 Stefania Puttini1
Stefania Puttini1 Isabelle Plaisance1Lucio Barile2
Isabelle Plaisance1Lucio Barile2 Elisabetta Cervio2Giuseppina Milano1,2
Elisabetta Cervio2Giuseppina Milano1,2 Paola Marcato3
Paola Marcato3 Thierry Pedrazzini1
Thierry Pedrazzini1 Giuseppe Vassalli1,2*
Giuseppe Vassalli1,2*- 1Cardiovascular Department, CHUV University Hospital, Lausanne, Switzerland
- 2Cardiocentro Ticino Foundation and Swiss Institute for Regenerative Medicine, Lugano, Switzerland
- 3Departments of Pathology, Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada
High aldehyde dehydrogenase (ALDHhi) activity has been reported in normal and cancer stem cells. We and others have shown previously that human ALDHhi cardiac atrial appendage cells are enriched with stem/progenitor cells. The role of ALDH in these cells is poorly understood but it may come down to the specific ALDH isoform(s) expressed. This study aimed to compare ALDHhi and ALDHlo atrial cells and to identify the isoform(s) that contribute to ALDH activity, and their functional role.
Methods and Results: Cells were isolated from atrial appendage specimens from patients with ischemic and/or valvular heart disease undergoing heart surgery. ALDHhi activity assessed with the Aldefluor reagent coincided with primitive surface marker expression (CD34+). Depending on their ALDH activity, RT-PCR analysis of ALDHhi and ALDHlo cells demonstrated a differential pattern of pluripotency genes (Oct 4, Nanog) and genes for more established cardiac lineages (Nkx2.5, Tbx5, Mef2c, GATA4). ALDHhi cells, but not ALDHlo cells, formed clones and were culture-expanded. When cultured under cardiac differentiation conditions, ALDHhi cells gave rise to a higher number of cardiomyocytes compared with ALDHlo cells. Among 19 ALDH isoforms known in human, ALDH1A3 was most highly expressed in ALDHhi atrial cells. Knocking down ALDH1A3, but not ALDH1A1, ALDH1A2, ALDH2, ALDH4A1, or ALDH8A1 using siRNA decreased ALDH activity and cell proliferation in ALDHhi cells. Conversely, overexpressing ALDH1A3 with a retroviral vector increased proliferation in ALDHlo cells.
Conclusions: ALDH1A3 is the key isoform responsible for ALDH activity in ALDHhi atrial appendage cells, which have a propensity to differentiate into cardiomyocytes. ALDH1A3 affects in vitro proliferation of these cells.
Introduction
In many studies, putative cardiac-resident stem and progenitor cells were isolated based on the expression of hematopoietic stem cell (HSC) markers, such as CD34, stem cell antigen-1 (Sca-1), and c-kit/CD117 (1–3). However, no single cell surface marker definitely identifies a specific entity of cardiac stem cells (4). Therefore, “functional” stem cell markers have been intensively searched for. It has been proposed that, irrespective of their lineage origin, stem cells may respond in similar ways to regulate self-renewal and differentiation, and that putative “stemness” genes may serve as universal stem cell markers (5). A widely used universal stem cell marker is aldehyde dehydrogenase (ALDH) (6–8). In tunicates, few pluripotential cells with high ALDH enzymatic activity (ALDHhi) were able to give rise to all somatic and reproductive cell lineages (9).
ALDH is a generic designation for a superfamily of NAD-dependent enzymes that catalyze the oxidation of aldehydes to acids. The ALDH family includes 19 members in human, which share some amino acid sequence similarity and certain functions while differing in intracellular localization, tissue distribution, and substrate specificity. ALDHs are localized in the cytoplasm, mitochondria or nucleus and participate in many biological processes including the detoxification of exogenous aldehydes and the metabolism of vitamin A, alcohol and reactive oxygen species (ROS) (7, 10). ALDH1A1 is a detoxifying enzyme responsible for oxidizing aldehydes to carboxylic acids (11). High-level ALDH1A1 expression regulates self-renewal and differentiation in HSCs and neural stem cells (12–14). ALDH1A1 has been implicated in chemotherapeutic resistance of HSCs via metabolization and detoxification of chemotherapeutics like cyclophosphamide (15). ALDH1A is the rate-limiting factor in the production of retinoic acid (RA) via oxidation of all-trans retinal and 9-cis-retinal (16–18). RA activates nuclear RA receptors (RARs) that control the transcription of genes with RA response elements (RAREs) in their promoters, thereby regulating stem cell functions (13, 19). Elevated activity of additional ALDH isoforms, namely ALDH1A2, ALDH1A3, ALDH1A7, ALDH2*2, ALDH3A1, ALDH4A1, ALDH5A1, ALDH6, and ALDH9A1, has been observed in normal and cancer stem cells (10, 20–25). It has been proposed that the role of ALDH as a stem cell marker may come down to the specific isoform(s) expressed (20). Thus, ALDH not only may be considered a stem cell marker, but also may well play functional roles in terms of self-renewal, differentiation, and/or expansion. It should be noted, however, that currently available commercial assays identifying ALDHhi cells as those actively metabolizing BODIPY-aminoacetaldehyde (Aldefluor®) (26) do not distinguish the specific ALDH isoforms (8).
In human, ALDH expression by HSCs has been evaluated as a predictor of hematopoietic recovery after peripheral stem cell mobilization (27) and a biomarker for umbilical cord blood potency (28). Both bone marrow and cord blood-derived ALDHhi cells have shown therapeutic potential in limb ischemia (29) and myocardial infarction models (30). In clinical trials, autologous bone marrow-derived ALDHhi cells did not improve functional or magnetic resonance outcomes in patients with peripheral artery disease (31). More encouraging results were reported in patients with ischemic heart failure (32). We were the first to isolate cardiac atrial appendage-derived progenitor cells based on ALDH activity (33, 34). Koninckx et al. (35) then reported that human ALDHhi cardiac atrial appendage stem cells (CASC) gave rise to cardiac cells and improved cardiac function upon injection into infarcted pig hearts. However, this study did not compare ALDHhi and ALDHlo cells nor did it define the specific ALDH isoform(s) expressed and their functional roles.
The present study aimed to compare human ALDHhi and ALDHlo atrial appendage cells both phenotypically and functionally, and to identify the specific ALDH isoform(s) expressed. ALDH1A3 was found to be the key isoform responsible for Aldefluor positivity in ALDHhi cells. Gain- and loss-of-function experiments revealed a role for ALDH1A3 in cell proliferation.
Materials and Methods
Cell Isolation and Flow Cytometric Analysis
Human right atrial appendage specimens were obtained from male and female patients (29–91 years old) who underwent cardiac surgery for ischemic and/or valvular heart disease through donation. The protocol received authorization from the University Hospital Ethics Committee and the Cantonal Ethics Committee Ethics Committee of Canton Vaud, Switzerland on research involving humans. Informed, written consent was obtained from the participants. In 3 patients (76–86 years old) who underwent left ventricular (LV) assist device implantation, tissue specimens were obtained from the LV apex. Immediately after their procurement, tissue specimens were kept on ice, minced, and digested in a buffer containing 0.45 mg/ml collagenase from Clostridium histolyticum and 0.1 mg/ml proteinase bacterial Type XXIV (both from Sigma Aldrich, St. Louis, MO, USA). Four rounds of enzymatic digestion were used. Freshly isolated cells were immediately reacted with Aldefluor (Stem Cell Technologies, Vancouver, BC, Canada) to identify ALDHhi cells. Briefly, 2 × 106 cells/mL were suspended in Aldefluor assay buffer containing the ALDH substrate BODIPYaminoacetaldehyde and incubated at 37°C for 45 min. For each sample, cell aliquots were incubated with or without 50 mM diethylaminobenzaldehyde (DEAB), an ALDH-specific inhibitor (36), and analyzed on a Gallios flow cytometer (Beckman Coulter, Indianapolis, IN, USA). The threshold used for the ALDHhi gate was 2.0% of DEAB-treated control cells. Dead cells and cells in the early-mid apoptosis were identified using DAPI and Annexin V apoptosis detection kit-APC (eBioscience; Thermo Fisher Scientific, Waltham, MA, USA), respectively. For flow cytometric analyses of surface marker expression, 1 × 105 cells/tube were incubated with Aldefluor and subsequently stained with marker-specific antibodies (Supplementary Table 1) for 30 min in the dark. Fluorescence-activated cell sorting (FACS) based on ALDH activity was performed on a MoFloAstrios cell sorter (Beckman Coulter). To prevent cross-contamination between ALDHhi and ALDHhi cells, sorting gates of these 2 populations were set up at least one log apart. The purity of sorted populations was reanalyzed using ALDHhi and ALDHlo cells and was shown to be greater than 95%. For expansion, cells were plated in gelatin-coated 6-well plates and cultured in expansion medium (3:1 DMEM 1g/l glucose/Medium 199 [Invitrogen, Carlsbad, CA, USA] supplemented with 10% horse serum [Serotec; BioRad Labs., Hercules, CA, USA], 5% fetal calf serum (FCS) [Serotec], 100 U/ml penicillin [Invitrogen], and 100 mg/ml streptomycin [Invitrogen]). Cells that remained in suspension after 24 h in culture were discarded, and adherent cells were expanded. In a subset of experiments, atrial appendage specimens were not dissociated using enzymatic techniques, but cultured ex vivo as primary tissue explants (37). Briefly, atrial tissue explants were cut into small pieces (≈1 mg) that were placed in gelatin-coated culture dishes (Corning; Thermo Fisher Scientific) and cultured in IMDM supplemented with 20% FCS, 100 U/ml penicillin and 100 mg/ml streptomycin. After 10 days, the small pieces of tissue were removed from culture dishes and the cell populations that had migrated out of them (outgrowth cells) were expanded.
