 Emiel P. C. van der Vorst
Emiel P. C. van der Vorst Renske J. de Jong3
Renske J. de Jong3 Marjo M. P. C. Donners
Marjo M. P. C. Donners
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med., 22 January 2018
Sec. Atherosclerosis and Vascular Medicine
Volume 5 - 2018 | https://doi.org/10.3389/fcvm.2018.00002
This article is part of the Research TopicExtracellular Vesicle-Mediated Processes in Cardiovascular DiseasesView all 11 articles
Extracellular vesicles (EVs) have emerged as a novel intercellular communication system. By carrying bioactive lipids, miRNAs and proteins they can modulate target cell functions and phenotype. Circulating levels of EVs are increased in inflammatory conditions, e.g., cardiovascular disease patients, and their functional contribution to atherosclerotic disease development is currently heavily studied. This review will describe how EVs can modulate vascular cell functions relevant to vascular inflammation and atherosclerosis, particularly highlighting the role of EV-associated proteolytic activity and effector proteins involved. Furthermore, we will discuss key questions and challenges, especially for EV-based therapeutics.
Extracellular vesicles (EVs) play a crucial physiological and pathophysiological role, as they have been identified as regulators of cell-to-cell communication (1).
Extracellular vesicles are small spherical vesicles, consisting of a lipid bilayer membrane encasing a small organelle-free cytosol, that are released by cells into the extracellular environment (2). It has been shown that most cell types can release EVs, originating from various subcellular membrane compartments (3). Nowadays, EVs are generally classified into three main classes, i.e., exosomes, microvesicles (MVs), and apoptotic bodies (3). Exosomes arise from intracellular compartments called multivesicular bodies (MVBs) and are released by an active process, leading to fusion of these MVBs with the plasma membrane (4). Exosomes typically have a size of 30–100 nm, i.e., representing the smallest subgroup of EVs, and are enriched for tetraspanins (CD9, CD63, and CD81) or other markers, such as flotillin and tumor susceptibility gene 101, which are often used to distinguish them from other populations of EVs (5). The second class of EVs is MVs, which are typically larger in size (ranging from 100 to 1,000 nm) and are produced by budding off directly from the plasma membrane in a process called microvesiculation (5). Microvesiculation involves the externalization of phosphatidylserine (PS) followed by cytoskeleton rearrangement and the formation of membrane curvatures (6, 7). Therefore, MVs membranes are also enriched in PS (detectable by Annexin A5) and the membrane composition resembles that of the parental cell (8). The third type of EVs is apoptotic bodies with a size of >1 μm. These vesicles are released from apoptotic cells through membrane blebbing and therefore contain apoptotic nuclear material (9). However, although the field is rapidly evolving, it is still quite challenging to specifically isolate, characterize, and classify the different populations of vesicles as discussed below.
Extracellular vesicles can cargo a large variety of biomolecules, such as various DNA, RNA, and microRNA species, bioactive lipids, and proteins. The latter include receptor ligands, by which EVs can interact with target cells (2), and proteolytically active enzymes, by which these vesicles can influence many cellular functions (10). This review will give a brief overview on how EVs can modulate vascular cell functions relevant to vascular inflammation and atherosclerosis, particularly highlighting the role of EV-associated proteolytic activity and effector proteins involved. Furthermore, we will discuss key questions and challenges, especially for EV-based therapeutics.
Recent years, great efforts have already been made to elucidate the role of EVs in cardiovascular diseases (CVDs), which is still the major cause of mortality worldwide. CVDs are mainly caused by atherosclerosis, a chronic inflammatory disease initiated by a continuous damage of the vascular endothelium leading to endothelial dysfunction (11). It has already been clearly shown that inflammation and endothelial injury augment the release of EVs (12, 13), generally reflecting the pro-inflammatory state of the parental cell. In addition, EVs influence thrombus formation which can occur after plaque rupture (3). Indeed, atherosclerotic lesions contain and release EVs, derived from leukocytes, platelets, smooth muscle cells (SMCs), and endothelial cells, during all stages of atherosclerosis development (14, 15) (Figure 1). As a consequence, patients with CVD mediated by endothelial damage show significantly elevated levels of circulating cell-derived EVs (16). This observation has therefore also been the starting point to investigate EVs as potential prognostic and diagnostic biomarkers. While most research has focused on the presence and function of MVs, also exosomes have been observed in human atherosclerotic lesions (17), although their functional roles remain largely unexplored.
