteoblast differentiation. J Biol Chem. (2008) 283:29119–25. doi: 10.1074/jbc.M801774200
32. Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E. Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect Tissue Res. (2003) 44(Suppl. 1):109–16. doi: 10.1080/03008200390152188
33. Kook SH, Heo JS, Lee JC. Crucial roles of canonical Runx2-dependent pathway on Wnt1-induced osteoblastic differentiation of human periodontal ligament fibroblasts. Mol Cell Biochem. (2015) 402:213–23. doi: 10.1007/s11010-015-2329-y
34. Raaz U, Schellinger IN, Chernogubova E, Warnecke C, Kayama Y, Penov K, et al. Transcription factor Runx2 promotes aortic fibrosis and stiffness in type 2 diabetes mellitus. Circ Res. (2015) 117:513–24. doi: 10.1161/CIRCRESAHA.115.306341
35. Houben E, Neradova A, Schurgers LJ, Vervloet M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: a systematic review. G Ital Nefrol. (2016) 33:gin/33.6.5.
36. Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation (2001) 104:1881–3. doi: 10.1161/CIRCULATIONAHA.107.743161
37. Li N, Cheng W, Huang T, Yuan J, Wang X, Song M. Vascular adventitia calcification and its underlying mechanism. PLoS ONE (2015) 10:e0132506. doi: 10.1371/journal.pone.0132506
38. Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, Kuppe C, et al. Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell (2016) 19:628–42. doi: 10.1016/j.stem.2016.08.001
39. Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, Shimohama S, et al. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc Natl Acad Sci USA. (2008) 105:15599–604. doi: 10.1073/pnas.0800612105
40. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature (2005) 437:270–4. doi: 10.1038/nature03940
41. Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science (2012) 337:59–64. doi: 10.1126/science.1222879
42. Zoltewicz JS, Ashique AM, Choe Y, Lee G, Taylor S, Phamluong K. Wnt signaling is regulated by endoplasmic reticulum retention. PLoS ONE (2009) 4:e6191. doi: 10.1371/journal.pone.0006191
43. Willert K Nusse R. Wnt proteins. Cold Spring Harb Perspect Biol. (2012) 4:a007864. doi: 10.1101/cshperspect.a007864
44. Damien Coudreuse HK. The making of Wnt: new insights into Wnt maturation, sorting and secretion. Development (2007) 134:3–12. doi: 10.1242/dev.02699
45. Gao C, Xiao G, Hu J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. (2014) 4:13. doi: 10.1186/2045-3701-4-13
46. Mill C and George SJ. Wnt signalling in smooth muscle cells and its role in cardiovascular disorders. Cardiovasc Res. (2012) 95:233–40. doi: 10.1093/cvr/cvs141
47. Holloway KR, Calhoun TN, Saxena M, Metoyer CF, Kandler EF, Rivera CA, et al. SIRT1 regulates Dishevelled proteins and promotes transient and constitutive Wnt signaling. Proc Natl Acad Sci USA. (2010) 107:9216–21. doi: 10.1073/pnas.0911325107
48. Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. (2005) 280:33132–40. doi: 10.1074/jbc.M500608200
49. Baschant U, Rauner M, Balaian E, Weidner H, Roetto A, Platzbecker U, et al. Wnt5a is a key target for the pro-osteogenic effects of iron chelation on osteoblast progenitors. Haematologica (2016) 101:1499–507. doi: 10.3324/haematol.2016.144808
50. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science (1996) 2721023–6.