Immunocytochemistry
Immunocytochemical staining was performed on cells cultured in gelatin-coated wells using the antibodies listed in Supplementary Table 2. Stained cells were mounted with Vectashield mounting medium containing DAPI (Vector Labs, Burlingame, CA, USA). Images were captured using an Axiovision (Carl Zeiss Jena, Germany) fluorescence microscope and a Nikon LSR Eclipse TE2000S inverted fluorescence microscope (Nikon, Tokyo, Japan).
Clonogenicity and Cell Proliferation Assays
To assess clonogenicity, atrial cells were seeded at limit dilution and single cells were identified under a light microscope. Clones derived from single cells were counted at later time points. To assess cell growth, cell numbers were counted at different time points after plating. The effect of DEAB on proliferation was assessed in outgrowth cells. Cell proliferation was measured using Click-iT® EdU Alexa Fluor® 488 imaging kit (Invitrogen), as per manufacturer's instructions. Briefly, cells were incubated with EdU (10 μM final concentration) for 2 h in expansion medium, fixed with paraformaldehyde 2%, permeabilized with 0.3% Triton, incubated with Alexafluor azide for 30 min in the dark, and mounted with Vectashield. EdU+ cells and total cells were counted under a fluorescence microscope, and percentages of EdU+ cells were calculated. Cell density was expressed using a microscopic score (range: 0–4) assigned by two independent investigators blind to experimental conditions. RA levels in cells were measured using Human Retinoic Acid ELISA Kit (MBS705877-96; ANAWA Biotrend, Wangen, Switzerland). The ability of the RA receptor agonist all-trans RA to replace ALDH in promoting cell growth was tested. Varying concentrations of all-trans RA (Sigma) were added to explant outgrowths cultured in the presence of 100 μM DEAB, and cell density was assessed under a light microscope.
RNA Isolation, Reverse Transcription, and Real-Time PCR
Total RNA was extracted using RNeasy isolation kit (Qiagen N V, Hilden, Germany). Reverse transcription was performed with QuantiTect Whole Transcriptome Kit (Qiagen) for RNA from freshly isolated cells, and with Verso cDNA Synthesis Kit (Thermo Fisher) for RNA from culture-expanded cells. For expression analyses of cardiac transcription factors, pluripotency-associated genes and cardiac differentiation markers, cDNA was used as a template for real-time PCR or relative quantitative PCR (qPCR) using specific probes and the ABI Prism 7500 Fast Real-Time PCR System (Applied Biosystems; Thermo Fisher). For expression analyses of ALDH isoforms, Fast SYBR®Green Master Mix (Life Technologies; Thermo Fisher) with gene-specific primers (Thermo Fisher; Supplementary Table 3) were used. Each reaction was carried out in triplicate. To calculate the relative expression levels, we used the 2−ΔΔCt-method. RPL27 mRNA was used as an endogenous control. Experiments linked to differentiation were done with Taqman probes using β2–microglobuline as a reference gene.
Western Blotting
Western blotting was performed on ALDHhi and ALDHlo sorted cells using rabbit anti-human ALDH1A3 antibody (Ab 129815, Abcam, Cambridge, UK).
Cardiomyogenic Differentiation
Atrial cells at culture-passage 2 were reacted with Aldefluor. ALDHhi and ALDHlo populations were sorted. Two protocols were used to induce cardiac differentiation. Using the first protocol (no NOTCH modulation), differentiation was induced by switching the cells to MEM alpha (Invitrogen) containing 2% horse serum, 1 mmol/l dexamethasone (Sigma-Aldrich), 50 mg/ml ascorbic acid (Sigma-Aldrich), 10 mmol/l b-glycerophosphate (Sigma-Aldrich), 100 U/ml penicillin, and 100 mg/ml streptomycin (differentiation medium). Because cells were spontaneously differentiating into smooth muscle cells using this protocol, a recently developed protocol involving sequential NOTCH activation and inhibition was applied (38). Cells were cultured on immobilized Delta-like1 (DLL1) ligand (R&D Systems, Minneapolis, MN, USA) during 24 h. After cell re-suspension, NOTCH pathway was inhibited by the inhibitor of gamma secretase, DAPT (Sigma-Aldrich). Cells were cultured under those conditions for 3 weeks in differentiation medium, and cells were then assessed for expression of cardiac-specific (Myh6, Myh7) and smooth muscle (SM)-specific (Myh11) genes using quantitative RT-PCR. Expression of cardiac-specific α-actinin and troponin I (cTnI), as well as SM myosin heavy chain (sm-MHC) was assessed immunocytochemically. Cells positive for α-actinin or cTnI, or both markers, and total cells were counted under the fluorescence microscope. Percentages of positive cells were calculated.
siRNA Silencing
Small interfering RNA (siRNA) was used to knockdown ALDH isoform expression in atrial cells. All siRNAs were obtained from Thermo Fisher. ALDHhi sorted cells seeded on 6 well-plates (50.000 cells/well) were allowed to adhere overnight. Transfection was performed with Lipofectamine RNAiMAX (Invitrogen), as per manufacturer's instructions. siALDH1A3 was tested at different concentrations. In subsequent experiments, siRNA specific for ALDH isoforms 1A1, 1A2, 1A3, 2A, 4A1, 8A1 was used at the 10 nM concentration. Percentages of ALDHhi cells were measured at 24 h post-transfection using the Aldefluor assay. EdU was added to cells at 24 h post-transfection, and EdU incorporation was assessed using Click-iT® EdU Alexa Fluor® 488 imaging kit.
ALDH1A1 and ALDH1A3 Overexpression
The retroviral vector pMSCVpuro with either ALDH1A1 or ALDH1A3 coding sequences inserted was described and validated previously (32). A retroviral vector with no insert was used to control for unspecific virus-related effects. ALDHlo sorted cells were transduced with retroviral supernatants in 6-well plates (50,000 cells/well), as described previously (32). Twenty-four hours post-transduction, cells were passaged and seeded at ≈50% density. EdU was added to the cells 24 h later. Cell proliferation was analyzed using Click-iT® EdU Alexa Fluor® 488 imaging kit.
Statistics
The data are presented as mean ± S.D. Student t-test was used to compare two normally distributed populations. The Mann-Whithney U test was used for nonparametric comparisons of two groups. Differences with probability values p < 0.05 were considered statistically significant.
Results
Identification of ALDHhi Cells
Freshly isolated, myocyte-depleted populations were isolated based on ALDH activity using the Aldefluor assay. ALDHhi gating was established by inhibiting ALDH activity using DEAB (Figure 1A). Freshly isolated cells from right atrial appendages contained an average of 21.5 + 12.4% ALDHhi cells (n = 86). In a few LV apex specimens available (n = 3), ALDHhi cells averaged 12.3 ± 2.1% of total cells. This finding demonstrated that the LV apex also contains ALDHhi cells; however, quantitative comparisons of atrial and LV cells were not performed due to the small LV sample size. A majority of ALDHhi atrial cells (73.1 + 20.2%) expressed the primitive surface marker CD34 (Figure 1B), which was confirmed immunocytochemically (Figure 1C), but not CD45 (<5%). There was no significant correlation between ALDHhi cell number and age (Figure 1D), gender, or ischemic vs. heart valve disease (data not shown). Because freshly isolated ALDHlo cells could not be expanded ex vivo, we used ALDHlo outgrowth cells (37) in a subset of experiments including direct comparisons with ALDHhi cells. In addition, outgrowth cells are of translational interest, as they give rise to cardiosphere-derived cells that have been tested clinically in patients after myocardial infarction (39). Representative gating sequences from FACS experiments with different populations, namely freshly isolated atrial cells, freshly isolated LV cells, expanded atrial cells, and outgrowth cells are shown in Figure 1E. In freshly isolated cells, but not in expanded ones and outgrowth cells, ALDHhi and ALDHlo cells were visualized as discrete populations, and the Aldefluor signal was incompletely inhibited by DEAB. By contrast, the Aldefluor signal was fully suppressed by DEAB in both expanded and outgrowth cells. These results suggest that ALDH activity in freshly isolated cells may be higher than in expanded and outgrowth cells. The frequency of ALDHhi cells within the outgrowth population was stable during 3 weeks but decreased thereafter (Figure 1F), suggesting that the pool of ALDHhi cells within cultured tissue explants may be exhausted at late time points.
