 Eva Jover
Eva Jover Marco Fagnano†
Marco Fagnano† Gianni Angelini
Gianni Angelini Paolo Madeddu
Paolo Madeddu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 06 November 2018
Sec. Atherosclerosis and Vascular Medicine
Volume 5 - 2018 | https://doi.org/10.3389/fcvm.2018.00155
This article is part of the Research Topic Exploring the Frontiers of Regenerative Cardiovascular Medicine View all 13 articles
Cardiovascular calcification is an independent risk factor and an established predictor of adverse cardiovascular events. Despite concomitant factors leading to atherosclerosis and heart valve disease (VHD), the latter has been identified as an independent pathological entity. Calcific aortic valve stenosis is the most common form of VDH resulting of either congenital malformations or senile “degeneration.” About 2% of the population over 65 years is affected by aortic valve stenosis which represents a major cause of morbidity and mortality in the elderly. A multifactorial, complex and active heterotopic bone-like formation process, including extracellular matrix remodeling, osteogenesis and angiogenesis, drives heart valve “degeneration” and calcification, finally causing left ventricle outflow obstruction. Surgical heart valve replacement is the current therapeutic option for those patients diagnosed with severe VHD representing more than 20% of all cardiac surgeries nowadays. Tissue Engineering of Heart Valves (TEHV) is emerging as a valuable alternative for definitive treatment of VHD and promises to overcome either the chronic oral anticoagulation or the time-dependent deterioration and reintervention of current mechanical or biological prosthesis, respectively. Among the plethora of approaches and stablished techniques for TEHV, utilization of different cell sources may confer of additional properties, desirable and not, which need to be considered before moving from the bench to the bedside. This review aims to provide a critical appraisal of current knowledge about calcific VHD and to discuss the pros and cons of the main cell sources tested in studies addressing in vitro TEHV.
Cardiovascular calcification (CVC) is an independent risk factor and an established predictor of adverse and disabling cardiovascular events (Figure 1) (1–3). Histopathological studies have demonstrated hydroxyapatite deposits in vulnerable atherosclerotic plaques (4) and aortic valves (5). No longer considered a passive age-related disease, CVC is identified as the active, progressive and multifactorial ectopic bone-like calcification of blood vessels, myocardium or heart valves, leading to the “degeneration”/deterioration and dysfunction of the affected tissue (5, 6). Although there is an overlap between the risk factors leading to atherosclerosis and valvular calcification, only 40–50% of patients diagnosed with atherosclerosis concomitantly develop calcific valvular heart disease (VHD), thus suggesting that VHD is an independent pathological entity (7, 8).
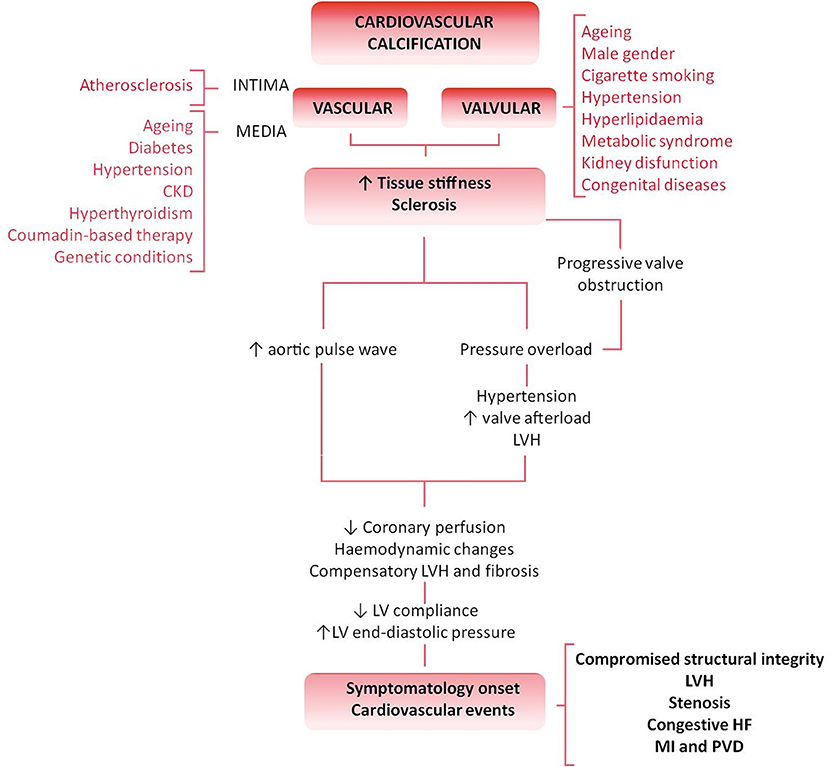
Figure 1. Differential pathology and clinical impact of valvular vs. vascular calcification flowchart. Cardiovascular calcification is an active and degenerative bone-like process affecting the cardiovascular tissues. Both vessels and valves show an athero-inflammatory background and, despite the commonalities and overlap of several risk factors (such as aging, hyperlipidaemia or kidney disease), both atherosclerosis and calcific VHD are two independent pathologic entities. The biological progression of the disease, tissue characteristics and clinical impact stand those differences. The result is the independent plaque rupture primary outcome found in the progression of VHD. An increased stiffness or sclerosis induces an increased aortic pulse wave, triggering hypertension, and a reduction in coronary perfusion. Besides, the pressure overload caused by a sclerotic pre-stadium and observed in the progression of the VHD leads to LV structural and hemodynamic changes. Symptomatology onset and calcification burden are poor prognosis predictors associated with multiple adverse cardiovascular complications, such as left ventricular hypertrophy (LVH), aortic valve stenosis, congestive heart failure (HF), ascending aorta aneurysm, myocardial infarction (MI), and peripheral vascular disease (PVD).
VHD is the third most common cardiovascular pathology after hypertension and coronary artery disease in developed nations (9). Specifically, aortic valve stenosis (AVS) is the most common primary valvulopathy because of either congenital malformation (such as bicuspid aortic valve or BAV) or senile “degeneration.” The result is an increased stiffness and impaired leaflet motion and calcification, lately leading to the left ventricle outflow obstruction (10). Moreover, aging, male gender, cigarette smoking, hypertension, hyperlipidaemia, metabolic syndrome or kidney dysfunction are frequent independent risk factors for calcific VHD and significantly impair the outcome and prognosis of patients (11). The progression of calcific VHD consists of a valve sclerosis prestadium affecting more than 25% of the general population over 65 years old and associated to a 50% increased cardiovascular risk over 5 years (2). The prevalence of calcification and stenosis is reported in ~2% or 2.5% in a population aged over 65 or 75 years, respectively, representing a major cause of morbidity and mortality in the elderly (11, 12). Stenotic aortic valves are also found in congenital bicuspid valves and may require valve replacement even two decades earlier than valves anatomically normal (13). VHD is predicted to become a new cardiovascular epidemic in the next 20 years because of the increase of life expectancy in industrialized nations (9, 12, 14). No specific pharmacological strategy has been developed to retard, halt or revert the progression of VHD. Valve replacement represents the gold standard method to treat VHD through either mechanical or biological prosthesis implantation (15), but it is not suitable or definitive for all patients. New therapeutic solutions are claimed from the clinic to overcome the limitations of current therapeutic options including the chronic oral anticoagulation required for mechanical valves implantation or the degeneration, calcification, and failure of the biological counterparts. A plethora of novel tissue engineering-based approaches has emerged promising a definitive solution. Between the two main tissue engineered heart valves (TEHV) approaches, in vitro TEHV may provide, among others, a “native-like” extracellular matrix (ECM) surrogate and promote a “physiologic-like” regeneration in a pathologic environment with a deteriorated reparative system. Implantation of those devices is appealing for pediatric patients with congenital VHD as it might circumvent the failure of growth, repair, and remodeling required after somatic growth. In this review, we assess the current knowledge in the clinical relevance and mechanisms of valvular calcification and critically discuss the benefits and limitations of different cell sources currently used for the development of in vitro TEHV.
Calcific VHD of anatomically normal valves is a slow and active process driving to degeneration and dysfunction, with a long preclinical and asymptomatic phase. The onset of symptomatology is a general sign of advanced and severe disease associated with a high event rate, rapid valve deterioration and malfunctioning, thus being a poor prognostic indicator and elective for valve replacement surgery (15). However, the management of patients with asymptomatic valve disease is challenging. The real prevalence of unsuspected VHD is unsure, and a significant proportion of patients remain asymptomatic and undiagnosed until late stages when the long-term benefits of intervention are ambiguous due to increased postoperative complications and further mortality (8, 14). Large European and North American observational studies have provided most of the valuable insights on the overall VHD prevalence and the effect on overall survival (8, 14, 16, 17). In 2001, the Euro Heart Survey study (8) evidenced “degeneration” as the dominant etiological cause of VHD, with AVS (43%), mitral regurgitation (32%), and aortic regurgitation (13%) representing the commonest forms of adult valvopathies. AVS progression occurring in up to 5% of elderly patients (11, 14) carries an 80% 5-year risk of developing heart failure, valve replacement requirement, or death (18). Moreover, a US population-based study in more than 28,000 adults demonstrated the age-dependent VHD prevalence, rising from 0.7% in subjects aged 18–44 to 13% in those over 75 years old (16), significantly impacting the survival rates and emphasizing its significance as a health care issue. A more recent publication showed that general population aged ≥60 years across 37 advanced economies (16.1 million people) has a whole prevalence of 4.5% VHD (2.8 and 13.1% in individuals aged 60–74 and ≥75 years, respectively) (19). Only in the UK, VHD might account for approximately 1 million people aged over 65 years, and trend predictions suggest a significant raise due to increased life expectancy and the continuum of population aging in industrialized countries. The degeneration of anatomically normal valves is more often and rapid in people over 70 years because of progressive fibrosis and calcification of the valve cusps (www.bcs.com). A population aged over 75 years is projected to rise around 50% by 2025 resulting in a substantial VHD impact (www.statistics.gov.uk) recently estimated in ≈331,300 new cases of severe aortic stenosis per year including 65,600 patients (19). Thereby, VHD may become the next imminent and real cardiac epidemic (9, 12, 20). Genetic background and structural valve differences due to congenital malformations, such as BAV may be considered separately and are not deeply discussed in this review.
The presence and extent of CVC are generally acknowledged as strong predictors of future adverse clinical events including cardiovascular and all-cause mortality (21–23). The latter is highlighted by the up to 73% all-cause survival rate reduction estimated in patients diagnosed with high coronary artery calcification score (21). Importantly, 5–20% of the atherosclerotic lesions contain calcium deposits (24, 25), and it is alarming the potential underestimation of affected tissues due to the presence of chondrogenic intermediates, asymptomatic phases, or the lack of more powerful calcification screening methods. Additionally, the extent of valvular calcification correlates with the severity of stenosis (26). Therefore, a comprehensive and early understanding of the cardiovascular risk associated with calcification is critical for patient management and long-term prognosis (15, 27, 28).
Echocardiography is the mainstay for diagnosis, assessment and follow-up of VHD (15). It allows the calculation of the continuity equation-based aortic valve area both for predicting the clinical outcome and for clinical decisions making as well as aortic jet velocity and leaflet calcification (5, 29). However, visualizing abnormal valve anatomy becomes difficult once severe calcification is established. Moreover, concomitant hypertension increases the systemic vascular resistance in addition to the valvular obstruction, thus imposing a double over-load on the left ventricle which may lead to underestimate the assessment of the stenosis severity (30).
Other imaging methods, notably cardiac magnetic resonance imaging (MRI) and coronary computed tomography (CCT), are used if echocardiographic imaging is not satisfactory. Three-dimensional time-resolved, phase contrast cardiac magnetic resonance, otherwise referred as 4-dimensional (4D) flow MRI, is an innovative and appealing method for studying cardiovascular diseases. Dataset integration of 4D-Flow MRI can be retrospectively quantified providing a comprehensive evaluation of complex secondary vascular parameters, such as mechanical wall shear stress (WSS) on the vessels and heart valves (31) but also flow energy loss and flow displacements (32, 33). BAV is frequently associated with the progression of ascending thoracic aorta aneurysm (AsAo). Intrinsic wall abnormalities cannot fully explain the differential aneurysm progression resulting from different aortic leaflet fusion patterns and asymmetry (34). Echocardiography findings have suggested that abnormal blood flow could potentially trigger those differences in AsAo progression. In the context of VHD, 4D-Flow MRI has demonstrated to be a powerful tool to determine the association of flow hemodynamic, especially in those situations in which eccentric systolic blood flow jets result in abnormal helical systolic flow. The latter has highlighted the potential application of 4D-Flow MRI to study the progression and stratify/predict the risk of AsAo development specially in BAV patients (34), while echocardiography is not a reliable method. Moreover, recent studies have demonstrated the association of WSS and aortic peak velocity with parameters of left ventricle remodeling, allowing to distinguish BAV patients with or without aortic stenosis or regurgitation (35). Post-operative follow-up of reparative surgery of tetralogy of Fallot is another potential application of 4D-Flow MRI (36). However, long acquisition times, lack of blood pressure determination, susceptibility to motion artifacts, poor spatial resolution and the need of massive data post-processing are the main drawbacks of this technique. In addition, aberrant hemodynamic changes are seen only in advanced stenotic VHD and that represents a limitation for hemodynamic analysis techniques. Earlier phases of the VHD, such as asymptomatic sclerosis pre-stadium of well-functional anatomically normal valves, may not be detected by echocardiography or 4D-Flow MRI. Complementary imaging techniques such as CCT may provide substantial information on the detection and risk assessment of VHD. Multi-slice CCT together with the implementation of new acquisition techniques including ECG synchronization, retrospective image reconstruction and application of algorithms such as Agatston score (37), permit a direct, real-time and easy assessment of calcium content in coronary arteries (38). It has substantially improved the detection of early CVC stages. The high sensitivity of CCT has improved the screening for CVC, evidencing a progressive increment on CVC in patients over 60 years, which is especially relevant in patients diagnosed with VHD. Moreover, CCT combined with coronary angiography (gold-standard for coronary lesion evaluation) has demonstrated a good correlation between coronary calcium content and coronary artery disease (39, 40). The suitability of CCT to screen early stages of valve calcification in sclerotic valves with no hemodynamic obstruction has been also demonstrated (41). Moreover, CCT screening has proved to be a superior and more trustable method than carotid-intima-media thickness or ankle-brachial index for identifying patients at high risk (42). Finally, MRI and CCT can also provide complementary information to improve assessment of the valve lesion and cardiac function to aid the timing of surgery and determine risk (43).
