
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 31 May 2017
Sec. Cardiovascular Genetics and Systems Medicine
Volume 4 - 2017 | https://doi.org/10.3389/fcvm.2017.00031
 Vasundhara Kain
Vasundhara Kain Ganesh V. Halade*
Ganesh V. Halade*
Diabetic cardiomyopathy (DCM) or diabetes-induced cardiac dysfunction is a direct consequence of uncontrolled metabolic syndrome and is widespread in US population and worldwide. Despite of the heterogeneous and distinct features of DCM, the clinical relevance of DCM is now becoming established. DCM progresses to pathological cardiac remodeling with the higher risk of heart attack and subsequent heart failure in diabetic patients. In this review, we emphasize lipid substrate quality and the phenotypic, metabolic, and biochemical stressors of DCM in the rodent and human pathophysiology. We discuss lipoxygenase signaling in the inflammatory pathway with multiple contributing and confounding factors leading to DCM. Additionally, emerging biochemical pathways are emphasized to make progress toward therapeutic advancement to treat DCM.
Cardiovascular disease (CVD) is the primary cause of death including substantial people suffering from obesity and type 2 diabetes. With an increased population of patients displaying metabolic syndrome, there are 17.3 million deaths per year, and this number is expected to increase to more than 23.6 million by 2030 (1, 2). Diabetes and obesity are primary metabolic triggers associated with imbalanced energy [fatty acids (FAs)] intake, which is an inherent part of the modern lifestyle. Obese people are often prone to or diagnosed with insulin resistance, pre-diabetes, impaired glucose tolerance, or type 2 diabetes. According to current statistics, nearly 29 million individuals in US have diabetes, and one in three adults has the pre-diabetic condition (3) and heart disease, and stroke is leading cause of disability, morbidity, and death among people with accelerated or uncontrolled type 2 diabetes. CVD is associated with ~65% of deaths related to diabetes (4) and has an adverse effect on left ventricle size, geometry, and function leading to diabetic cardiomyopathy (DCM).
Diabetic cardiomyopathy is a complex pathological as well as adaptive condition characterized by dysfunctional effects on the left ventricle and is developed by a combination of several metabolic disorders including prediabetes, hyperglycemia, insulin resistance (in type 2 diabetes), hypertension, and obesity (5). There are multiple factors and mechanisms including aging that aggravate the pathology of DCM. However, in the current context of cardiomyopathy, DCM is defined by presence of abnormal myocardial diastolic or systolic function in the presence of diabetes without known hypertension or coronary artery disease (6). It has been categorized and presented in different manners as it includes features of left ventricular (LV) hypertrophy, myocardial fibrosis, and myocardial energy dysregulation with differential degrees of myocardial biochemical, mechanical, or structural dysfunction. Due to its multifactorial origin and distinct pathophysiology, there are some controversies regarding the existence or non-existence of DCM. Clinically, DCM lacks classical features of a cardiomyopathy such as ventricular dilation and meaningful systolic dysfunction. From metabolism perspective, DCM is a combination of molecular myocardial abnormalities that lead to the development of myocardial dysfunction with co-existence of additional stressors such as obesity, hypertension, and coronary artery disease. In this review, we focus on the etiology that aggravates DCM in clinical and pre-clinical settings. We discuss pathophysiological and metabolic stressors that are prime contributors to DCM. In order to focus on the development and advancement of therapeutic targets in the DCM field, we asked three questions: (1) Do we need to treat the original conditions or metabolic abnormalities of DCM in order to reduce or prevent DCM and heart failure? (2) Do we need to develop a pharmacological lifestyle or targeted aggressive metabolomics approach to identify the biomarker signatures to treat DCM? (3) Does a combination of (1) and (2) allow adequate control of the original metabolic abnormality with focused treatment of DCM, particularly in an aging population?
In order to understand the pathophysiology of DCM and its etiology related to metabolic remodeling in the heart, we have highlighted the major confounding causes below and summarized experimental rodent models used to understand the dysregulation in the latter (Table 1), acknowledging that the scope of this review does not allow us to discuss them all.
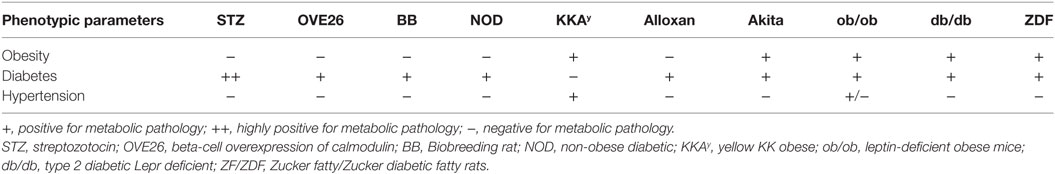
Table 1. Metabolic dysregulations in diabetic cardiomyopathy.
Obesity is characterized by a low-grade chronic inflammation phenotype with an excessive amount of body fat leading to heart disease, diabetes, and high blood pressure. More than 34.9%, almost 78.6 million of US adults are obese (7). Obese and overweight individuals are prone to insulin resistance and diabetes, accompanied frequently by LV eccentric or concentric hypertrophy (8). Obesity is characterized on the basis of body mass index (BMI) and has been proved to be associated with ventricular hypertrophy (9, 10). The association of obesity with systolic dysfunction and with the prevalence of diabetes makes obesity a confounding factor of DCM. The predicted body weight along with the excess fat mass accounts for a rise in LV stroke volume and stroke works due to accelerated heart rate. The Framingham study suggested that a BMI >30 kg/m2 is positively correlated with increased LV wall thickness, LV internal dimension in diastole, and LV mass (11). A number of studies have shown that visceral adipose tissue has higher levels of pro-inflammatory cytokines when associated with LV diastolic dysfunction (12). Studies performed on the isolated hearts of genetic animal models of obesity, such as Zucker fatty (ZF) rats or leptin-deficient obese mice, showed a depression in cardiac function (13, 14). However, a few studies also reported the normal function of the heart in obese rodent models (15, 16). As type of fat, duration, and dose consumed impacts LV structure, function, and healing in the myocardial infarction (MI)-induced model, the translational prospects of fat intake and intervention studies should be used with caution. This is exemplified by studies from Brainard and colleagues suggesting that a high-fat diet in the form of lard or milk before and after MI is insufficient to induce cardiac dysfunction, despite adiposity and impaired glucose disposal (17). The study showed that the signs of cardiac dysfunction are not visible in commonly used mouse model [type 2 diabetic Lepr-deficient (db/db) mice or streptozotocin (STZ)-treated wild-type mice] when subjected to pressure overload. But the db/db model showed depressed cardiac function when subjected to ischemia–reperfusion injury in comparison with non-diabetic mice (17). By contrast, when the obesity is superimposed on aging that is the trigger to develop non-resolving inflammation post-MI. Recent study by Lopez et al. highlighted that supplementation of n-6 FAs in aged mice resulted in higher levels of 12-S-hydroxyeicosatetraenoic acids (HETE) leading to an acute inflammatory response, further delaying healing in MI (18). These results are similar to the findings that showed higher levels of 12-HETE in individuals with stable angina (19). Studies of aging coupled with obesity have shown that the n-6 enriched fat diet creates lipid metabolites that are pro-inflammatory in nature (Figure 1). Aging mice fed a diet enriched in linoleic acid showed increased neutrophils in the gut and development of dysbiosis (20). Thus, these metabolites and their action in disease pathology such as pressure overload, post-MI healing, and overall cardiac remodeling will provide novel tools for drug discovery and target identification.
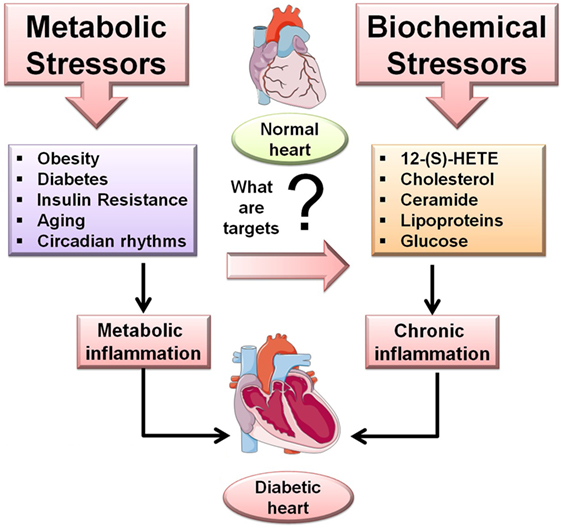
Figure 1. Overview of the metabolic and biochemical stressors in diabetic cardiomyopathy (DCM). Metabolic stressor such as obesity, diabetes, insulin resistance, aging, and circadian rhythms while the biochemical stressors 12-(S)-HETE, cholesterol, ceramide, lipoproteins, glucose trigger inflammation-mediated DCM.