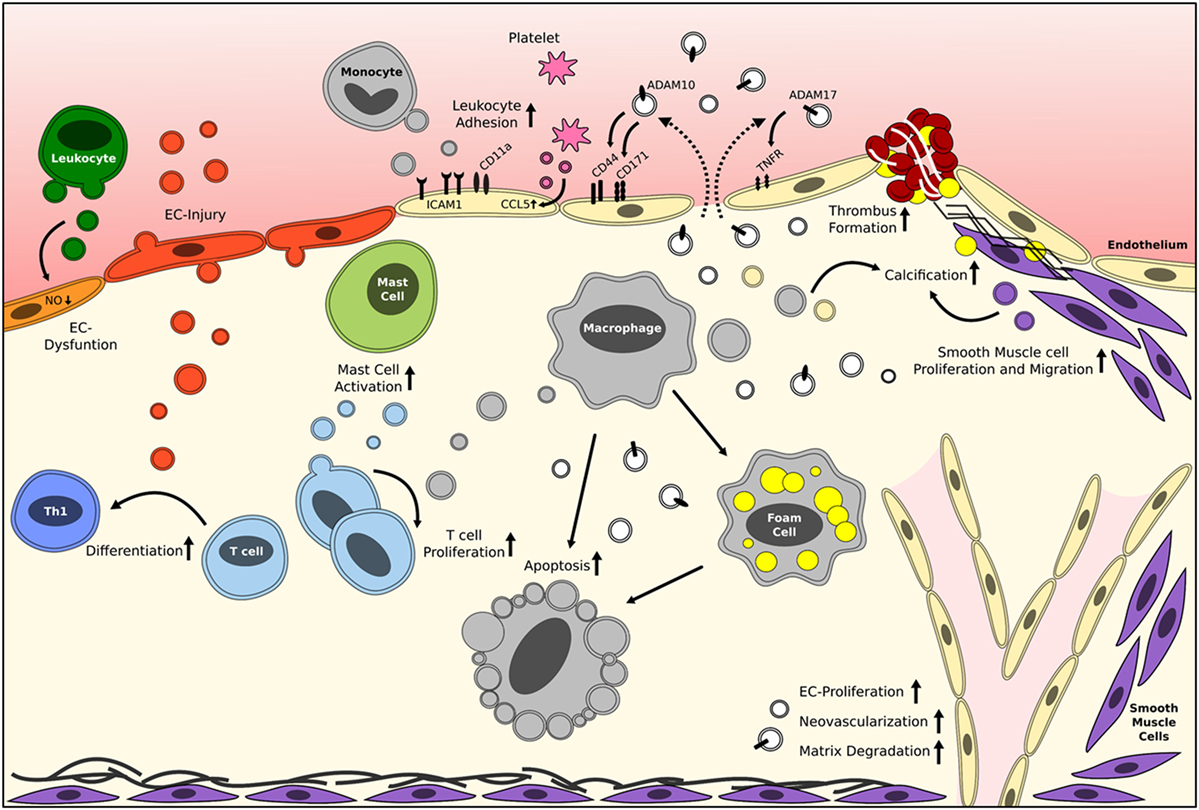
Figure 1. Reported roles of EVs in vascular inflammation and atherosclerosis. Brief schematic representation of the reported effects of circulating cell-derived and plaque-derived EVs on different processes in atherosclerosis development. The mentioned effector molecules are merely examples, and it should be noted that many more exist. White vesicles are of unknown origin/parental cell. EC, endothelial cell; EV, extracellular vesicle. Please refer to Table 1 for more detailed information.
Several in vitro studies clearly show that platelet and leukocyte-derived MVs from unstimulated cells increase the release of pro-inflammatory cytokines from endothelial cells and leukocytes, especially interleukin (IL)-6 and IL-8 (18, 19). Release of these cytokines will inherently promote monocyte adhesion to the endothelium and migration into the atherosclerotic lesions. MVs released from human atherosclerotic plaques were shown to increase the expression of endothelial adhesion molecules such as intercellular adhesion molecule 1 and monocyte adhesion molecule receptors, like CD11a, thereby further augmenting monocyte adhesion (18, 20). Furthermore, CCL5 (RANTES) is transferred from healthy platelet MVs to activated endothelial cells and can thereby enhance leukocyte adhesion (21). Endothelial and leukocyte MVs have also been shown to induce endothelial cell dysfunction, by decreasing the production of nitric oxide (NO) (22, 23). This is the result of an inhibition of the endothelial NO synthase and/or an increase in caveolin-1, increasing local oxidative stress (24, 25). Besides this mediator, MVs can act as potential markers of endothelial dysfunction, as nicely reviewed in Ref. (26). Together, these data clearly show that MVs, derived from various (vascular) cell types, can greatly influence the initiation of atherosclerosis development.
Microvesicles derived from macrophages and fibroblasts have also been implicated in later stages of lesion development, as they can stimulate foam cell formation by lipid/cholesterol uptake in macrophages (27). Furthermore, several reports have indicated that T cell-derived MVs can contribute to monocyte and macrophage apoptosis, via two proposed mechanisms (28, 29). The first mechanism involves the phagocytosis of MVs by monocytes and macrophages, leading to an increased cellular content of membrane phospholipids. These phospholipids are likely cleaved by phospholipase A2 into arachidonic acid, which will subsequently result in an increased amount of proapoptotic ceramides inside the cells (28, 29). The second mechanism involves MVs containing caspase-1 or caspase-3, which can induce target cell apoptosis (30, 31). Several studies have shown that MVs also play an important role in lymphocytes, as both human atherosclerotic plaque and in vitro generated dendritic cell MVs can stimulate T cell proliferation (32, 33). Most likely this influence is mediated by the presence of major histocompatibility complex class II presence on the MVs secreted from macrophages and dendritic cells (32). Furthermore, endothelial cell-derived MVs can promote lymphocyte differentiation toward a more proatherogenic T helper-1 phenotype as shown by priming of naive T cells with dendritic cells which were matured with endothelial MVs (34). On their turn, activated T cells release MVs that can induce mast cell activation, degranulation, and cytokine release (35). Mast cells are also present in the arterial wall, where they can contribute to atherosclerosis development (36).