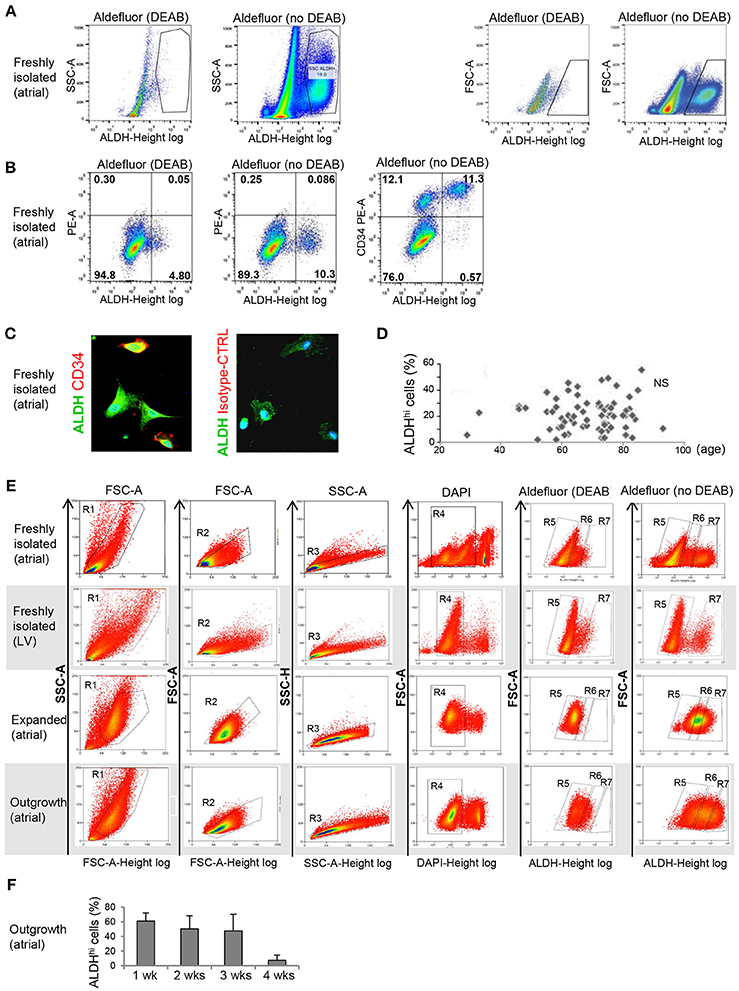
Figure 1. Flow cytometry analysis of ALDHhi atrial cells. (A) Freshly isolated cells. ALDHhi gating was established by inhibiting ALDH activity in Aldefluor-reacted cells with DEAB. Left panels: ALDH/side scatter (SSC-A) plots. Right panels: ALDH/forward scatter (FSC-A) plots. (B) Freshly isolated cells. Aldefluor activity and CD34 expression. (C) CD34 immunostaining (red); ALDH (green); staining with istotype-matched control antibody is shown. (D) ALDHhi cells (%) did not correlate with age. (E) FACS plots of different populations (top to bottom: freshly isolated atrial cells, freshly isolated LV cells, expanded atrial cells, atrial outgrowth cells). Gating sequences are shown. R1-gated cells in FSC-A/SSC-A plots are gated for homogeneity (R2, R3) and DAPI exclusion (R4). R5 and R7 gates define ALDHlo and ALDHhi cells, respectively. To prevent cross-contamination between ALDHhi and ALDHhi cells, R5 and R7 gates were set one log apart (i.e., R6-gated cells were discarded). (F) Percentages of ALDHhi outgrowth cells after 1–4 weeks of ex vivo tissue culture.
Freshly Isolated ALDHhi Cells Express CD34 Along With Mesenchymal and Endothelial Markers
Differential surface marker expression in ALDHhi and ALDHlo atrial cells has not been analyzed previously. Freshly isolated ALDHhi cells (Figure 2A) expressed the primitive marker CD34, platelet endothelial cell adhesion molecule-1 (PECAM-1)/CD31, and endoglin/CD105, a marker expressed by both endothelial and stromal progenitor cells. Thus, the ALDHhi population appeared to include both mesenchymal progenitors and differentiating endothelial progenitors. The ALDHhi population was enriched with cells expressing vascular cell adhesion molecule-1 (VCAM-1)/CD106 (38-fold increase). Small cell subsets expressed the common leukocyte antigen CD45 (<5%) and stem cell markers c-kit/CD117 (1.2 ± 0.9%) and prominin/CD133 (2.3 ± 3.1%). ALDHhi outgrowth cells lacked CD34 expression. Their surface marker profile did not significantly differ from that of ALDHlo cells (Figure 2B).
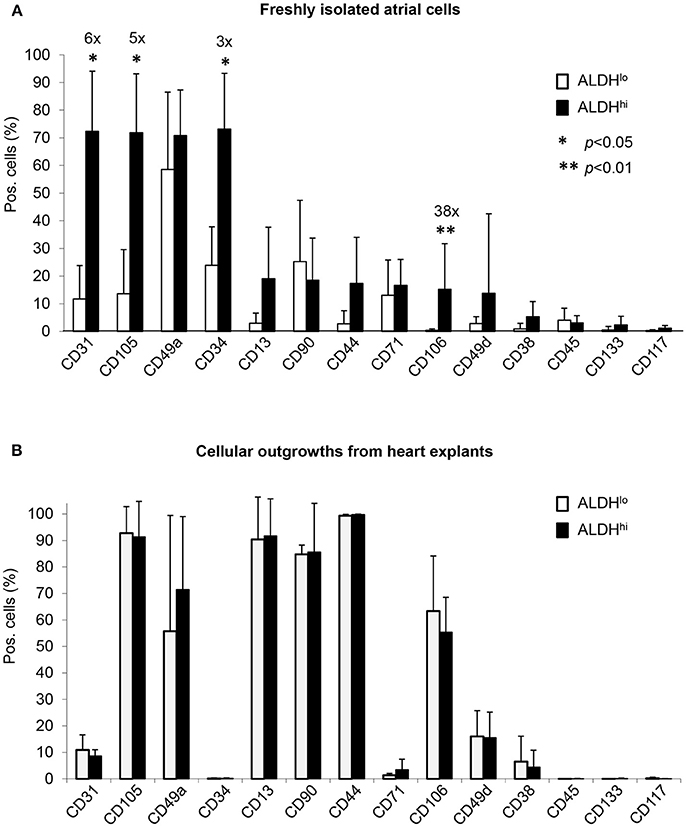
Figure 2. Flow cytometry analyses of cell-surface marker expression in ALDHhi and ALDHlo atrial cells. (A) Freshly isolated cells. Aldefluor-reacted cells were stained with antibodies to the indicated markers (see Supplementary Table 1). CD31+, CD105+, CD34+, and CD106+ cells (%) are significantly increased in ALDHhi cells relative to ALDHlo cells (fold-increases are indicated); *p < 0.05. (B) Outgrowth cells. ALDHhi cells lack CD34 expression and exhibit a marker profile similar to that of ALDHlo cells (n ≥ 5 per marker and group).
Differential Expression of Pluripotency Genes and Markers for Cardiac Lineages
We analyzed expression of pluripotency-associated genes and cardiac-specific markers as evidence of presence of primitive cells and cardiac progenitor cells, respectively, in atrial cell populations. Freshly isolated ALDHhi cells expressed pluripotency-associated genes (Oct4, Nanog) and cardiac-specific transcription factors (Nkx2.5, Mef2c, GATA4, Tbx5) at higher levels (3–5 orders of magnitude) than did ALDHlo cells (Figure 3A). Nkx2.5, GATA4, and Mef2c expression was confirmed immunocytochemically (Figure 3C). Similar albeit weaker differences (1–2 orders of magnitude) were found between ALDHhi and ALDHlo outgrowth cells (Figure 3B). These results suggest that the freshly isolated ALDHhi population may be enriched with primitive cells and cells committed to a cardiac fate.
