 Matthijs Steven Ruiter
Matthijs Steven Ruiter Maurizio Pesce
Maurizio Pesce- Cardiovascular Tissue Engineering Unit, Centro Cardiologico Monzino (IRCCS), Milan, Italy
Autologous saphenous veins are the most commonly used conduits in revascularization of the ischemic heart by coronary artery bypass graft surgery, but are subject to vein graft failure. The current mini review aims to provide an overview of the role of mechanotransduction signalling underlying vein graft failure to further our understanding of the disease progression and to improve future clinical treatment. Firstly, limitation of damage during vein harvest and engraftment can improve outcome. In addition, cell cycle inhibition, stimulation of Nur77 and external grafting could form interesting therapeutic options. Moreover, the Hippo pathway, with the YAP/TAZ complex as the main effector, is emerging as an important node controlling conversion of mechanical signals into cellular responses. This includes endothelial cell inflammation, smooth muscle cell proliferation/migration, and monocyte attachment/infiltration. The combined effects of expression levels and nuclear/cytoplasmic translocation make YAP/TAZ interesting novel targets in the prevention and treatment of vein graft disease. Pharmacological, molecular and/or mechanical conditioning of saphenous vein segments between harvest and grafting may potentiate targeted and specific treatment to improve long-term outcome.
Introduction
Autologous saphenous veins (SVs) are the most commonly used conduits in revascularization of the ischemic heart by coronary artery bypass graft surgery (CABG), due to their availability, accessibility for harvesting and easy handling when making anastomoses. Continuous improvements in surgical techniques have improved clinical outcome and short-term graft patency; on the other hand, the long term SV grafts have a cumulative 10 year patency of 60% due to graft disease (VGD), and are still outperformed by arterial grafts (1–6). In VGD, three subsequent phases are distinguished: thrombosis, which occurs typically within the first month, intima hyperplasia, which occurs up to 1 year after surgery, and finally atherosclerosis, which becomes predominant several years following CABG. SV graft patency is affected by many factors, ranging from surgical technique and SV preservation before implantation, to SV quality, patient comorbidities and medication, and is associated with quality of life (7–10). The role of mechanosensing in the progression of graft failure has been recognized and is predictive for later graft patency. However, the effects of individual factors remain hard to determine, due to the difficulty of addressing mechanical cues in experimental systems and in vivo (11, 12). In the circulation, blood vessels are exposed to several mechanical stimuli simultaneously, including shear stress on endothelial cells (ECs), luminal pressure and circumferential stretch, both of which exert their effect mainly on smooth muscle cells (SMCs), and longitudinal stretch. These conditions vary widely throughout the circulation, and regulate blood pressure, vascular permeability, and attraction and invasion of inflammatory cells. The interplay of all these factors determines the local vascular microenvironment, and dictates physiological and pathological remodelling of blood vessels. This is particularly relevant in VGD, when SVs are exposed to challenging conditions that are widely different from their native environment. Therefore, the current review aims to provide an overview of the role of the mechanobiology underlying VGD, with a focus on pathways participating in detection of physiologic and pathologic mechanical stress, in an attempt to further our understanding of the disease progression and to improve future clinical treatment.
Vein Harvest and Engraftment
Both the surgical procedure and the new hemodynamic conditions have pervasive consequences for structure as well as function of the SV. Harvesting of the vein inevitably results in denervation and loss of external blood supply through vasa vasorum, inducing ischemia and potentially changing vasomotor tone and potentiating vasospasm (13). Subsequently, high-pressure distension is routinely performed to check for leakage and to reduce vasospasm. Together with ischemia and reperfusion injury after engraftment, this causes damage and loss of ECs and SMCs through oxidative stress and cytotoxic activation (14–17). The extent of damage is dependent on the type of harvesting; endoscopic techniques are associated with a lower rate of leg infection, whereas open vein harvesting (OVH) seems to be superior in terms of long-term patency. However, results from different studies have not been conclusive (18–21). The same limitations are true for traditional versus no touch harvesting, where the no touch technique seems better for graft patency by (i) leaving the peri-adventitial tissue, (ii) providing physical support, and (iii) maintaining the integrity of both endothelial and adventitial cells (4, 22, 23). Vessel damage during harvest and engraftment complicates the SV recovery and adaptation capacity to the new conditions, and the combination of SV excision and arterialization results in increased vasoconstriction and reduced endothelium-mediated vasodilation (24, 25).
Adaptation to a New Hemodynamic Environment
Immediately after engraftment, SVs are challenged with hemodynamic conditions to which they need to adapt. Under native conditions, SVs experience low pressure loads (5–10 mmHg) and quasi-steady flow patterns with low shear stresses (0.1–0.6 Pa). By contrast, after CABG, SVs are subjected to high pulsatile pressure (120/80 mmHg) with a circumferential strain of 10–15%, a mean flow rate up to 250 ml/min, and a high wall shear stress (0.75–2.25 Pa) (26, 27). Opposite to arteries, SVs are anisotropic and become incompliant at high pressures (28). The increases in flow and shear stress together with wall tension, result in additional loss of ECs, damage to SMCs and extracellular matrix (ECM) alterations (29, 30). Stretching of SMCs disrupts actin bundles and results in a structure with scattered pores, followed by loss of SMC nuclei and actin filaments, inducing SMC proliferation (31). Subsequently, deposition of platelets and fibrin takes place, and leukocytes from the circulation infiltrate the vessel wall. Next, growth factors are released from platelets, SMCs and macrophages, which leads to SMC proliferation and migration to the intima, as well as ECM deposition, leading to intima hyperplasia (14, 29, 30).
Untangling Mechanical Factors
The new hemodynamic environment exposes the SV to various different mechanical factors simultaneously, which can act either synergistically or antagonistically. Mechanical static deformations and stresses, increased pulsatile deformations and stresses, and altered shear stress, require the vein graft to acquire an artery-like structure with geometric remodelling and wall stiffening, but also induce intima hyperplasia and inflammation, which may induce failure in the long term (32).
Exposure to a combination of increased flow, pressure and shear stress leads to rearrangement of SMCs, both ex vivo and in vivo (31, 33). To untangle the separate effects of flow and pressure, several ex vivo perfusion models have addressed single or combined mechanical factors on SV adaptation and pathophysiology. Under continuous laminar flow, an increase in flow reduces intimal hyperplasia, while an increase in pressure induces intimal accumulation. When flow and pressure increase simultaneously, apoptosis becomes apparent in the vessel wall after day 1, resulting in lower cell density and media thinning. Some ex vivo models find intima accumulation after 1 week, but this is not completely consistent between models (33–39).
