 Qinglong Meng
Qinglong Meng Carlos Ramírez-Palacios1,2
Carlos Ramírez-Palacios1,2 Hein J. Wijma
Hein J. Wijma Dick B. Janssen
Dick B. Janssen- 1Biotransformation and Biocatalysis, Groningen Biomolecular Sciences and Biotechnology Institute (GBB), University of Groningen, Groningen, Netherlands
- 2Molecular Dynamics, Groningen Biomolecular Sciences and Biotechnology Institute (GBB), University of Groningen, Groningen, Netherlands
Protein engineering is a powerful and widely applied tool for tailoring enzyme properties to meet application-specific requirements. An attractive group of biocatalysts are PLP-dependent amine transaminases which are capable of converting prochiral ketones to the corresponding chiral amines by asymmetric catalysis. The enzymes often display high enantioselectivity and accept various amine donors. Practical applications of these amine transaminases can be hampered by enzyme instability and by their limited substrate scope. Various strategies to improve robustness of amine transaminases and to redirect their substrate specificity have been explored, including directed evolution, rational design and computation-supported engineering. The approaches used and results obtained are reviewed in this paper, showing that different strategies can be used in a complementary manner and can expand the applicability of amine transaminases in biocatalysis.
1 Introduction
Transaminases are pyridoxal 5′-phosphate (PLP) dependent enzymes that catalyze the reversible transfer of an amino group from an amino donor (usually an amine or amino acid) to an amino acceptor (a ketone, aldehyde, or keto acid) (Scheme 1) (John, 1995). The transamination reaction follows a ping-pong bi-bi reaction mechanism that can be divided in two half-reactions, each consisting of several reversible chemical steps (Eliot and Kirsch, 2004). In the resting stage, the PLP cofactor is covalently bound to a conserved lysine residue via a Schiff base linkage. During the first reaction step, the linkage to the conserved lysine is replaced by a Schiff base linkage to the amino donor, forming the external aldimine intermediate. Via proton transfer steps the imine functionality is moved from the exocyclic carbonyl of the cofactor (C4' = N) to the amino donor carbon (Cα = N), yielding the ketimine intermediate. Following hydrolysis of the latter, the deaminated donor leaves as a carbonyl compound (a ketone, aldehyde, or keto acid) with formation of the PMP intermediate. In the second half-reaction this PMP intermediate reacts with the amino acceptor to first form a Schiff base linkage and the second ketimine intermediate. Subsequent steps in the second half-reaction are the reverse of the first half-reaction and lead to an external aldimine intermediate, which releases the aminated product during reformation of the internal aldimine by reaction with the conserved lysine. Thus, in the resting stage the PLP cofactor is covalently bound to the enzyme, but during the catalytic cycle it is covalently bound to the various forms of the substrate. The interaction between enzyme and cofactor during catalysis is based on hydrophobic packing of the PLP ring in a pocket surrounded by aromatic and other apolar groups, as well as by an electrostatic interaction between the pyridinium nitrogen and a conserved aspartate or glutamate in the enzyme and by binding of the PLP phosphate group in the so-called phosphate cup. The PLP cofactor is strongly bound to the enzyme via covalent or non-covalent interactions and stays in the active site throughout the catalytic cycle (Shin and Kim, 2002).
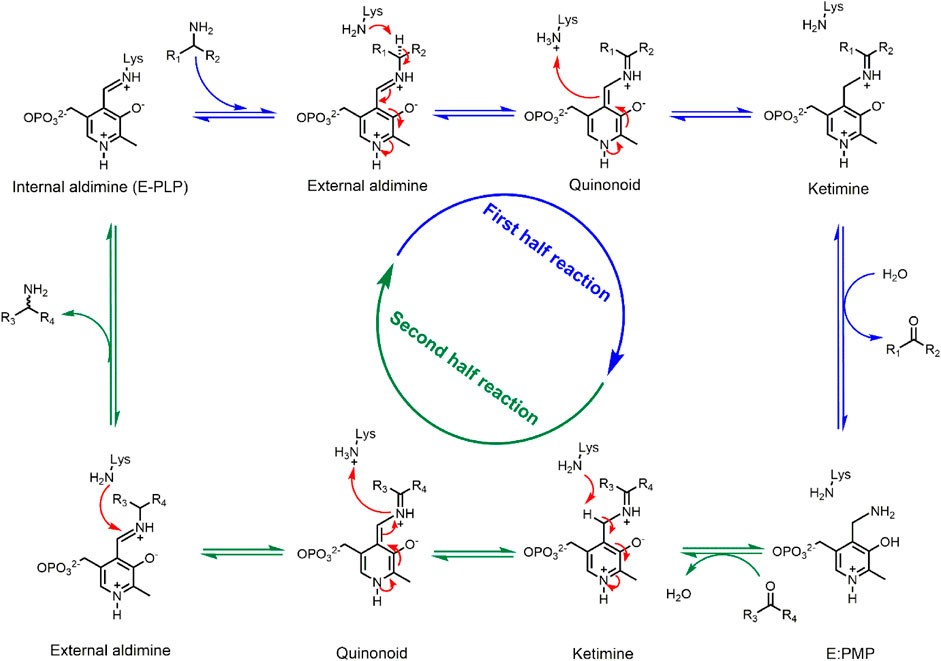
SCHEME 1. ATA-catalyzed transamination reaction. Blue arrows indicate the first half-reaction, green arrows indicate the second half-reaction. All reaction steps are reversible.
Transaminases have been grouped into six classes according to catalytic properties: L-aspartate transaminases (class I), L-alanine transaminases (class II), ω-transaminases (class III), D-amino acid transaminases and branched chain transaminases (class IV), L-serine transaminases (class V) and sugar transaminases (class VI) (Mehta et al., 1993). This grouping is somewhat confusing because the activity of these enzymes only partly overlaps with sequence- and structure-based classification. The latter classification correlates with grouping by PLP-enzyme fold type (Percudani and Peracchi, 2009). Most transaminases of class I, II, III and V belong to the fold-type I enzymes, which include the classical aspartate transaminase and several enzymes well explored in applied biocatalysis, such as the Vibrio fluvialis ω-transaminase (VfTA) (Shin and Kim, 1999). The fold-type I class-III ω-transaminases encompass different subgroups of enzymes, including amine transaminases (ATAs) and β-transaminases (Kelly et al., 2018). On the other hand, the class IV transaminases have a very different structure and belong to the group of PLP fold-type IV enzymes. The fold-type IV transaminases include the well-known D-alanine transaminase and several sequence-related ω-transaminases, including the enzyme from Arthrobacter sp. (ATA-117), the enzyme that was engineered for sitagliptin manufacture (Savile et al., 2010).
Transaminases that do not require the substrate to have a carboxylate group are of particular interest for the synthesis of chiral amines from prochiral ketones as the amino acceptor. These ATAs can use cheap or recyclable amino donors such as isopropylamine or alanine in the preparation of chiral amines via asymmetric synthesis from ketones (Shin and Kim, 2002). Many ATAs can produce chiral amines with high enantiomeric excess due to differences between the binding pockets that accommodate the substituents on the carbonyl carbon of a ketone acceptor (Shin and Kim, 2002). The high enantioselectivity makes ATAs attractive biocatalysts for the synthesis of pharmaceutical ingredients, agrochemicals and other fine chemicals where enantiopurity is desirable to avoid exposure to non-functional stereoisomers in the final product (Ghislieri and Turner, 2014). Another attractive point of ATAs is the existence of enantiocomplementary enzymes, i.e., ATAs that preferentially produce the (S) or (R)-amine. Enantiopreference in ATAs is mainly based on steric constraints, but the bulkier substituent usually has the highest CIP priority. Accordingly, fold-type IV ATAs accept D-alanine as donor and form (R)-amines, while L-alanine is used by fold-type I ATAs to synthesize (S)-enantiomers of chiral amines (Percudani and Peracchi, 2009). Most fold-type I (S)-selective ATAs are homodimeric or homotetrameric whereas fold-type IV enzymes usually are (R)-selective and homotetrameric. In both groups of transaminases, the active site is formed by residues from different subunits (Shin and Kim, 2002).
In view of the promising potential of transaminases in industrial amine synthesis, the catalytic properties of ATAs have been intensively studied on a wide range of amination reactions (Koszelewski et al., 2010; Mathew and Yun, 2012; Ferrandi and Monti, 2018; Gomm and O’Reilly, 2018; Patil et al., 2018). The results suggest that while biocatalytic transamination could be highly attractive, obstacles for its practical application are the poor stability of ATAs under harsh reaction conditions such as high temperature, the low tolerance to cosolvents, and the sensitivity to high substrate concentrations (Guo and Berglund, 2017). Different explanations for the observed instability of ATAs have been proposed. Börner et al. (2017b) found that the release from the binding site of the aminated cofactor PMP formed in the first half-reaction can trigger irreversible denaturation and consequently loss of activity. Subunit dissociation in multimeric ATAs could be another cause of inactivation, leading to local unfolding, cofactor loss, exposure of hydrophobic groups, and protein aggregation (Fernandez-Lafuente, 2009). Additionally, the limited substrate scope of ATAs is another obstacle in its practical application. In the dimeric class III ATAs, the active site is located at the interface between the monomers and usually comprises a distinct large and a small binding pocket (Shin and Kim, 2002). When a ketone or keto acid binds to the enzyme, a bulky substituent such as a phenyl group can only be accepted in the large binding pocket. Consequently, bulky amines bearing two large substituents cannot be produced by most native ATAs.
To address the challenges of stability and substrate scope that hamper the application of ATAs in industry, protein engineering efforts have been attempted. Protein engineering mainly follows one of three strategies: directed evolution, structure-based rational redesign, and computational redesign (Bornscheuer and Pohl, 2001). The availability of a protein structure and some knowledge on the structure-activity relationship promote rational design, while the existence of efficient screening methods with sufficient throughput promotes directed evolution (Slabu et al., 2017; Kelly et al., 2018; Rocha et al., 2022). In this review, various engineering strategies to improve the properties of ATAs along with their most prominent results are discussed, making emphasis on the interplay between different approaches and the exploitation of structural information.
2 Stabilization of transaminases
2.1 Enzyme stability
High enzyme robustness under process conditions and reusability over multiple catalytic cycles are a prerequisite for the industrial use of ATAs (Tufvesson et al., 2011; Bornscheuer et al., 2012). Additionally, desirable aspects relevant to the operational stability of ATAs are tolerance to high concentrations of reagents and products, compatibility with cosolvents used to increase substrate solubility, activity at elevated temperatures that enhance reaction rates, and compatibility with storage, immobilization, and recycling methods (Guo and Berglund, 2017). The necessary operational stability requirements can be satisfied by more stable variants discovered through microbiological screening and genome mining (Mathew et al., 2016), or by suppressing the effect of harsh process conditions through enzyme immobilization on solid supports (De Souza et al., 2016) and medium engineering (Chen et al., 2016). However, discovering new ATA variants that meet all aspects of operational stability is not always successful, and enzyme immobilization or medium engineering has a limited impact on improving stability. Protein engineering is a preferred strategy to increase enzyme stability as it allows to adapt an existing ATA to better suit process considerations. An overview of protein engineering strategies for ATA stabilization is provided in Table 1.
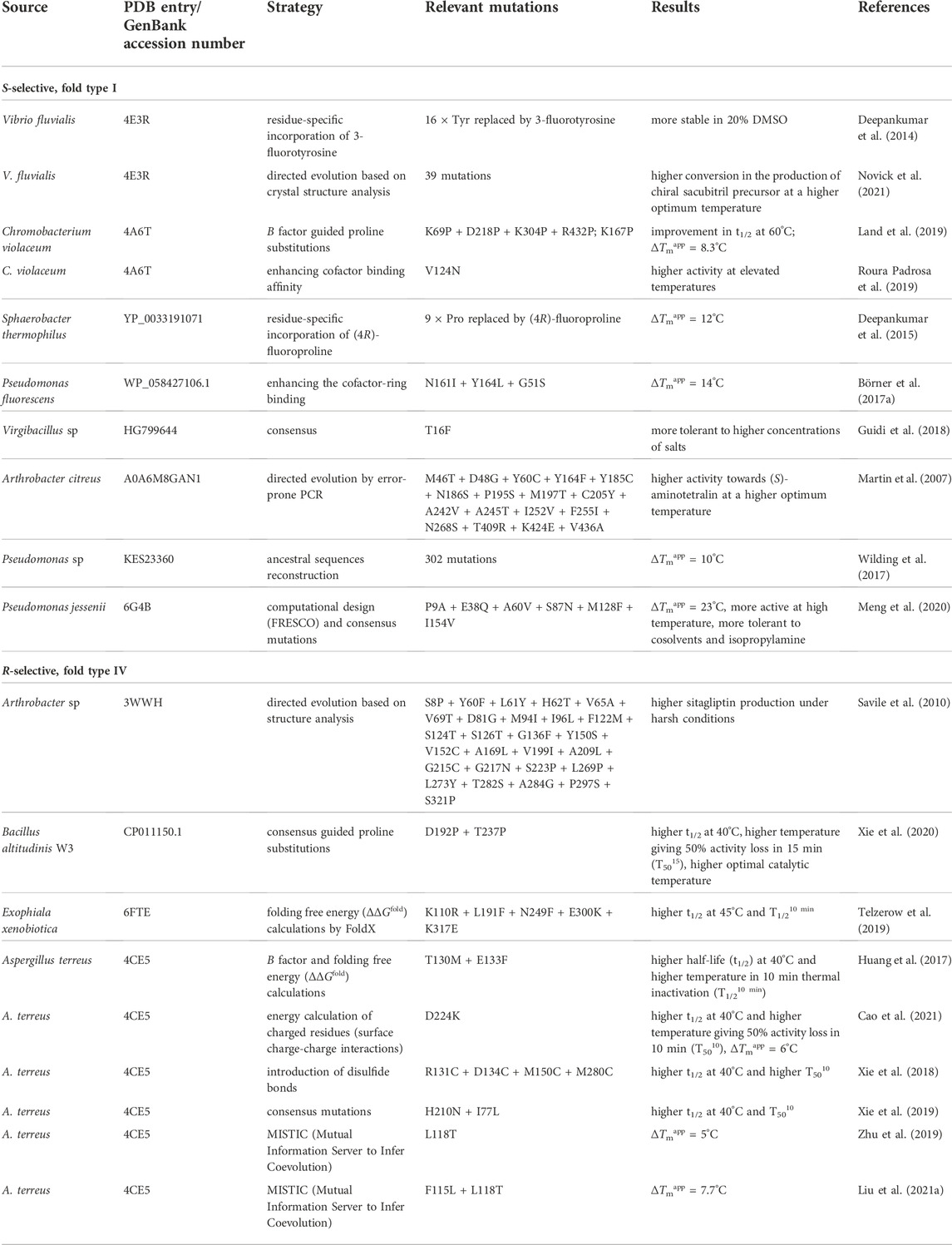
TABLE 1. Stabilization of ATAs by protein engineering.
2.2 Directed evolution
Directed evolution of enzymes involves multiple iterations of a four-step cycle: generation of a library of mutants with sufficient genetic diversity, gene expression in a suitable microbial host, screening of the library to discover improved variants, and sequencing to identify the beneficial mutations. This is a powerful strategy for enzyme stabilization, especially because no knowledge of the protein structure or the unfolding mechanism is needed (Kuchner and Arnold, 1997), and it has been used to engineer several ATAs (Turner, 2009). For example, a robust variant of (S)-selective ATA from Arthrobacter citreus (AcTA) was obtained after five rounds of error-prone PCR with screening at elevated temperature (30°C–60°C). The resulting enzyme carries 17 mutations (Table 1) and showed a 260-fold enhanced activity in the synthesis of (S)-aminotetralin at its higher optimum temperature (55°C) (Martin et al., 2007). Directed evolution can also be used to broaden the substrate scope. An (R)-selective ATA from Arthrobacter sp. (ATA-117) was engineered by 11 rounds of evolution with screening under challenging conditions (higher temperature, higher concentration of cosolvent, isopropylamine as donor, structurally demanding substrate) and a variant with 27 mutations was obtained (Table 1) (Savile et al., 2010). This robust variant produced a sitagliptin from prositagliptin ketone (yield 92%, e.e. > 99.95%) under harsh reaction conditions (45°C, 1 M isopropylamine as the amino donor, 200 g/L substrate, 50% DMSO as cosolvent). More details on the outstanding engineering campaign aimed at the production of sitagliptin, which is used for the treatment of diabetes mellitus type 2, are described below in the Expanding the substrate scope section. Recently, Novick et al. (2021) employed 11 rounds of directed evolution on the (S)-selective VfTA to improve the production of a chiral precursor of sacubitril (Table 1). The final variant displayed 90% conversion of 75 g/L substrate at an elevated optimal reaction temperature (58°C). Further information on strategies for substrate selectivity engineering is given below.
2.3 Phylogenetic approaches, consensus design, and ancestral sequence reconstruction
When high-throughput screening is not available, finding improved enzyme variants through random mutagenesis and experimental screening may be time- and resource-consuming due to the need for multiple iterations of mutagenesis and testing. Using sequence and structure information to guide the design of libraries can reduce the number of iterations and size of the required libraries. The notion that evolution favors the conservation of residues that are important for function and stability has led to the exploration of strategies using sequence alignments to improve protein stability (Steipe et al., 1994). Consensus mutagenesis employs multiple sequence alignments of homologous proteins to discover positions where the target protein deviates from the consensus sequence (Steipe et al., 1994). The alignments are used to calculate position-specific conservation scores and to derive phylogenetic trees (Thornton et al., 2003). Introduction of mutations that replace residues deviating from consensus by more common residues can improve stability (Nikolova et al., 1998).
