 Manna Li
Manna Li Jian Xu
Jian Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 09 August 2017
Sec. Atherosclerosis and Vascular Medicine
Volume 4 - 2017 | https://doi.org/10.3389/fcvm.2017.00051
Obesity is a worldwide epidemic that predisposes individuals to metabolic complications, such as type 2 diabetes mellitus and non-alcoholic fatty liver disease, all of which are related to an imbalance between food intake and energy expenditure. Identification of the pathogenic molecular mechanisms and effective therapeutic approaches are urgently needed. A well-accepted paradigm is that crosstalk between organs/tissues contributes to diseases. Endothelial dysfunction characterizes metabolic disorders and the related vascular complications. Over the past two decades, overwhelming studies have focused on mechanisms that lead to endothelial dysfunction. New investigations, however, have begun to appreciate the opposite direction of the crosstalk: endothelial regulation of metabolism, although the underlying mechanisms remain to be elucidated. This review summarizes the evidence that supports the concept of endothelial regulation of obesity and the associated insulin resistance in fat, liver, and skeletal muscles, the classic targets of insulin. Outstanding questions and future research directions are highlighted. Identification of the mechanisms of vascular endothelial regulation of metabolism may offer strategies for prevention and treatment of obesity and the related metabolic complications.
The triad of obesity, diabetes, and cardiovascular diseases (CVD) becomes a major public health threat because of the global shift toward an energy-rich and sedentary life style. The WHO predicts that overweight and obesity may soon become the most significant cause of poor health, in addition to malnutrition and infectious diseases (1). While half of the population in developed countries is overweight or obese, more than 15% of children in these countries are overweight or obese (1). Obesity and the associated metabolic complications, including metabolic syndrome and type 2 diabetes, contribute to cardiovascular morbidities and present a great threat to global human health (2). These complications account for more than 300,000 deaths per year in the U.S. (3, 4). Obesity results from an imbalance between energy intake and expenditure, which increases adipose tissue mass and ectopic fat accumulation. As a consequence, obesity leads to various defects, such as insulin resistance in fat, skeletal muscle, and liver, hepatosteatosis, pancreatic lipotoxicity, and eventually type 2 diabetes and other cardiovascular complications, including accelerated atherosclerosis (5). Identification of the pathogenic molecular mechanisms and effective therapeutic approaches are needed.
Despite decades of intense research, the major molecular pathogenesis behind complex metabolic diseases is unknown (6). A well-accepted paradigm is that cellular crosstalk in and between organs/tissues contributes to metabolic diseases (7–10). Endothelial dysfunction characterizes and contributes to the pathology of metabolic disorders and the related vascular complications (5, 11). Over the past two decades, overwhelming studies have focused on mechanisms that lead to endothelial dysfunction. The mechanisms include the reactive oxygen species-mediated eNOS uncoupling, loss of eNOS-derived NO bioavailability, hyperglycemia-promoted apoptosis of vascular endothelial cells (ECs) in diabetes, which ultimately leads to impaired endothelium-dependent vessel relaxation, a general biomarker of endothelial dysfunction (11–15). Emerging investigations, however, suggest that the opposite direction in the crosstalk, i.e., the endothelial regulation of metabolism, could be crucial, although the underlying mechanisms remain largely unknown. This review summarizes the emerging evidence that supports the concept of vascular endothelial regulation of obesity-associated insulin resistance in classic targets of insulin, including fat, liver, and skeletal muscles, and highlights the unanswered questions for future research directions. Advancement in understanding endothelial control of metabolism may provide novel approaches for intervention in obesity and obesity-related diseases.
Endothelial cells form the lining of all blood and lymphatic vessels within the vascular network. The adult human body contains more than one trillion ECs, which weigh more than 100 g and cover a surface area of more than 1,000 square meters (16). As such, ECs can be regarded as an organ with endothelium forming a dynamic interface with all other organs in the body (17). Normal ECs line capillaries and establish specialized vascular niches or tissue-specific endothelium. On receiving cues from a wide variety of cells and tissues, this “organ” maintains vascular homeostasis by regulating each component of the endothelial function (18, 19). The most important endothelial function is to control vasomotor tone. The healthy ECs constitutively generate eNOS-derived NO to modulate vascular smooth muscle relaxation, which properly regulates both nutrient trafficking and blood fluidity. A well-controlled EC-derived NO is also essential to other aspects of the endothelial function, such as the capability of maintaining an anticoagulant, antiplatelet, and fibrinolytic phenotype of the endothelium (5). It has been well established that loss of endothelial function facilitates the development of atherosclerosis (20). Therefore, endothelium operates as a complex organ to maintain whole-body homeostasis by fulfilling proper endothelial function, which can be impaired under disease conditions, such as diabetes and obesity.
Obesity contributes significantly to the development and progression of cardiovascular disease (21), which is believed to be mediated by oxidative stress, inflammation and endothelial dysfunction (22). Recent studies have highlighted the importance of metabolic pathways in ECs and suggested the therapeutic potential of targeting EC metabolism, since microcirculation is a key player in obesity-associated cardiovascular disease. Obesity elevates circulating free fatty acids and alters adipokines and inflammatory cytokines that are released from visceral and perivascular fat, which leads to structural and functional changes in microvasculature (23). Given the recognized differences in the metabolism of healthy ECs vs. diabetes-associated dysfunctional ECs, EC metabolism could be targeted for therapeutic benefit (24). For instance, by restoring diabetes-impaired glycolytic-influx and angiogenic response of the ECs, diabetic vasculopathies can be improved (24). Endothelial insulin resistance is associated with diabetic cardiovascular complications, including atherosclerosis (25). The role of endothelial insulin resistance in the development of obesity and systemic insulin resistance is not as straight-forward. Common treatments for obesity involving strategies to increase insulin sensitivity (26) have often been associated with improvement of endothelial function and/or endothelial insulin sensitivity (5). For instance, administration of metformin, which is known to enhance whole-body insulin sensitivity, improved endothelium-dependent vessel relaxation (27). Another excellent example is that physical activity has been found to modulate EC phenotype by overcoming obesity-induced endothelial dysfunction (28). However, emerging evidence support the concept of endothelial regulation of metabolism, e.g., through endothelial interactions with the insulin targets in obesity. Advancement in this research area may provide new strategies to prevent and treat obesity and obesity-related diseases.