51. Hedgepeth CM, Deardorff MA, Rankin K, Klein PS. Regulation of glycogen synthase kinase 3β and downstream Wnt signaling by axin. Mol Cell Biol. (1999) 19:7147–57. doi: 10.1128/MCB.19.10.7147
52. Tauriello DV, Maurice MM. The various roles of ubiquitin in Wnt pathway regulation. Cell Cycle (2010) 9:3700–9. doi: 10.4161/cc.9.18.13204
53. Ren DN, Chen J, Li Z, Yan H, Yin Y, Wo D, et al. LRP5/6 directly bind to Frizzled and prevent Frizzled-regulated tumour metastasis. Nat Commun. (2015) 6:6906. doi: 10.1038/ncomms7906
54. González-Sancho JM, Greer YE, Abrahams CL, Takigawa Y, Baljinnyam B, Lee KH, et al. Functional consequences of Wnt-induced dishevelled 2 phosphorylation in canonical and noncanonical Wnt signaling*. J Biol Chem. (2013) 288:9428–37. doi: 10.1074/jbc.M112.448480
55. Fagotto F, Gluck U, Gumbiner BM. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of beta-catenin. Curr Biol. (1998) 8:181–90. doi: 10.1016/S0960-9822(98)70082-X
56. Daniels D L, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol. (2005) 12:364–71. doi: 10.1038/nsmb912
57. Chocarro-Calvo A, García-Martínez JM, Ardila-González S, De la Vieja A, García-Jiménez C. Glucose-induced β-catenin acetylation enhances Wnt signaling in cancer. Mol Cell (2013) 49:474–86. doi: 10.1016/j.molcel.2012.11.022
58. Ono M, Lai KKY, Wu K, Nguyen C, Lin DP, Murali R, et al. Nuclear receptor/Wnt beta-catenin interactions are regulated via differential CBP/p300 coactivator usage. PLoS ONE (2018) 13:e0200714. doi: 10.1371/journal.pone.0200714
59. Gravesen E, Nordholm A, Mace M, Morevati M, Høgdall E, Nielsen C, et al. Effect of inhibition of CBP-coactivated β-catenin-mediated Wnt signalling in uremic rats with vascular calcifications. PLoS ONE (2018) 13:e0201936. doi: 10.1371/journal.pone.0201936
60. Yoon H, Shin SH, Shin DH, Chun YS, Park JW. Differential roles of Sirt1 in HIF-1alpha and HIF-2alpha mediated hypoxic responses. Biochem Biophys Res Commun. (2014) 444:36–43. doi: 10.1016/j.bbrc.2014.01.001
61. Tsaousi A, Mill C, George SJ. The Wnt pathways in vascular disease: lessons from vascular development. Curr Opin Lipidol. (2011) 22:350–7. doi: 10.1097/MOL.0b013e32834aa701
62. Simic P, Zainabadi K, Bell E, Sykes DB, Saez B, Lotinun S, et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol Med. (2013) 5:430–40. doi: 10.1002/emmm.201201606
63. Wu Q, Wang Y, Qian M, Qiao Y, Zou S, Chen C, et al. Sirt1 suppresses Wnt/betaCatenin signaling in liver cancer cells by targeting betaCatenin in a PKAalpha-dependent manner. Cell Signal. (2017) 37:62–73. doi: 10.1016/j.cellsig.2017.06.001
64. Liu S, Yang H, Hu B, Zhang M. Sirt1 regulates apoptosis and extracellular matrix degradation in resveratrol-treated osteoarthritis chondrocytes via the Wnt/beta-catenin signaling pathways. Exp Ther Med. (2017) 14:5057–62. doi: 10.3892/etm.2017.5165
65. Srisuttee R, Koh SS, Kim SJ, Malilas W, Boonying W, Cho IR, et al. Hepatitis B virus X (HBX) protein upregulates beta-catenin in a human hepatic cell line by sequestering SIRT1 deacetylase. Oncol Rep. (2012) 28:276–82. doi: 10.3892/or.2012.1798
66. Chen EEM, Zhang W, Ye CCY, Gao X, Jiang LLJ, Zhao TTF, et al. Knockdown of SIRT7 enhances the osteogenic differentiation of human bone marrow mesenchymal stem cells partly via activation of the Wnt/beta-catenin signaling pathway. Cell Death Dis. (2017) 8:e3042. doi: 10.1038/cddis.2017.429