The consensus design strategy was employed to improve the stability of the class III (R)-selective ATA from Aspergillus terreus (AtTA). The Consensus Finder tool (Jones et al., 2017) predicted towards-consensus mutations from alignments of 90 AtTA homologs retrieved from the NCBI non-redundant protein sequence database (Xie et al., 2019). Six substitutions towards the most prevalent amino acids were identified (I77L, Q97E and N245D in surface α-helices, H210N, G292D and I295V in surface loop regions). The most stable single mutant was H210N which showed a 3.3-fold increase in half-life at 40°C and a 4.6°C increase in the T5010 value, the temperature at which the enzyme loses 50% of its activity during 10 min of heat treatment. A combination of two single mutations (H210N + I77L) further stabilized AtTA, displaying a 6.1-fold increase in half-life at 40°C, and a 6.6°C increase in T5010. The stabilizing effect of mutation H210N was attributed to formation of a new hydrogen bond between Asn210 and Asp208. Mutation I77L likely reduces the energy of the folded state in comparison to wild-type, suggesting better interaction with adjacent residues.
The consensus approach was also employed to stabilize the (S)-selective fold-type I ATA from Virgibacillus sp. (Guidi et al., 2018). Residue- and position-specific conservation scores were calculated with the program ConSurf (Glaser et al., 2003), whereby positions with a high degree of conservation were considered to be important for catalytic function or stability. The alignment of the ATA with around 500 homologous sequences revealed that Phe16 was conserved among other ATAs, instead of the Thr present in the target enzyme. Thus, a single mutation T16F was introduced, producing a variant more stable in the presence of polar organic solvents such as DMSO and methanol and also tolerant to the presence of high concentrations of salts (KCl and NaCl). The crystal structure of the T16F variant (PDB 6FYQ) revealed that Phe16 facilitates dimerization, which is beneficial for the stability of the enzyme.
Xie et al. (2020) applied consensus design in combination with introduction of prolines at target positions to improve the stability of the (R)-selective ATA from Bacillus altitudinis W3. Based on sequence alignments between the wild-type enzyme and four potentially thermophilic transaminases, five residues were targeted for proline substitution because the expected rigidification of the protein can contribute to stability (Watanabe and Suzuki, 1998). The best variant D192P + T237P exhibited a 2.5-fold improvement in half-life at 40°C, a 6.3°C increase in T5015 (temperature at which the enzyme loses 50% of its activity during a 15 min heat treatment), and a 5°C increase in its optimal reaction temperature. The modeled structure of the variant D192P + T237P revealed that the improved stability could be attributed to a newly formed hydrogen bond between Pro192 and Gly189. Moreover, newly formed hydrophobic interactions including Pro192 with Leu249, Pro192 with Ala254, and Pro237 with Phe236 could also contribute to stability.
Phylogenetic information obtained via multiple sequence alignments is at the basis of ancestral sequence reconstruction (ASR). ASR can be used to infer the sequence of evolutionary ancient proteins, which are expected to have higher stabilities (Wheeler et al., 2016). Deviating residues in an extant protein are then replaced via site-directed mutagenesis, enhancing protein stability (Watanabe et al., 2006). ASR was applied this way to obtain a stable homolog of the (S)-selective ATA from Pseudomonas sp. (PsTA) (Wilding et al., 2017). The ancestral sequence was constructed based on the protein sequences of PsTA and the 192 closest homologs. From the entire phylogenetic tree, a subtree containing the sequences of PsTA and 47 other proteins was examined, and six nodes (67% average sequence identity with PsTA) were selected for gene synthesis. This gave variants with up to 10°C improvement in apparent melting temperature (Tmapp).
A similar phylogenetic approach makes use of the notion that functionally relevant residues may coevolve. A protein can be considered as a network of interacting residues, which can be seen as nodes in a graph (Aguilar et al., 2012). Links between residues (nodes) create a residue-residue coevolution network. Residues are said to coevolve when a mutation at one residue is accompanied by a mutation at an interacting residue (Dietrich et al., 2012). The Mutual Information Server To Infer Coevolution (MISTIC) (Simonetti et al., 2013) can be used to graphically represent coevolution information for protein families from multiple sequence alignments. Graphical representation of coevolution information can help identify coevolving residues to guide mutagenesis efforts. Zhu et al. used MISTIC to select positions where mutations might influence the stability of AtTA (Zhu et al., 2019). The eight positions identified from the MISTIC representation were examined by alanine scanning mutagenesis, from which mutant L118A was discovered to have an increased stability. Saturation mutagenesis at position 118 produced three extra stabilizing mutations (L118T, L118I and L118V). The best variant (L118T, ∆Tmapp = 5°C) exhibited the highest catalytic efficiency (kcat/KM) towards (R)-1-phenylethylamine. To further increase the stability of variant L118T, three residues (Phe115, Leu181 and Trp184) that have strong interactions with Leu118 in the coevolution network were selected for saturation mutagenesis (Liu C. Y. et al., 2021). A double mutant F115L + L118T was found to have higher stability (∆Tmapp = 7.7°C) than the single mutant L118T. Molecular dynamics (MD) simulations confirmed that the overall flexibility of mutant F115L + L118T was decreased compared with the wild-type AtTA and mutant L118T.
2.4 Rational design
The availability of extensive structure and sequence information on transaminases makes rational design for improving stability an attractive option. Inspection of 3D structures is useful to identify regions with suboptimal interactions or destabilizing features and, thus, detect positions to introduce stabilizing mutations. An established strategy to guide the introduction of stabilizing mutations is to promote the biophysical interactions that contribute to folding energy the energy. Some of the factors that contribute to protein stability are well known, e.g. close packing of hydrophobic side chains (Vlassi et al., 1999), the lack of unsatisfied hydrogen bond donors or acceptors, the presence of H-bonds and salt bridges between polar and charged groups in apolar environments (Marqusee and Sauer, 1994), entropic stabilization by residues with restricted conformational freedom in unfolded states (Berezovsky et al., 2005), the presence of polar and charged surface residues (Pokkuluri et al., 2002), and the presence of disulfide bonds (Betz, 1993). Strong binding of ligands also can prevent unfolding (Niesen et al., 2007). Rational design of more stable enzymes can also be based on an analysis of the process of unfolding, targeting kinetic stabilization by increasing stability the native conformation of early unfolding regions (Johnson et al., 2005). Reversible processes like local unfolding, subunit dissociation, or cofactor loss may initiate irreversible inactivation due to further unfolding, exposure of hydrophobic regions and their aggregation, or by (self)proteolytic cleavage (Fernandez-Lafuente, 2009; Börner et al., 2017b). Reducing the propensity to form conformations susceptible to irreversible inactivation therefore can contribute to improved stability.
2.5 Preventing cofactor loss
An important cause of inactivation in ATAs is the loss of the aminated cofactor (PMP) formed in the first half-reaction. Inactivation following cofactor loss has been well documented in PLP fold-type I ATAs (Börner et al., 2017b) and is likely to become especially relevant when high amino donor concentrations are used to shift the reaction equilibrium to product formation, which causes relatively high levels of the PMP-form of the enzyme, especially in case of a poor amino acceptor. To design stabilized variants of a tetrameric (S)-selective ATA from Pseudomonas fluorescens, the PLP binding site was explored and in total 32 residues around the active site, including residues forming the PLP pyridinium ring binding motif, were replaced by mutagenesis (Börner et al., 2017a). After screening, two robust variants (ATA-v1, mutations N161I + Y164L, ∆Tmapp = 11°C; ATA-v2, mutations N161I + Y164L + G51S, ∆Tmapp = 14°C) were obtained. The mutations N161I and Y164L stabilize the PLP-ring binding motif through hydrophobic contacts, and mutation Y164L anchors the PLP cofactor by restricting rotation of the hydroxyphenyl side chain of the conserved residue Tyr148, which interacts with the pyridine ring. The mutation G51S could increase stability by constraining loops close to the surface of the protein.
Mutations not directly contacting the PLP group may still reduce inactivation of ATAs by indirect effects. Roura Padrosa et al. (2019) found that replacing the Val in position 124 by an asparagine in the ATA from C. violaceum (CvTA) contributed to improved PLP binding even though the position is not part of the PLP-ring binding motif or phosphate binding cup. Mutant V124N showed higher stability than wild-type CvTA at elevated temperatures (35°C–65°C). The introduced asparagine is suitably positioned to establish a hydrogen bond with the conserved residue Asp259, which interacts with the PLP pyridinium nitrogen. These interactions could contribute to reduced loss of PMP from the intermediate formed in the first half-reaction, of which the importance was described by Börner et al. (2017b).
2.6 B factors
Crystallographic B factors provide an experimental measure of the protein dynamics in the crystal state, where residues with high B factors are more subject to thermal fluctuations or functional motions (Reetz et al., 2006; Augustyniak et al., 2012). Thus, regions in a protein structure with high B factors may indicate high sensitivity to local unfolding. If allowed by geometric constraints, introduction of a proline in a flexible region of a protein can be beneficial for stability (Watanabe and Suzuki, 1998). Analysis of B factors to improve thermostability was applied to several enzymes including CvTA (Land et al., 2019), whose half-life at 60°C was increased by 2.7-fold by the mutations K69P + D218P + K304P + R432P. Moreover, the single mutation K167P increased the Tmapp by 8.3°C. Intriguingly, mutation K69P is located at the interface of the two subunits, suggesting the mutation may play a role in preventing subunit dissociation.
2.7 Introduction of non-canonical amino acids
Introduction of non-canonical amino acids can be used to improve enzyme stability (Steiner et al., 2008). For example, incorporation of fluorinated amino acids produces minimal local changes in the protein structure yet may have large effects on global properties by influencing bond energies, charge distribution, hydrogen bonding, and steric interactions (Merkel and Budisa, 2012). When all 16 tyrosine residues of the (S)-selective VfTA were substituted with 3-fluorotyrosine, the resulting enzyme was more resistant to the presence of 20% DMSO giving a 2-fold higher production of (S)-1-phenylethylamine (Deepankumar et al., 2014). Similarly, substitution of 9 proline residues with (4R)-fluoroproline in an (S)-selective ATA from Sphaerobacter thermophilus resulted in an increase in Tmapp of 12°C (Deepankumar et al., 2015).
2.8 Disulfide bonds
The introduction of disulfide bonds is a widely applied strategy for protein stabilization (Betz, 1993). Finding positions where disulfide-bond forming cysteines may be introduced can be facilitated by algorithms such as Disulfide by Design (Craig and Dombkowski, 2013) and MODIP (Sowdhamini et al., 1989), which use geometric criteria to find suitable positions. Disulfide by Design can also incorporate B factor analysis to support design of disulfide bonds in flexible regions. Using both algorithms, disulfide bonds were introduced in AtTA to improve its thermostability (Xie et al., 2018). Among the seven mutants predicted, three particularly stable mutants were obtained (N25C + A28C, R131C + D134C, and M150C + M280C). The combination of two of the disulfide bonds (R131C + D134C and M150C + M280C) gave the highest stability improvement with a 4.6-fold increase in half-life (t1/2) at 40°C and a 5.5°C increase in T5010. MD simulations of the designed mutants showed a reduced flexibility of the surface loops when compared with the wild-type AtTA.
2.9 Computational design
The use of computational methods to improve enzyme stability has been motivated by an improvement of molecular mechanics methods to calculate energy changes upon mutations and by the development of more efficient search algorithms to try to find the energy minima of a given sequence (Kiss et al., 2013). For instance, energy calculations were used to identify mutations that increase the stability of AtTA (Huang et al., 2017). In this work, FoldX (Guerois et al., 2002) was used to calculate the effect of mutations in a stretch of six residues (from Gly129 to Asp134) of AtTA on the folding free energy (ΔΔG fold) of the enzyme. Mutations that led to a ΔΔG fold < 0 were selected for experimental verification. From the total of 19 potentially stabilizing mutants predicted, four stabilized mutants (T130M, T130F, E133F and D134L) were obtained. The best single mutant (T130M) displayed a 2.2-fold improvement of t1/2 at 40°C and increased the T1/210 min (defined as the temperature at which enzyme activity is reduced to 50% upon 10 min of incubation) by 3.5°C. The modeled structures of the four stabilized mutants showed that the mutations could introduce additional hydrophobic interactions with adjacent residues. After combination of the best single mutants, a double mutant (T130M + E133F) was constructed and shown to further increase the stability with a 3.3-fold improvement of half-life at 40°C and a 5°C higher T1/210 min.
The stability of AtTA was also improved via energy calculation of chargeable residues using a computational approach called enzyme thermal stability system (ETSS) (Cao et al., 2021). It is used for the analysis of surface charge-charge interaction using TK-SA model calculations (Zhang et al., 2014). The Tanford-Kirkwood (TK) model is to represent the electrostatic properties of the whole protein, and the introduction of solvent accessibility (SA) is used to refine the model. After analyzing surface charge-charge interactions of AtTA, four surface residues were targeted for mutagenesis. To switch the charge of residues to opposite or neutral, 13 mutants were tested and three of them (E133Q, D224K, and E253A) indeed displayed higher stability. Interestingly, residue Glu133 was also targeted in a stabilization study using B factor analysis and folding energy calculations, and mutation E133F indeed gave higher stability (Huang et al., 2017). The best mutant D224K showed a 4.2-fold increase in t1/2 at 40°C and a 6°C increase in both T5010 and Tmapp in comparison to the wild-type AtTA. Mutant D224K was studied by MD simulations and it was concluded that the new hydrogen bond interactions reduced the flexibility of α-helices 8 and 9, which greatly improved enzyme stability.
FoldX calculations were used in the stabilization of the (R)-selective fold-type IV ATA from Exophiala xenobiotica (ExTA) (Telzerow et al., 2019). The most stable variant (K110R + L191F + N249F + E300K + K317E) had a three times longer thermal inactivation half-life (t1/2) at 45°C and a 4.4°C improvement in T1/210 min. The increased stability of ExTA by the surface mutations K110R, N249F, E300K, and the interface mutation L191F could be attributed to the introduction of new cation-π interactions, while mutation K317E could have introduced an additional ionic interaction.
2.10 FRESCO
Our group developed a computational workflow called FRESCO (framework for rapid enzyme stabilization by computational libraries) for enzyme stability engineering (Figure 1) (Wijma et al., 2018). So far, nine different enzymes including an ATA from Pseudomonas jessenii (PjTA) have been successfully stabilized using FRESCO (Floor et al., 2014; Wijma et al., 2014; Wu et al., 2016; Arabnejad et al., 2017; Bu et al., 2018; Martin et al., 2018; Fürst et al., 2019; Aalbers et al., 2020; Meng et al., 2020). Briefly, potentially stabilizing mutations are identified by calculating the effect on folding energy of all possible point mutations, using Rosetta (Richter et al., 2011) and/or FoldX (Guerois et al., 2002). Mutations that are possibly stabilizing are examined by rapid short MD simulations, where mutations causing local instability are dismissed, and by visual inspection. These steps define a small library of promising variants that qualify for experimental verification.
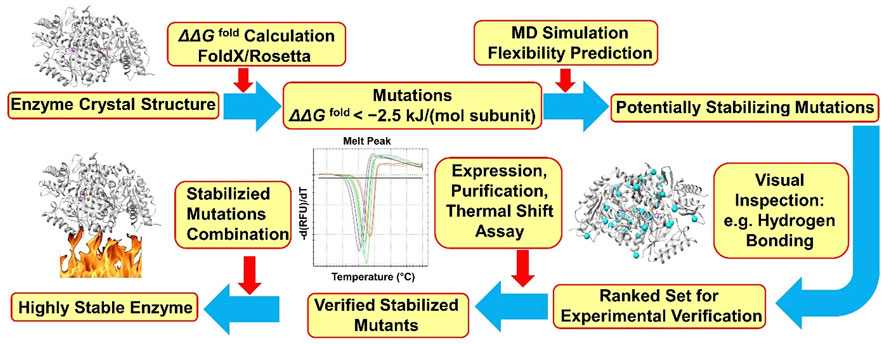
FIGURE 1. FRESCO workflow for PjTA stabilization. The protocol is described in detail by Wijma et al. (2018).
When applied to the fold-type I class III transaminase PjTA, 29 stabilizing single point mutations were discovered (∆Tm app ≥ 1°C) by FRESCO and two robust variants (PjTA-R4, ∆Tmapp = 18°C, and PjTA-R6, ∆Tmapp = 23°C) with four mutations and six mutations, respectively, were obtained by combining the most beneficial substitutions. These two stabilized variants were more active at high temperatures and more tolerant to cosolvents and to a high concentration of isopropylamine. Under harsh conditions (56°C, 20% DMSO and 1 M isopropylamine) with variant PjTA-R6, the analytical yield of (S)-1-phenylethylamine from acetophenone (100 mM) was 92% (e.e. > 99%). It is noticeable that an exceptionally high success rate (56%) was found for mutations that were predicted to stabilize the subunit interface, which indicated that preventing subunit dissociation was crucial for PjTA stability. Furthermore, the consensus approach contributed a stabilizing mutation in PjTA (A60V, ∆Tmapp = 4°C) that was not discovered by energy calculations yet displayed an additive effect on ∆Tmapp when combined into the final robust variant PjTA-R6. This is an example that shows that predictions by energy calculations and consensus analysis do not fully overlap.
