 Danielle Kamato1
Danielle Kamato1 Lyna Thach1Rebekah Bernard1
Lyna Thach1Rebekah Bernard1 Vincent Chan1Wenhua Zheng2,3Harveen Kaur4Margaret Brimble4
Vincent Chan1Wenhua Zheng2,3Harveen Kaur4Margaret Brimble4 Narin Osman1
Narin Osman1 Peter J. Little1*
Peter J. Little1*- 1Discipline of Pharmacy, Diabetes Complications Group, School of Medical Sciences, Health Innovations Research Institute, RMIT University, Bundoora, VIC, Australia
- 2State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Centre, Guangzhou, China
- 3Faculty of Health Sciences, University of Macau, Macau, China
- 4Department of Chemistry, University of Auckland, Auckland, New Zealand
G protein coupled receptors (GPCRs) are one of the major classes of cell surface receptors and are associated with a group of G proteins consisting of three subunits termed alpha, beta, and gamma. G proteins are classified into four families according to their α subunit; Gαi, Gαs, Gα12/13, and Gαq. There are several downstream pathways of Gαq of which the best known is upon activation via guanosine triphosphate (GTP), Gαq activates phospholipase Cβ, hydrolyzing phosphatidylinositol 4,5-biphosphate into diacylglycerol and inositol triphosphate and activating protein kinase C and increasing calcium efflux from the endoplasmic reticulum. Although G proteins, in particular, the Gαq/11 are central elements in GPCR signaling, their actual roles have not yet been thoroughly investigated. The lack of research of the role on Gαq/11 in cell biology is partially due to the obscure nature of the available pharmacological agents. YM-254890 is the most useful Gαq-selective inhibitor with antiplatelet, antithrombotic, and thrombolytic effects. YM-254890 inhibits Gαq signaling pathways by preventing the exchange of guanosine diphosphate for GTP. UBO-QIC is a structurally similar compound to YM-254890, which can inhibit platelet aggregation and cause vasorelaxation in rats. Many agents are available for the study of signaling downstream of Gαq/11. The role of G proteins could potentially represent a novel therapeutic target. This review will explore the range of pharmacological and molecular tools available for the study of the role of Gαq/11 in GPCR signaling.
Introduction
G protein coupled receptors (GPCRs) constitute the largest class of cell surface receptors. GPCR genes account for 5% of the human genome (1, 2). Of these receptors, all are seven membrane spanning receptors but not all are G protein binding but it is convenient to refer to the receptors as GPCRs. GPCRs also represent the largest and among the most efficacious class of therapeutic targets for diseases including cardiovascular disease, cancer, and asthma (1, 2). Many drugs have been developed based on GPCRs and these include some of the most important agents in human medicine, for example, in the treatment of asthma and hypertension (3). GPCRs are helical transmembrane receptors complemented by functional extracellular and intracellular loops (4). Within the GPCR superfamily, there have been five major families identified. They are the rhodopsin, secretin, glutamate, adhesion and frizzled/taste2 families (5). Most GPCRs contain seven helices and three intracellular loops; however, some members of the rhodopsin family may have eight helices and four intracellular loops (6). GPCRs bind hormones, neurotransmitters, or growth factors (7), which initiate a plethora of cellular responses. GPCRs are generally ligand activated but they can also bind to Gα-subunits in the absence of a ligand, a phenomenon known as receptor pre-coupling. GPCRs interact with their respective G proteins only upon receptor activation known as the collision coupling model or in the absence of agonist known as the pre-coupled receptor model (8).
Whereas protein tyrosine and serine/threonine kinase receptors have intrinsic catalytic activity, GPCRs do not have enzymatic activity per se but are linked to Gα proteins, which are GTPases, and mediate the signal transduction (9). G proteins of the α, β, and γ families provide the specificity and functionality of GPCRs.
G proteins are classified into four families according to their α subunit: Gi, Gs, G12/13, and Gq (Figure 1). The Gs and Gi families regulate adenylyl cyclase activity, while Gq activates phospholipase Cβ and G12/13 can activate small GTPase families (10). The Gq family consists of four members: Gq, G11, G14, and G15/16 (11, 12) and their respective α subunits are thus Gαq, Gα11, Gα14, and Gα15/16 (Figure 1).
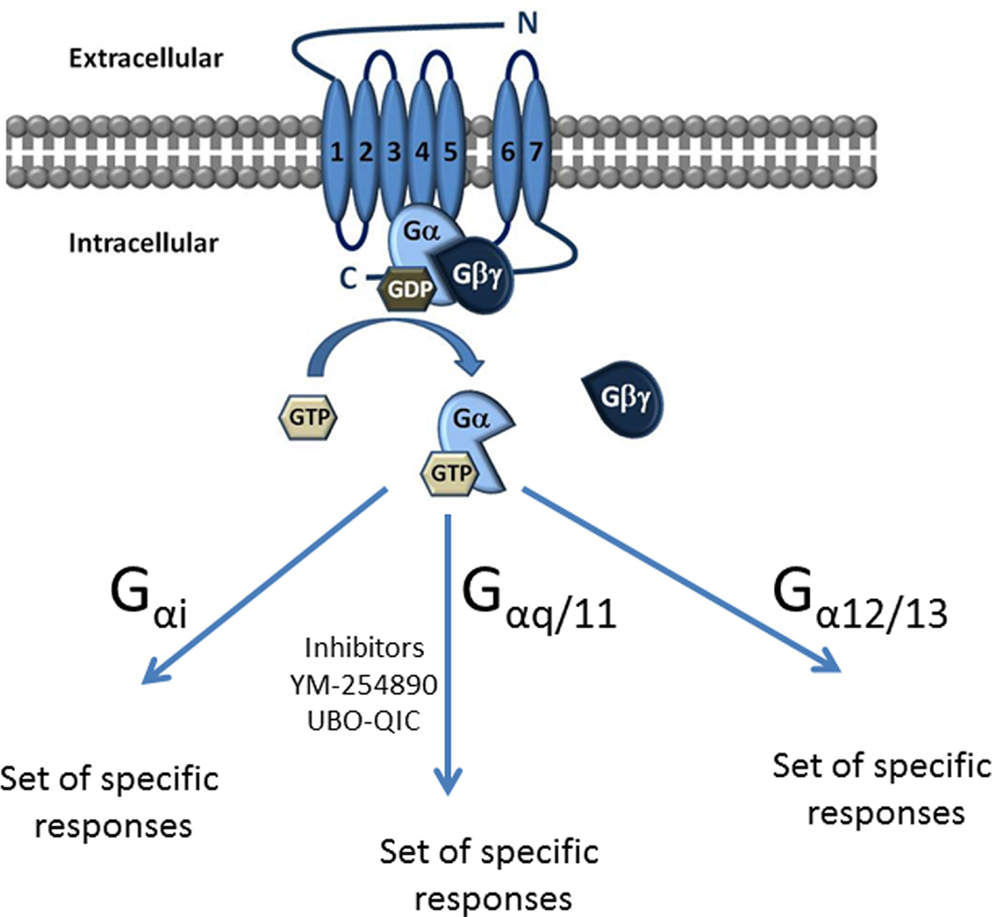
Figure 1. Classification of G proteins into four families according to their α subunit. The Gα subunit is made up of Gαs, Gαi, Gαq/11, and Gα12/13. Gαs and Gαi families regulate adenylyl cyclase activity, while Gαq activates PLC-β and the Gα12/13 can activate small GTPase families. The Gαq subunit is made up of four members, which include the Gαq, Gαq/11, Gαq/14, and Gαq15/16.
The role of G proteins in GPCR signaling has not been as intensively investigated as other aspects of GPCR signaling possibly due to the limited availability of convenient pharmacological tools. The most useful pharmacological agent has been the compound known as YM-254890, which is a cyclic depsipeptide isolated from the Chromobacterium sp. Initial studies indicated that this is a specific inhibitor of Gαq/11. YM-254890 has had variable availability and has not been available in recent times. As the importance of GPCR signaling in physiology and pathophysiology continues to grow, the potential importance of G proteins increases both for the fundamental cell biology and as potential therapeutic targets.
