 Adelina Ivanova
Adelina Ivanova Francesco Falcioni
Francesco Falcioni
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Catal. , 03 February 2022
Sec. Biocatalysis
Volume 1 - 2021 | https://doi.org/10.3389/fctls.2021.810779
Variability in the glycosylation profile of therapeutic monoclonal antibodies (mAbs), due to recombinant production technologies, leads to inconsistencies in effector functions and pharmacokinetic properties, both batch-to-batch and within single batches. It also poses regulatory concerns over the effectiveness of commercially available formulations. In vitro chemoenzymatic glycoengineering of variants displaying a homogeneous glycan profile is a trending strategy for ensuring consistent, controlled, and enhanced therapeutic performance, but reported successes are largely limited to small-scale applications. The major challenges for the industrial-scale introduction of the technique stem from the need for activated sugar donors, which can participate in undesired side reactions, and from the economic cost of the additional enzymatic steps and purification stages. While recent developments within the area address some of these obstacles, it appears that more effort is required in order to access the untapped potential of biocatalysis to enable the robust production of therapeutically superior constructs.
Monoclonal antibodies (mAbs) are established pharmaceutical modalities with rapidly expanding application in multiple therapeutic areas (Lin et al., 2015; Seeberger and Cummings, 2017; Li et al., 2020). In 2019, six out of the ten best-selling drugs world-wide were mAbs, treating macular degeneration, autoimmune conditions, and cancer (Urquhart, 2020). mAbs possess a conserved N-glycosylation site at Asparagine 297 (Asn297) in their crystallizable fragment (Fc), with its carbohydrate (glycan) identity dictating Fcγ-receptor (FcγR)- mediated pro- and anti-inflammatory activity and mAb stability (Reusch and Tejada, 2015). This N-glycosylation site is considered a critical quality attribute for mAbs that depend on Fc-mediated effector functions (Reason et al., 2014; Reusch and Tejada, 2015); its profound impact on mAb-dependent cell-mediated cytotoxicity (ADCC) and mAb-mediated inflammation has been studied in-depth (Batra and Rathore, 2016; Mimura et al., 2018). Changes in the available glycoforms (i.e. mAbs with identical protein component, but different Asn297 glycan composition) in therapeutic formulations can also affect the pharmacokinetics of mAbs, as exemplified in the C-type lectin clearance mechanism which is triggered by hypermannosylation (Goetze et al., 2011; Liu et al., 2013; Liu, 2015). Glycans on natural human antibodies are usually of the bi-antennary complex type (Figure 1). Reliable production of targeted homogenous glycoforms is thus of pivotal importance for consistent, optimal therapeutic performance and improved pharmacokinetic profile of such drugs (Jefferis, 2009).
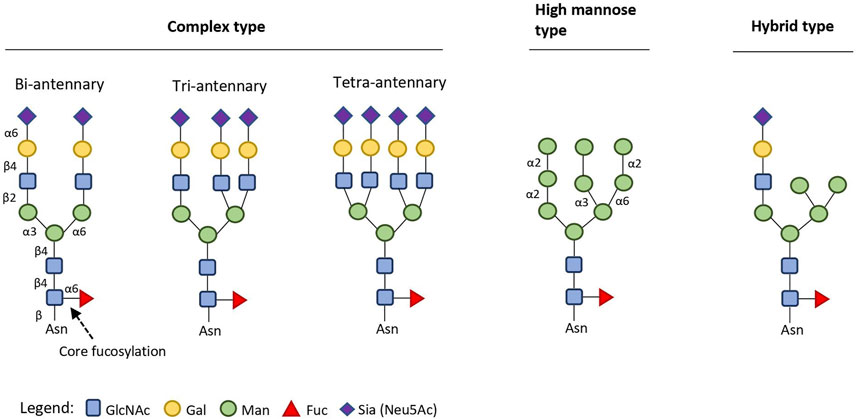
FIGURE 1. Structural types of endogenous eukaryotic N-linked (to Asn residues) glycans (2015–2017). Conserved glycosidic bonds are omitted for clarity. Complex type glycans can have up to six antennae (starting with a GlcNAc unit), each elongated with Gal-β-(1,4)-GlcNAc (LacNAc) repeats. Recommended standard symbols for monosaccharides and bond regiochemistry labelling are used throughout the review (Varki et al., 2015).
While mammalian systems like Chinese hamster ovary (CHO) and mouse myeloma (NS0, SP2/0) cells are the traditional gold standard for recombinant mAb production, these cell lines produce highly heterogenous glycoprofiles, reviewed in the literature (Batra and Rathore, 2016; Liu et al., 2016; Mastrangeli et al., 2019). Briefly, during incubation glycosylation is affected by changes in pH, availability of precursors, nutrients, growth factors and hormones; in subsequent extraction, glycosidases that are released by dead cells can catalyze partial to full degradation of the therapeutics’ glycan component in the culture medium. These processes result in the presence of more than 70 N-glycoforms identified in eight commercial therapeutic mAb formulations (Rosati et al., 2013; Song et al., 2014).
Therapeutics with homogenous glycoprofiles can be produced via total synthesis of the protein and glycan components and subsequent conjugation, utilizing mild chemo-selective ligation methods such as native chemical ligation and auxiliary-assisted ligation (Dawson et al., 1994; Dawson and Kent, 2000; Muir, 2003; Brik et al., 2006; Liu et al., 2006; Unverzagt and Kajihara, 2013). Despite the commendable progress in total synthetic approaches, the significant decrease in efficiency for proteins with over 50 residues limits the applicability in large-scale therapeutic manufacturing (Fernández-Tejada et al., 2015). Another approach to humanizing and homogenizing the protein glycoprofile is to extensively engineer the in vivo glycosylation pathways of the expression systems, such as in yeast, plant or mammalian cell lines, in order to access a restricted range of glycan types (Wang et al., 2019). Notable technologies in yeast and mammalian cells lines are summarized in Table 1, with similar developments underway in plant cell lines (Fischer et al., 2018; Montero-Morales and Steinkellner, 2018). However, in vivo glycoengineering usually does not lead to a completely homogeneous targeted glycoform – for example, the GlycoFi technology produces >90% homogenous glycoproteins with humanized complex N-glycans (Beck et al., 2010).