Over the past four decades, experimental and clinical research has elucidated the pathophysiology of CVC. Ectopic calcification is an active and tightly organized process, which recapitulates several molecular mechanisms orchestrating physiologic chondro/osteogenesis (44–46). Both the phenotypic trans-differentiation into osteo/chondroblast-like cells and the active ECM remodeling, including its mineralization, represent the two hallmarks of ectopic calcifications (47) (Figure 2). In particular, calcific VHD has been described as a multifactorial, complex and active heterotopic endochondral lamellar bone-like formation process, driving heart valve calcification, degeneration and dysfunction (5, 6) toward integration of ECM remodeling, osteogenesis, and angiogenesis. Heterotopic bone exhibits morphological and biochemical features of orthotopic bone, and it is capable of generating bone marrow (48). Higher remodeling rates have been reported in calcified valves than in physiologic bone formation (48) though, thus suggesting uncoupling of bone formation and resorption activities (49). However, histological observations in human specimens of calcific valves have evidenced an 83% prevalence of dystrophic calcification with only a 13% of active bone remodeling (5) and a 92% prevalence of microfractures, which are the main site of active bone remodeling. One could interpret valve calcification as a senile degeneration leading to cellular aging and death, and hydroxyapatite deposition on cellular degradation products rather than an active osteogenic-derived mineralization occurring on collagen and elastin fibers. Further research on this regard will significantly contribute to the understanding of the calcific VHD pathophysiology in the next years and it is currently cause of controversy. Observation of advanced end-stage phases in dystrophic human specimens could lead to misinterpretation of the underlying pathology in which a “preliminary” heterotopic bone-like tissue demonstrated by both in vitro and in vivo studies, might lately be replaced toward a misbalanced bone resorption (49) leading to an increased presence of dystrophic mineralisation.
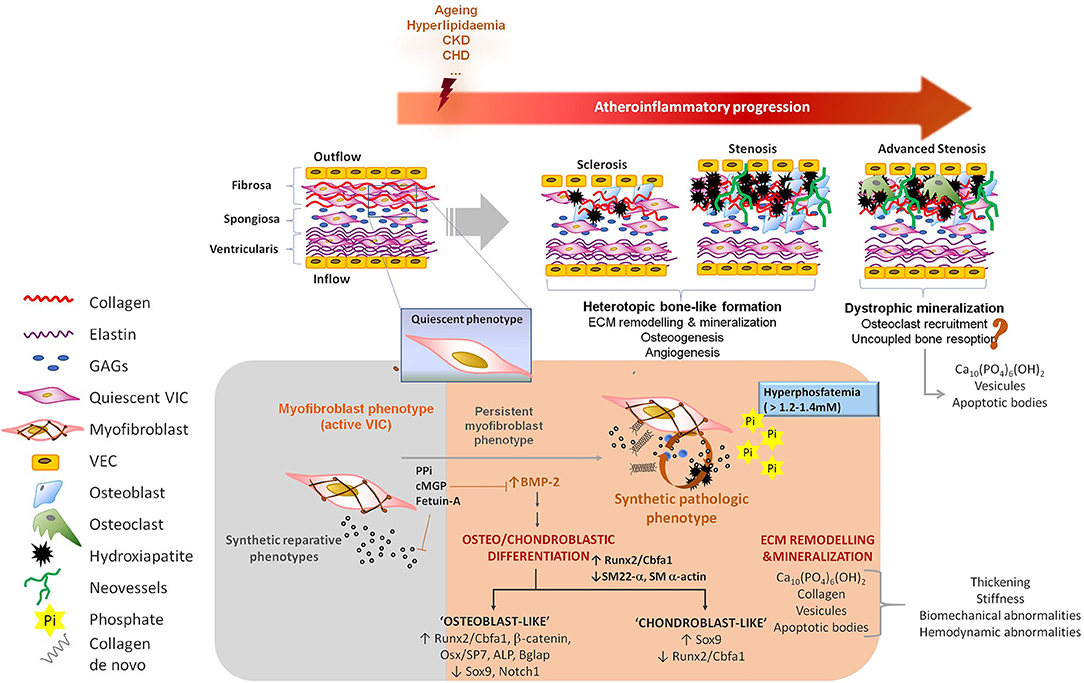
Figure 2. Pathophysiology of valve calcification. VICs are myofibroblast-like cells endowed with high plasticity, allowing them to participate in reparative, regenerative, and pathological processes. Underlying athero-inflammatory disease, aging as well as other clinical conditions such as chronic kidney disease or hyperlipidaemia, can trigger the acquisition of synthetic, proliferative and migratory phenotypes by VICs. These conditions are often associated with a reduction of defense mechanism against calcification. Two major features are recognized during valve calcification: (i) ECM remodeling, including the synthesis of a collagen-enriched matrix and its mineralization, and (ii) osteo/chondroblast differentiation of VICs. Osteogenic processes are associated with the abundant synthesis of collagen type-I and other ECM remodeling processes causing stiffness changes capable of perpetuate and extend the osteogenesis in the valve. A prestadium of sclerosis in well-functional valves finally leading to stenosis and obstruction in an active heterotopic bone-like formation process has been described. Observations in human samples also suggest the possibility of dystrophic mineralisation in advanced end-stages of calcific VHD. Pyrophosphate, osteopontin, osteoprotegerin, gamma-carboxylated matrix Gla protein and circulating fetuin-A are listed as physiologic inhibitors of vascular calcification; while hyperphosphatemia and hypercalcemia, expression of bone morphogenetic proteins or alkaline phosphatase and activation of osteo/chondroblast regulators (Runx2, Osterix, Wnt) are counted among the pro-calcifying elements orchestrating osteoblast-like transdifferentiation. PPi, pyrophosphate; cMGP, gamma-carboxylated matrix Gla protein; BMP-2, bone morphogenetic protein 2; ECM, extracellular matrix; CKD, chronic kidney disease; CHD, congestive heart disease; VIC, valve interstitial cells; VEC, valve endothelial cells; GAGs, glycosaminoglycans; Ca10(PO4)6(OH)2, hydroxyapatite; Pi, inorganic phosphate; ALP, alkaline phosphatase; Bglap, bone Gla protein or osteocalcin.
Mature valves have an avascular (50) trilaminar organization including the upper surface of the valve or fibrosa (outflow), the central spongiosa and the inflow-orientated ventricularis. Those layers differ with each other by distinct ECM organization, composition and mechanical properties [a detailed description of the valve anatomy and function has been provided by Schoen (44)]. Moreover, two major resident cells are found in the valve: valve endothelial cells (VEC) and valve interstitial cells (VIC). Although VECs are not fully characterized, differential phenotypes and expressional profiles have been identified (51, 52). Biological functions of the VEC may include regulation of permeability, mediate immune responses and establish a paracrine signaling with VICs (53). The VIC represents a heterogenous population of mesenchymal cell type which shares several commonalities with vascular smooth muscle cells (VSMC) and fibroblasts, acts as mechanical sensor thorough complex cell-to-ECM interactions and shows highly dynamic phenotype plasticity (54). The latter allows the VIC to contribute to the permanent ECM turnover and reparative processes guarantying the maintenance of the valve integrity and function (44), but also contributes to the development of valve stenosis (55, 56).
Calcific VHD is regarded as an active athero-inflammatory disease associated with a damaged endothelium and an unresolved immunological inflammation resulting from such insult. The pathogenic role of hyperlipidaemia in the valve was recognized toward the introduction of dyslipidaemia experimental models (57, 58). Early studies in human aortic valve lesions demonstrated the association among atherogenic oxidized low-density lipoproteins (oxLDL) risk factor and the expression of signaling molecules promoting osteogenic processes (57). Moreover, inflammation plays a key role on the pathogenesis of VHD with superimposed calcification (59). Numerous histological studies have suggested inflammation to trigger ECM remodeling, fibrosis, and valve thickening leading to the structural changes of VHD (60) and the subsequent differentiation of VICs into osteoblast-like phenotypes (55). Despite the commonalities and overlap of several risk factors, both atherosclerosis and calcific VHD are currently considered two independent pathologic entities mainly due to differences in the biological progression of the disease, tissue characteristics, clinical impact and resulting outcomes independent of plaque rupture (8). Among other differences, a CD8 T cell-based inflammatory background has been evidenced in valve calcification instead of the polyclonal lymphocytes reported in atherosclerotic lesions (59, 61).
The discovery of genetic modifications such as Notch1 mutations and their association with dysfunctional tissue structure of BAV and the spectrum of VHD has increased the current knowledge of these abnormalities through congenital cardiology (62). Signaling components of embryonic valvular development, such as Notch1 as well as bone morphogenetic protein (Bmp) members, transforming growth factor beta 1 (TGF-β1) or Wnt/β-catenin participate in the onset of AVS by contributing to ECM remodeling, osteogenesis and angiogenesis [further reviewed in (47, 63, 64)].
Several cell types have been involved in the development and progression of CVC and that includes vascular resident cells (VSMCs, VICs, or VECs) and circulating cells, such as mesenchymal stromal cells (MSCs), endothelial progenitor cells (EPCs) or calcifying vascular cells (CVC) (47). A dysfunctional valvular endothelium together with an imbalance between activators and natural inhibitors may promote the calcification of neighboring VICs (Figure 2) (65–67). Different VICs sub-populations have been identified in the heart valve and may differentially contribute to the pathology of calcific VHD (54). Multiple signaling molecules (such as Bmp2, TGF-b, Wnt/b-catenin, VEGF, or Notch1) are integrated in what resembles a pathologic post-natal recapitulation of fetal valvulogenesis, including the acquisition of quiescent-to-active phenotypes, an active ECM remodeling, cytokine release and promoting in situ osteoblast/chondroblast-like differentiation as an environmentally maladaptation (68, 69). Bmp2 is a strong morphogen inducing osteoblast-like phenotypes, and it plays a key role in the pathogenesis of VHD (47). Bmp2 signaling triggers nuclear translocation of Smad proteins and the activation of osteogenic-regulators such as Runx2/Cbfa1. Runx2 is an early master gene of osteoblast differentiation and chondroblast maturation during heterotopic endochondral bone formation (5, 6, 70). Active Runx2 can bind to the SP7 promoter to induce the expression of Osterix, a master transcription factor of differentiated osteoblasts. Both Runx2 and Osterix bind to BGLAP promoter to induce osteocalcin, a marker for differentiated osteoblasts, which contributes to maturation of the mineralised ECM and it is present in calcified heart valves (69). Moreover, ECM synthesis is induced directly by Osterix and its binding to the COL1A1 promoter or indirectly by Runx2 through the activation of ATF6 and subsequent COL1A1, COL1A2, and BGLAP expression (66, 67, 70). In addition, Osterix triggers the expression of alkaline phosphatase (ALP), a pyrophosphatase capable of releasing inorganic phosphate from PPi, thus inducing local hyperphosphatemia and PPi deprivation. ALP also inhibits osteopontin phosphorylation and thus its protective biological function. Finally, Wnt/β-catenin pathway mainly perpetuates osteoblastic phenotype by further induction of Runx2 and Osterix toward Bmp2-dependent signaling (57).
It is now appreciated that VECs, under certain circumstances, may undergo endothelial-to-mesenchymal transition, which is reminiscent of the early formation of the endocardial cushions (71, 72). The result might be an increase in the number of VICs susceptible to display an osteoblast phenotype (45). Dysfunctional VECs also manifest an altered secretome (73). VEC-derived nitric oxide (NO) is a regulator of Notch1 signaling in calcifying VICs (74), and a decreased Notch signaling has been found in AVC (45). Moreover, genetic studies in BAV have identified eNOS and Notch1 as candidate genes contributing to the valve anatomy and the development of VHD (62). Notch signaling leads to the cleavage and nuclear internalization of the Notch1 intracellular domain. One of the target genes of the Notch1 intracellular domain is the Hairy/enhancer-of-split (Hes)-related with YPRW motif (Hey) element, which is involved in early valve development. Both the nuclear location of the Notch1 intracellular domain and the expression of Hey1 are regulated by the VEC-derived NO (74). Furthermore, Notch1-dependent signaling is transduced through Bmp2/Runx2 axis, which directly regulates Sox9 in chondrogenesis and is an important mediator of AVS (75). Hey1 activated by Notch1 signaling forms a complex with Runx2/Cbfa1 and inhibits its transcriptional activation (64).
Pluripotent resident cells, EPCs, MSCs, and MSC-like pericytes have been found in calcified lesions suggesting a role of progenitor cells in the development and progression of ectopic calcification (45, 76, 77). The athero-inflammation associated with the release of multiple cytokines and chemokines may contribute to the recruitment of stem/progenitor cells into an environment whose homeostasis has been hampered by pro-calcifying factors and the depletion of physiologic calcification inhibitors. Finally, bone marrow (BM)-derived cells may contribute to replenishing the VIC population, modifying the proportion of VIC subpopulations yielding increased susceptibility to calcification. By using chimeric mice whose BM was repopulated with enhanced green fluorescent protein expressing total nucleated BM cells, Hajdu et al. documented the engraftment of BM-derived cells occurs as part of normal valve homeostasis (78).
Besides providing biomechanical support, valvular ECM participates in a plethora of biological functions, such as cell communication and differentiation. In addition, the ECM may contribute to ectopic CVC (74, 79, 80). Differentiation of VICs toward myofibroblast or osteoblast phenotype is highly dependent on the complex and unique VIC-to-ECM components interactions (81). Therefore, a loss of the valve ECM integrity causes malfunction and results in VHD. In line with this, propagation of the inflammation-dependent calcification of the heart valves is associated with the active ECM remodeling resulting from the proteolytic and synthetic activities of active macrophages, VICs and mast cells (82). Moreover, substrate stiffness elicits the myofibroblast activation of VICs which remaining persistent can lead to osteoblast differentiation although the exact molecular mechanisms remain unclear (83, 84).
Regulatory factors, such as thrombospondins, have been found characteristically up-regulated in calcific valves (47). Moreover, microstructural changes in collagen fiber number, width, length, density or alignment may regulate pathogenic processes compromising the mechanical properties of the valve, in particular, and most frequently at the level of the spongiosa, chondrogenic-like layer (80). Mechanistically, cartilage-specific ECM genes are downregulated in calcifying VICs because of Sox9 downregulation (74). Moreover, ECM influences VEC function and it is involved in VEC-to-myofibroblast transformation toward EMT processes (85).
Heart valves have a sparse vascularity at the proximal part (50) being considered mainly avascular. That valve avascularity is seemly abrogated in VHD (86), and the extent of neovascularisation correlates well with the burden of the disease (87). The expression of pro- and anti-angiogenic factors in stenotic valves or calcifying VICs (74, 88) has reinforced the idea that angiogenesis in the valve may promote calcific VHD, which calls for the use of modulators of angiogenesis in the therapy of valve degeneration (86, 88, 89). Accordingly, anti-angiogenic therapy has shown a protective effect on the valvular cusp endothelium (86). Immunohistochemical studies have revealed the co-localization of micro-vascularization with actively proliferating VICs, bone-related proteins, and heavy calcification (90). In addition, during calcific VHD, expression of osteonectin (pro-angiogenic and chondrogenic factor) and Lect1/chondromodulin-1 (Chm-I) (anti-angiogenic factor) is disrupted (74). In human calcified valves, the expression of vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs) and angiogenesis is concomitant with a downregulated expression of Chm-I.