One of the major and expanding areas of GPCR signaling is transactivation-dependent signaling (13) in which GPCRs transactivate protein tyrosine kinase (PTK) and protein serine/threonine kinase receptors (14–16). Transactivation greatly expands the roles of GPCRs in cell biology (13, 17–19). GPCR transactivation of PTK receptors was discovered in 1996, has been the subject of almost 200 publications, and has been recently reviewed (20). Our laboratory has recently extended the paradigm of GPCR to PTK receptor transactivation to include the transactivation of protein serine/threonine kinase receptors and specifically the protease-activated receptor (PAR)-1 and endothelin receptor (ETR)-mediated transactivation of the transforming growth factor (TGF)-β type I receptor (TGFBR1) also known as Activin-like Kinase (Alk)-V (15, 16, 21). There is very little information on the role of Gα proteins in GPCR transactivation signaling. There is a need for synthetic programs to provide new molecules with the pharmacological properties of YM-254890 and such programs will provide agents, which allow for a much broader range of studies on the role of G proteins in GPCR signaling. This review focuses on the role of Gαq/11 in GPCR signaling in the context that the availability of new tools will shortly lead to a large increase in studies in this area. The two targets of compound such as YM-254890 are Gαq and Gα11 – these two proteins are distinct gene products but they have an identical number of amino acids and essentially identical structures and functions. In this review, we refer to Gαq but most statements will also relate to Gα11 and only where differences are known and of significance will this distinction be drawn.
Gαq/11 Signaling
The responses to GPCR agonists and the conformational changes in the GPCR that are induced by ligand binding are transduced and then mediated by heterotrimeric G protein complexes. Consisting of three subunits α, β, and γ, their role is to transduce external stimuli into intracellular signaling cascades. Most of the specificity of signaling resides in the Gα subunit. In an inactivated state, the α subunit binds guanosine diphosphate (GDP); however, upon binding activation of the GPCR, GTPase activity is induced and promotes the exchange of bound GDP for guanosine triphosphate (GTP). The α subunit and βγ complex then dissociate from one another and interact with their associated effectors (22). In the most common signaling pathways, Gαq activates phospholipase Cβ (PLCβ), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) releasing diacylglycerol (DAG) and 1,4,5-inositol trisphosphate (IP3). DAG activates a number of isoforms of protein kinase C (PKC), whereas IP3 diffuses to the endoplasmic reticulum (ER) and binds to IP3 receptors on ligand-gated calcium channels on the surface of the ER leading to a massive release of calcium ions into the cytosol and subsequently in some cells, the opening of cell surface calcium channels leading to the influx of extracellular calcium (23). The calcium cycle continues with the uptake of calcium back into the ER by Ca ATPases.
In addition to this paradigm, it has been shown that RhoA is a mediator of calcium sensitization and is downstream of Gα signaling. Activation of the members of the Rho family is via GTP binding. The exchange of GDP for GTP on these proteins is controlled through guanine nucleotide exchange factors (GEFs), which catalyze the exchange of GDP for GTP (24). Activation of Rho-mediated signaling pathways can be indirectly mediated by GPCRs, integrins, or receptor tyrosine kinases. G proteins, Gα12 and Gα13, activate Rho by activation of a Rho GEF (25). It is only when RhoA is active that it can interact with and activate downstream effectors such as Rho kinase (ROCK).
Thrombin activation of PAR-1 involves both Gαq/11 and Gα12/13, which causes RhoA activation signaling downstream to stimulate ROCK and PKC-related kinase. RhoA activation coupled to Gαq/11 involves intracellular release of calcium involving the downstream activation of the two Rho-regulated protein kinases, which in turn regulates the contraction of actomysin and the formation of focal adherence in human endothelial cells. In the case of Gα12/13, RhoA is activated through GEFs such as p115 RhoGEF, PDZ-RhoGEF, or leukemia-associated RhoGEF. In the case of Gαq/11, it is suggested that the GEFs utilized may involve p63 Rho GEF or Trio; however, the specific GEFs involved in this signaling pathway are yet to be confirmed.
Overexpression of active Gα11 or stimulation of the m1 muscarinic acetylcholine receptor induces apoptosis in HeLa cells. Rho kinase and ROCK are stimulated due to the cleavage of activated caspase 3 during apoptosis. There have been several studies on the mechanisms involved in Gαq/11-induced apoptosis, which show that this phenomenon is cell- and context-dependent. In COS-7 and CHO cells, Gαq-induced apoptosis is dependent on PKC, and angiotensin II-induced myocyte apoptosis is dependent on the release of intracellular calcium suggesting the involvement of PLC pathway. The molecular mechanism of Gαq/11 induced apoptosis leading to the activation of Rho/ROCK is not clearly understood; however, some studies have shown that Gαq/11 signaling activated RhoA, which inhibited insulin-stimulated Akt phosphorylation in HeLa cells. In CHO cells, Gαq and Gα11 regulate actin cytoskeleton remodeling through the activation of ADP-ribosylation factor 6. Platelets stimulated with P2Y1 agonist leads to the activation of RhoA, this activation was inhibited by Gαq inhibitor YM-254890, indicating that RhoA activation downstream of purinergic (P2Y)-1 receptors requires Gαq stimulation (26).
Structure of Gαq
Gαq and Gα11 are distinct gene products but from the same chromosome (12). These two proteins have an identical number of amino acids and are functionally almost identical. However, the tissue distribution of the two isoforms is distinct (12). Gαq is a 359 amino acid protein comprising two domains: a helical domain and a GTPase binding domain. The GTPase domain is responsible for hydrolyzing GTP to GDP, as well as binding the Gβγ subunits, GPCRs, and other effectors. This domain is conserved between all members of the G protein superfamily (6). The GTPase domain contains three switch regions, which are flexible loops that change conformation when bound with GTP. The helical domain contains six α-helices, which encapsulates nucleotides in the protein core by forming a lid over the nucleotide-binding pocket. Of all G protein families identified, members of the Gαq family share the most amino acid sequence homology. In humans, Gα11, Gα14, and Gα16 share 90, 80, and 57% sequence similarities, respectively (27).
Functions of Gαq
Gαq plays a role in platelet aggregation. Bleeding time and resistance to thromboembolism are dramatically increased in Gαq-deficient mice compared to wild type (28). Gαq is also implicated in insulin-stimulated glucose transport (29). In 3T3–L1 adipocytes, Gαq is required for insulin-induced GLUT4 translocation and the stimulation of 2-deoxy-d-glucose uptake. Angiotensin II dose-dependently increases cell proliferation in smooth muscle cells and this is inhibited by the Gαq antagonist, GP-2A (30). Gαq/11 proteins are involved in HIV-1 envelope glycoprotein-dependent cell–cell fusion upstream of Rac-1 (31). Genetically modified mice studies suggest that receptors coupled to the Gαq play a role in the development of heart failure (32). Following treatment to activate Gαq in transgenic mice expressing a silent Gαq, the mice rapidly developed a dilated cardiomyopathy and heart failure. Transgenic mice expressing an inducible Gαq that cannot activate PLCβ do not develop heart failure. Thus, the activation of Gαq resulting in heart failure requires the activation of PLCβ (32).
Role of Gαq in the GPCR Transactivation of Kinase Receptors
There are now two major pathways of GPCR to cell surface receptor kinase transactivation – the well-established transactivation of PTK receptors, notably epidermal growth factor receptor (EGFR) and the recently identified transactivation of serine/threonine kinase receptors, specifically the TGFBR1 (14–16, 33). There is some information of the role of Gα proteins and thus Gαq in the transactivation of PTK receptors but nothing is known of the role of Gαq in the transactivation of serine/threonine kinase receptors.
G protein coupled receptors coupled to Gαq, such as bombesin receptor or Gαi proteins, such as M2 muscarinic acetylcholine receptor, expressed in COS-7 cells show increased EGFR tyrosine phosphorylation more than that resulting from Gαi coupled receptor stimulation. Cells transfected with Gαq-coupled GPCRs are unaffected by pertussis toxin while Gαi coupled receptors are, as expected, blocked by pertussis toxin treatment (34). Thus, EGFR transactivation may occur through both pertussis toxin-sensitive and -insensitive pathways. GPCR transactivation of serine/threonine kinase receptors and specifically TGFBR1 by both ETR and PAR-1 has been identified in vascular smooth muscle cells (VSMCs) but the role of Gαq in transactivation of TGFBR1 has not been reported (15, 16, 21).
Thus far, the biochemical mechanisms of GPCR to protein tyrosine and protein serine/threonine kinase receptors have been found to be completely distinct with, for example, the former involving MMPs and the latter being independent of MMPs (16). The transactivation of serine/threonine kinase but not tyrosine kinases involves the cytoskeleton (16). The independent signaling pathways have made it difficult to envisage a single potential therapeutic target for the inhibition of all GPCR transactivation signaling (18).
It will be interesting to investigate the role of Gαq proteins in tyrosine and serine/threonine kinase transactivation signaling as it has the potential to be a point of commonality in GPCR-mediated transactivation of cell surface protein tyrosine and serine/threonine kinase receptor signaling.