Furthermore, MVs have significant effects on plaque stability as they can influence SMC proliferation and migration, via protease-activated receptor interaction or various microRNAs (37, 38). In addition, plaque MVs can contribute to matrix degradation as they contain several active proteases (39), which will be discussed in more detail later. This influence on matrix degradation is also one of the mechanisms by which MVs could potentially contribute to intraplaque neovascularization. It has also been shown that human plaque MVs can increase endothelial proliferation, a crucial step in neovascularization, in vitro as well as in vivo in matrigel plugs (40). During human atherosclerosis development, intimal calcification occurs at different stages of lesion development (41). Moreover, endothelial, SMC, and macrophage-derived EVs are present at the sites of calcification (3), nicely reviewed in Ref. (42). EVs released from SMCs have the potential to stimulate calcification by these same SMCs, mediated by sortilin-dependent regulation of alkaline phosphatase trafficking (43). In addition, EVs enriched in bone morphogenetic protein 2 released from endothelial cells can promote calcification in vascular SMCs (44).
In the latest stages of atherosclerosis, i.e., plaque rupture and thrombosis, MVs can also play an important role. MVs/EVs carry various proteolytic factors that likely contribute to matrix degradation, as shown in cancer (45) and could thereby potentially also influence plaque destabilization. In addition, human plaque MVs have been shown to be particularly prothrombogenic (15). Plaque MVs can contribute to the coagulation pathway via two different pathways: the presence of tissue factor on the surface of MVs and the exposure of PS on the outer membrane layer (3, 46). In contrast to MVs, exosomes seem to have antithrombotic effects. Platelet aggregation was suppressed by platelet-derived exosomes by inhibiting platelet CD36 (47). The procoagulant role of MVs is more elaborately reviewed in Ref. (48).
Besides communication between different cells within an atherosclerotic plaque, it is generally assumed that EVs, as they are relatively stable, mediate cross talk with cells at relatively large distances. This is particularly relevant for CVDs, which is widely acknowledged to be a systemic disease, and the basis for the “vulnerable patient concept” (49, 50). Indeed, it has already been long recognized that clinical symptoms in CVD patients (e.g., myocardial infarction or stroke) are often followed by secondary CVD events. Moreover, CVDs are often associated with several comorbidities, e.g., diabetes, chronic kidney disease, non-alcoholic steatohepatitis, small cerebral vessel disease, and heart failure. It is likely, yet it remains to be determined, that EVs play a crucial role in this systemic intercellular communication.
Extracellular vesicles are known to carry a large amount of bioactive molecules, including proteins/enzymes. Still, relatively little is known on the influence of various (atherogenic) stimuli on EV composition and thus EV function. Proteomic analysis recently identified several proteolytical enzymes in EVs, such as the cell surface-bound sheddases a disintegrin and metalloproteinases (ADAMs), soluble ADAMs with thrombospondin motifs (ADAMTSs), as well as cell surface-bound and soluble matrix metalloproteinases (MMPs) (51).
A disintegrin and metalloproteinases are involved in ectodomain shedding of various transmembrane proteins, thereby regulating cell adhesion, migration, and cell–cell communication (52). ADAM10 and ADAM17 are the best studied members of this family. ADAM17 is considered the primary enzyme for shedding of tumor necrosis factor (TNF), and its receptors (TNFR1 and 2), and the epidermal growth factor receptor ligands (53). On the other hand, ADAM10 is physiologically critical for Notch signaling via receptor cleavage (54). ADAMs have been reported to mediate various exosome/MV functions, e.g., by cleavage of EV surface molecules, releasing them as soluble factors in the target cell microenvironment. Indeed, ADAM17 is present on MVs released from atherosclerotic lesions and shown to cleave pro-TNF from these vesicles, which could provide a means to locally release pro-inflammatory mediators at large distances from the cell/site from which the MVs are released (39). In addition, plaque MVs have been shown to increase the shedding of TNF and its receptor (TNFR) from the surface of endothelial cells in an ADAM17-dependent manner (39), further supporting a role for ADAM17+ MVs in the regulation of (systemic) vascular inflammation.
Little is known on the role of other EV-associated ADAMs in relation to atherosclerosis. In exosomes, secreted from ovarian carcinoma cells, especially ADAM10 has been shown to be crucially involved in the cleavage of CD171 (L1) and CD44 (55), two important cell adhesion molecules. Cleavage did not only occur in the released exosomes but also already in the earlier phases of vesicle formation in the endosomal compartment. ADAM17 is also able to cleave CD171, although this occurs only at the cell surface demonstrating that different ADAMs are involved in distinct cellular compartments (55), and thus potentially in different EV populations. Other ADAMs such as ADAM15 (56), have also been identified in exosomes. Tumor cell-derived exosomes, enriched in ADAM15, display a high binding affinity for integrin αvβ3 and suppress cell adhesion, migration and growth (56). Exosomes derived from macrophages have also been shown to express ADAM15 and demonstrate described tumor inhibitory effects (56). The functional contribution of ADAM proteases in EVs to CVD disease progression, however, remains to be determined.
ADAMs with thrombospondin motifs are relatively comparable to ADAMs, but have thrombospondin-like motifs instead of transmembrane and cytoplasmic domains and are therefore generally secreted as soluble proteins (45). A large subgroup of ADAMTSs is known as aggrecanases, because they can proteolytically cleave proteoglycans and are involved in cartilage degradation (57). This degradation of cartilage by aggrecanases has been associated with the progression of arthritis (58). Recently, it has been shown that rheumatoid synovial fibroblasts secrete MVs containing aggrecanase activity, most likely mediated by ADAMTS1, ADAMTS4, or ADAMTS5 (59). Synovial fluids in rheumatoid arthritis also contain T cell- and monocyte-derived MVs, which can induce the synthesis of several MMPs in fibroblasts (60). Considering the role of various ADAMTS proteases in inflammation and vascular biology (61, 62), it is likely that EV-associated ADAMTSs are implicated in CVD. However, there are no clear indications for such a role of ADAMTSs in EVs in other pathologies, such as CVDs, yet.