Exposure of SVs to arterial shear stress under low pressure induced expression and activity of matrix metalloproteinase (MMP)−2 and MMP-9, tissue inhibitor of metalloproteinase-1, as well as expression of plasminogen activator inhibitor-1 and osteopontin. Venous marker Ephrin B4 on the other hand, was attenuated under arterial shear stress (40, 41). Endothelial cells harbour a multitude of sensors on their apical, junctional, and basal surfaces to sense mechanical signals from the blood, which have key roles during developmental, physiological, and pathological processes. Mechanosensing molecules include junctional proteins, receptor kinases, integrins, focal adhesions, G-proteins and G-protein-coupled receptors, ion carriers, actin cytoskeleton, primary cilia, and the glycocalyx, enabling the vasculature to respond to changing demands within seconds or during the course of several days. The pathways through which the different sensors transduce the physical signals remains elusive, but integrins seem to play an important role. The mechanisms of shear stress sensing and transduction are reviewed extensively elsewhere (42–44). The effects of shear stress are mainly mediated by ECs. However, exposure of SMCs to shear stress cannot be excluded, due to increased pressure and fenestration, and in particular due to endothelial denudation early after engraftment (45). What is important to distinguish in the context of VGD, is the difference between the effects of laminar flow and disturbed flow on the ECs. In fact, laminar flow results in EC alignment, stress fiber formation, low proliferation, and elevated Krüppel-like Factor 2 expression, whereas NF-κB activation, expression of adhesion molecules ICAM-1 and VCAM-1, high EC turnover, and production of reactive oxygen species are the result of disturbed flow (43). SMCs are sensitive to deformations and strain induced by arterial pressure. Increases in pressure are associated with elevated vasoconstriction and attenuated nitric oxide-mediated dilatation (46). Veins perfused for 7 days under high pressure exhibited enhanced expression of transforming growth factor (TGF)-β1 and upregulation of microRNAs-138/200b/200 c, suppression of tissue inhibitor of metallo-protease-1, and equal or more intima hyperplasia than veins exposed to low pressure (40, 47). In vitro straining of isolated human cells has revealed that cyclic stretch of SMCs induces the number of cells and DNA synthesis, collagen, fibronectin and TGF-β expression, and MMP2 activity, while reducing αSMA and p27kip1 expression, resulting in a proliferative SMC phenotype (29, 48–50). These effects were detected in cells derived from SVs, but absent in cells from the internal thoracic artery (48, 49). Moreover, cellular responses are specific for the type of strain, indicated by Asanuma et al., who found that static but not cyclic stretch induced MMP-9 mRNA levels and MMP-2 mRNA, protein content and secretion (51). Interestingly, in rat SMCs, cyclic strain attenuated SMC proliferation and upregulated VEGF secretion, which could be beneficial for re-endothelialisation of SV grafts (52).
The role of adventitial cells in the development of VGD is acknowledged in animal models (53, 54), but remains elusive in the human pathology (55, 56). Ex vivo, CABG-like oxygen levels, i.e., high luminal oxygen levels, and deprivation of adventitial oxygen due to severing of the vasa vasorum, induced proliferation of adventitial microvasculature even before SMC proliferation, and resulted in neovascularization (57, 58). The adventitia of human SVs harbours CD34-positive cells, which display multilineage differentiation capacities, and were able to induce neovascularization in mice (59, 60). Study of the effects of mechanical factors on adventitial cell function is needed to elucidate the relevance of these cells in physiologic and pathologic remodelling in VGD. Key experimental findings in mechanotransduction relevant for VGD are summarized in Table 1.
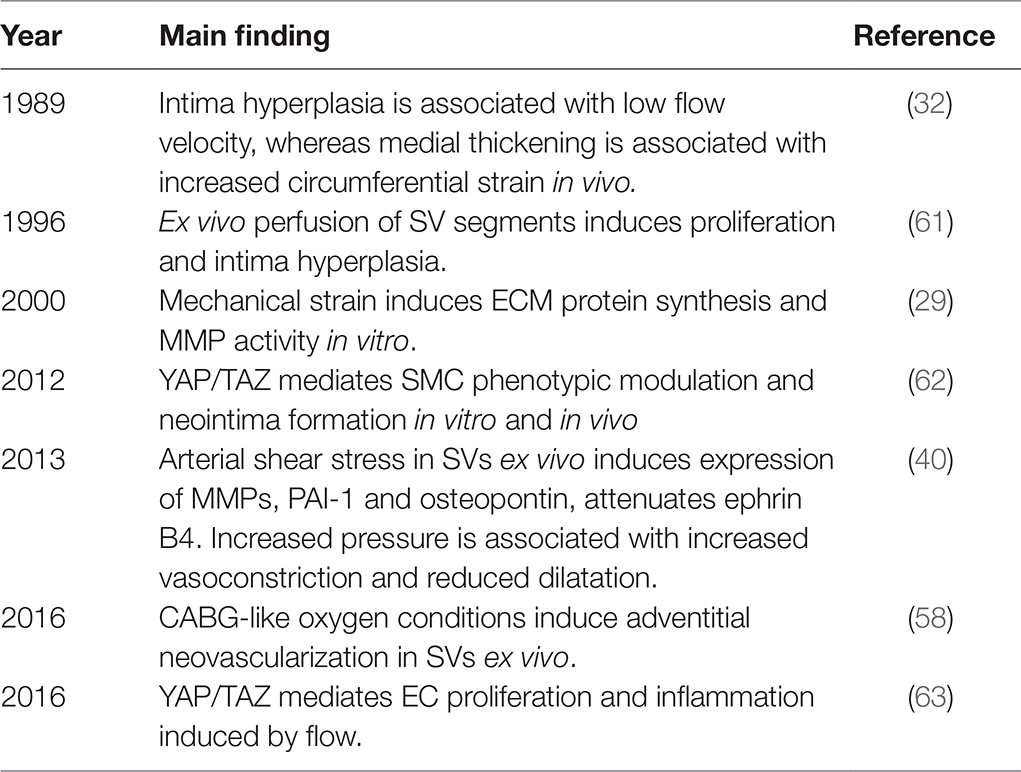
Table 1. Key experimental findings of mechanotransduction relevant for VGD.
As in all vascular remodelling events, inflammatory mediators are involved in VGD. Both systemic and local inflammatory markers are associated with SV remodeling and VGD (64–69). Since these factors are, at the moment, only indirectly connected to mechanosensing, they fall beyond the scope of this review.
YAP/TAZ Signalling
The Hippo pathway is a growth control signalling pathway first described in the context of developmental biology, but has since emerged as an important factor in uncontrolled cell growth in cancer. Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) are the main downstream effectors of the Hippo pathway regulating cell survival, proliferation and apoptosis. Activity of the YAP/TAZ complex is confined to cells exposed to stiffness of the surrounding extracellular matrix, localization at edges and curvatures, or cells undergoing mechanical straining (70). YAP and TAZ exert their effect by nuclear translocation and binding to transcriptional enhancer associate domain (TEAD), which induces YAP target gene transcription. Currently, evidence is accumulating that YAP and TAZ are involved in vascular remodelling and cardiovascular disease, such as pulmonary hypertension, atherosclerosis and restenosis (71). This may also implicate a role for YAP/TAZ in VGD.
In ECs, YAP is a transcriptional regulator that transduces signals from VE-cadherin mediated endothelial cell-cell contact to the nucleus. ECs exposed to disturbed flow, display YAP/TAZ activation and nuclear translocation with subsequent target genes upregulation, including ankyrin repeat domain 1 (ANKRD1), cysteine-rich angiogenic inducer 61 (CYR61), and connective tissue growth factor (CTGF), resulting in increased proliferation and decreased apoptosis of ECs. On the other hand, laminar flow suppressed YAP/TAZ, with downregulation of YAP target genes ANKRD1, CYR61, CTGF, baculoviral IAP repeat-containing 5, and angiopoietin-2. This suggests an atheroprotective role of the Hippo-kinase pathway that antagonizes the YAP/TAZ nuclear translocation by enhancing their phosphorylation levels (63, 72–75). In mice, EC-specific YAP/TAZ deletion led to a hyper-pruned vascular network, disrupted barrier integrity and reduced neovascularization (76). In accordance, YAP has been demonstrated to be critical in retinal angiogenesis (77, 78). Moreover, in vivo blockade of YAP/TAZ translation significantly suppressed endothelial inflammation and the size of atherosclerotic lesions in mice (79). YAP/TAZ inhibition in ECs suppressed c-Jun N-terminal kinase (JNK) signalling and downregulated expression of pro-inflammatory genes, reducing monocyte attachment and infiltration (79). In zebrafish, YAP nuclear translocation was shown to be transient, shuttling in or out of the nucleus within 10 min after starting or stopping flow. Interestingly, in this model, laminar flow induced nuclear translocation of YAP, even though this was during blood vessel formation (80).
