Abstract
Hydrogenase-mediated reduction of flavin mononucleotide by H2 is exploited to enable cleaner application of nitroreductase enzymes for reduction of aromatic nitro functional groups. This turns the overall reaction into a biocatalytic hydrogenation. Use of flavin-containing nitroreductases in industrial biotechnology typically relies upon NADH or NADPH as reductant, together with glucose dehydrogenase and glucose as a regeneration system for the reduced nicotinamide cofactor, with 3 equivalents of the carbon-intensive glucose required for a single 6-electron nitro to amine conversion. We show here that reduced flavin mononucleotide is an alternative reductant for nitroreductases, and by combining this with H2-driven recycling of reduced flavin, we avoid glucose, thereby enabling atom-efficient biocatalytic nitro reductions. We compare this biocatalytic system, via green chemistry metrics, to existing strategies for biocatalytic nitro-group reductions, particularly with respect to replacing glucose with H2 gas. We take steps towards demonstrating industrial viability: we report an overexpression system for E. coli hydrogenase 1, giving a 12-fold improvement in enzyme yield; we show a reaction in which the hydrogenase exhibits > 26,000 enzyme turnovers; and we demonstrate reasonable solvent tolerance of the hydrogenase and flavin reduction system which would enable reaction intensification.
Introduction
Biocatalysis has become embedded in pharmaceutical and fine chemical manufacturing methodologies, particularly for the reduction of unsaturated bonds via hydride transfer from the nicotinamide cofactor NADH, or its more expensive phosphorylated derivative, NADPH. Regeneration of these cofactors during industrial biocatalytic chemical synthesis is typically achieved using glucose dehydrogenase (GDH) with glucose as reductant. This sacrificial use of the C6H12O6 sugar for transfer of a single hydride (H−) to the nicotinamide cofactor gives reductive biocatalysis a poor atom-economy. This has led us to explore biocatalytic systems which use H2 as an atom-economical reductant, and we have previously developed an NADH recycling system based around hydrogenase and NAD+-reductase enzyme moieties co-immobilised on a carbon support for H2-driven NADH supply to a variety of NADH-dependent reductases (Reeve et al., 2017; Poznansky et al., 2021). The use of H2 as reductant, turns the overall biocatalytic process into a hydrogenation reaction equivalent to that catalysed by metal catalysts such as palladium on carbon (Pd/C). We are now looking to extend the reach of H2-driven reductive processes in industrial biocatalysis. Two enzyme types that have been added more recently to the set of commercially-available enzymes for chemical synthesis are the “old yellow enzyme” (OYE) ene reductases (Scholtissek et al., 2017) and the nitroreductases “NR” (Roldán et al., 2008), both of which use a flavin organocatalytic site for substrate reduction. The OYE ene reductases catalyse asymmetric reduction of activated alkenes, particularly enones (having a C=C bond conjugated to a ketone). The nitroreductases catalyse reduction of aromatic nitro-groups. These enzymes are generally denoted as NAD(P)H-dependent, and are usually supplied with reducing equivalents in the form of the reduced nicotinamide cofactors, which in turn are recycled with the GDH/glucose system (Figure 1A). However it has now been established that the OYE ene reductases will alternatively accept electrons from the reduced hydroquinone form of flavins (denoted FMNH2 for FMN). Reduced flavin for supply to ene reductases has been recycled by electrochemical or photochemical means, (Knaus et al., 2016; Zhang and Hollmann, 2018) or using H2 gas and an NAD+-reducing hydrogenase (Al-Shameri et al., 2020). We made a further surprising finding that the robust hydrogenase 1 from Escherichia (E.) coli (Hyd1) has non-native activity for reduction of FAD or FMN to their hydroquinone level and provides a convenient flavin recycling system for supply of FADH2 or FMNH2 to OYE ene reductases (Joseph Srinivasan et al., 2021).
FIGURE 1
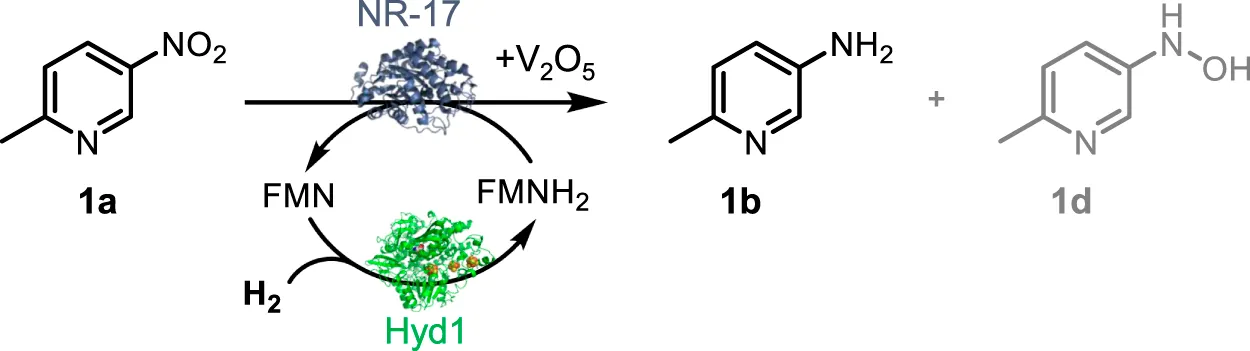
We hypothesised that the flavin-containing nitroreductases might also be operated in a much more atom-efficient way by using hydrogenase/H2 to provide exogenous reduced flavin as a reductant, eliminating the need for glucose-driven NAD(P)H recycling (Scheme 1). As in the OYE ene reductases, the bound prosthetic flavin is fairly exposed at the surface of the protein in nitroreductases (Supplementary Figure S1), enabling access to varied reductants (Roldán et al., 2008). In this study, we use the reduction of 2-methyl-5-nitropyridine (1a) to 6-methylpyridin-3-amine (1b) as a model reaction to explore whether catalysis by a range of commercial nitro reductases can be supported by FMNH2, with hydrogenase/H2 as the recycling system for reduced flavin. All nitroreductase enzymes tested showed activity, confirming that it is possible to run biocatalytic nitro-group reductions as hydrogenation reactions with FMNH2 as the electron donor.