Obesity results from accumulation of white adipose tissue (29, 30). Adipose tissue is a central component for whole-body energy homeostasis. Insulin resistance in adipose tissue is presented as impaired insulin-stimulated glucose transport and blunted inhibition of lipolysis. However, adipocytes present a selective insulin resistance: while the insulin-promoted glucose transporter-4 trafficking is impaired, its impact on Forkhead box O-1 nuclear exclusion is preserved (31).
Responding to caloric intake, adipose tissue expands. People who are prone to deposit visceral but not subcutaneous adipose tissue have a higher risk of metabolic disease (32). Although normal subcutaneous adipose tissue expands with more capillary network than visceral tissue, this capability reduced with obesity and insulin resistance, suggesting that lack of angiogenesis in subcutaneous adipose tissue links to metabolic disease (33). The inability of peripheral adipose tissue to store excess energy has been reported in general population with insulin resistance (34, 35).
Linage tracing experiments demonstrated that the vascular endothelium of the adipose tissue gives rise to both white and brown fat cells (36), presumably through a Zfp423 (a multi zinc-finger transcription factor)-dependent pathway (37), suggesting a potential clinical use of EC-derived pre-adipocytes. In line with this, the capillary-derived beige adipocytes, when implanted in mice, improved metabolic homeostasis (38). Clearly, these data suggest that the plasticity and function of adipose tissue can both be regulated by proliferation/differentiation of stem cells and transformation of mature adipocytes by proper stimuli (39). It remains unknown, however, whether EC-derived factors are among these stimuli.
Adipose tissue angiogenesis and vascular functions have long been associated with obesity, adipose metabolism, and insulin sensitivity (40–42). Angiogenesis and adipogenesis are tightly coupled during development (43) and postnatal fat expansion (44). In vitro studies have shown that vascular-derived signals, e.g., endothelial-VEGF, directly affect the proliferation and differentiation of the surrounding pre-adipocytes, suggesting that angiogenesis can be a therapeutic target for obesity and metabolic diseases (45). Pharmacological inhibition of angiogenesis has been reported in vivo for the first time to reduce fat mass in distinct obesity models, likely through different vasculature-dependent mechanisms (46). However, the physiological consequences of modulating angiogenic activity seem to be context-dependent: gain of function studies show that overexpression of VEGF-A improves metabolic profiles, which maintains tissue function during the early phase of diet-induced obesity (44). In contrast, reducing vascularization promotes dysfunctional adipose cell death which may reverse obesity by improving metabolic outcomes in established obesity (47). Similar beneficial results were reported in nanoparticles-targeting adipose tissue transformation and angiogenesis, which prevented obesity in HFD-fed mice (48). It is unclear whether any EC-derived factors are involved in these studies.
Adipose stromal cells (ASCs) are therapeutically potent progenitor cells because they possess properties of pericytes. ASCs in combination with EC establish functional multilayer vessels. Transforming growth factor (TGF)-β is secreted by EC (49) and is able to induce αSMA expression in ASC, a marker of smooth muscle cell differentiation. A recent study found that ECs initiate this differentiation program in co-cultured ASC and propagate the program in distant ASC by induction of Activin A but not TGF-β (50, 51). Activin A is a secreted protein and a member of the TGF-β family that has pleiotropic effects on regulating apoptosis, proliferation, and cell differentiation (52). How ECs induce Activin A in ASC is yet to be determined (53).
Endocrine fibroblast growth factors (FGFs), such as FGF21, FGF15/19, and FGF23, are critical for maintaining whole-body homeostasis, because they operates in inter-organ endocrine signaling pathways that govern glucose, bile acid, and lipid metabolism (54, 55). FGF21 has gained much attention because of its favorable pharmacological properties in glucose and lipid metabolism (56). FGF21 is expressed in the liver, pancreas, adipose, and skeletal muscle, which can be altered by different stimuli, such as starvation or overfeeding (57). The role of endogenous FGF21 has just emerged (58). A recent study in HFD-fed mice (59) implicated that endothelial-derived FGF21 may contribute to improved metabolic profiles in mice lacking the autophagic protein LC3, although confirmative evidence is needed (e.g., demonstration of FGF21-dependency using FGF21-blocking antibodies (60) or endothelial FGF21 knockout mice). The role of autophagic proteins in the regulation of adipose tissue is not fully understood (61). If the role of LC3 is supported, it would be interesting and important to determine whether this feature also applies to other EC-derived FGFs.
MicroRNAs recently link ECs to adipocyte function in diabetes and CVD (62, 63). One example is miR-181b, which was downregulated in adipose ECs from mice fed an HFD (64). Downregulation of miR-181b induced insulin resistance and low-grade inflammation in adipose tissue, which can be reversed by genetic delivery of miR-181b. Mechanistically, miR-181b targets the PH domain and leucine-rich repeat protein phosphatase-2 in ECs and, thus, improves eNOS-NO signaling. As such, adipose tissue ECs promote glucose uptake in adipocytes in a paracrine manner (64). miR-181b also decreased protein levels of vascular cell adhesion molecule and intercellular adhesion molecule, consistent with previous studies on systemic delivery of miR-181b (65, 66). Other miRNAs, such as miR-126, have been shown to preserve normal endothelial function, likely via blocking unwanted endothelial activation (67). These findings highlight the role of adipose ECs in the development of obesity-induced insulin resistance and the potential of using miRNAs as a tool to modulate EC function (68).
The skeletal muscle is one of the major target organs of insulin, essential for insulin-induced glucose uptake (69). Insulin resistance in skeletal muscle is manifested as a decrease in glucose transport and a decline in muscle glycogen synthesis in response to circulating insulin. Insulin sensitivity is decreased in myocytes obtained from obese individuals, or cultured myocytes in the presence of adipocyte-derived lipids (70), supporting the concept that accumulation of excess lipids or their metabolic derivatives causes decreased insulin signaling in skeletal muscle (71, 72). Insulin promotes the glucose uptake by increasing blood flow and recruiting perfused capillaries in skeletal muscle. Insulin signaling in vascular endothelium may control its own delivery to skeletal muscle and other tissues (73), although it remains to be determined whether this mechanism is central to systemic insulin sensitivity (74).