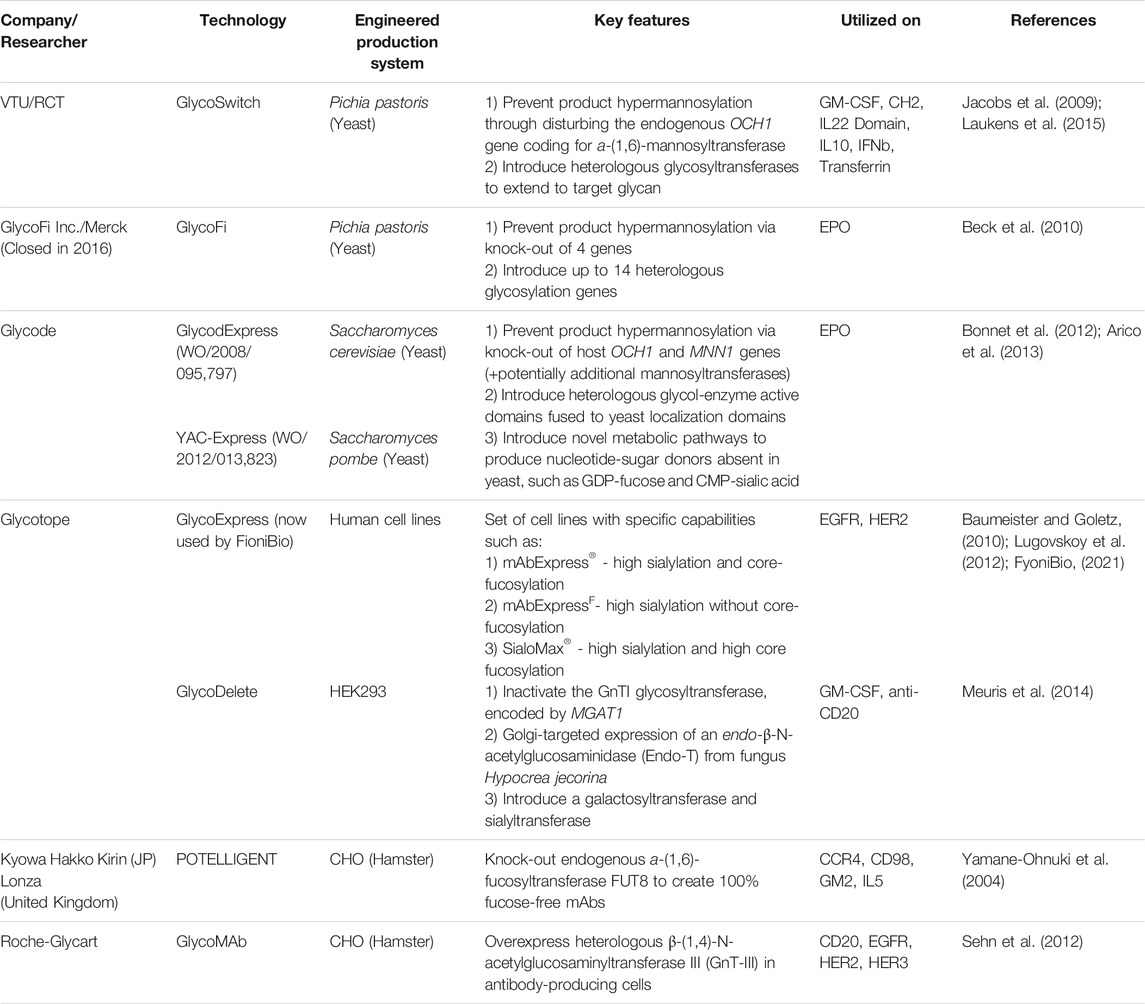
TABLE 1. Technologies for in vivo glycoengineering of yeast and mammalian protein expression systems.
In vitro chemoenzymatic glycoengineering technologies have emerged as a way to circumvent the primary limitation of in vivo engineering, namely the requirement for divergent optimization of expression systems for each particular protein therapeutic and respective glycan type (via extensive gene knock-out or knock-in of specific glycosyltransferases) (Tang et al., 2017). Chemoenzymatic glycoengineering is a two-step process, whereby the native heterogeneous glycans are trimmed off to a single GlcNAc/Fuc-α-(1,6)-GlcNAc unit using wild-type endo-N-acetylglucosaminidases (ENGases) (Fairbanks, 2017), with subsequent biocatalytic en bloc homogenous glycosylation (Figure 2A) (Chao et al., 2020). The ENGases (glycosyl hydrolase families GH85 and GH18) selectively cleave the diacetylchitobiose core [GlcNAc-β-(1,4)-GlcNAc] of N-linked glycans between the two GlcNAc residues (Maley et al., 1989; Karamanos et al., 1995) via a substrate-assisted mechanism which proceeds through an oxazolinium ion intermediate (Figure 2B) (Fujita et al., 2001; Umekawa et al., 2010). The (-1) 2-acetamido group of the substrate acts as a putative nucleophile, while the enzyme provides general acid/base catalysis and confers proper substrate orientation through the action of a pair of catalytic residues.