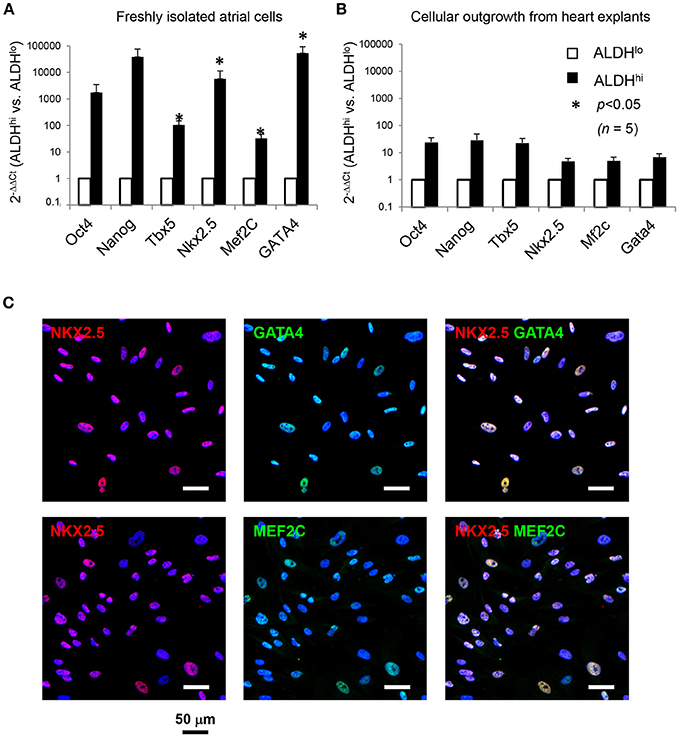
Figure 3. Expression of pluripotency-associated genes (Oct4, Nanog) and cardiac-specific transcription factors (Nkx2.5, Mef2c, GATA4, Tbx5). (A) Quantitative RT-PCR analyses of freshly isolated atrial cells (n = 5/group). (B) Outgrowth cells (n = 5/group). 2−ΔΔCt values for ALDHhi vs. ALDHlo cells are shown. (C). Freshly isolated ALDHhi cells. Immunostaining for Nkx2.5 (red), GATA4 (green), and Mef2c (green); DAPI nuclear counterstaining (blue). Nkx2.5/GATA4 and Nkx2.5/Mef2c double stainings are shown.
Freshly Isolated ALDHhi Cells Are Clonogenic and Selectively Expandable
Freshly isolated ALDHhi cells selectively formed clones and could be expanded ex vivo. In principle, this might be due to selective resistance to apoptosis induced by the isolation procedure. After DAPI exclusion, virtually no ALDHhi cells but part of the ALDHlo population were Annexin V-positive (Figure 4A) suggesting they were in the early-mid apoptosis. The clonogenicity index was 0.27 ± 0.09% in ALDHhi cells, 0% in ALDHlo cells, and 0.03 ± 0.01% in the bulk (unsorted) population (Figure 4B). Freshly isolated ALDHhi cells grew faster than the bulk population. Similarly, ALDHhi outgrowth cells grew faster than ALDHlo ones (Figure 4C). Expanded ALDHhi cells lost ALDH activity over time (e.g., the frequency of ALDHhi cells in the progeny of freshly isolated ALDHhi cells after 13 culture-passages was 32%). These observations suggest that high ALDH activity may be required for the initial survival and growth of freshly isolated cells, but not in established cell cultures, reflecting cell adaptation to in vitro conditions.
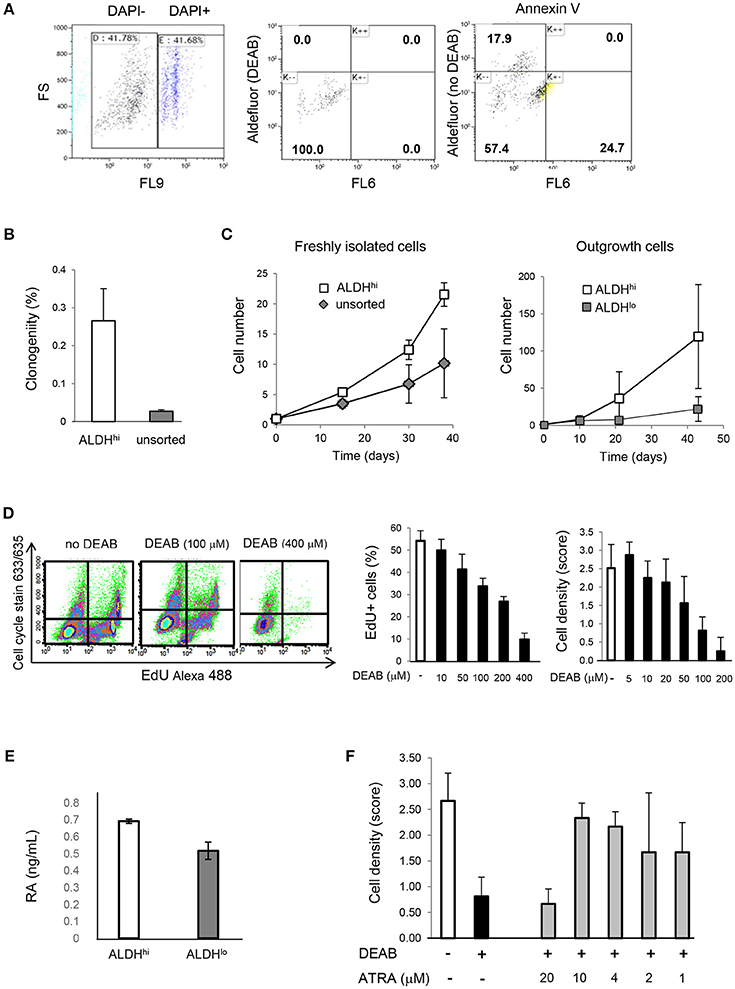
Figure 4. Cell viability, clonogenicity, cell growth, intracellular RA levels, effects of DEAB and all-trans RA. (A) Freshly isolated cells. Flow cytometry analysis of DAPI, Aldefluor (with and without DEAB), and Annexin V (right panel). Part of ALDHlo cells are Annexin V-positive (K-+). (B) Freshly isolated ALDHhi cells and unsorted cells. Clonogenic analysis (n = 3/group). (C) In vitro growth kinetics. Left panel: freshly isolated cells; right panel: outgrowth cells (n = 3/group in both panels). (D) Dose-dependent effects of DEAB on EdU incorporation and cell density. (E) RA concentrations in ALDHhi and ALDHlo cells measured by ELISA. (F) Dose-dependent effects of all-trans RA (ATRA) on cell density in the presence of DEAB (100 μM).
All-Trans RA Reverses DEAB-Mediated Cell Growth Suppression
To assess the effect of ALDH on cell growth, explant outgrowths were cultured with or without DEAB at varying concentrations. DEAB reduced proliferation in outgrowth cells in a dose-dependent manner (Figure 4D), as determined using an EdU incorporation assay. Because ALDH1 naturally synthesizes RA, we measured RA concentrations in ALDHhi and ALDHlo cells. Not surprisingly, we found higher RA levels in the former (Figure 4E). We then tested the ability of all-trans RA to replace ALDH in promoting cell growth. all-trans RA (10 μM) reversed cell growth suppression mediated by DEAB (Figure 4F). These results suggest that ALDH might promote atrial cell growth through RA, at least in part.
ALDHhi Cells Have Higher Propensity to Produce Mature Cardiomyocytes Than ALDHlo Cells
Having shown that the ALDHhi population is enriched with stem/progenitor cells, we addressed its cardiomyogenic potential. Two differentiation protocols, with or without Notch modulation, were used. Under differentiation conditions with no Notch-modulation, both ALDHhi and ALDHlo cells expressed marked levels of MYH11 gene, while the cardiomyocyte markers MYH6 and MYH7 were not induced (Figure 5A). Both populations massively differentiated into smooth muscle cells, as illustrated by smMHC immunostaining; less than 2% of cells stained positive for cardiomyocyte-specific marker α-actinin (Figure 5B). By contrast, under Notch-modulated conditions both ALDHlo and ALDHhi cells expressed significant levels of cardiomyocyte-specific MYH6 and MYH7 (Figure 5A) and α-actinin (Figures 5B,D). Cardiac troponin-I (TNNI) was also identified in differentiated cells. Interestingly, under Notch-modulated conditions MYH6 and MYH7 gene expression was more pronounced in ALDHhi than ALDHlo cells. Similarly, the number of α-actinin/TNNI double positive cells derived from ALDHhi cells was significantly higher than that derived from ALDHlo cells (Figures 5C,D). These results indicated that the differentiated ALDHhi cells were more terminally differentiated than ALDHlo cells, consistent with a higher propensity of ALDHhi cells to differentiate into cardiomyocytes.