In addition to shear-sensing ECs, YAP is implicated in SMC function, where YAP levels are consistently associated with proliferation, migration and a synthetic phenotype. In patient material, YAP was markedly lower in the aortic wall of patients with ascending aortic aneurysms compared with healthy aortic samples, while downregulation of YAP in SMCs was associated with ECM disorders of the media (81). In animal models of arterial injury, it was demonstrated that YAP levels are elevated in response to injury, correlating with a synthetic SMC phenotype (62, 82). In cultured rat cells, YAP knockdown impaired SMC proliferation and enhanced the expression of contractile SMC genes by upregulating myocardin expression (82). Overexpression of YAP induced SMC proliferation and migration, whereas genetic deletion of YAP attenuated the injury-induced phenotypic switch in SMCs and attenuated neointima formation in mice (62). In accordance, inhibition of YAP/TAZ function with RNA interference or Verteporfin significantly reduced vascular SMC proliferation (83). In breast cancer cell lines, the effect of YAP on proliferation is at least partially mediated by p27kip1, but this pathway has not yet been confirmed in vascular (mal)adaptation (84).
In addition to ECs and SMCs, the YAP/TAZ pathway is involved in the cellular response to stretch in mesenchymal stem cells (85). Evidence suggests that YAP can expand stem cell pools and induce reprogramming of differentiated cells to stem cell-like progenitor phenotypes (86). Finally, YAP is linked to platelet function, since platelet-released thromboxane A2 (TXA2) induces YAP activation, facilitating wound healing in response to vascular injury (87). Together, these studies indicate the broad role of YAP signalling in the mechanosensing of vascular tissue relevant in the development of VGD, and may provide an opportunity for therapeutic intervention. However, it is still to be unveiled how mechanical signals lead to changes in YAP and TAZ localization, in the cell cytoplasm under pathologic conditions (88–90). On the other hand, very recent findings suggest that force generation by docking stress fibres to focal adhesion contacts and translation of this tension to nuclear deformation and opening of the nuclear pores promote YAP nuclear translocation, thus connecting directly mechanosensing and YAP-dependent transcriptional circuitries (91, 92).
Therapeutic Opportunities
As discussed elsewhere, the problem of SV graft patency remains a clinical priority (93, 94). A first improvement of clinical outcome may be obtained by limiting the initial damage to the SV. As discussed above, comparisons of open vs. endoscopic isolation, and traditional vs. no touch harvesting have not been conclusive, but are very relevant for future clinical practice. Open, no touch harvesting might be the least damaging technique, but this has to be confirmed by future research. However, a reduction of damage is not likely to prevent VGD completely. Therefore, additional therapy remains necessary with the clear primary outcome to increase the longevity of the vein grafts, and prevent re-interventions such as redo graft implantation or graft stenting, two risky procedures.
Since SMC hyperplasia is a hallmark of VGD, it seems worthwhile investigating the cell cycle. To support this notion, there are promising results from a single nucleotide polymorphism in the p27Kip1 gene. p27Kip1, together with other cyclin-dependent kinase inhibitors, is upregulated during vascular repair and negatively regulates growth of vascular SMCs in vivo (95, 96). The p27(kip1)−838AA genotype is associated with a reduction in coronary artery in-stent restenosis and improved patency of lower extremity bypass grafts, through inhibition of both SMCs and fibroblasts (55, 97, 98). Intervention in the cell cycle may be an interesting approach to inhibit SMC proliferation in VGD, and experience may be drawn from drug-eluting stent research (99).
Due to the complex nature of VGD, schematically represented in Figure 1, identification of therapies that target multiple cell types and/or are upstream from multiple processes, are most likely to lead to a robust inhibition of thrombosis, intima hyperplasia and/or atherosclerosis. Experimental evidence from vascular pathology, injury and adaptation suggests several options. Nuclear receptor Nur77 has demonstrated effects across multiple cell types in atherosclerosis, where it inhibits lesion formation by reducing SMC proliferation, reduces MMPs and attenuates macrophage accumulation and infiltration (100–103). In a rabbit model of in-stent restenosis, Nur77 activator 6-mercaptopurine (6-MP) inhibited intima hyperplasia, although this effect was not observed in a porcine model (104, 105). In addition, Nur77 enhances EC survival and lowers vasoconstrictor endothelin-1 (106, 107). Interestingly, Nur77 levels were elevated both by mechanical stretch of isolated venous SMCs, and by arterial pressure in vein segments, while activation of these receptors reduced SMC proliferation and elevated expression of contractile SMC markers (48). In contrast, in a rat carotid interposition model, Nur77 inhibition reduced intima hyperplasia (108). Therefore, more research is needed to establish the role and therapeutic potential of Nur77 in VGD.
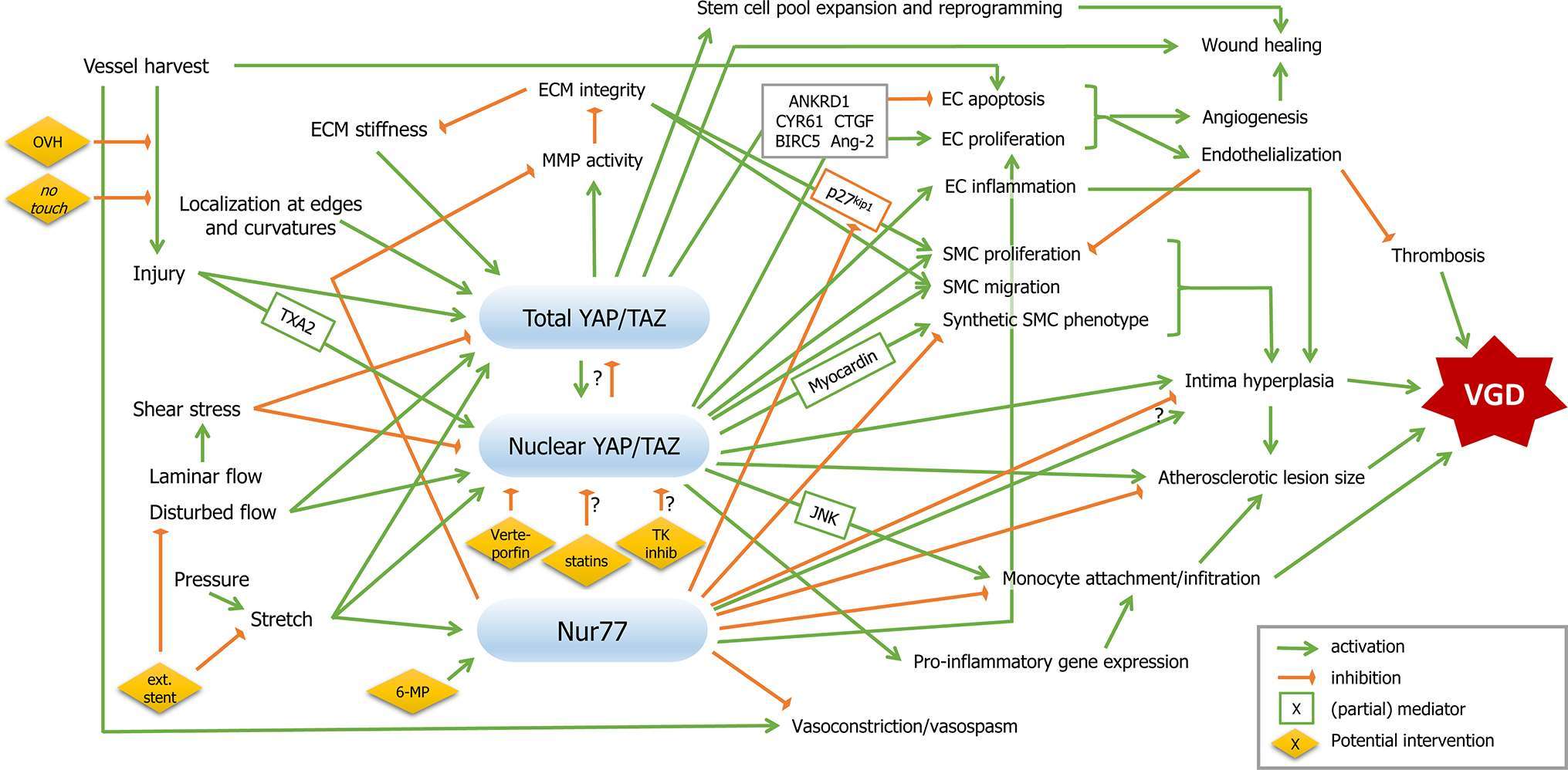
Figure 1. Mechanotransduction and therapeutic targets in coronary vein graft disease. Mechanical factors are depicted on the left, vein graft disease on the right, and signalling pathways described in this review in between. Signalling nodes are marked in blue. Green arrows indicate activation, orange inhibition. Boxed factors are (partial) mediators. Potential therapeutic interventions are presented in yellow diamonds. Abbreviations are explained in the main text.