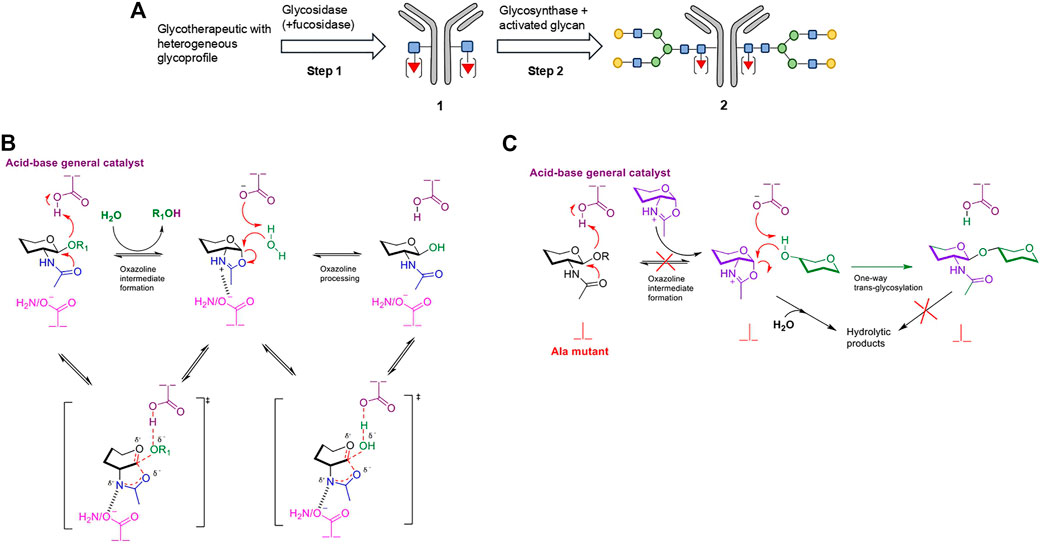
FIGURE 2. Strategy for chemoenzymatic N-glycoengineering of mAbs using an ENGase glycosidase-glycosynthase cascade. (A) Overview of the strategy using an example bi-antennary complex type N-glycan. Step 1 involves the trimming of the heterogenous glycoprofile with a wild-type ENGase. If desired, the accompanying use of a fucosidase can remove core-fucosylation. In Step 2, the deglycosylated mAb 1 is re-glycosylated via en block addition of activated donor glycans by a glycosynthase to form the homogenously glycosylated species 2. (B) Substrate-assisted mechanism of wild-type ENGases, proceeding via an oxazolinium ion intermediate. The enzyme provides only general acid-base catalysis and confers proper orientation of the putative nucleophilic 2-acetamido group through the Asn (GH85 family)/Asp (GH18 family) analogous residues. (C) Mechanism of ENGase glycosynthase, where Asn/Asp has been mutated to a catalytically inactive Ala as an example. The highly activated oxazoline substrate, which is externally supplied, is still subject to unproductive hydrolysis in aqueous environments. However, the product cannot be re-hydrolyzed back as the oxazoline intermediate cannot be reformed by the mutant enzyme. Non-reacting hydroxyl groups have been omitted for clarity.
When the concept was originally introduced, the second step was to be catalyzed by glycosyltransferases (Witte et al., 1997), or by stimulating the transglycosylation activity possessed by some wild-type ENGases (Vic et al., 1996; Vetere et al., 2000; Balogh et al., 2004; Rising et al., 2006; Heidecke et al., 2008; Rising et al., 2008; Umekawa et al., 2008; Wei et al., 2008; Parsons et al., 2009). Following their construction in 1998 (Mackenzie et al., 1998), glycosynthases overtook as the preferred catalyst. Prevention of oxazolinium ion formation in engineered ENGase-based glycosynthases can be obtained by two mutagenesis strategies, with the resulting mutants successfully processing externally supplied, pre-formed oxazolines as donors (Figure 2C). The more common and reliable strategy relies on mutation of the functionally equivalent Asn (GH85 family)/Asp (GH18 family) residues, responsible for correct substrate orientation, to a catalytically inert counterpart (like Ala) (Umekawa et al., 2008). The second approach, which employs mutation of the general acid-base catalytic residue, has successfully generated only one synthetically competent enzyme (Endo-A E173Q/H glycosynthase from Arthrobacter protophormiae), with non-performance for other ENGases (namely Endo-M from Mucor hiemalis) (Umekawa et al., 2010; Wang et al., 2019). The general inability of glycosynthases to re-hydrolyze the synthetic product in an aqueous environment leads to significantly improved yields compared to the use of wild-type glycosidases. They also have a more relaxed substrate specificity compared to glycosyltransferases and utilize easily accessible and less expensive sugar donors (Tang et al., 2017).
Chemoenzymatic glycoengineering affords homogenous glycoforms in high yields and in a chemo-, regio- and stereo-specific manner, which are all crucial in large-scale therapeutic production (Trincone and Giordano, 2006; Fairbanks, 2019). Reactions proceed in water under mild conditions, offering an attractive sustainable alternative to traditional organic synthesis. Multiple studies have demonstrated the utility of the glycosidase-glycosynthase cascade in producing homogenized constructs of commercially available mAb modalities, which possess enhanced activity and targeted properties (Supplementary Table S1). An extensive review of glycoprotein modification via chemoenzymatic methods by Li and Wang highlights the development of the technique to-date and further examples of its applicability (Li and Wang, 2018).
However, the technique has remained largely limited to preparative scale despite claims that it provides the optimal route towards homogenous N-glycosylated mAbs (Tomabechi et al., 2013; Lin et al., 2015), and no analysis of its applicability to industrial gram-scale therapeutic production is available. This review addresses this gap, identifying the challenges and potential solutions for large-scale implementation, with the goal of enhancing current therapeutic formulations. It considers the required components for chemoenzymatic glycoengineering: wild-type glycosidase with appropriate trimming specificity, activated extended glycan donor, glycosynthase with selected donor and acceptor substrate scope, and how these can be accessed selectively and in high yields to design an efficient manufacturing process.
There are three key steps when considering the required glycan donors for large-scale therapeutics with homogeneous glycoprofile: availability of the pure glycans in gram quantities, an efficient chemical activation method to produce suitable glycosynthase substrates, and selective and high-yielding protein glycosylation. All three aspects have been extensively developed for N-glycosylation. For implementation into therapeutic manufacturing, the entirety of the process must comply with therapeutic manufacturing standards and be sustainable and cost-effective.
Gram quantities of pure N-glycans can be accessed either through total chemical synthesis or via enzymatic modification of naturally available oligosaccharide precursors (Fairbanks, 2018; Chao et al., 2020). In total chemical synthesis, two major approaches have been predominantly applied: the epimerization approach and the Crich direct β-mannosylation, reviewed by Fairbanks (Fairbanks, 2018). Total chemical synthesis provides the advantage to access asymmetric glycans and add selective unnatural tags as handles for further chemical transformation (Lin et al., 2015). However, it involves multiple steps, complex orthogonal protecting group strategies, and heavy organic solvent use, which altogether have a negative impact on the process sustainability.