67. Monteserin-Garcia J, Al-Massadi O, Seoane LM, Alvarez CV, Shan B, Stalla J (2017). Sirt1 inhibits the transcription factor CREB to regulate pituitary growth hormone synthesis. Fed Am Soc Exp Biol. (2013) 27:1561–71. doi: 10.1096/fj.12-220129
68. Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Fact. (2004) 22:233–41. doi: 10.1080/08977190412331279890
69. Jimi E, Hirata S, Shin M, Yamazaki M, Fukushima H. Molecular mechanisms of BMP-induced bone formation: cross-talk between BMP and NF-κB signaling pathways in osteoblastogenesis. Jap Dent Sci Rev. (2010) 46:33–42. doi: 10.1016/j.jdsr.2009.10.003
70. Badimon L, Borrell-Pages M. Wnt signaling in the vessel wall. Curr Opin Hematol. (2017) 24:230–9. doi: 10.1097/MOH.0000000000000336
71. Rong S, Zhao X, Jin X, Zhang Z, Chen L, Zhu Y, et al. Vascular calcification in chronic kidney disease is induced by bone morphogenetic protein-2 via a mechanism involving the Wnt/beta-catenin pathway. Cell Physiol Biochem. (2014) 34:2049–60. doi: 10.1159/000366400
72. Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res. (2003) 18:1842–53. doi: 10.1359/jbmr.2003.18.10.1842
73. Zhou N, Li Q, Lin X, Hu N, Liao JY, Lin LB, et al. BMP2 induces chondrogenic differentiation, osteogenic differentiation and endochondral ossification in stem cells. Cell Tissue Res. (2016) 366:101–11. doi: 10.1007/s00441-016-2403-0
74. Wright V, Peng H, Usas A, Young B, Gearhart B, Cummins J, et al. BMP4-expressing muscle-derived stem cells differentiate into osteogenic lineage and improve bone healing in immunocompetent mice. Mol Ther. (2002) 6:169–78. doi: 10.1006/mthe.2002.0654
75. Wang P, Wang Y, Tang W, Wang X, Pang Y, Yang S, et al. Bone morphogenetic protein-9 enhances osteogenic differentiation of human periodontal ligament stem cells via the JNK pathway. PLoS ONE (2017) 12:e0169123. doi: 10.1371/journal.pone.0169123
76. Yu PB, Deng DY, Beppu H, Hong CC, Lai C, Hoyng SA, et al. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J Biol Chem. (2008) 283:3877–88. doi: 10.1074/jbc.M706797200
77. Zhang X, Arnott JA, Rehman S, Delong WG Jr Sanjay A, Safadi FF, et al. Src is a major signaling component for CTGF induction by TGF-β1 in osteoblasts. J Cell Physiol. (2010) 224:691–701. doi: 10.1002/jcp.22173
78. Xu L. Regulation of smad activities. Biochim Biophys Acta (2006) 1759:503–13. doi: 10.1016/j.bbaexp.2006.11.001
79. Lee SJ, Jeong JY, Oh CJ, Park S, Kim Y, Kim J, et al. Pyruvate dehydrogenase kinase 4 promotes vascular calcification via SMAD1/5/8 phosphorylation. Sci Rep. (2015) 5:16577. doi: 10.1038/srep16577
80. Li X, Yang HY, Giachelli CM. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis (2008) 199:271–7. doi: 10.1016/j.atherosclerosis.2007.11.031
81. Guerrero F, Herencia C, Almadén Y, Martínez-Moreno JM, Montes de Oca A, Rodriguez-Ortiz ME, et al. TGF-β prevents phosphate-induced osteogenesis through inhibition of BMP and Wnt/β-catenin pathways. PLoS ONE (2018) 9:e89179. doi: 10.1371/journal.pone.0089179
82. Zhang M, Sara JD, Wang F, Liu LP, Su LX, Zhe J, et al. Increased plasma BMP-2 levels are associated with atherosclerosis burden and coronary calcification in type 2 diabetic patients. Cardiovasc Diabetol. (2015) 14:64. doi: 10.1186/s12933-015-0214-3
83. Wang Y, Shan J, Yang W, Zheng H, Xue S. High mobility group box 1 (HMGB1) mediates high-glucose-induced calcification in vascular smooth muscle cells of saphenous veins. Inflammation (2013) 36:1592–604.