Revisiting the physiology of bone formation, one could speculate that valve angiogenesis is not the cause, but the consequence of the osteogenesis perpetuation described for endochondral bone formation or that may provide of support to the thickened tissue produced de novo and resulting in a hypoxic microenvironment requiring of oxygen supply (50). An angiogenic switch of cartilage allows neovascular invasion and triggers the replacement of cartilage by bone (91). Resting chondrocytes become active and proliferative to differentiate into pre-hypertrophic chondrocytes, which can then secrete the cartilage matrix (92). Angiogenic factors such as TGF-β or VEGF are normally expressed in the cartilage and Chm-I has been proposed as an inhibitor during avascular phases of chondrogenesis. Chm-I plays divergent biological functions including chondrocyte growth and angiogenesis inhibition, stimulation of osteoblast proliferation and differentiation with a reduction of ALP activity (92, 93) and contribution to bone remodeling (92). Accordingly, Chm-I expression is upregulated by Sox9 during chondrogenesis in the avascular cartilage, but it is not present in the late hypertrophic and calcified zones leading to final osteogenesis (88, 91). In line with this, Chm-I deficient mice showed a significant increase in bone mineral density and lowered resorption (92). Moreover, the basal cartilage-like profile of the normal and mature valve is lost during AVS, though in vitro assays have shown an early up-regulation of Sox9 followed by Runx2 and ALP up-regulation (94). This may indicate an early chondroblast intermediate stage before the down-regulation of Sox9 and Cmh-I. Noteworthy, angiogenic factors and abundant vascularization have been mostly co-localized in late-stage heavy calcified plaques (87, 94) with the presence of osteopontin and osteocalcin suggesting a mature mineralised ECM (90, 95) but also coinciding with the thickest remodeled tissue. Importantly, VEGF induces osteoblast proliferation and differentiation and osteoclast recruitment (96) but also inhibits calcification of ovine VICs in the presence of particular ECM compositions (97), highlighting again the regulatory importance of the ECM in modulating the action of growth factors.
According to another theory, VHD recapitulates the signaling pathways that control developmental valvulogenesis (98). For instance, Bmp2, canonical Wnt, TGF-β1, and Notch signaling occurs during the endothelial-to-mesenchymal transition (99, 100) which may induce a myofibroblast phenotype on VECs and the subsequent calcification if the signaling network activation persists in time.
Several pharmacological attempts have been made for establishing a medical treatment of CVC. The regression observed by in vivo calcification models suggests the existence of endogenous mechanisms capable of dismantling the extremely insoluble and stable calcium phosphate deposits (101). Potential strategies to revert CVC have been proposed during the last few years and reviewed by O'Neill et al. (101). Preliminary evidence suggests a beneficial effect of treating calcific VHD, but frequently in association with bone mass weakening (101). Therefore, preventing or reverting the ectopic bone-like formation in the cardiovascular territory may boost bone resorption and increase the risk of fractures, which represents a serious concern for extensive use in the elderly population. To date, surgical valve replacement (SVR) represents the only available therapeutic approach for treating VHD.
Valve replacement, specifically aortic valve replacement, represents the second commonest cardiovascular surgical procedure (102) and accounts for 10 to 20% of all cardiac surgical procedures in the US (9). A 26% increase in the number of patients undergoing aortic SVR was calculated over a 5-year period comprising 2004–2009 in Great Britain and Ireland (103). It is anticipated that the number of patients requiring SVR will be 2.93-fold increased by 2050, in less than 50 years' time. Refusing to undergo SVR is associated with poor prognosis, a significant morbidity (104, 105) and >12-fold the risk of mortality (105). More than half of the patients will die within the next 12–18 months of symptom onset (106). Risk factors, co-morbidities and patient denial are common exclusion criteria for valve replacement. According to a recent survey, about 40% of patients with severe symptomatic VHD and 70% of patients with asymptomatic VHD were not eligible for SVR (19). This heterogeneous population require therefore alternative approaches.
The introduction of SVR has improved the outcome of patients with VHD. Mechanical or biological prosthesis are the two main options for current SVR (107). Mechanical valves last longer and are still the gold standard for patients under 60 years (108), but may come with a high inherent risk of thrombosis and therefore a requirement for chronic oral anticoagulation, based on coumadin derivatives. More frequent use of biological prostheses (mainly porcine or bovine-derived), introduction of minimally-invasive implantation techniques, and better control of risk factors and complications (109, 110) have considerably improved the clinical outcome of people undergoing SVR (111). Geometrical, nano-structural and material features of the bioprosthetic valve are more similar to the native tissue. Moreover, recent bioprosthetic valve improvements have significantly lowered the age for recommended mechanical valve replacement (111, 112). Nevertheless, biological prosthesis has a relatively poor long-term durability; thus, it does not provide a definitive cure. Instead, owing to the progressive deterioration and failure of current valve substitutes, native VHD is traded for “bioprosthetic valve disease,” which entails expensive treatments, hospital readmission and reintervention (110). Structural prosthetic valve deterioration represents a major limit for durability, independently the substitute is a homograft or xenograft. Several factors contribute to this phenomenon. Animal-derived prostheses, now prevalently used because of the shortage of human valve substitutes, are subjected to decellularization procedures to prevent recipient's immune response, and are cross-linked with glutaraldehyde, to provide tensile strength and elasticity, and render them further non-immunogenic. However, improvements in pliability and tolerogenicity come at a price. In fact, elimination of VICs, which synthetize ECM proteins and possess contractile properties, deprive the valves of their unique function in such a mechanically demanding environment and makes prostheses more susceptible to degeneration. Moreover, residual fragments of devitalized VICs and VECs may act as hydroxyapatite nucleation sites and induce activation of immune responses. Atherosclerotic processes also participate in prosthetic valve remodeling, with initial accumulation of oxLDL, followed by monocyte recruitment, generation of a pro-inflammatory milieu, collagen disruption and osteogenic differentiation of resident endothelial cells (ECs) and precursor cells recruited from the circulation (113, 114). Damage of the ECM is cumulative: calcium deposits enlarge and merge, forming nodules that interfere with the bioprosthesis function. Manufacture protocols preserving ECM integrity and encouraging in vivo recellularization prolong durability (115, 116). Anti-calcifying agents are also effective (117). However, ECM disorganization and degradation remains the ultimate limiting factor in durability (118).
Pediatric or adolescent patients diagnosed with congenital valve diseases are specially challenging. The risk of prosthetic valve failure becomes relevant in these populations with a 10% rate of failure within 4 years after implantation (117) and usage of mechanical valves linked to chronic oral anticoagulation does not fit with their active lifestyle. Failure of somatic growth, repair and remodeling are also common problems of both mechanical and biological prosthesis. The ideal valve prosthesis has yet to be developed.
Landmark experimental and clinical work has demonstrated the potential of tissue engineering, which combines cells from the body with template materials, to guide the somatic growth of new tissue and correction of organ defects (119). Application of this approach has been proposed to improve the durability of cardiac prostheses and thereby optimize long-term outcomes in patients with congenital or acquired valve defects. Therefore, Tissue Engineering of Heart Valves (TEHV) has emerged as a valuable alternative for definitive treatment of VHD promising to overcome either the chronic oral anticoagulation or the time-dependent deterioration and reintervention of current mechanical or biological prosthesis, respectively and to offer a valve substitute capable to grow in a “physiologic-like” manner. In the past few decades, two main strategies have been developed to generate TEHVs. The underpinning concept for both TEHVs approaches is that patient's own cells will generate a viable and physiologically competent tissue able to withstand hemodynamic forces before (in vitro) or after (in situ) implantation. Briefly, in vitro TEHV uses various types of autologous cells, including stem/progenitor cells, that are expanded in culture, seeded on decellularized biological (120, 121) or synthetic scaffolds (122, 123) (see below), and may be conditioned in a bioreactor to ensure fast and competent “natural-like” matrix production before implantation (124). The underlying concept is that in vitro incorporation of cells shall confer prosthetic grafts with the characteristics of a living tissue that remodel in a physiologic manner and concert with cardiac and whole-body needs, withstanding the impact of degeneration and calcification. Implantation of in vitro TEHV is an appealing alternative for pediatric patients with congenital VHD requiring of SVR even two decades earlier than VHD patients with anatomically normal valves. On the other hand, in situ TEHV, aims to create an acellular biodegradable scaffold which gradually transforms into a living valve by recruiting endogenous cells upon orthotopic implantation (125–127). An interesting combination of the in vitro and in situ approaches is represented by tissue-engineered matrixes (TEMs), which are usually made of autologous vascular cell- or fibroblast-derived ECM/fibrin gel sheets undergoing a decellularization process before implantation. TEMs are supposed to provide a more natural substrate for homing of the recipient's cells (128, 129). A similar strategy to produce a natural ECM graft is the in vivo TEHV by which synthetic non-degradable molds are implanted at sub-cutaneous level and expected to produce a collagen-rich, non-immunogenic, harvestable, and implantable fibrotic capsule (108). In the in vitro procedures and TEM, a balance between the extent of decellularization and conservation of the native properties of the ECM must be reached to avoid undesired alterations of biomechanical and hydrodynamic properties. The in situ approach is instead totally reliant on the endogenous capacity of the hosting organism to mobilize and incorporate the right cells, which may be negated by a disease-associated alteration in cell behavior (130–133). The two main strategies, in vitro vs. in situ, are briefly summarized in Table 1.
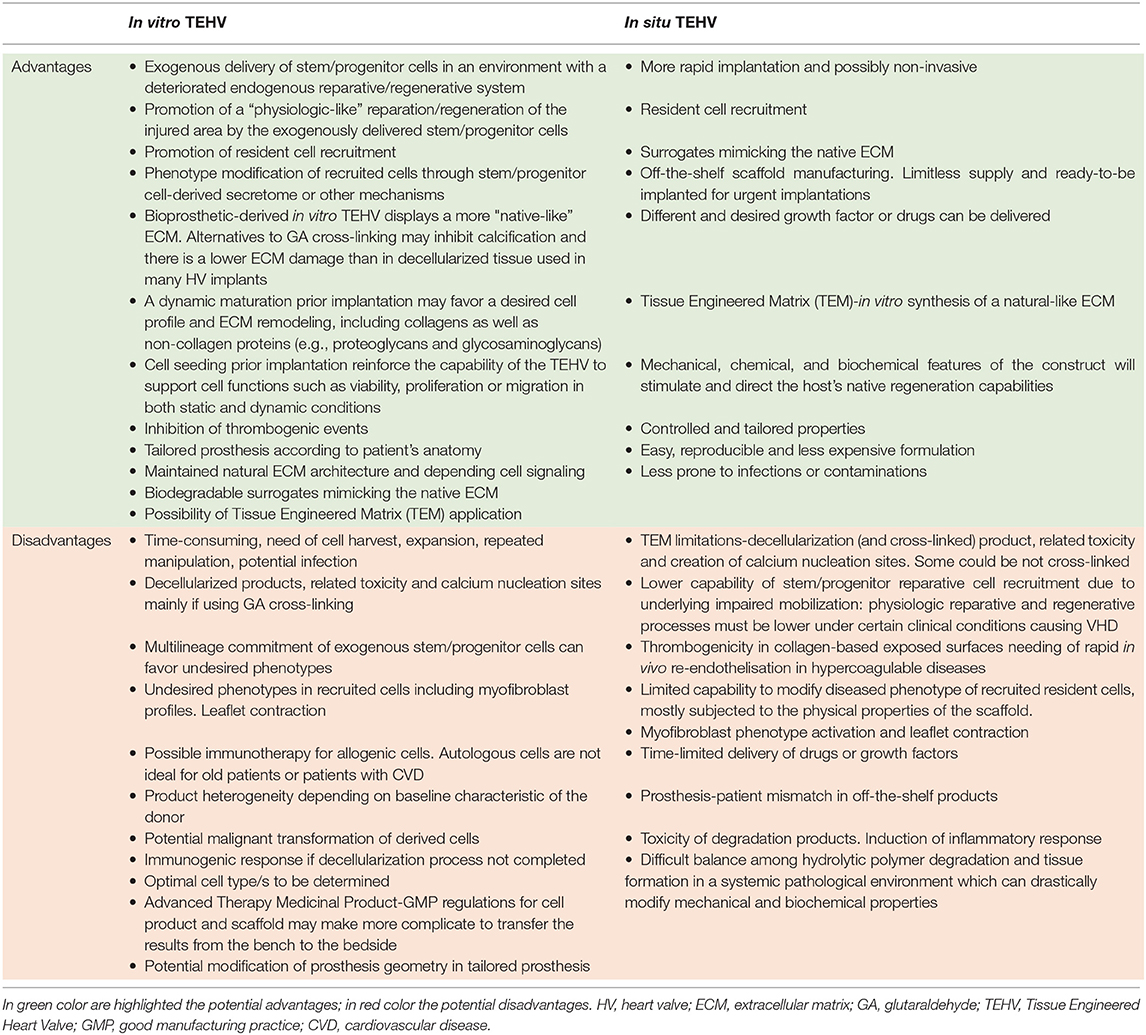
Table 1. Advantages and disadvantages of in vitro and in situ TEHV.
A three-dimensional scaffold and the correct choice of cells are the cornerstone elements to consider generating a living valve substitute. A plethora of approaches and techniques have been established on TEHV and that has been recently and extensively reviewed elsewhere (134–136). Unique valve mechanobiology features and implications in the development and design of TEHV has been nicely reviewed by Schoen (137). Furthermore, utilization of different cell sources may confer of additional properties to the valve substitute which may, or may not, be desirable in the VHD environment. The cell of choice to be seeded in in vitro TEHV should sense and perform optimal adaptative responses to environmental changes. All that must be considered before moving from the bench to the bedside and is further discussed below.
Intuitively, the best scaffold/graft to comply with all the requirements of TEHV would be the native aortic valve-derived ECM or similar biological composites. Commercially available bioprostheses are being currently tested as cell carriers thanks to significant improvements in decellularization protocols, which include novel cross-linking procedures to increase pliability while avoiding calcification (138). For instance, glutaraldehyde fixation replaced by heparin has shown to ameliorate valve prosthetic calcification rates probably by blocking calcium phospholipid binding sites as well as to inhibit thrombosis (108, 139). Other cross-linking alternatives are the reduction of free amine groups and targeting free aldehydes by using reducing agents capable of forming Schiff bases which may allow for glycosaminoglicans and elastin stabilization, avoid collagen deformation and inhibition of calcium binding (138).
As anticipated above, the typical in vitro approach is to seed cells on a scaffold, and induce differentiation and ECM synthesis. However, it has been demonstrated that respiring cells on the scaffold periphery and the size of the scaffold can restrict oxygen and nutrients availability at the center of the tissue, leading to areas of necrosis and degeneration(140). Different procedures have been proposed to circumvent this problem, including mechanical compression (141) and flow perfusion (142). The implementation of dynamic systems, such as use of bioreactors, before implantation into the recipient host may give better results than static systems and considerably contribute to maintain viability of the three-dimensional TEHVs supporting its maturation. Mature grafts/scaffolds might be easier to integrate into the recipient's heart and to acquire the definitive native features of a living valve (143–145) (Figure 3). However, dynamic culture conditions can also negatively impact cell differentiation and tissue formation. Since two intermediates products are combined in the final Investigational Medicinal Product [definition provided in Directive 2001/20/EC, Article 2 (d)], it is vital the latter is checked for quality and quantity before implantation.