Complex N-glycans can also be isolated from natural precursors, such as sialylglycopeptide (SGP) 3 (Figure 3A). An optimized procedure for the production of SGP from dried egg yolk powder (as opposed to entire eggs) yields ≈0.8 mg of SGP per 1 g of egg yolk powder (Liu et al., 2017). The glycan from SGP can be truncated in situ with a mix of specific exo-glycosidases (like sialidase, β-(1.4)-galactosidase, etc.) to its desired composition; it can then be directly released from the peptide component via enzymatic cleavage catalyzed by wild-type Endo-M (Figure 3B) (Huang et al., 2009; Sun et al., 2014; Lin et al., 2015), or it can be divergently chemically functionalized for further modifications prior to Endo-M release, making it a versatile starting material for glycan isolation (Tang et al., 2016). The works by Kajihara, and more recently Boons, constitute further advances, allowing the production of truncated or tri- and tetra-antennary complex N-glycans (symmetrical or asymmetrical), which are otherwise inaccessible (especially at a large scale) via total synthetic approaches, from SGP (Maki et al., 2016; Liu et al., 2019).
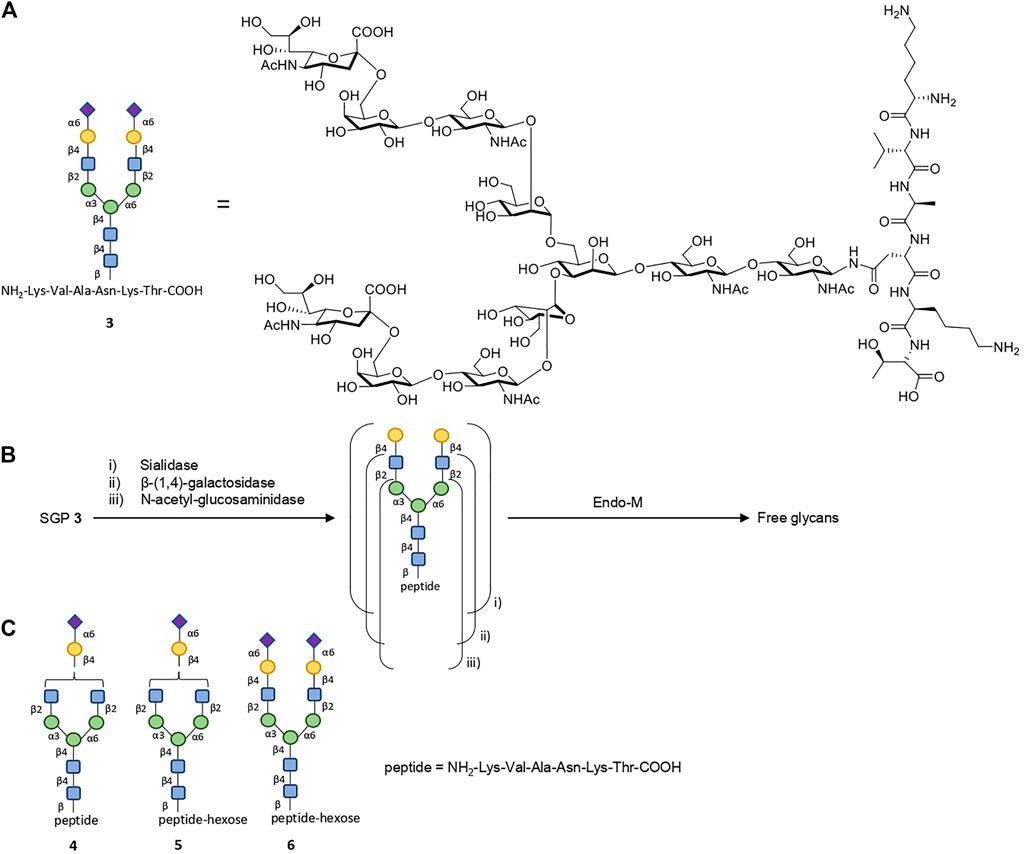
FIGURE 3. Isolation of complex glycans from SGP. (A) Structure of SGP 3 and its according cartoon representation. (B) SGP can be trimmed with one or several exo-glycosidases to the desired glycan composition (i, ii or iii). The glycan component is then released from the peptide using wild-type Endo-M catalysis. (C) When isolated from dried egg yolk powder, SGP contains about ∼10% impurities as species 4-6, as observed on the chromatogram (Liu et al., 2017).
SGP can be easily separated from its readily available precursor, with the caveat of incomplete homogeneity of the isolated SGP, which contains ≈10% impurities as species 4–6 (Figure 3C) (Liu et al., 2017). Impurity 6 is of little concern as the peptide component of SGP is cleaved off regardless of the additional hexose moiety. Structures 4 and 5, however, may be relevant if the targeted glycan is of the complex bi-antennary type, in which case SGP needs to be further purified. Using preparative hydrophilic interaction chromatography (HiLiC), the authors obtained homogenous SGP 3 in gram quantities, making this purification feasible for accessing pure N-glycans as part of a large-scale chemoenzymatic glycoengineering technology, albeit not very efficient. Whilst accessing SGP from egg-yolk may be cost-effective, its sustainability is questionable: the Process Mass Intensity (PMI), one of the standard green chemistry metrics, is bound to be very poor, due to the very low abundance of SGP within the egg yolk powder and the enormous amounts of waste associated, represented by a PMI contribution of 1,250 by the starting material alone.
Conversely, high-mannose type N-glycans can be isolated from soybean agglutinin, which is prepared from soybean flour (Wang et al., 2004). Sequential digestion with Pronase and Endo-A, and subsequent chromatographic purification affords homogenous Man9GlcNAc glycans for further activation. Glycans with lower level of mannosylation (Man5-and Man6GlcNAc) were prepared by the same methodology from chicken ovalbumin.
In many ways, the approach to obtain pure glycans resembles that for many natural products: in the early stages of the demand, there is a competition between extraction from natural sources and chemical total synthesis. Isolation from natural sources - normally by-products of the agricultural industry - can be very cost-effective because of the extremely low cost of the natural source, but the low content of the desired compounds make the process very wasteful and historically, if the demand increases, eventually it cannot be sustained by extraction alone. On the other hand, chemical synthesis begins its journey with a much less efficient process but strives to incrementally improve efficiency and diversity of products, allowing researchers to tap into the therapeutic potential of unnatural glycan structures.