Matrix metalloproteinases are a family of zinc-dependent endopeptidases, which are also crucial to extracellular matrix degradation and cleavage of surface proteins. It has already been shown that EVs released from mouse melanoma cells and human colorectal carcinoma cells have gelatinolytic and collagenolytic activity, indicating the presence of active MMPs (63, 64). Indeed, more recently several MMPs have been detected in EVs derived from tumor cells (45). Interestingly, there is also a positive correlation between the quantity of shed vesicles, the amount of vesicle bound lytic enzymes and the in vitro invasive capability of different human cancer cell lines (65). Since MMPs also play a role in CVD (66), a role of MMPs in EVs in CVD can be expected but has surprisingly not been evaluated so far.
Targeting EVs seems like a promising novel therapeutic option, where EVs containing RNA, DNA, or proteins involved in disease pathogenesis can be blocked. Blockage of EVs and especially the delivery of their cargo to the target cell can be achieved in various ways, e.g., by inhibiting the vesicle release, uptake or formation [reviewed by El Andaloussi et al. (67)]. Vesicle formation can be suppressed by inhibiting crucial cellular compartments, for instance by ceramide or syndecan proteoglycans blockage. Furthermore, the release of vesicles can be blocked by inhibiting GTPases, which are needed for the fusion of MVBs with the plasma membrane. In addition, EVs could be used as therapeutic delivery tools. For this purpose, both endogenously produced EVs and EVs, which are deliberately packaged with specific components can be used (68). For example, a recent proof of concept study has shown that EVs could deliver specific siRNA to mouse brains (69). In the context of CVD, a recent study has shown that in vitro generated endothelial EVs could reduce atherosclerosis formation by the transfer of miRNAs (38). The first clinical trials using EVs have also already been started in the field of antitumor immunotherapy. Two separate phase I trials used Good Manufacturing Practice compatible protocols to isolate EVs from dendritic cells and could show a good feasibility and safety of EV administration in patients (70). The phase II trial that followed unfortunately did not give the expected positive outcomes, but combined these results clearly show the therapeutic potential of EVs.
In addition to their therapeutic use, EVs could also be used as biomarkers as they are also found in several body fluids, such as blood (71) and urine (72), making them easily accessible for prognostic or diagnostic purposes. Emphasizing the prognostic potential, it has already been shown that Cystatin C, Serpin F2, and CD14 MV levels correlate with an increased risk for cardiovascular event and mortality (73). In addition, miRNA content of EVs has already been clearly linked with disease outcome (74). More details about the clinical potential of EVs and their use as biomarkers are recently elaborately reviewed in Ref. (75).
The field of EV research is rapidly progressing, although the EV research complexity and challenges are still considerable (76). EVs represent a very heterogeneous population, both in size and composition. This has led to some confusing and variable nomenclature, although as described before some consensus has already been achieved. Another major difficulty is the presence of non-EV components in preparations of EVs, which have comparable features (77). Currently, various isolation methods are used to isolate EV subtypes, such as differential (ultra)centrifugation, density gradient centrifugation, size exclusion chromatography, and immunocapture. All of these methods result in EV preparations of different composition and especially purity. For example, ultracentrifugation not only pellets EVs but also protein aggregates, while lipoproteins have similar size and density as EVs and are therefore often co-isolated (78). Recently, more confounding factors of ultrafiltration and protein analysis have been identified (79). Since, these different methods have not yet been tested side by side on a single EV sample, reliable quantitative comparisons regarding recovery and purity are difficult. Another interesting point that needs consideration is the influence of medication on EVs. For example, several antiplatelets agents, such as aspirin, can inhibit platelet activation and the related release of MVs (3). Antihypertensive agents have also been shown to reduce circulating platelet- and monocyte-derived MVs (3). In addition, statin therapy influences the composition of endothelial MVs (80). Besides the variety in contaminating factors in the different isolation methods, another major limitation is the unstandardized and often inadequate reporting on the specific methods used. Previously, the International Society for Extracellular Vesicles already introduced the minimal information for studies on EVs guidelines (81). More recently, to further improve the reliability of EV-related data/publications an international consortium developed the EV-TRACK (transparent reporting and centralizing knowledge in EV research) platform (82). This platform urges researchers to report more specific and detailed parameters which are necessary to fully interpret the obtained data and compare different studies. In addition, a recent review gives some methodological guidelines to study EVs (83). All these efforts clearly show the intention to standardize EV procedures, which will also be necessary to advance this research field toward comparable/supportive studies, crucial to pave the way toward clinical trials.
In the context of CVD and in particular atherosclerosis, a large variety of risk factors and contributing factors have already been identified and are currently targeted to treat this pathology, such as inflammatory molecules and lipids/lipoproteins. Although EVs have already been shown to be of crucial importance in the modulation of vascular inflammation and atherosclerosis (Table 1), at least in vitro, little is known on their therapeutic potential for CVD. Moreover, there are still several major limitations that should be overcome, such as detailed characterization and isolation procedures. Therefore, more preclinical studies are necessary before attempting to translate this research field to human medicine. In conclusion, EVs are promising targets for vascular inflammation and atherosclerosis and future research will further elucidate the full potential of such vesicles in disease prognosis, diagnosis and therapy.