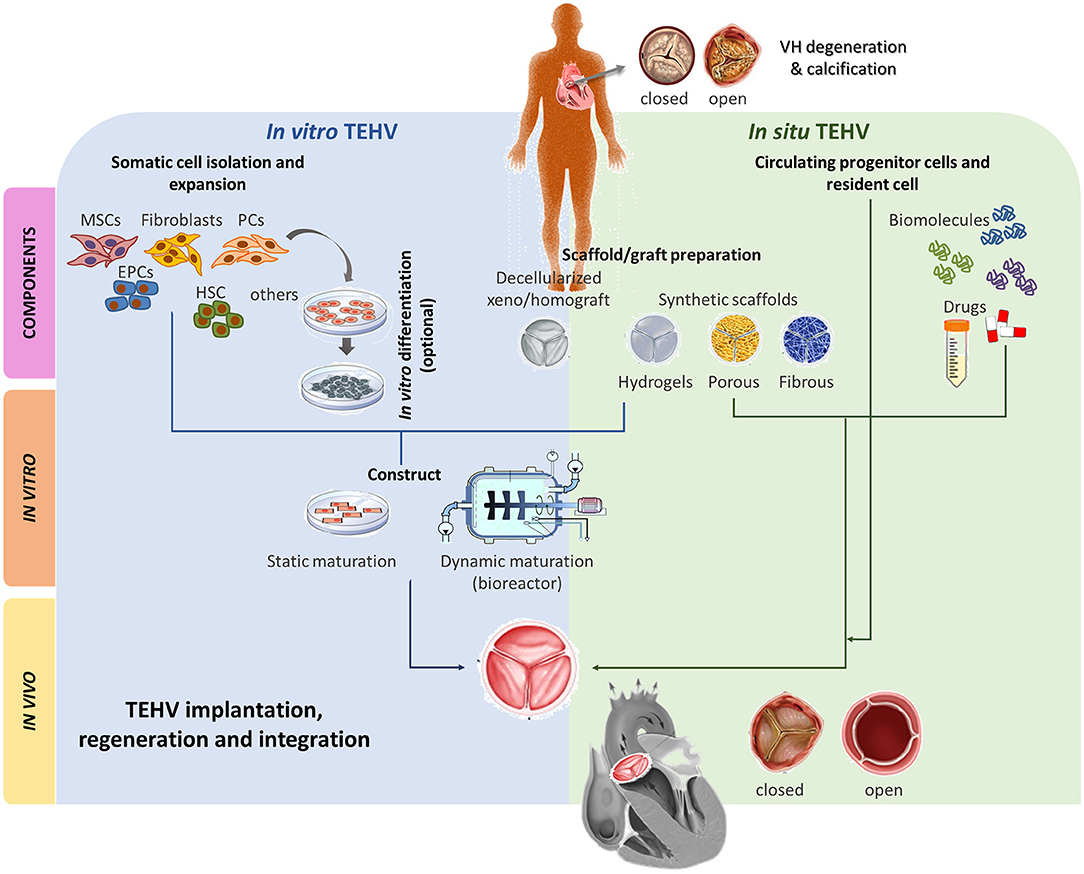
Figure 3. Main TEHV approaches comparison. Two main TEHV strategies are being explored; in vitro and in situ. In vitro TEHV consist of the combination of, ideally, autologous cells (e.g., MSCs, fibroblasts, EPCs, HSC, PCs or others) that may, or may not, be previously differentiated into the target cell. Those cells are seeded on a scaffold or graft preparation and statically or dynamically matured prior orthotopic implantation. On the other hand, in situ TEHV is based on the use of an acellular scaffold/graft capable to recruit endogenous cells which will remodel and integrate. Both in vitro and in situ meet at the scaffold/graft preparation step. Scaffold/grafts can be based on decellularized xeno/homografts or synthetic totally/partially biodegradable materials (hydrogels, porous, or fibrous). Both approaches can be enriched with biomolecules or drugs stimulating desired cell recruitment and phenotypes or inhibiting deleterious events such as calcification, angiogenesis, etc of the TEHV. After implantation, the TEHV must be able to regenerate and integrate into the recipient host and be native-like functional. MSCs, mesenchymal stromal cells; EPCs, endothelial progenitor cells; HSC, hematopoietic stem cells; PCs, pericytes.
Cells represent the biological component of the TEHV, e.g., the ones supposed to confer living properties. The optimal cell type for valve engineering should be non-immunogenic and able to maintain its specialized function or gain such a specialization through differentiation. Autologous cells are the first choice, but they show significant dysfunctions if obtained from old patients or patients with cardiovascular diseases (146), while allogenic cells might be immunogenic (147). Induced pluripotent stem cells (iPSCs) generated by reprogramming somatic cells would be the ultimate solution for patient-tailored therapy but there are still several concerns (148). Hence, differentiated cells or progenitor cells, including VICs, VECs, MSCs, BM-mononuclear cells (MNCs), fibroblasts or EPCs, remain a safer option thus far. We will go through some examples (144, 149–155) here which are further summarized in Table 2.
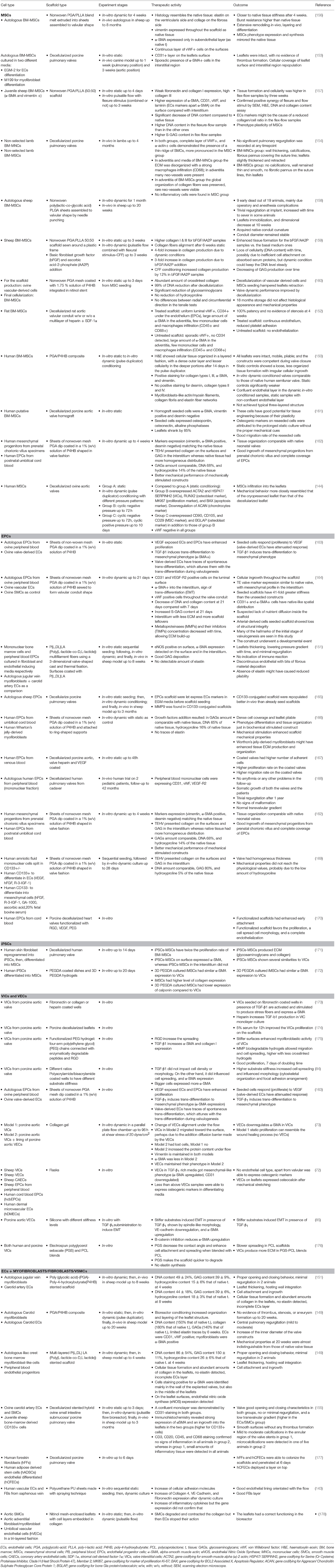
Table 2. Different cell type candidates for in vitro TEHV.
Two main scopes are followed for graft repopulation: (i) to recreate the internal biologic environment of a valve and (ii) to provide them with an EC coverage. Short-term follow-up studies in sheep and primates showed the potential advantage of repopulating the core of scaffolds/grafts with VICs or MSCs (156, 179–181). However, other aspects such as the prone differentiation into myofibroblast or osteoblast phenotypes must be considered and is discussed below for the main cell sources explored so far.
MSC with different origins seems to be a consistent choice for the TEHVs cellularization since the VIC represents a heterogenous population of cells sharing a mesenchymal ancestor. Moreover, the VIC shares phenotypic commonalities with VSMC and fibroblasts, that could be achieved by MSC differentiation as confirmed by antigenic expression (21, 22, 156, 157). In addition, MSCs are easy to be harvested and expanded in vitro, and there are multiple tissue sources (e.g., bone marrow, adipose tissue, peripheral blood, umbilical cord blood, umbilical cord, and placenta or amniotic fluid). Both animal and human studies support the immunoprivileged state of the MSC and evidences their unique immunomodulatory characteristics. Accordingly, the MSC is nowadays the preferred cell of choice for in vitro TEHV and the several studies in animal models account for that (147, 182). Since MSCs are progenitor stem cells able to differentiate in all the valvular cell phenotypes, they can overcome the issue of primary cells harvested from old and sick patients (147). Finally, unlike other stem cells, MSCs do not develop teratomas and there are not ethical concerns as for the embryonic stem cells (ESCs) (183).
MSC-bioengineered valves differentiated through conditioning in biomimetic and dynamic environments have shown physiologic profiles in terms of ECM composition (e.g., higher amounts of collagen type I and III), mechanical properties and VIC/myofibroblast markers expression (147, 183). These studies have also shown the influence of chemical, flow, and flexural stimuli on the cell phenotype expression and synthesis capabilities, also demonstrating an enhanced outcome of their synergic action. Moreover, MSC preserve their phenotype plasticity, being able to express endothelial or mesenchymal markers in response to different biochemical and mechanical stimuli (157). No evidence of glycosaminoglycans synthesis has been demonstrated by MSCs, but that issue could be circumvented by an additional stimulation with concomitant insulin and hypoxic conditioning (184). In vivo experiments performed on rat, sheep, and canine models have confirmed the positive in vitro outcomes. MSCs differentiation and different secretion of ECM components were mirroring the native structure (156). Cell labeling of implanted cells has also suggest their active collaboration in tissue regeneration (152, 153). However, performance issues, such as regurgitation and leaflets mobility restriction, have been found in some MSC-bioengineered substitutes (158).
Animal model, mainly pig and sheep, can give some help to test the valve ability to withstand some aspect of the immune reaction and calcification. Sheep model are preferred because of their valve anatomy similarity with humans, and the slower growth pace compared to the pigs. Moreover, juvenile sheep models represent the worst-case scenario to evaluate calcification because of the high level of calcium and phosphorous in the serum. However, they do not consider all the peculiarity of the human immune system. For instance, sheep have reduced platelet activity than the humans (185). Therefore, many in vivo studies ended up in failures when translated to clinical practice (186, 187). Comparison studies attempted to determine the superiority of available cells products, in particular, MSCs vs. other cell types, such as BM-mononuclear cells (MNCs) or CD133+ aortic-derived cells (144, 154, 188). An interesting report from Vincentelli et al. (154) compared the efficacy of MSC- or MNC-engineered TEHVs implanted in lambs. Both cell groups promoted the re-endothelization of the TEHV through recruitment by the recipient's ECs after 4 months implantation. However, MNC-seeded valves caused leaflet thickening, retraction, inflammation and calcification, while the MSC-seeded valves displayed a αSMA+ cellularization with no signs of calcification.
Controversial results have been observed in humans and the therapeutic use of MSCs (188). Modest or null benefit has been documented in clinical trials using BM-MSCs (189, 190). In the context of VHD, experiments are mostly limited to in vitro, static or dynamic, TEHV cellularization (185, 186). The experimental results of human in vitro studies are aligned with the animal-derived MSCs in vitro models. Dynamic culture enhanced the construct mechanical properties, which were comparable to the native valves; tissue formation and organization; endothelialization; and native-like markers patterns (150). Different mechanical stimuli, or different intensity of them, promote several MSC behaviors such as migration or differentiation (144). For example, media enriched with VEGF and high shear stress leads to endothelial phenotype differentiation (191, 192). Concerns also surround the stability of the acquired phenotypes and the potential unwanted fibrotic overgrowth causing retraction and regurgitation of the TEHV. In fact, osteogenic markers (such as alkaline phosphatases, osteopontin, and osteonectin) have been found expressed in the implanted graft, suggesting the susceptibility to prosthetic valve calcification (132, 147). Pro-osteogenic cells may influence resident VICs to acquire similar properties, thus raising concerns about their transplantation into pro-calcifying environments.
Similarly to the MSCs, the endothelial progenitor cells (EPCs) have broaden differentiation potential and can be supplied by non-surgical procedures, since they can be isolated from peripheral and umbilical cord blood (147). EPCs are particularly interesting because of their capability to differentiate into EC and to contribute to vascular regeneration and development as well as to neovascularization processes after limb or myocardial ischemia (193). Moreover, EPCs can undergo EMT processes to acquire a VIC-like phenotype under determinate stimuli (e.g., growth factors such as TGFβ1 or mechanical conditioning) and therefore offering the possibility of a complete valve regeneration with a single cell type (163, 194, 195). However, EPCs may also contribute to different pathological stages including cancer and diabetes (196, 197) and the ideal antigenic profile remains controversial (165).
In vitro and in vivo experiments have demonstrated the ability of the EPCs to colonize the whole TEHV. Importantly, EPCs express both endothelial and mesenchymal lineage markers (CD31 and α-SMA, respectively), spatially arranged in a native-like fashion with a concomitant expression of MMPs and TIMPs, suggesting an active remodeling which recapitulates a developmental-like process (198). The result is a higher mechanical performance than the one achieved by other cell sources (164). No calcification or thrombi were noticed in all the reported studies. Furthermore, EPCs have been implanted on valve leaflets in animal models leading to a reduced infection and graft failure (199). However, the lack of leaflet pliability and discontinuous endothelialization raise concerns about the EPC-based bioengineered devices (151).
In vitro studies conducted on human-derived EPCs show similar results to those find in vitro and in vivo using animal sources. Human EPCs derived from umbilical cord blood have been co-seeded with Wharton's Jelly-derived myofibroblasts. Biochemical and mechanical stimulations are also necessary to promote the desired native-like mechanical organization, phenotype determination, and functionalization (166, 167, 170). Human EPCs harvested from umbilical cord blood have been also combined with prenatally harvested chorionic villus-derived MSCs to provide a tissue engineered prosthesis for pediatric patients. A good phenotype and mechanical properties of the resulting prosthetic valve was achieved (162).
Clinical studies of re-endothelialization have confirmed the feasibility of correcting pulmonary valve defects using allografts engineered with vein-derived autologous ECs or EPCs (200). To the best of our knowledge, the only one human in vivo study took place in Republic of Moldova in 2002 and was published on Circulation in 2006. Two pediatric patients affected by tetralogy of Fallot, underwent a pulmonary valve replacement with decellularized human pulmonary valves which have been repopulated by autologous MNCs from their peripheral blood. After valve recellularization, the cells were characterized as EPCs. Throughout the follow-up duration (3.5 years), the patients recovered well. Themselves and their prosthetic valves had a somatic growth, and there was no complication whatsoever. Only a trivial regurgitation was reported (168).
In some cases, adult stem cells are not enough proliferative due to diseases or patient old age. A good alternative might be the iPSC, which are autologous reprogrammed fibroblasts, able to differentiate in MSCs and ECs. Using this cell type, ethical or need for compatible stem cells issues are avoided. Simpson et al. managed to reprogram skin fibroblast into iPSC, and then produce iPSC -derived MSCs (iPSC -MSCs) and iPSC -derived ECs (iPSC -ECs). iPSC -MSCs were seeded on decellularized human pulmonary valves, resulting in valve repopulation and ECM production (171). Compared with MSCs, the iPSC -MSCs have higher proliferation potential and have some expression pattern similarities with VICs (172). However, there are still safety concerns about IPCSs. Recent reports have emphasized the pitfalls of iPSC technology, including the potential for genetic and epigenetic abnormalities, tumorigenicity, and immunogenicity (148).
TEHV seeded with human mitral or aortic VICs can generate a valvular tissue with mechanical properties similar to the naive human aortic valve (176) while retaining native antigenic expression (201, 202). Similar results have been reported for human VICs isolated from sclerotic valves (146). However, several models using native VICs and VECs to produce in vitro TEHV aim to model the pathological development of prosthetic degeneration, calcification and fibrosis by mimicking native-like environments rather than to design prosthetic solutions (203, 204). Other studies have used native resident cells to establish tissue engineering and cell culture protocols (147, 176, 205). For instance, VICs seeded on heparin-coated wells in presence of TGF-β1 undergo to activation into myofibroblast phenotypes with enhanced synthetic and contractile activity, producing stress fibers and expressing α-SMA, all of them typical markers of active VICs (173). Mechanical properties of the scaffolds are also tested on native resident cells to better understand the mechanobiology of the VIC. Stiffer surfaces enhance their myofibroblastic activity, their density, and spreading (84). Conducting those studies is especially relevant to develop methods capable to obtain a temporary myofibroblast phenotype. Myofibroblasts are known to exert wound healing function but its persistence may promote fibrosis and calcification causing prosthetic valve disease (81, 83) as well as prosthetic retraction and regurgitation due to an excessive contractibility (206).