However, one can envision that the true long-term solution to the supply of pure glycans will come from synthetic biology, where a whole-cell microorganism will be engineered to produce the desired compound from cheap agricultural waste in much higher yields and homogeneity than those naturally occurring in egg yolk or soybean flour. A similar path was followed for insulin manufacturing, which moved from pancreatic extraction to recombinant production in either Escherichia coli or Saccharomyces cerevisiae (Walsh, 2005; Baeshen et al., 2014). Subsequent chemical modification of the biotechnologically obtained glycan may offer access to semisynthetic glycans with unique chemical handles, in analogy to current approaches for accessing semisynthetic penicillins (Deng et al., 2016).
The glycosylation step via glycosynthase catalysis relies on chemical activation of the donor glycans into oxazolines, which mimic the enzyme-activated intermediate. Initially, oxazoline formation was carried out on per-acetylated glycans in dichloroethane with bromotrimethylsilane (TMSBr) and BF3Et2O in the presence of 2,4,6-collidine; the activated species was deprotected with catalytic amounts of MeONa in methanol (Umekawa et al., 2008; Huang et al., 2009). This procedure was revolutionized by Shoda et al., who developed and optimized a one-step synthetic strategy for oxazoline formation using 2-chloro-1,3-dimethyl-1H-imidazolium chloride (DMC, 7) as a dehydrating agent (Scheme 1) (Noguchi et al., 2009). A recent review by Fairbanks highlights the utility and significance of DMC for the carbohydrate field, as it enables selective activation of the anomeric centre of unprotected carbohydrates without the need for any protecting-group chemistry (Fairbanks, 2021). Shoda then further developed 2-chloro-1,3-dimethyl-1H-benzimidazol-3-ium chloride (CDMBI, 8); CDMBI-based oxazoline formation was deemed more advantageous as the by-product 1,3-dimethyl-1H-benzimidazol-3-ium (DMBI, 9) precipitates upon reaction completion due to high hydrophobicity and can be readily removed by filtration; the filtrate containing the oxazoline is suitable for direct addition to the glycosynthase-catalyzed reaction without further purification (Noguchi et al., 2012). Nevertheless, DMC remains the routinely utilized reagent for oxazoline formation, as it is commercially available and relatively inexpensive, so one would envision that any large-scale oxazoline production would rely exclusively on DMC.
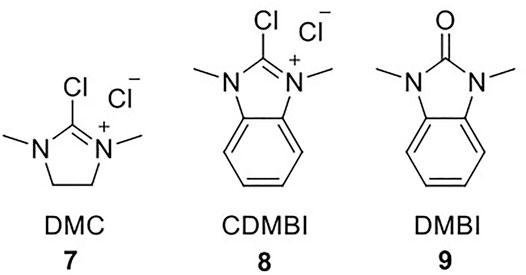
SCHEME 1. Structures of DMC 7, CDMBI 8 and DMBI 9.
However, oxazoline use in chemoenzymatic glycoengineering is still sub-optimal due to several challenges. As an externally-supplied mimic of the natural catalytic intermediate, the highly activated oxazoline moiety is readily hydrolyzed by the water molecules in the enzymatic active site (Umekawa et al., 2008). While the hydrolyzed free glycan can be recovered from the reaction and re-formed into the oxazoline (Wang and Amin, 2014), this unproductive hydrolytic conversion imposes the use of the corresponding donor in excess to achieve competitive glycosylation yields. The excess of oxazoline required increases significantly for folded protein targets and mAbs, due to entropy limitations and reduced access to the target peptide site; even in conditions with high oxazoline excess, glycosylation of more than a single site may not proceed to completion (Wang et al., 2020). In addition, the high concentration of the reactive oxazoline species in the reaction mixture leads to non-enzymatic, chemical “glycation” of the mAb target, first reported by Davis (Parsons et al., 2016). Further investigation showed that when GlcNAc-oxazoline was added to the model peptide Ac-L-Ala-L-Lys-L-Ala-NH2 under traditional glycosylation conditions (50 mM phosphate buffer of pH 8.0) but in the absence of a glycosynthase, there was evident chemical ligation of the glycan chain onto the single Lys residue (Figure 4) (Wang et al., 2016). Incubation of up to 15 h resulted in 90% yield of the glycated side-product. As expected, such glycation was not observed in the control reaction with GlcNAc instead of GlcNAc-oxazoline, confirming that it is the highly activated oxazoline moiety which drives Lys-glycan association.
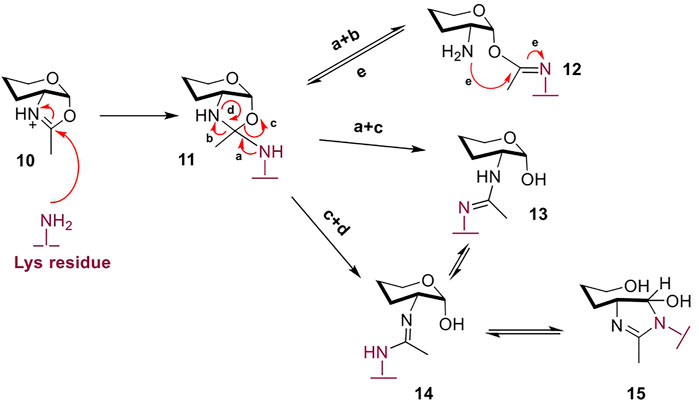
FIGURE 4. The excess oxazoline during glycosylation, especially of complex multimeric protein targets like mAbs, stimulates non-enzymatic ligation of the glycan to Lys residues in the protein therapeutic – termed “glycation”. According to the proposed mechanism, species 12 and 13 may be kinetic products, but with longer incubation equilibrate to products 14 and 15 (Wang et al., 2016). Non-reacting hydroxyl groups are omitted for clarity.