SCHEME 1
Six-electron reduction of the nitro group to an amine proceeds via a series of partially reduced intermediates (see Scheme 1). The application of nitroreductases is often to prepare the hydroxylamine (d in Scheme 1A; see Nguyen-Tran et al., 2014; Pitsawong et al., 2014; Miller et al., 2018), and use of nitroreductases to produce the corresponding amine product is typically incomplete. Bornadel et al. reported a process to overcome this using V2O5 as co-catalyst, aiding the disproportionation of the hydroxylamine intermediate, and hence eventually favouring the amine product (Bisagni et al., 2020; Bornadel et al., 2021). Addition of 2 mol% V2O5 was found to yield the aniline product, b in Scheme 1A. A further complication has been found when GDH/glucose is used as the cofactor recycling system, in that glucose reacts to form N-glucoside as a common side-product (c in Scheme 1A). The resulting gluconolactone also necessitates continual pH monitoring and either adjustment with base or high buffer levels. Furthermore, with 3 equivalents of glucose needed for the nitro to amine conversion, the cost of glucose becomes significant in the overall reaction economics. The disadvantages of having glucose in the system are all overcome by the hydrogenase/H2 flavin recycling system we demonstrate here, which results in no net pH change and avoids by-products and complications that result from glucose. The hydrogenase/H2 flavin recycling system is used here in conjunction with the V2O5 additive to obtain high levels of conversion to the pure amine product.
Industrial scale catalytic hydrogenation of nitroarenes is a favourable method to produce the corresponding anilines (Orlandi et al., 2016), taking advantage of the benefits of H2 as the reductant. Two predominant challenges with catalytic nitro hydrogenations centre on preventing the reduction of other reducible functional groups in the reactant, and promoting complete reduction to the corresponding amine (hence avoiding accumulation of hydroxylamine or other side products). Metal catalysts that use H2 have been developed to address these selectivity problems (reviewed in Orlandi et al., 2016; recent examples include Goyal et al., 2020; Zhang et al., 2021; Zubar et al., 2021; Fu et al., 2022), though the cost and/or toxicity of the metals is problematic in commercial applications. Compared with metal catalysts, the nitroreductase system shows much more functional group selectivity (e.g., no reduction of nitrile, alkene, or aryl chloride; Bisagni et al., 2020), enabling late-stage generation of aromatic amines within complex molecules. Therefore, developing an H2-driven system that avoids toxic metals and takes advantage of the nitroreductase selectivity would be beneficial.
To be viable for application in industrial biotechnology, the hydrogenase for H2-driven flavin recycling must be active, robust and relatively easy to express. In previous work coupling Hyd1 to OYE ene reductase, we showed stability of Hyd1 over >5.5 days, with TTN of 20,200 during flavin recycling, suggesting promise for utility in biotechnological applications ((Joseph Srinivasan et al., 2021). Hyd1 is also very tolerant to O2 (Lukey et al., 2010; Reeve et al., 2017), and we have previously used Hyd1 in catalysis systems run on the bench without strict control over O2 levels (Rowbotham et al., 2020; Poznansky et al., 2021). Achieving high expression levels of hydrogenase is challenging due to the accessory proteins needed to assemble the complex bimetallic active site and the fact that the enzyme is natively membrane-associated. We report here an over-expression system for Hyd1 yielding >10-fold increase in expression of the enzyme compared to native expression levels in E. coli, suggesting promise for Hyd1 in biotechnology.
The results presented here confirm that the NADPH cofactor typically used as reductant for flavin-containing nitroreductases may be exchanged for FMNH2, and that Hyd1 represents a viable recycling system for cleaner operation of nitroreductases with H2-driven flavin recycling. We calculate the E factors to compare the waste generated by the glucose/GDH/NADPH system versus our H2/Hyd1/FMN method, then explore solvent tolerance of Hyd1 up to 50% loading of dimethyl sulfoxide (DMSO) or acetonitrile. Finally, we discuss how these aspects combined with the Hyd1 overexpression system help to bridge the gap toward a viable process scale-up route.