Molecular and Pharmacological Regulation of G Proteins
G proteins in cells can be effectively knocked down utilizing a molecular approach and this has allowed for detailed studies of the function of various G proteins and their interactions. Classic experimental approaches assume that the intervention is specific and does not alter other parameters that would impact on the experimental result of the index intervention. This is not always the situation and is certainly not the reliable paradigm in the case of the regulation of G proteins. Gilman and colleagues (35) demonstrated that knocking down Gβ proteins resulted in a compensatory increase in both the effector, adenylyl cyclase and even the GPCR, being the β2-adrenergic receptor. Results of knock down interventions are also not always reciprocal – the knock down of one G protein may lead to a compensatory increase in another G protein family member but the reverse or reciprocal phenomenon may not occur (35). Thus, the knock down of Gαq and Gα11 in HeLa cells increased the accumulation of Gαi and Gα0 but the reciprocal response did not occur (35).
Gα and β proteins exist in approximately equal mass stoichiometry in most cells. This occurs primarily because Gβ proteins stabilize bound Gα proteins with the corollary that free Gα proteins are degraded. However, Gα proteins are subject to palmitylation and myristoylation and these processes may bind Gα proteins to the cell membrane and stabilize the proteins (36). A consequence of the role of post-translational regulation on stability and the cellular levels of G proteins is that the relationship between mRNA and protein levels may be perturbed. Higher mRNA levels may lead to increased expression of the G protein, but if it is orphaned and free the protein may be degraded providing for high level of mRNA and in the presence of low levels of protein.
Molecular approaches to the up- and down-regulation of target proteins are a major component of modern mechanistic studies of cell biology. However, as exemplified above, alteration of target protein levels may result in compensatory changes in other components of a system and the perturbation might not provide the expected result. Pharmacological approaches nullify the activity or function of a target protein without in most cases altering the level of the target protein. If there is greater availability of G protein inhibitors such as YM-254890 or alternative new tools, then it will be interesting to determine if blocking a Gα protein results in any changes in the level of other G proteins within the cell. Such studies are currently underway in our laboratory.
Pharmacology of Gαq Inhibitors
YM-254890
The compound known as YM-254890, a cyclic depsipeptide isolated from the Chromobacterium sp. QS3666, is a specific Gαq inhibitor. YM-254890 has been shown to inhibit ADP-induced platelet aggregation, which is mediated via GPCRs, P2Y1, and P2Y12 (37). These receptors are associated with the Gαq and Gαi signaling pathways, respectively. YM-254890 has no effect on the P2Y12 signal transduction pathway, indicating that the compound has some specificity for Gαq. It was also shown to inhibit Gαq-coupled GPCR signaling by inhibiting calcium mobilization in P2Y2-expressing C6-15 cells but not cAMP accumulation (38).
YM-254890 inhibits the signal transduction of Gαq by inhibiting the exchange of GDP for GTP preventing the activation of the G protein, rather than receptor-Gαq interactions (38). When bound to GDP, the non-polar side chains of YM-254890 form hydrogen bonds with the Switch I region; however, this is a conformation that cannot be maintained when bound with GTP (39). Aside from antiplatelet activity, by electrically inducing carotid artery thrombosis in rodents, YM-254890 was also shown to have antithrombotic and thrombolytic effects (40).
YM-254890 was discovered and developed by Yamaguichi Pharmaceuticals, Japan; Yamaguichi subsequently became the property of Astellas Pharmaceuticals, Japan. YM-254890 was made available to researchers 10 years ago and a small number of interesting studies were published. The initial results indicated that YM-254890 is a useful tool for investigating Gαq/11-coupled receptor signaling and the physiological roles of Gαq/11. For example, Gαq knockout mice have lower blood pressure than appropriate controls (41). This indicates some potential for a Gαq inhibitor to be an anti-hypertensive agent and accordingly YM-254890 has not been provided to researchers presumably because of such identified commercial value. As discussed above, molecular approaches in this area, for example, G protein knock down can lead to rebound increases in other G proteins with unexpected results. Accordingly, it is understood that a number of groups are undertaking programs for the synthesis of compounds related to YM-254890 and it is likely that the availability of potent and specific Gαq/11 inhibitors would greatly expand activity and knowledge in this area and answer important questions such as the role of Gαq/11 in GPCR transactivation signaling of protein kinase receptors.
UBO-QIC/FR300359
FR300359, henceforth referred to as UBO–QIC, is also, like YM-254890 a cyclic depsipeptide; it is isolated from the Ardisia crenata sims plant (42). UBO-QIC is structurally very similar to YM-254890 and not surprisingly shows similar pharmacological activity. UBO-QIC inhibits platelet aggregation in rabbits in vitro and causes dose-related hypotension in anesthetized normotensive rodents, which is consistent with the effect on blood pressure in Gαq knock down mice (41, 43). The blood pressure lowering effect was attributed to the ability of UBO-QIC to partially mediate nitric oxide release from endothelial cells and inhibit calcium migration caused by voltage-dependent and receptor-operated channels (44).
Since the discovery of UBO-QIC as a Gαq antagonist, there have been limited studies showing its use. In HEK cells transfected with TRPV4, PAR-2-mediated intracellular calcium release was abolished by UBO-QIC when compared to control; however, extracellular calcium influx through the TRPV4 ion channel was unaffected thus showing that PAR-2 coupling to TRPV4 is not mediated by Gαq signaling (45). There have been no studies directly comparing the activity of YM-254890 and UBO-QIC possibly because of the linked variability of the former compound whereas at the time of preparing this review, UBO–QIC is commercially available.
The Peptide Antagonist GP-2A
In 2004, Tanski et al. (30) discovered a competitive Gαq inhibitor, G Protein antagonist-2A, also known as GP-2A. GP-2A is a peptide that selectively inhibits the action of Gαq by M1 muscarinic cholinergic receptors. The signaling pathway of Gαq and its role in cell proliferation with rat pulmonary artery smooth muscle cells were studied. Angiotensin II-mediated proliferation, PLCβ activation, and Erk1/2 phosphorylation were inhibited by more than 50% in the presence of GP-2A (30). The EGFR can be activated by EGF to generate an intracellular signaling pathway leading to the phosphorylation of several downstream effector proteins such as Erk1/2 (46). Tanski and colleagues have evaluated angiotensin II (as specific Gαq agonist) to effectively reduce Erk1/2 activation mediated by PLCβ via Gαq in the presence of GP-2A by showing its association with the phosphorylation of Erk1/2 in rat pulmonary artery smooth muscle cells (30). This study provides a strong foundation for our laboratory research as we can further investigate the possibility of this downstream signaling pathway to see whether or not GP-2A can act on other GPCR agonists such as thrombin to effectively respond similarly via Gαq in other smooth muscle cell types such as human VSMCs.
Other Pharmacological Tools for Evaluating the Role of Gαq in GPCR Signaling
It is also possible to indirectly assess the role of Gαq in GPCR signaling by analyzing downstream events through the use of inhibitors (Figure 2). For example, as detailed above GPCR ligand engagement activates Gαq which in turn activates phospholipase C leading to the catalysis of PIP2 and the release of DAG and IP3. There are inhibitors of PLC–β including U73122, and its inactive analog U-73343 is available to use as a control compound. These compounds have been widely used (47–51) although they are not considered to be especially useful and specific agents. The antibiotic, neomycin, can also be used as a PLCβ inhibitor in that it binds to the target substrate, PIP2 and inhibits the action of PLCβ to release DAG and IP3, which can be assessed as a reduction in IP3 accumulation or increased free intracellular calcium (23). As always with pharmacological approaches, it is likely that the use of multiple approaches can provide the best information on the role of Gαq in GPCR signaling (Table 1).