Xie et al. (2019) also demonstrated that it is possible to enhance transaminase stability by combining individual stabilizing mutations. By combining the best mutations of AtTA obtained from consensus analysis (H210N + I77L) and introducing a disulfide bond (M150C + M280C) a more stable variant was obtained. The improved enzyme showed a 16.6-fold increase in t1/2, an 11.8°C higher T5010 and a 2.8-fold increase of the catalytic efficiency (kcat/KM) with (R)-1-phenylethylamine.
2.11 Summary and perspectives of stability engineering
Different protein engineering strategies, often supported by sequence alignment and structural information, have been used to stabilize ATAs with many successful examples. These strategies include directed evolution, consensus design, rational design, and computational design, and are increasingly being used in combination with one another (Huang et al., 2017; Xie et al., 2019; Meng et al., 2020). Several examples show that mutations that improve stability can be predicted and/or explained by known biophysical principles of protein stability. These stabilizing principles are diverse but often affect local events (e.g. unfolding of flexible regions, subunit- or cofactor dissociation) that are followed by irreversible events (aggregation).
The results also indicate that some stabilizing mutations will be missed if only a single approach is used. For instance, AtTA was stabilized via five different approaches including consensus design, rational design and computational design, and it appeared that the stabilizing mutations obtained from each approach were mostly different (Table 1). Combining one or more strategies may be more efficient in finding stabilized mutants, e.g. using both consensus and computational design (folding energy calculations) (Meng et al., 2020), or both consensus and rational design (introduction of disulfide bonds) (Xie et al., 2019). Machine learning is increasingly integrated in protein engineering; it predicts structures and effects of mutations via processing a large amount of experimental data (Yang et al., 2019; Mazurenko et al., 2020). This potentially improves prediction accuracies and was reported to predict thermostabilities of variants of AtTA obtained previously by consensus analysis (Xie et al., 2019; Jia L. L. et al., 2021). However, at present there are no reports of stability improvement of ATAs via machine learning methods that match the achievements of directed evolution and structure-based strategies discussed above.
3 Expanding the substrate scope
3.1 Steric hindrance
The substrate range of ATAs is limited by steric hindrance in the active site binding pockets (Shin and Kim, 2002). Genome mining, screening of microbial cultures, and protein engineering are commonly employed to obtain ATAs with a desired substrate range, both for application in kinetic resolution of racemic mixtures and for asymmetric synthesis by amination of ketones (Koszelewski et al., 2010). Different protein engineering strategies have been applied to widen the substrate scope and allow acceptance of non-natural compounds for the preparation of chiral amines, non-proteinogenic amino acids and chiral amino alcohols (Table 2). Chemicals accepted as substrates or produced by engineered ATAs are summarized in Figure 2.
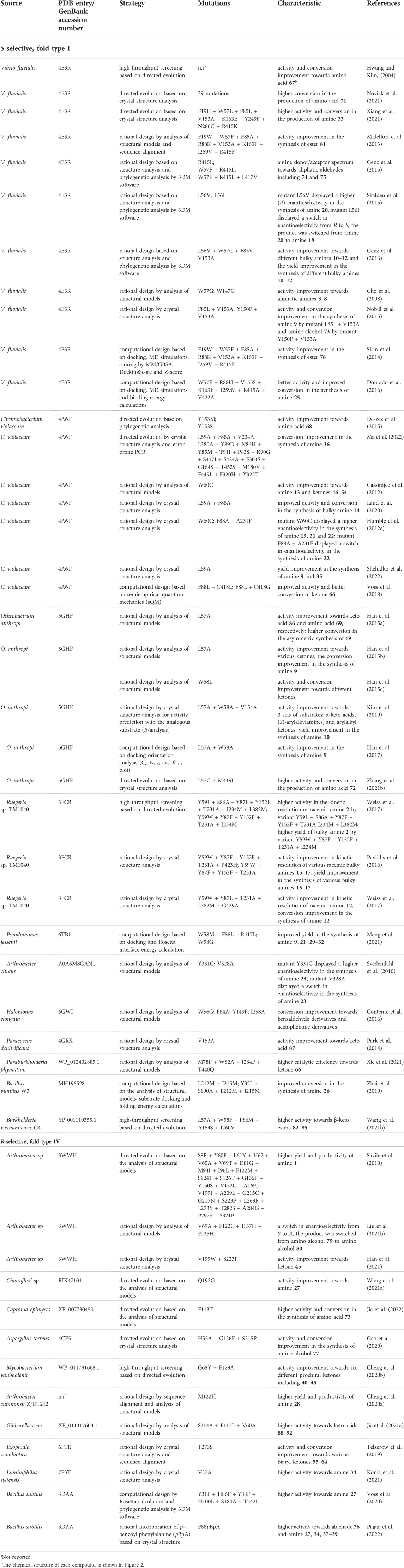
TABLE 2. Protein engineering of ATAs for improved activity or enantioselectivity.
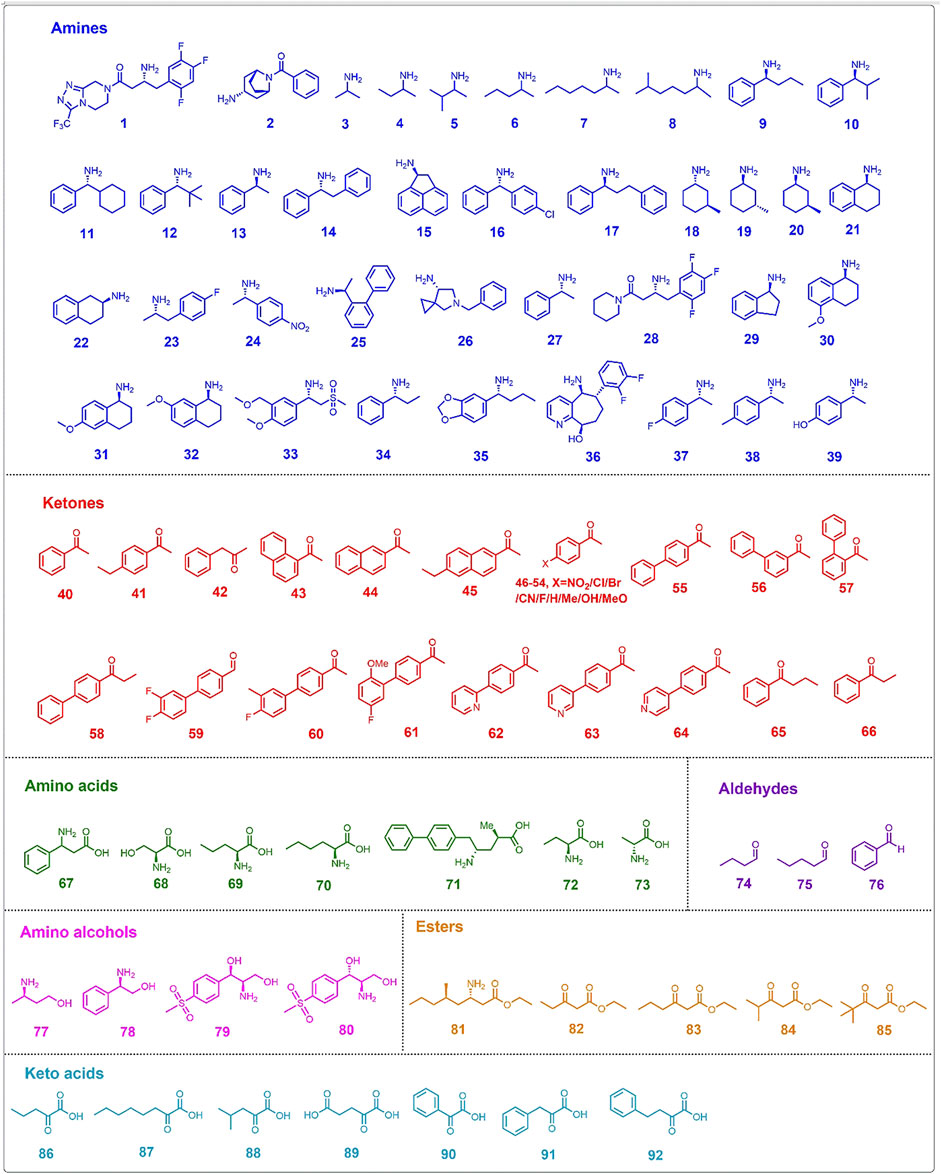
FIGURE 2. Compounds converted or produced by engineered ATAs.
3.2 Directed evolution with high-throughput screening
Directed evolution is a well-stablished approach in protein engineering, widely used to expand the substrate scope of ATAs. For efficient screening in directed evolution, several high-throughput methods have been developed, most often based on conversion-associated changes in fluorescence or color (Figure 3) (Hwang and Kim, 2004; Green et al., 2014; Weiss et al., 2014; Deszcz et al., 2015; Cheng et al., 2020b). For example, a blue complex is formed in the reaction of a CuSO4/MeOH solution with an α-amino acid, allowing spectrophotometric detection at 595 nm of the α-amino acid in 96-well microplates (Figure 3D). This detection method was used to screen an error-prone directed evolution library of VfTA for variants with improved activity towards amino acid 67 (Figure 2), which gave an improved enzyme with 3-fold higher activity than the wild-type enzyme (Hwang and Kim, 2004).
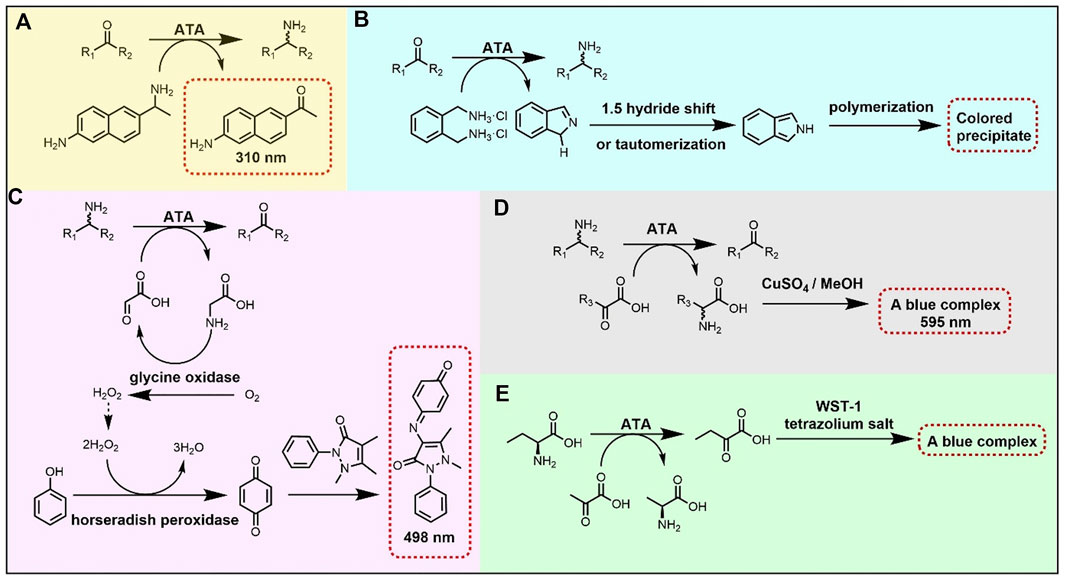
FIGURE 3. High-throughput methods for screening activity of ATAs. (A) Ketone conversion assay based on formation of a fluorescent product from the amino donor 6-amino-α-methyl-2-naphthalenemethanamine. (B) Ketone conversion assay based on deamination of the donor ortho-xylylenediamine and further conversion to a polymeric precipitate. (C) Coupled assay for amine conversion based on detection of glycine with glycine oxidase and horseradish peroxidase. (D) Amine conversion assay based on formation of a blue complex of an amino acid with copper. (E) Amino acid conversion assay based on detection of ketoacid formation with a tetrazolium salt.
Another high-throughput method to detect transaminase activity is based on the conversion of the amino donor 6-amino-α-methyl-2-naphthalenemethanamine to 2-acetyl-6-aminonaphthalene, which gives a strong fluorescence (Figure 3A). Since the assay screens for conversion of the amino donor, it can be used to measure amination of various ketones. This fluorogenic screening assay was used during the directed evolution of an (R)-selective ATA from Mycobacterium vanbaalenii, which led to the discovery of mutants with improved activity toward ketones 40-45 (Figure 2) (Cheng et al., 2020b). The work followed a knowledge-gaining strategy, where each position in the sequence was tested by saturation mutagenesis until a beneficial mutation was discovered at that position. The variant G68Y + F129A showed a 10–100-fold higher activity in the amination of the six ketones. Ala129 created more space in the small binding pocket, and Tyr68 caused new π-π interactions with the substrate.
The directed evolution of the (S)-selective ATA from Ruegeria sp. TM1040 to obtain activity with the bulky amine 2 (Figure 2) included a high-throughput screening assay that employed glycine oxidase and horseradish peroxidase (Weiss et al., 2014, 2016). The bicyclic compound 2 is challenging because of the bridged rings, and cannot be synthesized with the wild-type enzyme. Mutant Y59W + T231A was found in a previous study to be active with various bulky amines (Pavlidis et al., 2016) and was taken as the starting point to search for beneficial mutations by error-prone PCR. In the screening assay, amine 2 and glyoxylate react to form glycine which is used by glycine oxidase to generate hydrogen peroxide. Hydrogen peroxide is consumed by horseradish peroxidase (Figure 3C) to form benzoquinone which produces a red quinone imine dye (498 nm) by condensation. Using this assay, a 7-fold mutant (Table 2) having a high activity in the kinetic resolution of racemic amine 2 was finally obtained. Another variant (Y59W + Y87F + Y152F + T231A + I234M) gave 75% conversion of the corresponding ketone to amine (e.e. > 99.5%) with the use of isopropylamine as the amino donor.
Wang Y. et al. (2021) performed directed evolution on an (S)-selective ω-transaminase from Burkholderia vietnamiensis G4 (HBV) to aminated β-keto esters 82–85 (Figure 2), which was not possible with available ω-transaminases. A homology model of the wild-type HBV was created using the crystal structures of VfTA and an ω-transaminase from Paracococcus denitrificans with which it shares ca. 51% sequence identity. Residues within 5 Å from the active site were individually subjected to saturation mutagenesis. The mutant libraries were screened with a colorimetric assay (Figure 3B) (Green et al., 2014), in which an intensively colored polymer is formed in the reaction with ortho-xylylenediamine acting as the amino donor in the ATA reaction. The cyclic imine formed after deamination of ortho-xylylenediamine reorganizes into a stable isoindole which forms a colored polymer. Single hits from the mutant library were combined and a final variant bearing five mutations was obtained (Table 2). The five mutations created more space in the binding pockets, thereby improving the activity with the four β-keto esters (82–85).
3.3 Structure-inspired directed evolution
Directed evolution can be made more efficient by using structural information to guide library design (Arnold, 2018). Examination of structural models of enzyme-substrate complexes can reveal positions in the protein structure that likely influence substrate binding or reactivity by improving steric or electronic interactions. The goal is to increase the number of beneficial mutations in a library, thereby reducing the size of the search space required to find good variants.
A prominent example of structure-inspired directed evolution is the engineering of an (R)-selective ATA from Arthrobacter sp. (ATA-117) for the production of sitagliptin (amine 1, Figure 2) from prositagliptin ketone (Savile et al., 2010). A homology model of ATA-117 was constructed based on the crystal structure of three transaminases: a D-amino acid transaminase from Bacillus sp. YM-1 (PDB 3DAA), a branched-chain amino acid transaminase from T. thermophilus (PDB 1WRV), and a branched-chain amino acid transaminase from E. coli (PDB 1AGE). Computational docking of prositagliptin ketone in the modeled structure of ATA-117 identified residues in the small binding pocket causing steric hindrance with the substrate and residues in the large binding pocket that may cause unfavorable interactions. The large binding pocket was first reshaped to accommodate a truncated version of prositagliptin ketone [tetrahydro-triazolo (4,3-α)pyrazine]. Next, the small binding pocket was redesigned to make more space for the trifluorophenyl group of prositagliptin ketone. After 11 rounds of localized saturation mutagenesis and screening, a variant with 27 mutations (Table 2) was obtained that converted 200 g/L prositagliptin ketone to amine with 92% yield under optimal conditions. The overall yield of sitagliptin was increased and a remarkable 53% improvement in volumetric productivity (kg/L per day) was achieved in comparison to the then-current rhodium-catalyzed industrial process. In a subsequent work, Liu Q. et al. (2021) took variant ATA-117 as the template for engineering a new variant with switched product enantioselectivity from amino alcohol 79 to 80 (Figure 2). After four rounds of evolution, variant V69A + F122C + I157H + F225H was found and it had inverted enantiopreference from E = 9 (1S,2R) of 1-aryl-2-amino alcohol 79 to E = 12 (1R,2R) of amino alcohol 80. When the enzyme was tested in a cascade with an engineered transketolase, the results indicated that amino alcohol 80 could be obtained in 76% yield with >99% e.e.