Materials and Methods
Buffer salts (Sigma-Aldrich), FAD (disodium salt, ≥98%, Cayman Chemical Company), and FMN (monosodium salt dihydrate, Applichem Panreac) were all used as-received. 2-Methyl-5-nitropyridine (1a, 95%) and 6-methylpyridin-3-amine (1b, 97%) were purchased from Alfa Aesar. The hydroxyl amine 1d used for the 1H NMR spectroscopy standard was synthesised from 1a using published procedures (Feng et al., 2016; see Supplementary information for details). All aqueous solutions were prepared with deoxygenated MilliQ water (Millipore, 18 MΩcm). All experiments were carried out in a glovebox (Glove Box Technology Ltd.) under a protective N2 atmosphere (O2 < 3 ppm).
UV-visible spectra were recorded by a Cary 60 spectrophotometer using a quartz cuvette (path length 1 cm, cell volume 1 mL, Hellma). The buffer was used to take a baseline scan. In some of the experiments, there was a uniform shift of the baseline across the entire spectral region (200–800 nm), which was corrected for during data processing. GC-FID data were obtained using a TRACE 1310 instrument (Thermo Fisher Scientific), with a CP-Chirasil-Dex CB column (Agilent), 25 m length, 0.25 mm diameter, 0.25 μm (film thickness), fitted with a guard of 10 m deactivated fused silica of the same diameter. 1H NMR spectra were taken at room temperature using a Bruker Avance III HD nanobay (400 MHz) instrument. 10% v/v 2H2O was added to allow for field locking. Reactant and product peaks were determined by comparison against commercial standards. Mass spectrometry was performed on an Agilent 6120 bench-top single quadrupole in direct infusion mode (connected to an Agilent 1260 IsoPump and CTC Analytics 2777 Sample Manager).
The hydrogenase genes were expressed according to the over-expression protocol described in Overexpression System for Hydrogenase 1 (vide infra) with details reported in Supplementary Section S2.1. Over-expressed Hyd1 was purified using a Ni-NTA chromatography step followed by size exclusion enrichment. The activity of the over-expressed Hyd1 was verified using a spectrophotometric assay measuring the hydrogenase-mediated reduction of methylene blue in a H2-saturated solution. Quantities of Hyd1 used in catalysis here are given in mU, which was determined using our previously described FMN reduction assay (Joseph Srinivasan et al., 2021), wherein 1 mU = 1 μmol FMN min−1.
Nitroreductases (NR-4, NR-5, NR-14, NR-17, and NR-24) were provided by Johnson Matthey in the lyophilised form and used without further purification.
General Procedure for Biocatalytic H2-Driven Nitro Reduction via Flavin Recycling
Using a syringe and needle, 300 μL of H2-saturated potassium phosphate buffer (100 mM, pH 7.0, 25°C) was transferred to a centrifuge tube (Eppendorf, 0.5 mL) that contained 1 mM FMN, 2 mM V2O5 and 10 mM 1a in DMSO (5 vol%). A portion of this solution (approx. 0.1 mL) was used to transfer the designated quantity of Hyd1 and NR into the reaction tube in sequence via a needle and syringe. The lid of the centrifuge tube was pierced once with a needle, capped, and placed in the Büchi Tinyclave pressure vessel which was then charged to 1 bar H2. The pressure vessel was then removed from the glovebox and placed in a water bath that maintained a temperature of 35°C. The setup was covered with aluminium foil to exclude any light. See analysis details in Supplementary Material (GC-FID: section 2.2; 1H NMR spectroscopy: section 2.3).
General Procedure for Measuring Hyd1 Activity for FMN Recycling in the Presence of Cosolvent
As previously described (Joseph Srinivasan et al., 2021), activity for Hyd1 and H2-driven FMN or FAD reduction was performed using UV-vis spectroscopy. The concentration of FMN was directly calculated based on the absorbance at λ = 445 nm (ε = 12.50 mM−1 cm−1) and FAD based on the absorbance at λ = 450 nm (ε = 11.30 mM−1 cm−1). During the linear phase of the reaction, the decrease in [oxidised flavin] over time was determined in order to calculate the hydrogenase activity (μmol min−1 mg−1).
The indicated volume of Tris-HCl buffer (50 mM, pH 8.0) was added to a UV-visible quartz cuvette, which was placed in the cell holder and a baseline was recorded using the UV-visible spectrophotometer. A solution of 0.1 mM flavin in Tris-HCl buffer (50 mM, pH 8.0) and the indicated volume% of organic co-solvent was next prepared in the cuvette, which was then capped with a rubber septum that was pierced with two needles to provide a gas inlet and outlet. An H2-line was then connected and H2 was bubbled through the flavin solution via the inlet needle for 10 min. The needle was then moved up to the headspace through which a continuous H2 flow was supplied. About 0.4 mL of the flavin solution was then used to transfer a designated quantity of Hyd1 into the cuvette using a syringe and needle, and the needle and syringe rinsed by drawing solution in and out of the cuvette. The assay was carried out by taking one scan (200–800 nm) every 30 s over 30 min.