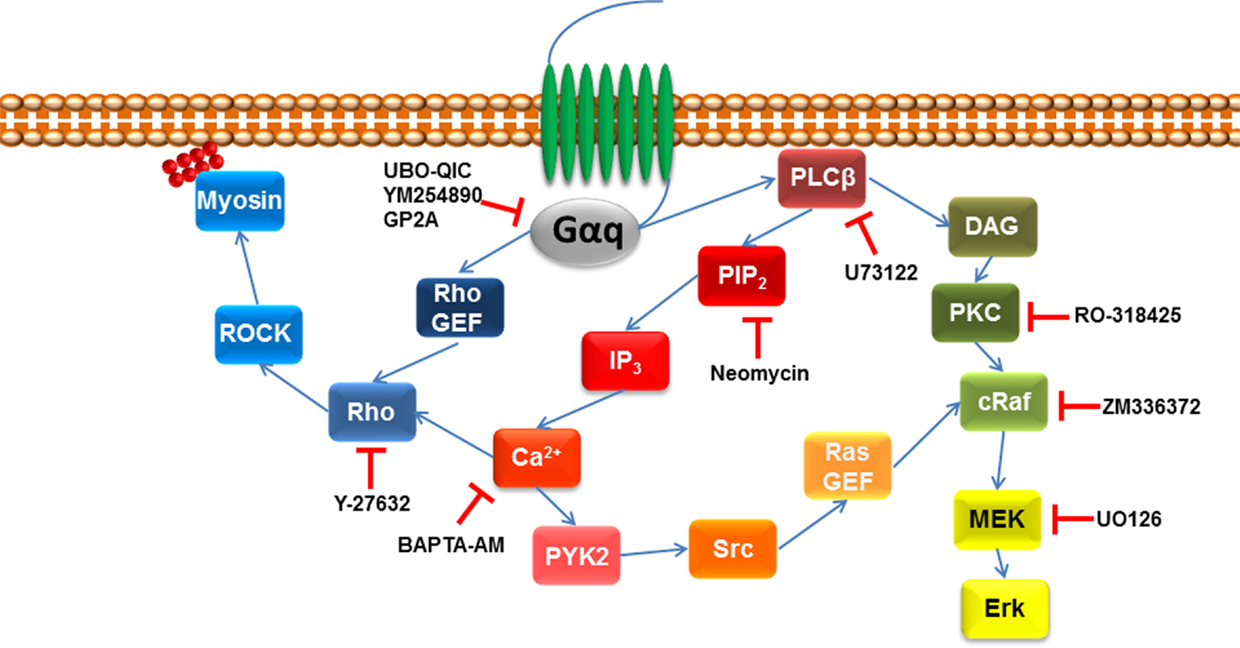
Figure 2. Pharmacological agents to inhibit downstream signaling intermediated of Gαq. Once GPCR is activated by its agonist, Gαq signaling activates phospholipase C β (PLCβ), which leads to the hydrolysis of phosphatidylinositol 4,5-biphosphate (PIP2) and diacylglycerol (DAG). The former leads to initiate the release of 1,4,5-inositol tris phosphate (IP3) initiating calcium release, activating protein tyrosine kinase 2 (PYK2), which leads to proto-oncogene tyrosine protein kinase (Src) activating Ras guanine nucleotide exchange factor (Ras GEF), which leads to the activation of MAPK signaling. MAPK signaling pathway can also be downstream of DAG that activates protein kinase C (PKC), which leads to the activation of MAPK signaling. Gαq signaling can also go indirect of PLCβ by activating Rho GEF leading to the activation of the Rho/ROCK signaling pathway.
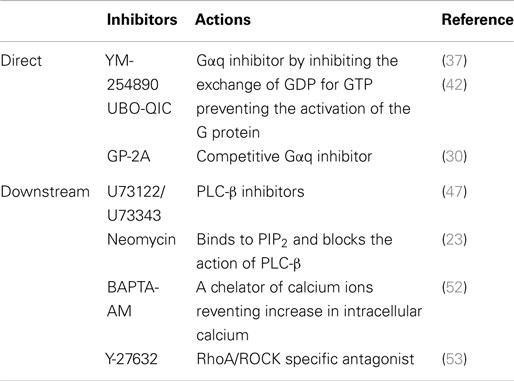
Table 1. Pharmacological tools used as inhibitors of Gαq or its downstream signaling intermediates.
U73122 and Its Inactive Analog U-73343
To maximize our knowledge of Gαq, it is possible to examine the downstream role of Gαq in GPCR by assessing the inhibitors of PLC, U73122, and its inactive analog U73343. U73122 and its analog U73343 were used to show the effect of human platelet calcium signaling and protein tyrosine phosphorylation in the presence of thrombin, collagen, and thapsigargin (47). U73122 showed complete inhibition of calcium signaling in the presence of this agonist, which was generated via the activation of PLC specifically the β and γ isoforms (47, 54). U73343 did not show any calcium inhibitory effect via the activation of PLC but rather showed the calcium inhibitory effect via the upstream activation of cPLA2 in the presence of thapsigargin and collagen (47). This provides a clear indication that U73343 has minimal activity as a PLC inhibitor.
The study also investigated the role of Gαq in the transactivation of PTK receptors to show that platelets, stimulated by thrombin increased protein tyrosine phosphorylation. In the presence of U73122, the phosphorylation of tyrosine kinase was abolished (47). As mentioned earlier, Gαq are prominent in signaling in VSMCs. From the recent study, it is possible that we can replicate this investigation in other cell type to further study the PTK and perhaps the serine/threonine kinase transactivation pathway in the presence of U73122.
Neomycin and Its Potential Role for Gαq Signaling Studies
As mentioned above, another agent, which can further be investigated, is neomycin, a PLC inhibitor. Our laboratory has previously reported that neomycin strongly inhibits the formation of IP3 in rat aortic smooth muscle cells in the presence of endothelin, an agonist that influences contraction in smooth muscle (23). Endothelin acts via specific ET-A receptors, which are coupled to PLC to stimulate calcium mobilization (23). ETR is coupled to PLC via G proteins (55) and its activation acts on the cardiac muscle where it binds to ryanodine located on the SR, which releases calcium mobilization within the cardiac muscle cell (56). As known, the signaling pathways of GPCRs through G proteins contribute to various functions in different cell types such as the contraction of blood vessels and are involved in many diseases such as cancer and cardiovascular disease (16, 57). In unpublished data, we have found that neomycin has a dose-dependent inhibition of thrombin-mediated release of intracellular calcium in human VSMCs. Further investigation is required to understand its pharmacological signaling pathway via Gαq.
The Action of Intracellular Calcium Ion to Study the Role of Gαq in GPCR Signaling
The activities of calcium ions on the inhibitory action of calcium channel blockers and its impact on atherogenesis in the regulation of proteoglycan biosynthesis in human VSMCs was studied via the role of ionomycin and bis (2-amionophenyl) ethyleneglycol-N,N,N′,N′-tetraacetic acid, tetraacetoxymethyl ester (BAPTA-AM) (58).
Ionomycin is a calcium ionophore, which elevates intracellular calcium (58). Radioactive sulfate incorporation into proteoglycans was unaffected by ionomycin, providing support that calcium regulation is not involved in proteoglycan synthesis (58).
Similarly, BAPTA-AM, a chelator of calcium ions, which prevents an elevation of intracellular calcium by acting as a calcium buffer (52) had no effect on proteoglycan synthesis (58). Agonists, TGF-β, and ET-1 stimulated BAPTA-AM to decrease sulfate incorporation into proteoglycans. The interpretation therefore concluded that there were no effects on calcium ion stimulation hence intracellular calcium does not play a role in VSMC proteoglycan synthesis (58).
Y-27632 – A RhoA/ROCK Inhibitor
Y-27632 is a widely used specific inhibitor of RhoA/ROCK family of protein kinases (53). The ROCK family of kinases is involved in Rho-induced formation of actin-stress fibers and focal adhesion as well as the down-regulation of myosin light chain (MLC) phosphatases. Deng et al. (59) examined the role of the PLC calcium pathway and Rho Kinase in PAR-1-mediated CCL2 release. Rho kinase activation is mediated by a Gαq-PLC-calcium-dependent PKC pathway to release thrombin-mediated CCL2. Thrombin-induced phosphorylation of MLC was inhibited by PLC calcium and calcium-dependent PKC inhibitors. Q94 a PAR-1 selective Gαq antagonist abolished thrombin-mediated MLC phosphorylation. Subsequent experiments showed that blockade of Rho kinase signaling is not essential for CCL2 protein production but is important in the release of CCL2 from the cell, as thrombin-mediated CCL2 levels are inhibited by Y-27632.
Having provided evidence that cytoskeletal rearrangement is involved in thrombin-mediated transactivation of TGFBR1, ROCK inhibitor Y-27632 was used to study the role of ROCK in the transactivation signaling pathway. Y-27632 inhibited the downstream product of ROCK-phosphor-Ezrin, Radixin, and Moesin. The ROCK inhibitor abolished the thrombin-mediated increase in phosphoSmad2, indicating that Y-27632 inhibits the activity of ROCK and ROCK is involved in the thrombin-mediated transactivation of TGFBR1. To evaluate the role of ROCK signaling in cardiac contractility, hearts were treated with Y-27632. This lead to a significant inhibition in the peak of pressure of non-transfected hearts but no reduction in basal contractility in hearts overexpressed with α1A-adrenergic receptor signifying that ROCK pathways play an important physiological role in maintaining baseline contractility (60).