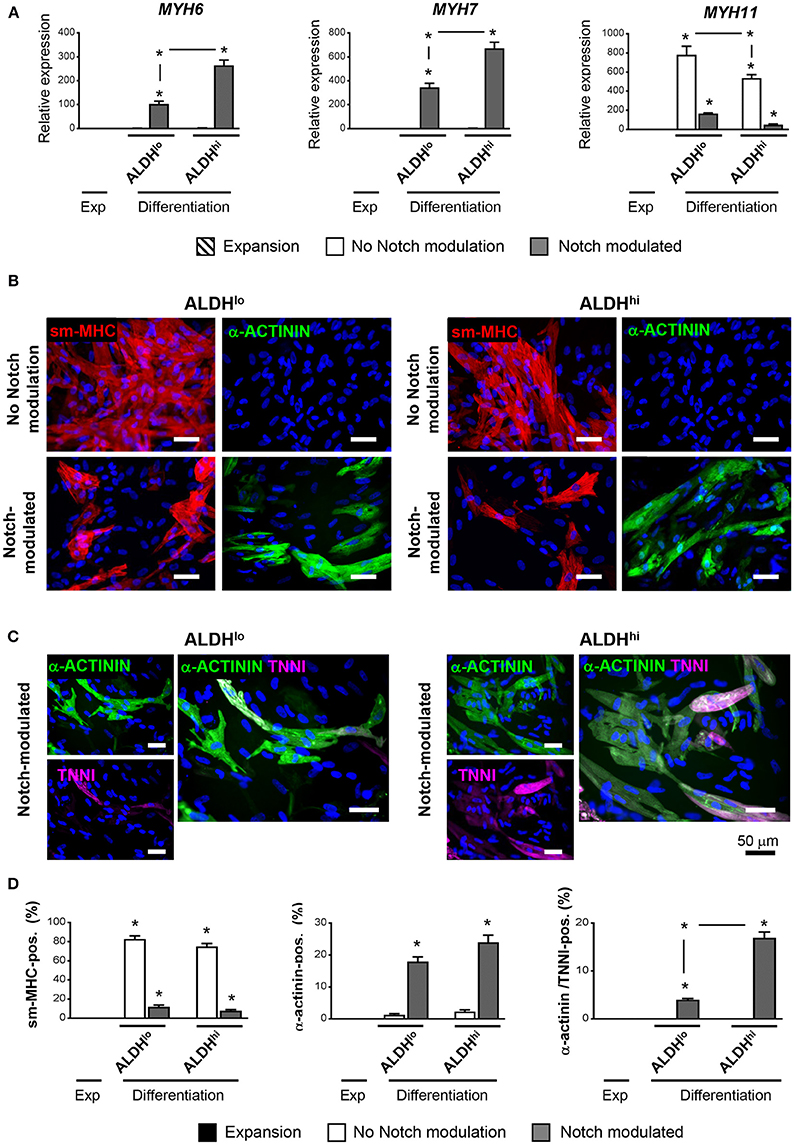
Figure 5. Cardiac differentiation of ALDHhi and ALDHlo atrial cells using a Notch modulation-based protocol. (A) real-time RT-PCR expression analyses of cardiac-specific (Myh6, Myh7) and smooth muscle-specific genes (Myh11) under expansion (Exp) and differentiation conditions, with or without Notch modulation. 2−ΔCtvalues are shown. One example of two independent experiments performed in triplicates is shown; *p < 0.05 vs. cells in expansion or between the indicated conditions. (B) Immunostaining of ALDHhi and ALDHlo cells for smooth musche-myosin heavy chain (smMHC) and cardiac-specific α-actinin, with or without Notch modulation. (C) Immunostaining of ALDHhi and ALDHlo cells for cardiac-specific α-actinin and troponin I (cTNNI) under Notch-modulated conditions. (D) Quantitative analyses of smMHC, α-actinin, and α-actinin/cTNNI positive cells; *p < 0.05 vs. cells in expansion (a minimum of 2000 cells in 30 different fields at 40x magnification were analyzed for quantification).
ALDH1A3 Is the Most Highly Expressed ALDH Isoform in Atrial Cells
The specific ALDH isoforms expressed in ALDHhi atrial cells have not been identified previously. We therefore analyzed the ALDH isoform expression profiles of ALDHhi and ALDHlo cells. ALDH1A3 was the most highly expressed isoform in freshly isolated ALDHhi cells, as well as the most highly overexpressed isoform in ALDHhi relative to ALDHlo cells (≈8-fold increase; Figure 6A). ALDH1A3 protein levels in ALDHhi cells were higher than in ALDHlo cells, as evidenced by Western blot analysis (Figure 6B). Similar findings were observed in outgrowth cells (≈6-fold increase in ALDH1A3 mRNA levels in ALDHhi vs. ALDHlo cells; Figure 6C). For comparison, a subset of 10 ALDH isoforms selected based on published data (40) were measured in atrial total tissue, and ALDH2 was found to be the most highly expressed isoform (Figure 6D). Thus, ALDH1A3 is the key isoform expressed in ALDHhi atrial cells.
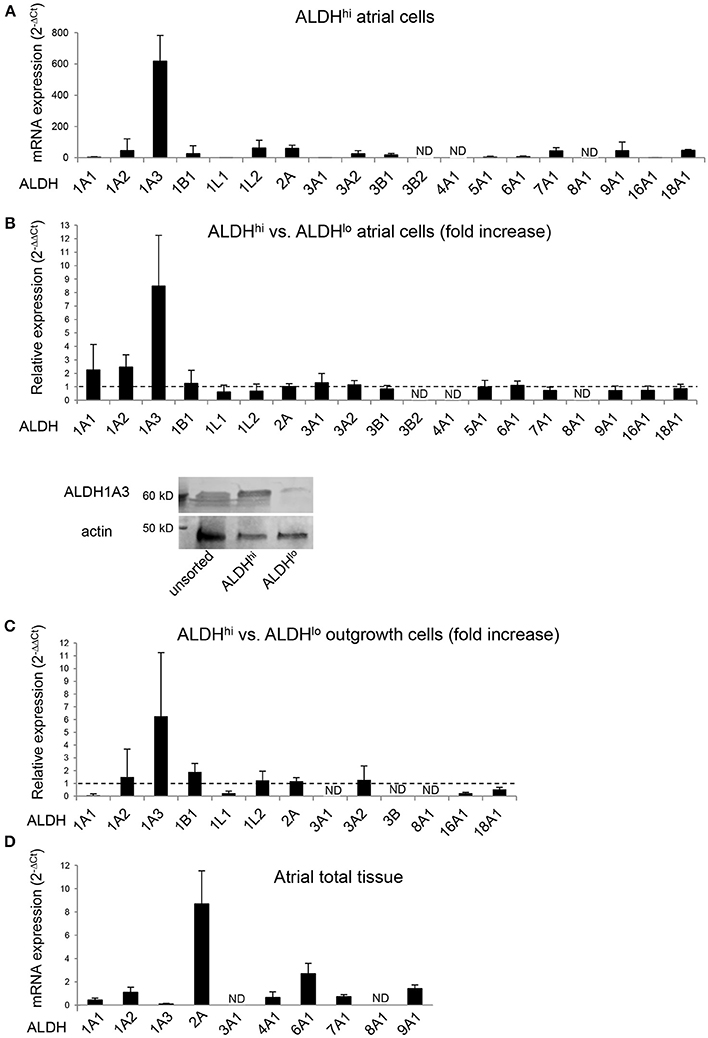
Figure 6. RT-PCR analysis of ALDH isoform gene expression in different populations. (A) ALDHhi cells isolated using enzymatic techniques. Upper panel: ALDH isoform expression (2−ΔCt values; n = 4 patients). Lower panel: Relative expression in ALDHhi vs. ALDHlo cells (2−ΔΔCt values; n = 4). (B) Western blot showing ALDH1A3 protein expression in unsorted, ALDHhi and ALDHlo atrial cells. (C) Outgrowth cells. Relative expression in ALDHhi vs. ALDHlo cells is shown (n = 3). (D) Total atrial tissue (2−ΔCt values; n = 3). ND, not determined (2−ΔΔCt values that could not be calculated).
ALDH1A3 siRNA Reduces ALDH Activity and in Vitro Cell Proliferation
To address the contributions of individual ALDH isoforms to ALDH activity in ALDHhi cells, we examined whether altering their expression levels would affect ALDH activity. Isoform-specific siRNAs were transfected into ALDHhi sorted cells, which were then analyzed using the Aldefluor assay. siALDH1A3 reduced the percentage of ALDHhi cells in a dose-dependent manner up to ≈90% (Figures 7A,B). On the other hand, siALDH1A1, siALDH1A2, siALDH2A, siALDH4A1, and siALDH8A1 (10 nM) did not significantly affect percentages of ALDHhi cells (Figure 7C). To address the role of ALDH1A3 in cell proliferation, siALDH1A3 (10 nM) was transfected into ALDHhi sorted cells and proliferation was assessed using EdU incorporation kit. siALDH1A3 reduced the number of EdU+ cells by ≈40% (Figure 7D).