Results and Discussion
Overexpression System for Hydrogenase 1
Traditional methods for the synthesis and isolation of Hyd1 have relied on chromosomal expression, resulting in yields unsuitable for processes at higher scales (Lukey et al., 2010). We report an over-expression system for Hyd1, which is a breakthrough that makes hydrogenase-catalysed processes more industrially viable. The E. coli [NiFe] Hyd1 is a membrane-bound multi-subunit enzyme natively expressed during anaerobic stationary growth. The mature enzyme consists of two structural subunits and a cytochrome c protein which is bound to the periplasmic membrane. The [NiFe] bimetallic active site is synthesised by several pleiotropic enzymes, and incorporated into the apo-enzyme aided by additional Hyd1-specific enzymes. Additionally, Hyd1 harbours three iron-sulfur clusters (FeS), whose synthesis is regulated inside the cells by metabolic signals. Taken together, the biosynthesis of Hyd1 is a complex process involving multiple proteins whose expression is internally regulated (Sargent, 2016).
The complex biosynthesis of Hyd1 prevents a simple over-expression strategy, and heterologous expression systems for [NiFe] hydrogenases are uncommon. Weyman et al. developed a system to heterologously express [NiFe] hydrogenases from Alteromonas macleodii and Thiocapsa roseopersicina in E. coli. This method consisted of expressing the structural genes of the enzyme alongside eight additional genes known to be involved in [NiFe] co-factor biosynthesis and enzyme maturation within a suitable E. coli strain (Weyman et al., 2011). To develop an over-expression system for the homologous Hyd1, we took a similar approach (see details in section 2.1, Supplementary Information). Succinctly, the genes known to be involved in the biosynthesis and maturation of the enzyme were amplified from a Hyd1-producing E. coli strain and cloned inside plasmids with inducible promoters. The plasmids were transformed into a strain of E. coli lacking the large subunit of each of the native hydrogenases. Active enzymes were recovered from anaerobically-grown cells in which protein over-expression was induced at the stationary phase.
The yield of Hyd1 was consistently around 0.6 mg of protein per litre of culture. In comparison, the yield of chromosomally-expressed Hyd1 was around 0.05 mg of protein per litre of culture, therefore the over-expression system provided a 12-fold improvement in enzyme yield. Specific activity of 143 ± 9 μmol of H2 min−1 (mg of enzyme)−1 was measured on three independent hydrogenase-mediated methylene blue reduction assays. This result is consistent with the previously reported activity of chromosomally expressed Hyd1 (153 ± 18 μmol of H2 min−1 mg Hyd1−1; Brooke et al., 2017).
Demonstration of a Clean, Complete Nitro Reduction Using Hyd1/H2/FMN With Nitroreductases “NR”
Improvements to our Hyd1 production inspired us to extend its application in H2-driven cofactor recycling. Here we examine the use of Hyd1 to supply reduced FMN to NR for selective nitroreductions, which is likely to have a significant impact on minimising waste, even greater than for the alkene reductions which we have previously demonstrated with H2-driven flavin recycling (Joseph Srinivasan et al., 2021). Whereas the 2-electron alkene bond reductions require ≥1 equivalent of glucose for NAD(P)H recycling, nitro reduction to the corresponding amine requires ≥3 equivalents since it is a 6-electron reduction. Furthermore, the need for glucose results in build-up of gluconolactone (thus high buffer levels are required to mitigate pH change) and the N-glucoside by product 1c (Bornadel et al., 2021, see structure in Scheme 1) which complicates product analysis and purification. Therefore the need to find alternative processes for biocatalytic nitro reduction is that much more important.
We first tested our H2/Hyd1/FMN system with a suite of NRs (provided by Johnson Matthey) to determine if any of the enzymes would accept electrons from externally supplied FMNH2 in place of NADPH, as we had observed with the OYEs (Joseph Srinivasan et al., 2021). Model nitro aromatic substrate 1a was selected because it was previously shown to reduce to 1b using various NRs with the glucose/GDH/NADP+ cofactor recycling system (Bornadel et al., 2021). Here, 3.95 mU Hyd1 was mixed with 1 mM FMN, 10 mM 1a, 2 mM V2O5, and 0.3 mg of the various NR in potassium phosphate buffer (250 mM, pH 7) with 5 vol% DMSO at 35 °C (see Section S2.2 for more detail). The reactions were compared for conversion of 1a using GC-FID (see Supplementary Figure S2 and the associated description for details). Encouragingly, all the nitroreductases tested showed significant conversion of the nitro substrate when supplied with reduced flavin, and to our knowledge this is the first demonstration of operating nitroreductases for preparing aniline products in the absence of NAD(P)H. The results in Table 1 show that use of each NR led to similar conversion rates, thus we opted to continue with NR-17 for assessing the catalyst system further.