Gα Gene Knockdown Using siRNA
Despite the very large number of GPCRs, there are relatively few studies that have used the potential of Gαq/11 gene knockdown by siRNA to explore their roles in the signaling cascades. One of the first reported gene knockdown studies of Gα proteins was the knockdown of Gαq and Gα11 gene expression using siRNA in HeLa cells (35). This work demonstrated an absolute requirement of Gαq/11 to stimulate histamine-mediated phospholipase C activity. Silencing of Gαq or Gα11 caused indistinguishable phenotypes, loss of half of histamine-stimulated PLC activity, despite the fact that concentrations of Gα11 exceed those of Gαq by 10-fold. No compensatory increases of either Gαq or Gα11 were observed following loss of either protein. Loss of Gαq or Gα11 did cause increased accumulation of Gαi and Gαo (35). A study characterizing the Gα subunits required for PAR-1-mediated endothelial cell permeability showed that both Gαq and Gα11 were necessary for thrombin to increase permeability while the need for Gα12/13 was less. Both protein subunit families contributed significantly to RhoA activation by thrombin (61). Knockdown of Gαq/11 in human pulmonary artery smooth muscle cells alters but does not prevent hypoxia-induced mitogenic factor-mediated calcium release demonstrating that Gαq/11 contributes to hypoxia-induced PLC signaling pathway (62). Using siRNA knockdown of Gαq or Gαs in human prostate epithelial cells, GPCR melatonin receptor MTNR1A has a dual requirement of Gαq and Gαs receptor coupling for effective MTNR1A signal transduction (63). In HEK cells expressing high levels of thyrotropin-releasing hormone receptor 2, knockdown of Gαq/11 reduces persistent agonist-induced signaling by 82% and suggests that Gαq/11 is a required component of the activated receptor signaling pathway (64). Clearly, there is considerable scope to use siRNA technology more often as a very useful tool in delineating the importance of Gα proteins in GPCR signaling.
Gα Knockout Mice and Gα Protein Mutants and Chimeras
Other molecular approaches to investigate the varied roles of Gα proteins include generating Gα protein knockout mice or overexpressing a variety of Gα mutants and chimeras in cell lines and examining their effects in different cellular contexts.
Over the last decade, the most widely used Gα knockout model is the Gαq/11 knockout mouse. Gαq/11-deficient fibroblasts from these knockout mice have been used to study a large number of GPCR signaling pathways. These studies have demonstrated that Gαq/11-deficient mouse fibroblasts expressing bradykinin B (2) receptor require both Gαi and Gαq/11 for effective bradykinin-mediated stimulation of the Erk cascade (65). Gαq/11 knockout mouse fibroblasts expressing GPCR α1b-adrenoreceptor protein or fusion proteins consisting of the α1b-adrenoreceptor and wild-type Gαq/11 or palmitolyation-resistant Gαq/11 mutants reveal agonist-mediated receptor/Gαq/11-coordinated release of the βγ complex (66). Expressing fusion proteins consisting of the GPCR α1b-adrenoreceptor with various Gα mutants results in altered receptor contact domain residues and enables the identification of key agonist and antagonist receptor contact sites that are necessary for α1b-adrenoreceptor activation. An aromatic group four amino acids before the carboxy terminus in Gαq/11 provides maximal α1b-adrenoreceptor activation information (67). Mouse embryonic fibroblasts from double knockout Gαq/11 and β-arrestin mice demonstrated that kisspeptin activation of GPCR 54 regulates the hypothalamic–pituitary–gonadal axis in reproductive function and that GPCR 54 has a co-dependency of both the Gαq/11 and β-arrestin pathways in a time-dependent manner to regulate Erk and localize pErk to the nucleus for downstream gene expression (68). Knock-in of ETR type A or type B in Gαq/11-deficient mice showed differential craniofacial development is based on specific ETR Gαq and Gαq/11 requirements (69). Clearly, the Gαq/11 knockout mouse has proven itself a reliable and useful tool in the study of Gα protein-mediated signaling.
An alternative approach to exploring Gα protein signals has been to co-express Gα protein mutants or chimeras in different cell types. Gαq chimeric mutants containing Gαi or Gαo tails co-expressed in COS-7 cells with opioid receptors and stimulated with opioid agonist are insensitive to pertussis toxin catalyzed ADP-ribosylation demonstrating an inability of Gαi or Gαo tails to serve as pertussis toxin substrates (70). Gi-coupled opioid receptors increase Gαq signals as demonstrated by the co-expression of constitutively active Gα mutants in COS-7 cells and requires activated phospholipase beta and Gβγ dimers (71).
A considerable number of studies have explored membrane localization of Gα proteins in different cellular contexts and described a diversity of requirements. N-terminal sequence-mutated Gα proteins expressed in HEK293 cells are unable to localize to the plasma membrane due to their inability to bind to Gβγ or attach myristate and palmitate (72). Mutated Gαq and Gαs proteins deficient in Gβγ-binding and co-expressed with different β(1–5) or γ2/3 subunits show that Gβγ and Gα proteins promote membrane localization of the other and requires palmitoylation (73). Defects in plasma membrane localization of Gαs occur when four N-terminal basic residues are mutated to glutamine; however, mutation of nine basic residues in Gαq is required. Gβγ co-expression partially rescues localization indicating that the characteristics of the N-terminal residues of Gαs and Gαq are critical in membrane localization of these proteins (74). Using co-expressed constitutively active Gαq or Gαq/12, the activation of ETRs was shown to mediate the binding of Gαq or Gαq/12 in caveolae to enable the downstream activation of Erk1/2 (75).
Gα protein-mediated signaling studies across a wide variety of GPCRs dominate the literature using Gα protein mutants or chimeras. Constitutively active Gαq, Gαq12, or Gαq13 mutants transfected into Jurkat cells co-expressing GPCR muscarinic cholinergic receptor subtypes demonstrated a requirement for Gαq13 to activate downstream transcription factor serum response factor. However, the M1 subtype also required Gαq/11 and calcium when regulator proteins RGS2 and RGS4 were co-transfected that demonstrates a unique pathway for the M1 receptor (76). Gαq/11 (Y356D) mutation results in altered GPCR α1B-adrenoreceptor contact domain and abolishes receptor function, however, does not affect ligand binding (67). Studies using constitutively active mutants Gαq (Q209L) and Gαq/13 (Q226L) demonstrate that Gαq activates rat brain phospholipase D1; however, Gαq/13 inhibits its activity (77). Gαq deletion mutants were used to demonstrate that Gαq mediates down-regulation of the vesicle-associated GPCR vesicular monoamine transmitter transporter VMAT2 activity in platelets (78). Expression of a constitutively active Gαq (R183C) mutant inhibited the expression of ezrin–radixin–moesin-binding phosphoprotein 50 and subsequent internalization of GPCR thromboxane A(2) beta receptor independently of PLC and PKC pathways (79). Specific Gα peptides and dominant negative Gα mutants were used to demonstrate the ability of α-thrombin to activate different effectors via Gα, Gβγ, and Gαi2, respectively, in Chinese hamster embryonic fibroblasts and thereby regulate the activation of the PI3kinase/Akt pathway (80). Molecular modeling and testing GST–fusion proteins of Gαq mutants–GPCR kinase complexes revealed a critical residue Gαq Pro185 at the interface with GPCR kinase 2 with residues Gαq K77, L78, Q81, and R92 also playing key interactive roles (81). Constitutively active Gαq, Gαq/12, and Gαq/13 overexpressed in human astrocytoma cells increased agonist-activated thromboxane A2 receptor-mediated IL-6 production while mutated Gαq and Gαq/13 overexpression blocks IL-6 production (82). Using both constitutively active and dominant negative Gαq subunit expression showed that in neuroblastoma cells Gαq elicits a rapid signal at the plasma membrane (83). Expression of constitutively active Gαq (Q209L) mutant inhibits Ras and the PI3K/Akt pathway; however, minimal effects are seen on the Ras/Raf/MEK/Erk signaling pathway (84). Gαq mutants that cannot bind Gβγ are unable to be stimulated by the mitogenic Pasteurellosis multocida toxin (PMT) demonstrating the requirement of cohesive Gαq/Gβγ signaling for this toxin activation pathway (85). Expressing GTPase-deficient Gαq mutant in the human adrenal cell line H295R depolarizes the two-pore loop potassium channel TASK and thereby increases aldosterone secretion (86). Chimeric G proteins have been used to determine Gα responses from orphan GPCRs with unknown Gα coupling partners. A luciferase reporter system with a chimera that contains promoter elements that drive Gs, Gq, and G12 signals and another chimera with promoters to drive Gi signals revealed neuromedin U receptor 1 activating Gq, neuromedin U receptor 2 activating Gi, and luteinizing hormone receptor activating Gq and Gs proteins (87).