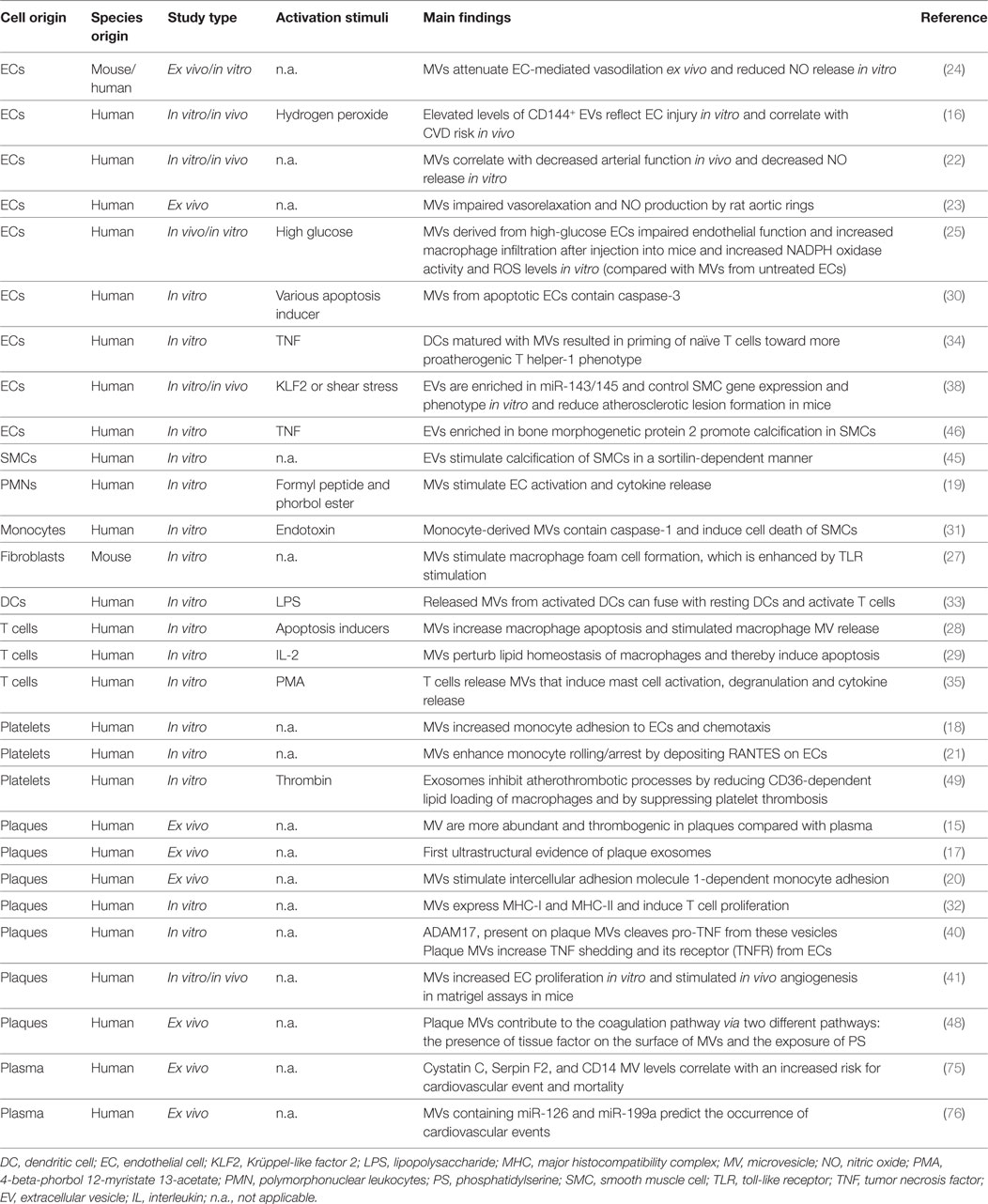
Table 1. Summarizing described studies supporting the role of EVs in vascular inflammation and atherosclerosis.
EV and RJ: drafting the manuscript. MD: concept and design; drafting the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, although no other collaboration, with one of the authors MD.
This work was supported by the DZHK (German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research); Project 81X2600244.
1. Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet (2012) 21:R125–34. doi:10.1093/hmg/dds317
2. Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracel Vesicles (2014) 3(1):24641. doi:10.3402/jev.v3.24641
3. Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol (2017) 14:259–72. doi:10.1038/nrcardio.2017.7
4. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol (2013) 200:373–83. doi:10.1083/jcb.201211138
5. Chistiakov DA, Orekhov AN, Bobryshev YV. Extracellular vesicles and atherosclerotic disease. Cell Mol Life Sci (2015) 72:2697–708. doi:10.1007/s00018-015-1906-2
6. Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol (2011) 31:15–26. doi:10.1161/ATVBAHA.109.200956
7. Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci (2010) 123:1603–11. doi:10.1242/jcs.064386
8. Camussi G, Deregibus MC, Bruno S, Grange C, Fonsato V, Tetta C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res (2011) 1:98–110.