TABLE 1
| Entry | Nitroreductase | Conversion to 1b (%)a | ||
|---|---|---|---|---|
| 3 h | 6 h | 8 h | ||
| 1 | NR-4 | 10 | 43 | 70 |
| 2 | NR-5 | 9 | 46 | 73 |
| 3 | NR-14 | 10 | 45 | 74 |
| 4 | NR-17 | 13 | 51 | 75 |
| 5 | NR-24 | 12 | 50 | 77 |
Screening different NRs with H2-driven FMN recycling for converting 1a to 1b.
Reaction conditions: 300 µL scale, 0.18 mg/mL Hyd1 (3.95 mU), 1 mg/mL NR, 1 mM FMN, 10 mM 1a, 2 mM V2O5 in potassium phosphate buffer (100 mM, pH 7), 5 vol% DMSO at 35°C in a pressure vessel (1 bar H2).
GC conversion to 1b (calculated using Supplementary Eq. S1; see details in Supplementary Section S2.2).
We next evaluated systematic elimination of specific components in order to understand the importance of the components. We examined the ability of these catalyst systems to reduce 1a completely to 1b, aiming to avoid reaction intermediates (e.g., 1d) and side-products in the final mixture (results summarised in entries 1–4, Table 2). All reactions were run with the components marked (✓) at 35°C in a pressure vessel (1 bar H2) using 10 mM 1a in potassium phosphate buffer (100 mM, pH 7.0) that contained 5 vol% DMSO for substrate solubility. After mixing for 20 h, reactions were taken for 1H NMR spectroscopy analysis in order to quantify compound composition in the reaction mixture. See analysis details in Supplementary Material section 2.3. Figure 2 shows the aromatic portion of 1H NMR spectra taken from each control reaction (full spectra and diagnostic peak integrations are shown in Supplementary Figures S3–S6, Supplementary Material). These are compared against commercial standards of 1a and 1b, as well as 1d which was synthesised from 1a using hydrazine and Rh/C (Feng et al., 2016) to provide a 0.7: 1 ratio of 1d and 1b.
TABLE 2
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Hyd1 | NR-17 | FMN | V2O5 | Product composition (%)a | ||
| 1a | 1b | 1d | |||||
| 1b | ✓ | ✓ | ✓ | — | Present | Present | Present |
| 2 | — | ✓ | ✓ | ✓ | 100 | 0 | 0 |
| 3 | ✓ | ✓ | - | ✓ | 20 | 80 | 0 |
| 4 (full reaction) | ✓ | ✓ | ✓ | ✓ | 0 | 100 | 0 |
| 5 (full reaction, 20 mM 1a)c | ✓ | ✓ | ✓ | ✓ | Traced | 96 | Traced |
Evaluation of the necessity for each component in the H2-driven enzymatic nitroreduction.
Reaction conditions: 300 μL scale, 10 mM 1a, potassium phosphate buffer (100 mM, pH 7.0), 5 vol% DMSO at 35°C in a pressure vessel (1 bar H2) for 20 h; in the presence or absence of 1 mM FMN, 0.18 mg/mL Hyd1 (3.95 mU), 1 mg/mL NR-17, 2 mM V2O5.
Determined using 1H NMR spectroscopy (see Supplementary Figures S3–S6).
Entry 1 resulted in a complex mixture of products such that percent compositions could not be determined (see spectrum, Figure 2).
The full reaction that used 20 mM 1a (entry 5) used 0.22 mg/mL Hyd1, NR-14 in place of NR-17, and was stopped after 19 h instead of 20 h, and taken for analysis by mass spectrometry in addition to 1H NMR spectroscopy.
Mass spectrometry showed peaks totaling < 4% combined of 1a and 1d (see Supplementary Figure S7).
FIGURE 2
Table 2, entry 1 shows that, in the absence of the V2O5 disproportionation co-catalyst, the hydroxyl amine 1d accumulates in addition to production of some 1b after 20 h. Product compositions were not calculated due to the complex mixture of unidentified side-products (see spectrum in Figure 2). The results shown in entry 2 show that, in the absence of Hyd1, no conversion was observed, which confirms the necessity for our FMN-reducing system for the biocatalytic reduction of 1a. In entry 3, when no FMN was added there was 80% conversion to 1b and 20% 1a left unreacted after mixing for 20 h. In this control, it is possible that FMN was available in solution after dissociation from the NR-17 active site, or that it was present from the original enzyme prep (Bisagni et al., 2020). Importantly, at the 6 h time point, the NMR conversion to 1b was only 10%, whereas the full reaction (described in entry 4) provided 1b at 49% conversion after 6 h (not tabulated). This highlights a nearly 5-fold improvement in catalyst activity when 1 mM FMN is added.
Ultimately, when every component was present, 100% conversion to 1b was obtained after reacting for 20 h (entry 4). The “full reaction” conditions shown in entry 4 were repeated in duplicate and conversions at 3 and 6 h showed a maximum standard deviation of only 2.1% indicating good reproducibility (see Supplementary Table S3 in the Supplementary Material for results).
When the starting concentration of 1a was increased to 20 mM, the “full reaction” conditions led to 96% conversion to amine 1b. This product was taken for analysis by mass spectrometry to confirm the chemical composition (Supplementary Figure S8 in the Supplementary Material), which revealed < 4% impurities attributed primarily to 1a and trace amounts of 1d. The Hyd1 total turnover number (TTN) in this case was 26,100 (accounting for the Hyd1 completing three turnovers in order to convert each 1a to 1b).