Type 2 diabetes is defined as a metabolic syndrome with a combination of insulin resistance and defective insulin secretion by pancreatic β-cells. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) study was important in showing the role of hyperglycemia in micro- and macro-vascular diabetes-mediated complications (21, 22). Impaired insulin sensitivity results from obesity and physical inactivity or genetic susceptibility (23, 24). Currently, 39 million Americans are diabetic, predisposing them to cardiovascular risk. Clinical data have demonstrated that diabetes is an independent risk factor for cardiovascular complications. In both men and women, diabetes has been observed to be one of the primary causes of DCM and subsequent heart failure over the last three decades (5, 25). The development of diabetes with LV dysfunction was clinically evident 20 years earlier (26). Many clinical studies have shown that more than 60% of diabetic patients compared with well controlled subjects are diagnosed with early and mild ventricular diastolic dysfunction (27, 28). Studies have confirmed that DCM is not an unusual condition but caused by low-grade non-resolving inflammation, uncontrolled hyperglycemia (glucotoxicity), or hyperlipidemia (lipotoxicity). These factors mainly contribute to metabolic syndrome, leading to cardiac dysfunction and increasing the risk of MI or recurrent MI (29). Glucotoxicity refers to uncontrolled hyperglycemia and subsequent metabolic changes triggered by excess sugar or carbohydrate (30). Lipotoxicity is more complex and diverse because of the essential nature of FAs, but excessive intake in aging may lead to bone marrow adiposity and non-resolving inflammation post-MI (31–35). The consequences of glucotoxicity or lipotoxicity considering the metabolic health and physical inactivity of a diverse human population are key challenges in the prevention and treatment of DCM (36). The Framingham Heart Study showed that the frequency of heart failure is doubled in diabetic men and quintupled in diabetic women in comparison with age-matched control subjects. Clinical studies have shown that diabetic subjects have a high risk of cardiac failure accompanied with systolic dysfunction and LV hypertrophy (37, 38). Number of surveys and studies have shown a higher prevalence of diastolic dysfunction in diabetic patients (39, 40). Rodent and clinical studies in diabetes setting have shown functional and structural alterations in myocardium tissue. Studies performed in type 1 diabetic model, i.e., STZ-induced diabetic mouse, and rodents with type 2 diabetes, i.e., ZF rats or db/db mice, have shown diastolic dysfunction (Table 2) (13, 41). In fat and fructose-feeding studies, serum uric acid levels led to an increase in cardiac tissue xanthine oxidase activity, which was temporally related to an increase in body weight, fat mass, and insulin resistance without changes in blood pressure, when these mice were subjected to excess fat (46%) and fructose (17.5%) for 16 weeks. The high-fat and fructose diet led to cardiomyocyte hypertrophy, oxidative stress, interstitial fibrosis with impaired diastolic relaxation, and macrophage polarization toward a pro-inflammatory phenotype (42). Recent studies in many models have shown increases in levels of the 12/15-lipoxygenase (LOX) pro-inflammatory intermediate metabolite 12-HETE (43, 44). Insulin resistance is the prime risk factor to initiate the defects in insulin signaling pathways and glucose transport to cells. The resistance of the body toward insulin results in increased production of insulin in the pancreas leading to hyperinsulinemia (45). An association between insulin resistance and heart failure was noted a century ago and later traced to metabolic alteration (46).
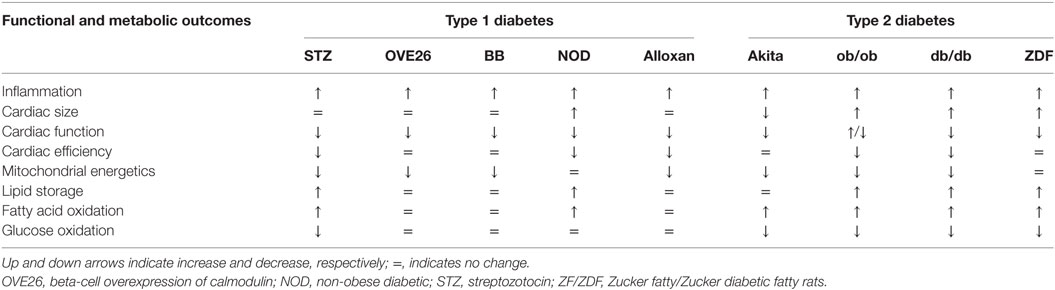
Table 2. Biochemical dysregulations in diabetic cardiomyopathy.
Studies have shown that insulin resistance is a common factor in patients with non-ischemic cardiomyopathy compared with control populations, which exclude pre-existing diabetes patients (47). A study of 1,187 Swedish people showed that heart failure could be anticipated in the patients with insulin resistance without any prior heart failure history, excluding all other factors (48). Lack of insulin response can lead to inactivation of cellular pathways such as AKT inactivation, reduced nitric oxide production, and increased apoptosis with alterations in myocardial structure (49–51). Insulin resistance is now considered to be a cardiometabolic disorder predisposing people to both CVD and diabetes. Metabolic risk factors in patients with insulin resistance are atherogenic dyslipidemia, hypertension, glucose intolerance, and a prothrombotic state (52). Further, McGavock et al., by using proton magnetic resonance spectroscopy, have shown that triglyceride accumulation occurs in human myocardium in association with diabetes mellitus and insulin resistance, much earlier than the symptoms of heart failure develop (53). Thus, the accumulation of fat and abundance of FAs is associated with an impaired cardiac efficiency and lipotoxicity.
Elevated blood pressure exerted on the vessel wall is commonly referred to as hypertension. Hypertension is a well-known risk factor for dilated hypertrophy, heart failure, and stroke. About 50% of ischemic strokes are caused by hypertension with a high-risk factor for hemorrhagic stroke. The risk ratio of MI doubles when the diastolic pressure is 94 mm Hg and systolic pressure is 140 mm Hg (54). Untreated hypertension is an add-on factor for DCM, as it leads to the rapid advancement of mild subclinical DCM to the clinically visible diastolic dysfunction and then later systolic dysfunction (55). Studies have shown that the hypertensive-diabetic rats have greatest relative cardiac hypertrophy and increased interstitial fibrosis compared with only diabetic or hypertensive animals. The combination of hypertension and diabetes mellitus led to myocardial degeneration similar as observed in human patients (56). Increased blood pressure is linked with sodium intake, hyperlipidemia, insulin resistance, impaired glucose tolerance, obesity, pre-diabetes, and diabetes and can lead to pulmonary edema and heart attack. The progression of DCM leads to activation of the renin-angiotensin system, which leads to an increase in oxidative damage with cell apoptosis and necrosis in the heart thereby increasing interstitial fibrosis (57). The potent vasoconstrictor endothelin-1 (ET-1) isolated from endothelial cells has inotropic, chemotactic, and mitogenic properties, activating the renin-angiotensin-aldosterone system to increase blood pressure. Many in vitro studies have highlighted that high glucose activates ET-1, which mediates cardiomyocyte hypertrophy via mitogen-activated protein kinase (MAPK) activation (58). Genetic deletion or pharmacological inhibition of ET-1 results in salt-sensitive hypertension (59). Depending upon its presence in either medulla or cortex, the renal ET-1 has different effects on blood pressure. The upregulation of cortical ET-1 expression has been well demonstrated in various hypertensive models via an increase in renal vascular resistance and a reduction in glomerular filtration rate (60, 61). Further, activation of the glomerular ETA receptor leads to hypertension by enhancing production of monocyte chemoattractant protein-1 and other pro-inflammatory factors, sequestering macrophages and lymphocytes, thereby increasing sodium reabsorption (62). Clinically, it has been observed that mortality of patients suffering from MI is greater in hypertensive patients, as showed in the PROCAM study (63). Hypertension, when it coexists with diabetes, doubles the risk of cardiac failure. Other trials, 4S, CARE, and LIPID, which included patients with MI and angina pectoris, showed a 23–45% incidence of hypertension (64, 65, 166). Recent studies suggest the involvement of 12/15-LOX in the murine models of experimental hypertension by altering macrophage functions (66). Patients with essential hypertension were observed to have higher levels of 12-HETE and 12/15-LOX protein compared with control subjects (67). A randomized, double-blinded, and controlled clinical trial with patients having peripheral arterial disease (75% hypertensive) showed that patients who consumed a diet including 30 g of milled flaxseed (n-3-fatty acid-α-linolenic acid) for 6 months displayed significant reductions in systolic (−10 mm Hg) and diastolic (−7 mm Hg) blood pressure (68). The plasma of FlaxPAD (Flaxseed for Peripheral Arterial Disease) patients exhibited significant decreases in 5,6-, 8,9-, 11,12-, and 14,15-dihydroxyeicosatrienoic acid and 9,10- and 12,13-dihydroxyoctadecenoic acid versus controls. The study showed that inhibition of soluble epoxide hydrolase contributed to the antihypertensive effects and could be one of the pharmacological targets (68).
This review further discusses biochemical mechanisms such inflammation, 12/15-LOX signaling, FA oxidation, and lipotoxicity that are directed toward increasing our understanding of novel ways for the prevention and treatment of cardiomyopathy.