Similar to amine 1, amine 36 is a highly bulky amine, thus a substrate walking strategy was employed to obtain a transaminase for its synthesis. The amine is used for the synthesis of Rimegepant, a drug for the treatment of migraines. A previously constructed mutant of the (S)-selective CvTA displayed activity towards the truncated substrate analogue ((R)-9-hydroxy-6,7,8,9-tetrahydro-5H- cyclohepta [b]pyridin-5-one) and was selected as a template to obtain variants with improved synthesis of amine 36 (Figure 2) (Ma et al., 2022). Initially, several bulky residues shaping the substrate binding site were mutated to alanine. Using variants improved with the model substrate as the template, in total 10 rounds of mutagenesis and screening were applied to improve activity in the synthesis of amine 36, mainly targeting a flexible loop at the substrate entrance tunnel but also using random mutagenesis by error-prone PCR and site-directed mutagenesis to combine mutations. The best variant carried 19 mutations and gave 99% conversion in the production of amine 36 with >99.5% e.e. (Table 2). Scale up to kg amounts was demonstrated (Ma et al., 2022).
Structure-inspired directed evolution was also applied to redesign the well-known (S)-selective VfTA to improve the production of amino acid 71 (Figure 2) (Novick et al., 2021). The product is a chiral precursor to the antihypertensive agent sacubitril which is a component of the heart failure drug Entresto. A variant of VfTA (ATA-217) containing 17 mutations was selected as the template after screening a kit which includes 24 ATA variants. Variant ATA-217 showed trace activity in the synthesis of amino acid 71. Based on docking studies with the quinonoid intermediate of 71, 96 residues were targeted for saturation mutagenesis. After 11 rounds of evolution a variant with 39 mutations (Table 2) was obtained. This variant showed a 5 × 105-fold increase in activity and in a reaction mixture containing 75 g/L substrate gave 90% conversion to amino acid 71 (e.e. > 99.8%).
The aromatic amine 33 (Figure 2) is the precursor of Apremilast, a selective inhibitor of phosphodiesterase 4 and of tumor necrosis factor-α with anti-inflammatory activity. The desired (S)-enantiomer can be produced via transaminase-catalyzed kinetic resolution (Xiang et al., 2021). The VfTA variant required for this reaction was obtained by six rounds of evolution, with mutations mainly introduced in the small substrate-binding pocket in an iterative manner. The work resulted in a variant with eight mutations (Table 2) and an > 400-fold increase in activity with respect to the wild-type VfTA. The work again demonstrates the remarkable evolvability of the V. fluvialis enzyme; it can be engineered by directed evolution to acquire activity with very different substrates (Table 2).
The (S)-selective ATA from Ochrobactrum anthropi (OaTA) was redesigned by saturation mutagenesis of six residues near the active site to improve the conversion to amino acid 72 (Figure 2) (Zhang Z. et al., 2021). The top three single mutants with the highest activity were selected and after combination of their substitutions, the best double mutant (L57C + M419I) gave 94% conversion to amino acid 72 (e.e. > 90%). Visual inspection of docked complexes of the α-ketoacid with the mutant suggested that mutation L57C improved the interaction between the amino group of PMP and the carbonyl oxygen of the substrate, and that a new hydrogen bond between the carbonyl oxygen of the substrate and PMP was formed due to the interaction between the ethyl group of the substrate and Ile419. The higher activity was also attributed to new hydrophobic interactions, i.e. between Ala230, Ile419, and the ethyl group of the substrate.
To obtain an ATA that can catalyze the synthesis of amino alcohol 77 (Figure 2), which is an intermediate in the synthesis of the new anti-HIV drug dolutegravir, activity-determining residues were identified from molecular docking using the crystal structure of an (R)-selective ATA from Aspergillus terreus (AtTA) (PDB 4CE5). Four influential positions close to the substrate tunnel and large binding pocket were separately targeted for site-saturation mutagenesis yielding several improved variants (Gao et al., 2020). After combination of mutations, the best variant (H55A + G126F + S215P) converted 20 and 50 g/L substrate to amino alcohol 77, with 90.8% and 79.1% conversion, respectively, whereas the conversion with the wild-type enzyme was below 5%. The mutation H55A facilitates substrate binding by enlarging the active site entrance tunnel. Mutation G126F is located in a loop that forms the outer end of the entrance tunnel and that could be pushed outward due to the bulkier side chain of Phe126, thereby enlarging the tunnel. Mutation S215P improves enzyme stability by introducing new van der Waals interactions with the substrate.
Using a similar strategy in which structural information was used to restrict directed evolution to specific positions, the (R)-selective ATAs from Chloroflexi bacterium (CbTA) and Capronia epimyces (CeTA) were engineered to improve the activity towards amine 27 and D-alanine (73), respectively (Figure 2) (Wang C. et al., 2021; Jia et al., 2022). Structures with docked substrate were analyzed and five residues in the small binding pocket of CbTA and seven residues in the large binding pocket of CeTA were targeted by saturation mutagenesis. The best variant of CbTA carried mutation Q192G which created more space in the small binding pocket and displayed 9.8-fold higher activity than the wild-type enzyme in the deamination of amine 27. In assays with D-alanine, the F113T variant of CeTA gave the highest activity and conversion in the synthesis of D-alanine reached 95%.
3.4 Phylogenetic analysis supporting directed evolution
Database mining and phylogenetic analysis offer powerful tools to support the design of libraries for discovery of enzymes with desired activities (Kelly et al., 2020). Such methods can also be employed to guide engineering of the activities of ATAs with α-amino acids (Deszcz et al., 2015). The sequence of CvTA was aligned with 28 related transaminases including serine:pyruvate α-transaminase from Sulfolobus solfataricus to compare their active site residues and a phylogenetic tree was constructed. In total 14 positions in CvTA were selected for saturation mutagenesis based on the diversity in aligned sequences, and several different mutations at three positions (Trp60, Phe88 and Tyr153) gave higher activity with serine (68, Figure 2) as an amino donor to produce hydroxypyruvate. Activity was determined via a high-throughput colorimetric screening assay, in which a blue color was formed by a reaction between the tetrazolium salt WST-1 and 2-hydroxypyruvate (Figure 3E). The catalytic efficiencies (kcat/KM) towards serine of the two most active mutants (Y153M and Y153S) were increased by 65-fold and 15-fold, respectively.
3.5 Structure-inspired rational design
Structural inspection of ATAs, often combined with sequence- or structure-based alignments with ATAs that have related properties, can be used for designing small sets of mutants. When a crystal structure is not available, homology modelling is often used to generate a 3D model of the protein. An example is the work of Cho et al. (2008), who built a homology model of VfTA using the structure of a 2,2-dialkylglycine decarboxylase (PDB 1DGE), before the crystal structure of VfTA (Midelfort et al., 2013) was determined. Analysis of the modelled structure pointed at two active-site hotspot positions for mutations that could improve the activity towards amines 3–8 (Figure 2). The homology model of VfTA predicted that two positions (W57 and W157) located in the large binding pocket would cause steric hindrance with the molecules of interest. Thus, two single mutants, W57G and W157G, were expressed and tested for activity towards the set of compounds. While mutation of position 57 to glycine produced a remarkable increase in activity, mutation of position 147 barely altered the substrate scope of VfTA. Determination of the crystal structure of VfTA in 2013 (PDB 4E3R) showed that while residue W57 is located at the predicted position, W147 is nowhere near the active site of the enzyme (Midelfort et al., 2013). In another study, homology modelling of VfTA was based on two templates (1D7R and 2EO5), subsequently performing substrate docking in combination with sequence alignment to identify 14 positions as targets for mutagenesis (Midelfort et al., 2013). A focused library was constructed from which less than 450 variants needed to be screened to find mutants with improved activity in the synthesis of ester 81 (Figure 2), a key intermediate in the synthesis of imagabalin which is used for treatment of generalized anxiety disorders (Birch et al., 2011). The best variant contained eight mutations (Table 2) and exhibited a 60-fold higher activity. During the course of their research, the crystal structure of VfTA (PDB 4E3R) was solved (Midelfort et al., 2013) and five key positions in the small binding pocket which could cause steric hindrance were selected for mutagenesis to improve the yield of amine 9 and amino alcohol 78 (Figure 2) (Nobili et al., 2015). The mutant F85L + V153A gave the best yield (53%) in the synthesis of amine 9 (e.e. = 98%), whereas the highest yield (60%, e.e. = 98%) in the synthesis of amino alcohol 78 was obtained by mutant Y150F + V153A.
The commercially available 3DM bioinformatics platform builds and uses databases of structurally related proteins to support protein engineering. Structure-based sequence alignments that reflect functional relationships and networks in a defined protein superfamily are used to select positions and diversity for substitutions (Kuipers et al., 2010). The 3DM platform has been used to understand and improve activity of VfTA towards aliphatic aldehydes (74 and 75, Figure 2) (Genz et al., 2015). A library containing 1,200 variants with mutations at four positions potentially involved in steric hindrance (Leu56, Trp57, Arg415 and Leu417) in the large binding pocket was investigated and six promising mutants were discovered and characterized in more detail. The results showed that in three mutants (R415L, W57F+R415L and W57F + R415L + L417V) the amino acceptor spectrum had shifted towards aliphatic aldehydes. Mutations W57F and R415L created more space, whereas mutation R415L also increased the hydrophobicity of the large binding pocket to accept hydrophobic groups like the alkyl moiety of aliphatic aldehydes. The 3DM platform was also used for VfTA engineering aimed at improving the synthesis of bulky amines [10, 11 and 12 (Figure 2)] which are not accepted by wild-type VfTA (Genz et al., 2016). Seven residues that sterically constrain the substrate binding pocket were selected for mutagenesis. After screening, a variant was identified (L56V + W57C + F85V+V153A) that gave an improved yield of (R)-amine in the synthesis of 10, 11 and 12.
The (S)-selective OaTA was engineered to improve activity towards various ketones, using homology models to select the target positions (Han et al., 2015b; 2015c). First, influential positions were identified by alanine scanning. Two hotspots (Leu57 and Trp58) in the large binding pocket were targeted, because Trp58 causes steric hindrance with the phenyl group of aromatic ketones, whereas removal of Leu57 may allow reorientation of the phenyl group of aromatic ketones in the large binding pocket, thereby enabling the methyl group of such an acetophenone-like compound to move into a spacious region of the small binding pocket. Site-directed mutagenesis at these two positions gave two improved variants, L57A and W58L, which exhibited higher activity towards various ketones with the use of isopropylamine as the amino donor. In the asymmetric synthesis of amine 9 (Figure 2) by mutant L57A, conversion reached 79% (e.e. > 99%) whereas no conversion was detected with the wild-type OaTA. A similar approach was used to improve OaTA for the production of non-proteinogenic amino acids (Han et al., 2015a). The same best mutant L57A displayed a 48-fold higher activity towards keto acid 86 (Figure 2) with (S)-1-phenylethylamine as the amino donor and a 56-fold higher activity towards L-norvaline (69, Figure 2) using propanal as amino acceptor. In the asymmetric synthesis of 69 by mutant L57A, the conversion was drastically enhanced up to 99.3% (e.e. > 99.9%), while the wild-type OaTA only gave 23% conversion. To enhance the efficiency of combining mutations after the identification of improved single mutants, a method called R-analysis was employed to predict if adding a mutation in OaTA will have a positive (R > 1) or negative (0 < R < 1) effect on activity with a set of structurally related target substrates including α-keto acids, (S)-arylalkylamines and arylalkyl ketones (Kim et al., 2019). The cosubstrate was fixed. Based on the crystal structure of OaTA (PDB 5GHF) (Han et al., 2017), nine residues in the active site were selected for alanine scanning, and three mutants (L57A, W58A and V154A) were identified with higher activities towards target substrates. On the basis of predictions by R-analysis, the mutations were expected to show additive effects on enzyme properties and by combining the substitutions a final variant (L57A + W58A + V154A) with significantly higher activity towards the three sets of substrates was obtained. It allowed the synthesis of bulky amine 10 (Figure 2) with a 45.6% yield (e.e. > 99%). The three corresponding positions of VfTA (Leu56, Trp57 and Val153) were also targeted for redesign to improve the conversion to amine 11, and the mutations apparently indeed relieved steric hindrance in the active site (Genz et al., 2016).
In the absence of a crystal structure of CvTA, a homology model of the enzyme was used to improve the activity in the conversion of amine 13 or ketones 46–54 (Figure 2) (Cassimjee et al., 2012). The structures of VfTA (Midelfort et al., 2013) and an (S)-selective ATA from Arthrobacter citreus (AcTA) (Svedendahl et al., 2010) served as templates for modeling. A key residue in the large binding pocket that could interfere with substrate binding is Trp60 and therefore it was replaced. Mutation W60C increased the activity of CvTA towards amine 13 by 29-fold, whereas the activity increased up to 5-fold in the amination of 4′-substituted acetophenones (46–54). Afterward, the crystal structure of CvTA (PDB 4A6T) was solved (Humble et al., 2012b) and used to improve the activity for the production of bulky amine 14 (Figure 2) (Land et al., 2020). Specifically, to enhance the acceptance of a phenyl group of 14 in the small binding pocket, residues Leu57 and Phe88 were mutated to smaller amino acids. The results showed that increasing the size of the small pocket in variant L57A + F88A gave a 30-fold higher activity in the kinetic resolution of racemic amine 14 with nearly 50% yield (e.e. > 99%). Position 59 of CvTA is also a hotspot to redesign in the synthesis of amine 9 and 35 (Figure 2) and the excellent yields with >99% e.e. of these two amines were achieved by the variant L59A (Sheludko et al., 2022). The corresponding mutant L57A of OaTA also gave a significant conversion (>99% e.e.) to amine 9 (Han et al., 2015b).
The crystal structure of CvTA was used as the template for building a homology model of an (S)-selective ATA from Halomonas elongate that was used in experiments aimed at improving conversion of para-, meta- and ortho-mono-substituted benzaldehydes and acetophenones (Contente et al., 2016). In the active site, four residues suspected to cause steric hindrance were targeted, i.e. Trp56 (large binding pocket), Phe84 (between two pockets), Tyr149 (large binding pocket) and Ile258 (large binding pocket). Mutations W56G, F84A and Y149F were found to relieve steric hindrance and mutation I258A may facilitate side-chain rotation of Trp56 creating more space for substrate binding. Similarly, a homology model of an (S)-selective ATA from Paraburkholderia phymatum was used to discover mutations that could enhance the activity towards ketone 66 (Figure 2) (Xie et al., 2021). In the large binding pocket of the active site, four residues including Met78, Trp82, Ile284, and Thr440 were replaced. To relieve the steric hindrance, mutant W82A was constructed. An aromatic interaction was introduced by mutations M78F and I284F which was expected to be beneficial for binding the phenyl group of ketone 66 in the large binding pocket. Mutation T440Q was anticipated to increase hydrophobicity which would also improve binding affinity of 66. The final quadruple variant M78F + W82A + I284F + T440Q gave the highest catalytic efficiency (>470-fold higher than wild-type), and the conversion of 66 was improved from 1.3% to 94.4%.
The ATA from Ruegeria sp. TM1040 (PDB 3FCR) was redesigned to improve the synthesis of the bulky amines 12, 15, 16, and 17 (Figure 2) (Pavlidis et al., 2016). The wild-type enzyme had no activity in the synthesis of racemic amine 15 and only minor activities were detected with racemic amines 16 and 17. Mutants were designed and tested from the structural analysis of residues forming the binding site. Sequence alignment with an (S)-selective ATA from Silicibacter pomeroyi suggested that mutations Y59W and T213A could increase activity towards bulky amines, as the same mutations had improved catalytic activity in kinetic resolution assays of the S. pomeroyi ATA (Steffen-Munsberg et al., 2013). Additionally, three residues, namely Tyr87 (small binding pocket), Tyr152 (PLP binding via a stacking interaction) and Pro423 (in a loop at the entrance of the active site) were examined and mutants (Y87F, Y152F and P423H) were tested. Mutation Y87F was expected to create more space by removing polar interactions of the tyrosine hydroxyl group with the substrate. Mutation P423H makes the loop more flexible and allows better binding of aromatic compounds in the active site. Three more positions were modified and the best variant (Y59W + Y87F + Y152F + T231A + P423H) exhibited a noticeable activity improvement in the kinetic resolution of racemic amines 15, 16 and 17, and a remarkable 8900-fold higher activity towards racemic amine 17. Mutation P423H was later eliminated because while it improves activity it does so at the cost of stability. The variant Y59W + Y87F + Y152F + T231A still exhibited outstanding performance in the asymmetric synthesis of bulky amines 15, 16 and 17.
The ATA from Ruegeria TM1040 was also engineered for asymmetric synthesis of amine 12 (Weiss et al., 2017). In this case, the starting point was the variant Y59W + T231A, for which no detectable activity was found with racemic amine 12. An improved variant with three additional mutations (Y59W + Y87L + T231A + L382M + G429A) in comparison to the template was obtained by error-prone PCR and subsequent site-saturation mutagenesis with prescreening for active clones with the glycine oxidase assay mentioned above. The five-fold mutant had improved activity in the kinetic resolution of racemic amine 12. In asymmetric synthesis, conversions reached 100% (e.e. > 99%) and 71% (e.e. > 99%) when driving the reaction with the amino donors L-alanine and isopropylamine, respectively.