In order to benchmark the waste-minimisation that the H2/Hyd1/FMN system provides, we quantified E factor and compared it to the reported glucose/GDH/NADP+ system when 20 mM 1a was used (Bornadel et al., 2021). E factor = masswaste/massproduct, and we tallied the different components shown in Table 3. Solvent and H2 were not counted in these “simple” E factor calculations, which is the most useful metric when a process is still in early stages of development (Sheldon, 2017).
TABLE 3
| Entry | Nitro reduction method | Masswaste componentsa | Waste (mg) | Mass1b (mg) | E factor |
|---|---|---|---|---|---|
| 1 | Glucose/GDH and NADP+ to supply NR-14 (from Bornadel et al. (2021) in the Supporting Information) | Potassium phosphate (250 mM) | 19.6 | 0.85 | 37 |
| V2O5 (2 mM) | 0.2 | ||||
| NADP+ (1 mM), glucose (100 mM) | 0.35 | ||||
| GDH | 9 | ||||
| NR-14, | 0.5 | ||||
| unreacted 1a | 1.75 | ||||
| side products 1c, 1d and | 0.070.160.18 | ||||
 | |||||
| 2 | H2/Hyd1 and FMN to supply NR-14 (this work) | Potassium phosphate (100 mM), | 4.7 | 0.65 | 8 |
| V2O5 (2 mM) | 0.11 | ||||
| FMN (1 mM) | 0.14 | ||||
| Hyd1 | 0.07 | ||||
| NR-14, | 0.3 | ||||
| unreacted 1a | 0.01 | ||||
| side product 1d | 0.02 |
E factor comparison of the nitroreductase reactions using either glucose/GDH/NADP+ or H2/Hyd1/FMN.
Waste components were calculated based on pH 7.0 potassium phosphate buffer (concentration indicated) and on reported conversions to the various side products. In entry 2, masses of 1a, 1d, and 1b were calculated based on 1, 3 and 96% conversions, respectively (based on mass spectrometry).
Entry 1 shows that the glucose/GDH/NADP+ method has significant waste contribution from the high buffer concentration (250 mM, 19.6 mg, 62% of the total masswaste), however the high buffer levels were required in order to mitigate the pH change that results from the generation of the gluconolactone by-product. The glucose also added significant mass to the waste (9 mg, 28% overall masswaste). Combined with the other waste contributors, the E factor = 37. Alternatively, entry two shows that the H2/Hyd1/FMN system provides 1b with the E factor = 8, which represents a >4-fold improvement thanks to the avoidance of glucose. This system has yet to be optimised for E factor, and we anticipate that further lowered buffer levels are likely to be well-tolerated. For example, our previous FMN recycling was shown at 50 mM buffer (Joseph Srinivasan et al., 2021), and we have shown operation of Hyd1 in NADH recycling in un-buffered water (Reeve et al., 2017). Further improvements to E factor and overall reaction intensity are anticipated with higher substrate loadings, which would rely on increasing substrate concentration and likely the percentage of co-solvent.
Demonstration of Solvent Tolerance for Hyd1 Flavin Recycling
We demonstrated here that Hyd1 activity for reduction of the flavins FAD and FMN is well-retained in the presence of up to 50 volume% of co-solvent (DMSO and MeCN, Figure 3). The specific activity for Hyd1 is higher with FMN than FAD. In DMSO, the flavin reductions show an initial drop-off in specific activity as the cosolvent is introduced up to about 5%, but specific activity then remains fairly constant as the cosolvent concentration is increased further. Considering that the nitroreduction reactions run in this study contained 5 vol% DMSO, it is likely that up to 40% DMSO would not impact the Hyd1 FMN reduction activity. Similarly, the Hyd1-catalysed FMN reduction also appears to maintain fairly consistent activity between 5 and 50 vol% MeCN. This solvent tolerance demonstration adds promise for successful intensification of our system for converting 1a to 1b at higher substrate concentrations. It may also be vital to future work in which we will extend the substrate scope to nitroaromatic compounds that might otherwise be insoluble in the aqueous solution (e.g., those already demonstrated with NRs using glucose/GDH/NADPH in Bisagni et al., 2020).
FIGURE 3
Conclusion
In conclusion, we have shown that reduced FMN, recycled in a H2-mediated process by E. coli Hyd1, is a viable reductant for a range of commercial nitroreductases. When used in combination with V2O5 co-catalyst, complete reduction of an aromatic nitro compound to the corresponding amine product is achievable, giving a significantly improved E factor compared to the glucose-driven NADPH-dependent process, even without any optimisation towards this end. The ability to over-express Hyd1 in E. coli, its tolerance to solvents DMSO and MeCN up to 50%, and a TTN >26,000 suggest that the Hyd1 flavin recycling system may be compatible with applications in industrial biotechnology. Future work in this area will focus on determining the purity requirements of the flavin cofactor, in addition to process intensification and confirmation that the previously reported NR substrate scope is retained with this system.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
All authors helped to design experiments. SJS, MR, and PT conducted experiments. All authors analysed data, discussed the results and assisted in preparation of the manuscript. SC, HR, and KV supervised the project and wrote the paper.