84. Rabadi MM, Xavier S, Vasko R, Kaur K, Goligorksy MS, Ratliff BB. High mobility group box 1 is a novel deacetylation target of Sirtuin1. Kidney Int. (2015) 87:95–108. doi: 10.1038/ki.2014.217
85. Hwang JS, Choi HS, Ham SA, Yoo T, Lee WJ, Paek K S, et al. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci Rep. (2015) 5:15971. doi: 10.1038/srep15971
86. Martinez-Moreno JM, Munoz-Castaneda JR, Herencia C, Oca AM, Estepa JC, Canalejo R, et al. In vascular smooth muscle cells paricalcitol prevents phosphate-induced Wnt/beta-catenin activation. Am J Physiol Renal Physiol. (2012) 303:F1136–44. doi: 10.1152/ajprenal.00684.2011
87. Lévy L, Wei Y, Labalette C, Wu Y, Renard AC, Buendia MA, et al. Acetylation of β-catenin by p300 regulates β-catenin-Tcf4 interaction. Mol Cell Biol. (2004) 24:3404–14. doi: 10.1128/MCB.24.8.3404-3414.2004
88. Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH, et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem. (2006) 281:16502–11. doi: 10.1074/jbc.M512494200
89. Cohen ED, Tian Y, Morrisey EE. Wnt signaling: an essential regulator of cardiovascular differentiation, morphogenesis and progenitor self-renewal. Development (2008) 135:789–98. doi: 10.1242/dev.016865
90. Zhong Z, Ethen NJ, Williams BO. WNT signaling in bone development and homeostasis. Wiley Interdiscip Rev Dev Biol. (2014) 3:489–500. doi: 10.1002/wdev.159
91. Rahman MS, Akhtar N, Jamil HM, Banik RS, Asaduzzaman SM. TGF-β/BMP signaling and other molecular events: regulation of osteoblastogenesis and bone formation. Bone Res. (2015) 3:15005. doi: 10.1038/boneres.2015.5
92. Manolagas SC, Almeida M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol. (2007) 21:2605–14. doi: 10.1210/me.2007-0259
93. Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol. (2012) 4:535–40. doi: 10.1101/cshperspect.a007906
94. Nakashima A, Katagiri T, Tamura M. Cross-talk between Wnt and Bone Morphogenetic Protein 2 (BMP-2) signaling in differentiation pathway of C2C12 myoblasts. J Biol Chem. (2005) 280:37660–8. doi: 10.1074/jbc.M504612200
95. Zhang S, Xiao Z, Luo J, He N, Mahlios J, Quarles LD. Dose-dependent effects of Runx2 on bone development. J Bone Miner Res. (2009) 24:1889–904. doi: 10.1359/jbmr.090502
96. Bodine PV, Stauffer B, Ponce-de-Leon H, Bhat RA, Mangine A, Seestaller-Wehr LM, et al. A small molecule inhibitor of the Wnt antagonist secreted frizzled-related protein-1 stimulates bone formation. Bone (2009) 44:1063–8. doi: 10.1016/j.bone.2009.02.013
97. Sabbagh Y, O'Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. (2009) 20:2348–58. doi: 10.1681/ASN.2009050559
98. Lv W, Guan L, Zhang Y, Yu S, Cao B, Ji Y. Sclerostin as a new key factor in vascular calcification in chronic kidney disease stages 3 and 4. Int Urol Nephrol. (2016) 48:2043–50. doi: 10.1007/s11255-016-1379-8
99. Cai T, Sun D, Duan Y, Wen P, Dai C, Yang J, et al. WNT/beta-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. (2016) 345:206–17. doi: 10.1016/j.yexcr.2016.06.007
100. Sebastian A, Loots GG. Transcriptional control of Sost in bone. Bone (2017) 96:76–84. doi: 10.1016/j.bone.2016.10.009
101. Perez-Campo FM, Santurtun A, Garcia-Ibarbia C, Pascual MA, Valero C, Garces C, et al. Osterix and RUNX2 are transcriptional regulators of sclerostin in human bone. Calcif Tissue Int. (2016) 99:302–9. doi: 10.1007/s00223-016-0144-4
102. Cohen-Kfir E, Artsi H, Levin A, Abramowitz E, Bajayo A, Gurt I, et al. Sirt1 is a regulator of bone mass and a repressor of sost encoding for sclerostin, a bone formation inhibitor. Endocrinology (2018) 152:4514–24. doi: 10.1210/en.2011-1128
103. D'Onofrio N, Servillo L, Balestrieri ML. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. (2018) 28:711–32. doi: 10.1089/ars.2017.7178
104. Chong ZZ, Wang S, Shang Y C, Maiese K. Targeting cardiovascular disease with novel SIRT1 pathways. Fut Cardiol. (2012) 8:89–100. doi: 10.2217/fca.11.76
105. Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. (2014) 35:146–54. doi: 10.1016/j.tips.2013.12.004