Endothelial nitric oxide synthase (eNOS)-derived NO, the major endothelial-derived vasodilator, is central to endothelial regulation of insulin sensitivity in skeletal muscle, by stimulating blood vessel relaxation (in vascular smooth muscle cells) and perfusion (in skeletal muscles) (75). In addition, eNOS-derived NO regulates Akt signaling in skeletal muscle, through a cGMP-PI3K-dependent pathway (76). Interestingly, rapid formation of capillary ECs occurred in rat skeletal muscle after exposure to insulin (77), supporting the notion that insulin is a vascular hormone (78). However, the normal response of skeletal muscle capillary to NO is impaired in obesity-induced insulin resistance (79). Physical training appears to be able to improve it, likely because both endurance training and high intensity training with intervals increased eNOS protein contents specifically in the endothelium of capillaries and arterioles of skeletal muscle in previously sedentary lean and obese young men (80).
There is evidence indicating that insulin delivery to skeletal muscle interstitium through the ECs is rate-limiting in insulin-stimulated glucose uptake in mice (74, 81) and humans (82). This process seems to be impaired by insulin resistance in type 2 diabetes and obesity (83). Mechanistically, damage to the endothelial glycocalyx barrier in skeletal muscle is believed to be an early event, as shown in mice fed an HFD (84). A recent study showed that endothelial insulin resistance also plays a pivotal role in the regulation of glucose uptake by the skeletal muscle (83). Mechanistically, HFD-impaired insulin signaling in EC downregulated insulin-induced eNOS phosphorylation and attenuated capillary recruitment and insulin delivery, which, in turn, reduced glucose uptake by the skeletal muscle (83). Conversely, restoration of the insulin-induced eNOS phosphorylation in ECs normalized capillary recruitment and insulin delivery, which restored glucose uptake by the skeletal muscle (81).
Fatty acids represent a key energy source that is used by a number of tissues, which must be tightly controlled to avoid lipotoxicity induced by an excess of unoxidized fatty acids (85). Obesity often causes over-accumulation of lipids in non-adipose tissues (e.g., skeletal muscle), which can trigger a toxic reaction and impair the function of the tissues, contributing to insulin resistance (86). Previous studies have identified endothelium as a key regulator of fatty acid transport, mediated by a complex signaling pathway including VEGF-B, PPARγ, and Apelin (87). In a recent study, endothelial Fcγ receptor IIB activation was shown to blunt insulin delivery to skeletal muscle, causing insulin resistance in mice (88). A study found that the capillary endothelial fatty acid binding proteins 4 and 5 (FABP4/5) are required for fatty acid uptake in heart and skeletal muscle, organs that possess muscle-type continuous capillary (89). In contrast, the function of an EC-derive mitogen activated protein kinase kinase kinase kinase 4 (MAP4K4) has opposing functions in blood and lymphatic ECs: while MAP4K4 is required for lymphatic vascular integrity, blood EC-MAP4K4 induces insulin resistance by impairing vascular function in obesity (90).
The liver is responsible for maintaining fasting glucose levels. Excessive accumulation of lipids in the liver impairs hepatic responsiveness to insulin, leading to elevated levels of glucose and insulin in the circulation, eventually chronic hyperinsulinemia (91). Insulin resistance in the liver is selective in that insulin fails to suppress gluconeogenesis but continues to boost fatty acid synthesis (92). This uncoupling of insulin-mediated glucose and lipid metabolism will ultimately lead to hyperglycemia and hypertriglyceridemia, likely due to the unique activities of the mechanistic target of rapamycin complex (93) and the serine–threonine protein kinase (94) in hepatic lipogenesis. The role of endothelial regulation of insulin resistance in the liver has just begun to emerge.
Obesity and insulin resistance can cause NAFLD, which is characterized by liver metabolic dysregulation, such as hyperlipidemia (95). Recent epidemiology of NAFLD and its connection with CVD suggest that blocking endothelial dysfunction would be beneficial (96), since endothelial function is impaired in patients with NAFLD (97).
Liver sinusoidal endothelial cells are highly specialized ECs, which form the wall of liver sinusoids and represent up to 20% of liver cells (98). LSECs are well known for their crucial roles in liver regeneration (99–101) and chronic liver diseases associated with obesity (e.g., fibrosis) (102). LSECs maintain hepatic stellate cells and hepatocytes in quiescence at basal condition but induce hepatocyte proliferation and angiogenesis during regeneration and undergo morphological and functional changes during fibrosis (103). It is believed that LSECs accomplish these by interacting with other hepatic cell types to maintain liver homeostasis. Since healthy LSECs facilitate the rapid transfer of fatty acids formed from cholesteryl esters to parenchymal cells, LSECs injury contributes to NAFLD (104). LSECs injury has been associated with NAFLD progression, where NAFLD may alter LSECs-mediated transport of nutrients, lipids, and lipoproteins. A recent study in choline-deficient, l-amino acid-defined, and high-fat diet models that mimic human NAFLD indicated that LSEC injury functions as “gatekeeper” in the progression from simple steatosis to the early non-alcoholic steatohepatitis stage, and LSEC injury may activate Kupffer cells and hepatic stellate cells, which in turn results in chronic liver injuries (105). In this regard, LSECs operate as a traffic-director at the crossroad of regeneration and fibrosis (106). It is yet to be determined whether LSEC-derived factors are required for this function.
Iron overload often accompanies NAFLD. However, the key molecules involved in NAFLD-associated iron dysregulation have not been fully elucidated. A recent study found that LSEC secreted a bone morphogenetic protein-binding endothelial regulator that inhibited BMP-SMAD signaling in hepatocytes and reduced hepcidin protein expression (107). Fatty liver is the hepatic component of insulin resistance before developing NAFLD (108). In an attempt to identify physiological consequences of mice missing mucin-type O-glycans that are highly expressed in vascular ECs, a recent study found that the pups developed fatty liver disease, due to disconnected portal vein and intestinal lymphatic systems, leading to chylomicron deposition (109).