Funding
This research was supported financially by the awards from European Research Council (BiocatSusChem ERC-2018-CoG 819580 to KAV), Biotechnology and Biological Sciences Research Council (BBSRC, BB/R018413/1 and an award from E3B NIBB BB/S009787/1 to KAV), and Engineering and Physical Sciences Research Council (EPSRC, IB Catalyst EP/N013514/1 to KAV and HAR).
Acknowledgments
We are grateful to Beatriz Dominguez (Johnson Matthey) for supplying nitroreductases and for helpful discussions about the catalyst and substrates. We thank Frank Sargent (Newcastle University) for his gift of the E. coli strains FTD147(DE3)J, MC4100, and FTH004 and for helpful discussion. We are grateful to Jiaao Wei (U. Oxford) for help with characterisation of the overexpressed Hyd1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2022.906694/full#supplementary-material
References
1
Al-ShameriA.WillotS. J.-P.PaulC. E.HollmannF.LauterbachL. (2020). H2 as a Fuel for Flavin- and H2O2-Dependent Biocatalytic Reactions. Chem. Commun.56, 9667–9670. 10.1039/d0cc03229h
2
BisagniS.BornadelA.DominguezB.LepaihJ.PushpanathA.SlabuI.et al (2020). Method of Reducing Aromatic Nitro Compounds. Patent WO2020128434A1.
3
BornadelA.BisagniS.PushpanathA.SlabuI.LepaihJ.CherneyA. H.et al (2021). Process Development and Protein Engineering Enhanced Nitroreductase-Catalyzed Reduction of 2-Methyl-5-Nitro-Pyridine. Org. Process Res. Dev.25, 648–653. 10.1021/acs.oprd.0c00464
4
BrookeE. J.EvansR. M.IslamS. T. A.RobertsG. M.WehlinS. A. M.CarrS. B.et al (2017). Importance of the Active Site “Canopy” Residues in an O2-Tolerant [NiFe]-Hydrogenase. Biochemistry56, 132–142. 10.1021/acs.biochem.6b00868
5
FengP.LeeK. N.LeeJ. W.ZhanC.NgaiM.-Y. (2016). Access to a New Class of Synthetic Building Blocks via Trifluoromethoxylation of Pyridines and Pyrimidines. Chem. Sci.7, 424–429. 10.1039/C5SC02983J
6
FuH.ZhangH.YangG.LiuJ.XuJ.WangP.et al (2022). Highly Dispersed Rhodium Atoms Supported on Defect-Rich Co(OH)2 for the Chemoselective Hydrogenation of Nitroarenes. New J. Chem.46, 1158–1167. 10.1039/D1NJ04936D
7
GoyalV.SarkiN.SinghB.RayA.PoddarM.BordoloiA.et al (2020). Carbon-Supported Cobalt Nanoparticles as Catalysts for the Selective Hydrogenation of Nitroarenes to Arylamines and Pharmaceuticals. ACS Appl. Nano Mat.3, 11070–11079. 10.1021/acsanm.0c02254
8
Joseph SrinivasanS.ClearyS. E.RamirezM. A.ReeveH. A.PaulC. E.VincentK. A. (2021). E. C Nickel‐Iron Hydrogenase 1 Catalyses Non‐Native Reduction of Flavins: Demonstration for Alkene Hydrogenation by Old Yellow Enzyme Ene‐Reductases. Angew. Chem. Int. Ed.60, 13824–13828. 10.1002/anie.202101186
9
KnausT.PaulC. E.LevyC. W.De VriesS.MuttiF. G.HollmannF.et al (2016). Better Than Nature: Nicotinamide Biomimetics that Outperform Natural Coenzymes. J. Am. Chem. Soc.138, 1033–1039. 10.1021/jacs.5b12252
10
LukeyM. J.ParkinA.RoesslerM. M.MurphyB. J.HarmerJ.PalmerT.et al (2010). How Escherichia coli is Equipped to Oxidize Hydrogen under Different Redox Conditions. J. Biol. Chem.285, 3928–3938. 10.1074/jbc.M109.067751
11
MillerA.-F.ParkJ.FergusonK.PitsawongW.BommariusA. (2018). Informing Efforts to Develop Nitroreductase for Amine Production. Molecules23, 211–233. 10.3390/molecules23020211
12
Nguyen-TranH.-H.ZhengG.-W.QianX.-H.XuJ.-H. (2014). Highly Selective and Controllable Synthesis of Arylhydroxylamines by the Reduction of Nitroarenes with an Electron-Withdrawing Group Using a New Nitroreductase BaNTR1. Chem. Commun.50, 2861–2864. 10.1039/C3CC48590K
13
OrlandiM.BrennaD.HarmsR.JostS.BenagliaM. (2016). Recent Developments in the Reduction of Aromatic and Aliphatic Nitro Compounds to Amines. Org. Process Res. Dev.22, 430–445. 10.1021/acs.oprd.6b00205
14