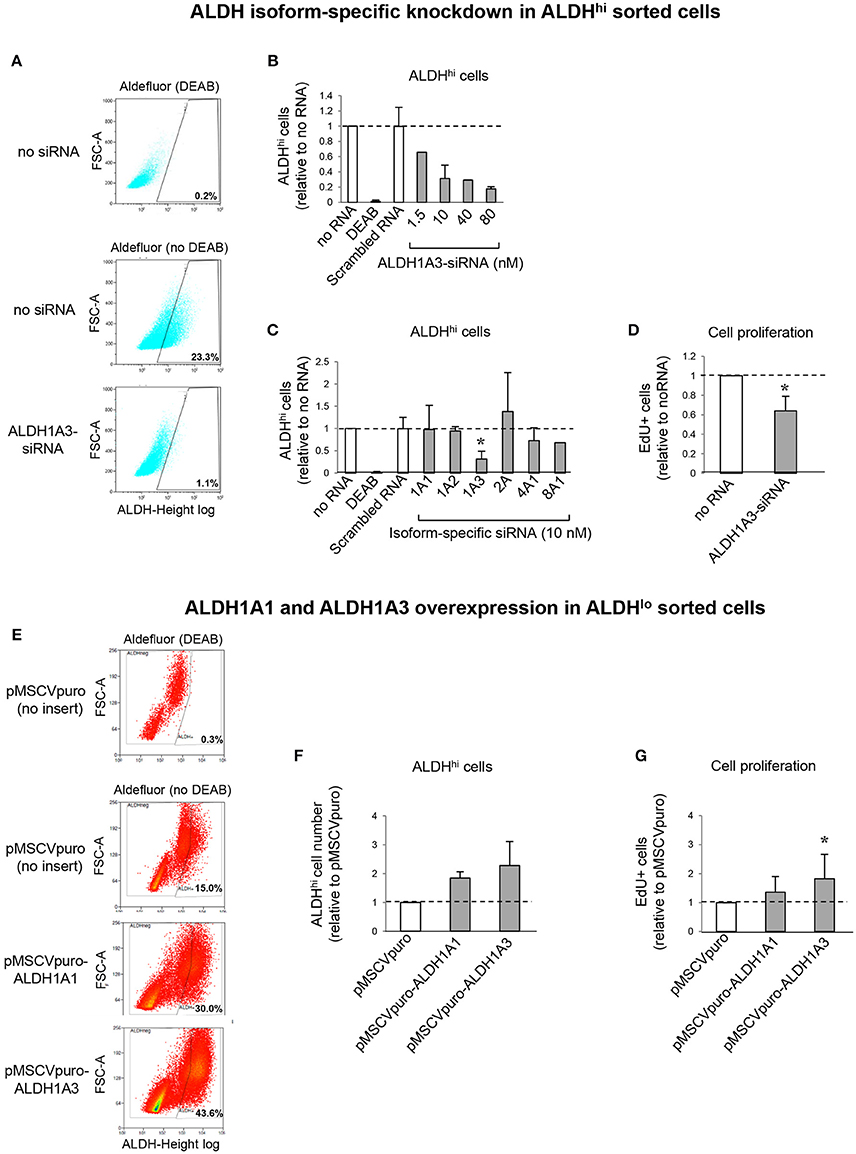
Figure 7. ALDH isoform-specific loss- and gain-of-function studies. (A) Flow cytometry plots of ALDHhi sorted, Aldefluor-reacted cells (with or w/o DEAB) after siALDH1A3 transfection or mock transfection (no siRNA). (B) siALDH1A3 dose-response study. ALDHhi cell numbers after siALDH1A3 transfection using the indicated concentrations relative no siRNA. (C) ALDHhi cell numbers after siALDH-isoform-specific transfection (10 nM) relative to no siRNA). (D) EdU+ cell numbers (%) after siALDH1A3 transfection (10 nM) relative no siRNA. (E) Flow cytometry plots of ALDHlo sorted, Aldefluor-reacted cells (with or w/o DEAB) transduced with pMSCVpuro-ALDH1A1 or pMSCVpuro-ALDH1A3 retroviral vectors, or control vector with no insert (pMSCVpuro). (F) ALDHhi cell numbers after transduction with pMSCVpuro-ALDH1A1 or pMSCVpuro-ALDH1A3 relative to control vector. (G) EdU+ cell numbers (%) after transduction with pMSCVpuro-ALDH1A1 or pMSCVpuro-ALDH1A3 relative to control vector; *p < 0.05.
ALDH1A3 Overexpression Increases in Vitro Cell Proliferation
Having shown that ALDH1A3 knockdown inhibits ALDHhi cell proliferation, we overexpressed this isoform in ALDHlo cells. Retrovirus-mediated ALDH1A3 gene transduction into ALDHlo sorted cells enhanced both ALDH activity (Figures 7E,F) and cell proliferation (Figure 7G) compared with cells infected with control retrovirus with no insert. ALDH1A1 gene transduction also induced a minor increase in Aldefluor-positive cells. Collectively, these results indicate that ALDH1A3 promotes in vitro proliferation of atrial-derived cells.
Discussion
We and others have shown previously that high ALDH activity defines a human cardiac atrial appendage cell population enriched with stem and progenitor cells that have cardiomyogenic potential (33, 35). ALDHhi activity coincides with primitive surface marker (CD34+) as well as mesenchymal and endothelial progenitor marker expression. Regarding the co-expression of CD34 and CD31, CD34+ endothelial progenitor-like cells have been described previously (41). Interestingly, the ALDHhi population is significantly enriched with VCAM1+ cells. Moreover, ALDHhi cells differentially express both pluripotency genes and markers for more established cardiac lineages including Nkx2.5, consistent with them being enriched with both primitive cells and cells committed to a cardiac fate. VCAM1+Nkx2.5+ cells have been identified previously as a discrete lineage of myocardial committed cells during early human cardiovascular development (42). These findings raise the question as to whether the VCAM1+Nkx2.5+ subset of the adult ALDHhi atrial population consists of myocardial committed progenitor cells. These results suggest that ALDHhi atrial cells may be enriched with cells that have cardiomyogenic potential. To address this aspect, we used a novel differentiation protocol based on Notch modulation (38). Compared with ALDHlo cells, ALDHhi cells produced higher numbers of mature cardiomyocytes, as evidenced by higher expression levels of cardiac specific genes (MYH6, MYH7) and higher frequencies of cardiac-specific α-actinin/troponin I double-positive cells. These results indicate that the ALDHhi population is enriched with cardiac progenitor cells. In vivo lineage tracing studies addressing the role of ALDHhi cardiac cells are still needed.
Freshly isolated ALDHhi cells, but not ALDHlo cells, include a subset of clonogenic cells and can be expanded. Annexin V-staining suggests that ALDHhi cells may be more apoptosis-resistant than ALDHlo ones. Higher apoptosis-resistance of ALDHhi vs. ALDHlo cells has been reported in various cancer stem cells (43–45). Silencing ALDH1A by siRNA or shRNA in human melanoma cancer stem cells led to cell cycle arrest, apoptosis and decreased cell viability in vitro and reduced tumorigenesis in vivo (45). In our study, the theoretical possibility that low ALDH activity in a majority of freshly isolated cells is a result of apoptosis, rather than its cause, cannot be ruled out. Nevertheless, DAPI/Annexin-V double-negative ALDHlo cells that were not in the early-mid apoptosis still failed to grow in vitro, suggesting that high ALDH activity might be required in the initial cell culture stages. On the other hand, both the ALDHlo subset of the ALDHhi progeny (i.e., ALDHhi cells having lost their ALDH activity over time in vitro) and ALDHlo outgrowth cells could be expanded, albeit with slower growth kinetics compared with ALDHhi cells. This observation suggests that high ALDH activity may not be required in established cell cultures possibly due to cell adaptation mechanisms.