Any solution for large-scale oxazoline use in chemoenzymatic glycoengineering must address all these issues simultaneously, optimizing selective glycosylation yields and minimizing unproductive hydrolysis or side glycation. Reduction of unproductive oxazoline hydrolysis has been attempted via the inclusion of organic co-solvents such as dimethylsulfoxide (DMSO) or acetone, which decrease effective water concentration; however, they also negatively impact synthetic reaction rates or enzyme stability (Heidecke et al., 2009). Lowering the pH of the reaction buffer to 6.5 reduces non-enzymatic glycan ligation to Lys residues, but it also lowers targeted glycosylation efficiency due to the instability of the oxazoline moiety at acidic pH (Manabe et al., 2019). Successful suppression of side-reactions without negative impact on protein glycosylation yields was achieved with the following conditions: 10% w/w glycosynthase loading, and step-wise oxazoline addition (Parsons et al., 2016; Manabe et al., 2019). The required strict control over peak oxazoline concentration, alongside the high enzymatic loading, can be challenging for industrial-scale application.
We propose that the ideal solution to this issue could be achieved in the future by a combination of enzyme and process engineering: in absence of glycosynthase, the activated oxazoline is stable to chemical hydrolysis under the typical assay conditions. Therefore, enzyme engineering could be aimed at reducing the enzyme-catalyzed hydrolysis of the oxazoline, favoring the glycosynthase reaction over the unproductive hydrolysis. This improvement should reduce the overall excess of oxazoline required. Conversely, the critical oxazoline concentration at process scale can be controlled to maximize the productive glycosynthase reaction over the undesired competing oxazoline hydrolysis and Lys glycation. This can be achieved by carefully designed engineering solutions, such as fed-batch or continuous reactor with appropriate process analytics; flow biocatalysis has also emerged as an interesting technology to operate at controlled concentration of reactive chemicals (amongst other benefits) and we envision it will become prominent in the future of chemoenzymatic glycosylation (Britton et al., 2018).
Another complementary approach to manage the oxazoline problem is in situ substrate formation, as described below.
A novel approach to resolving the issues associated with excess oxazoline is utilizing a one-pot glycosylation strategy that confers activated species formation in situ; this has the added benefit of bypassing oxazoline purification. The strategy was initially developed by the Huang group, which converted in situ the glycan from SGP to the corresponding oxazoline in order to homogenously glycosylate Fuc-α-(1,6)-GlcNAc-Fc trastuzumab with Endo-S D233Q glycosynthase (from Streptococcus pyogenes) (Figure 5A) (Tang et al., 2016). As discussed, Endo-M can be combined with additional exo-glycosidases in order to trim the glycan further to the desired composition prior to acceptor addition. Minimizing the peak oxazoline concentration and glycosylation incubation times reduces unproductive hydrolysis and avoids non-enzymatic glycation. The authors report that the activity of the glycosynthase Endo-S D233Q is not affected by the presence of the 1,3-dimethyl-2-imidazolidin-2-one (DMI) hydrolysate of DMC (Tang et al., 2016), which is in disagreement with Shoda’s previous findings that DMI has a notable inhibitory effect on enzymatic catalysts (Noguchi et al., 2012).
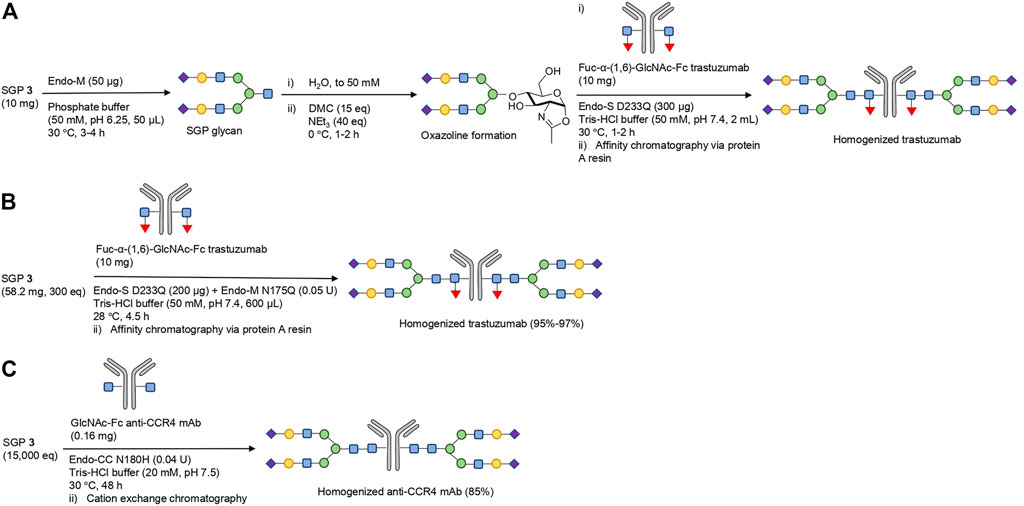
FIGURE 5. Strategies for one-pot glycosylation of deglycosylated therapeutic glycoproteins. (A) Fuc-α-(1,6)-GlcNAc Fc trastuzumab was homogenously glycosylated with the sialylated bi-antennary complex type glycan from SGP from SGP via a one-pot strategy with in situ oxazoline formation and Endo-S D233Q glycosynthase catalysis (Tang et al., 2016). (B) One-pot glycosylation of Fuc-α-(1,6)-GlcNAc Fc trastuzumab, utilizing the complementary activity of Endo-M N175Q and Endo-S D233Q variants (Iwamoto et al., 2018). It is hypothesized that the Endo-M mutant releases the activated glycan from SGP, which is then conjugated to the mAb by the variants of Endo-S. Complementarity arises from the fact that Endo-M N175Q does not recognize the fucosylated mAb site for glycosylation. The nature of the suggested activated glycan intermediate is unclear. (C) One-pot glycosylation of core-fucose deficient GlcNAc-Fc anti-CCR4 mAb by Endo-CC N180H (Manabe et al., 2018). The transformation was performed with a single enzymatic catalyst, but required long incubation time and very high SGP excess to achieve high glycosylation yields.
A step further in one-pot glycosylation strategies is circumventing chemically-induced oxazoline formation altogether: the Hasegawa group reported a successful transfer of the sialylated bi-antennary complex type glycan moiety from SGP to Fuc-α-(1,6)-GlcNAc-Fc trastuzumab via the complementary activity of two glycosynthases (Figure 5B) (Iwamoto et al., 2018). One glycosynthase (Endo-M N175Q) converts the glycan from SGP to an active intermediate (not yet identified), while the second glycosynthase (derived from Endo-S) catalyzes the selective glycosylation of the mAb with this activated SGP-derived glycan species. The glycosylation efficiency was excellent, at 95% with Endo-S D233Q, but could be increased even further to 97% via utilizing either the double mutant Endo-S D233Q/E350Q or the glycan-Asn as a starting material (instead of full-length SGP peptide). While the precise mechanism of the transformation is unclear, it implies some level of retained hydrolytic capability by Endo-M N175Q which enables glycan release from the peptide component of SGP.