Park et al. reshaped the small binding pocket of the (S)-selective ATA from Paracoccus denitrificans (PdTA, PDB 4GRX) to synthesize non-proteinogenic amino acids with long alkyl chains (Park et al., 2014). Binding of the carboxylic group of L-α-amino acids occurs in the large binding pocket, whereas the alkyl substituent is accommodated in the small binding pocket. Structural alignment of the five residues forming the small binding pocket of PdTA (Phe19, Phe85, Ser118, Tyr150, Val153 and Phe321) showed that Val153 is a key residue that confers the non-canonical substrate specificity of PdTA. When an L-α-amino acid bearing an alkyl chain forms the Michaelis complex with the enzyme, Val153 can move back from the small binding pocket allowing it to accommodate the carboxylic group of the L-α-amino acid. Excavation of the small binding pocket via the V153A mutation mutation was found to promote accommodation of preferentially long alkyl chains as in 69 and 70 (Figure 2). The V153A mutation also resulted in a 4-fold activity improvement in the amination of keto acid 87.
In the amination of a series of keto acids (88–92, Figure 2), the modeled structure of an (R)-selective ATA from Gibberella zeae was constructed based on the crystal structure of an ATA from Nectria haematococca (PDB 4CMD) and selected as a template in the enzyme design (Jia D. X. et al., 2021). To create more space in the binding pocket and the substrate entrance tunnel, five residues were targeted for alanine scanning. After further rational design and mutation combination, the variant S214A + F113L + V60A displayed the best activities towards those keto acids (88–92). Another (R)-selective ATA from Arthrobacter cumminsii ZJUT212 (AcATA) was engineered to improve the production of sitagliptin intermediate (amine 28) (Figure 2) (Cheng et al., 2020a). A sequence alignment with the final improved variant ATA117-Rd11 (PDB 3WWJ) which was useful for sitagliptin production was used to suggest mutations (Savile et al., 2010; Guan et al., 2015). In total, 23 different residues were identified in the alignment sequence and 14 of these harbored mutations that were beneficial for activity towards pro-sitagliptin ketone. These 14 substitutions were then introduced in AcATA to construct a variant AcATA M1. Because of the high sequence identity with AcATA M1, ATA117-Rd11 was selected as a template to construct a homology model for docking simulations. In total 13 active site residues were targeted for alanine scanning and three beneficial positions (Met122, Thr134, and Gln155) in AcATA M1 were identified for saturation mutagenesis. The best variant M122H displayed >50-fold higher activity towards pro-sitagliptin ketone compared with the wild type AcATA, and the conversion of 50 g/L substrate to amine 28 reached 92% (e.e. > 99%). MD simulations showed that mutation M122H lengthened the substrate cavity, which could facilitate the large substrate moving in and out of the binding pocket, hence facilitate substrate binding.
The fold-type Ⅳ ATA from Exophiala xenobiotica (ExTA, PDB 6FTE) can naturally aminate various biaryl ketones (55–64, Figure 2), but was further engineered to improve its catalytic activity (Telzerow et al., 2019). After crystal structure determination, six highly conserved residues in the small binding pocket (Val60, Phe113, Thr273, Thr274 and Ala275) were identified as potential producers of steric hindrance with biaryl ketones, from which three variants were constructed (T273S, T273S + T274S, and T273S + T274S + A275G) and tested. The best variant (T273S) displayed the desired improved activity with various biaryl ketones, reaching >99% conversion in three cases (e.e. > 99%). The crystal structure of an (R)-selective ATA from Arthrobacter sp. (PDB 3WWH) (Guan et al., 2015) was used by Han et al. for docking simulations to find mutants with enhanced activity towards ketone 43 (Figure 2) (Han et al., 2021). Ten residues around the active site were first selected for alanine scanning. The three positions that showed the highest activities upon mutation to alanine (Gly136, Val199 and Ser223) were selected for saturation mutagenesis. The single mutants G136I, V199W and S223P were most active in the conversion of 43. The combination of two mutations to form the double mutant V199W + S223P further increased the activity. Another example of the use of alanine scanning was performed by Konia et al., who engineered an (R)-selective ATA from Luminiphilus syltensis using the crystal structure (PDB 7P3T) to improve activity towards amine 34 (Figure 2). Amine 34 is too bulky to be accepted by the wild-type enzyme due to the size of the substituent (Konia et al., 2021). Alanine scanning of three residues in the small binding pocket that may cause clashes with 34 yielded mutant V37A with increased activity.
Incorporation of non-canonical amino acids has been widely used in protein engineering to improve and introduce a new catalytic activity (Drienovská and Roelfes, 2020; Pagar et al., 2021). This rational strategy was applied to expand the substrate scope of ATAs and an (R)-selective ATA that was previously engineered from a D-amino acid transaminase was selected as a scaffold (Voss et al., 2020; Pagar et al., 2022). The active site of this template enzyme is highly hydrophobic, mainly because of three phenylalanine residues (Phe31, Phe86, and Phe88). Therefore, displacement of phenylalanine analogs with higher hydrophobicity potentially increases the enzyme activity towards the hydrophobic substrate. After screening phenylalanine analogs, replacement of Phe88 by p-benzoyl phenylalanine was found to give gave the best activity towards amine 27 (Figure 2). Furthermore, the mutation F88pBpA also highly improved the activity towards aldehyde 76 (amine 27 was set as amino donor) and the activity in the deamination of amine 34, 37, 38, and 39 (pyruvate was set as amino acceptor) (Figure 2).
Structure-inspired rational design has not only been used to expand the substrate scope of ATAs but also to improve or even switch the enantioselectivity. For example, the enantioselectivity of VfTA was improved in the amination of (S)-3-methylcyclohexanone (Skalden et al., 2015). The work targeted the active site residue Leu56 as its side chain interacts with the methyl substituent of the external aldimines formed by reaction of PLP with amine 18 and amine 19 (Figure 2). In an asymmetric synthesis reaction using L-alanine as the amino donor, the enantiomeric excess of the produced (R)-amine 20 (Figure 2) was enhanced by up to 66%, with a diastereomeric composition of 83% amine 20 and 17% amine 18. The relevant mutation was L56V. Mutation L56I switched the selectivity from amine 21 to amine 18 as the product, with 70% enantiomer excess of the (S)-amine and a diastereomeric composition of 15% 20 and 85% amine 18.
The enantioselectivity of CvTA has also been fine-tuned via mutagenesis (Humble et al., 2012a). To enhance the S preference in the production of amines 13, 21 and 22 (Figure 2) mutation W60C was constructed and tested, displaying an up to 15-fold increase in E value. Mutation W60C created more space to accommodate the ketone substrate in a pro-(S) conformation. This mutation was also found to be beneficial for activity with amine 13 and ketones 46–54 (Cassimjee et al., 2012). Another designed variant, F88A + A231F, even switched the enantiopreference from S (E = 3.9) to R (E = 63) in the amine 22 synthesis. Mutation F88A enlarges the small binding pocket to fit the large group of the substrate, and mutation A231F reduces the space of the large binding pocket to selectively fit the small group of the substrate.
Another example of increasing or switching the enantioselectivity of AcTA concerns the asymmetric synthesis of amine 23 (Figure 2) (Svedendahl et al., 2010). In the phosphate-group binding cup, three residues (Glu326, Val328 and Tyr331) at an active site loop were substituted and five mutants (E326D, V328A, Y331C, E326D + Y331C and V328A + Y331C) were identified. Mutants E326D, Y331C and E326D + Y331C showed increased (S)-product selectivity using isopropylamine as the amino donor, and the best variant (Y331C) displayed an improvement from 98% to >99.5% e.e. The other two mutants (V328A and V328A + Y331C) switched the enantioselectivity and mutant V328A gave the (R)-enantiomer of 23 with 58% e.e. All five mutants retained (S) preference with >99.5% e.e. in the synthesis of amine 24 (Figure 2).
3.6 Computer-aided rational design
In recent years, several computational methods have been used to support rational design aimed at tailoring the substrate scope of ATAs (Sirin et al., 2014; Dourado et al., 2016; Han et al., 2017; Voss et al., 2018; Zhai et al., 2019; Meng et al., 2021). An approach often used is the rational selection of a restricted number of positions at which mutations may occur that may improve a desired activity. Mutants are designed rationally, and modeling in atomic detail is used to examine their enzyme-substrate complexes, including substrate-enzyme interactions and the occurrence of reactive poses. Mutations at these positions are then examined by various computational tools or discovered by an appropriate search algorithm that in silico scans sequence- and conformational space for beneficial variants, e.g. on binding. We refer to these approaches as computation-supported engineering. The methods used in computation-aided rational design of ATAs include molecular docking, MD simulations, Monte-Carlo search algorithms, and quantum mechanical (QM) calculations. The Rosetta package, developed by D. Baker and coworkers (Richter et al., 2011) uses a Monte Carlo search algorithm and an energy-based scoring function to discover enzyme variants that have a redesigned active site with improved ligand binding properties. Substrate is docked in a user-defined constrained reactive position that based on mechanistic insight or QM calculations is predicted to represent a reactive conformation (often called a near attack conformation). Then, selected amino acids around the substrate are varied by identity and conformation during a Monte Carlo search that produces designs with optimized scores (lowest energy). These designs are expected to show improved activity, and are tested in the lab after ranking by visual inspection or by further computational tools such as docking- or MD simulations.
Sirin et al. developed a computational protocol to predict the catalytic activity of VfTA by using a combination of docking and MD simulations. Three scoring schemes (MM/GBSA, DockingScore, and Z-score) (Lyne et al., 2006; Sastry et al., 2013), all based on substrate binding energies, were used to assess the fitness of the variants towards the synthesis of ester 81 (Figure 2) (Sirin et al., 2014). From the calculated binding energies, a library of 89 mutants was constructed of which 63 were found to be active. The success rates of the three scoring schemes were 75% (MM/GBSA), 70% (DockingScore) and 80% (Z-score). The best computationally designed mutant carried eight mutations (Table 2) and showed a 60-fold increase in activity, in agreement with the prediction of this mutant as one of the top-ranking candidates.
VfTA engineering supported by computational analysis was also pursued by Dourado et al. (2016) to improve the activity in the synthesis of amine 12 (Figure 2). The approach included docking simulations of both the quinonoid intermediate and the corresponding ketone of amine 12 in complex with a set of VfTA variants. The complexes of promising candidates were used for MD simulations. Several triple mutants were identified after the first two rounds in silico screening, and after that a further round of screening by coevolution network analysis gave variants with 4–7 mutations. Mutants were evaluated for production of bulky amine 12, for which wild-type VfTA has no activity. Eight residues (Trp57, Lys163, Ala228, Glu257, Val258, Ile259, Val422, Arg415) in the large binding pocket and five residues (Gly55, Leu56, His83, Arg88 and Val153) in the small binding pocket were targeted with the aim of creating more space in the binding pockets and reducing their overall charge to enhance intermolecular interactions with the hydrophobic substrate. From multiple rounds of computational evaluation and mutant selection, 113 variants were identified and tested experimentally. After three rounds of in silico screening, the best variant carried seven rationally combined mutations (Table 2). It showed more than 1716-fold increase in activity and reached 42% conversion (e.e. > 99%) in the synthesis of bulky amine 12.
Another ATA that was redesigned with the support of in silico methods is the (S)-selective ATA from Bacillus pumilus W3 (Zhai et al., 2019). The aim was to improve activity for the production of bulky amine 26 (Figure 2) which is a building block of the antibiotic sitafloxacin, used in the treatment of Buruli ulcer. First, a homology model was constructed using the related ATA from Geoglobus acetivorans (51% sequence similarity) as a template. The modelled structure was then used for substrate docking and four residues (Tyr32, Lys155, Ile215 and Thr252) around the active site were selected. The effect of point mutations at these positions on folding energies of enzyme-substrate complexes (ΔΔG fold) were calculated by PoPMuSiC-2.1, a web server to predict the effect of mutations on thermostability (Dehouck et al., 2011). Since the mutations are around the active site, increased stability may correlate with increased substrate binding and eight mutations with ΔΔG fold < 0 were selected for laboratory testing. After testing combinations, the best mutants (L212M + I215M and Y32L + S190A + L212M + I215M) displayed 77% and 79% conversion (e.e. > 99%), respectively, of amine 26.
Han et al. attempted to solve the question of why ω-TAs show lower activities towards ketones than towards α-keto acids and aldehydes, and based on their discoveries improved the activity of OaTA towards ketones (Han et al., 2017). Via molecular docking they found that ketones typically form non-productive binding complexes: the dihedral angle between the plane of Oβ, Cα, and NPMP and of Oβ, Cα and Cβ must be close to 90° and the Cα−NPMP distance to be below 2.5 Å for the nucleophilic attack of the PMP amine on the ketone in the acceptor-amination half-reaction (Radisky and Koshland, 2002; Oliveira et al., 2011). The orientations of the ketone substrate in the complex were estimated via docking, and the results were used to computationally score a set of OaTA variants. The best variant, carrying two mutations in the large binding pocket (L57A + W58A), was found to have a 105-fold higher activity in the production of amine 9 (Figure 2) without loss of product enantioselectivity. An L57A mutant was previously obtained by rational design and gave a 110-fold activity improvement in the synthesis of amine 9, indicating that rational design based on structural information predicted a similar mutation as computational design based on the docking orientation analysis (Han et al., 2015b).
Voss et al. explored a computational method to improve the activity of CvTA in the synthesis of amine 9 from ketone 65 (Figure 2) (Voss et al., 2018). Specifically, substrate docking and MD simulations were first conducted using the crystal structure. This showed that an energy barrier between conformations with substrate in the entrance tunnel and in the active site hindered movement of substrate to a reactive position. The contribution of flanking residues to energy barriers between conformations were calculated using quantum mechanics and used to select positions for in silico alanine or glycine scanning mutagenesis. This way, 10 single or double mutants with lower computed interaction energies of reactive conformations were selected. To decide which amino acid would be most beneficial at the four most influential positions (Trp60, Phe88, Ser121 and Cys418), sequence and structure alignments were used and the three most frequent amino acids at each of the four selected positions were selected. The 216 variants were ranked computationally according to the lowest interaction energies and minimal distance of ketone 65 to the catalytic site, and 11 top-ranking variants were selected for the experimental verification. The two best variants (F88L + C418G and F88L + C418L) were found to exhibit higher activity and better conversion in the synthesis of amine 9. On the basis of structural analysis, mutations F88L and C419L were proposed to create more space in the small binding pocket and the entrance tunnel, thereby improving the acceptance of ketone 65. To achieve amination of ketone 65 by engineered VfTA, the corresponding F85A mutant was also used to provide more space in the small binding pocket (Nobili et al., 2015).
A D-amino acid transaminase (DATA) was engineered to gain the activity towards amine 27 (Figure 2) via a computational strategy including rational design, Rosetta calculations and 3DM analysis (Voss et al., 2020). Since the starting enzyme is a (R)-amino acid aminotransferase, the small carboxylate binding site (also called P-site, since it points in the same direction as the phosphate binding cup) needs to be modified and the Y88E mutation was chosen for that because the corresponding R97E mutation introduced some 27 synthesis activity in the homologous AT from Aspergillus fumigatus. To further optimize the O-site, the ligand PLP-amine 27 complex was docked into DATA (PDB 3DAA) and nine positions around the ligand were targeted for optimization by Rosetta design. Based on Rosetta scores and contribution of the ligand, five top-scoring mutants were selected for the experimental verification. No improved activity towards amine 27 was observed among those five variants, whereas a variant that showed the acceptance of amine 27 was selected for further study because some mutations in this variant could be detrimental in the second half of transamination. 3DM software was applied for sequence alignment and residues with low frequency among the aligned sequences were removed. Finally, only one computationally selected mutation was avoided to obtain the best variant (Table 2) which gave a similar activity towards amine 27 as natural (R)-selective ATAs. In the best variant, the larger and more hydrophobic binding pockets both contributed to better accept amine 27.
Our group explored a computational method employing the Rosetta interface energy as the main metric in a single dock-and-design step to redesign transaminase activity (Figure 4) (Meng et al., 2021). The template was a thermostable PjTA variant (PjTA-R6, PDB 6TB1) (Meng et al., 2020) that displayed no detectable activity in the synthesis of the six enantiopure bulky amines 9, 21, 29, 30, 31, and 32 (Figure 2). The modeled ligands were the external aldimine intermediates. Complexes of the ligands with the PjTA-R6 scaffold were obtained by Rosetta, visually inspected for reactive conformations, and the Rosetta interface energies were used to score and rank the designs. Seven residues in the large binding pocket and one residue in the small binding pocket were targeted. Six separately designed small libraries composed of 7–18 variants each (40 variants in total) were constructed with 1–6 mutations and the majority (97%) of the variants exhibited activity in the production of the desired (S)-amine with excellent enantioselectivity (e.e. > 99%). Moreover, a good correlation (r2 = 0.75–0.96) was obtained between Rosetta enzyme-external aldimine interface energy and the experimental yield, suggesting Rosetta interface energy is a reliable metric to detect improved variants among a large number of primary designs generated by Rosetta search algorithm. The best-performing mutant in the synthesis of amine 9 was a triple mutant W58M + F86L + R417L. Notably, the best variants derived from VfTA, CvTA and OaTA for production of amine 9 all possessed a corresponding mutation: F85L, F88L, and W58A, respectively (Figure 5) (Nobili et al., 2015; Han et al., 2017; Voss et al., 2018). In case of PjTA, the single mutant W58G displayed the best performance for synthesis of structurally similar bulky amines and the best variant of OaTA for the synthesis of amine 21 and 29 contained the corresponding mutation W58L (Han et al., 2015c).