ALDH isoform expression analysis identified ALDH1A3 as both the most highly expressed isoform in ALDHhi atrial cells and the main differentially expressed isoform in ALDHhi vs. ALDHlo cells. Similar results were found in both freshly isolated and outgrowth cells. Moreover, ALDHhi cells showed higher ALDH1A3 protein levels than ALDHlo cells. siALDH1A3 transfected into ALDHhi cells reduced the number of cells that remained ALDHhi by ≈90%, whereas knocking down ALDH1A1, ALDH1A2, ALDH2A, ALDH4A1, or ALDH8A1 did not significantly affect it. These findings establish ALDH1A3 as the key isoform responsible for ALDH activity in ALDHhi cells. siALDH1A3 transfected into ALDHhi cells reduced their proliferation. Specific ALDH inhibition by DEAB likewise suppressed cell proliferation. Conversely, retrovirus-mediated ALDH1A3 gene transduction into ALDHlo cells promoted it. ALDH1A1 overexpression induced a similar albeit weaker effect, suggesting that exogenous ALDH1A1 may replace the naturally expressed ALDH1A3 isoform in this respect.
ALDH1A is the rate-limiting factor in the production of RA (16–18). We found higher RA levels in ALDHhi atrial cells than in ALDHlo cells. Moreover, the RA receptor agonist, all-trans RA, replaced ALDH in promoting atrial cell growth in the presence of DEAB. In breast cancer cell lines, ALDH1A3 influenced cancer progression via differential RA signaling (44). In human melanoma cancer stem cells, ALDH1A contributed to the stemness through RA-dependent and -independent pathways (45). In HSCs, all-trans RA reversed DEAB-mediated effects on cell expansion and differentiation, suggesting that the ability of ALDH to generate RA is important in determining HSC fate (13). Of note, the catalytic activity of ALDH1A3 for all-trans retinal may be 10-fold higher than that of ALDH1A1 (46). Moreover, ALDH1A3 is transcriptionally activated by all-trans RA in human epidermal keratinocytes, which raises the theoretical possibility of a positive feedback loop between ALDH1A3 and RA in some cell types (47). Our results are consistent with, but do not firmly establish, RA-dependent ALDH effects on atrial cell growth.
Functional roles of ALDH1A3 in stem/progenitor cells are poorly understood. ALDH1A3 promotes neural progenitor cell self-renewal through the regulation of ALDH1A3 pre-mRNA 3′-end processing by RNA-binding protein Sam68 (Khdrbs1). Decreases in ALDH1A3 expression and activity resulted in decreased clonogenicity and reduced the pool of proliferating neural progenitor cells (48). Several studies showed that ALDH1A3 is a cancer stem cell gene. ALDH1A3 expression was detected in 78% of small-sized Ki-67+ proliferating progenitor cells in the human breast cancer cell line HCC1937 (43). Most recently, ALDH1A3 was shown to affect colon cancer in vitro proliferation and invasion depending on CXCR4 status (49). In neuroblastoma cell lines expressing high ALDH1A3 levels, ALDH1A3 knockout via CRISPR/Cas9 gene editing reduced clonogenicity and mediated cell type-dependent inhibition of self-renewal properties of tumor inducing cells (50). Collectively, these findings indicate that ALDH1A3 play functional roles in both normal and cancer stem/progenitor cells.
In conclusion, our results suggest that the ALDHhi population isolated from human cardiac atrial appendages may be enriched with stem/progenitor cells showing cardiomyogenic potential, and that ALDH1A3 is the key ALDH isoform responsible for ALDH activity in these cells, affecting their in vitro proliferation.
Author Contributions
Substantial contributions to the conception or design of the work (GV and SP) or the acquisition, analysis, or interpretation of data for the work (SP, IP, LB, EC, GM, PM, TP, GV), Drafting the work or revising it critically for important intellectual content (SP, IP, GV), Final approval of the version to be published, Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved (SP, IP, LB, EC, GM, PM, TP, GV).
Funding
This study was supported by grants by the Swiss National Science Foundation (310030_140485), the Swiss Heart Foundation, and the Fondation Vaudoise de Cardiologie (Lausanne) to GV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Albert Spicher, Ph.D., carried out the initial characterization of ALDHhi cells and initial ALDH isoform expression analyses. Ludwig von Segesser, M.D., Piergiorgio Tozzi, M.D., Matthias Kirsch, M.D., Enrico Ferrari, M.D., Patrick Ruchat, M.D., Javier Orrit, M.D., Lars Niclauss, M.D., Philipp Gersbach, M.D., and Dominique Delay, M.D., procured heart tissue specimens from patients undergoing heart surgery. Sara Bolis, R.A., Sintia Winkler, Ph.D., and Andreia Marques Ribeiro, Ph.D., provided expert assistance in cell sorting. Darko Maric, Ph.D., and Andrée Porret provided expert assistance in retrovirus production and siRNA transfection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2018.00090/full#supplementary-material
References
1. Sidney LE, Branch MJ, Dunphy SE, Dua HS, Hopkinson A. Concise review: evidence for CD34 as a common marker for diverse progenitors. Stem Cells (2014) 32:1380–9. doi: 10.1002/stem.1661
2. Tateishi K, Ashihara E, Takehara N, Nomura T, Honsho S, Nakagami T, et al. Clonally amplified cardiac stem cells are regulated by Sca-1 signaling for efficient cardiovascular regeneration. J Cell Sci. (2007) 120:1791–800. doi: 10.1242/jcs.006122
3. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell (2003) 114:763–76.
4. Vicinanza C, Aquila I, Scalise M, Cristiano F, Marino F, Cianflone E, et al. Adult cardiac stem cells are multipotent and robustly myogenic: c-kit expression is necessary but not sufficient for their identification. Cell Death Differ. (2017) 24:2101–16. doi: 10.1038/cdd.2017.130
5. Cai J, Weiss ML, Rao MS. In search of “stemness”. Exp Hematol. (2004) 32:585–98. doi: 10.1016/j.exphem.2004.03.013
6. Moreb JS. Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther. (2008) 3:237–46. doi: 10.2174/157488808786734006
7. Balber AE. Concise review: aldehyde dehydrogenase bright stem and progenitor cell populations from normal tissues: characteristics, activities, and emerging uses in regenerative medicine. Stem Cells (2011) 29:570–5. doi: 10.1002/stem.613
8. Xu X, Chai S, Wang P, Zhang C, Yang Y, Wang K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett (2015) 369:50–7. doi: 10.1016/j.canlet.2015.08.018
9. Laird DJ, De Tomaso AW, Weissman IL. Stem cells are units of natural selection in a colonial ascidian. Cell (2005) 123:1351–60. Erratum in: Cell (2006) 124:647–8. doi: 10.1016/j.cell.2005.10.026
10. Jackson B, Brocker C, Thompson DC, Black W, Vasiliou K, Nebert DW, et al. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum Genomics (2011) 5:283–303. doi: 10.1186/1479-7364-5-4-283
11. Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. (2008) 4:697–720. doi: 10.1517/17425255.4.6.697
12. Kastan MB, Schlaffer E, Russo JE, Colvin OM, Civin CI, Hilton J. Direct demonstration of elevated aldehyde dehydrogenase in human hematopoietic progenitor cells. Blood (1990) 74:1945–50.
13. Chute JP, Muramoto GG, Whitesides J, Colvin M, Safi R, Chao NJ, et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA (2006) 103:11707–12. doi: 10.1073/pnas.0603806103
14. Corti S, Locatelli F, Papadimitriou D, Donadoni C, Del Bo R, Crimi M, et al. Transplanted ALDHhiSSClo neural stem cells generate motor neurons and delay disease progression of nmd mice, an animal model of SMARD1. Hum Mol Genet (2006) 15:167–87. doi: 10.1093/hmg/ddi446
15. Hilton J. Role of aldehyde dehydrogenase in cyclophosphamide resistant L1210 leukemia. Cancer Res. (1984) 445:156.
16. Zhao D, McCaffery P, Ivins KJ, Neve RL, Hogan P, Chin WW, et al. Molecular identification of a major retinoic-acid-synthesizing enzyme, a retinaldehyde-specific dehydrogenase. Eur J Biochem (1996) 240:15–22.
17. Elizondo G, Corchero J, Sterneck E, Gonzalez FJ. Feedback inhibition of the retinaldehyde dehydrogenase gene ALDH1 by retinoic acid through retinoic acid receptor alpha and CCAAT/enhancer-binding protein beta. J Biolog Chem. (2000) 275:39747–353. doi: 10.1074/jbc.M004987200
18. Black W, Vasiliou V. The aldehyde dehydrogenase gene superfamily resource center. Hum Genomics (2009) 136:e142. doi: 10.1186/1479-7364-4-2-136
19. Collins SJ. Retinoic acid receptors, hematopoiesis and leukemogenesis. Curr Opin Hematol. (2008) 15:346–51. doi: 10.1097/MOH.0b013e3283007edf
20. Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle (2011) 10:1378–84. doi: 10.4161/cc.10.9.15486
21. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
22. Burger PE, Gupta R, Xiong X, Ontiveros CS, Salm SN, Moscatelli D, et al. High aldehyde dehydrogenase activity: a novel functional marker of murine prostate stem/progenitor cells. Stem Cells (2009) 27:2220–8. doi: 10.1002/stem.135
23. Marcato P, Dean C, Pan D, Araslanova R, Gillis M, Joshi M, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells (2011) 29:32–45. doi: 10.1002/stem.563
24. Singh S, Arcaroli J, Chen Y, Thompson DC, Messersmith W, Jimeno A, et al. ALDH1B1 is crucial for colon tumorigenesis by modulating Wnt/beta-Catenin, notch and PI3K/Akt signaling pathways. PLoS ONE (2015) 10:e0121648. doi: 10.1371/journal.pone.0121648
25. Yan J, De Melo J, Cutz JC, Aziz T, Tang D. Aldehyde dehydrogenase 3A1 associates with prostate tumorigenesis. Br J Cancer (2014) 110:2593–603. doi: 10.1038/bjc.2014.201
26. Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Luderman S, et al. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci USA. (1999) 96:9118–23.