Our understanding of the endothelial layer has progressed significantly since its historical view as an inert layer of cells that serve as the inner lining of the circulatory system (110, 111). Now more than ever, the endothelium is demonstrated to regulate physiologic and pathologic processes by cross-talking with its residing organs. Over the past two decades, most research has focused on cellular and molecular mechanisms of endothelial dysfunction in disease conditions, such as diabetes (14, 112). Recent studies have looked into differences in the metabolism of healthy vs. diabetes in ECs, aiming at targeting endothelial metabolism for therapeutic benefit (24, 113). This review focuses on recent studies regarding the endothelial regulation of obesity-associated insulin resistance (Figure 1). The reviewed evidence supports an essential role for ECs in regulating the metabolic function of adipose tissues, skeletal muscle, and liver. However, except for the proposed regulation by EC-derived factors, other modes of regulation appear to be tissue-dependent. For instance, angiogenesis seems to be prominent in adipose tissue, consistent with its role in adipogenesis (21, 114). In contrast, blood vessel barrier function favors targeting the skeletal muscle, while LSEC intrahepatic interaction dominates in the liver (Figure 1). The difference may be ascribed to the distinct role of each organ/tissue in whole-body metabolism and the heterogeneity of the tissue-residing ECs. This concept may well apply to endothelial interaction with other type of cells not discussed here, such as inflammatory cells [e.g., macrophages (115)] and hypothalamic cells [e.g., hypothalamic-adipocyte axis (116) and brain (117)], which eventually affect insulin targets. The integrative conclusions of these studies present a picture of ECs interacting directly with cells of the classic insulin targets, through differential mechanisms, implicating organ- and pathway-specific imbalanced insulin actions (118). However, there are considerable unanswered questions that merit future investigations.
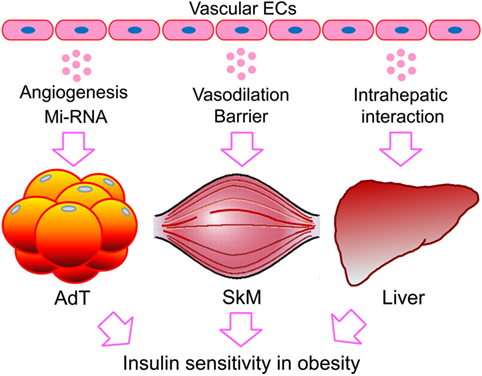
Figure 1. Endothelial regulation of obesity-associated insulin resistance in fat, skeletal muscle, and the liver. While direct interaction with ECs or EC-derived factors is expected to be shared by the classic targets of insulin, the other regulatory mechanisms could be tissue dependent. The preference in mode of action may reflect the distinct role of each organ/tissue in whole-body metabolism and the heterogeneity of the residing ECs. ECs, endothelial cells; miRNA, microRNA; AdTs, adipose tissues; SkM, skeletal muscle.
(1) While tissue- or organ-specific gene expression [e.g., tissue-specific promoters (119)] has been identified and widely used in pre-clinical studies, only global endothelial makers are currently available. To date, there are few reports on markers that may be unique to a tissue type [e.g., gene expression profile of microvascular ECs specific to the liver (120)] or to a vascular bed [e.g., a transcriptional factor restricted to arterial-ECs (121)]. Given the recognized phenotypic and functional heterogeneity in the endothelium, can we identify endothelial factors specific to their sites (e.g., tissues/organs or vascular beds) so that site-specific ECs could be manipulated for proof-of-concept studies and drug development?
(2) Phenotypic heterogeneity is a central feature of the endothelium, referring to differences in the location of the conduit vessels (e.g., arteries vs. veins) and their functions in serving specific organs (e.g., adipose tissue and skeletal muscle) (110, 111). In microvasculature, the diversity of the endothelium contributes to their specialized functions in regulating permeability, leukocyte trafficking, and hemostasis, the key features of endothelial function (122). How is this heterogeneity linked to the interactions between ECs and the classic targets of insulin discussed here?
(3) Endothelial cell-derived factors can be essential in maintaining organ homeostasis and regeneration. Could endothelium itself also operate as a rate-limiting apparatus [e.g., through its barrier function (123)] that regulates other hormones (e.g., IGFs and FGFs) in the same fashion as insulin for their delivery to targeted tissues or relevant locations?
(4) Severe impairment of endothelial function eventually promotes CVD. Non-invasive assessment of conduit vascular endothelial function is possible both in adults (124) and children (125) with metabolic disorders [e.g., using reactive hyperthermia and flow-mediated dilatation (126)]. In the same token, can endothelial regulation of metabolism be assessed in vivo? Or can specific parameters in this regard be identified by global assessment of gene expression profiles, epigenetic markers, and transcriptional factors?
(5) Over the past two decades, many experimental or therapeutic drugs that improve CVD risk factors also improved endothelial functions (127). Do these drugs modulate the crosstalk between ECs and the cells in their residing tissues? Or is this crosstalk an unrecognized mode of action, which should be considered in designing new and better drugs in the future?
In conclusion, the mechanisms underlying the endothelial-specific effects discussed here remain to be fully determined. Further translational studies to determine the clinical relevance of endothelial regulation of metabolism will provide greater insights that may ultimately lead to therapeutic advances against the increasing burden of the obesity pandemic and the associated metabolic disorders.
JX contributed to the conception. JX, ML, and MQ wrote the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by National Institutes of Health Grants (R01HL-130845), COBRE Program of the National Institute of General Medical Sciences (5P20GM104934, Project 2), a Beginning-Grant-in-Aid Award (14BGIA20030027) from the American Heart Association, a Junior Faculty Award (1-12-JF-58) from the American Diabetes Association, and Research Awards (HR11-200, HR14-062, and HR17-046) from the Oklahoma Center for the Advancement of Science and Technology (all to JX).