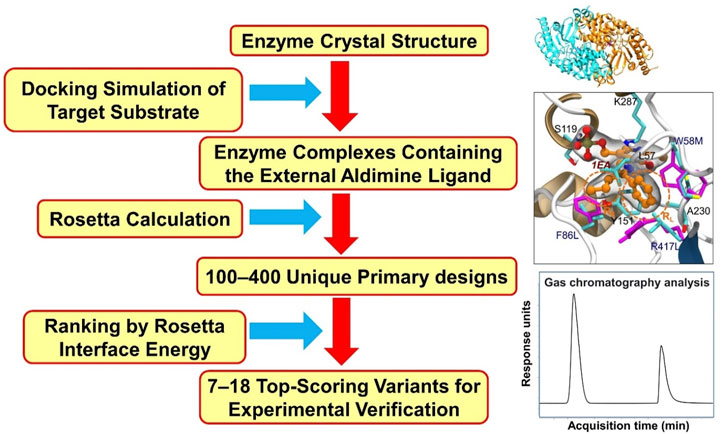
FIGURE 4. The workflow of the computational redesign of PjTA-R6 to expand the enzyme substrate scope.
FIGURE 5. Schematic view of the large and small binding pocket of three class Ⅲ, fold-type I ATAs bearing the external aldimine intermediate [PLP-Schiff base with (S)-1-phenylethylamine]. Residues around the blue circle belong to the large binding pocket, and the residues around the red circle belong to the small binding pocket. The catalytic lysine is also shown. The residues of VfTA, CvTA, OaTA, and PjTA are in orange, green, magenta, and grey, respectively.
3.7 Conserved active site topology
A common strategy of the aforementioned studies aimed at redesigning the substrate scope of ATAs is to increase the size of the two binding pockets by replacing bulky side chains with smaller residues and promoting hydrophobic interactions between substrate and enzyme by replacing polar with hydrophobic residues. Following this approach, based on steric and polarity considerations, hotspots in the binding pockets have been identified as target to improve variants. The binding pocket residues most commonly substituted in engineered variants of the class III ATAs VfTA, CvTA, OaTA, and PjTA are summarized in Figure 5. The key residues can easily be translated among the four enzymes because of structural similarities between class III ATAs. Some of the key residues are as follows. The switching Arg415 (VfTA)/Arg417 (PjTA) is both polar and bulky, and modification of catalytic selectivity by creating more space in the large binding pocket and reducing its polarity via four different mutations (R415K, R415F, R415L and R415A) has been reported (Midelfort et al., 2013; Sirin et al., 2014; Genz et al., 2015; Dourado et al., 2016; Meng et al., 2021; Xiang et al., 2021). Residue Phe85 (VfTA)/Phe88 (CvTA)/Phe86 (PjTA), located in the small binding pocket, is frequently targeted and mutated to smaller residues like alanine, valine or leucine to relieve the steric hindrance limiting acceptance of bulky substituents (Midelfort et al., 2013; Sirin et al., 2014; Nobili et al., 2015; Genz et al., 2016; Voss et al., 2018; Land et al., 2020; Meng et al., 2021; Novick et al., 2021; Xiang et al., 2021; Sheludko et al., 2022). Another key residue in the small binding pocket is Val153 (VfTA)/Val154 (OaTA) and two smaller residues (alanine and serine) were tested (Midelfort et al., 2013; Sirin et al., 2014; Nobili et al., 2015; Dourado et al., 2016; Genz et al., 2016; Kim et al., 2019; Novick et al., 2021; Xiang et al., 2021). A key residue from the large binding pocket often responsible for steric hindrance with substrate is Trp57 (VfTA)/Trp60 (CvTA)/Trp58 (OaTA/PjTA), and mutations to smaller residues such as glycine, alanine, leucine, phenylalanine, and cysteine have been successful in reducing steric interference (Cho et al., 2008; Cassimjee et al., 2012; Midelfort et al., 2013; Sirin et al., 2014; Han et al., 2015c, 2017; Genz et al., 2015, 2016; Dourado et al., 2016; Kim et al., 2019; Meng et al., 2021; Novick et al., 2021; Xiang et al., 2021). Moreover, a conserved leucine found in the large pocket has been targeted in three ATAs: Leu56 (VfTA)/Leu59 (CvTA)/Leu57 (OaTA). Again, replacement by smaller residues (alanine, cysteine and valine) broadened the substrate scope (Han et al., 2015b; 2015a; Genz et al., 2016; Han et al., 2017; Kim et al., 2019; Land et al., 2020; Zhang Z. et al., 2021; Ma et al., 2022). Among three ATAs, the same position in the large binding pocket with different residues was targeted: Leu417 (VfTA)/Cys418 (CvTA)/Met419 (OaTA). Mutations including L417V, C418L, C418G, and M419I were constructed to create more space for substrate binding (Genz et al., 2015; Voss et al., 2018; Zhang Z. et al., 2021). The key residues mentioned are routinely found to help accommodate non-natural substrates in ATAs, and therefore are primary targets for mutagenesis when redesigning the substrate scope of homologous ATAs.
3.8 Conclusions and perspectives
Rational design is as frequently used for tailoring the substrate scope of ATAs as directed evolution employing random mutant libraries because of the well understood catalytic mechanism, the availability of crystal structures, and the advantages of rational design in terms of reducing the screening effort needed for finding improved variants. When small sets of mutants are sufficient to find desired variants, no development of screening assays is needed and work can be done with real substrates instead of surrogate substrates that are selected because they facilitate screening. On the other hand, the comprehensiveness of rational design is lower because only residues surrounding the substrate in the active site or entrance tunnel or otherwise are expected to be important for performance will be examined by mutagenesis. Furthermore, only a restricted set of combinations of substitutions is tested. Fortunately, computational design algorithms such as Rosetta can search a much larger sequence space than on-screen inspection or even directed evolution. Computational modeling can predict mutations in binding pockets as well as in entrance tunnels, including mutations that influence transitions between conformations, create space to accommodate bulky non-natural substrates, and predict occupancy of reactive conformations. The use of complementary algorithms that offer orthogonal in silico screening, including MD and QM, can increase the reliability of predictions. Challenges in the field of computational design are achieving higher accuracy to facilitate selection of designs with the best catalytic properties, avoiding false positives such as variants that do not well express, covering catalytic cycles that consist of multiple chemical steps and conformational changes, and better description of aromatic interactions and interactions that are influenced by polarization effects.
Currently, machine learning algorithms are a hardly explored alternative for computational redesign of transaminases. The only work we are aware of where machine learning is used for transaminases, is that of Jia L. L. et al. (2021), in which machine learning is used to predict enzyme stability. Machine learning applications already implemented in other enzyme systems are prediction of binding energies (Ellingson et al., 2020) or kcat (Li et al., 2022), generation of artificial enzymes (Strokach et al., 2021), enzyme classification (Li et al., 2018), binding site prediction (Zhao et al., 2020), protein scaffolding (Wang et al., 2022), and de novo protein design (Anishchenko et al., 2021). More thorough reviews on the subject are published (Feehan et al., 2021; Ovchinnikov and Huang, 2021). It is to be expected that such methodologies will also be applied in the field of transaminase engineering.
The release of AlphaFold, which is a machine learning pipeline to predict 3D structures of proteins from the sequence alone (Jumper et al., 2021), is a major step forward that brings great benefits to protein engineering of enzymes with no reported crystal structure. Structure-based and computation-supported redesign to expand the substrate scope of ATAs, and enzymes in general, has until recently been limited by the availability of high-resolution crystal structures. A few examples where homology modelling was used to obtain ATA structures to be employed in protein engineering campaigns are mentioned in the text above and in Table 2. However, the accuracy of such structures depends on the availability of a good template, i.e., a close relative of the enzyme in question (Bohnuud et al., 2017). For example, mutation W147G of VfTA had little effect on the substrate range, even though a homology model predicted it to be near the substrate-binding site (Cho et al., 2008). This negative result was explained by the later crystal structure which revealed that W147 was not near the active site (Midelfort et al., 2013). Remarkably, the structures produced by AlphaFold are of near-experimental resolution, with most proteins solved with an accuracy of 0.96 Å (C-α RMSD). AlphaFold multimer, which predicts the structure of homo- or heteromers (Evans et al., 2022) is of particular importance for ATAs because many are dimers or tetramers with binding sites at the interface formed by residues from two subunits. Another application of for AlphaFold would be to discover the substrate scope of enzymes, which may be used to find ATAs with a desired substrate range in genomic databases, instead of engineering. Other methodologies, including algorithms for stability prediction and definition of search space for mutagenesis, can be refined with the help of AlphaFold-produced structures (Zhang Y. et al., 2021). A recent report indicates that an AlphaFold-generated structure can be of sufficient quality to allow structure-based design since the predicted protein-ligand complexes were indistinguishable from their experimentally-determined counterparts (Wong et al., 2022). In June 2022, the AlphaFold team released 200 million protein structures (https://alphafold.ebi.ac.uk/), which essentially covers all catalogued proteins, significantly increasing the number of protein structures available.
Author contributions
QM drafted the first version of the complete manuscript in consultation with DJ, CR added information about computational methods and edited the text. HW and DJ edited the text.
Funding
The research of HW was supported by the Dutch Ministry of Economic Affairs through BE-Basic, grant FS02.005.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aalbers, F. S., Fürst, M. J. L. J., Rovida, S., Trajkovic, M., Rubén Gómez Castellanos, J., Bartsch, S., et al. (2020). Approaching boiling point stability of an alcohol dehydrogenase through computationally-guided enzyme engineering. Elife 9, e54639. doi:10.7554/eLife.54639
Aguilar, D., Oliva, B., and Marino Buslje, C. (2012). Mapping the mutual information network of enzymatic families in the protein structure to unveil functional features. PLoS One 7, e41430. doi:10.1371/journal.pone.0041430
Anishchenko, I., Pellock, S. J., Chidyausiku, T. M., Ramelot, T. A., Ovchinnikov, S., Hao, J., et al. (2021). De novo protein design by deep network hallucination. Nature 600, 547–552. doi:10.1038/s41586-021-04184-w
Arabnejad, H., Lago, M. D., Jekel, P. A., Floor, R. J., Thunnissen, A. M. W. H., Terwisscha Van Scheltinga, A. C., et al. (2017). A robust cosolvent-compatible halohydrin dehalogenase by computational library design. Protein Eng. Des. Sel. 30, 173–187. doi:10.1093/protein/gzw068
Arnold, F. H. (2018). Directed evolution: Bringing new chemistry to life. Angew. Chem. Int. Ed. 57, 4143–4148. doi:10.1002/anie.201708408
Augustyniak, W., Brzezinska, A. A., Pijning, T., Wienk, H., Boelens, R., Dijkstra, B. W., et al. (2012). Biophysical characterization of mutants of Bacillus subtilis lipase evolved for thermostability: Factors contributing to increased activity retention. Protein Sci. 21, 487–497. doi:10.1002/pro.2031
Berezovsky, I. N., Chen, W. W., Choi, P. J., and Shakhnovich, E. I. (2005). Entropic stabilization of proteins and its proteomic consequences. PLoS Comput. Biol. 1, e47. doi:10.1371/journal.pcbi.0010047
Betz, S. F. (1993). Disulfide bonds and the stability of globular proteins. Protein Sci. 2, 1551–1558. doi:10.1002/pro.5560021002
Birch, M., Challenger, S., Crochard, J., Fradet, D., Jackman, H., Luan, A., et al. (2011). A simplified process for the manufacture of imagabalin hydrochloride (PD-0332334), an α2δ-ligand for the treatment of generalised anxiety disorder. Org. Process Res. Dev. 15, 1358–1364. doi:10.1021/op2002326
Bohnuud, T., Luo, L., Wodak, S. J., Bonvin, A. M. J. J., Weng, Z., Vajda, S., et al. (2017). A benchmark testing ground for integrating homology modeling and protein docking. Proteins. 85, 10–16. doi:10.1002/prot.25063
Börner, T., Rämisch, S., Bartsch, S., Vogel, A., Adlercreutz, P., and Grey, C. (2017a). Three in one: Temperature, solvent and catalytic stability by engineering the cofactor-binding element of amine transaminase. ChemBioChem 18, 1482–1486. doi:10.1002/cbic.201700236
Börner, T., Rämisch, S., Reddem, E. R., Bartsch, S., Vogel, A., Thunnissen, A. M. W. H., et al. (2017b). Explaining operational instability of amine transaminases: Substrate-induced inactivation mechanism and influence of quaternary structure on enzyme-cofactor intermediate stability. ACS Catal. 7, 1259–1269. doi:10.1021/acscatal.6b02100
Bornscheuer, U. T., Huisman, G. W., Kazlauskas, R. J., Lutz, S., Moore, J. C., and Robins, K. (2012). Engineering the third wave of biocatalysis. Nature 485, 185–194. doi:10.1038/nature11117
Bornscheuer, U. T., and Pohl, M. (2001). Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 5, 137–143. doi:10.1016/S1367-5931(00)00182-4
Bu, Y., Cui, Y., Peng, Y., Hu, M., Tian, Y., Tao, Y., et al. (2018). Engineering improved thermostability of the GH11 xylanase from Neocallimastix patriciarum via computational library design. Appl. Microbiol. Biotechnol. 102, 3675–3685. doi:10.1007/s00253-018-8872-1
Cao, J. R., Fan, F. F., Lv, C. J., Wang, H. P., Li, Y., Hu, S., et al. (2021). Improving the thermostability and activity of transaminase from Aspergillus terreus by charge-charge interaction. Front. Chem. 9, 664156. doi:10.3389/fchem.2021.664156
Cassimjee, K. E., Humble, M. S., Land, H., Abedi, V., and Berglund, P. (2012). Chromobacterium violaceum ω-transaminase variant Trp60Cys shows increased specificity for (S)-1-phenylethylamine and 4′-substituted acetophenones, and follows Swain-Lupton parameterisation. Org. Biomol. Chem. 10, 5466–5470. doi:10.1039/c2ob25893e
Chen, S., Land, H., Berglund, P., and Humble, M. S. (2016). Stabilization of an amine transaminase for biocatalysis. J. Mol. Catal. B Enzym. 124, 20–28. doi:10.1016/j.molcatb.2015.11.022
Cheng, F., Chen, X. L., Li, M. Y., Zhang, X. J., Jia, D. X., Wang, Y. J., et al. (2020a). Creation of a robust and R-selective ω-amine transaminase for the asymmetric synthesis of sitagliptin intermediate on a kilogram scale. Enzyme Microb. Technol. 141, 109655. doi:10.1016/j.enzmictec.2020.109655
Cheng, F., Chen, X. L., Xiang, C., Liu, Z. Q., Wang, Y. J., and Zheng, Y. G. (2020b). Fluorescence-based high-throughput screening system for R-ω-transaminase engineering and its substrate scope extension. Appl. Microbiol. Biotechnol. 104, 2999–3009. doi:10.1007/s00253-020-10444-y
Cho, B. K., Park, H. Y., Seo, J. H., Kim, J., Kang, T. J., Lee, B. S., et al. (2008). Redesigning the substrate specificity of ω-aminotransferase for the kinetic resolution of aliphatic chiral amines. Biotechnol. Bioeng. 99, 275–284. doi:10.1002/bit.21591
Contente, M. L., Planchestainer, M., Molinari, F., and Paradisi, F. (2016). Stereoelectronic effects in the reaction of aromatic substrates catalysed by Halomonas elongata transaminase and its mutants. Org. Biomol. Chem. 14, 9306–9311. doi:10.1039/c6ob01629d
Craig, D. B., and Dombkowski, A. A. (2013). Disulfide by design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinforma. 14, 346. doi:10.1186/1471-2105-14-346
De Souza, S. P., Junior, I. I., Silva, G. M. A., Miranda, L. S. M., Santiago, M. F., Leung-Yuk Lam, F., et al. (2016). Cellulose as an efficient matrix for lipase and transaminase immobilization. RSC Adv. 6, 6665–6671. doi:10.1039/c5ra24976g