PitsawongW.HobenJ. P.MillerA.-F. (2014). Understanding the Broad Substrate Repertoire of Nitroreductase Based on its Kinetic Mechanism. J. Biol. Chem.289, 15203–15214. 10.1074/JBC.M113.547117
15
PoznanskyB.ClearyS. E.ThompsonL. A.ReeveH. A.VincentK. A. (2021). Boosting the Productivity of H2-Driven Biocatalysis in a Commercial Hydrogenation Flow Reactor Using H2 from Water Electrolysis. Front. Chem. Eng.3, 35. 10.3389/FCENG.2021.718257
16
ReeveH. A.AshP. A.ParkH.HuangA.PosidiasM.TomlinsonC.et al (2017). Enzymes as Modular Catalysts for Redox Half-Reactions in H2-Powered Chemical Synthesis: From Biology to Technology. Biochem. J.474, 215–230. 10.1042/BCJ20160513
17
RoldánM. D.Pérez-ReinadoE.CastilloF.Moreno-ViviánC. (2008). Reduction of Polynitroaromatic Compounds: The Bacterial Nitroreductases. FEMS Microbiol. Rev.32, 474–500. 10.1111/J.1574-6976.2008.00107.X
18
RowbothamJ. S.RamirezM. A.LenzO.ReeveH. A.VincentK. A. (2020). Bringing Biocatalytic Deuteration into the Toolbox of Asymmetric Isotopic Labelling Techniques. Nat. Commun.11, 1454. 10.1038/s41467-020-15310-z
19
SargentF. (2016). The Model [NiFe]-Hydrogenases of Escherichia coli. Adv. Microb. Physiol.68, 433–507. 10.1016/BS.AMPBS.2016.02.008
20
ScholtissekA.TischlerD.WestphalA.van BerkelW.PaulC. (2017). Old Yellow Enzyme-Catalysed Asymmetric Hydrogenation: Linking Family Roots with Improved Catalysis. Catalysts7, 130. 10.3390/CATAL7050130
21
SheldonR. A. (2017). The E Factor 25 Years on: The Rise of Green Chemistry and Sustainability. Green Chem.19, 18–43. 10.1039/c6gc02157c
22
WeymanP. D.VargasW. A.ChuangR.-Y.ChangY.SmithH. O.XuQ. (2011). Heterologous expression of Alteromonas macleodii and Thiocapsa roseopersicina [NiFe] hydrogenases in Escherichia coli. Microbiology157, 1363–1374. 10.1099/mic.0.044834-0
23
ZhangP.XuS.WangY.ZhangW.LiW.WeiC.et al (2021). Fabrication of Pd/Mg2P2O7 via a Struvite-Template Way from Wastewater and Application as Chemoselective Catalyst in Hydrogenation of Nitroarenes. Chem. Eur. J.27, 10666–10676. 10.1002/CHEM.202100684
24
ZhangW.HollmannF. (2018). Nonconventional Regeneration of Redox Enzymes-A Practical Approach for Organic Synthesis?Chem. Commun.54, 7281–7289. 10.1039/c8cc02219d
25
ZubarV.DewanjiA.RuepingM. (2021). Chemoselective Hydrogenation of Nitroarenes Using an Air-Stable Base-Metal Catalyst. Org. Lett.23, 2742–2747. 10.1021/acs.orglett.1c00659
Summary
Keywords
biocatalytic hydrogenation, flavin recycling, nitro-reduction, hydrogenation, industrial biotechnology, nitroreductase, cofactor recycling, overexpression
Citation
Ramirez MA, Joseph Srinivasan S, Cleary SE, Todd PMT, Reeve HA and Vincent KA (2022) H2-Driven Reduction of Flavin by Hydrogenase Enables Cleaner Operation of Nitroreductases for Nitro-Group to Amine Reductions. Front. Catal. 2:906694. doi: 10.3389/fctls.2022.906694
Received
28 March 2022
Accepted
18 May 2022
Published
17 June 2022
Volume
2 - 2022
Edited by
Frank Hollmann, Delft University of Technology, Netherlands
Reviewed by
Dirk Tischler, Ruhr University Bochum, Germany
Santosh A Misal, National Institutes of Health (NIH), United States
Gao-Wei Zheng, East China University of Science and Technology, China
Updates
Copyright
© 2022 Ramirez, Joseph Srinivasan, Cleary, Todd, Reeve and Vincent.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah E. Cleary, sarah@hydregenoxford.com; Kylie A. Vincent, kylie.vincent@chem.ox.ac.uk
†Present Address: Miguel A. Ramirez, HydRegen, Centre for Innovation and Enterprise, Oxford, United KingdomSarah E. Cleary, HydRegen, Centre for Innovation and Enterprise, Oxford, United KingdomHolly A. Reeve, HydRegen, Centre for Innovation and Enterprise, Oxford, United Kingdom
‡These authors have contributed equally to this work
This article was submitted to Biocatalysis, a section of the journal Frontiers in Catalysis
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.