Similar capability to process full-length SGP in mAb glycosylation was observed with the N180H glycosynthase variant of Endo-CC from Coprinopsis cinerea (Figure 5C) (Manabe et al., 2018). Core-fucose deficient anti-CCR4 mAb was deglycosylated by wild-type Endo-S, and then re-engineered with homogenous sialylated bi-antennary complex type glycosylation pattern in 85% yield via one-pot glycosylation strategy with Endo-CC N180H and SGP. One significant drawback of both these processes is that SGP is required in hundreds of molar equivalents excess to drive high yields of mAb remodeling, significantly impacting the efficiency and sustainability (Iwamoto et al., 2018; Manabe et al., 2018).
On paper, one-pot glycosylation without chemical oxazoline formation looks like a simplified process with minimized side reactions, but it presently requires significant further optimization in order to become an industrially viable technology. Given that the process envisions the use of two separate glycosynthases, potentially both capable of catalyzing every reaction in the process (including unproductive hydrolyses and transglycosylation) it is speculative whether it will be possible to fine tune both catalyst by enzyme engineering to selectively enable only the desired step and avoid any side reactions.
Incorporating chemoenzymatic glycoengineering in the production of glycoengineered mAb therapeutics may appear challenging due to the need to accommodate multiple enzymes and additional reaction and purification steps into the overall process. The task is further complicated by the increasing choice of both wild-type and engineered enzymes. However, through careful analysis of the available enzymatic panels and strategies, the technology can be streamlined to access optimal glycoforms with minimal divergence.
Successful implementation of chemoenzymatic glycoengineering relies upon the selection of wild-type glycosidase and engineered glycosynthase enzymes with the appropriate specificity to match the synthetic target. The major advance in mAb glycoengineering came with the discovery of Endo-S, which hydrolyses solely the core complex glycans on humanized, fully folded IgG mAbs, with marginal activity on hybrid type and high-mannose type glycans (Fairbanks, 2017). It is traditionally used across the literature for chemoenzymatic remodeling of mAbs (Fan et al., 2012; Huang et al., 2012; Lin et al., 2015; Manabe et al., 2018; Manabe et al., 2019) due to its unique specificity and high activity on native, folded IgGs (Collin and Olsén, 2001; Du et al., 2020). A recent replacement is Endo-S2, an endo-glycosidase from the specific serotype M49 of Streptococcus pyogenes which shares only 37% identity with Endo-S (Sjögren et al., 2013). Endo-S2 can successfully hydrolyze all major types of N-glycans, making it more appropriate for streamlined process integration as it can deglycosylate mAbs from various production systems (Sjogren et al., 2015). In multiple cases, the wild-type endo-glycosysidase is utilized in combination with a α-(1,6)-fucosidase, which removes core fucosylation from the protein-attached GlcNAc moiety (Huang et al., 2012; Lin et al., 2015; Li et al., 2016; Giddens et al., 2018). An interesting improvement, offering process simplification, is to bypass the in vitro deglycosylation step by co-expressing the endo-glycosidase in the therapeutic’s production system. This has been successfully demonstrated in Nicotiana benthamiana plant cells, which co-expressed rituximab and Endo-H (Bennett et al., 2018). Endo-H was targeted to the endoplasmic reticulum (ER), where the newly formed mAb is initially decorated with high-mannose type glycans that are readily hydrolyzed by Endo-H. Endo-H co-expression did not affect rituximab production yields, and the deglycosylated GlcNAc-Fc mAb was readily recovered via a combination of protein A chromatography and cation exchange chromatography for further in vitro glycosylation.
The success of the second step in chemoenzymatic glycoengineering relies on the careful selection of an appropriate glycosynthase. The glycosynthase must possess not only the desired acceptor (therapeutic) specificity, like in deglycosylation, but also recognize the according glycan donor. A further complication is that the substrate specificity of glycosynthases is not always the same as the wild-type enzyme, and can vary for the different mutants. For example, wild-type Endo-D from Streptococcus pneumoniae hydrolyses core-fucosylated glycans, whereas its N322Q glycosynthase mutant favors the core-fucose deficient proteins as acceptors (Fan et al., 2012). In the case of Endo-M glycosynthases, the N175A and N175Q mutants act solely on non-fucosylated acceptors, while the N175Q/W251N double mutant can successfully glycosylate both fucosylated and non-fucosylated acceptors (Umekawa et al., 2010).
The logical approach to this heterogeneity is to investigate and characterize a broader spectrum of glycosynthases, both by expanding the spectrum of wild-type enzymes and the number of engineered mutants for a given enzyme. The ultimate goal is to provide a number of well-characterized catalysts, ranging from generally applicable to specifically tailored, to cover the broadest possible panel of donors and acceptors.
Indeed, Endo-S2 mutants are emerging as the optimal choice for mAb homogenous glycosylation due to their broad donor and acceptor scopes, which surpass the capabilities of Endo-S glycosynthases. Endo-S2 D184M can glycosylate both core-fucosylated and non-fucosylated acceptors with all major types of N-glycans (Li et al., 2016). Further mutagenesis studies resulted in a broad panel of glycosynthase-like Endo-S2 mutants (T138D, T138E, T138F, T138H, T138K, T138L, T138M, T138N, T138Q, T138R, T138V, T138W, D182Q, D226Q, T227Q, and T228Q), which afford access to an expanded glycan donor set, including tri-antennary variants (Shivatare et al., 2018). Ability to utilize multi-antennary glycan donors is a sought-after improvement, as such donors are generally not accepted by the ENGases; a notable exception is Endo-F3 (from Flavobacterium meningosepticum) mutants D165A and D165Q, whose activity is however limited to fucosylated IgGs (Giddens et al., 2016). The use of Endo-S2 mutants in glycoprotein remodeling was patented by CHO Pharma Inc., a Taiwan-based start-up company, as part of their propriety technology for mAb glycoprofile homogenization (Lin et al., 2018).