1. Korner J, Aronne LJ. The emerging science of body weight regulation and its impact on obesity treatment. J Clin Invest (2003) 111:565–70. doi:10.1172/JCI17953
5. Xu J, Zou MH. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation (2009) 120:1266–86. doi:10.1161/CIRCULATIONAHA.108.835223
6. Meng Q, Makinen VP, Luk H, Yang X. Systems biology approaches and applications in obesity, diabetes, and cardiovascular diseases. Curr Cardiovasc Risk Rep (2013) 7:73–83. doi:10.1007/s12170-012-0280-y
7. Shimobayashi M, Hall MN. Making new contacts: the motor network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol (2014) 15:155–62. doi:10.1038/nrm3757
8. Lu B, Bridges D, Yang Y, Fisher K, Cheng A, Chang L, et al. Metabolic crosstalk: molecular links between glycogen and lipid metabolism in obesity. Diabetes (2014) 63:2935–48. doi:10.2337/db13-1531
9. Dumortier O, Hinault C, Van Obberghen E. MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metab (2013) 18:312–24. doi:10.1016/j.cmet.2013.06.004
10. Fessler MB. Liver X receptor: crosstalk node for the signaling of lipid metabolism, carbohydrate metabolism, and innate immunity. Curr Signal Transduct Ther (2008) 3:75–81. doi:10.2174/157436208784223170
11. Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab (2013) 17:20–33. doi:10.1016/j.cmet.2012.11.012
12. Mauricio MD, Aldasoro M, Ortega J, Vila JM. Endothelial dysfunction in morbid obesity. Curr Pharm Des (2013) 19:5718–29. doi:10.2174/1381612811319320007
13. Elsheikh E, Younoszai Z, Otgonsuren M, Hunt S, Raybuck B, Younossi ZM. Markers of endothelial dysfunction in patients with non-alcoholic fatty liver disease and coronary artery disease. J Gastroenterol Hepatol (2014) 29:1528–34. doi:10.1111/jgh.12549
14. Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord (2010) 11:61–74. doi:10.1007/s11154-010-9134-4
15. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease – a 30th anniversary update. Acta Physiol (Oxf) (2017) 219:22–96. doi:10.1111/apha.12646
16. Jaffe EA. Cell biology of endothelial cells. Hum Pathol (1987) 18:234–9. doi:10.1016/S0046-8177(87)80005-9
17. Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemost (2005) 3:1392–406. doi:10.1111/j.1538-7836.2005.01328.x
18. Gross PL, Aird WC. The endothelium and thrombosis. Semin Thromb Hemost (2000) 26:463–78. doi:10.1055/s-2000-13202
19. Wagner DD, Frenette PS. The vessel wall and its interactions. Blood (2008) 111:5271–81. doi:10.1182/blood-2008-01-078204
20. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation (2004) 109:III27–32. doi:10.1161/01.CIR.0000131515.03336.f8
21. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell (2014) 156:20–44. doi:10.1016/j.cell.2013.12.012
22. Avogaro A, de Kreutzenberg SV. Mechanisms of endothelial dysfunction in obesity. Clin Chim Acta (2005) 360:9–26. doi:10.1016/j.cccn.2005.04.020
23. Sorop O, Olver TD, van de Wouw J, Heinonen I, van Duin RW, Duncker DJ, et al. The microcirculation: a key player in obesity-associated cardiovascular disease. Cardiovasc Res (2017). 113:1035–45. doi:10.1093/cvr/cvx093
24. de Zeeuw P, Wong BW, Carmeliet P. Metabolic adaptations in diabetic endothelial cells. Circ J (2015) 79:934–41. doi:10.1253/circj.CJ-15-0230
25. Kanter JE, Bornfeldt KE. Evidence stacks up that endothelial insulin resistance is a culprit in atherosclerosis. Circ Res (2013) 113:352–4. doi:10.1161/CIRCRESAHA.113.301998
26. Haas JT, Biddinger SB. Dissecting the role of insulin resistance in the metabolic syndrome. Curr Opin Lipidol (2009) 20:206–10. doi:10.1097/MOL.0b013e32832b2024
27. Mather KJ, Verma S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol (2001) 37:1344–50. doi:10.1016/S0735-1097(01)01129-9
28. Bender SB, Laughlin MH. Modulation of endothelial cell phenotype by physical activity: impact on obesity-related endothelial dysfunction. Am J Physiol Heart Circ Physiol (2015) 309:H1–8. doi:10.1152/ajpheart.00177.2015
29. Schinzari F, Tesauro M, Cardillo C. Endothelial and perivascular adipose tissue abnormalities in obesity-related vascular dysfunction: novel targets for treatment. J Cardiovasc Pharmacol (2017) 69:360–8. doi:10.1097/FJC.0000000000000469
30. Scherer PE, Hill JA. Obesity, diabetes, and cardiovascular diseases: a compendium. Circ Res (2016) 118:1703–5. doi:10.1161/CIRCRESAHA.116.308999
31. Gonzalez E, Flier E, Molle D, Accili D, McGraw TE. Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc Natl Acad Sci U S A (2011) 108:10162–7. doi:10.1073/pnas.1019268108
32. Britton KA, Fox CS. Ectopic fat depots and cardiovascular disease. Circulation (2011) 124:e837–41. doi:10.1161/CIRCULATIONAHA.111.077602
33. Gealekman O, Guseva N, Hartigan C, Apotheker S, Gorgoglione M, Gurav K, et al. Depot-specific differences and insufficient subcutaneous adipose tissue angiogenesis in human obesity. Circulation (2011) 123:186–94. doi:10.1161/CIRCULATIONAHA.110.970145
34. Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet (2017) 49:17–26. doi:10.1038/ng0217-317c
35. Ledoux S, Queguiner I, Msika S, Calderari S, Rufat P, Gasc JM, et al. Angiogenesis associated with visceral and subcutaneous adipose tissue in severe human obesity. Diabetes (2008) 57:3247–57. doi:10.2337/db07-1812
36. Tran KV, Gealekman O, Frontini A, Zingaretti MC, Morroni M, Giordano A, et al. The vascular endothelium of the adipose tissue gives rise to both white and brown fat cells. Cell Metab (2012) 15:222–9. doi:10.1016/j.cmet.2012.01.008