An interesting approach, which might be beneficial in terms of both prevention and repair, is external stenting of SV grafts. Both in vitro and ex vivo, external stents consistently attenuated intima hyperplasia (109–112). In accordance, reduction of neointima formation was reported in a clinical study after 1 year, however without significantly affecting SVG failure rate (113). These responses are mediated by both strain and shear stress; clinically, oscillatory shear rate was lower in the stented group, and correlated with the development of diffuse intima hyperplasia, whereas ex vivo work implicated wall tension, which was lower in the stented group, attenuating intimal hyperplasia, medial SMC apoptosis and subsequent medial fibrosis (110, 114). Additional clinical results are needed to evaluate the efficacy of this promising treatment.
As discussed in the previous section, YAP levels and nuclear translocation play a central role in the translation of mechanical signals into cellular responses, not only in shear-sensing ECs and strain-sensitive SMCs, but also with a link to ECM components, stem cells and even the blood. Inhibition of YAP/TAZ signalling inhibits EC inflammation and SMC proliferation, and is associated with a contractile SMC phenotype and attenuation of atherosclerosis, which are all central elements in VGD. For therapeutic application of YAP inhibition, cancer research has identified several potent inhibitors, including Verteporfin, tyrosine kinase (TK) inhibitors and statins (115, 116). Simvastatin was associated with reduced neointima formation, which could partially be explained by inhibition of SMC proliferation and migration, and lowering of LDL levels and MMP-9 activity (117–119). In the CASCADE trial, statin therapy was associated with decreased intima accumulation after 1 year (120). The relation between statins and YAP however, was not investigated in these studies. Further research should shed light on the feasibility and efficacy of YAP inhibition in the prevention of VGD.
Regardless of the therapeutic target selected for prevention of VGD, CABG offers a unique opportunity for topical delivery of pharmacological, molecular or mechanical therapy. Between harvest and grafting, the SV segment is maintained in a physiological solution. Ideally, for the patient this time is kept as short as possible, but can still allow a window for treatment, allowing exposure to specific drugs or gene expression interfering strategies, e.g., siRNAs, antagoMIRs or agoMIRs (121). For example, agomiR-33 and adenovirus-mediated microRNA-21 gene transfer attenuated neointima formation in rat vein grafts (122, 123). Conversely, Smooth Muscle Enriched Long Noncoding RNA (SMILR) knockdown inhibited cell proliferation in vitro (124). As a different approach, more complex treatments can be considered, with a dedicated easy-to-use bioreactor, allowing mechanical preconditioning of the SV, exposure to different molecules/drugs on the luminal and adventitial side (58), or combinations thereof. For preconditioning or molecular/pharmacological pre-treatment, one could consider intervening in a mechanical event, in the cell cycle, in potential cell phenotypic modulation, in paracrine signalling, or in other processes. Depending on the putative target, the optimal delivery modality has to be devised. Pharmacological pretreatment seems a feasible approach and, from a technical point of view, is probably the least challenging method. It remains to be confirmed whether inhibition of YAP/TAZ signalling in this stage is beneficial. In this view, administration of Verteporfin, statins and TK inhibitors would provide a feasible and promising approach. In addition, oral statin treatment may be beneficial after CABG.
Conclusions
Clinical therapy to prevent or attenuate the development of VGD are necessary to improve long-term SV patency. Refinements in harvesting and grafting techniques can limit the damage, but are not sufficient to prevent VGD. A better understanding of the mechanotransduction signalling pathways underlying this condition may give rise to the development novel therapeutic strategies. Due to the complex nature of VGD, upstream targets that affect multiple mechanisms are most likely to form a robust therapy. Inhibition of YAP/TAZ signalling, responsive to both shear stress and mechanical strain, reduces not only EC inflammation and SMC proliferation and migration, but also monocyte attachment and infiltration, and may therefore be an effective therapy for VGD. Pharmacological and/or mechanical conditioning of the SV between harvest and grafting should be considered, since it allows for targeted and specific treatment.
Author Contributions
MR analyzed the literature and wrote the manuscript. MP wrote sections of the manuscript and critically reviewed the manuscript. Both authors contributed to manuscript conception and revision, and both read and approved the submitted version.
Funding
The present work was supported by a Health Research Grant (Ricerca Finalizzata: RF-2011-02346867).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MF and handling Editor declared their shared affiliation.
References
1. Motwani JG, Topol EJ. Aortocoronary saphenous vein graft disease: pathogenesis, predisposition, and prevention. Circulation (1998) 97(9):916–31. doi: 10.1161/01.CIR.97.9.916
2. Owens CD. Adaptive changes in autogenous vein grafts for arterial reconstruction: clinical implications. J Vasc Surg (2010) 51(3):736–46. doi: 10.1016/j.jvs.2009.07.102
3. Walts AE, Fishbein MC, Matloff JM, Thrombosed MJM. Thrombosed, ruptured atheromatous plaques in saphenous vein coronary artery bypass grafts: ten years' experience. Am Heart J (1987) 114(4 Pt 1):718–23. doi: 10.1016/0002-8703(87)90780-0
4. Sen O, Gonca S, Solakoglu S, Dalcik H, Dalcik C, Ozkara A. Comparison of conventional and no-touch techniques in harvesting saphenous vein for coronary artery bypass grafting in view of endothelial damage. Heart Surg Forum (2013) 16(4):177–83. doi: 10.1532/HSF98.20131006
5. Hwang HY, Lee KH, Han JW, Kim KB. Equivalency of saphenous vein and arterial composite grafts: 5-year angiography and midterm clinical follow-up. Ann Thorac Surg (2016) 102(2):580–8. doi: 10.1016/j.athoracsur.2016.02.017
6. Shi WY, Tatoulis J, Newcomb AE, Rosalion A, Fuller JA, Buxton BF. Is a third arterial conduit necessary? Comparison of the radial artery and saphenous vein in patients receiving bilateral internal thoracic arteries for triple vessel coronary disease. Eur J Cardiothorac Surg (2016) 50(1):53–60. doi: 10.1093/ejcts/ezv467
7. Bas M, Luther B, Knopf A, Suvorava T, Kojda G. Preservation of endothelial and smooth muscle function of human saphenous vein transplants. Exp Clin Transplant (2016) 14(1):86–92.