A priori, more interesting for therapeutic applications would be the VEC given the phenotypic peculiarities mentioned above. Those unique properties allow the VEC to provide an antithrombogenic surface, replenish the VIC population toward EMT processes and regulate VIC phenotype, in response to mechanical and biochemical stimuli. Indeed, Butcher et al. described the capability of the VEC to maintain quiescence of bioengineered VICs (73). However, difficult harvest and their degeneration can lead to valve failure due to neo-vascularization, infiltration of inflammatory agents, or lipid deposition, amongst the many (207). In addition, VECs undergoing to EMT can express osteogenic markers, not reported in other EC populations (72, 85). Although VECs can trigger valve dysfunction, providing an endothelial lining is a main concern when designing TEHV. That justify the use of primary endothelial cells (ECs) or EPCs in many studies (147).
A way to attempt to reproduce the valvular structure is through using cell types whose lineage is close to VICs and VECs including ECs, fibroblasts (FBs) and VSCM. Sequential seeding of FBs and human adipose-MSC-derived endothelial cells were able to colonize and penetrate into animal decellularized heart valves (177). However, activation of FBs and VSMC into myofibroblasts has been also reported leading to expression of cytokines, and scaffold degradation and contraction (143, 178). Incorporation of ECs lining seems to stop it (178). Those approaches are getting us close to understand the mechanisms to switch off and on undesired phenotypes by controlling the mechanical and biochemical signals of the designed TEHV (208). In line with in vitro results, TEHV bioengineered with myofibroblasts and ECs in sheep for 8–20 weeks was associated with leaflet thickening and moderate regurgitation (149, 151). Incipient calcification and regurgitation have been also found in BM-derived SMC valve substitutes implanted in sheep (155).
The utility of using native resident cells stands also in establishing the suitability of scaffolds designated for in situ TEHV and their capability to accommodate the recruitment of resident cells and a proper phenotype.
There are other cell products with potential application in the in vitro TEHV with minor or not assessment so far. For example, two fractions have been differentially characterized among the progenitor cells isolated from the amniotic fluid. It has been shown that CD133+ fraction of the mononuclear cells in the amniotic fluid can acquire endothelial phenotype, whereas the CD133− fraction can differentiate into myofibroblast-like cells. Those cells are especially relevant for the preparation of cellularized valves before birth (169). An unexplored alternative for in vitro TEHV in the adult is the suitability of heterogeneous perivascular stem/progenitor cells described in the vascular niche by our group and others and considered native ancestors of heterogenous MSCs (179, 209). The therapeutic potential of those perivascular cells in the cardiovascular regenerative medicine has been already demonstrated (210–212). Moreover, adventitial perivascular progenitor cells (APC) derived from cardiac surgery saphenous vein leftovers have properties, which make them a potential candidate for regenerative medicine (145), including the suitability for cellularization of xenografts (213) and application into myocardial ischemic models (214–216). The latter has shown the superiority of the APC to keep their specialized function upon implantation without acquiring undesired phenotypes (214). Importantly, intramyocardially transplanted APCs did not induce calcification, in contrast with BM-MSCs. It remains unclear if these properties are peculiar to APCs. In vivo and in vitro studies demonstrated the capability of other pericytes to contribute to the pathogenesis of vascular calcification toward osteogenesis and angiogenesis promotion from the adventitial vasa vasorum and the intimal layer (217). However, no intact perivascular coat has been described yet around the new vessels irrigating the growing of the advanced plaque-like tissue (87) and BM-MSC-derived endothelial cells and adventitial Sca1+ cells, rather than derived from adventitial vasa vasorum, have been described in association with atheromatous plaque progression (218, 219). Further evidences are needed to state that the APC is a cell of choice for in vitro TEHV. Adding new cell sources may bear the risk of adding more approaches to the several techniques and approaches found in the literature.
In order to improve graft durability, additional aspects must be considered. (A) Cell-graft interactions. These features are inherent to the cells, but also depend on proper interactions between the “right cell” and “right prosthesis.” For instance, VEGF significantly inhibits the formation of calcium nodules when ovine VICs are grown on collagen, fibronectin, and laminin (97), while may confer osteoblast-like phenotypes using other substrates, suggesting that providing “right” specific ECM and/or growth factors may protect VICs from calcification and degeneration. Combining cells and prostheses already available in a clinical format may provide the means for swift exploitation. Thus it may be advantageous to test them first. (B) Cell accessibility and scalability. Tissues that are easily accessible as a source of candidate cell products represent the ideal solution. However, thanks to advances in cardiac imaging, it is now possible to obtain tissue specimens for cardiac cell harvesting with minimally invasive procedures. Additionally, expansion and storage protocols of various cell types are well established, thus allowing potential use of diverse cell populations for TE. (C) Tissue specificity. It is thought that progenitor cells and differentiated cells maintain an epigenetic memory of the source tissue. In line with this concept, cardiac progenitor cells, VICs and VECs may represent the logic solution for disease conditions that require reparative cardiomyogenesis or valve replacements. (D) Paracrine activity. As discussed above, cells seeded onto the graft/scaffold represent a source of biomolecules, favoring re-endothelialization, new native-like ECM (138). In addition, the presence of cells can decrease the degradation rate of the constituent scaffold ECM resulting in enhanced preservation of its mechanical properties (176) and eventually against prosthetic calcification. (E) Cell retention on the implanted scaffolds. This important aspect has not been extensively assessed, because of the difficulties in tracking cells incorporated into the graft. A study investigating TEHVs made by autologous canine BM-MSCs, seeded on allogenic or porcine-derived xenogeneic pulmonary valves demonstrated cell retention of 1 and 3 weeks, respectively (153). It remains uncertain whether the pathologic and pro-calcifying environment found in the aortic wall contiguous to the prosthetic valve implantation site may affect the retention and “right” phenotype preservation of cells used for TEHV and that needs to be studied.
A wide range of approaches is still being explored in the manufacture of TEVHs, based on established technologies and novel cutting-edge techniques. Due to many patients targeted by TE for substitution of cardiac valves, the financial volume for these technologies/products is substantial. A market forecast for tissue engineered products indicates the total value will surpass $4.8 billion by 2028.
Several publications with promising in vivo and in vitro results have underestimated the effects of the “minor outcomes” reported and that could lead to valve substitute degeneration in a next generation of the current “biologic prosthetic valve disease.” Active native-like ECM deposition and even valvulogenesis-like events must be desirable during the process of valve substitute production, but those must be abolished thereafter to avoid excessive fibrosis, contraction, retraction, degeneration, and calcification of the valve substitute. On this regard, the ideal cell type of choice has yet to be determined and more research is needed to provide the best therapeutic alternative to both adult and congenital VHD. Besides, results from experimental modeling performed with resident native cells seeded on different types of scaffolds, show that scaffold compositions or designs still need to be substantially improved to achieve the correct cell behavior in a diseased environment. Further research on this regard, combined with a better knowledge of the pathology, including the factors triggering myofibroblast phenotype perpetuation, osteoclast recruitment in the calcific valve or the exquisite behavior of the VEC, will significantly contribute to successfully develop valve substitutes. Innovative technologies are required to meet specific, quantitative standards of safety and performance. Similar standards will have to be developed to enable routine clinical use and customized fabrication of TEHVs. While a large number of options have been tested in animal models, more work is warranted before the use of TEHVs can be proposed as a better therapeutic option than available prostheses.
EJ reviewed the literature, drafted the manuscript, and prepared the figures and tables. MF reviewed the literature, drafted the manuscript, and prepared the tables. GA critically revised the manuscript. PM reviewed the literature, drafted, and critically revised the manuscript. All the authors have approved the final submission of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
EJ holds a postdoctoral grant funded by British Heart Foundation (BHF) (Grant number PG/15/95/31853). MF also holds a PhD studentship funded by BHF. The Bristol Heart Institute is part of the BHF Centre of Excellence in Vascular Regeneration.
1. Madhavan MV, Tarigopula M, Mintz GS, Maehara A, Stone GW, Genereux P. Coronary artery calcification: pathogenesis and prognostic implications. J Am Coll Cardiol. (2014) 63:1703–14. doi: 10.1016/j.jacc.2014.01.017
2. Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. (1999) 341:142–7. doi: 10.1056/NEJM199907153410302
3. Maganti K, Rajamannan N. Slowing the progression of aortic stenosis. Curr Treat Options Cardiovasc Med. (2008) 10:18–26. doi: 10.1007/s11936-008-0003-3
4. Cheng GC, Loree HM, Kamm RD, Fishbein MC, Lee RT. Distribution of circumferential stress in ruptured and stable atherosclerotic lesions. A structural analysis with histopathological correlation. Circulation (1993) 87:1179–87. doi: 10.1161/01.CIR.87.4.1179
5. Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation (2001) 103:1522–8. doi: 10.1161/01.CIR.103.11.1522
6. Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, et al. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. (2006) 47:1707–12. doi: 10.1016/j.jacc.2006.02.040
7. Otto CM. Calcific aortic stenosis–time to look more closely at the valve. N Engl J Med. (2008) 359:1395–8. doi: 10.1056/NEJMe0807001
8. Iung B, Baron G, Butchart EG, Delahaye F, Gohlke-Barwolf C, Levang OW, et al. A prospective survey of patients with valvular heart disease in Europe: the Euro heart survey on valvular heart disease. Eur Heart J. (2003) 24:1231–43. doi: 10.1016/S0195-668X(03)00201-X
9. Iung B, Vahanian A. Epidemiology of valvular heart disease in the adult. Nat Rev Cardiol. (2011) 8:162–72. doi: 10.1038/nrcardio.2010.202
10. O'Brien KD. Epidemiology and genetics of calcific aortic valve disease. J Investig Med. (2007) 55:284–91. doi: 10.2310/6650.2007.00010
11. Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular health study. J Am Coll Cardiol. (1997) 29:630–4. doi: 10.1016/S0735-1097(96)00563-3
12. Supino PG, Borer JS, Preibisz J, Bornstein A. The epidemiology of valvular heart disease: a growing public health problem. Heart Fail Clin. (2006) 2:379–93. doi: 10.1016/j.hfc.2006.09.010
13. Rajamannan NM. Bicuspid aortic valve disease: the role of oxidative stress in Lrp5 bone formation. Cardiovasc Pathol. (2011) 20:168–76. doi: 10.1016/j.carpath.2010.11.007
14. Lindroos M, Kupari M, Heikkila J, Tilvis R. Prevalence of aortic valve abnormalities in the elderly: an echocardiographic study of a random population sample. J Am Coll Cardiol. (1993) 21:1220–5. doi: 10.1016/0735-1097(93)90249-Z
15. Rosenhek R, Zilberszac R, Schemper M, Czerny M, Mundigler G, Graf S, et al. Natural history of very severe aortic stenosis. Circulation (2010) 121:151–6. doi: 10.1161/CIRCULATIONAHA.109.894170
16. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet (2006) 368:1005–11. doi: 10.1016/S0140-6736(06)69208-8
17. Marciniak A, Glover K, Sharma R. Cohort profile: prevalence of valvular heart disease in community patients with suspected heart failure in UK. BMJ Open (2017) 7:e012240. doi: 10.1136/bmjopen-2016-012240
18. Otto CM, Burwash IG, Legget ME, Munt BI, Fujioka M, Healy NL, et al. Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome. Circulation (1997) 95:2262–70. doi: 10.1161/01.CIR.95.9.2262
19. De Sciscio P, Brubert J, De Sciscio M, Serrani M, Stasiak J, Moggridge GD. Quantifying the shift toward transcatheter aortic valve replacement in low-risk patients: a meta-analysis. Circ Cardiovasc Qual Outcomes (2017) 10:e003287. doi: 10.1161/CIRCOUTCOMES.116.003287
20. d'Arcy JL, Prendergast BD, Chambers JB, Ray SG, Bridgewater B. Valvular heart disease: the next cardiac epidemic. Heart (2011) 97:91–3. doi: 10.1136/hrt.2010.205096
21. Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension (2001) 38:938–42. doi: 10.1161/hy1001.096358
22. McClelland RL, Jorgensen NW, Budoff M, Blaha MJ, Post WS, Kronmal RA, et al. 10-year coronary heart disease risk prediction using coronary artery calcium and traditional risk factors: derivation in the MESA (multi-ethnic study of atherosclerosis) with validation in the HNR (Heinz Nixdorf Recall) study and the DHS (Dallas Heart Study). J Am Coll Cardiol. (2015) 66:1643–53. doi: 10.1016/j.jacc.2015.08.035
23. Budoff MJ, McClelland RL, Nasir K, Greenland P, Kronmal RA, Kondos GT, et al. Cardiovascular events with absent or minimal coronary calcification: the Multi-Ethnic Study of Atherosclerosis (MESA). Am Heart J. (2009) 158:554–61. doi: 10.1016/j.ahj.2009.08.007
24. Seemayer TA, Thelmo WL, Morin J. Cartilaginous transformation of the aortic valve. Am J Clin Pathol. (1973) 60:616–20. doi: 10.1093/ajcp/60.5.616
25. Hunt JL, Fairman R, Mitchell ME, Carpenter JP, Golden M, Khalapyan T, et al. Bone formation in carotid plaques: a clinicopathological study. Stroke (2002) 33:1214–9. doi: 10.1161/01.STR.0000013741.41309.67