9. Castejon OJ, Arismendi GJ. Nerve cell death types in the edematous human cerebral cortex. J Submicrosc Cytol Pathol (2006) 38:21–36.
10. Chistiakov DA, Chekhonin VP. Extracellular vesicles shed by glioma cells: pathogenic role and clinical value. Tumour Biol (2014) 35:8425–38. doi:10.1007/s13277-014-2262-9
11. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation (2002) 105:1135–43. doi:10.1161/hc0902.104353
12. Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol (2011) 31:27–33. doi:10.1161/ATVBAHA.110.218123
13. Libby P. Changing concepts of atherogenesis. J Intern Med (2000) 247:349–58. doi:10.1046/j.1365-2796.2000.00654.x
14. Bobryshev YV, Killingsworth MC, Orekhov AN. Increased shedding of microvesicles from intimal smooth muscle cells in athero-prone areas of the human aorta: implications for understanding of the predisease stage. Pathobiology (2013) 80:24–31. doi:10.1159/000339430
15. Leroyer AS, Isobe H, Lesèche G, Castier Y, Wassef M, Mallat Z, et al. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Col Cardiol (2007) 49:772–7. doi:10.1016/j.jacc.2006.10.053
16. Koga H, Sugiyama S, Kugiyama K, Watanabe K, Fukushima H, Tanaka T, et al. Elevated levels of VE-cadherin-positive endothelial microparticles in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol (2005) 45:1622–30. doi:10.1016/j.jacc.2005.02.047
17. Perrotta I, Aquila S. Exosomes in human atherosclerosis: an ultrastructural analysis study. Ultrastruct Pathol (2016) 40:101–6. doi:10.3109/01913123.2016.1154912
18. Barry OP, Pratico D, Savani RC, FitzGerald GA. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest (1998) 102:136–44. doi:10.1172/JCI2592
19. Mesri M, Altieri DC. Endothelial cell activation by leukocyte microparticles. J Immunol (1998) 161:4382–7.
20. Rautou PE, Leroyer AS, Ramkhelawon B, Devue C, Duflaut D, Vion AC, et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ Res (2011) 108:335–43. doi:10.1161/CIRCRESAHA.110.237420
21. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol (2005) 25:1512–8. doi:10.1161/01.ATV.0000170133.43608.37
22. Amabile N, Guérin AP, Leroyer A, Mallat Z, Nguyen C, Boddaert J, et al. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol (2005) 16:3381–8. doi:10.1681/ASN.2005050535
23. Brodsky SV, Zhang F, Nasjletti A, Goligorsky MS. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circ Physiol (2004) 286:H1910–5. doi:10.1152/ajpheart.01172.2003
24. Densmore JC, Signorino PR, Ou J, Hatoum OA, Rowe JJ, Shi Y, et al. Endothelium-derived microparticles induce endothelial dysfunction and acute lung injury. Shock (2006) 26:464–71. doi:10.1097/01.shk.0000228791.10550.36
25. Jansen F, Yang X, Franklin BS, Hoelscher M, Schmitz T, Bedorf J, et al. High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc Res (2013) 98:94–106. doi:10.1093/cvr/cvt013
26. Liu ML, Williams KJ. Microvesicles: potential markers and mediators of endothelial dysfunction. Curr Opin Endocrinol Diabetes Obes (2012) 19:121–7. doi:10.1097/MED.0b013e32835057e9
27. Keyel PA, Tkacheva OA, Larregina AT, Salter RD. Coordinate stimulation of macrophages by microparticles and TLR ligands induces foam cell formation. J Immunol (2012) 189:4621–9. doi:10.4049/jimmunol.1200828
28. Distler JH, Huber LC, Hueber AJ, Reich CF III, Gay S, Distler O, et al. The release of microparticles by apoptotic cells and their effects on macrophages. Apoptosis (2005) 10:731–41. doi:10.1007/s10495-005-2941-5
29. Huber LC, Jüngel A, Distler JH, Moritz F, Gay RE, Michel BA, et al. The role of membrane lipids in the induction of macrophage apoptosis by microparticles. Apoptosis (2007) 12:363–74. doi:10.1007/s10495-006-0622-7
30. Abid Hussein MN, Nieuwland R, Hau CM, Evers LM, Meesters EW, Sturk A. Cell-derived microparticles contain caspase 3 in vitro and in vivo. J Thromb Haemost (2005) 3:888–96. doi:10.1111/j.1538-7836.2005.01240.x
31. Sarkar A, Mitra S, Mehta S, Raices R, Wewers MD. Monocyte derived microvesicles deliver a cell death message via encapsulated caspase-1. PLoS One (2009) 4:e7140. doi:10.1371/journal.pone.0007140
32. Mayr M, Grainger D, Mayr U, Leroyer AS, Leseche G, Sidibe A, et al. Proteomics, metabolomics, and immunomics on microparticles derived from human atherosclerotic plaques. Circ Cardiovasc Genet (2009) 2:379–88. doi:10.1161/CIRCGENETICS.108.842849
33. Obregon C, Rothen-Rutishauser B, Gitahi SK, Gehr P, Nicod LP. Exovesicles from human activated dendritic cells fuse with resting dendritic cells, allowing them to present alloantigens. Am J Pathol (2006) 169:2127–36. doi:10.2353/ajpath.2006.060453
34. Angelot F, Seillès E, Biichlé S, Berda Y, Gaugler B, Plumas J, et al. Endothelial cell-derived microparticles induce plasmacytoid dendritic cell maturation: potential implications in inflammatory diseases. Haematologica (2009) 94:1502–12. doi:10.3324/haematol.2009.010934
35. Shefler I, Salamon P, Reshef T, Mor A, Mekori YA. T cell-induced mast cell activation: a role for microparticles released from activated T cells. J Immunol (2010) 185:4206–12. doi:10.4049/jimmunol.1000409
36. Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev (2006) 86:515–81. doi:10.1152/physrev.00024.2005