8. Une D, Kulik A, Voisine P, Le May M, Ruel M. Correlates of saphenous vein graft hyperplasia and occlusion 1 year after coronary artery bypass grafting: analysis from the CASCADE randomized trial. Circulation (2013) 128(11 Suppl 1):S213–18. doi: 10.1161/CIRCULATIONAHA.112.000328
9. Samano N, Geijer H, Liden M, Fremes S, Bodin L, Souza D. The no-touch saphenous vein for coronary artery bypass grafting maintains a patency, after 16 years, comparable to the left internal thoracic artery: a randomized trial. J Thorac Cardiovasc Surg (2015) 150(4):880–8. doi: 10.1016/j.jtcvs.2015.07.027
10. Hess CN, Lopes RD, Gibson CM, Hager R, Wojdyla DM, Englum BR, et al. Saphenous vein graft failure after coronary artery bypass surgery: insights from PREVENT IV. Circulation (2014) 130(17):1445–51. doi: 10.1161/CIRCULATIONAHA.113.008193
11. Bryan AJ, Angelini GD. The biology of saphenous vein graft occlusion: etiology and strategies for prevention. Curr Opin Cardiol (1994) 9(6):641–9.
12. Gasper WJ, Owens CD, Kim JM, Hills N, Belkin M, Creager MA, et al. Thirty-day vein remodeling is predictive of midterm graft patency after lower extremity bypass. J Vasc Surg (2013) 57(1):9–18. doi: 10.1016/j.jvs.2012.06.098
13. Loesch A, Dashwood MR. On the sympathetic innervation of the human greater saphenous vein: relevance to clinical practice. Curr Vasc Pharmacol (2009) 7(1):58–67. doi: 10.2174/157016109787354150
14. Lardenoye JH, de Vries MR, Löwik CW, Xu Q, Dhore CR, Cleutjens JP, et al. Accelerated atherosclerosis and calcification in vein grafts: a study in APOE*3 Leiden transgenic mice. Circ Res (2002) 91(7):577–84. doi: 10.1161/01.RES.0000036901.58329.D7
15. Osgood MJ, Hocking KM, Voskresensky IV, Li FD, Komalavilas P, Cheung-Flynn J, et al. Surgical vein graft preparation promotes cellular dysfunction, oxidative stress, and intimal hyperplasia in human saphenous vein. J Vasc Surg (2014) 60(1):202–11. doi: 10.1016/j.jvs.2013.06.004
16. Kwei S, Stavrakis G, Takahas M, Taylor G, Folkman MJ, Gimbrone MA, et al. Early adaptive responses of the vascular wall during venous arterialization in mice. Am J Pathol (2004) 164(1):81–9. doi: 10.1016/S0002-9440(10)63099-4
17. Zhao J, Andreasen JJ, Yang J, Rasmussen BS, Liao D, Gregersen H. Manual pressure distension of the human saphenous vein changes its biomechanical properties-implication for coronary artery bypass grafting. J Biomech (2007) 40(10):2268–76. doi: 10.1016/j.jbiomech.2006.10.014
18. Ad N, Henry L, Hunt S, Holmes S, Burton N, Massimiano P, et al. Endoscopic versus direct vision for saphenous vein graft harvesting in coronary artery bypass surgery. J Cardiovasc Surg (2011) 52(5):739–48.
19. Lopes RD, Hafley GE, Allen KB, Ferguson TB, Peterson ED, Harrington RA, et al. Endoscopic versus open vein-graft harvesting in coronary-artery bypass surgery. N Engl J Med (2009) 361(3):235–44. doi: 10.1056/NEJMoa0900708
20. Ouzounian M, Hassan A, Buth KJ, Macpherson C, Ali IM, Hirsch GM, et al. Impact of endoscopic versus open saphenous vein harvest techniques on outcomes after coronary artery bypass grafting. Ann Thorac Surg (2010) 89(2):403–8. doi: 10.1016/j.athoracsur.2009.09.061
21. van Diepen S, Brennan JM, Hafley GE, Reyes EM, Allen KB, Ferguson TB, et al. Endoscopic harvesting device type and outcomes in patients undergoing coronary artery bypass surgery. Ann Surg (2014) 260(2):402–8. doi: 10.1097/SLA.0000000000000377
22. Souza DS, Johansson B, Bojö L, Karlsson R, Geijer H, Filbey D, et al. Harvesting the saphenous vein with surrounding tissue for CABG provides long-term graft patency comparable to the left internal thoracic artery: results of a randomized longitudinal trial. J Thorac Cardiovasc Surg (2006) 132(2):373–8. doi: 10.1016/j.jtcvs.2006.04.002
23. Sepehripour AH, Jarral OA, Shipolini AR, Mccormack DJ. Does a 'no-touch' technique result in better vein patency? Interact Cardiovasc Thorac Surg (2011) 13(6):626–30. doi: 10.1510/icvts.2011.281998
24. Davies MG, Klyachkin ML, Svendsen E, Hagen PO. A comparative study of endothelium-derived relaxing factor-mediated relaxation and smooth muscle cell function in arterial and venous vein bypass grafts. Cardiovasc Surg (1996) 4(2):150–60. doi: 10.1016/0967-2109(96)82306-4
25. Wilson YG, Davies AH, Southgate K, Currie IC, Sheffield E, Baird RN, et al. Vein quality influences neointimal hyperplasia in an organ culture model of human saphenous vein. Eur J Vasc Endovasc Surg (1997) 13(6):557–62. doi: 10.1016/S1078-5884(97)80064-0
26. Bouten CV, Dankers PY, Driessen-Mol A, Pedron S, Brizard AM, Baaijens FP. Substrates for cardiovascular tissue engineering. Adv Drug Deliv Rev (2011) 63(4-5):221–41. doi: 10.1016/j.addr.2011.01.007
27. Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA (1999) 282(21):2035–42. doi: 10.1001/jama.282.21.2035
28. Tai NR, Salacinski HJ, Edwards A, Hamilton G, Seifalian AM. Compliance properties of conduits used in vascular reconstruction. Br J Surg (2000) 87(11):1516–24. doi: 10.1046/j.1365-2168.2000.01566.x
29. O'Callaghan CJ, Williams B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: role of TGF-beta(1). Hypertension (2000) 36(3):319–24. doi: 10.1161/01.HYP.36.3.319
30. Owens CD, Wake N, Jacot JG, Gerhard-Herman M, Gaccione P, Belkin M, et al. Early biomechanical changes in lower extremity vein grafts-distinct temporal phases of remodeling and wall stiffness. J Vasc Surg (2006) 44(4):740–6. doi: 10.1016/j.jvs.2006.06.005
31. Liu SQ, Fung YC. Changes in the organization of the smooth muscle cells in rat vein grafts. Ann Biomed Eng (1998) 26(1):86–95. doi: 10.1114/1.52
32. Dobrin PB, Littooy FN, Endean ED. Mechanical factors predisposing to intimal hyperplasia and medial thickening in autogenous vein grafts. Surgery (1989) 105(3):393–400.
33. Piola M, Prandi F, Bono N, Soncini M, Penza E, Agrifoglio M, et al. A compact and automated ex vivo vessel culture system for the pulsatile pressure conditioning of human saphenous veins. J Tissue Eng Regen Med (2016) 10(3):E204–15. doi: 10.1002/term.1798
34. Miyakawa AA, Dallan LA, Lacchini S, Borin TF, Krieger JE. Human saphenous vein organ culture under controlled hemodynamic conditions. Clinics (2008) 63(5):683–8. doi: 10.1590/S1807-59322008000500018
35. Piola M, Ruiter M, Vismara R, Mastrullo V, Agrifoglio M, Zanobini M, et al. Full mimicking of coronary hemodynamics for ex-vivo stimulation of human saphenous veins. Ann Biomed Eng (2017) 45(4):884–97. doi: 10.1007/s10439-016-1747-7
36. Gusic RJ, Myung R, Petko M, Gaynor JW, Gooch KJ. Shear stress and pressure modulate saphenous vein remodeling ex vivo. J Biomech (2005) 38(9):1760–9. doi: 10.1016/j.jbiomech.2004.10.030
37. Porter KE, Nydahl S, Dunlop P, Varty K, Thrush AJ, London NJ. The development of an in vitro flow model of human saphenous vein graft intimal hyperplasia. Cardiovasc Res (1996) 31(4):607–14. doi: 10.1016/S0008-6363(95)00230-8
38. Saucy F, Probst H, Alonso F, Bérard X, Déglise S, Dunoyer-Geindre S, et al. Ex vivo pulsatile perfusion of human saphenous veins induces intimal hyperplasia and increased levels of the plasminogen activator inhibitor 1. Eur Surg Res (2010) 45(1):50–9. doi: 10.1159/000318602
39. Voisard R, Ramiz E, Baur R, Gastrock-Balitsch I, Siebeneich H, Frank O, et al. Pulsed perfusion in a venous human organ culture model with a Windkessel function (pulsed perfusion venous HOC-model). Med Sci Monit (2010) 16(11):CR523–9.