27. Gunduz E, Demirel G, Bal C, Gulbas Z. Evaluation of mobilized peripheral stem cells according to CD34 and aldehyde dehydrogenase expression and effect of SSC(lo) ALDH(br) cells on hematopoietic recovery. Cytotherapy (2010) 12:1006–12. doi: 10.3109/14653249.2010.509393
28. Shoulars K, Noldner P, Troy JD, Cheatham L, Parrish A, Gentry T, et al. Development and validation of a rapid, aldehyde dehydrogenase bright-based cord blood potency assay. Blood (2016) 127:2346–54. doi: 10.1182/blood-2015-08-666990
29. Capoccia BJ, Robson DL, Levac KD, Maxwell DJ, Hohm SA, Neelamkavil MJ, et al. Revascularization of ischemic limbs after transplantation of human bone marrow cells with high aldehyde dehydrogenase activity. Blood (2009) 113:5340–51. doi: 10.1182/blood-2008-04-154567
30. Sondergaard CS, Hess DA, Maxwell DJ, Weinheimer C, Rosová I, Creer MH, et al. Human cord blood progenitors with high aldehyde dehydrogenase activity improve vascular density in a model of acute myocardial infarction. J Transl Med. (2010) 8:24. doi: 10.1186/1479-5876-8-24
31. Perin EC, Murphy MP, March KL, Bolli R, Loughran J, Yang PC, et al. Cardiovascular cell therapy research network (CCTRN). Evaluation of cell therapy on exercise performance and limb perfusion in peripheral artery disease: The CCTRN PACE Trial (Patients with intermittent claudication injected with ALDH bright cells). Circulation (2017) 135:1417–28. doi: 10.1161/CIRCULATIONAHA.116.025707
32. Perin EC, Silva GV, Zheng Y, Gahremanpour A, Canales J, Patel D, et al. Randomized, double-blind pilot study of transendocardial injection of autologous aldehyde dehydrogenase-bright stem cells in patients with ischemic heart failure. Am Heart J (2012) 163:415–21. doi: 10.1016/j.ahj.2011.11.020
33. Spicher A, Meinhardt A, Roehrich ME, Vassalli G. Phenotypic characterization of murine and human cardiac-resident progenitor cells isolated on basis of aldehyde-dehydrogenase activity. Circulation (2007) 116:II−167–II−168.
34. Roehrich ME, Spicher A, Milano G, Vassalli G. Characterization of cardiac-resident progenitor cells expressing high aldehyde dehydrogenase activity. Biomed Res Int. (2013) 2013:503047. doi: 10.1155/2013/503047
35. Koninckx R, Daniëls A, Windmolders S, Mees U, Macianskiene R, Mubagwa K, et al. The cardiac atrial appendage stem cell: a new and promising candidate for myocardial repair. Cardiovasc Res. (2013) 97:413–23. doi: 10.1093/cvr/cvs427
36. Morgan CA, Parajuli B, Buchman CD, Dria K, Hurley TD. N,N-diethylaminobenzaldehyde (DEAB) as a substrate and mechanism-based inhibitor for human ALDH isoenzymes. Chem Biol Interact. (2015) 234:18–28. doi: 10.1016/j.cbi.2014.12.008
37. Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. (2004) 95:911–21. doi: 10.1161/01.RES.0000147315.71699.51
38. Plaisance I, Perruchoud S, Fernandez-Tenorio M, Gonzales C, Ounzain S, Ruchat P, et al. Cardiomyocyte lineage specification in adult human cardiac precursor cells via modulation of enhancer-associated long noncoding RNA expression. J Am Coll Cardiol Basic Trans Sci. (2016) 1:472–93. doi: 10.1016/j.jacbts.2016.06.008
39. Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet (2012) 379:895–904. doi: 10.1016/S0140-6736(12)60195-0
40. Alnouti Y, Klaassen CD. Tissue distribution, ontogeny, and regulation of aldehyde dehydrogenase (Aldh) enzymes mRNA by prototypical microsomal enzyme inducers in mice. Tox Sci. (2008) 101:51–64. doi: 10.1093/toxsci/kfm280
41. Ferreras C, Cole CL, Urban K, Jayson GC, Avizienyte E. Segregation of late outgrowth endothelial cells into functional endothelial CD34- and progenitor-like CD34+ cell populations. Angiogenesis (2015) 18:47–68. doi: 10.1007/s10456-014-9446-1
42. Skelton RJP, Costa M, Anderson DJ, Bruveris F, Finnin BW, Koutsis K, et al. SIRPA, VCAM1 and CD34 identify discrete lineages during early human cardiovascular development. Stem Cell Res. (2014) 13:172–9. doi: 10.1016/j.scr.2014.04.016
43. Kashii-Magaribuchi K, Takeuchi R, Haisa Y, Sakamoto A, Itoh A, Izawa Y, et al. Induced expression of cancer stem cell markers ALDH1A3 and Sox-2 in hierarchical reconstitution of apoptosis-resistant human breast cancer cells. Acta Histochem Cytochem. (2016) 49:149–58. doi: 10.1267/ahc.16031
44. Marcato P, Dean CA, Liu RZ, Coyle KM, Bydoun M, Wallace M, et al. Aldehyde dehydrogenase 1A3 influences breast cancer progression via differential retinoic acid signaling. Mol Oncol. (2015) 9:17–31. doi: 10.1016/j.molonc.2014.07.010
45. Luo Y, Dallaglio K, Chenc Y, Robinson WA, Robinson SE, McCarter MD, et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells (2012) 30:2100–13. doi: 10.1002/stem.1193
46. Sima A, Parisotto M, Mader S, Bhat PV. Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates. Biochim Biophys Acta (2009) 1790:1660–4. doi: 10.1016/j.bbagen.2009.09.004
47. Koenig U, Amatschek S, Mildner M, Eckhart L, Tschachler E. Aldehyde dehydrogenase 1A3 is transcriptionally activated by all-trans-retinoic acid in human epidermal keratinocytes. Biochem Biophys Res Commun. (2010) 400:207–11. doi: 10.1016/j.bbrc.2010.08.035
48. La Rosa P, Bielli P, Compagnucci C, Cesari E, Volpe E, Farioli Vecchioli S, et al. Sam68 promotes self-renewal and glycolytic metabolism in mouse neural progenitor cells by modulating Aldh1a3 pre-mRNA 3′-end processing. Elife (2016) 5:e20750. doi: 10.7554/eLife.20750
49. Feng H, Liu Y, Bian X, Zhou F, Liu Y. ALDH1A3 affects colon cancer in vitro proliferation and invasion depending on CXCR4 status. Br Cancer (2018) 118:224–32. doi: 10.1038/bjc.2017.363
Keywords: aldehyde dehydrogenase, ALDH1A3, cardiac progenitor cell, stem cell, heart
Citation: Puttini S, Plaisance I, Barile L, Cervio E, Milano G, Marcato P, Pedrazzini T and Vassalli G (2018) ALDH1A3 Is the Key Isoform That Contributes to Aldehyde Dehydrogenase Activity and Affects in Vitro Proliferation in Cardiac Atrial Appendage Progenitor Cells. Front. Cardiovasc. Med. 5:90. doi: 10.3389/fcvm.2018.00090
Received: 05 February 2018; Accepted: 25 June 2018;
Published: 24 July 2018.
Edited by:
Rajesh Katare, University of Otago, New ZealandReviewed by:
Daniele Torella, Università degli studi Magna Græcia di Catanzaro, ItalyParul Dixit, Bristol Heart Institute, United Kingdom
Copyright © 2018 Puttini, Plaisance, Barile, Cervio, Milano, Marcato, Pedrazzini and Vassalli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Vassalli, Z2l1c2VwcGUudmFzc2FsbGlAY2FyZGlvY2VudHJvLm9yZw==