Potential of Gαq as a Therapeutic Target
Gαq as a protein has several functions, which are valuable therapeutically. The GTPase activity, which hydrolyzes bound GTP to GDP, is an enzyme action that can be targeted. The binding of GDP and GTP are potential targets in the same manner in which the ATP binding site is target of many drugs inhibiting kinases (88). The ligand-activated GPCR acts as a GEF, which stimulates the exchange of GDP for GTP on the Gα peptide and this could be targeted. Furthermore, the protein contains a switch mechanism and this can be targeted as it is the target of the YM class of inhibitors (39). So, it is both theoretically possible and has been demonstrated that Gαq can be exploited as a drug target.
The consequences of targeting signaling molecules have theoretical limitations based on the role of such targets in normal physiology but also conceptually there may be situations, pathophysiology, in which the activity of Gαq is elevated or enhanced and presents itself as a target. Such situations are common in therapeutics but in most cases can only be established experimentally.
As discussed above, inhibition of Gαq/11 using YM-254890 has demonstrated anti-platelet aggregation, antithrombotic, and thrombolytic properties in a rat model of carotid artery thrombosis (40). Therefore, compounds that inhibit Gαq/11 could show enormous potential in the treatment of thrombotic conditions such as thrombotic stroke and myocardial infarction in humans. Additionally, a number of recent studies have also implicated a role for Gαq/11 in a range of metabolic conditions such as obesity and type 2 diabetes (89, 90). Activation of Gαq results in pronounced increases in blood glucose levels in a mouse model (89), thus, compounds that inhibit Gαq could also show promise as a future treatment option for type 2 diabetes.
In the important cardiovascular context of hypertension, Gαq knockout mice have reduced blood pressure (41) and YM-254890 has demonstrated some anti-hypertensive properties (40). Although there are many effective anti-hypertensive agents currently available, there are also many subjects with medication-resistant hypertension, which does require a niche for new therapies although it is unclear if a Gαq/11 inhibitor would be suitable to consider for such a niche.
Conclusion
G protein coupled receptor signaling is a major area of cell biology and therapeutics. The functioning of the seven transmembrane GPCR has been one of the most intensively studied areas of protein function. GPCRs signal through G proteins of the α and βγ subtypes where most of the signaling specificity is determined by the Gα protein. For the Gα protein family, these signaling pathways include the well-known PLC, PKC, and IP3 pathways and the lesser appreciated Rho/ROCK pathway. For multiple reasons, mostly the limited availability of pharmacological agents, which inhibit G protein function, the role of G proteins in GPCR signaling has been severely under-studied relative to the intense activity around the GPCRs. This is true for the Gαq proteins, which are the subject of this review but also for other G proteins. Given the broad involvement of GPCRs in cellular functioning, this is a major deficit in cellular signaling studies and potentially more importantly in the search for new drug targets. The recently expanding area of GPCR signaling is that of transactivation-dependent signaling in which GPCR transactivation of protein tyrosine and protein serine/threonine kinase cell surface receptors enormously expands the range of activities associated with the respective GPCRs. The potential role of G proteins and Gα proteins in particular in GPCR transactivation signaling is one very interesting area to be explored. It is likely that there are programs of chemical synthesis underway to synthesize inhibitors of Gαq proteins and these will increase the availability of inhibitors and also with the importance of this area hopefully lead to new studies, which produce a range of agents, some of which may be useful in in vivo studies. It is hoped that such studies may provide insights into the potential role of Gαq in disease processes and reveal the extent to which such inhibitors may represent novel therapeutic agents in a range of conditions from cancer to cardiovascular disease.
Author Contributions
PL, NO, VC, and WZ conceived the focus of the review, wrote, and edited the paper. HK and MB provided chemical insight about cyclic depsipeptide. DK, LT, and RB contributed expertise with preparation of the manuscript and the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Health and Medical Research Council Project Grant 2012–2014 (#1022800) (PL and NO). We thank the Ministry of Foreign Experts of the Government of the People’s Republic of China for support by way of a High End Professor (Education) Award through Zhongshan (Sun Yat-sen) University (WZ).
References
1. Morris AJ, Malbon CC. Physiological regulation of G protein-linked signaling. Physiol Rev (1999) 79:1373–430.
2. McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G-protein signaling: back to the future. Cell Mol Life Sci (2005) 62:551–77. doi: 10.1007/s00018-004-4462-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Insel PA, Tang CM, Hahntow I, Michel MC. Impact of GPCRs in clinical medicine: monogenic diseases, genetic variants and drug targets. Biochim Biophys Acta (2007) 1768:994–1005. doi:10.1016/j.bbamem.2006.09.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Wess J. G-protein-coupled receptors: molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. FASEB J (1997) 11:346–54.
5. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol (2003) 63:1256–72. doi:10.1124/mol.63.6.1256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol (2008) 9:60–71. doi:10.1038/nrm2299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Neer EJ, Clapham DE. Roles of G protein subunits in transmembrane signalling. Nature (1988) 333:129–34. doi:10.1038/333129a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Hein P, Bunemann M. Coupling mode of receptors and G proteins. Naunyn Schmiedebergs Arch Pharmacol (2009) 379:435–43. doi:10.1007/s00210-008-0383-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Wedegaertner PB, Wilson PT, Bourne HR. Lipid modifications of trimeric G proteins. J Biol Chem (1995) 270:503–6. doi:10.1074/jbc.270.2.503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Neves SR, Ram PT, Iyengar R. G protein pathways. Science (2002) 296:1636–9. doi:10.1126/science.1071550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Strathmann M, Simon MI. G protein diversity: a distinct class of alpha subunits is present in vertebrates and invertebrates. Proc Natl Acad Sci U S A (1990) 87:9113–7. doi:10.1073/pnas.87.23.9113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, Simon MI. Characterization of G-protein alpha subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. Proc Natl Acad Sci U S A (1991) 88:10049–53. doi:10.1073/pnas.88.22.10049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Kamato D, Rostum MA, Bernard R, Piva TJ, Mantri N, Guidone D, et al. The expansion of GPCR transactivation-dependent signalling to include serine/threonine kinase receptors represents a new cell signalling frontier. Cell Mol Life Sci (2015) 72(4):799–808. doi:10.1007/s00018-014-1775-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature (1996) 379:557–60. doi:10.1038/379557a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Burch ML, Ballinger ML, Yang SN, Getachew R, Itman C, Loveland K, et al. Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor beta type I receptor. J Biol Chem (2010) 285:26798–805. doi:10.1074/jbc.M109.092767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Burch ML, Getachew R, Osman N, Febbraio MA, Little PJ. Thrombin mediated proteoglycan synthesis utilizes both protein tyrosine kinase and serine/threonine kinase receptor transactivation in vascular smooth muscle cells. J Biol Chem (2013) 288(10):7410–9. doi:10.1074/jbc.M112.400259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Little PJ, Burch ML, Al-Aryahi S, Zheng W. The paradigm of g protein receptor transactivation: a mechanistic definition and novel example. ScientificWorldJournal (2011) 11:709–14. doi:10.1100/tsw.2011.75