26. Ferda J, Linhartova K, Kreuzberg B. Comparison of the aortic valve calcium content in the bicuspid and tricuspid stenotic aortic valve using non-enhanced 64-detector-row-computed tomography with prospective ECG-triggering. Eur J Radiol. (2008) 68:471–5. doi: 10.1016/j.ejrad.2007.09.011
27. Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP 3rd, Guyton RA, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American college of cardiology/American heart association task force on practice guidelines. J Am Coll Cardiol. (2014) 63:2438–88. doi: 10.1016/j.jacc.2014.02.537
28. Maganti K, Rigolin VH, Sarano ME, Bonow RO. Valvular heart disease: diagnosis and management. Mayo Clin Proc. (2010) 85:483–500. doi: 10.4065/mcp.2009.0706
29. Rosenhek R, Binder T, Porenta G, Lang I, Christ G, Schemper M, et al. Predictors of outcome in severe, asymptomatic aortic stenosis. N Engl J Med. (2000) 343:611–7. doi: 10.1056/NEJM200008313430903
30. Kadem L, Rieu R, Dumesnil JG, Durand LG, Pibarot P. Flow-dependent changes in Doppler-derived aortic valve effective orifice area are real and not due to artifact. J Am Coll Cardiol. (2006) 47:131–7. doi: 10.1016/j.jacc.2005.05.100
31. Stankovic Z, Allen BD, Garcia J, Jarvis KB, Markl M. 4D flow imaging with MRI. Cardiovasc Diagn Ther. (2014) 4:173–92. doi: 10.3978/j.issn.2223-3652.2014.01.02
32. Burris NS, Hope MD. 4D flow MRI applications for aortic disease. Magn Reson Imaging Clin N Am. (2015) 23:15–23. doi: 10.1016/j.mric.2014.08.006
33. Itatani K, Miyazaki S, Furusawa T, Numata S, Yamazaki S, Morimoto K, et al. New imaging tools in cardiovascular medicine: computational fluid dynamics and 4D flow MRI. Gen Thorac Cardiovasc Surg. (2017) 65:611–21. doi: 10.1007/s11748-017-0834-5
34. Hope MD, Hope TA, Crook SE, Ordovas KG, Urbania TH, Alley MT, et al. 4D flow CMR in assessment of valve-related ascending aortic disease. JACC Cardiovasc Imaging (2011) 4:781–7. doi: 10.1016/j.jcmg.2011.05.004
35. Geiger J, Rahsepar AA, Suwa K, Powell A, Ghasemiesfe A, Barker AJ, et al. 4D flow MRI, cardiac function, and T1 -mapping: association of valve-mediated changes in aortic hemodynamics with left ventricular remodeling. J Magn Reson Imaging (2018) 48:121–31. doi: 10.1002/jmri.25916
36. Vasanawala SS, Hanneman K, Alley MT, Hsiao A. Congenital heart disease assessment with 4D flow MRI. J Magn Reson Imaging. (2015) 42:870–86. doi: 10.1002/jmri.24856
37. Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M Jr, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. (1990) 15:827–32. doi: 10.1016/0735-1097(90)90282-T
38. Sandfort V, Bluemke DA. CT calcium scoring. History, current status and outlook. Diagn Interv Imaging (2017) 98:3–10. doi: 10.1016/j.diii.2016.06.007
39. Masuda Y, Naito S, Aoyagi Y, Yamada Z, Uda T, Morooka N, et al. Coronary artery calcification detected by CT: clinical significance and angiographic correlates. Angiology (1990) 41:1037–47. doi: 10.1177/000331979004101203
40. Timins ME, Pinsk R, Sider L, Bear G. The functional significance of calcification of coronary arteries as detected on CT. J Thorac Imaging (1991) 7:79–82. doi: 10.1097/00005382-199112000-00010
41. Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and lung and blood institute aortic stenosis working group. Executive summary: calcific aortic valve disease-2011 update. Circulation (2011) 124:1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767
42. Mahabadi AA, Mohlenkamp S, Moebus S, Dragano N, Kalsch H, Bauer M, et al. The Heinz Nixdorf Recall study and its potential impact on the adoption of atherosclerosis imaging in European primary prevention guidelines. Curr Atheroscler Rep. (2011) 13:367–72. doi: 10.1007/s11883-011-0199-7
43. Budoff MJ, Takasu J, Katz R, Mao S, Shavelle DM, O'Brien KD, et al. Reproducibility of CT measurements of aortic valve calcification, mitral annulus calcification, and aortic wall calcification in the multi-ethnic study of atherosclerosis. Acad Radiol. (2006) 13:166–72. doi: 10.1016/j.acra.2005.09.090
44. Schoen FJ. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation (2008) 118:1864–80. doi: 10.1161/CIRCULATIONAHA.108.805911
45. Liu X, Xu Z. Osteogenesis in calcified aortic valve disease: from histopathological observation towards molecular understanding. Prog Biophys Mol Biol. (2016) 122:156–61. doi: 10.1016/j.pbiomolbio.2016.02.002
46. Duer MJ, Friscic T, Proudfoot D, Reid DG, Schoppet M, Shanahan CM, et al. Mineral surface in calcified plaque is like that of bone: further evidence for regulated mineralization. Arterioscler Thromb Vasc Biol. (2008) 28:2030–4. doi: 10.1161/ATVBAHA.108.172387
47. Leopold JA. Cellular mechanisms of aortic valve calcification. Circ Cardiovasc Interv. (2012) 5:605–14. doi: 10.1161/CIRCINTERVENTIONS.112.971028
48. Sawyer JR, Myers MA, Rosier RN, Puzas JE. Heterotopic ossification: clinical and cellular aspects. Calcif Tissue Int. (1991) 49:208–15. doi: 10.1007/BF02556120
49. New SE, Aikawa E. Cardiovascular calcification: an inflammatory disease. Circ J. (2011) 75:1305–13. doi: 10.1253/circj.CJ-11-0395
50. Weind KL, Ellis CG, Boughner DR. Aortic valve cusp vessel density: relationship with tissue thickness. J Thorac Cardiovasc Surg. (2002) 123:333–40. doi: 10.1067/mtc.2002.119696
51. Butcher JT, Penrod AM, Garcia AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler Thromb Vasc Biol. (2004) 24:1429–34. doi: 10.1161/01.ATV.0000130462.50769.5a
52. Butcher JT, Tressel S, Johnson T, Turner D, Sorescu G, Jo H, et al. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: influence of shear stress. Arterioscler Thromb Vasc Biol. (2006) 26:69–77. doi: 10.1161/01.ATV.0000196624.70507.0d
53. Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. (2011) 108:1510–24. doi: 10.1161/CIRCRESAHA.110.234237
54. Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. (2007) 171:1407–18. doi: 10.2353/ajpath.2007.070251
55. Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. (2004) 13:841–7.
56. Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation (2001) 104:2525–32. doi: 10.1161/hc4601.099489
57. Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation (2005) 112(9 Suppl):I229–34. doi: 10.1161/01.CIRCULATIONAHA.104.524306
58. Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, et al. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation (2002) 105:2660–5. doi: 10.1161/01.CIR.0000017435.87463.72
59. Wu HD, Maurer MS, Friedman RA, Marboe CC, Ruiz-Vazquez EM, Ramakrishnan R, et al. The lymphocytic infiltration in calcific aortic stenosis predominantly consists of clonally expanded T cells. J Immunol. (2007) 178:5329–39. doi: 10.4049/jimmunol.178.8.5329
60. Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation (2007) 115:377–86. doi: 10.1161/CIRCULATIONAHA.106.654913
61. Stemme S, Rymo L, Hansson GK. Polyclonal origin of T lymphocytes in human atherosclerotic plaques. Lab Invest. (1991) 65:654–60.
62. Rusanescu G, Weissleder R, Aikawa E. Notch signaling in cardiovascular disease and calcification. Curr Cardiol Rev. (2008) 4:148–56. doi: 10.2174/157340308785160552
63. Yetkin E, Waltenberger J. Molecular and cellular mechanisms of aortic stenosis. Int J Cardiol. (2009) 135:4–13. doi: 10.1016/j.ijcard.2009.03.108
64. Hakuno D, Kimura N, Yoshioka M, Fukuda K. Molecular mechanisms underlying the onset of degenerative aortic valve disease. J Mol Med. (2009) 87:17–24. doi: 10.1007/s00109-008-0400-9
65. Merryman WD, Youn I, Lukoff HD, Krueger PM, Guilak F, Hopkins RA, et al. Correlation between heart valve interstitial cell stiffness and transvalvular pressure: implications for collagen biosynthesis. Am J Physiol Heart Circ Physiol. (2006) 290:H224–31. doi: 10.1152/ajpheart.00521.2005
66. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. (2006) 99:1044–59. doi: 10.1161/01.RES.0000249379.55535.21
67. Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation (2008) 117:2938–48. doi: 10.1161/CIRCULATIONAHA.107.743161
68. Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation (2006) 113:1344–52. doi: 10.1161/CIRCULATIONAHA.105.591768
69. Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation (2003) 107:2181–4. doi: 10.1161/01.CIR.0000070591.21548.69
70. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. (2011) 109:697–711. doi: 10.1161/CIRCRESAHA.110.234914
71. Paranya G, Vineberg S, Dvorin E, Kaushal S, Roth SJ, Rabkin E, et al. Aortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro. Am J Pathol. (2001) 159:1335–43. doi: 10.1016/S0002-9440(10)62520-5
72. Wylie-Sears J, Aikawa E, Levine RA, Yang JH, Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscler Thromb Vasc Biol. (2011) 31:598–607. doi: 10.1161/ATVBAHA.110.216184
73. Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. (2006) 12:905–15. doi: 10.1089/ten.2006.12.905
74. Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. (2013) 60:27–35. doi: 10.1016/j.yjmcc.2013.04.001
75. Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. (2009) 47:828–34. doi: 10.1016/j.yjmcc.2009.08.008
76. Tanaka K, Sata M, Fukuda D, Suematsu Y, Motomura N, Takamoto S, et al. Age-associated aortic stenosis in apolipoprotein E-deficient mice. J Am Coll Cardiol. (2005) 46:134–41. doi: 10.1016/j.jacc.2005.03.058
77. Leskela HV, Satta J, Oiva J, Eriksen H, Juha R, Korkiamaki P, et al. Calcification and cellularity in human aortic heart valve tissue determine the differentiation of bone-marrow-derived cells. J Mol Cell Cardiol. (2006) 41:642–9. doi: 10.1016/j.yjmcc.2006.07.014
78. Hajdu Z, Romeo SJ, Fleming PA, Markwald RR, Visconti RP, Drake CJ. Recruitment of bone marrow-derived valve interstitial cells is a normal homeostatic process. J Mol Cell Cardiol. (2011) 51:955–65. doi: 10.1016/j.yjmcc.2011.08.006
79. Jover E, Silvente A, Marin F, Martinez-Gonzalez J, Orriols M, Martinez CM, et al. Inhibition of enzymes involved in collagen cross-linking reduces vascular smooth muscle cell calcification. FASEB J. 2018:fj201700653R. doi: 10.1096/fj.201700653R
80. Hutson HN, Marohl T, Anderson M, Eliceiri K, Campagnola P, Masters KS. Calcific aortic valve disease is associated with layer-specific alterations in collagen architecture. PLoS ONE (2016) 11:e0163858. doi: 10.1371/journal.pone.0163858
81. Rodriguez KJ, Masters KS. Regulation of valvular interstitial cell calcification by components of the extracellular matrix. J Biomed Mater Res A. (2009) 90:1043–53. doi: 10.1002/jbm.a.32187
82. New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. (2011) 108:1381–91. doi: 10.1161/CIRCRESAHA.110.234146
83. Kloxin AM, Benton JA, Anseth KS. In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials (2010) 31:1–8. doi: 10.1016/j.biomaterials.2009.09.025
84. Quinlan AM, Billiar KL. Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation. J Biomed Mater Res A. (2012) 100:2474–82. doi: 10.1002/jbm.a.34162
85. Zhong A, Mirzaei Z, Simmons CA. The roles of matrix stiffness and ss-catenin signaling in endothelial-to-mesenchymal transition of aortic valve endothelial cells. Cardiovasc Eng Technol. (2018) 9:158–67. doi: 10.1007/s13239-018-0363-0
86. Chalajour F, Treede H, Ebrahimnejad A, Lauke H, Reichenspurner H, Ergun S. Angiogenic activation of valvular endothelial cells in aortic valve stenosis. Exp Cell Res. (2004) 298:455–64. doi: 10.1016/j.yexcr.2004.04.034
87. Collett GD, Canfield AE. Angiogenesis and pericytes in the initiation of ectopic calcification. Circ Res. (2005) 96:930–8. doi: 10.1161/01.RES.0000163634.51301.0d
88. Yoshioka M, Yuasa S, Matsumura K, Kimura K, Shiomi T, Kimura N, et al. Chondromodulin-I maintains cardiac valvular function by preventing angiogenesis. Nat Med. (2006) 12:1151–9. doi: 10.1038/nm1476
89. Soini Y, Salo T, Satta J. Angiogenesis is involved in the pathogenesis of nonrheumatic aortic valve stenosis. Hum Pathol. (2003) 34:756–63. doi: 10.1016/S0046-8177(03)00245-4
90. Rajamannan NM, Nealis TB, Subramaniam M, Pandya S, Stock SR, Ignatiev CI, et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation (2005) 111:3296–301. doi: 10.1161/CIRCULATIONAHA.104.473165
91. Hiraki Y, Shukunami C. Chondromodulin-I as a novel cartilage-specific growth-modulating factor. Pediatr Nephrol. (2000) 14:602–5. doi: 10.1007/s004670000339
92. Nakamichi Y, Shukunami C, Yamada T, Aihara K, Kawano H, Sato T, et al. Chondromodulin I is a bone remodeling factor. Mol Cell Biol. (2003) 23:636–44. doi: 10.1128/MCB.23.2.636-644.2003
93. Mori Y, Hiraki Y, Shukunami C, Kakudo S, Shiokawa M, Kagoshima M, et al. Stimulation of osteoblast proliferation by the cartilage-derived growth promoting factors chondromodulin-I and -II. FEBS Lett. (1997) 406:310–4. doi: 10.1016/S0014-5793(97)00291-3
94. Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, et al. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS ONE (2011) 6:e27743. doi: 10.1371/journal.pone.0027743
95. Perrotta I, Moraca FM, Sciangula A, Aquila S, Mazzulla S. HIF-1alpha and VEGF: immunohistochemical profile and possible function in human aortic valve stenosis. Ultrastruct Pathol. (2015) 39:198–206. doi: 10.3109/01913123.2014.991884
96. Engsig MT, Chen QJ, Vu TH, Pedersen AC, Therkidsen B, Lund LR, et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. (2000) 151:879–89. doi: 10.1083/jcb.151.4.879
97. Gwanmesia P, Ziegler H, Eurich R, Barth M, Kamiya H, Karck M, et al. Opposite effects of transforming growth factor-beta1 and vascular endothelial growth factor on the degeneration of aortic valvular interstitial cell are modified by the extracellular matrix protein fibronectin: implications for heart valve engineering. Tissue Eng Part A. (2010) 16:3737–46. doi: 10.1089/ten.tea.2010.0304
98. Shworak NW. Angiogenic modulators in valve development and disease: does valvular disease recapitulate developmental signaling pathways? Curr Opin Cardiol. (2004) 19:140–6. doi: 10.1097/00001573-200403000-00013
99. Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. (2004) 166:359–67. doi: 10.1083/jcb.200403050
100. Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. (2005) 243:287–335. doi: 10.1016/S0074-7696(05)43005-3
101. O'Neill WC, Lomashvili KA. Recent progress in the treatment of vascular calcification. Kidney Int. (2010) 78:1232–9. doi: 10.1038/ki.2010.334
102. Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation (2005) 111:920–5. doi: 10.1161/01.CIR.0000155623.48408.C5
103. Dunning J, Gao H, Chambers J, Moat N, Murphy G, Pagano D, et al. Aortic valve surgery: marked increases in volume and significant decreases in mechanical valve use–an analysis of 41,227 patients over 5 years from the Society for Cardiothoracic Surgery in Great Britain and Ireland National database. J Thorac Cardiovasc Surg. (2011) 142:776–82e3. doi: 10.1016/j.jtcvs.2011.04.048
104. Perera S, Wijesinghe N, Ly E, Devlin G, Pasupati S. Outcomes of patients with untreated severe aortic stenosis in real-world practice. N Z Med J. (2011) 124:40–8.