Deepankumar, K., Nadarajan, S. P., Mathew, S., Lee, S. G., Yoo, T. H., Hong, E. Y., et al. (2015). Engineering transaminase for stability enhancement and site-specific immobilization through multiple noncanonical amino acids incorporation. ChemCatChem 7, 417–421. doi:10.1002/cctc.201402882
Deepankumar, K., Shon, M., Nadarajan, S. P., Shin, G., Mathew, S., Ayyadurai, N., et al. (2014). Enhancing thermostability and organic solvent tolerance of ω-transaminase through global incorporation of fluorotyrosine. Adv. Synth. Catal. 356, 993–998. doi:10.1002/adsc.201300706
Dehouck, Y., Kwasigroch, J. M., Gilis, D., and Rooman, M. (2011). PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinforma. 12, 151. doi:10.1186/1471-2105-12-151
Deszcz, D., Affaticati, P., Ladkau, N., Gegel, A., Ward, J. M., Hailes, H. C., et al. (2015). Single active-site mutants are sufficient to enhance serine:pyruvate α-transaminase activity in an ω-transaminase. FEBS J. 282, 2512–2526. doi:10.1111/febs.13293
Dietrich, S., Borst, N., Schlee, S., Schneider, D., Janda, J. O., Sterner, R., et al. (2012). Experimental assessment of the importance of amino acid positions identified by an entropy-based correlation analysis of multiple-sequence alignments. Biochemistry 51, 5633–5641. doi:10.1021/bi300747r
Dourado, D. F. A. R., Pohle, S., Carvalho, A. T. P., Dheeman, D. S., Caswell, J. M., Skvortsov, T., et al. (2016). Rational design of a (S)-selective-transaminase for asymmetric synthesis of (1S)-1-(1, 1’-biphenyl-2-yl) ethanamine. ACS Catal. 6, 7749–7759. doi:10.1021/acscatal.6b02380
Drienovská, I., and Roelfes, G. (2020). Expanding the enzyme universe with genetically encoded unnatural amino acids. Nat. Catal. 3, 193–202. doi:10.1038/s41929-019-0410-8
Eliot, A. C., and Kirsch, J. F. (2004). Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415. doi:10.1146/annurev.biochem.73.011303.074021
Ellingson, S. R., Davis, B., and Allen, J. (2020). Machine learning and ligand binding predictions: A review of data, methods, and obstacles. Biochimica Biophysica Acta - General Subj. 1864, 129545. doi:10.1016/j.bbagen.2020.129545
Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A. W., Green, T., et al. (2022). Protein complex prediction with AlphaFold-Multimer. bioRxiv. doi:10.1101/2021.10.04.463034
Feehan, R., Montezano, D., and Slusky, J. S. G. (2021). Machine learning for enzyme engineering, selection and design. Protein Eng. Des. Sel. 34, gzab019–10. doi:10.1093/protein/gzab019
Fernandez-Lafuente, R. (2009). Stabilization of multimeric enzymes: Strategies to prevent subunit dissociation. Enzyme Microb. Technol. 45, 405–418. doi:10.1016/j.enzmictec.2009.08.009
Ferrandi, E. E., and Monti, D. (2018). Amine transaminases in chiral amines synthesis: Recent advances and challenges. World J. Microbiol. Biotechnol. 34, 13. doi:10.1007/s11274-017-2395-2
Floor, R. J., Wijma, H. J., Colpa, D. I., Ramos-Silva, A., Jekel, P. A., Szymański, W., et al. (2014). Computational library design for increasing haloalkane dehalogenase stability. ChemBioChem 15, 1660–1672. doi:10.1002/cbic.201402128
Fürst, M. J. L. J., Boonstra, M., Bandstra, S., and Fraaije, M. W. (2019). Stabilization of cyclohexanone monooxygenase by computational and experimental library design. Biotechnol. Bioeng. 116, 2167–2177. doi:10.1002/bit.27022
Gao, X., Zhang, X., Zhu, N., Mou, Y., Zhang, H., Liu, X., et al. (2020). Reshaping the substrate binding region of (R)-selective ω-transaminase for asymmetric synthesis of (R)-3-amino-1-butanol. Appl. Microbiol. Biotechnol. 104, 3959–3969. doi:10.1007/s00253-020-10539-6
Genz, M., Melse, O., Schmidt, S., Vickers, C., Dörr, M., van den Bergh, T., et al. (2016). Engineering the amine transaminase from Vibrio fluvialis towards branched-chain substrates. ChemCatChem 8, 3199–3202. doi:10.1002/cctc.201601007
Genz, M., Vickers, C., van den Bergh, T., Joosten, H. J., Dörr, M., Höhne, M., et al. (2015). Alteration of the donor/acceptor spectrum of the (S)-amine transaminase from Vibrio fluvialis. Int. J. Mol. Sci. 16, 26953–26963. doi:10.3390/ijms161126007
Ghislieri, D., and Turner, N. J. (2014). Biocatalytic approaches to the synthesis of enantiomerically pure chiral amines. Top. Catal. 57, 284–300. doi:10.1007/s11244-013-0184-1
Glaser, F., Pupko, T., Paz, I., Bell, R. E., Bechor-Shental, D., Martz, E., et al. (2003). ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164. doi:10.1093/bioinformatics/19.1.163
Gomm, A., and O’Reilly, E. (2018). Transaminases for chiral amine synthesis. Curr. Opin. Chem. Biol. 43, 106–112. doi:10.1016/j.cbpa.2017.12.007
Green, A. P., Turner, N. J., and O’Reilly, E. (2014). Chiral amine synthesis using ω-transaminases: An amine donor that displaces equilibria and enables high-throughput screening. Angew. Chem. Int. Ed. 53, 10714–10717. doi:10.1002/anie.201406571
Guan, L. J., Ohtsuka, J., Okai, M., Miyakawa, T., Mase, T., Zhi, Y., et al. (2015). A new target region for changing the substrate specificity of amine transaminases. Sci. Rep. 5, 10753. doi:10.1038/srep10753
Guerois, R., Nielsen, J. E., and Serrano, L. (2002). Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 320, 369–387. doi:10.1016/S0022-2836(02)00442-4
Guidi, B., Planchestainer, M., Contente, M. L., Laurenzi, T., Eberini, I., Gourlay, L. J., et al. (2018). Strategic single point mutation yields a solvent- and salt-stable transaminase from Virgibacillus sp. in soluble form. Sci. Rep. 8, 16441. doi:10.1038/s41598-018-34434-3
Guo, F., and Berglund, P. (2017). Transaminase biocatalysis: Optimization and application. Green Chem. 19, 333–360. doi:10.1039/c6gc02328b
Han, R., Cao, X., Fang, H., Zhou, J., and Ni, Y. (2021). Structure-based engineering of ω-transaminase for enhanced catalytic efficiency toward (R)-(+)-1-(1-naphthyl)ethylamine synthesis. Mol. Catal. 502, 111368. doi:10.1016/j.mcat.2020.111368
Han, S. W., Kim, J., Cho, H. S., and Shin, J. S. (2017). Active site engineering of ω-transaminase guided by docking orientation analysis and virtual activity screening. ACS Catal. 7, 3752–3762. doi:10.1021/acscatal.6b03242
Han, S. W., Park, E. S., Dong, J. Y., and Shin, J. S. (2015a). Active-site engineering of ω-transaminase for production of unnatural amino acids carrying a side chain bulkier than an ethyl substituent. Appl. Environ. Microbiol. 81, 6994–7002. doi:10.1128/AEM.01533-15
Han, S. W., Park, E. S., Dong, J. Y., and Shin, J. S. (2015b). Expanding substrate specificity of ω-transaminase by rational remodeling of a large substrate-binding pocket. Adv. Synth. Catal. 357, 2712–2720. doi:10.1002/adsc.201500239
Han, S. W., Park, E. S., Dong, J. Y., and Shin, J. S. (2015c). Mechanism-guided engineering of ω-transaminase to accelerate reductive amination of ketones. Adv. Synth. Catal. 357, 1732–1740. doi:10.1002/adsc.201500211
Huang, J., Xie, D. F., and Feng, Y. (2017). Engineering thermostable (R)-selective amine transaminase from Aspergillus terreus through in silico design employing B-factor and folding free energy calculations. Biochem. Biophys. Res. Commun. 483, 397–402. doi:10.1016/j.bbrc.2016.12.131
Humble, M. S., Cassimjee, K. E., Abedi, V., Federsel, H.-J., and Berglund, P. (2012a). Key amino acid residues for reversed or improved enantiospecificity of an ω-transaminase. ChemCatChem 4, 1167–1172. doi:10.1002/cctc.201100487
Humble, M. S., Cassimjee, K. E., Hãkansson, M., Kimbung, Y. R., Walse, B., Abedi, V., et al. (2012b). Crystal structures of the Chromobacterium violaceum ω-transaminase reveal major structural rearrangements upon binding of coenzyme PLP. FEBS J. 279, 779–792. doi:10.1111/j.1742-4658.2012.08468.x
Hwang, B. Y., and Kim, B. G. (2004). High-throughput screening method for the identification of active and enantioselective ω-transaminases. Enzyme Microb. Technol. 34, 429–436. doi:10.1016/j.enzmictec.2003.11.019
Jia, D., Wang, F., Zhao, R., Gu, B., Peng, C., Jin, L., et al. (2022). Engineering novel (R)-selective transaminase for efficient symmetric synthesis of D-alanine. Appl. Environ. Microbiol. 88, e0006222. doi:10.1128/aem.00062-22
Jia, D. X., Peng, C., Li, J. L., Wang, F., Liu, Z. Q., and Zheng, Y. G. (2021a). Redesign of (R)-omega-transaminase and its application for synthesizing amino acids with bulky side chain. Appl. Biochem. Biotechnol. 193, 3624–3640. doi:10.1007/s12010-021-03616-7
Jia, L. L., Sun, T. T., Wang, Y., and Shen, Y. (2021b). A machine learning study on the thermostability prediction of (R)-ω-selective amine transaminase from Aspergillus terreus. Biomed. Res. Int. 2021, 1–7. doi:10.1155/2021/2593748
John, R. A. (1995). Pyridoxal phosphate-dependent enzymes. Biochimica Biophysica Acta - Protein Struct. Mol. Enzym. 1248, 81. doi:10.1016/0167-4838(95)00025-p
Johnson, S. M., Wiseman, R. L., Sekijima, Y., Green, N. S., Adamski-Werner, S. L., and Kelly, J. W. (2005). Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Acc. Chem. Res. 38, 911–921. doi:10.1021/ar020073i
Jones, B. J., Lim, H. Y., Huang, J., and Kazlauskas, R. J. (2017). Comparison of five protein engineering strategies for stabilizing an α/β-Hydrolase. Biochemistry 56, 6521–6532. doi:10.1021/acs.biochem.7b00571
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kelly, S. A., Mix, S., Moody, T. S., and Gilmore, B. F. (2020). Transaminases for industrial biocatalysis: Novel enzyme discovery. Appl. Microbiol. Biotechnol. 104, 4781–4794. doi:10.1007/s00253-020-10585-0
Kelly, S. A., Pohle, S., Wharry, S., Mix, S., Allen, C. C. R., Moody, T. S., et al. (2018). Application of ω-transaminases in the pharmaceutical industry. Chem. Rev. 118, 349–367. doi:10.1021/acs.chemrev.7b00437
Kim, H. G., Han, S. W., and Shin, J. S. (2019). Combinatorial mutation analysis of ω-transaminase to create an engineered variant capable of asymmetric amination of isobutyrophenone. Adv. Synth. Catal. 361, adsc.201900184–2606. doi:10.1002/adsc.201900184
Kiss, G., Çelebi-Ölçüm, N., Moretti, R., Baker, D., and Houk, K. N. (2013). Computational enzyme design. Angew. Chem. Int. Ed. 52, 5700–5725. doi:10.1002/anie.201204077
Konia, E., Chatzicharalampous, K., Drakonaki, A., Muenke, C., Ermler, U., Tsiotis, G., et al. (2021). Rational engineering of Luminiphilus syltensis (R)-selective amine transaminase for the acceptance of bulky substrates. Chem. Commun. 57, 12948–12951. doi:10.1039/d1cc04664k
Koszelewski, D., Tauber, K., Faber, K., and Kroutil, W. (2010). ω-Transaminases for the synthesis of non-racemic α-chiral primary amines. Trends Biotechnol. 28, 324–332. doi:10.1016/j.tibtech.2010.03.003
Kuchner, O., and Arnold, F. H. (1997). Directed evolution of enzyme catalysts. Trends Biotechnol. 15, 523–530. doi:10.1016/S0167-7799(97)01138-4
Kuipers, R. K., Joosten, H. J., Van Berkel, W. J. H., Leferink, N. G. H., Rooijen, E., Ittmann, E., et al. (2010). 3DM: Systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins 78, 2101–2113. doi:10.1002/prot.22725
Land, H., Campillo-Brocal, J. C., Svedendahl Humble, M., and Berglund, P. (2019). B-factor guided proline substitutions in Chromobacterium violaceum amine transaminase: Evaluation of the proline rule as a method for enzyme stabilization. ChemBioChem 20, 1297–1304. doi:10.1002/cbic.201800749
Land, H., Ruggieri, F., Szekrenyi, A., Fessner, W., and Berglund, P. (2020). Engineering the active site of an (S)-selective amine transaminase for acceptance of doubly bulky primary amines. Adv. Synth. Catal. 362, 812–821. doi:10.1002/adsc.201901252
Li, F., Yuan, L., Lu, H., Li, G., Chen, Y., Engqvist, M. K. M., et al. (2022). Deep learning-based k cat prediction enables improved enzyme-constrained model reconstruction. Nat. Catal. 5, 662–672. doi:10.1038/s41929-022-00798-z
Li, Y., Wang, S., Umarov, R., Xie, B., Fan, M., Li, L., et al. (2018). DEEPre: Sequence-based enzyme EC number prediction by deep learning. Bioinformatics 34, 760–769. doi:10.1093/bioinformatics/btx680
Liu, C. Y., Cecylia Severin, L., Lyu, C. J., Zhu, W. L., Wang, H. P., Jiang, C. J., et al. (2021a). Improving thermostability of (R)-selective amine transaminase from Aspergillus terreus by evolutionary coupling saturation mutagenesis. Biochem. Eng. J. 167, 107926. doi:10.1016/j.bej.2021.107926
Liu, Q., Xie, X., Tang, M., Tao, W., Shi, T., Zhang, Y., et al. (2021b). One-pot asymmetric synthesis of an aminodiol intermediate of florfenicol using engineered transketolase and transaminase. ACS Catal. 11, 7477–7488. doi:10.1021/acscatal.1c01229
Lyne, P. D., Lamb, M. L., and Saeh, J. C. (2006). Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 49, 4805–4808. doi:10.1021/jm060522a
Ma, Y., Jiao, X., Wang, Z., Mu, H., Sun, K., Li, X., et al. (2022). Engineering a transaminase for the efficient synthesis of a key intermediate for rimegepant. Org. Process Res. Dev. 26, 1971–1977. doi:10.1021/acs.oprd.1c00376
Marqusee, S., and Sauer, R. T. (1994). Contributions of a hydrogen bond/salt bridge network to the stability of secondary and tertiary structure in λ repressor. Protein Sci. 3, 2217–2225. doi:10.1002/pro.5560031207
Martin, A. R., DiSanto, R., Plotnikov, I., Kamat, S., Shonnard, D., and Pannuri, S. (2007). Improved activity and thermostability of (S)-aminotransferase by error-prone polymerase chain reaction for the production of a chiral amine. Biochem. Eng. J. 37, 246–255. doi:10.1016/j.bej.2007.05.001
Martin, C., Ovalle Maqueo, A., Wijma, H. J., and Fraaije, M. W. (2018). Creating a more robust 5-hydroxymethylfurfural oxidase by combining computational predictions with a novel effective library design. Biotechnol. Biofuels 11, 56. doi:10.1186/s13068-018-1051-x
Mathew, S., Nadarajan, S. P., Chung, T., Park, H. H., and Yun, H. (2016). Biochemical characterization of thermostable ω-transaminase from Sphaerobacter thermophilus and its application for producing aromatic β- and γ-amino acids. Enzyme Microb. Technol. 87, 52–60. –88. doi:10.1016/j.enzmictec.2016.02.013
Mathew, S., and Yun, H. (2012). ω-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal. 2, 993–1001. doi:10.1021/cs300116n
Mazurenko, S., Prokop, Z., and Damborsky, J. (2020). Machine learning in enzyme engineering. ACS Catal. 10, 1210–1223. doi:10.1021/acscatal.9b04321
Mehta, P. K., Hale, T. I., and Christen, P. (1993). Aminotransferases: Demonstration of homology and division into evolutionary subgroups. Eur. J. Biochem. 214, 549–561. doi:10.1111/j.1432-1033.1993.tb17953.x
Meng, Q., Capra, N., Palacio, C. M., Lanfranchi, E., Otzen, M., Van Schie, L. Z., et al. (2020). Robust ω-transaminases by computational stabilization of the subunit interface. ACS Catal. 10, 2915–2928. doi:10.1021/acscatal.9b05223
Meng, Q., Ramírez-Palacios, C., Capra, N., Hooghwinkel, M. E., Thallmair, S., Rozeboom, H. J., et al. (2021). Computational redesign of an ω-transaminase from Pseudomonas jessenii for asymmetric synthesis of enantiopure bulky amines. ACS Catal. 11, 10733–10747. doi:10.1021/acscatal.1c02053
Merkel, L., and Budisa, N. (2012). Organic fluorine as a polypeptide building element: In vivo expression of fluorinated peptides, proteins and proteomes. Org. Biomol. Chem. 10, 7241–7261. doi:10.1039/c2ob06922a
Midelfort, K. S., Kumar, R., Han, S., Karmilowicz, M. J., McConnell, K., Gehlhaar, D. K., et al. (2013). Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng. Des. Sel. 26, 25–33. doi:10.1093/protein/gzs065