40. Berard X, Déglise S, Alonso F, Saucy F, Meda P, Bordenave L, et al. Role of hemodynamic forces in the ex vivo arterialization of human saphenous veins. J Vasc Surg (2013) 57(5):1371–82. doi: 10.1016/j.jvs.2012.09.041
41. Model LS, Hall MR, Wong DJ, Muto A, Kondo Y, Ziegler KR, et al. Arterial shear stress reduces eph-b4 expression in adult human veins. Yale J Biol Med (2014) 87(3):359–71.
42. Chistiakov DA, Sobenin IA, Orekhov AN. Vascular extracellular matrix in atherosclerosis. Cardiol Rev (2013) 21(6):270–88. doi: 10.1097/CRD.0b013e31828c5ced
43. Givens C, Tzima E. Endothelial mechanosignaling: does one sensor fit all? Antioxid Redox Signal (2016) 25(7):373–88. doi: 10.1089/ars.2015.6493
44. Urner S, Kelly-Goss M, Peirce SM, Lammert E. Mechanotransduction in blood and lymphatic vascular development and disease. Adv Pharmacol (2018) 81:155–208. doi: 10.1016/bs.apha.2017.08.009
45. Davies PF. Mechanical sensing mechanisms: shear stress and endothelial cells. J Vasc Surg (1991) 13(5):729–31. doi: 10.1016/0741-5214(91)90364-Z
46. Momin A, Sharabiani MT, Wendler O, Angelini GD, Desai J. Arterial blood pressure and vascular function in human saphenous vein. Perfusion (2015) 30(3):233–8. doi: 10.1177/0267659114540021
47. Prandi F, Piola M, Soncini M, Colussi C, D'Alessandra Y, Penza E, et al. Adventitial vessel growth and progenitor cells activation in an ex vivo culture system mimicking human saphenous vein wall strain after coronary artery bypass grafting. PLoS ONE (2015) 10(2):e0117409. doi: 10.1371/journal.pone.0117409
48. de Waard V, Arkenbout EK, Vos M, Mocking AI, Niessen HW, Stooker W, et al. TR3 nuclear orphan receptor prevents cyclic stretch-induced proliferation of venous smooth muscle cells. Am J Pathol (2006) 168(6):2027–35. doi: 10.2353/ajpath.2006.050932
49. Predel HG, Yang Z, von Segesser L, Turina M, Bühler FR, Lüscher TF. Implications of pulsatile stretch on growth of saphenous vein and mammary artery smooth muscle. Lancet (1992) 340(8824):878–9.
50. Stanley AG, Patel H, Knight AL, Williams B. Mechanical strain-induced human vascular matrix synthesis: the role of angiotensin II. J Renin Angiotensin Aldosterone Syst (2000) 1(1):32–5. doi: 10.3317/jraas.2000.007
51. Asanuma K, Magid R, Johnson C, Nerem RM, Galis ZS. Uniaxial strain upregulates matrix-degrading enzymes produced by human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol (2003) 284(5):H1778–84. doi: 10.1152/ajpheart.00494.2002
52. Schad JF, Meltzer KR, Hicks MR, Beutler DS, Cao TV, Standley PR. Cyclic strain upregulates VEGF and attenuates proliferation of vascular smooth muscle cells. Vascular Cell (2011) 3:21. doi: 10.1186/2045-824X-3-21
53. Chen Y, Wong MM, Campagnolo P, Simpson R, Winkler B, Margariti A, et al. Adventitial stem cells in vein grafts display multilineage potential that contributes to neointimal formation. Arterioscler Thromb Vasc Biol (2013) 33(8):1844–51. doi: 10.1161/ATVBAHA.113.300902
54. Xie Y, Potter CMF, Le Bras A, Nowak WN, Gu W, Bhaloo SI, et al. Leptin induces Sca-1+progenitor cell migration enhancing neointimal lesions in vessel-injury mouse models. Arterioscler Thromb Vasc Biol (2017) 37(11):2114–27. doi: 10.1161/ATVBAHA.117.309852
55. Kenagy RD, Kikuchi S, Chen L, Wijelath ES, Stergachis AB, Stamatoyannopoulos J, et al. A single nucleotide polymorphism of cyclin-dependent kinase inhibitor 1B (p27) associated with human vein graft failure affects growth of human venous adventitial cells but not smooth muscle cells. J Vasc Surg (2018) 67(1):309–17. doi: 10.1016/j.jvs.2016.12.113
56. Majesky MW, Dong XR, Hoglund V, Mahoney WM, Daum G. The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol (2011) 31(7):1530–9. doi: 10.1161/ATVBAHA.110.221549
57. Westerband A, Crouse D, Richter LC, Aguirre ML, Wixon CC, James DC, et al. Vein adaptation to arterialization in an experimental model. J Vasc Surg (2001) 33(3):561–9. doi: 10.1067/mva.2001.112230
58. Piola M, Prandi F, Fiore GB, Agrifoglio M, Polvani G, Pesce M, et al. Human saphenous vein response to trans-wall oxygen gradients in a novel ex vivo conditioning platform. Ann Biomed Eng (2016) 44(5):1449–61. doi: 10.1007/s10439-015-1434-0
59. Gubernator M, Slater SC, Spencer HL, Spiteri I, Sottoriva A, Riu F, et al. Epigenetic profile of human adventitial progenitor cells correlates with therapeutic outcomes in a mouse model of limb ischemia. Arterioscler Thromb Vasc Biol (2015) 35(3):675–88. doi: 10.1161/ATVBAHA.114.304989
60. Campagnolo P, Cesselli D, Al Haj Zen A, Beltrami AP, Kränkel N, Katare R, et al. Human adult vena saphena contains perivascular progenitor cells endowed with clonogenic and proangiogenic potential. Circulation (2010) 121(15):1735–45. doi: 10.1161/CIRCULATIONAHA.109.899252
61. Porter KE, Varty K, Jones L, Bell PR, London NJ. Human saphenous vein organ culture: a useful model of intimal hyperplasia? Eur J Vasc Endovasc Surg (1996) 11(1):48–58. doi: 10.1016/S1078-5884(96)80134-1
62. Wang X, Hu G, Gao X, Wang Y, Zhang W, Harmon EY, et al. The induction of yes-associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arterioscler Thromb Vasc Biol (2012) 32(11):2662–9. doi: 10.1161/ATVBAHA.112.254730
63. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA (2016) 113(41):11525–30. doi: 10.1073/pnas.1613121113
64. Momin A, Melikian N, Wheatcroft SB, Grieve D, John LC, El Gamel A, et al. The association between saphenous vein endothelial function, systemic inflammation, and statin therapy in patients undergoing coronary artery bypass surgery. J Thorac Cardiovasc Surg (2007) 134(2):335–41. doi: 10.1016/j.jtcvs.2006.12.064
65. Pintucci G, Saunders PC, Gulkarov I, Sharony R, Kadian-Dodov DL, Bohmann K, et al. Anti-proliferative and anti-inflammatory effects of topical MAPK inhibition in arterialized vein grafts. Faseb J (2006) 20(2):398–400. doi: 10.1096/fj.05-4114fje
66. Ozaki CK. Cytokines and the early vein graft: strategies to enhance durability. J. Vasc. Surg. (2007) 45(Suppl A):A92–8. doi: 10.1016/j.jvs.2007.02.032
67. Schepers A, Pires NM, Eefting D, de Vries MR, van Bockel JH, Quax PH. Short-term dexamethasone treatment inhibits vein graft thickening in hypercholesterolemic ApoE3Leiden transgenic mice. J Vasc Surg (2006) 43(4):809–15. doi: 10.1016/j.jvs.2005.11.019