37. Brousseau C, Morissette G, Fortin JP, Marceau F, Petitclerc E. Tumor cells expressing tissue factor influence the migration of smooth muscle cells in a catalytic activity-dependent way. Can J Physiol Pharmacol (2009) 87:694–701. doi:10.1139/y09-063
38. Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol (2012) 14:249–56. doi:10.1038/ncb2441
39. Canault M, Leroyer AS, Peiretti F, Lesèche G, Tedgui A, Bonardo B, et al. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am J Pathol (2007) 171:1713–23. doi:10.2353/ajpath.2007.070021
40. Leroyer AS, Rautou PE, Silvestre JS, Castier Y, Lesèche G, Devue C, et al. CD40 ligand+ microparticles from human atherosclerotic plaques stimulate endothelial proliferation and angiogenesis a potential mechanism for intraplaque neovascularization. J Am Coll Cardiol (2008) 52:1302–11. doi:10.1016/j.jacc.2008.07.032
41. Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol (2014) 34:724–36. doi:10.1161/ATVBAHA.113.302642
42. Krohn JB, Hutcheson JD, Martinez-Martinez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol (2016) 594:2895–903. doi:10.1113/JP271338
43. Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest (2016) 126:1323–36. doi:10.1172/JCI80851
44. Buendía P, Montes de Oca A, Madueño JA, Merino A, Martín-Malo A, Aljama P, et al. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J (2015) 29:173–81. doi:10.1096/fj.14-249706
45. Shimoda M, Khokha R. Metalloproteinases in extracellular vesicles. Biochim Biophys Acta (2017) 1864:1989–2000. doi:10.1016/j.bbamcr.2017.05.027
46. Mallat Z, Hugel B, Ohan J, Lesèche G, Freyssinet JM, Tedgui A. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation (1999) 99:348–53. doi:10.1161/01.CIR.99.3.348
47. Srikanthan S, Li W, Silverstein RL, McIntyre TM. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro-atherothombotic cellular functions. J Thromb Haemost (2014) 12:1906–17. doi:10.1111/jth.12712
48. Owens AP III, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res (2011) 108:1284–97. doi:10.1161/CIRCRESAHA.110.233056
49. Naghavi M, Falk E, Hecht HS, Jamieson MJ, Kaul S, Berman D, et al. From vulnerable plaque to vulnerable patient – part III: executive summary of the screening for heart attack prevention and education (SHAPE) task force report. Am J Cardiol (2006) 98:2H–15H. doi:10.1016/j.amjcard.2006.03.002
50. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Circulation (2003) 108:1664–72. doi:10.1161/01.CIR.0000087480.94275.97
51. Shimoda M, Khokha R. Proteolytic factors in exosomes. Proteomics (2013) 13:1624–36. doi:10.1002/pmic.201200458
52. van der Vorst EP, Keijbeck AA, de Winther MP, Donners MM. A disintegrin and metalloproteases: molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis (2012) 224:302–8. doi:10.1016/j.atherosclerosis.2012.04.023
53. Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. An essential role for ectodomain shedding in mammalian development. Science (1998) 282:1281–4. doi:10.1126/science.282.5392.1281
54. Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet (2002) 11:2615–24. doi:10.1093/hmg/11.21.2615
55. Stoeck A, Keller S, Riedle S, Sanderson MP, Runz S, Le Naour F, et al. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem J (2006) 393:609–18. doi:10.1042/BJ20051013
56. Lee HD, Koo BH, Kim YH, Jeon OH, Kim DS. Exosome release of ADAM15 and the functional implications of human macrophage-derived ADAM15 exosomes. FASEB J (2012) 26:3084–95. doi:10.1096/fj.11-201681
57. Tortorella MD, Burn TC, Pratta MA, Abbaszade I, Hollis JM, Liu R, et al. Purification and cloning of aggrecanase-1: a member of the ADAMTS family of proteins. Science (1999) 284:1664–6. doi:10.1126/science.284.5420.1664
58. Lohmander LS, Neame PJ, Sandy JD. The structure of aggrecan fragments in human synovial fluid. Evidence that aggrecanase mediates cartilage degradation in inflammatory joint disease, joint injury, and osteoarthritis. Arthritis Rheum (1993) 36:1214–22. doi:10.1002/art.1780360906
59. Lo Cicero A, Majkowska I, Nagase H, Di Liegro I, Troeberg L. Microvesicles shed by oligodendroglioma cells and rheumatoid synovial fibroblasts contain aggrecanase activity. Matrix Biol (2012) 31:229–33. doi:10.1016/j.matbio.2012.02.005
60. Distler JH, Jüngel A, Huber LC, Seemayer CA, Reich CF III, Gay RE, et al. The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles. Proc Natl Acad Sci U S A (2005) 102:2892–7. doi:10.1073/pnas.0409781102
61. Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol (2015) 16:113. doi:10.1186/s13059-015-0676-3
62. Salter RC, Ashlin TG, Kwan AP, Ramji DP. ADAMTS proteases: key roles in atherosclerosis? J Mol Med (2010) 88:1203–11. doi:10.1007/s00109-010-0654-x
63. Murayama T, Kataoka H, Koita H, Nabeshima K, Koono M. Glycocalyceal bodies in a human rectal carcinoma cell line and their interstitial collagenolytic activities. Virchows Arch B Cell Pathol Incl Mol Pathol (1991) 60:263–70. doi:10.1007/BF02899556
64. Zucker S, Wieman JM, Lysik RM, Wilkie DP, Ramamurthy N, Lane B. Metastatic mouse melanoma cells release collagen-gelatin degrading metalloproteinases as components of shed membrane vesicles. Biochim Biophys Acta (1987) 924:225–37. doi:10.1016/0304-4165(87)90091-2
65. Ginestra A, La Placa MD, Saladino F, Cassarà D, Nagase H, Vittorelli ML. The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res (1998) 18:3433–7.