Inflammation is an essential biological process to restore normal tissue homeostasis after injury. The state of inflammation is mediated by upregulation of multiple signaling pathways, such as NF-κB, c-Jun NH2-terminal kinase, or p38-MAPK associated with insulin resistance, which have profound roles in diabetic complications (69, 70). Thus, overactive inflammation is unifying component of many chronic diseases eventually leading to heart failure. Since in early 1950s, several reports validates that chronic inflammation is key hallmark signature in congestive heart failure pathology. Levine et al., in 1990, documented a positive correlation between TNF-α and chronic heart failure (71). Direct correlation between many chemokines/cytokines and heart failure suggests large oxidative products, which are key regulators of the inflammatory process, exerting both pro- and anti-inflammatory effects (72, 73). The lipid-metabolizing enzymes, including different members of the LOX family, are major players in the pathogenesis of heart failure. LOXs are enzymes that metabolize polyunsaturated FAs (74). The differential prostaglandins are produced by the cyclooxygenase pathway and leukotrienes through the LOX pathway, with availability of arachidonic acid substrate leading to overactive inflammation during the healing process in heart failure pathology. Arachidonic acid serves as the substrate for LOX pathway that facilitates interaction with 5-, 12-, and 15-LOX. Within the family of LOXs, 12- and 15-LOX (referred as together 12/15-LOX) forms a subgroup of phylogenetically closely related enzymes that are highly, but not exclusively, expressed in distinct monocyte-derived cells (74). 12/15-LOX is often referred to as “leukocyte-type” 12-LOX, and has orthologs in other species such as human 15-LOX and rabbit 15-LOX-1 (75). Murine and human 12/15-LOX differ in their enzymatic activity and in their position during arachidonic acid oxygenation resulting in the predominant generation of 12-(S)-HETE by murine 12/15-LOX and 15-HETE by human 12/15-LOX (15-LOX-1), respectively (76). The pro-inflammatory role of 12/15-LOX has been evidenced in failing hearts (44, 77). Wen and colleagues demonstrated that 12/15-LOX products, i.e., 12(S)-HpETE (12(S)-hydroperoxyeicosatetraenoic acid) and 12(S)-HETE, are pro-inflammatory in nature and stimulate TNF-α and IL-6 expression in macrophages as a particular effect of the 12/15-LOX products. A recent report by Suzuki et al. demonstrated that the TNF-α and collagen markers were elevated with increases in 12/15-LOX expression in the STZ-induced diabetic heart (44). By contrast, 12/15-LOX knockout mice showed suppressed levels of TNF-α and collagen markers with improved cardiac function. These authors also demonstrated that administration of a 12/15-LOX inhibitor (CDC) suppressed the TNF-α levels associated with high blood glucose levels in vitro (44).
The beta-oxidation of FAs is the primary mechanism for the heart to produce energy. Under resting conditions, FAs cover more than 70% of the cardiac energy needs by meeting this demand through the tricarboxylic acid cycle and electron transport chain. The excess of FAs is stored in adipose tissue. During obesity and diabetes, the adipose tissue increases in size that overspills free FAs leading to imbalance between energy demand and supply (78). An imbalance of FA oxidation or glucose oxidation leads to changes in cardiac mitochondrial metabolic energy that leads to ventricular dysfunction and to a decrease in cardiac performance. The impaired FA oxidation or increase in FA uptake in diabetes leads to an intramyocardial lipid overload (79). Myocardial abundance triggers defects in insulin signaling and activation of peroxisome proliferator-activated receptor-α (PPAR-α)/PGC-1 (80). The changes in FA oxidation lead to lipotoxicity and ceramide accumulation in cardiac tissue. Wu et al. have demonstrated that the deletion of the gene encoding aryl hydrocarbon nuclear translocator (ARNT) in the liver/pancreas leads to a diabetic phenotype with twofold increase in FA oxidation. The deletion of ARNT leads to an increase in the expression of PPAR-α and its target genes (81). FA oxidation is a major contributor of carbon substrates to ATP generation in the adult heart. However, the heart has a unique ability of metabolic flexibility and utilizes glucose, lactate, ketones, and amino acids (82). The heart has the capacity to selectively use substrate based on availability and pathophysiological state. It is well documented that glucose and lactate are preferred by the fetal heart; however, lipids are the predominant fuel in the adult heart. Animal models of cardiac hypertrophy are observed to switch their substrate metabolism, recapitulating the “fetal metabolic profile” utilizing carbohydrates as primary energy sources (83, 84). Cardiac hypertrophy models show the consistent appearance of fetal gene expression, which is considered prime trigger in the pathological remodeling of the heart.
Altered FA utilization and intramyocardial lipid accumulation are key major factors in the pathogenesis of DCM. The metabolic preference of the diabetic heart toward glucose leads to an increase in FAs, which leads to lipid accumulation resulting in lipotoxicity. Clinical studies suggest that congenital lipodystrophy, a rare disease causing accumulation of lipids in non-adipose tissue, is one of the causes of premature cardiomyopathy (85). In diabetic and obese animal models, accumulation of triglycerides in cardiomyocytes is often associated with impaired contractile function (86). Further, rat models of obesity have shown that defects in the leptin receptor result in an excess of fat overload in non-adipose tissues resulting in lipotoxicity (87). A study in obese Zucker rats have shown that PPAR-α-regulated genes play a crucial role in FA oxidation but become impaired in the failing heart, suggesting that metabolic dysregulation due to triglyceride overload and gene expression alteration leads to contractile dysfunction. It has also been reported that intake of unsaturated FAs increases low-density lipoprotein (LDL), triacylglycerols, and Lp(a) lipoprotein, and decreases high-density lipoprotein with a reduction in LDL cholesterol particle size, which leads to alteration in serum lipid profiles, thereby doubling the risk of CVD (88). Further, the consumption of FAs increases inflammation altering the prostaglandin balance. This impairment in the activity of desaturase (the enzyme converting linoleic acid to arachidonic acid and other n-6 polyunsaturated FAs) confers a high-risk of CVD (89–91).
Dysregulation of myocardium FAs use for energy source results into glucotoxicity in hyperglycemia setting developing progressive myocardial fibrosis due to glycosylation, cross-linking, and accumulation of extracellular matrix proteins (92). There is a reduction in glucose transporter expression, which inhibits glucose translocation from plasma membrane to the cell in DCM. During ischemia, PPAR’s inhibits insulin’s action that reduces the rates of glycolysis and pyruvate oxidation resulting in shutdown of glucose metabolism to the hexosamine biosynthesis pathway (93–95). The inappropriate hexosamine biosynthesis metabolism leads to the production of reactive oxygen species (ROS) and the increased formation of intracellular advanced glycosylation end-products (AGEs). AGEs and hexosamine biosynthesis impacts the sarco-endoplasmic reticulum Ca2+-ATPase and the Ca2+ release channel, ryanodine receptor 2, leading to abnormal cardiac relaxation and contractility (96, 97). Wang et al. showed a novel role of active heparanase in modulating cardiac metabolism via cross talk between endothelial cell and cardiomyocyte to increase lipoprotein lipase secretion after hyperglycemia (98). The study showed that high glucose is a common stimulus for latent heparanase secretion from the endothelial cells and promotes its uptake into the cardiomyocyte (99). Presence of latent form of heparanase in the cardiomyocyte leads to the significant shift in the expression of apoptosis-targeted genes, providing an acute cardioprotective effect indicating diversified roles of heparanase in DCM and heart failure pathology (99).
Although gender-specific studies have shown that females tend to develop cardiac complications 10–15 years later than men (100), it has also been proven that women with diabetes and hypertension have a greater risk of developing CVD (101, 102). Clinical reports have also shown that if CVD presents at a younger age in women, it is more detrimental (103). The female heart tolerates stress, such as ischemic insults, better than the male heart. In diabetes, the estrogen in females may interact with certain risk factors, which may be deleterious to overall cardiac function. A study published by Peters and colleagues showed a sex-specific risk of stroke conferred by diabetes. Meta-analysis conducted for 64 cohorts including 775,385 individuals who did not show any signs of cardiac problems at baseline determined that 12,539 individuals experienced fatal or non-fatal stroke events (104). The circadian clock allows the body to adjust metabolic cycles according to the shift in day and night. These circadian oscillations impact physiological parameters of a cardiovascular function, thermoregulation, lipid, and glucose metabolism. Studies have shown that circadian misalignment accelerates diabetes due to disruption of glucose-stimulated insulin secretion and beta-cell loss (105). Two transcription factors, CLOCK and BMAL1, transcriptionally control the circadian clock by binding to E-boxes of target genes upon heterodimerization (106, 107). In the heart, BMAL1 regulates substrate utilization, FA and glucose metabolism, and the PI3K/AKT/GSK3β signaling axis. The cardiomyocyte-specific knockout of BMAL1 leads to age-onset cardiomyopathy, reducing lifespan with impaired FA and glucose metabolism (108). Our group has shown that genetic disruption of BMAL1 results in diastolic dysfunction exacerbating extracellular matrix remodeling, with the increase in expression of a pro-inflammatory gene profile signifying early cardiac aging in mice (109). Aging, diabetes, and hypertension have similar effects on heart dysfunction, resulting in LV hypertrophy and stiffness. Aging leads to an increase in cardiovascular stiffness contributing to the development of fibrosis. Fibrosis increases collagen cross-linking due to the formation of AGEs (110). Rodent studies have shown that at 16 weeks of age, structural and functional changes are observed in both the heart and kidney, in the Zucker diabetic fatty model of type 2 diabetes. Aging is widely impacted by the diverse intake of dietary ingredients (111). Both insulin resistance and hyperglycemia can occur with an increase in n-6 FAs, which further magnifies the inflammatory response in heart failure in aging (112). Clinically, with advancing age, the risk to people with the normal systolic function of having heart failure is 50%. However, diastolic dysfunction occurrence is higher in women with hypertensive heart disease and diabetes.