68. Cooley BC, Nevado J, Mellad J, Yang D, Hilaire CS, Negro A, et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med (2014) 6(227):227ra34. doi: 10.1126/scitranslmed.3006927
69. Sharony R, Pintucci G, Saunders PC, Grossi EA, Baumann FG, Galloway AC, et al. Matrix metalloproteinase expression in vein grafts: role of inflammatory mediators and extracellular signal-regulated kinases-1 and -2. Am J Physiol Heart Circ Physiol (2006) 290(4):H1651–59. doi: 10.1152/ajpheart.00530.2005
70. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell (2013) 154(5):1047–59. doi: 10.1016/j.cell.2013.07.042
71. He J, Bao Q, Yan M, Liang J, Zhu Y, Wang C, et al. The role of Hippo/yes-associated protein signalling in vascular remodelling associated with cardiovascular disease. Br J Pharmacol (2017). doi: 10.1111/bph.13806
72. Xu S, Koroleva M, Yin M, Jin ZG. Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells. Transl Res (2016) 176:18–28. doi: 10.1016/j.trsl.2016.05.003
73. Maleszewska M, Vanchin B, Harmsen MC, Krenning G. The decrease in histone methyltransferase EZH2 in response to fluid shear stress alters endothelial gene expression and promotes quiescence. Angiogenesis (2016) 19(1):9–24. doi: 10.1007/s10456-015-9485-2
74. Chitragari G, Shalaby SY, Sumpio BJ, Sumpio BE. Effect of pulsatile and continuous flow on yes-associated protein. Int J Angiol (2014) 23(3):183–6. doi: 10.1055/s-0034-1376865
75. Tsuneki M, Madri JA. CD44 influences fibroblast behaviors via modulation of cell-cell and cell-matrix interactions, affecting survivin and hippo pathways. J Cell Physiol (2016) 231(3):731–43. doi: 10.1002/jcp.25123
76. Kim J, Kim YH, Kim J, Park DY, Bae H, Lee DH, et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest (2017) 127(9):3441–61. doi: 10.1172/JCI93825
77. Choi HJ, Kwon YG. Roles of YAP in mediating endothelial cell junctional stability and vascular remodeling. BMB Rep (2015) 48(8):429–30. doi: 10.5483/BMBRep.2015.48.8.146
78. Choi HJ, Zhang H, Park H, Choi KS, Lee HW, Agrawal V, et al. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat Commun (2015) 6:6943. doi: 10.1038/ncomms7943
79. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature (2016):579–582. doi: 10.1038/nature20602
80. Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, et al. Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev Cell (2017) 40(6):523–36. doi: 10.1016/j.devcel.2017.02.019
81. Li H, Jiang W, Ren W, Guo D, Guo J, Wang X, et al. Downregulation of the yes-associated protein is associated with extracellular matrix disorders in ascending aortic aneurysms. Stem Cells Int (2016) 2016:1–8. doi: 10.1155/2016/6786184
82. Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem (2012) 287(18):14598–605. doi: 10.1074/jbc.M111.329268
83. Kimura TE, Duggirala A, Smith MC, White S, Sala-Newby GB, Newby AC, et al. The Hippo pathway mediates inhibition of vascular smooth muscle cell proliferation by cAMP. J Mol Cell Cardiol (2016) 90:1–10. doi: 10.1016/j.yjmcc.2015.11.024
84. Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J, et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget (2015) 6(19):17685–97. doi: 10.18632/oncotarget.3779
85. Driscoll TP, Cosgrove BD, Heo SJ, Shurden ZE, Mauck RL. Cytoskeletal to nuclear strain transfer regulates YAP signaling in mesenchymal stem cells. Biophys J (2015) 108(12):2783–93. doi: 10.1016/j.bpj.2015.05.010
86. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. (2010) 24(11):1106–18. doi: 10.1101/gad.1903310
87. Feng X, Liu P, Zhou X, Li MT, Li FL, Wang Z, et al. Thromboxane A2 activates YAP/TAZ protein to induce vascular smooth muscle cell proliferation and migration. J Biol Chem (2016) 291(36):18947–58. doi: 10.1074/jbc.M116.739722
88. Dupont S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp Cell Res (2016) 343(1):42–53. doi: 10.1016/j.yexcr.2015.10.034
89. Fulford A, Tapon N, Ribeiro PS, Upstairs RPS. Upstairs, downstairs: spatial regulation of Hippo signalling. Curr Opin Cell Biol (2017) 51:22–32. doi: 10.1016/j.ceb.2017.10.006
90. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev (2014) 94(4):1287–312. doi: 10.1152/physrev.00005.2014
91. Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell (2017) 171(6):1397–410. doi: 10.1016/j.cell.2017.10.008
92. Elosegui-Artola A, Oria R, Chen Y, Kosmalska A, Pérez-González C, Castro N, et al. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat Cell Biol (2016) 18(5):540–8. doi: 10.1038/ncb3336
93. de Vries MR, Simons KH, Jukema JW, Braun J, Quax PH. Vein graft failure: from pathophysiology to clinical outcomes. Nat Rev Cardiol (2016) 13(8):451–70. doi: 10.1038/nrcardio.2016.76
94. Newby AC. Coronary vein grafting: the flags keep waving but the game goes on. Cardiovasc Res (2013) 97(2):193–4. doi: 10.1093/cvr/cvs351
95. Tanner FC, Boehm M, Akyürek LM, San H, Yang ZY, Tashiro J, et al. Differential effects of the cyclin-dependent kinase inhibitors p27(Kip1), p21(Cip1), and p16(Ink4) on vascular smooth muscle cell proliferation. Circulation (2000) 101(17):2022–5. doi: 10.1161/01.CIR.101.17.2022
96. Tanner FC, Meier P, Greutert H, Champion C, Nabel EG, Lüscher TF. Nitric oxide modulates expression of cell cycle regulatory proteins: a cytostatic strategy for inhibition of human vascular smooth muscle cell proliferation. Circulation (2000) 101(16):1982–9. doi: 10.1161/01.CIR.101.16.1982
97. Conte MS, Owens CD, Belkin M, Creager MA, Edwards KL, Gasper WJ, et al. A single nucleotide polymorphism in the p27(Kip1) gene is associated with primary patency of lower extremity vein bypass grafts. J Vasc Surg (2013) 57(5):1179–85. doi: 10.1016/j.jvs.2012.11.040
98. Sedding DG, Seay U, Fink L, Heil M, Kummer W, Tillmanns H, et al. Mechanosensitive p27Kip1 regulation and cell cycle entry in vascular smooth muscle cells. Circulation (2003) 108(5):616–22. doi: 10.1161/01.CIR.0000079102.08464.E2
99. Costa MA, Simon DI. Molecular basis of restenosis and drug-eluting stents. Circulation (2005) 111(17):2257–73. doi: 10.1161/01.CIR.0000163587.36485.A7
100. Arkenbout EK, de Waard V, van Bragt M, van Achterberg TA, Grimbergen JM, Pichon B, et al. Protective function of transcription factor TR3 orphan receptor in atherogenesis: decreased lesion formation in carotid artery ligation model in TR3 transgenic mice. Circulation (2002) 106(12):1530–5.