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Kamato D, Burch ML, Osman N, Zheng W, Little PJ. Therapeutic implications of endothelin and thrombin G-protein-coupled receptor transactivation of tyrosine and serine/threonine kinase cell surface receptors. J Pharm Pharmacol (2013) 65:465–73. doi:10.1111/j.2042-7158.2012.01577.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Little PJ. GPCR responses in vascular smooth muscle can occur predominantly through dual transactivation of kinase receptors and not classical Galphaq protein signalling pathways. Life Sci (2013) 92:951–6. doi:10.1016/j.lfs.2013.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Liebmann C. EGF receptor activation by GPCRs: an universal pathway reveals different versions. Mol Cell Endocrinol (2011) 331:222–31. doi:10.1016/j.mce.2010.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Little PJ, Burch ML, Getachew R, Al-Aryahi S, Osman N. Endothelin-1 stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by endothelin receptor transactivation of the transforming growth factor-[beta] type I receptor. J Cardiovasc Pharmacol (2010) 56:360–8. doi:10.1097/FJC.0b013e3181ee6811
22. Berman DM, Gilman AG. Mammalian RGS proteins: barbarians at the gate. J Biol Chem (1998) 273:1269–72. doi:10.1074/jbc.273.3.1269
23. Little PJ, Neylon CB, Tkachuk VA, Bobik A. Endothelin-1 and endothelin-3 stimulate calcium mobilization by different mechanisms in vascular smooth muscle. Biochem Biophys Res Commun (1992) 183:694–700. doi:10.1016/0006-291X(92)90538-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. Rho and Rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res (1999) 84:1186–93. doi:10.1161/01.RES.84.10.1186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem (2007) 282:7833–43. doi:10.1074/jbc.M608288200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Jin J, Mao Y, Thomas D, Kim S, Daniel JL, Kunapuli SP. RhoA downstream of G(q) and G(12/13) pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem Pharmacol (2009) 77:835–44. doi:10.1016/j.bcp.2008.11.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Hubbard KB, Hepler JR. Cell signalling diversity of the Gqalpha family of heterotrimeric G proteins. Cell Signal (2006) 18:135–50. doi:10.1016/j.cellsig.2005.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature (1997) 389:183–6. doi:10.1038/38284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Imamura T, Ishibashi K, Dalle S, Ugi S, Olefsky JM. Endothelin-1-induced GLUT4 translocation is mediated via Galpha(q/11) protein and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. J Biol Chem (1999) 274:33691–5. doi:10.1074/jbc.274.47.33691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Tanski WJ, Roztocil E, Hemady EA, Williams JA, Davies MG. Role of Galphaq in smooth muscle cell proliferation. J Vasc Surg (2004) 39:639–44. doi:10.1016/j.jvs.2003.10.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Harmon B, Ratner L. Induction of the Galpha(q) signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J Virol (2008) 82:9191–205. doi:10.1128/JVI.00424-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Fan G, Jiang YP, Lu Z, Martin DW, Kelly DJ, Zuckerman JM, et al. A transgenic mouse model of heart failure using inducible Galpha q. J Biol Chem (2005) 280:40337–46. doi:10.1074/jbc.M506810200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J (1997) 16:7032–44. doi:10.1093/emboj/16.23.7032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Koch WJ, Hawes BE, Allen LF, Lefkowitz RJ. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc Natl Acad Sci U S A (1994) 91:12706–10. doi:10.1073/pnas.91.26.12706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Krumins AM, Gilman AG. Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J Biol Chem (2006) 281:10250–62. doi:10.1074/jbc.M511551200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Hwang JI, Choi S, Fraser ID, Chang MS, Simon MI. Silencing the expression of multiple Gbeta-subunits eliminates signaling mediated by all four families of G proteins. Proc Natl Acad Sci U S A (2005) 102:9493–8. doi:10.1073/pnas.0503503102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Taniguchi M, Nagai K, Arao N, Kawasaki T, Saito T, Moritani Y, et al. YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J Antibiot (Tokyo) (2003) 56:358–63. doi:10.7164/antibiotics.56.358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, et al. A novel Galphaq/11-selective inhibitor. J Biol Chem (2004) 279:47438–45. doi:10.1074/jbc.M408846200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K, et al. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci U S A (2010) 107:13666–71. doi:10.1073/pnas.1003553107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kawasaki T, Taniguchi M, Moritani Y, Hayashi K, Saito T, Takasaki J, et al. Antithrombotic and thrombolytic efficacy of YM-254890, a G q/11 inhibitor, in a rat model of arterial thrombosis. Thromb Haemost (2003) 90:406–13. doi:10.1267/THRO03030406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med (2008) 14:64–8. doi:10.1038/nm1666
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Miyamae S. Influence of magnesium and extracellular calcium reduction on ouabain-treated sinoatrial node cells in rabbit heart. Pharmacol Toxicol (1989) 65:192–7. doi:10.1111/j.1600-0773.1989.tb01155.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Fujioka M, Koda S, Morimoto Y, Biemann K. Structure of Fr900359, a cyclic depsipeptide from Ardisia crenata sims. J Org Chem (1988) 53:2820–5. doi:10.1021/Jo00247a030
44. Zaima K, Deguchi J, Matsuno Y, Kaneda T, Hirasawa Y, Morita H. Vasorelaxant effect of FR900359 from Ardisia crenata on rat aortic artery. J Nat Med (2013) 67:196–201. doi:10.1007/s11418-012-0644-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Grace MS, Lieu T, Darby B, Abogadie FC, Veldhuis N, Bunnett NW, et al. The tyrosine kinase inhibitor bafetinib inhibits PAR2-induced activation of TRPV4 channels in vitro and pain in vivo. Br J Pharmacol (2014) 171:3881–94. doi:10.1111/bph.12750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Akhtar S, Yousif MH, Chandrasekhar B, Benter IF. Activation of EGFR/ERBB2 via pathways involving ERK1/2, P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from ischemia-reperfusion injury. PLoS One (2012) 7:e39066. doi:10.1371/journal.pone.0039066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Heemskerk JW, Farndale RW, Sage SO. Effects of U73122 and U73343 on human platelet calcium signalling and protein tyrosine phosphorylation. Biochim Biophys Acta (1997) 1355:81–8. doi:10.1016/S0167-4889(96)00113-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Mogami H, Lloyd Mills C, Gallacher DV. Phospholipase C inhibitor, U73122, releases intracellular Ca2+, potentiates Ins(1,4,5)P3-mediated Ca2+ release and directly activates ion channels in mouse pancreatic acinar cells. Biochem J (1997) 324(Pt 2):645–51.
49. Bosch RR, Patel AM, Van Emst-De Vries SE, Smeets RL, De Pont JJ, Willems PH. U73122 and U73343 inhibit receptor-mediated phospholipase D activation downstream of phospholipase C in CHO cells. Eur J Pharmacol (1998) 346:345–51. doi:10.1016/S0014-2999(98)00070-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Wilsher NE, Court WJ, Ruddle R, Newbatt YM, Aherne W, Sheldrake PW, et al. The phosphoinositide-specific phospholipase C inhibitor U73122 (1-(6-((17beta-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione) spontaneously forms conjugates with common components of cell culture medium. Drug Metab Dispos (2007) 35:1017–22. doi:10.1124/dmd.106.014498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Klein RR, Bourdon DM, Costales CL, Wagner CD, White WL, Williams JD, et al. Direct activation of human phospholipase C by its well known inhibitor u73122. J Biol Chem (2011) 286:12407–16. doi:10.1074/jbc.M110.191783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Tsien RY, Rink TJ. Neutral carrier ion-selective microelectrodes for measurement of intracellular free calcium. Biochim Biophys Acta (1980) 599:623–38. doi:10.1016/0005-2736(80)90205-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, et al. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol (2000) 57:976–83.
54. Macmillan D, McCarron JG. The phospholipase C inhibitor U-73122 inhibits Ca(2+) release from the intracellular sarcoplasmic reticulum Ca(2+) store by inhibiting Ca(2+) pumps in smooth muscle. Br J Pharmacol (2010) 160:1295–301. doi:10.1111/j.1476-5381.2010.00771.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Eguchi S, Hirata Y, Imai T, Marumo F. Endothelin receptor subtypes are coupled to adenylate cyclase via different guanyl nucleotide-binding proteins in vasculature. Endocrinology (1993) 132:524–9. doi:10.1210/en.132.2.524
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Kuemmerle JF, Murthy KS, Makhlouf GM. Agonist-activated, ryanodine-sensitive, IP3-insensitive Ca2+ release channels in longitudinal muscle of intestine. Am J Physiol (1994) 266:C1421–31.
57. Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) (2007) 190:9–19. doi:10.1111/j.1365-201X.2007.01693.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Survase S, Ivey ME, Nigro J, Osman N, Little PJ. Actions of calcium channel blockers on vascular proteoglycan synthesis: relationship to atherosclerosis. Vasc Health Risk Manag (2005) 1:199–208. doi:10.2147/VHRM.S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Deng X, Mercer PF, Scotton CJ, Gilchrist A, Chambers RC. Thrombin induces fibroblast CCL2/JE production and release via coupling of PAR1 to Galphaq and cooperation between ERK1/2 and Rho kinase signaling pathways. Mol Biol Cell (2008) 19:2520–33. doi:10.1091/mbc.E07-07-0720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Yu ZY, Tan JC, McMahon AC, Iismaa SE, Xiao XH, Kesteven SH, et al. RhoA/ROCK signaling and pleiotropic alpha1A-adrenergic receptor regulation of cardiac contractility. PLoS One (2014) 9:e99024. doi:10.1371/journal.pone.0099024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Gavard J, Gutkind JS. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Galpha12/13 and Galpha11/q. J Biol Chem (2008) 283:29888–96. doi:10.1074/jbc.M803880200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Fan C, Su Q, Li Y, Liang L, Angelini DJ, Guggino WB, et al. Hypoxia-induced mitogenic factor/FIZZ1 induces intracellular calcium release through the PLC-IP(3) pathway. Am J Physiol Lung Cell Mol Physiol (2009) 297:L263–70. doi:10.1152/ajplung.90416.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Shiu SY, Pang B, Tam CW, Yao KM. Signal transduction of receptor-mediated antiproliferative action of melatonin on human prostate epithelial cells involves dual activation of Galpha(s) and Galpha(q) proteins. J Pineal Res (2010) 49:301–11. doi:10.1111/j.1600-079X.2010.00795.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Boutin A, Allen MD, Neumann S, Gershengorn MC. Persistent signaling by thyrotropin-releasing hormone receptors correlates with G-protein and receptor levels. FASEB J (2012) 26:3473–82. doi:10.1096/fj.12-207860
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Blaukat A, Barac A, Cross MJ, Offermanns S, Dikic I. G protein-coupled receptor-mediated mitogen-activated protein kinase activation through cooperation of Galpha(q) and Galpha(i) signals. Mol Cell Biol (2000) 20:6837–48. doi:10.1128/MCB.20.18.6837-6848.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Stevens PA, Pediani J, Carrillo JJ, Milligan G. Coordinated agonist regulation of receptor and G protein palmitoylation and functional rescue of palmitoylation-deficient mutants of the G protein G11alpha following fusion to the alpha1b-adrenoreceptor: palmitoylation of G11alpha is not required for interaction with beta*gamma complex. J Biol Chem (2001) 276:35883–90. doi:10.1074/jbc.M103816200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Liu S, Carrillo JJ, Pediani JD, Milligan G. Effective information transfer from the alpha 1b-adrenoceptor to Galpha 11 requires both beta/gamma interactions and an aromatic group four amino acids from the C terminus of the G protein. J Biol Chem (2002) 277:25707–14. doi:10.1074/jbc.M201015200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Szereszewski JM, Pampillo M, Ahow MR, Offermanns S, Bhattacharya M, Babwah AV. GPR54 regulates ERK1/2 activity and hypothalamic gene expression in a Galpha(q/11) and beta-arrestin-dependent manner. PLoS One (2010) 5:e12964. doi:10.1371/journal.pone.0012964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Sato T, Kawamura Y, Asai R, Amano T, Uchijima Y, Dettlaff-Swiercz DA, et al. Recombinase-mediated cassette exchange reveals the selective use of Gq/G11-dependent and -independent endothelin 1/endothelin type A receptor signaling in pharyngeal arch development. Development (2008) 135:755–65. doi:10.1242/dev.012708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Joshi SA, Fan KP, Ho VW, Wong YH. Chimeric Galpha(q) mutants harboring the last five carboxy-terminal residues of Galpha(i2) or Galpha(o) are resistant to pertussis toxin-catalyzed ADP-ribosylation. FEBS Lett (1998) 441:67–70. doi:10.1016/S0014-5793(98)01527-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Chan JS, Lee JW, Ho MK, Wong YH. Preactivation permits subsequent stimulation of phospholipase C by G(i)-coupled receptors. Mol Pharmacol (2000) 57:700–8. doi:10.1124/mol57.4.700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Evanko DS, Thiyagarajan MM, Wedegaertner PB. Interaction with Gbetagamma is required for membrane targeting and palmitoylation of Galpha(s) and Galpha(q). J Biol Chem (2000) 275:1327–36. doi:10.1074/jbc.275.2.1327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Evanko DS, Thiyagarajan MM, Siderovski DP, Wedegaertner PB. Gbeta gamma isoforms selectively rescue plasma membrane localization and palmitoylation of mutant Galphas and Galphaq. J Biol Chem (2001) 276:23945–53. doi:10.1074/jbc.M101154200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Crouthamel M, Thiyagarajan MM, Evanko DS, Wedegaertner PB. N-terminal polybasic motifs are required for plasma membrane localization of Galpha(s) and Galpha(q). Cell Signal (2008) 20:1900–10. doi:10.1016/j.cellsig.2008.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Chahdi A, Sorokin A. The role of beta(1)Pix/caveolin-1 interaction in endothelin signaling through Galpha subunits. Biochem Biophys Res Commun (2010) 391:1330–5. doi:10.1016/j.bbrc.2009.12.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Lin K, Wang D, Sadee W. Serum response factor activation by muscarinic receptors via RhoA. Novel pathway specific to M1 subtype involving calmodulin, calcineurin, and Pyk2. J Biol Chem (2002) 277:40789–98. doi:10.1074/jbc.M202745200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Xie Z, Ho WT, Spellman R, Cai S, Exton JH. Mechanisms of regulation of phospholipase D1 and D2 by the heterotrimeric G proteins G13 and Gq. J Biol Chem (2002) 277:11979–86. doi:10.1074/jbc.M109751200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Holtje M, Winter S, Walther D, Pahner I, Hortnagl H, Ottersen OP, et al. The vesicular monoamine content regulates VMAT2 activity through Galphaq in mouse platelets. Evidence for autoregulation of vesicular transmitter uptake. J Biol Chem (2003) 278:15850–8. doi:10.1074/jbc.M212816200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Rochdi MD, Parent JL. Galphaq-coupled receptor internalization specifically induced by Galphaq signaling. Regulation by EBP50. J Biol Chem (2003) 278:17827–37. doi:10.1074/jbc.M210319200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Goel R, Phillips-Mason PJ, Gardner A, Raben DM, Baldassare JJ. Alpha-thrombin-mediated phosphatidylinositol 3-kinase activation through release of Gbetagamma dimers from Galphaq and Galphai2. J Biol Chem (2004) 279:6701–10. doi:10.1074/jbc.M308753200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Day PW, Tesmer JJ, Sterne-Marr R, Freeman LC, Benovic JL, Wedegaertner PB. Characterization of the GRK2 binding site of Galphaq. J Biol Chem (2004) 279:53643–52. doi:10.1074/jbc.M401438200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Obara Y, Kurose H, Nakahata N. Thromboxane A2 promotes interleukin-6 biosynthesis mediated by an activation of cyclic AMP-response element-binding protein in 1321N1 human astrocytoma cells. Mol Pharmacol (2005) 68:670–9. doi:10.1124/mol.105.012922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Vasudevan N, Kia HK, Hadjimarkou M, Koibuchi N, Chin WW, Forrest D, et al. Retinoid-related receptor (ROR) alpha mRNA expression is altered in the brain of male mice lacking all ligand-binding thyroid hormone receptor (TR) isoforms. Endocrine (2005) 26:25–32. doi:10.1385/ENDO
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Ballou LM, Chattopadhyay M, Li Y, Scarlata S, Lin RZ. Galphaq binds to p110alpha/p85alpha phosphoinositide 3-kinase and displaces Ras. Biochem J (2006) 394:557–62. doi:10.1042/BJ20051493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Preuss I, Kurig B, Nurnberg B, Orth JH, Aktories K. Pasteurella multocida toxin activates Gbetagamma dimers of heterotrimeric G proteins. Cell Signal (2009) 21:551–8. doi:10.1016/j.cellsig.2008.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Brenner T, Nizri E, Irony-Tur-Sinai M, Hamra-Amitay Y, Wirguin I. Acetylcholinesterase inhibitors and cholinergic modulation in myasthenia gravis and neuroinflammation. J Neuroimmunol (2008) 20(1–202):121–7. doi:10.1016/j.jneuroim.2008.05.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Wang CJ, Hsu SH, Hung WT, Luo CW. Establishment of a chimeric reporting system for the universal detection and high-throughput screening of G protein-coupled receptors. Biosens Bioelectron (2009) 24:2298–304. doi:10.1016/j.bios.2008.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol (2013) 20:146–59. doi:10.1016/j.chembiol.2012.12.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Li JH, Jain S, McMillin SM, Cui Y, Gautam D, Sakamoto W, et al. A novel experimental strategy to assess the metabolic effects of selective activation of a G(q)-coupled receptor in hepatocytes in vivo. Endocrinology (2013) 154:3539–51. doi:10.1210/en.2012-2127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kimple ME, Neuman JC, Linnemann AK, Casey PJ. Inhibitory G proteins and their receptors: emerging therapeutic targets for obesity and diabetes. Exp Mol Med (2014) 46:e102. doi:10.1038/emm.2014.40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: G proteins, GPCR, cell signaling, therapeutic targets, transactivation
Citation: Kamato D, Thach L, Bernard R, Chan V, Zheng W, Kaur H, Brimble M, Osman N and Little PJ (2015) Structure, function, pharmacology, and therapeutic potential of the G protein, Gα/q,11. Front. Cardiovasc. Med. 2:14. doi: 10.3389/fcvm.2015.00014
Received: 03 December 2014; Accepted: 11 March 2015;
Published online: 24 March 2015.
Edited by:
Ian Megson, University of the Highlands and Islands, UKCopyright: © 2015 Kamato, Thach, Bernard, Chan, Zheng, Kaur, Brimble, Osman and Little. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Little, Discipline of Pharmacy, School of Medical Sciences, RMIT University, PO Box 71, Bundoora, VIC 3083, Australia e-mail: peter.little@rmit.edu.au