Diabetic cardiomyopathy leads to structural and functional changes in myocardium by activation of signaling pathways, i.e., altered calcium signaling, increased ROS, ceramides, hexosamines, advanced glycation end-products, inflammatory signaling, and changes in many transcriptional regulators along with 12/15-LOX, which contribute toward the pathogenesis as shown in Figure 2 (113). Being a multifactorial disease, the crosstalk between immune cell dysregulation, cardiomyocytes, endothelial cells, and fibroblasts leads to the impairment of several signaling pathways leading to the etiology of DCM discussed above. The MAPK family is essential for cardiac survival, and MAPK signaling impairment has been well studied in diabetic tissue (114, 115). STZ-induced diabetic rats have shown upregulated p-38-MAPK activity (116). The diabetic myocardium mediates apoptosis via ASK1, a MAPKKK signaling molecule (117). FoxO (forkhead box-containing protein, O subfamily-3, -4) proteins are major targets for the maintenance of cardiac function and stress responsiveness by regulating cardiac growth, insulin signaling, and glucose metabolism in the heart (118–120). Also, several pathways including mammalian target of rapamycin, microRNAs, Pim-1 (proviral integration site for moloney murine leukemia virus-1), endoplasmic reticulum stress, and unfolded protein responses are dysregulated in DCM (121).
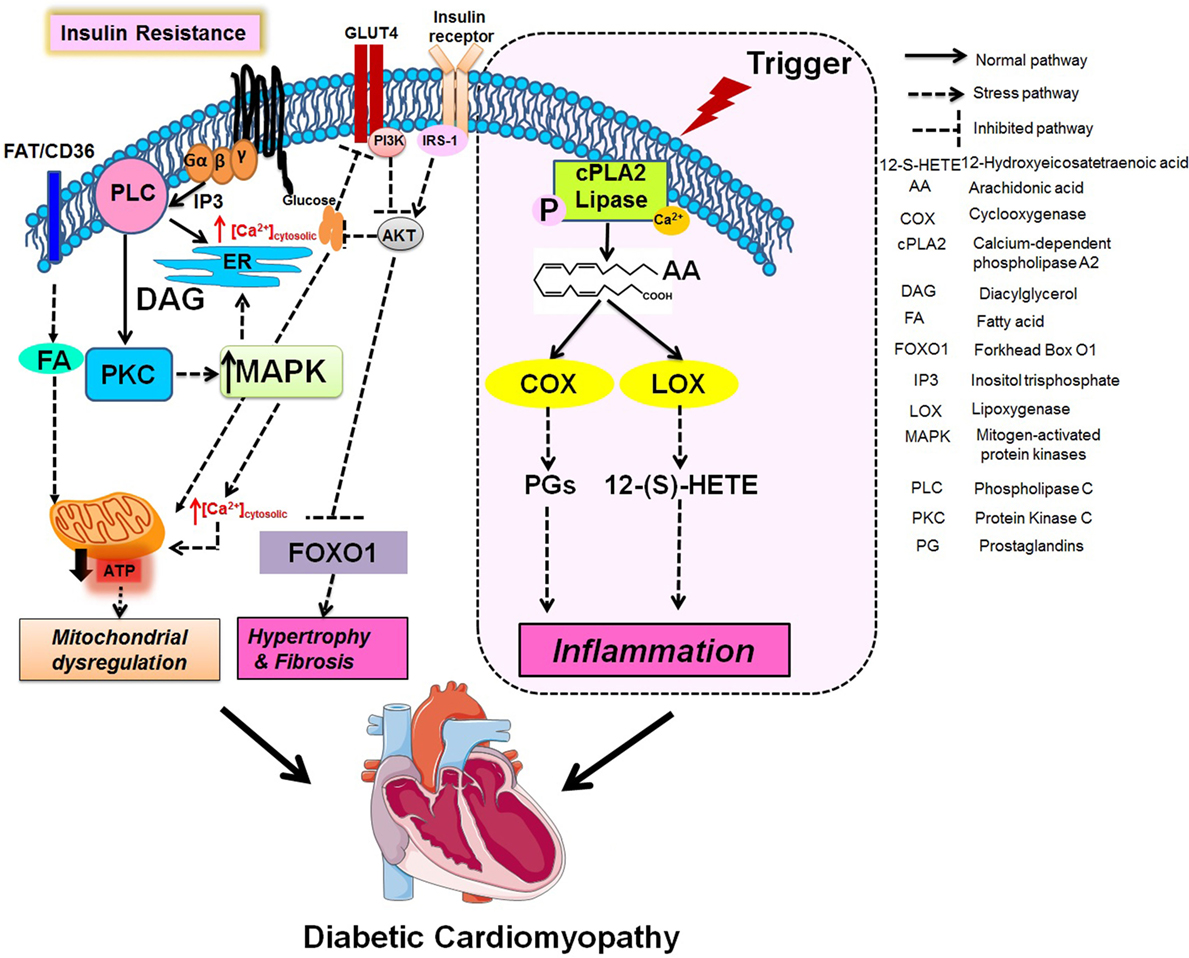
Figure 2. Integration of metabolic and fatty acids (FAs) metabolizing enzymes pathway in mitochondrial dysregulation, fibrotic hypertrophy, and chronic inflammation.
Being a multifactorial disease; currently, there is no specific therapy for DCM. Table 3 describes established rodent models to study DCM for novel treatment. Since insulin resistance is one of the leading causes of the pathogenesis of cardiomyopathy, insulin signaling is one of the targets. Anti-diabetic drugs, such as metformin, act on the metabolic target AMPK and may confer cardiovascular benefit (122, 123). Similarly, maintaining a proper diet and exercise can reduce the risk of diabetes and CVDs (124). Compounds modulating free FA metabolisms, such as perhexiline, trimetazidine, ranolazine, and amiodarone, have been shown to reduce lipotoxicity (125). The resveratrol-activated NAD-dependent protein deacetylase Sirt1 is a potent target as it plays a role in lowering blood glucose and increasing insulin sensitivity (126). Cardiac excitation–contraction coupling and insulin signaling dysregulation can also be improved by cell-based and genetic ablation therapy, which are among the robust strategies for treating CVDs (127). With the emergence of potent pro-inflammatory and anti-inflammatory roles of arachidonic acid metabolites such as 20-HETE, 12-HETE, and soluble epoxide hydrolase in diabetes and CVD (77, 128, 129), lipid mediators and their enzymes are among the potent future therapeutic targets to protect against the initiation and progression of DCM. The underlying concept “targeted or aggressive metabolomic” approach used by West et al. to the targeted metabolic profiling of cardiac tissue in dilative cardiomyopathy can be one the promising tool to delineate the DCM in future (130). With the rapid evolvement of the targeted metabolomics that aims to measure the endogenous metabolites in a cell or body fluid providing the functional readout. All the changes in the functional reads outs, such as shifts in the homeostasis of key lipids, carbohydrates, or amino acids, are associated with genetic variants. The first genome-wide association study with metabolomics (KORA study) using the quantitative measurement of 363 metabolites in serum of 284 male participants (131). The study found an association of single nucleotide polymorphisms with considerable differences in the metabolic homeostasis. The study found four genetic variants in genes coding for enzymes (FADS1, LIPC, SCAD, and MCAD) where the corresponding metabolic phenotype (metabolite) clearly matches the biochemical pathways in which these enzymes are active. The study clearly links genetic polymorphisms induce changes in the metabolic make-up of the human population opening a whole new field for personalized health care based on a combination of genotyping and metabolic characterization. The adaptation of sedentary lifestyle and eating habits has led to the increase in DCM about 5% of the global population (132). A blinded randomized personally tailored dietary intervention with consistent alterations to gut microbiota resulted in significantly lower postprandial responses (133). Accurate personal dietary recommendation using personal and microbiome features will be valuable in lowering the risk of DCM and associated inflammatory, metabolic, and neoplastic multifactorial disorders and can be effective clinical decision-making scheme.

Table 3. Established rodent diabetic cardiomyopathy models for the development of novel therapy.