106. Tomayko EJ, Cachia AJ, Chung HR, Wilund KR. Resveratrol supplementation reduces aortic atherosclerosis and calcification and attenuates loss of aerobic capacity in a mouse model of uremia. J Med Food (2014) 17:278–83. doi: 10.1089/jmf.2012.0219
107. Mattison J, Wang M, Bernier M, Zhang J, Park SS, Maudsley S, et al. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. (2014) 20:183–190. doi: 10.1016/j.cmet.2014.04.018
108. Zhong Z, Zylstra-Diegel CR, Schumacher CA, Baker JJ, Carpenter AC, Rao S, et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc Natl Acad Sci USA. (2012) 109:E2197–204. doi: 10.1073/pnas.1120407109
109. Feng G, Zheng K, Song D, Xu K, Huang D, Zhang Y, et al. SIRT1 was involved in TNF-alpha-promoted osteogenic differentiation of human DPSCs through Wnt/beta-catenin signal. In Vitro Cell Dev Biol Anim. (2016) 52:1001–11. doi: 10.1007/s11626-016-0070-9
110. Shao J S, Cheng S L, Pingsterhaus J M, Charlton-Kachigian N, Loewy A P, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest. (2005) 115:1210–20. doi: 10.1172/JCI24140
111. Mellini P, Valente S, Mai A. Sirtuin modulators: an updated patent review (2012 - 2014). Expert Opin Ther Pat. (2015) 25:5–15. doi: 10.1517/13543776.2014.982532
112. Liu X, Hu D, Zeng Z, Zhu W, Zhang N, Yu H, et al. SRT1720 promotes survival of aged human mesenchymal stem cells via FAIM: a pharmacological strategy to improve stem cell-based therapy for rat myocardial infarction. Cell Death Dis. (2017) 8:e2731. doi: 10.1038/cddis.2017.107
113. Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken E M, et al. SRT1720 improves survival and healthspan of obese mice. Sci Rep. (2011) 1:70. doi: 10.1038/srep00070
114. Singh C K, Liu X, Ahmad N. Resveratrol, in its natural combination in whole grape, for health promotion and disease management. Ann N Y Acad Sci. (2015) 1348:150–60. doi: 10.1111/nyas.12798
115. Cao H, Ou J, Chen L, Zhang Y, Szkudelski T, Delmas D, et al. Dietary polyphenols and type 2 diabetes: human study and clinical trials. Crit Rev Food Sci Nutr. (2018) 1–19. doi: 10.1080/10408398.2018.1492900
116. Arcanjo NMO, Luna C, Madruga M S, Estevez M. Antioxidant and pro-oxidant actions of resveratrol on human serum albumin in the presence of toxic diabetes metabolites: glyoxal and methyl-glyoxal. Biochim Biophys Acta (2018) 1862:1938–47. doi: 10.1016/j.bbagen.2018.06.007
117. Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol. (2016) 17:679–690. doi: 10.1038/nrm.2016.93
118. Conti V, Forte M, Corbi G, Russomanno G, Formisano L, Landolfi A, et al. Sirtuins: possible clinical implications in cardio and cerebrovascular diseases. Curr Drug Targets (2017) 18:473–84. doi: 10.2174/1389450116666151019095903
119. Aditya R, Kiran AR, Varma DS, Vemuri R, Gundamaraju R. A review on SIRtuins in diabetes. Curr Pharm Des. (2017) 23:2299–307. doi: 10.2174/1381612823666170125153334
120. Noh RM, Venkatasubramanian S, Daga S, Langrish J, Mills NL, Lang NN, et al. Cardiometabolic effects of a novel SIRT1 activator, SRT2104, in people with type 2 diabetes mellitus. Open Heart (2017) 4:e000647. doi: 10.1136/openhrt-2017-000647
Keywords: SIRT1, vascular calcification, Wnt, β-catenin, diabetes, calcifying vascular cells
Citation: Bartoli-Leonard F, Wilkinson FL, Langford-Smith AWW, Alexander MY and Weston R (2018) The Interplay of SIRT1 and Wnt Signaling in Vascular Calcification. Front. Cardiovasc. Med. 5:183. doi: 10.3389/fcvm.2018.00183
Received: 07 September 2018; Accepted: 04 December 2018;
Published: 18 December 2018.
Edited by:
Dwight A. Towler, University of Texas Southwestern Medical Center, United StatesReviewed by:
Patricia B. Maguire, University College Dublin, IrelandCopyright © 2018 Bartoli-Leonard, Wilkinson, Langford-Smith, Alexander and Weston. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ria Weston, Ui5XZXN0b25AbW11LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.