105. Kojodjojo P, Gohil N, Barker D, Youssefi P, Salukhe TV, Choong A, et al. Outcomes of elderly patients aged 80 and over with symptomatic, severe aortic stenosis: impact of patient's choice of refusing aortic valve replacement on survival. QJM (2008) 101:567–73. doi: 10.1093/qjmed/hcn052
106. American College of Cardiology/American Heart Association Task Force on Practice G, Society of Cardiovascular A, Society for Cardiovascular A, Interventions, Society of Thoracic S, Bonow RO, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): developed in collaboration with the society of cardiovascular anesthesiologists: endorsed by the Society for cardiovascular angiography and interventions and the society of thoracic surgeons. Circulation (2006) 114:e84–231. doi: 10.1161/CIRCULATIONAHA.106.176857
107. Butcher JT, Mahler GJ, Hockaday LA. Aortic valve disease and treatment: the need for naturally engineered solutions. Adv Drug Deliv Rev. (2011) 63:242–68. doi: 10.1016/j.addr.2011.01.008
108. Fioretta ES, Dijkman PE, Emmert MY, Hoerstrup SP. The future of heart valve replacement: recent developments and translational challenges for heart valve tissue engineering. J Tissue Eng Regen Med. (2016). 12:e323–35. doi: 10.1002/term.2326
109. Schmitt CA. Interventional cardiology: percutaneous pulmonary valve implantation: 1-year safety and efficacy reported in German study. Nat Rev Cardiol. (2011) 8:186. doi: 10.1038/nrcardio.2011.21
110. Pibarot P, Dumesnil JG. Prosthetic heart valves: selection of the optimal prosthesis and long-term management. Circulation (2009) 119:1034–48. doi: 10.1161/CIRCULATIONAHA.108.778886
111. Rahimtoola SH. Choice of prosthetic heart valve in adults an update. J Am Coll Cardiol. (2010) 55:2413–26. doi: 10.1016/j.jacc.2009.10.085
112. Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP 3rd, Fleisher LA, et al. 2017 AHA/ACC Focused Update of the 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. J Am Coll Cardiol. (2017) 70:252–89. doi: 10.1016/j.jacc.2017.03.011
113. Shetty R, Pibarot P, Audet A, Janvier R, Dagenais F, Perron J, et al. Lipid-mediated inflammation and degeneration of bioprosthetic heart valves. Eur J Clin Invest. (2009) 39:471–80. doi: 10.1111/j.1365-2362.2009.02132.x
114. Gossl M, Khosla S, Zhang X, Higano N, Jordan KL, Loeffler D, et al. Role of circulating osteogenic progenitor cells in calcific aortic stenosis. J Am Coll Cardiol. (2012) 60:1945–53. doi: 10.1016/j.jacc.2012.07.042
115. Honge JL, Funder J, Hansen E, Dohmen PM, Konertz W, Hasenkam JM. Recellularization of aortic valves in pigs. Eur J Cardiothorac Surg. (2011) 39:829–34. doi: 10.1016/j.ejcts.2010.08.054
116. Konertz W, Dohmen PM, Liu J, Beholz S, Dushe S, Posner S, et al. Hemodynamic characteristics of the Matrix P decellularized xenograft for pulmonary valve replacement during the Ross operation. J Heart Valve Dis. (2005) 14:78–81.
117. Schoen FJ, Levy RJ. Calcification of tissue heart valve substitutes: progress toward understanding and prevention. Ann Thorac Surg. (2005) 79:1072–80. doi: 10.1016/j.athoracsur.2004.06.033
118. Sacks MS, Schoen FJ. Collagen fiber disruption occurs independent of calcification in clinically explanted bioprosthetic heart valves. J Biomed Mater Res. (2002) 62:359–71. doi: 10.1002/jbm.10293
119. O'Brien FJ. Biomaterials and scaffolds for tissue engineering. Mater Today (2011) 14:88–95. doi: 10.1016/S1369-7021(11)70058-X
120. Zafar F, Hinton RB, Moore RA, Baker RS, Bryant R 3rd, Narmoneva DA, et al. Physiological growth, remodeling potential, and preserved function of a novel bioprosthetic tricuspid valve: tubular bioprosthesis made of small intestinal submucosa-derived extracellular matrix. J Am Coll Cardiol. (2015) 66:877–88. doi: 10.1016/j.jacc.2015.06.1091
121. Ruiz CE, Iemura M, Medie S, Varga P, Van Alstine WG, Mack S, et al. Transcatheter placement of a low-profile biodegradable pulmonary valve made of small intestinal submucosa: a long-term study in a swine model. J Thorac Cardiovasc Surg. (2005) 130:477–84. doi: 10.1016/j.jtcvs.2005.04.008
122. Weber B, Dijkman PE, Scherman J, Sanders B, Emmert MY, Grunenfelder J, et al. Off-the-shelf human decellularized tissue-engineered heart valves in a non-human primate model. Biomaterials (2013) 34:7269–80. doi: 10.1016/j.biomaterials.2013.04.059
123. Syedain Z, Reimer J, Lahti M, Berry J, Johnson S, Tranquillo RT. Tissue engineering of acellular vascular grafts capable of somatic growth in young lambs. Nat Commun. (2016) 7:12951. doi: 10.1038/ncomms12951
124. Sacks MS, Schoen FJ, Mayer JE. Bioengineering challenges for heart valve tissue engineering. Annu Rev Biomed Eng. (2009) 11:289–313. doi: 10.1146/annurev-bioeng-061008-124903
125. Kluin J, Talacua H, Smits AI, Emmert MY, Brugmans MC, Fioretta ES, et al. In situ heart valve tissue engineering using a bioresorbable elastomeric implant - From material design to 12 months follow-up in sheep. Biomaterials (2017) 125:101–17. doi: 10.1016/j.biomaterials.2017.02.007
126. Capulli AK, Emmert MY, Pasqualini FS, Kehl D, Caliskan E, Lind JU, et al. JetValve: rapid manufacturing of biohybrid scaffolds for biomimetic heart valve replacement. Biomaterials (2017) 133:229–41. doi: 10.1016/j.biomaterials.2017.04.033
127. Serruys PW, Miyazaki Y, Katsikis A, Abdelghani M, Leon MB, Virmani R, et al. Restorative valve therapy by endogenous tissue restoration: tomorrow's world? Reflection on the EuroPCR 2017 session on endogenous tissue restoration. EuroIntervention (2017) 13:AA68–77. doi: 10.4244/EIJ-D-17-00509
128. Reimer J, Syedain Z, Haynie B, Lahti M, Berry J, Tranquillo R. Implantation of a tissue-engineered tubular heart valve in growing lambs. Ann Biomed Eng. (2017) 45:439–51. doi: 10.1007/s10439-016-1605-7
129. Lu T, Li Y, Chen T. Techniques for fabrication and construction of three-dimensional scaffolds for tissue engineering. Int J Nanomedicine (2013) 8:337–50. doi: 10.2147/IJN.S38635
130. Slater SC, Carrabba M, Madeddu P. Vascular stem cells-potential for clinical application. Br Med Bull. (2016) 118:127–37. doi: 10.1093/bmb/ldw017
131. Rocco KA, Maxfield MW, Best CA, Dean EW, Breuer CK. In vivo applications of electrospun tissue-engineered vascular grafts: a review. Tissue Eng Part B Rev. (2014) 20:628–40. doi: 10.1089/ten.teb.2014.0123
132. Cheung DY, Duan B, Butcher JT. Current progress in tissue engineering of heart valves: multiscale problems, multiscale solutions. Expert Opin Biol Ther. (2015) 15:1155–72. doi: 10.1517/14712598.2015.1051527
133. Shinoka T, Miyachi H. Current status of tissue engineering heart valve. World J Pediatr Congenit Heart Surg. (2016) 7:677–84. doi: 10.1177/2150135116664873
134. Zhang X, Xu B, Puperi DS, Wu Y, West JL, Grande-Allen KJ. Application of hydrogels in heart valve tissue engineering. J Long Term Eff Med Implants (2015) 25:105–34. doi: 10.1615/JLongTermEffMedImplants.2015011817
135. Zhu AS, Grande-Allen KJ. Heart valve tissue engineering for valve replacement and disease modeling. Curr Opin Biomed Eng. (2018) 5:7. doi: 10.1016/j.cobme.2017.12.006
136. Kheradvar A, Groves EM, Dasi LP, Alavi SH, Tranquillo R, Grande-Allen KJ, et al. Emerging trends in heart valve engineering: part I. Solutions for future. Ann Biomed Eng. (2015) 43:833–43. doi: 10.1007/s10439-014-1209-z
137. Schoen FJ. Morphology, Clinicopathologic correlations, and mechanisms in heart valve health and disease. Cardiovasc Eng Technol. (2018) 9:126–40. doi: 10.1007/s13239-016-0277-7
138. Mosier J, Nancy N, Parker K, Simpson CL. Calcification of Biomaterials and Diseased States (2018). doi: 10.5772/intechopen.71594
139. Yang M, Lin YH, Shi WP, Shi HC, Gu YJ, Shu YS. Surface heparin treatment of the decellularized porcine heart valve: effect on tissue calcification. J Biomed Mater Res B Appl Biomater. (2017) 105:400–5. doi: 10.1002/jbm.b.33490
140. Tristan C, Silke G, Kilian M, Malcolm D, O'Connor AJ, Geoffrey W, et al. Modelling oxygen diffusion and cell growth in a porous, vascularizing scaffold for soft tissue engineering applications. Chem Eng Sci. (2005) 60:4924–34. doi: 10.1016/j.ces.2005.03.051
141. Wernike E, Li Z, Alini M, Grad S. Effect of reduced oxygen tension and long-term mechanical stimulation on chondrocyte-polymer constructs. Cell Tissue Res. (2008) 331:473–83. doi: 10.1007/s00441-007-0500-9
142. Volkmer E, Drosse I, Otto S, Stangelmayer A, Stengele M, Kallukalam BC, et al. Hypoxia in static and dynamic 3D culture systems for tissue engineering of bone. Tissue Eng Part A. (2008) 14:1331–40. doi: 10.1089/ten.tea.2007.0231
143. Aleksieva G, Hollweck T, Thierfelder N, Haas U, Koenig F, Fano C, et al. Use of a special bioreactor for the cultivation of a new flexible polyurethane scaffold for aortic valve tissue engineering. Biomed Eng Online (2012) 11:92. doi: 10.1186/1475-925X-11-92
144. Converse GL, Buse EE, Neill KR, McFall CR, Lewis HN, VeDepo MC, et al. Design and efficacy of a single-use bioreactor for heart valve tissue engineering. J Biomed Mater Res B Appl Biomater. (2017) 105:249–59. doi: 10.1002/jbm.b.33552
145. Avolio E, Caputo M, Madeddu P. Stem cell therapy and tissue engineering for correction of congenital heart disease. Front Cell Dev Biol. (2015) 3:39. doi: 10.3389/fcell.2015.00039
146. Poggio P, Sainger R, Branchetti E, Grau JB, Lai EK, Gorman RC, et al. Noggin attenuates the osteogenic activation of human valve interstitial cells in aortic valve sclerosis. Cardiovasc Res. (2013) 98:402–10. doi: 10.1093/cvr/cvt055
147. Jana S, Tranquillo RT, Lerman A. Cells for tissue engineering of cardiac valves. J Tissue Eng Regen Med. (2016) 10:804–24. doi: 10.1002/term.2010
148. Okano H, Nakamura M, Yoshida K, Okada Y, Tsuji O, Nori S, et al. Steps toward safe cell therapy using induced pluripotent stem cells. Circ Res. (2013) 112:523–33. doi: 10.1161/CIRCRESAHA.111.256149
149. Hoerstrup SP, Sodian R, Daebritz S, Wang J, Bacha EA, Martin DP, et al. Functional living trileaflet heart valves grown in vitro. Circulation (2000) 102(19 Suppl 3):III44–9. doi: 10.1161/01.CIR.102.suppl_3.III-44
150. Hoerstrup SP, Kadner A, Melnitchouk S, Trojan A, Eid K, Tracy J, et al. Tissue engineering of functional trileaflet heart valves from human marrow stromal cells. Circulation (2002) 106(12 Suppl 1):I143–50. doi: 10.1161/01.cir.0000032872.55215.05
151. Schmidt D, Dijkman PE, Driessen-Mol A, Stenger R, Mariani C, Puolakka A, et al. Minimally-invasive implantation of living tissue engineered heart valves: a comprehensive approach from autologous vascular cells to stem cells. J Am Coll Cardiol. (2010) 56:510–20. doi: 10.1016/j.jacc.2010.04.024
152. Zhou J, Ye X, Wang Z, Liu J, Zhang B, Qiu J, et al. Development of decellularized aortic valvular conduit coated by heparin-SDF-1alpha multilayer. Ann Thorac Surg. (2015) 99:612–8. doi: 10.1016/j.athoracsur.2014.09.001
153. Kim SS, Lim SH, Hong YS, Cho SW, Ryu JH, Chang BC, et al. Tissue engineering of heart valves in vivo using bone marrow-derived cells. Artif Organs (2006) 30:554–7. doi: 10.1111/j.1525-1594.2006.00258.x
154. Vincentelli A, Wautot F, Juthier F, Fouquet O, Corseaux D, Marechaux S, et al. In vivo autologous recellularization of a tissue-engineered heart valve: are bone marrow mesenchymal stem cells the best candidates? J Thorac Cardiovasc Surg. (2007) 134:424–32. doi: 10.1016/j.jtcvs.2007.05.005
155. Boldt J, Lutter G, Pohanke J, Fischer G, Schoettler J, Cremer J, et al. Percutaneous tissue-engineered pulmonary valved stent implantation: comparison of bone marrow-derived CD133+-cells and cells obtained from carotid artery. Tissue Eng Part C Methods (2013) 19:363–74. doi: 10.1089/ten.tec.2012.0078
156. Sutherland FW, Perry TE, Yu Y, Sherwood MC, Rabkin E, Masuda Y, et al. From stem cells to viable autologous semilunar heart valve. Circulation (2005) 111:2783–91. doi: 10.1161/CIRCULATIONAHA.104.498378
157. Engelmayr GC Jr, Sales VL, Mayer JE Jr, Sacks MS. Cyclic flexure and laminar flow synergistically accelerate mesenchymal stem cell-mediated engineered tissue formation: Implications for engineered heart valve tissues. Biomaterials (2006) 27:6083–95. doi: 10.1016/j.biomaterials.2006.07.045
158. Gottlieb D, Kunal T, Emani S, Aikawa E, Brown DW, Powell AJ, et al. In vivo monitoring of function of autologous engineered pulmonary valve. J Thorac Cardiovasc Surg. (2010) 139:723–31. doi: 10.1016/j.jtcvs.2009.11.006
159. Ramaswamy S, Gottlieb D, Engelmayr GC Jr, Aikawa E, Schmidt DE, Gaitan-Leon DM, et al. The role of organ level conditioning on the promotion of engineered heart valve tissue development in-vitro using mesenchymal stem cells. Biomaterials (2010) 31:1114–25. doi: 10.1016/j.biomaterials.2009.10.019
160. Dijkman PE, Driessen-Mol A, Frese L, Hoerstrup SP, Baaijens FP. Decellularized homologous tissue-engineered heart valves as off-the-shelf alternatives to xeno- and homografts. Biomaterials (2012) 33:4545–54. doi: 10.1016/j.biomaterials.2012.03.015
161. Knight RL, Booth C, Wilcox HE, Fisher J, Ingham E. Tissue engineering of cardiac valves: re-seeding of acellular porcine aortic valve matrices with human mesenchymal progenitor cells. J Heart Valve Dis. (2005) 14:806–13.