Niesen, F. H., Berglund, H., and Vedadi, M. (2007). The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221. doi:10.1038/nprot.2007.321
Nikolova, P. V., Henckel, J., Lane, D. P., and Fersht, A. R. (1998). Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. U. S. A. 95, 14675–14680. doi:10.1073/pnas.95.25.14675
Nobili, A., Steffen-Munsberg, F., Kohls, H., Trentin, I., Schulzke, C., Höhne, M., et al. (2015). Engineering the active site of the amine transaminase from Vibrio fluvialis for the asymmetric synthesis of aryl-alkyl amines and amino alcohols. ChemCatChem 7, 757–760. doi:10.1002/cctc.201403010
Novick, S. J., Dellas, N., Garcia, R., Ching, C., Bautista, A., Homan, D., et al. (2021). Engineering an amine transaminase for the efficient production of a chiral sacubitril precursor. ACS Catal. 11, 3762–3770. doi:10.1021/acscatal.0c05450
Oliveira, E. F., Cerqueira, N. M. F. S. A., Fernandes, P. A., and Ramos, M. J. (2011). Mechanism of formation of the internal aldimine in pyridoxal 5′-phosphate-dependent enzymes. J. Am. Chem. Soc. 133, 15496–15505. doi:10.1021/ja204229m
Ovchinnikov, S., and Huang, P. S. (2021). Structure-based protein design with deep learning. Curr. Opin. Chem. Biol. 65, 136–144. doi:10.1016/j.cbpa.2021.08.004
Pagar, A. D., Jeon, H., Khobragade, T. P., Sarak, S., Giri, P., Lim, S., et al. (2022). Non-canonical amino acid-based engineering of (R)-amine transaminase. Front. Chem. 10, 839636. doi:10.3389/fchem.2022.839636
Pagar, A. D., Patil, M. D., Flood, D. T., Yoo, T. H., Dawson, P. E., and Yun, H. (2021). Recent advances in biocatalysis with chemical modification and expanded amino acid alphabet. Chem. Rev. 121, 6173–6245. doi:10.1021/acs.chemrev.0c01201
Park, E. S., Park, S. R., Han, S. W., Dong, J. Y., and Shin, J. S. (2014). Structural determinants for the non-canonical substrate specificity of the ω-transaminase from Paracoccus denitrificans. Adv. Synth. Catal. 356, 212–220. doi:10.1002/adsc.201300786
Patil, M. D., Grogan, G., Bommarius, A., and Yun, H. (2018). Recent advances in ω-transaminase-mediated biocatalysis for the enantioselective synthesis of chiral amines. Catalysts 8, 254. doi:10.3390/catal8070254
Pavlidis, I. V., Weib, M. S., Genz, M., Spurr, P., Hanlon, S. P., Wirz, B., et al. (2016). Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat. Chem. 8, 1076–1082. doi:10.1038/nchem.2578
Percudani, R., and Peracchi, A. (2009). The B6 database: A tool for the description and classification of vitamin B6-dependent enzymatic activities and of the corresponding protein families. BMC Bioinforma. 10, 273. doi:10.1186/1471-2105-10-273
Pokkuluri, P. R., Raffen, R., Dieckman, L., Boogaard, C., Stevens, F. J., and Schiffer, M. (2002). Increasing protein stability by polar surface residues: Domain-wide consequences of interactions within a loop. Biophys. J. 82, 391–398. doi:10.1016/S0006-3495(02)75403-9
Radisky, E. S., and Koshland, D. E. (2002). A clogged gutter mechanism for protease inhibitors. Proc. Natl. Acad. Sci. U. S. A. 99, 10316–10321. doi:10.1073/pnas.112332899
Reetz, M. T., Carballeira, J. D., and Vogel, A. (2006). Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew. Chem. Int. Ed. 45, 7745–7751. doi:10.1002/anie.200602795
Richter, F., Leaver-Fay, A., Khare, S. D., Bjelic, S., and Baker, D. (2011). De novo enzyme design using Rosetta3. PLoS One 6, e19230. doi:10.1371/journal.pone.0019230
Rocha, R. A., Speight, R. E., and Scott, C. (2022). Engineering enzyme properties for improved biocatalytic processes in batch and continuous flow. Org. Process Res. Dev. 26, 1914–1924. doi:10.1021/acs.oprd.1c00424
Roura Padrosa, D., Alaux, R., Smith, P., Dreveny, I., López-Gallego, F., and Paradisi, F. (2019). Enhancing PLP-binding capacity of class-III ω-transaminase by single residue substitution. Front. Bioeng. Biotechnol. 7, 282. doi:10.3389/fbioe.2019.00282
Sastry, G. M., Inakollu, V. S. S., and Sherman, W. (2013). Boosting virtual screening enrichments with data fusion: Coalescing hits from two-dimensional fingerprints, shape, and docking. J. Chem. Inf. Model. 53, 1531–1542. doi:10.1021/ci300463g
Savile, C. K., Janey, J. M., Mundorff, E. C., Moore, J. C., Tam, S., Jarvis, W. R., et al. (2010). Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329, 305–309. doi:10.1126/science.1188934
Sheludko, Y. V., Slagman, S., Gittings, S., Charnock, S. J., Land, H., Berglund, P., et al. (2022). Enantioselective synthesis of pharmaceutically relevant bulky arylbutylamines using engineered transaminases. Adv. Synth. Catal. 364, 2972–2981. doi:10.1002/adsc.202200403
Shin, J. S., and Kim, B. G. (1999). Asymmetric synthesis of chiral amines with ω-transaminase. Biotechnol. Bioeng. 65, 206–211. Available at:. doi:10.1002/(SICI)1097-0290(19991020)65:2%3C206:AID-BIT11%3E3.0.CO;2-9
Shin, J. S., and Kim, B. G. (2002). Exploring the active site of amine:pyruvate aminotransferase on the basis of the substrate structure-reactivity relationship: How the enzyme controls substrate specificity and stereoselectivity. J. Org. Chem. 67, 2848–2853. doi:10.1021/jo016115i
Simonetti, F. L., Teppa, E., Chernomoretz, A., Nielsen, M., and Marino Buslje, C. (2013). Mistic: Mutual information server to infer coevolution. Nucleic Acids Res. 41, 8–14. doi:10.1093/nar/gkt427
Sirin, S., Kumar, R., Martinez, C., Karmilowicz, M. J., Ghosh, P., Abramov, Y. A., et al. (2014). A computational approach to enzyme design: Predicting ω-aminotransferase catalytic activity using docking and MM-GBSA scoring. J. Chem. Inf. Model. 54, 2334–2346. doi:10.1021/ci5002185
Skalden, L., Peters, C., Dickerhoff, J., Nobili, A., Joosten, H. J., Weisz, K., et al. (2015). Two subtle amino acid changes in a transaminase substantially enhance or invert enantiopreference in cascade syntheses. ChemBioChem 16, 1041–1045. doi:10.1002/cbic.201500074
Slabu, I., Galman, J. L., Lloyd, R. C., and Turner, N. J. (2017). Discovery, engineering, and synthetic application of transaminase biocatalysts. ACS Catal. 7, 8263–8284. doi:10.1021/acscatal.7b02686
Sowdhamini, R., Srinivasan, N., Shoichet, B., Santi, D. V., Ramakrishnan, C., and Balaram, P. (1989). Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis. Protein Eng. Des. Sel. 3, 95–103. doi:10.1093/protein/3.2.95
Steffen-Munsberg, F., Vickers, C., Thontowi, A., Schätzle, S., Meinhardt, T., Humble, S., et al. (2013). Revealing the structural basis of promiscuous amine transaminase activity. ChemCatChem 5, 154–157. doi:10.1002/cctc.201200545
Steiner, T., Hess, P., Bae, J. H., Wiltschi, B., Moroder, L., and Budisa, N. (2008). Synthetic biology of proteins: Tuning GFPs folding and stability with fluoroproline. PLoS One 3, e1680. doi:10.1371/journal.pone.0001680
Steipe, B., Schiller, B., Plückthun, A., and Steinbacher, S. (1994). Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240, 188–192. doi:10.1006/jmbi.1994.1434
Strokach, A., Becerra, D., Corbi-Verge, C., Perez-Riba, A., and Kim, P. M. (2021). Computational generation of proteins with predetermined three-dimensional shapes using ProteinSolver. Star. Protoc. 2, 100505. doi:10.1016/j.xpro.2021.100505
Svedendahl, M., Branneby, C., Lindberg, L., and Berglund, P. (2010). Reversed enantiopreference of an ω-transaminase by a single-point mutation. ChemCatChem 2, 976–980. doi:10.1002/cctc.201000107
Telzerow, A., Paris, J., Håkansson, M., González-Sabín, J., Ríos-Lombardía, N., Schürmann, M., et al. (2019). Amine transaminase from Exophiala xenobiotica - crystal structure and engineering of a Fold IV transaminase that naturally converts biaryl ketones. ACS Catal. 9, 1140–1148. doi:10.1021/acscatal.8b04524
Thornton, J. W., Need, E., and Crews, D. (2003). Resurrecting the ancestral steroid receptor: Ancient origin of estrogen signaling. Science 301, 1714–1717. doi:10.1126/science.1086185
Tufvesson, P., Lima-Ramos, J., Jensen, J. S., Al-Haque, N., Neto, W., and Woodley, J. M. (2011). Process considerations for the asymmetric synthesis of chiral amines using transaminases. Biotechnol. Bioeng. 108, 1479–1493. doi:10.1002/bit.23154
Turner, N. J. (2009). Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 5, 567–573. doi:10.1038/nchembio.203
Vlassi, M., Cesareni, G., and Kokkinidis, M. (1999). A correlation between the loss of hydrophobic core packing interactions and protein stability 1 1Edited by A. R. Fersht. J. Mol. Biol. 285, 817–827. doi:10.1006/jmbi.1998.2342
Voss, M., Das, D., Genz, M., Kumar, A., Kulkarni, N., Kustosz, J., et al. (2018). In silico based engineering approach to improve transaminases for the conversion of bulky substrates. ACS Catal. 8, 11524–11533. doi:10.1021/acscatal.8b03900
Voss, M., Xiang, C., Esque, J., Nobili, A., Menke, M. J., André, I., et al. (2020). Creation of (R)-amine transaminase activity within an α-amino acid transaminase scaffold. ACS Chem. Biol. 15, 416–424. doi:10.1021/acschembio.9b00888
Wang, C., Tang, K., Dai, Y., Jia, H., Li, Y., Gao, Z., et al. (2021a). Identification, characterization, and site-specific mutagenesis of a thermostable ω-transaminase from Chloroflexi bacterium. ACS Omega 6, 17058–17070. doi:10.1021/acsomega.1c02164
Wang, J., Lisanza, S., Juergens, D., Tischer, D., Watson, J. L., Anishchenko, I., et al. (2022). Scaffolding protein functional sites using deep learning. Science 394, 387–394. doi:10.1126/science.abn2100
Wang, Y., Feng, J., Dong, W., Chen, X., Yao, P., Wu, Q., et al. (2021b). Improving catalytic activity and reversing enantio-specificity of ω-transaminase by semi-rational engineering en route to chiral bulky β-amino esters. ChemCatChem 13, 3396–3400. doi:10.1002/cctc.202100503
Watanabe, K., Ohkuri, T., Yokobori, S., and Yamagishi, A. (2006). Designing thermostable proteins : Ancestral mutants of 3-isopropylmalate dehydrogenase designed by using a phylogenetic tree. J. Mol. Biol. 355, 664–674. doi:10.1016/j.jmb.2005.10.011
Watanabe, K., and Suzuki, Y. (1998). Protein thermostabilization by proline substitutions. J. Mol. Catal. B Enzym. 4, 167–180. doi:10.1016/S1381-1177(97)00031-3
Weiss, M. S., Pavlidis, I. V., Spurr, P., Hanlon, S. P., Wirz, B., Iding, H., et al. (2017). Amine transaminase engineering for spatially bulky substrate acceptance. ChemBioChem 18, 1022–1026. doi:10.1002/cbic.201700033
Weiss, M. S., Pavlidis, I. V., Spurr, P., Hanlon, S. P., Wirz, B., Iding, H., et al. (2016). Protein-engineering of an amine transaminase for the stereoselective synthesis of a pharmaceutically relevant bicyclic amine. Org. Biomol. Chem. 14, 10249–10254. doi:10.1039/c6ob02139e
Weiss, M. S., Pavlidis, I. V., Vickers, C., Hohne, M., and Bornscheuer, U. T. (2014). Glycine oxidase based high-throughput solid-phase assay for substrate profiling and directed evolution of (R)- and (S)-selective amine transaminases. Anal. Chem. 86, 11847–11853. doi:10.1021/ac503445y
Wheeler, L. C., Lim, S. A., Marqusee, S., and Harms, M. J. (2016). The thermostability and specificity of ancient proteins. Curr. Opin. Struct. Biol. 38, 37–43. doi:10.1016/j.sbi.2016.05.015
Wijma, H. J., Floor, R. J., Jekel, P. A., Baker, D., Marrink, S. J., and Janssen, D. B. (2014). Computationally designed libraries for rapid enzyme stabilization. Protein Eng. Des. Sel. 27, 49–58. doi:10.1093/protein/gzt061
Wijma, H. J., Fürst, M. J. L. J., and Janssen, D. B. (2018). A computational library design protocol for rapid improvement of protein stability: FRESCO. Methods Mol. Biol. 1685, 69–85. doi:10.1007/978-1-4939-7366-8_5
Wilding, M., Peat, T. S., Kalyaanamoorthy, S., Newman, J., Scott, C., and Jermiin, L. S. (2017). Reverse engineering: Transaminase biocatalyst development using ancestral sequence reconstruction. Green Chem. 19, 5375–5380. doi:10.1039/c7gc02343j
Wong, F., Krishnan, A., Zheng, E. J., Stark, H., Manson, A. L., Earl, A. M., et al. (2022). Benchmarking AlphaFold -enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 18, e11081. doi:10.15252/msb.202211081
Wu, B., Wijma, H. J., Song, L., Rozeboom, H. J., Poloni, C., Tian, Y., et al. (2016). Versatile peptide C-terminal functionalization via a computationally engineered peptide amidase. ACS Catal. 6, 5405–5414. doi:10.1021/acscatal.6b01062
Xiang, C., Wu, S., and Bornscheuer, U. T. (2021). Directed evolution of an amine transaminase for the synthesis of an Apremilast intermediate via kinetic resolution. Bioorg. Med. Chem. 43, 116271. doi:10.1016/j.bmc.2021.116271
Xie, D. F., Fang, H., Mei, J. Q., Gong, J. Y., Wang, H. P., Shen, X. Y., et al. (2018). Improving thermostability of (R)-selective amine transaminase from Aspergillus terreus through introduction of disulfide bonds. Biotechnol. Appl. Biochem. 65, 255–262. doi:10.1002/bab.1572
Xie, D. F., Yang, J. X., Lv, C. J., Mei, J. Q., Wang, H. P., Hu, S., et al. (2019). Construction of stabilized (R)-selective amine transaminase from Aspergillus terreus by consensus mutagenesis. J. Biotechnol. 293, 8–16. doi:10.1016/j.jbiotec.2019.01.007
Xie, Y., Xu, F., Yang, L., Liu, H., Xu, X., Wang, H., et al. (2021). Engineering the large pocket of an (S)-selective transaminase for asymmetric synthesis of (S)-1-amino-1-phenylpropane. Catal. Sci. Technol. 11, 2461–2470. doi:10.1039/d0cy02426k
Xie, Z., Zhai, L., Meng, D., Tian, Q., Guan, Z., Cai, Y., et al. (2020). Improving the catalytic thermostability of Bacillus altitudinis W3 ω-transaminase by proline substitutions. Biotech 10, 323. doi:10.1007/s13205-020-02321-2
Yang, K. K., Wu, Z., and Arnold, F. H. (2019). Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694. doi:10.1038/s41592-019-0496-6
Zhai, L., Yang, S., Lai, Y., Meng, D., Tian, Q., Guan, Z., et al. (2019). Effect of residue substitution via site-directed mutagenesis on activity and steroselectivity of transaminase BpTA from Bacillus pumilus W3 for sitafloxacin hydrate intermediate. Int. J. Biol. Macromol. 137, 732–740. doi:10.1016/j.ijbiomac.2019.07.027
Zhang, L., Tang, X., Cui, D., Yao, Z., Gao, B., Jiang, S., et al. (2014). A method to rationally increase protein stability based on the charge-charge interaction, with application to lipase LipK107. Protein Sci. 23, 110–116. doi:10.1002/pro.2388
Zhang, Y., Li, P., Pan, F., Liu, H., Hong, P., Liu, X., et al. (2021a). Applications of AlphaFold beyond protein structure prediction. bioRxiv. doi:10.1101/2021.11.03.467194
Zhang, Z., Liu, Y., Zhao, J., Li, W., Hu, R., Li, X., et al. (2021b). Active-site engineering of ω-transaminase from Ochrobactrum anthropi for preparation of L-2-aminobutyric acid. BMC Biotechnol. 21, 55. doi:10.1186/s12896-021-00713-7
Zhao, J., Cao, Y., and Zhang, L. (2020). Exploring the computational methods for protein-ligand binding site prediction. Comput. Struct. Biotechnol. J. 18, 417–426. doi:10.1016/j.csbj.2020.02.008
Keywords: aminotransferase, enantiopure amine, protein design, thermostability, substrate specificity
Citation: Meng Q, Ramírez-Palacios C, Wijma HJ and Janssen DB (2022) Protein engineering of amine transaminases. Front. Catal. 2:1049179. doi: 10.3389/fctls.2022.1049179
Received: 20 September 2022; Accepted: 01 November 2022;
Published: 22 November 2022.
Edited by:
Andreas Schneider, University of Stuttgart, GermanyReviewed by:
Elaine O'Reilly, University College Dublin, IrelandIoannis V. Pavlidis, University of Crete, Greece
Copyright © 2022 Meng, Ramírez-Palacios, Wijma and Janssen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dick B. Janssen, ZC5iLmphbnNzZW5AcnVnLm5s