Application of novel imaging technology and the use of biochemical markers (protein, enzymes, and metabolites) in clinical and pre-clinical models indicate that metabolic and biochemical stressors promote heart dysfunction in the diabetes setting. With the advancement of current research programs in pre-clinical and clinical models, it is possible to determine whether the DCM is of a dilated, hypertrophy, non-compaction or restrictive type, or of a combination of these. Whether the distinct target is at the enzymatic (LOX), mitochondria centric, ion-channel related, or defective chemokine-cytokine signaling level. Thus, additional studies with the major emphasis on metabolic and biochemical stressors in the advancement of heart function and dysfunction will determine future treatment(s). Despite the high mortality in diabetes patients due to heart failure, a number of questions regarding what triggers DCM or amplifies progressive cardiomyopathy remain unclear; therefore, urgent research on novel therapies is needed to meet the demand of personalized and precise medicine in the twenty-first century (165).
GH conceptualized the outline, edited the review, and approved for submission. VK prepared the first draft and edited the input from GH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors acknowledge the support from National Institutes of Health (NIH)-NCCIH (formerly known as NCCAM) AT006704 and HL132989 to GH and American Heart Association postdoctoral fellowship POST31000008 to VK.
1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2015 update: a report from the American Heart Association. Circulation (2014) 131(4):e29–322. doi: 10.1161/CIR.0000000000000152
2. Ma W, Wu JH, Wang Q, Lemaitre RN, Mukamal KJ, Djousse L, et al. Prospective association of fatty acids in the de novo lipogenesis pathway with risk of type 2 diabetes: the Cardiovascular Health Study. Am J Clin Nutr (2015) 101(1):153–63. doi:10.3945/ajcn.114.092601
3. Sheet CF. CDC-Diabetes Fact Sheet. Atlanta, GA: US Department of Health and Human Services (2014).
5. Voulgari C, Papadogiannis D, Tentolouris N. Diabetic cardiomyopathy: from the pathophysiology of the cardiac myocytes to current diagnosis and management strategies. Vasc Health Risk Manag (2010) 6:883–903. doi:10.2147/VHRM.S11681
6. Litwin SE. Diabetes and the heart: is there objective evidence of a human diabetic cardiomyopathy? Diabetes (2013) 62(10):3329–30. doi:10.2337/db13-0683
7. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA (2014) 311(8):806–14. doi:10.1001/jama.2014.732
8. Galderisi M, Anderson KM, Wilson PW, Levy D. Echocardiographic evidence for the existence of a distinct diabetic cardiomyopathy (the Framingham Heart Study). Am J Cardiol (1991) 68(1):85–9. doi:10.1016/0002-9149(91)90716-X
9. Eguchi K, Boden-Albala B, Jin Z, Rundek T, Sacco RL, Homma S, et al. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. Am J Cardiol (2008) 101(12):1787–91. doi:10.1016/j.amjcard.2008.02.082
10. Abächerli R, Zhou L, Schmid J-J, Kobza R, Niggli B, Frey F, et al. Correlation relationship assessment between left ventricular hypertrophy voltage criteria and body mass index in 41,806 Swiss conscripts. Ann Noninvasive Electrocardiol (2009) 14(4):381–8. doi:10.1111/j.1542-474X.2009.00330.x
11. Alpert MA. Obesity cardiomyopathy: pathophysiology and evolution of the clinical syndrome. Am J Med Sci (2001) 321(4):225–36. doi:10.1097/00000441-200104000-00003
12. Wu CK, Huang YT, Lin HH, Yang CY, Lien YC, Lee JK, et al. Dissecting the mechanisms of left ventricular diastolic dysfunction and inflammation in peritoneal dialysis patients. PLoS One (2013) 8(5):e62722. doi:10.1371/journal.pone.0062722
13. Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes (2003) 52(2):434–41. doi:10.2337/diabetes.52.2.434
14. Severson DL. Diabetic cardiomyopathy: recent evidence from mouse models of type 1 and type 2 diabetes. Can J Physiol Pharmacol (2004) 82(10):813–23. doi:10.1139/y04-065
15. Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, et al. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology (2003) 144(8):3483–90. doi:10.1210/en.2003-0242
16. Lavie CJ, Alpert MA, Arena R, Mehra MR, Milani RV, Ventura HO. Impact of obesity and the obesity paradox on prevalence and prognosis in heart failure. JACC Heart Fail (2013) 1(2):93–102. doi:10.1016/j.jchf.2013.01.006
17. Brainard RE, Watson LJ, Demartino AM, Brittian KR, Readnower RD, Boakye AA, et al. High fat feeding in mice is insufficient to induce cardiac dysfunction and does not exacerbate heart failure. PLoS One (2013) 8(12):e83174. doi:10.1371/journal.pone.0083174
18. Lopez EF, Kabarowski J, Ingle KA, Kain V, Barnes S, Crossman DK, et al. Obesity superimposed on aging magnifies inflammation and delays the resolving response following myocardial infarction. Am J Physiol Heart Circ Physiol (2014) 00604:02014. doi:10.1152/ajpheart.00604.2014
19. Fernandez Peralbo MA, Priego-Capote F, Galache-Osuna JG, Luque de Castro MD. Targeted analysis of omega-6-derived eicosanoids in human serum by SPE-LC-MS/MS for evaluation of coronary artery disease. Electrophoresis (2013) 34(19):2901–9. doi:10.1002/elps.201200603
20. Ghosh S, Molcan E, DeCoffe D, Dai C, Gibson DL. Diets rich in n-6 PUFA induce intestinal microbial dysbiosis in aged mice. Br J Nutr (2013) 110(3):515–23. doi:10.1017/S0007114512005326
21. Writing Group for the DCCT/EDIC Research Group, Orchard TJ, Nathan DM, Zinman B, Cleary P, Brillon D, et al. Association between 7 years of intensive treatment of type 1 diabetes and long-term mortality. JAMA (2015) 313(1):45–53. doi:10.1001/jama.2014.16107
22. Fahrmann ER, Adkins L, Loader CJ, Han H, Rice KM, Denvir J, et al. Severe hypoglycemia and coronary artery calcification during the diabetes control and complications trial/epidemiology of diabetes interventions and complications (DCCT/EDIC) study. Diabetes Res Clin Pract (2015) 107(2):280–9. doi:10.1016/j.diabres.2014.10.007
23. Gerich JE. The genetic basis of type 2 diabetes mellitus: impaired insulin secretion versus impaired insulin sensitivity. Endocr Rev (1998) 19(4):491–503. doi:10.1210/edrv.19.4.0338
24. Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, et al. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation (1999) 100(10):1134–46. doi:10.1161/01.CIR.100.10.1134
25. Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA (1979) 241(19):2035–8. doi:10.1001/jama.1979.03290450033020
26. Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart (2007) 93(9):1137–46. doi:10.1136/hrt.2003.025270
27. Struthers AD, Morris AD. Screening for and treating left-ventricular abnormalities in diabetes mellitus: a new way of reducing cardiac deaths. Lancet (2002) 359(9315):1430–2. doi:10.1016/S0140-6736(02)08358-7
28. Bertoni AG, Tsai A, Kasper EK, Brancati FL. Diabetes and idiopathic cardiomyopathy: a nationwide case-control study. Diabetes Care (2003) 26(10):2791–5. doi:10.2337/diacare.26.10.2791
29. Hajsadeghi S, Chitsazan M, Chitsazan M, Haghjoo M, Babaali N, Norouzzadeh Z, et al. Metabolic syndrome is associated with higher wall motion score and larger infarct size after acute myocardial infarction. Res Cardiovasc Med (2015) 4(1):e25018. doi:10.5812/cardiovascmed.25018
30. Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab (2003) 16(1):5–22. doi:10.1515/JPEM.2003.16.1.5
31. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J (2004) 18(14):1692–700. doi:10.1096/fj.04-2263com
32. Halade GV, Rahman MM, Williams PJ, Fernandes G. High fat diet-induced animal model of age-associated obesity and osteoporosis. J Nutr Biochem (2010) 21(12):1162–9. doi:10.1016/j.jnutbio.2009.10.002
33. Halade GV, El Jamali A, Williams PJ, Fajardo RJ, Fernandes G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp Gerontol (2011) 46(1):43–52. doi:10.1016/j.exger.2010.09.014
34. Laakso M. Heart in diabetes: a microvascular disease. Diabetes Care (2011) 34(Suppl 2):S145–9. doi:10.2337/dc11-s209
35. Halade GV, Kain V, Black LM, Prabhu SD, Ingle KA. Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging (Albany NY) (2016) 8(11):2611–34. doi:10.18632/aging.101077
36. Ussher JR. The role of cardiac lipotoxicity in the pathogenesis of diabetic cardiomyopathy. Expert Rev Cardiovasc Ther (2014) 12(3):345–58. doi:10.1586/14779072.2014.891939
37. Bella JN, Devereux RB, Roman MJ, Palmieri V, Liu JE, Paranicas M, et al. Separate and joint effects of systemic hypertension and diabetes mellitus on left ventricular structure and function in American Indians (the Strong Heart Study). Am J Cardiol (2001) 87(11):1260–5. doi:10.1016/S0002-9149(01)01516-8
38. Ilercil A, Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Welty TK, et al. Relationship of impaired glucose tolerance to left ventricular structure and function: the Strong Heart Study. Am Heart J (2001) 141(6):992–8. doi:10.1067/mhj.2001.115302
39. Poirier P, Bogaty P, Garneau C, Marois L, Dumesnil JG. Diastolic dysfunction in normotensive men with well-controlled type 2 diabetes: importance of maneuvers in echocardiographic screening for preclinical diabetic cardiomyopathy. Diabetes Care (2001) 24(1):5–10. doi:10.2337/diacare.24.1.5
40. Redfield MM, Jacobsen SJ, Burnett JC Jr, Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA (2003) 289(2):194–202. doi:10.1001/jama.289.2.194
41. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Ui JY, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology (2005) 146(12):5341–9. doi:10.1210/en.2005-0938
42. Jia G, Habibi J, Bostick BP, Ma L, DeMarco VG, Aroor AR, et al. Uric acid promotes left ventricular diastolic dysfunction in mice fed a Western diet. Hypertension (2015) 65(3):531–9. doi:10.1161/HYPERTENSIONAHA.114.04737
43. Tersey SA, Maier B, Nishiki Y, Maganti AV, Nadler JL, Mirmira RG. 12-lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol Cell Biol (2014) 34(19):3735–45. doi:10.1128/MCB.00157-14
44. Suzuki H, Kayama Y, Sakamoto M, Iuchi H, Shimizu I, Yoshino T, et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes (2015) 64(2):618–30. doi:10.2337/db13-1896
45. Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest (2000) 106(4):453–8. doi:10.1172/JCI10762
46. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol (1974) 34(1):29–34. doi:10.1016/0002-9149(74)90089-7
47. Witteles RM, Tang WH, Jamali AH, Chu JW, Reaven GM, Fowler MB. Insulin resistance in idiopathic dilated cardiomyopathy: a possible etiologic link. J Am Coll Cardiol (2004) 44(1):78–81. doi:10.1016/j.jacc.2004.03.037
48. Ingelsson E, Sundstrom J, Arnlov J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA (2005) 294(3):334–41. doi:10.1001/jama.294.20.2578-b
49. Asbun J, Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol (2006) 47(4):693–700. doi:10.1016/j.jacc.2005.09.050
50. Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci (2001) 26(11):657–64. doi:10.1016/S0968-0004(01)01958-2
51. Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci (2001) 114(Pt 16):2903–10.
52. Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C; American Heart Associationet al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation (2004) 109(3):433–8. doi:10.1161/01.CIR.0000112379.88385.67
53. McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, et al. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation (2007) 116(10):1170–5. doi:10.1161/CIRCULATIONAHA.106.645614
54. O’Donnell CJ, Ridker PM, Glynn RJ, Berger K, Ajani U, Manson JE, et al. Hypertension and borderline isolated systolic hypertension increase risks of cardiovascular disease and mortality in male physicians. Circulation (1997) 95(5):1132–7. doi:10.1161/01.CIR.95.5.1132
55. Russo C, Jin Z, Homma S, Rundek T, Elkind MS, Sacco RL, et al. Effect of diabetes and hypertension on left ventricular diastolic function in a high-risk population without evidence of heart disease. Eur J Heart Fail (2010) 12(5):454–61. doi:10.1093/eurjhf/hfq022
56. Factor SM, Bhan R, Minase T, Wolinsky H, Sonnenblick EH. Hypertensive-diabetic cardiomyopathy in the rat: an experimental model of human disease. Am J Pathol (1981) 102(2):219–28.
57. Liu X, Suzuki H, Sethi R, Tappia PS, Takeda N, Dhalla NS. Blockade of the renin–angiotensin system attenuates sarcolemma and sarcoplasmic reticulum remodeling in chronic diabetes. Ann N Y Acad Sci (2006) 1084(1):141–54. doi:10.1196/annals.1372.003
58. Chen S, Khan ZA, Karmazyn M, Chakrabarti S. Role of endothelin-1, sodium hydrogen exchanger-1 and mitogen activated protein kinase (MAPK) activation in glucose-induced cardiomyocyte hypertrophy. Diabetes Metab Res Rev (2007) 23(5):356–67. doi:10.1002/dmrr.689
59. Pollock DM, Pollock JS. Evidence for endothelin involvement in the response to high salt. Am J Physiol Renal Physiol (2001) 281(1):F144–50.
60. LaMarca B, Speed J, Fournier L, Babcock SA, Berry H, Cockrell K, et al. Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: effect of tumor necrosis factor-alpha blockade. Hypertension (2008) 52(6):1161–7. doi:10.1161/HYPERTENSIONAHA.108.120881
61. George EM, Granger JP. Linking placental ischemia and hypertension in preeclampsia: role of endothelin 1. Hypertension (2012) 60(2):507–11. doi:10.1161/HYPERTENSIONAHA.112.194845
62. Hepworth WB, Seegmiller RE, Carey JC. Thoracic volume reduction as a mechanism for pulmonary hypoplasia in chondrodystrophic mice. Pediatr Pathol (1990) 10(6):919–29. doi:10.3109/15513819009064727
63. Assmann G, Schulte H. The Prospective Cardiovascular Munster (PROCAM) study: prevalence of hyperlipidemia in persons with hypertension and/or diabetes mellitus and the relationship to coronary heart disease. Am Heart J (1988) 116(6 Pt 2):1713–24. doi:10.1016/0002-8703(88)90220-7
64. Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial Investigators. N Engl J Med (1996) 335(14):1001–9. doi:10.1056/NEJM199610033351401
65. Pedersen TR, Kjekshus J, Berg K, Haghfelt T, Faergeman O, Faergeman G, et al. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). 1994. Atheroscler Suppl (2004) 5(3):81–7. doi:10.1016/j.atherosclerosissup.2004.08.027
66. Kriska T, Cepura C, Magier D, Siangjong L, Gauthier KM, Campbell WB. Mice lacking macrophage 12/15-lipoxygenase are resistant to experimental hypertension. Am J Physiol Heart Circ Physiol (2012) 302(11):H2428–38. doi:10.1152/ajpheart.01120.2011
67. Gonzalez-Nunez D, Claria J, Rivera F, Poch E. Increased levels of 12(S)-HETE in patients with essential hypertension. Hypertension (2001) 37(2):334–8. doi:10.1161/01.HYP.37.2.334
68. Caligiuri SP, Aukema HM, Ravandi A, Guzman R, Dibrov E, Pierce GN. Flaxseed consumption reduces blood pressure in patients with hypertension by altering circulating oxylipins via an alpha-linolenic acid-induced inhibition of soluble epoxide hydrolase. Hypertension (2014) 64(1):53–9. doi:10.1161/HYPERTENSIONAHA.114.03179
69. Lorenzo O, Picatoste B, Ares-Carrasco S, Ramirez E, Egido J, Tunon J. Potential role of nuclear factor kappaB in diabetic cardiomyopathy. Mediators Inflamm (2011) 2011:652097. doi:10.1155/2011/652097
70. Li G, Barrett EJ, Barrett MO, Cao W, Liu Z. Tumor necrosis factor-alpha induces insulin resistance in endothelial cells via a p38 mitogen-activated protein kinase-dependent pathway. Endocrinology (2007) 148(7):3356–63. doi:10.1210/en.2006-1441
71. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med (1990) 323(4):236–41. doi:10.1056/NEJM199007263230405
72. Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation (2001) 104(22):2673–8. doi:10.1161/hc4601.099485
73. Markworth JF, Vella L, Lingard BS, Tull DL, Rupasinghe TW, Sinclair AJ, et al. Human inflammatory and resolving lipid mediator responses to resistance exercise and ibuprofen treatment. Am J Physiol Regul Integr Comp Physiol (2013) 305(11):R1281–96. doi:10.1152/ajpregu.00128.2013
74. Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem (1999) 274(34):23679–82. doi:10.1074/jbc.274.34.23679
75. Kuhn H, Thiele BJ. The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett (1999) 449(1):7–11. doi:10.1016/S0014-5793(99)00396-8
76. Kuhn H. Lipoxygenases in the cardiovascular system. Circ Res (2004) 94(12):1527–9. doi:10.1161/01.RES.0000134763.72053.50
77. Kayama Y, Minamino T, Toko H, Sakamoto M, Shimizu I, Takahashi H, et al. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J Exp Med (2009) 206(7):1565–74. doi:10.1084/jem.20082596
78. Pellegrinelli V, Carobbio S, Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia (2016) 59(6):1075–88. doi:10.1007/s00125-016-3933-4
79. Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol (2014) 171(8):2080–90. doi:10.1111/bph.12475
80. Ahmed W, Ziouzenkova O, Brown J, Devchand P, Francis S, Kadakia M, et al. PPARs and their metabolic modulation: new mechanisms for transcriptional regulation? J Intern Med (2007) 262(2):184–98. doi:10.1111/j.1365-2796.2007.01825.x
81. Wu R, Chang HC, Khechaduri A, Chawla K, Tran M, Chai X, et al. Cardiac-specific ablation of ARNT leads to lipotoxicity and cardiomyopathy. J Clin Invest (2014) 124(11):4795–806. doi:10.1172/JCI76737
82. Kolwicz SC, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of the cardiomyocte. Circ Res (2013) 113(5):603–16. doi:10.1161/CIRCRESAHA.1113.302095
83. Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta (1994) 1213(3):263–76. doi:10.1016/0005-2760(94)00082-4
84. Barger PM, Kelly DP. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci (1999) 318(1):36–42. doi:10.1016/S0002-9629(15)40570-1
85. Krahmer N, Farese RV, Walther TC. Balancing the fat: lipid droplets and human disease. EMBO Mol Med (2013) 5(7):905–15. doi:10.1002/emmm.201100671
86. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev (2008) 88(2):389–419. doi:10.1152/physrev.00017.2007
87. Wende AR, Abel ED. Lipotoxicity in the Heart. Biochim Biophys Acta (2010) 1801(3):311–9. doi:10.1016/j.bbalip.2009.09.023
88. Wanders AJ, Brouwer IA, Siebelink E, Katan MB. Effect of a high intake of conjugated linoleic acid on lipoprotein levels in healthy human subjects. PLoS One (2010) 5(2):e9000. doi:10.1371/journal.pone.0009000
89. Stoffel W, Holz B, Jenke B, Binczek E, Gunter RH, Kiss C, et al. Delta6-desaturase (FADS2) deficiency unveils the role of omega3- and omega6-polyunsaturated fatty acids. EMBO J (2008) 27(17):2281–92. doi:10.1038/emboj.2008.156
90. Le CH, Mulligan CM, Routh MA, Bouma GJ, Frye MA, Jeckel KM, et al. Delta-6-desaturase links polyunsaturated fatty acid metabolism with phospholipid remodeling and disease progression in heart failure. Circ Heart Fail (2014) 7(1):172–83. doi:10.1161/CIRCHEARTFAILURE.113.000744
91. Stoffel W, Hammels I, Jenke B, Binczek E, Schmidt-Soltau I, Brodesser S, et al. Obesity resistance and deregulation of lipogenesis in Delta6-fatty acid desaturase (FADS2) deficiency. EMBO Rep (2014) 15(1):110–20. doi:10.1002/embr.201338041
92. Dinko S, Jasmina V, Jwari A, Edward DF. Collagen cross-link breakers: a beginning of a new era in the treatment of cardiovascular changes associated with aging, diabetes, and hypertension. Curr Drug Targets Cardiovasc Haematol Disord (2004) 4(1):97–101. doi:10.2174/1568006043481347
93. Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation (2005) 112(17):2686–95. doi:10.1161/CIRCULATIONAHA.105.554360
94. Davidoff AJ. Convergence of glucose- and fatty acid-induced abnormal myocardial excitation–contraction coupling and insulin signalling. Clin Exp Pharmacol Physiol (2006) 33(1–2):152–8. doi:10.1111/j.1440-1681.2006.04343.x
95. Folmes CDL, Clanachan AS, Lopaschuk GD. Fatty acids attenuate insulin regulation of 5′-AMP-activated protein kinase and insulin cardioprotection after ischemia. Circ Res (2006) 99(1):61–8. doi:10.1161/01.RES.0000229656.05244.11
96. Fülöp N, Marchase RB, Chatham JC. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res (2007) 73(2):288–97. doi:10.1016/j.cardiores.2006.07.018
97. Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer ÜD, et al. Diabetes increases formation of advanced glycation end products on sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes (2004) 53(2):463–73. doi:10.2337/diabetes.53.2.463
98. Wang Y, Sun W, Du B, Miao X, Bai Y, Xin Y, et al. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-κB. Am J Physiol Heart Circ Physiol (2013) 304(4):H567–78. doi:10.1152/ajpheart.00650.2012
99. Wang F, Jia J, Lal N, Zhang D, Chiu AP, Wan A, et al. High glucose facilitated endothelial heparanase transfer to the cardiomyocyte modifies its cell death signature. Cardiovasc Res (2016) 112(3):656–68. doi:10.1093/cvr/cvw211
100. Lowel H, Meisinger C, Heier M, Hormann A, Kuch B, Gostomzyk J, et al. [Sex specific trends of sudden cardiac death and acute myocardial infarction: results of the population-based KORA/MONICA-Augsburg register 1985 to 1998]. Dtsch Med Wochenschr (2002) 127(44):2311–6. doi:10.1055/s-2005-858241
101. Barrett-Connor EL, Cohn BA, Wingard DL, Edelstein SL. Why is diabetes mellitus a stronger risk factor for fatal ischemic heart disease in women than in men? The Rancho Bernardo Study. JAMA (1991) 265(5):627–31. doi:10.1001/jama.265.5.627
102. Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet (2004) 364(9438):937–52. doi:10.1016/S0140-6736(04)17018-9
103. Maas A, Appelman YEA. Gender differences in coronary heart disease. Neth Heart J (2010) 18(12):598–602. doi:10.1007/s12471-010-0841-y
104. Peters SA, Huxley RR, Woodward M. Diabetes as a risk factor for stroke in women compared with men: a systematic review and meta-analysis of 64 cohorts, including 775,385 individuals and 12,539 strokes. Lancet (2014) 383(9933):1973–80. doi:10.1016/S0140-6736(14)60040-4
105. Gale JE, Cox HI, Qian J, Block GD, Colwell CS, Matveyenko AV. Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J Biol Rhythms (2011) 26(5):423–33. doi:10.1177/0748730411416341
106. Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science (1998) 280(5369):1564–9. doi:10.1126/science.280.5369.1564
107. Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci U S A (1998) 95(10):5474–9. doi:10.1073/pnas.95.10.5474
108. Young ME, Brewer RA, Peliciari-Garcia RA, Collins HE, He L, Birky TL, et al. Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms (2014) 29(4):257–76. doi:10.1177/0748730414543141
109. Ingle KA, Kain V, Goel M, Prabhu SD, Young ME, Halade GV. Cardiomyocyte-specific Bmal1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. Am J Physiol Heart Circ Physiol (2015) 309(11):H1827–36. doi:10.1152/ajpheart.00608.2015
110. Susic D, Varagic J, Ahn J, Frohlich ED. Collagen cross-link breakers: a beginning of a new era in the treatment of cardiovascular changes associated with aging, diabetes, and hypertension. Curr Drug Targets Cardiovasc Haematol Disord (2004) 4(1):97–101. doi:10.2174/1568006043481347
111. Baynes J, Murray DB. Cardiac and renal function are progressively impaired with aging in Zucker diabetic fatty type II diabetic rats. Oxid Med Cell Longev (2009) 2(5):328–34. doi:10.4161/oxim.2.5.9831
112. Lopez EF, Kabarowski JH, Ingle KA, Kain V, Barnes S, Crossman DK, et al. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. Am J Physiol Heart Circ Physiol (2015) 308(4):H269–80. doi:10.1152/ajpheart.00604.2014
113. Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA. Diabetic cardiomyopathy: mechanisms and therapeutic targets. Drug Discov Today Dis Mech (2010) 7(2):e135–43. doi:10.1016/j.ddmec.2010.08.001
114. Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol (1999) 31(8):1429–34. doi:10.1006/jmcc.1999.0979
115. Ho FM, Liu SH, Liau CS, Huang PJ, Lin-Shiau SY. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH(2)-terminal kinase and caspase-3. Circulation (2000) 101(22):2618–24. doi:10.1161/01.CIR.101.22.2618
116. Dunlop ME, Muggli EE. Small heat shock protein alteration provides a mechanism to reduce mesangial cell contractility in diabetes and oxidative stress. Kidney Int (2000) 57(2):464–75. doi:10.1046/j.1523-1755.2000.00866.x
117. Thandavarayan RA, Watanabe K, Ma M, Veeraveedu PT, Gurusamy N, Palaniyandi SS, et al. 14-3-3 protein regulates Ask1 signaling and protects against diabetic cardiomyopathy. Biochem Pharmacol (2008) 75(9):1797–806. doi:10.1016/j.bcp.2008.02.003
118. Ferdous A, Battiprolu PK, Ni YG, Rothermel BA, Hill JA. FoxO, autophagy, and cardiac remodeling. J Cardiovasc Transl Res (2010) 3(4):355–64. doi:10.1007/s12265-010-9200-z
119. Ni YG, Berenji K, Wang N, Oh M, Sachan N, Dey A, et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation (2006) 114(11):1159–68. doi:10.1161/CIRCULATIONAHA.106.637124
120. Ni YG, Wang N, Cao DJ, Sachan N, Morris DJ, Gerard RD, et al. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci U S A (2007) 104(51):20517–22. doi:10.1073/pnas.0610290104
121. Battiprolu PK, Lopez-Crisosto C, Wang ZV, Nemchenko A, Lavandero S, Hill JA. Diabetic cardiomyopathy and metabolic remodeling of the heart. Life Sci (2013) 92(11):609–15. doi:10.1016/j.lfs.2012.10.011
122. Rojas LB, Gomes MB. Metformin: an old but still the best treatment for type 2 diabetes. Diabetol Metab Syndr (2013) 5(1):6. doi:10.1186/1758-5996-5-6
123. Triggle CR, Ding H. Cardiovascular impact of drugs used in the treatment of diabetes. Ther Adv Chronic Dis (2014) 5(6):245–68. doi:10.1177/2040622314546125
124. Diabetes Prevention Program Research Group, Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet (2009) 374(9702):1677–86. doi:10.1016/S0140-6736(09)61457-4
125. Horowitz JD, Chirkov YY, Kennedy JA, Sverdlov AL. Modulation of myocardial metabolism: an emerging therapeutic principle. Curr Opin Cardiol (2010) 25(4):329–34. doi:10.1097/HCO.0b013e328339f191
126. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature (2009) 460(7255):587–91. doi:10.1038/nature08197
127. Haddad GE. Gene therapy for diabetic cardiomyopathy: a new approach for a difficult problem. Mol Ther (2006) 13(5):835–8. doi:10.1016/j.ymthe.2006.03.013
128. Elshenawy OH, Anwar-Mohamed A, El-Kadi AO. 20-Hydroxyeicosatetraenoic acid is a potential therapeutic target in cardiovascular diseases. Curr Drug Metab (2013) 14(6):706–19. doi:10.2174/1389200211314060007
129. Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, et al. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res (2010) 51(12):3481–90. doi:10.1194/jlr.M006007
130. West JA, Beqqali A, Ament Z, Elliott P, Pinto YM, Arbustini E, et al. A targeted metabolomics assay for cardiac metabolism and demonstration using a mouse model of dilated cardiomyopathy. Metabolomics (2016) 12(3):59. doi:10.1007/s11306-016-0956-2
131. Gieger C, Geistlinger L, Altmaier E, Hrabé de Angelis M, Kronenberg F, Meitinger T, et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet (2008) 4(11):e1000282. doi:10.1371/journal.pgen.1000282
132. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care (2004) 27(5):1047–53. doi:10.2337/diacare.27.5.1047
133. Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, et al. Personalized nutrition by prediction of glycemic responses. Cell (2015) 163(5):1079–94. doi:10.1016/j.cell.2015.11.001
134. Flarsheim CE, Grupp IL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol Heart Circ Physiol (1996) 271(1):H192–202.
135. Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM Jr, Klein JB, et al. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab (2004) 287(5):E896–905. doi:10.1152/ajpendo.00047.2004
136. Ghosh S, Pulinilkunnil T, Yuen G, Kewalramani G, An D, Qi D, et al. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am J Physiol Heart Circ Physiol (2005) 289(2):H768–76. doi:10.1152/ajpheart.00038.2005
137. Nemoto O, Kawaguchi M, Yaoita H, Miyake K, Maehara K, Maruyama Y. Left ventricular dysfunction and remodeling in streptozotocin-induced diabetic rats. Circ J (2006) 70(3):327–34. doi:10.1253/circj.70.327
138. Ares-Carrasco S, Picatoste B, Benito-Martín A, Zubiri I, Sanz AB, Sánchez-Niño MDS, et al. Myocardial fibrosis and apoptosis, but not inflammation, are present in long-term experimental diabetes. Am J Physiol Heart Circ Physiol (2009) 297(6):H2109–19. doi:10.1152/ajpheart.00157.2009
139. Epstein PN, Overbeek PA, Means AR. Calmodulin-induced early-onset diabetes in transgenic mice. Cell (1989) 58(6):1067–73. doi:10.1016/0092-8674(89)90505-9
140. Liang Q, Carlson EC, Donthi RV, Kralik PM, Shen X, Epstein PN. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes (2002) 51(1):174–81. doi:10.2337/diabetes.51.1.174
141. Ye G, Donthi RV, Metreveli NS, Epstein PN. Cardiomyocyte dysfunction in models of type 1 and type 2 diabetes. Cardiovasc Toxicol (2005) 5(3):285–92. doi:10.1385/CT:5:3:285
142. Song Y, Du Y, Prabhu SD, Epstein PN. Diabetic cardiomyopathy in OVE26 mice shows mitochondrial ROS production and divergence between in vivo and in vitro contractility. Rev Diabet Stud (2007) 4(3):159–68. doi:10.1900/RDS.2007.4.159
143. Li Y, Ma J, Zhu H, Singh M, Hill D, Greer PA, et al. Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes (2011) 60(11):2985–94. doi:10.2337/db10-1333
144. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes (2011) 60(6):1770–8. doi:10.2337/db10-0351
145. Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol (1992) 51:285–322. doi:10.1016/S0065-2776(08)60490-3
146. Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabó E, Szabó C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes (2002) 51(2):514–21. doi:10.2337/diabetes.51.2.514
147. Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol (2002) 283(3):H976–82. doi:10.1152/ajpheart.00088.2002
148. Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, et al. Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J Clin Invest (2004) 114(10):1493–503. doi:10.1172/JCI200419992
149. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol (2005) 23:447–85. doi:10.1146/annurev.immunol.23.021704.115643
150. Tang C, Kanter JE, Bornfeldt KE, Leboeuf RC, Oram JF. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J Lipid Res (2010) 51(7):1719–28. doi:10.1194/jlr.M003525
151. Dong B, Qi D, Yang L, Huang Y, Xiao X, Tai N, et al. TLR4 regulates cardiac lipid accumulation and diabetic heart disease in the nonobese diabetic mouse model of type 1 diabetes. Am J Physiol Heart Circ Physiol (2012) 303(6):H732–42. doi:10.1152/ajpheart.00948.2011
152. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372(6505):425–32. doi:10.1038/372425a0
153. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395(6704):763–70. doi:10.1038/27376
154. Naveilhan P, Svensson L, Nyström S, Ekstrand AJ, Ernfors P. Attenuation of hypercholesterolemia and hyperglycemia in ob/ob mice by NPY Y2 receptor ablation. Peptides (2002) 23(6):1087–91. doi:10.1016/S0196-9781(02)00042-6
155. Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes (2004) 53(12):3201–8. doi:10.2337/diabetes.53.12.3201
156. Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes (2004) 53(9):2366–74. doi:10.2337/diabetes.53.9.2366
157. Barouch LA, Gao D, Chen L, Miller KL, Xu W, Phan AC, et al. Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ Res (2006) 98(1):119–24. doi:10.1161/01.RES.0000199348.10580.1d
158. Van Den Bergh A, Vanderper A, Vangheluwe P, Desjardins F, Nevelsteen I, Verreth W, et al. Dyslipidaemia in type II diabetic mice does not aggravate contractile impairment but increases ventricular stiffness. Cardiovasc Res (2008) 77(2):371–9. doi:10.1093/cvr/cvm001
159. Li L, Hua Y, Dong M, Li Q, Smith DT, Yuan M, et al. Short-term lenalidomide (Revlimid) administration ameliorates cardiomyocyte contractile dysfunction in ob/ob obese mice. Obesity (2012) 20(11):2174–85. doi:10.1038/oby.2012.106
160. Kobayashi K, Forte TM, Taniguchi S, Ishida BY, Oka K, Chan L. The db/db mouse, a model for diabetic dyslipidemia: molecular characterization and effects of western diet feeding. Metabolism (2000) 49(1):22–31. doi:10.1016/S0026-0495(00)90588-2
161. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Xia XH, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes (2007) 56(10):2457–66. doi:10.2337/db07-0481
162. Yue P, Arai T, Terashima M, Sheikh AY, Cao F, Charo D, et al. Magnetic resonance imaging of progressive cardiomyopathic changes in the db/db mouse. Am J Physiol Heart Circ Physiol (2007) 292(5):H2106–18. doi:10.1152/ajpheart.00856.2006
163. Shen E, Li Y, Li Y, Shan L, Zhu H, Feng Q, et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes (2009) 58(10):2386–95. doi:10.2337/db08-0617
164. Mori J, Patel VB, Alrob OA, Basu R, Altamimi T, DesAulniers J, et al. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail (2014) 7(2):327–39. doi:10.1161/CIRCHEARTFAILURE.113.000672
165. Pulinilkunnil T, Kienesberger PC, Nagendran J, Sharma N, Young ME, Dyck JRB. Cardiac-specific adipose triglyceride lipase overexpression protects from cardiac steatosis and dilated cardiomyopathy following diet-induced obesity. Int J Obes (2014) 38(2):205–15. doi:10.1038/ijo.2013.103
166. Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med (1998) 339(19):1349–57. doi:10.1056/NEJM199811053391902
Keywords: cardiomyopathy, diabetes, fatty acids, inflammation, hypertension, cardiac remodeling
Citation: Kain V and Halade GV (2017) Metabolic and Biochemical Stressors in Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 4:31. doi: 10.3389/fcvm.2017.00031
Received: 31 January 2017; Accepted: 28 April 2017;
Published: 31 May 2017
Edited by:
George W. Booz, University of Mississippi Medical Center School of Dentistry, United StatesReviewed by:
Lisandra E. De Castro Bras, East Carolina University, United StatesCopyright: © 2017 Kain and Halade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ganesh V. Halade, Z2FuZXNoaGFsYWRlQHVhYm1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.