162. Schmidt D, Mol A, Breymann C, Achermann J, Odermatt B, Gossi M, et al. Living autologous heart valves engineered from human prenatally harvested progenitors. Circulation (2006) 114(1 Suppl):I125–31. doi: 10.1161/CIRCULATIONAHA.105.001040
163. Dvorin EL, Wylie-Sears J, Kaushal S, Martin DP, Bischoff J. Quantitative evaluation of endothelial progenitors and cardiac valve endothelial cells: proliferation and differentiation on poly-glycolic acid/poly-4-hydroxybutyrate scaffold in response to vascular endothelial growth factor and transforming growth factor beta1. Tissue Eng. (2003) 9:487–93. doi: 10.1089/107632703322066660
164. Sales VL, Mettler BA, Engelmayr GC Jr, Aikawa E, Bischoff J, Martin DP, et al. Endothelial progenitor cells as a sole source for ex vivo seeding of tissue-engineered heart valves. Tissue Eng Part A (2010) 16:257–67. doi: 10.1089/ten.tea.2009.0424
165. Wang D, Li LK, Dai T, Wang A, Li S. Adult stem cells in vascular remodeling. Theranostics (2018) 8:815–29. doi: 10.7150/thno.19577
166. Schmidt D, Mol A, Odermatt B, Neuenschwander S, Breymann C, Gossi M, et al. Engineering of biologically active living heart valve leaflets using human umbilical cord-derived progenitor cells. Tissue Eng. (2006) 12:3223–32. doi: 10.1089/ten.2006.12.3223
167. Ye X, Wang H, Zhou J, Li H, Liu J, Wang Z, et al. The effect of Heparin-VEGF multilayer on the biocompatibility of decellularized aortic valve with platelet and endothelial progenitor cells. PLoS ONE (2013) 8:e54622. doi: 10.1371/journal.pone.0054622
168. Cebotari S, Lichtenberg A, Tudorache I, Hilfiker A, Mertsching H, Leyh R, et al. Clinical application of tissue engineered human heart valves using autologous progenitor cells. Circulation (2006) 114(1 Suppl):I132–7. doi: 10.1161/CIRCULATIONAHA.105.001065
169. Schmidt D, Achermann J, Odermatt B, Breymann C, Mol A, Genoni M, et al. Prenatally fabricated autologous human living heart valves based on amniotic fluid derived progenitor cells as single cell source. Circulation (2007) 116(11 Suppl):I64–70. doi: 10.1161/CIRCULATIONAHA.106.681494
170. Zhou J, Ding J, Nie B, Hu S, Zhu Z, Chen J, et al. Promotion of adhesion and proliferation of endothelial progenitor cells on decellularized valves by covalent incorporation of RGD peptide and VEGF. J Mater Sci Mater Med. (2016) 27:142. doi: 10.1007/s10856-016-5750-1
171. Simpson DL, Wehman B, Galat Y, Sharma S, Mishra R, Galat V, et al. Engineering patient-specific valves using stem cells generated from skin biopsy specimens. Ann Thorac Surg. (2014) 98:947–54. doi: 10.1016/j.athoracsur.2014.04.075
172. Nachlas ALY, Li S, Jha R, Singh M, Xu C, Davis ME. Human iPSC-derived mesenchymal stem cells encapsulated in PEGDA hydrogels mature into valve interstitial-like cells. Acta Biomater. (2018) 71:235–46. doi: 10.1016/j.actbio.2018.02.025
173. Cushing MC, Liao JT, Anseth KS. Activation of valvular interstitial cells is mediated by transforming growth factor-beta1 interactions with matrix molecules. Matrix Biol. (2005) 24:428–37. doi: 10.1016/j.matbio.2005.06.007
174. Cushing MC, Jaeggli MP, Masters KS, Leinwand LA, Anseth KS. Serum deprivation improves seeding and repopulation of acellular matrices with valvular interstitial cells. J Biomed Mater Res A. (2005) 75:232–41. doi: 10.1002/jbm.a.30412
175. Benton JA, Fairbanks BD, Anseth KS. Characterization of valvular interstitial cell function in three dimensional matrix metalloproteinase degradable PEG hydrogels. Biomaterials (2009) 30:6593–603. doi: 10.1016/j.biomaterials.2009.08.031
176. Sant S, Iyer D, Gaharwar AK, Patel A, Khademhosseini A. Effect of biodegradation and de novo matrix synthesis on the mechanical properties of valvular interstitial cell-seeded polyglycerol sebacate-polycaprolactone scaffolds. Acta Biomater. (2013) 9:5963–73. doi: 10.1016/j.actbio.2012.11.014
177. Granados M, Morticelli L, Andriopoulou S, Kalozoumis P, Pflaum M, Iablonskii P, et al. Development and characterization of a porcine mitral valve scaffold for tissue engineering. J Cardiovasc Transl Res. (2017) 10:374–90. doi: 10.1007/s12265-017-9747-z
178. Alavi SH, Kheradvar A. A hybrid tissue-engineered heart valve. Ann Thorac Surg. (2015) 99:2183–7. doi: 10.1016/j.athoracsur.2015.02.058
179. Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell (2008) 3:301–13. doi: 10.1016/j.stem.2008.07.003
180. Feng J, Mantesso A, De Bari C, Nishiyama A, Sharpe PT. Dual origin of mesenchymal stem cells contributing to organ growth and repair. Proc Natl Acad Sci USA. (2011) 108:6503–8. doi: 10.1073/pnas.1015449108
181. Murray IR, West CC, Hardy WR, James AW, Park TS, Nguyen A, et al. Natural history of mesenchymal stem cells, from vessel walls to culture vessels. Cell Mol Life Sci. (2014) 71:1353–74. doi: 10.1007/s00018-013-14626
182. Sousa BR, Parreira RC, Fonseca EA, Amaya MJ, Tonelli FM, Lacerda SM, et al. Human adult stem cells from diverse origins: an overview from multiparametric immunophenotyping to clinical applications. Cytometry (2014) 85:43–77. doi: 10.1002/cyto.a.22402
183. Hilfiker A, Kasper C, Hass R, Haverich A. Mesenchymal stem cells and progenitor cells in connective tissue engineering and regenerative medicine: is there a future for transplantation? Langenbecks Arch Surg. (2011) 396:489–97. doi: 10.1007/s00423-011-0762-2
184. Balguid A, Mol A, van Vlimmeren MA, Baaijens FP, Bouten CV. Hypoxia induces near-native mechanical properties in engineered heart valve tissue. Circulation (2009) 119:290–7. doi: 10.1161/CIRCULATIONAHA.107.749853
185. Nachlas ALY, Li S, Davis ME. Developing a clinically relevant tissue engineered heart valve-a review of current approaches. Adv Healthc Mater. (2017) 6. doi: 10.1002/adhm.201700918
186. Fioretta ES, von Boehmer L, Motta SE, Lintas V, Hoerstrup SP, Emmert MY. Cardiovascular tissue engineering: from basic science to clinical application. Exp Gerontol. (2018) doi: 10.1016/j.exger.2018.03.022. [Epub ahead of print].
187. Ali ML, Kumar SP, Bjornstad K, Duran CM. The sheep as an animal model for heart valve research. Cardiovasc Surg. (1996) 4:543–9. doi: 10.1016/0967-2109(95)00142-5
188. VeDepo MC, Detamore MS, Hopkins RA, Converse GL. Recellularization of decellularized heart valves: progress toward the tissue-engineered heart valve. J Tissue Eng. (2017) 8:2041731417726327. doi: 10.1177/2041731417726327
189. Gyongyosi M, Wojakowski W, Lemarchand P, Lunde K, Tendera M, Bartunek J, et al. Meta-Analysis of Cell-based CaRdiac stUdiEs (ACCRUE) in patients with acute myocardial infarction based on individual patient data. Circ Res. (2015) 116:1346–60. doi: 10.1161/CIRCRESAHA.116.304346
190. Liu B, Duan CY, Luo CF, Ou CW, Sun K, Wu ZY, et al. Effectiveness and safety of selected bone marrow stem cells on left ventricular function in patients with acute myocardial infarction: a meta-analysis of randomized controlled trials. Int J Cardiol. (2014) 177:764–70. doi: 10.1016/j.ijcard.2014.11.005
191. Yuan L, Sakamoto N, Song G, Sato M. High-level shear stress stimulates endothelial differentiation and VEGF secretion by human mesenchymal stem cells. Cell Mol Bioeng. (2013) 6:220–9. doi: 10.1007/s12195-013-0275-x
192. Pankajakshan D, Kansal V, Agrawal DK. In vitro differentiation of bone marrow derived porcine mesenchymal stem cells to endothelial cells. J Tissue Eng Regen Med. (2013) 7:911–20. doi: 10.1002/term.1483
193. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science (1997) 275:964–7. doi: 10.1126/science.275.5302.964
194. Sales VL, Engelmayr GC Jr, Mettler BA, Johnson JA Jr, Sacks MS, Mayer JE Jr. Transforming growth factor-beta1 modulates extracellular matrix production, proliferation, and apoptosis of endothelial progenitor cells in tissue-engineering scaffolds. Circulation (2006) 114(1 Suppl):I193–9. doi: 10.1161/CIRCULATIONAHA.105.001628
195. Bischoff J, Aikawa E. Progenitor cells confer plasticity to cardiac valve endothelium. J Cardiovasc Transl Res. (2011) 4:710–9. doi: 10.1007/s12265-011-9312-0
196. Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. (2012) 2:a006692. doi: 10.1101/cshperspect.a006692
197. Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. (1999) 85:221–8. doi: 10.1161/01.RES.85.3.221
198. Jordan JE, Williams JK, Lee SJ, Raghavan D, Atala A, Yoo JJ. Bioengineered self-seeding heart valves. J Thorac Cardiovasc Surg. (2012) 143:201–8. doi: 10.1016/j.jtcvs.2011.10.005
199. Hibino N, McGillicuddy E, Matsumura G, Ichihara Y, Naito Y, Breuer C, et al. Late-term results of tissue-engineered vascular grafts in humans. J Thorac Cardiovasc Surg. (2010) 139:431–6, 6 e1–2. doi: 10.1016/j.jtcvs.2009.09.057
200. Dohmen PM, Lembcke A, Holinski S, Pruss A, Konertz W. Ten years of clinical results with a tissue-engineered pulmonary valve. Ann Thorac Surg. (2011) 92:1308–14. doi: 10.1016/j.athoracsur.2011.06.009
201. Xue Y, Yatsenko T, Patel A, Stolz DB, Phillippi JA, Sant V, et al. PEGylated poly(ester amide) elastomer scaffolds for soft tissue engineering. Polym Adv Technol. (2017) 28:1097–106. doi: 10.1002/pat.4002
202. Mohsen DK, Kakarougkas A, Allam NK. Recent advances on electrospun scaffolds as matrices for tissue-engineered heart valves. Materials Today Chemistry (2017) 5:11–23. doi: 10.1016/j.mtchem.2017.05.001
203. Wang X, Lee J, Ali M, Kim J, Lacerda CMR. Phenotype transformation of aortic valve interstitial cells due to applied shear stresses within a microfluidic chip. Ann Biomed Eng. (2017) 45:2269–80. doi: 10.1007/s10439-017-1871-z
204. Porras AM, Westlund JA, Evans AD, Masters KS. Creation of disease-inspired biomaterial environments to mimic pathological events in early calcific aortic valve disease. Proc Natl Acad Sci USA. (2018) 115:E363–71. doi: 10.1073/pnas.1704637115
205. Masoumi N, Larson BL, Annabi N, Kharaziha M, Zamanian B, Shapero KS, et al. Electrospun PGS:PCL microfibers align human valvular interstitial cells and provide tunable scaffold anisotropy. Adv Healthc Mater. (2014) 3:929–39. doi: 10.1002/adhm.201300505
206. Parvin Nejad S, Blaser MC, Santerre JP, Caldarone CA, Simmons CA. Biomechanical conditioning of tissue engineered heart valves: too much of a good thing? Adv Drug Deliv Rev. (2016) 96:161–75. doi: 10.1016/j.addr.2015.11.003
207. Tao G, Kotick JD, Lincoln J. Heart valve development, maintenance, and disease: the role of endothelial cells. Curr Top Dev Biol. (2012) 100:203–32. doi: 10.1016/B978-0-12-387786-4.00006-3
208. Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS ONE (2012) 7:e39969. doi: 10.1371/journal.pone.0039969
209. Crisan M, Corselli M, Chen WC, Peault B. Perivascular cells for regenerative medicine. J Cell Mol Med. (2012) 16:2851–60. doi: 10.1111/j.1582-4934.2012.01617.x
210. Geevarghese A, Herman IM. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res. (2014) 163:296–306. doi: 10.1016/j.trsl.2014.01.011
211. Avolio E, Madeddu P. Discovering cardiac pericyte biology: From physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vascul Pharmacol. (2016) 86:53–63. doi: 10.1016/j.vph.2016.05.009
212. Gaceb A, Barbariga M, Ozen I, Paul G. The pericyte secretome: Potential impact on regeneration. Biochimie (2018). doi: 10.1016/j.biochi.2018.04.015. [Epub ahead of print].
213. Avolio E, Rodriguez-Arabaolaza I, Spencer HL, Riu F, Mangialardi G, Slater SC, et al. Expansion and characterization of neonatal cardiac pericytes provides a novel cellular option for tissue engineering in congenital heart disease. J Am Heart Assoc. (2015) 4:e002043. doi: 10.1161/JAHA.115.002043
214. Katare R, Riu F, Mitchell K, Gubernator M, Campagnolo P, Cui Y, et al. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132. Circ Res. (2011) 109:894–906. doi: 10.1161/CIRCRESAHA.111.251546
215. Avolio E, Meloni M, Spencer HL, Riu F, Katare R, Mangialardi G, et al. Combined intramyocardial delivery of human pericytes and cardiac stem cells additively improves the healing of mouse infarcted hearts through stimulation of vascular and muscular repair. Circ Res. (2015) 116:e81–94. doi: 10.1161/CIRCRESAHA.115.306146
216. Alvino VV, Fernandez-Jimenez R, Rodriguez-Arabaolaza I, Slater S, Mangialardi G, Avolio E, et al. Transplantation of allogeneic pericytes improves myocardial vascularization and reduces interstitial fibrosis in a swine model of reperfused acute myocardial infarction. J Am Heart Assoc. (2018) 7:e006727. doi: 10.1161/JAHA.117.006727
217. Bujan J, Bellon JM, Sabater C, Jurado F, Garcia-Honduvilla N, Dominguez B, et al. Modifications induced by atherogenic diet in the capacity of the arterial wall in rats to respond to surgical insult. Atherosclerosis (1996) 122:141–52. doi: 10.1016/0021-9150(95)05727-7
218. Hu Y, Zhang Z, Torsney E, Afzal AR, Davison F, Metzler B, et al. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J Clin Invest. (2004) 113:1258–65. doi: 10.1172/JCI19628
Keywords: valve heart disease, calcification, tissue engineering heart valves, in vitro, heterotopic bone formation
Citation: Jover E, Fagnano M, Angelini G and Madeddu P (2018) Cell Sources for Tissue Engineering Strategies to Treat Calcific Valve Disease. Front. Cardiovasc. Med. 5:155. doi: 10.3389/fcvm.2018.00155
Received: 17 May 2018; Accepted: 10 October 2018;
Published: 06 November 2018.
Edited by:
Joshua D. Hutcheson, Florida International University, United StatesReviewed by:
Mark C. Blaser, Brigham and Women's Hospital, United StatesCopyright © 2018 Jover, Fagnano, Angelini and Madeddu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Madeddu, bWRwcm1AYnJpc3RvbC5hYy51aw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.