101. Bonta PI, Matlung HL, Vos M, Peters SL, Pannekoek H, Bakker EN, et al. Nuclear receptor Nur77 inhibits vascular outward remodelling and reduces macrophage accumulation and matrix metalloproteinase levels. Cardiovasc Res (2010) 87(3):561–8. doi: 10.1093/cvr/cvq064
102. Bonta PI, van Tiel CM, Vos M, Pols TW, van Thienen JV, Ferreira V, et al. Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler Thromb Vasc Biol (2006) 26(10):2288–94. doi: 10.1161/01.ATV.0000238346.84458.5d
103. Hamers AA, Vos M, Rassam F, Marinković G, Marincovic G, Kurakula K, et al. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res (2012) 110(3):428–38. doi: 10.1161/CIRCRESAHA.111.260760
104. Ruiter MS, van Golde JM, Schaper NC, Stehouwer CD, Huijberts MS. Diabetes impairs arteriogenesis in the peripheral circulation: review of molecular mechanisms. Clin Sci (2010) 119(6):225–38. doi: 10.1042/CS20100082
105. Ruiter MS, van Tiel CM, Doornbos A, Marinković G, Strang AC, Attevelt NJ, et al. Stents eluting 6-mercaptopurine reduce neointima formation and inflammation while enhancing strut coverage in rabbits. PLoS ONE (2015) 10(9):e0138459. doi: 10.1371/journal.pone.0138459
106. Qin Q, Chen M, Yi B, You X, Yang P, Sun J. Orphan nuclear receptor Nur77 is a novel negative regulator of endothelin-1 expression in vascular endothelial cells. J Mol Cell Cardiol (2014) 77:20–8. doi: 10.1016/j.yjmcc.2014.09.027
107. Yoo YG, Na TY, Yang WK, Kim HJ, Lee IK, Kong G, et al. 6-Mercaptopurine, an activator of Nur77, enhances transcriptional activity of HIF-1alpha resulting in new vessel formation. Oncogene (2007) 26(26):3823–34. doi: 10.1038/sj.onc.1210149
108. Liu K, Zhou W, Chen H, Pan H, Sun X, You Q. Autologous vein graft stenosis inhibited by orphan nuclear receptor Nur77-targeted siRNA. Vascul Pharmacol (2015) 73:64–70. doi: 10.1016/j.vph.2015.08.008
109. Ji Q, Mei Y, Wang X, Feng J, Cai J, Sun Y, et al. Short-term effects of double-layer autologous vein graft on restraint of excessive distension and alleviation of neointimal hyperplasia in a porcine saphenous vein graft model. Heart Vessels (2011) 26(2):190–5. doi: 10.1007/s00380-010-0034-z
110. Longchamp A, Alonso F, Dubuis C, Allagnat F, Berard X, Meda P, et al. The use of external mesh reinforcement to reduce intimal hyperplasia and preserve the structure of human saphenous veins. Biomaterials (2014) 35(9):2588–99. doi: 10.1016/j.biomaterials.2013.12.041
111. Vijayan V, Shukla N, Johnson JL, Gadsdon P, Angelini GD, Smith FC, et al. Long-term reduction of medial and intimal thickening in porcine saphenous vein grafts with a polyglactin biodegradable external sheath. J Vasc Surg (2004) 40(5):1011–9. doi: 10.1016/j.jvs.2004.08.047
112. Webb CM, Orion E, Taggart DP, Channon KM, di Mario C. OCT imaging of aorto-coronary vein graft pathology modified by external stenting: 1-year post-surgery. Eur Heart J Cardiovasc Imaging (2016) 17(11):1290–5. doi: 10.1093/ehjci/jev310
113. Taggart DP, Ben Gal Y, Lees B, Patel N, Webb C, Rehman SM, et al. A randomized trial of external stenting for saphenous vein grafts in coronary artery bypass grafting. Ann Thorac Surg (2015) 99(6):2039–45. doi: 10.1016/j.athoracsur.2015.01.060
114. Meirson T, Orion E, di Mario C, Webb C, Patel N, Channon KM, et al. Flow patterns in externally stented saphenous vein grafts and development of intimal hyperplasia. J Thorac Cardiovasc Surg (2015) 150(4):871–9. doi: 10.1016/j.jtcvs.2015.04.061
115. Oku Y, Nishiya N, Shito T, Yamamoto R, Yamamoto Y, Oyama C, et al. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio (2015) 5:542–9. doi: 10.1016/j.fob.2015.06.007
116. Wang C, Zhu X, Feng W, Yu Y, Jeong K, Guo W, et al. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am J Cancer Res (2016) 6(1):27–37.
117. Porter KE, Loftus IM, Peterson M, Bell PR, London NJ, Thompson MM. Marimastat inhibits neointimal thickening in a model of human vein graft stenosis. Br J Surg (1998) 85(10):1373–7. doi: 10.1046/j.1365-2168.1998.00888.x
118. Porter KE, Naik J, Turner NA, Dickinson T, Thompson MM, London NJ. Simvastatin inhibits human saphenous vein neointima formation via inhibition of smooth muscle cell proliferation and migration. J Vasc Surg (2002) 36(1):150–7. doi: 10.1067/mva.2002.122029
119. Porter KE, Turner NA. Statins for the prevention of vein graft stenosis: a role for inhibition of matrix metalloproteinase-9. Biochem Soc Trans (2002) 30(2):120–6. doi: 10.1042/bst0300120
120. Kulik A, Voisine P, Mathieu P, Masters RG, Mesana TG, Le May MR, et al. Statin therapy and saphenous vein graft disease after coronary bypass surgery: analysis from the CASCADE randomized trial. Ann Thorac Surg (2011) 92(4):1284–91. doi: 10.1016/j.athoracsur.2011.04.107
121. Ranjzad P, Salem HK, Kingston PA. Adenovirus-mediated gene transfer of fibromodulin inhibits neointimal hyperplasia in an organ culture model of human saphenous vein graft disease. Gene Ther (2009) 16(9):1154–62. doi: 10.1038/gt.2009.63
122. Huang K, Bao H, Yan ZQ, Wang L, Zhang P, Yao QP, et al. MicroRNA-33 protects against neointimal hyperplasia induced by arterial mechanical stretch in the grafted vein. Cardiovasc Res (2017) 113(5):cvw257–97. doi: 10.1093/cvr/cvw257
123. Wang XW, Zhang C, Lee KC, He XJ, Lu ZQ, Huang C, et al. Adenovirus-mediated gene transfer of microRNA-21 sponge inhibits neointimal hyperplasia in rat vein grafts. Int J Biol Sci (2017) 13(10):1309–19. doi: 10.7150/ijbs.20254
Keywords: vein graft disease, saphenous vein, CABG, mechanobiology, mechanotransduction, YAP, shear stress, strain
Citation: Ruiter MS and Pesce M (2018). Mechanotransduction in Coronary Vein Graft Disease. Front. Cardiovasc. Med. 5:20. doi: 10.3389/fcvm.2018.00020
Received: 20 December 2017; Accepted: 22 February 2018;
Published: 14 March 2018
Edited by:
Rajesh Katare, University of Otago, New ZealandReviewed by:
Praphulla C. Shukla, Indian Institute of Technology Kharagpur, IndiaXuechong Hong, Boston Children's Hospital, Harvard University, United States
Martin Fronius, University of Otago, New Zealand
Copyright © 2018 Ruiter and Pesce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Pesce, maurizio.pesce@ccfm.it