66. Azevedo A, Prado AF, Antonio RC, Issa JP, Gerlach RF. Matrix metalloproteinases are involved in cardiovascular diseases. Basic Clin Pharmacol Toxicol (2014) 115:301–14. doi:10.1111/bcpt.12282
67. El Andaloussi S, Mäger I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov (2013) 12:347–57. doi:10.1038/nrd3978
68. Danielson KM, Das S. Extracellular vesicles in heart disease: excitement for the future? Exosomes Microvesicles (2014) 2. doi:10.5772/58390
69. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol (2011) 29:341–5. doi:10.1038/nbt.1807
70. Lener T, Gimona M, Aigner L, Börger V, Buzas E, Camussi G, et al. Applying extracellular vesicles based therapeutics in clinical trials – an ISEV position paper. J Extracell Vesicles (2015) 4:30087. doi:10.3402/jev.v4.30087
71. Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol (2005) 17:879–87. doi:10.1093/intimm/dxh267
72. Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A (2004) 101:13368–73. doi:10.1073/pnas.0403453101
73. Kanhai DA, Visseren FL, van der Graaf Y, Schoneveld AH, Catanzariti LM, Timmers L, et al. Microvesicle protein levels are associated with increased risk for future vascular events and mortality in patients with clinically manifest vascular disease. Int J Cardiol (2013) 168:2358–63. doi:10.1016/j.ijcard.2013.01.231
74. Jansen F, Yang X, Proebsting S, Hoelscher M, Przybilla D, Baumann K, et al. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J Am Heart Assoc (2014) 3:e001249. doi:10.1161/JAHA.114.001249
75. Jansen F, Nickenig G, Werner N. Extracellular vesicles in cardiovascular disease: potential applications in diagnosis, prognosis, and epidemiology. Circ Res (2017) 120:1649–57. doi:10.1161/CIRCRESAHA.117.310752
76. Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell (2016) 164:1226–32. doi:10.1016/j.cell.2016.01.043
77. György B, Módos K, Pállinger E, Pálóczi K, Pásztói M, Misják P, et al. Detection and isolation of cell-derived microparticles are compromised by protein complexes resulting from shared biophysical parameters. Blood (2011) 117:e39–48. doi:10.1182/blood-2010-09-307595
78. Yuana Y, Levels J, Grootemaat A, Sturk A, Nieuwland R. Co-isolation of extracellular vesicles and high-density lipoproteins using density gradient ultracentrifugation. J Extracell Vesicles (2014) 3:23262. doi:10.3402/jev.v3.23262
79. Vergauwen G, Dhondt B, Van Deun J, De Smedt E, Berx G, Timmerman E, et al. Confounding factors of ultrafiltration and protein analysis in extracellular vesicle research. Sci Rep (2017) 7:2704. doi:10.1038/s41598-017-02599-y
80. Zu L, Ren C, Pan B, Zhou B, Zhou E, Niu C, et al. Endothelial microparticles after antihypertensive and lipid-lowering therapy inhibit the adhesion of monocytes to endothelial cells. Int J Cardiol (2016) 202:756–9. doi:10.1016/j.ijcard.2015.10.035
81. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles (2014) 3:26913. doi:10.3402/jev.v3.26913
82. EV-TRACK Consortium, Van Deun J, Mestdagh P, Agostinis P, Akay Ö, Anand S, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods (2017) 14:228–32. doi:10.1038/nmeth.4185
Keywords: extracellular vesicles, vascular inflammation, atherosclerosis, proteolytic activity, challenges
Citation: van der Vorst EPC, de Jong RJ and Donners MMPC (2018) Message in a Microbottle: Modulation of Vascular Inflammation and Atherosclerosis by Extracellular Vesicles. Front. Cardiovasc. Med. 5:2. doi: 10.3389/fcvm.2018.00002
Received: 01 October 2017; Accepted: 03 January 2018;
Published: 22 January 2018
Edited by:
Rory R. Koenen, Maastricht University, NetherlandsReviewed by:
Bernhard H. Rauch, University of Greifswald, GermanyCopyright: © 2018 van der Vorst, de Jong and Donners. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emiel P. C. van der Vorst, ZW1pZWwudmFuX2Rlcl92b3JzdEBtZWQudW5pLW11ZW5jaGVuLmRl;
Marjo M. P. C. Donners, bWFyam8uZG9ubmVyc0BtYWFzdHJpY2h0dW5pdmVyc2l0eS5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.