37. Gupta RK, Mepani RJ, Kleiner S, Lo JC, Khandekar MJ, Cohen P, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab (2012) 15:230–9. doi:10.1016/j.cmet.2012.01.010
38. Min SY, Kady J, Nam M, Rojas-Rodriguez R, Berkenwald A, Kim JH, et al. Human ‘brite/beige’ adipocytes develop from capillary networks, and their implantation improves metabolic homeostasis in mice. Nat Med (2016) 22:312–8. doi:10.1038/nm.4031
39. Cinti S. Ucp1 protein: the molecular hub of adipose organ plasticity. Biochimie (2017) 134:71–6. doi:10.1016/j.biochi.2016.09.008
40. Cao Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab (2013) 18:478–89. doi:10.1016/j.cmet.2013.08.008
41. Lemoine AY, Ledoux S, Larger E. Adipose tissue angiogenesis in obesity. Thromb Haemost (2013) 110:661–8. doi:10.1160/TH13-01-0073
42. Corvera S, Gealekman O. Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim Biophys Acta (2014) 1842:463–72. doi:10.1016/j.bbadis.2013.06.003
43. Voros G, Maquoi E, Demeulemeester D, Clerx N, Collen D, Lijnen HR. Modulation of angiogenesis during adipose tissue development in murine models of obesity. Endocrinology (2005) 146:4545–54. doi:10.1210/en.2005-0532
44. Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, et al. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A (2012) 109:5874–9. doi:10.1073/pnas.1200447109
45. Cao Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov (2010) 9:107–15. doi:10.1038/nrd3055
46. Rupnick MA, Panigrahy D, Zhang CY, Dallabrida SM, Lowell BB, Langer R, et al. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A (2002) 99:10730–5. doi:10.1073/pnas.162349799
47. Kolonin MG, Saha PK, Chan L, Pasqualini R, Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nat Med (2004) 10:625–32. doi:10.1038/nm1048
48. Xue Y, Xu X, Zhang XQ, Farokhzad OC, Langer R. Preventing diet-induced obesity in mice by adipose tissue transformation and angiogenesis using targeted nanoparticles. Proc Natl Acad Sci U S A (2016) 113:5552–7. doi:10.1073/pnas.1603840113
49. Merrilees MJ, Sodek J. Synthesis of TGF-beta 1 by vascular endothelial cells is correlated with cell spreading. J Vasc Res (1992) 29:376–84. doi:10.1159/000158954
50. Merfeld-Clauss S, Lupov IP, Lu H, Feng D, Compton-Craig P, March KL, et al. Adipose stromal cells differentiate along a smooth muscle lineage pathway upon endothelial cell contact via induction of activin A. Circ Res (2014) 115:800–9. doi:10.1161/CIRCRESAHA.115.304026
51. Merfeld-Clauss S, Lupov IP, Lu H, March KL, Traktuev DO. Adipose stromal cell contact with endothelial cells results in loss of complementary vasculogenic activity mediated by induction of activin A. Stem Cells (2015) 33:3039–51. doi:10.1002/stem.2074
52. Namwanje M, Brown CW. Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb Perspect Biol (2016) 8:1–55. doi:10.1101/cshperspect.a021881
53. Martin KA, Hirschi KK. The magic touch: endothelial cells muscle-up adipose. Circ Res (2014) 115:752–4. doi:10.1161/CIRCRESAHA.114.305030
54. Degirolamo C, Sabba C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov (2016) 15:51–69. doi:10.1038/nrd.2015.9
55. Markan KR, Potthoff MJ. Metabolic fibroblast growth factors (FGFs): mediators of energy homeostasis. Semin Cell Dev Biol (2016) 53:85–93. doi:10.1016/j.semcdb.2015.09.021
56. Kharitonenkov A, Adams AC. Inventing new medicines: the FGF21 story. Mol Metab (2014) 3:221–9. doi:10.1016/j.molmet.2013.12.003
57. Angelin B, Larsson TE, Rudling M. Circulating fibroblast growth factors as metabolic regulators – a critical appraisal. Cell Metab (2012) 16:693–705. doi:10.1016/j.cmet.2012.11.001
58. Fisher FM, Maratos-Flier E. Understanding the physiology of FGF21. Annu Rev Physiol (2016) 78:223–41. doi:10.1146/annurev-physiol-021115-105339
59. Li M, Qian M, Xu J. Lc3 blockade improves metabolic profiles in mice fed a high-fat diet. Diabetes (2017) 66(Suppl 1):A545. doi:10.2337/db17-1871-2152
60. Kharitonenkov A, DiMarchi R. FGF21 revolutions: recent advances illuminating FGF21 biology and medicinal properties. Trends Endocrinol Metab (2015) 26:608–17. doi:10.1016/j.tem.2015.09.007
61. Zhang Y, Zeng X, Jin S. Autophagy in adipose tissue biology. Pharmacol Res (2012) 66:505–12. doi:10.1016/j.phrs.2012.09.004
62. Zhang Y, Sun X, Icli B, Feinberg MW. Emerging roles for microRNAs in diabetic microvascular disease: novel targets for therapy. Endocr Rev (2017) 38:145–68. doi:10.1210/er.2016-1122
63. Icli B, Feinberg MW. MicroRNAs in dysfunctional adipose tissue: cardiovascular implications. Cardiovasc Res (2017). 113:1024–34. doi:10.1093/cvr/cvx098
64. Sun X, Lin J, Zhang Y, Kang S, Belkin N, Wara AK, et al. MicroRNA-181b improves glucose homeostasis and insulin sensitivity by regulating endothelial function in white adipose tissue. Circ Res (2016) 118:810–21. doi:10.1161/CIRCRESAHA.115.308166
65. Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, et al. MicroRNA-181b regulates nf-kappab-mediated vascular inflammation. J Clin Invest (2012) 122:1973–90. doi:10.1172/JCI61495
66. Sun X, He S, Wara AK, Icli B, Shvartz E, Tesmenitsky Y, et al. Systemic delivery of microRNA-181b inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ Res (2014) 114:32–40. doi:10.1161/CIRCRESAHA.113.302089
67. Fish JE, Cybulsky MI. Taming endothelial activation with a microRNA. J Clin Invest (2012) 122:1967–70. doi:10.1172/JCI63818
68. Yar S, Chang HC, Ardehali H. The good neighbor: coping with insulin resistance by modulating adipose tissue endothelial cell function. Circ Res (2016) 118:776–8. doi:10.1161/CIRCRESAHA.116.308338
69. Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med (1990) 322:223–8. doi:10.1056/NEJM199001253220403
70. Kovalik JP, Slentz D, Stevens RD, Kraus WE, Houmard JA, Nicoll JB, et al. Metabolic remodeling of human skeletal myocytes by cocultured adipocytes depends on the lipolytic state of the system. Diabetes (2011) 60:1882–93. doi:10.2337/db10-0427
71. Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab (2008) 7:45–56. doi:10.1016/j.cmet.2007.10.013
72. Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A (2007) 104:12587–94. doi:10.1073/pnas.0705408104
73. Barrett EJ, Eggleston EM, Inyard AC, Wang H, Li G, Chai W, et al. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia (2009) 52:752–64. doi:10.1007/s00125-009-1313-z
74. Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab (2011) 13:294–307. doi:10.1016/j.cmet.2011.01.018
75. Dora KA. Endothelial-smooth muscle cell interactions in the regulation of vascular tone in skeletal muscle. Microcirculation (2016) 23:626–30. doi:10.1111/micc.12322
76. Drenning JA, Lira VA, Soltow QA, Canon CN, Valera LM, Brown DL, et al. Endothelial nitric oxide synthase is involved in calcium-induced Akt signaling in mouse skeletal muscle. Nitric Oxide (2009) 21:192–200. doi:10.1016/j.niox.2009.08.001
77. Holmang A, Jennische E, Bjorntorp P. Rapid formation of capillary endothelial cells in rat skeletal muscle after exposure to insulin. Diabetologia (1996) 39:206–11. doi:10.1007/BF00403964
78. Scherrer U, Sartori C. Insulin as a vascular and sympathoexcitatory hormone: implications for blood pressure regulation, insulin sensitivity, and cardiovascular morbidity. Circulation (1997) 96:4104–13. doi:10.1161/01.CIR.96.11.4104
79. Chadderdon SM, Belcik JT, Bader L, Peters DM, Kievit P, Alkayed NJ, et al. Temporal changes in skeletal muscle capillary responses and endothelial-derived vasodilators in obesity-related insulin resistance. Diabetes (2016) 65:2249–57. doi:10.2337/db15-1574
80. Cocks M, Wagenmakers AJ. The effect of different training modes on skeletal muscle microvascular density and endothelial enzymes controlling no availability. J Physiol (2016) 594:2245–57. doi:10.1113/JP270329
81. Kubota T, Kubota N, Kadowaki T. The role of endothelial insulin signaling in the regulation of glucose metabolism. Rev Endocr Metab Disord (2013) 14:207–16. doi:10.1007/s11154-013-9242-z
82. Barrett EJ, Wang H, Upchurch CT, Liu Z. Insulin regulates its own delivery to skeletal muscle by feed-forward actions on the vasculature. Am J Physiol Endocrinol Metab (2011) 301:E252–63. doi:10.1152/ajpendo.00186.2011
83. Wang H, Liu Z, Li G, Barrett EJ. The vascular endothelial cell mediates insulin transport into skeletal muscle. Am J Physiol Endocrinol Metab (2006) 291:E323–32. doi:10.1152/ajpendo.00047.2006
84. Eskens BJ, Leurgans TM, Vink H, Vanteeffelen JW. Early impairment of skeletal muscle endothelial glycocalyx barrier properties in diet-induced obesity in mice. Physiol Rep (2014) 2:e00194. doi:10.1002/phy2.194
85. Kusminski CM, Shetty S, Orci L, Unger RH, Scherer PE. Diabetes and apoptosis: lipotoxicity. Apoptosis (2009) 14:1484–95. doi:10.1007/s10495-009-0352-8
86. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest (2011) 121:2094–101. doi:10.1172/JCI45887
87. Mehrotra D, Wu J, Papangeli I, Chun HJ. Endothelium as a gatekeeper of fatty acid transport. Trends Endocrinol Metab (2014) 25:99–106. doi:10.1016/j.tem.2013.11.001
88. Tanigaki K, Chambliss KL, Yuhanna IS, Sacharidou A, Ahmed M, Atochin DN, et al. Endothelial Fcgamma receptor IIB activation blunts insulin delivery to skeletal muscle to cause insulin resistance in mice. Diabetes (2016) 65:1996–2005. doi:10.2337/db15-1605
89. Iso T, Maeda K, Hanaoka H, Suga T, Goto K, Syamsunarno MR, et al. Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle. Arterioscler Thromb Vasc Biol (2013) 33:2549–57. doi:10.1161/ATVBAHA.113.301588
90. Roth Flach RJ, DiStefano MT, Danai LV, Senol-Cosar O, Yawe JC, Kelly M, et al. Map4k4 impairs energy metabolism in endothelial cells and promotes insulin resistance in obesity. Am J Physiol Endocrinol Metab (2017):ajendo.00037.2017. doi:10.1152/ajpendo.00037.2017
91. Reccia I, Kumar J, Akladios C, Virdis F, Pai M, Habib N, et al. Non-alcoholic fatty liver disease: a sign of systemic disease. Metabolism (2017) 72:94–108. doi:10.1016/j.metabol.2017.04.011
92. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab (2008) 7:95–6. doi:10.1016/j.cmet.2007.12.009
93. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A (2010) 107:3441–6. doi:10.1073/pnas.0914798107
94. Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR, et al. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab (2011) 14:516–27. doi:10.1016/j.cmet.2011.09.001
95. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology (2012) 142:1592–609. doi:10.1053/j.gastro.2012.04.001
96. Federico A, Dallio M, Masarone M, Persico M, Loguercio C. The epidemiology of non-alcoholic fatty liver disease and its connection with cardiovascular disease: role of endothelial dysfunction. Eur Rev Med Pharmacol Sci (2016) 20:4731–41. Available from: http://www.europeanreview.org/article/11778
97. Fan Y, Wei F, Zhou Y, Zhang H. Association of non-alcoholic fatty liver disease with impaired endothelial function by flow-mediated dilation: a meta-analysis. Hepatol Res (2016) 46:E165–73. doi:10.1111/hepr.12554
98. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol (2017) 66:212–27. doi:10.1016/j.jhep.2016.07.009
99. Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature (2010) 468:310–5. doi:10.1038/nature09493
100. Moniaux N, Faivre J. Key role of sinusoidal endothelial cells in the triggering of liver regeneration. J Hepatol (2011) 55:488–90. doi:10.1016/j.jhep.2011.02.005
101. Gamble J, Vadas M, McCaughan G. Sinusoidal endothelium is essential for liver regeneration. Hepatology (2011) 54:731–3. doi:10.1002/hep.24455
102. Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature (2014) 505:97–102. doi:10.1038/nature12681
103. Kostallari E, Shah VH. Angiocrine signaling in the hepatic sinusoids in health and disease. Am J Physiol Gastrointest Liver Physiol (2016) 311:G246–51. doi:10.1152/ajpgi.00118.2016
104. Nagelkerke JF, van Berkel TJ. Rapid transport of fatty acids from rat liver endothelial to parenchymal cells after uptake of cholesteryl ester-labeled acetylated LDL. Biochim Biophys Acta (1986) 875:593–8. doi:10.1016/0005-2760(86)90081-0
105. Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, et al. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest (2015) 95:1130–44. doi:10.1038/labinvest.2015.95
106. Huebert RC, Shah VH. Sinusoidal endothelial cells direct traffic at the intersection of regeneration and fibrosis. Hepatology (2014) 60:754–6. doi:10.1002/hep.27116
107. Hasebe T, Tanaka H, Sawada K, Nakajima S, Ohtake T, Fujiya M, et al. Bone morphogenetic protein-binding endothelial regulator of liver sinusoidal endothelial cells induces iron overload in a fatty liver mouse model. J Gastroenterol (2017) 52:341–51. doi:10.1007/s00535-016-1237-6
108. Perseghin G. The role of non-alcoholic fatty liver disease in cardiovascular disease. Dig Dis (2010) 28:210–3. doi:10.1159/000282088
109. Fu J, Gerhardt H, McDaniel JM, Xia B, Liu X, Ivanciu L, et al. Endothelial cell O-glycan deficiency causes blood/lymphatic misconnections and consequent fatty liver disease in mice. J Clin Invest (2008) 118:3725–37. doi:10.1172/JCI36077
110. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res (2007) 100:174–90. doi:10.1161/01.RES.0000255690.03436.ae
111. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res (2007) 100:158–73. doi:10.1161/01.RES.0000255691.76142.4a
112. Roberts AC, Porter KE. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab Vasc Dis Res (2013) 10:472–82. doi:10.1177/1479164113500680
113. Missiaen R, Morales-Rodriguez F, Eelen G, Carmeliet P. Targeting endothelial metabolism for anti-angiogenesis therapy: a pharmacological perspective. Vascul Pharmacol (2017) 90:8–18. doi:10.1016/j.vph.2017.01.001
114. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov (2016) 15:639–60. doi:10.1038/nrd.2016.75
115. Verdeguer F, Aouadi M. Macrophage heterogeneity and energy metabolism. Exp Cell Res (2017). doi:10.1016/j.yexcr.2017.03.043
116. Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, et al. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab (2011) 14:324–38. doi:10.1016/j.cmet.2011.06.020
117. Emanuel AL, Meijer RI, Muskiet MH, van Raalte DH, Eringa EC, Serne EH. Role of insulin-stimulated adipose tissue perfusion in the development of whole-body insulin resistance. Arterioscler Thromb Vasc Biol (2017) 37:411–8. doi:10.1161/ATVBAHA.116.308670
118. Kubota T, Kubota N, Kadowaki T. Imbalanced insulin actions in obesity and type 2 diabetes: key mouse models of insulin signaling pathway. Cell Metab (2017) 25:797–810. doi:10.1016/j.cmet.2017.03.004
119. Koch F, Fenouil R, Gut M, Cauchy P, Albert TK, Zacarias-Cabeza J, et al. Transcription initiation platforms and gtf recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol (2011) 18:956–63. doi:10.1038/nsmb.2085
120. Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell (2013) 26:204–19. doi:10.1016/j.devcel.2013.06.017
121. Aranguren XL, Agirre X, Beerens M, Coppiello G, Uriz M, Vandersmissen I, et al. Unraveling a novel transcription factor code determining the human arterial-specific endothelial cell signature. Blood (2013) 122:3982–92. doi:10.1182/blood-2013-02-483255
122. Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, et al. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A (2003) 100:10623–8. doi:10.1073/pnas.1434429100
123. Kolka CM, Bergman RN. The barrier within: endothelial transport of hormones. Physiology (Bethesda) (2012) 27:237–47. doi:10.1152/physiol.00012.2012
124. Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the international brachial artery reactivity task force. J Am Coll Cardiol (2002) 39:257–65. doi:10.1016/S0735-1097(01)01746-6
125. Hudgins LC, Annavajjhala V, Kovanlikaya A, Frank MD, Solomon A, Parker TS, et al. Non-invasive assessment of endothelial function in children with obesity and lipid disorders. Cardiol Young (2016) 26:532–8. doi:10.1017/S1047951115000657
126. Kelm M. Flow-mediated dilatation in human circulation: diagnostic and therapeutic aspects. Am J Physiol Heart Circ Physiol (2002) 282:H1–5.
Keywords: endothelial function, adipose, liver, skeletal muscle, obesity, insulin resistance, diabetes, metabolism
Citation: Li M, Qian M and Xu J (2017) Vascular Endothelial Regulation of Obesity-Associated Insulin Resistance. Front. Cardiovasc. Med. 4:51. doi: 10.3389/fcvm.2017.00051
Received: 30 June 2017; Accepted: 27 July 2017;
Published: 09 August 2017
Edited by:
Hong Chen, Boston Children’s Hospital, United StatesReviewed by:
Jun Yu, Temple University, United StatesCopyright: © 2017 Li, Qian and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Xu, amlhbi14dUBvdWhzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.