A caveat to in vitro chemoenzymatic glycoengineering is that the deglycosylated glycotherapeutic intermediate must be meticulously purified from the wild-type glycosidase used in heterogenous glycan trimming, since any residual contamination will result in re-hydrolysis of the final product generated in Step 2 (Li et al., 2018). Separating the products from the unreacted starting material and wild-type glycosidase is generally a difficult task, but is more readily achieved in the case of mAbs, where protein A column can be utilized for affinity chromatography (Hober et al., 2007). If the deglycosylation step is performed in vitro, easy separation of the glycosidase from the target protein can be readily achieved through glycosidase immobilization on solid support. Immobilizing both enzymes in the two-step glycoengineering cascade allows glycoprotein remodeling without intermediate purification, which reduces solvent usage, allows catalyst recycling, and simplifies product isolation compared to solution-phase reactions. It is also compatible with advantageous flow reactor designs.
Recently, there have been two significant reports of glycosidase/glycosynthase immobilization. In the first example, wild-type Endo-S2 and its D184M mutant were covalently immobilized on functionalized agarose (Figure 6A) (Li et al., 2018). The enzymes were successfully applied to the homogenization of commercial trastuzumab with bi-antennary complex type glycans. Following fast deglycosylation with wild-type Endo-S2, trastuzumab was eluted from the agarose-loaded column, alongside the released free N-glycans. The eluent was directly applied onto the second, glycosynthase-loaded column, together with the corresponding oxazoline, and homogenously glycosylated trastuzumab was obtained after 1–2 h incubation at ambient temperature.
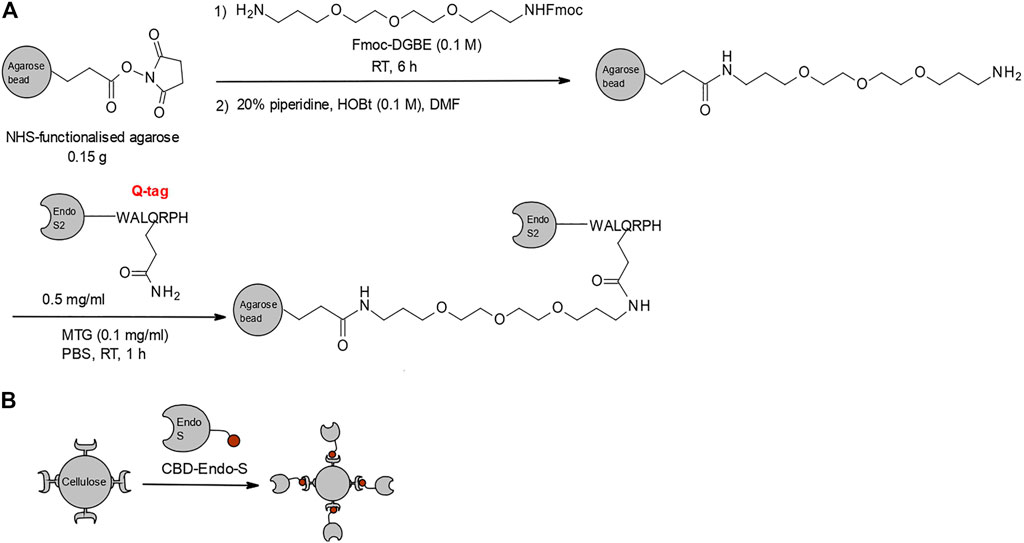
FIGURE 6. Glycosidase/glycosynthase immobilization strategies. (A) Strategy for covalent immobilization of Endo-S2 and its glycosynthase mutants on functionalized agarose by microbial trans-glutaminase (MTG) via a fused Q-tag (Li et al., 2018). (B) Second-generation immobilization strategy, utilizing a fusion of Endo-S and glycosynthase mutants to a cellulose-binding domain (CBD) affinity tag (Zhao et al., 2019).
It was reported that immobilized Endo-S2 could be re-used for several runs without notable loss of activity, but there was lack of specific data on the recycling capability and enzymatic stability over time (Li et al., 2018). A potential drawback of this strategy is that the pre-purification of the glycosynthase for trans-glutaminase-mediated immobilization on the support limits the scale up of this specific strategy. A second-generation immobilization strategy resolved this limitation by fusing Endo-S and its D233A and D233Q mutants to a cellulose-binding domain (CBD), which acts as an affinity tag and affords one-step purification and immobilization on cellulose, with respective loading efficiency of 86, 81 and 90% under optimal conditions (Figure 6B) (Zhao et al., 2019). Addressing the previous gap in data about the properties of the immobilized enzymes, this study concluded that Endo-S and its mutants could be stored at 4°C for over 30 days and re-used for more than five catalytic cycles without significant loss of activity. Alongside the demonstrated efficiency of commercial rituximab deglycosylation (1 h at 37°C under constant shaking) and subsequent re-glycosylation with azide-tagged oxazoline (1.5 h at 30°C, quantitative glycosylation of 5 mg/ml mAb), the proven stability and recyclability of the immobilized enzyme suggest that solid-phase glycan remodeling via CBD-immobilized enzymes could be an industrially viable process for gram-scale modifications, with lower additional costs over its solution-phase counterpart.
Even if the specific support and immobilization strategy may ultimately prove not convenient, enzyme immobilization is a well-established field with numerous cost-effective and precedented strategies for use at process scale, and finding a suitable strategy for glycosynthase immobilization should not prove difficult: plenty of fast and effective immobilization systems exist (e.g., the simple His-tag/Ni column), which can afford purification/enrichment and immobilization of a desired enzymatic catalyst in a single step (Tischer and Kasche, 1999; Zhou et al., 2017). A great benefit to the development and adoption of chemoenzymatic approaches to glycan remodeling will be the commercial availability of the broad-spectrum enzyme panel envisioned in Target-based selection of enzymes in an immobilized and ready-to-screen format.
The diversity of available glycan structures may pose a significant barrier to the viable industrial production of homogenously glycosylated mAbs, as distinct synthetic protocols would be required to access such vast array of donors. Although a long-term synthetic solution may be provided by synthetic biology and an array of glycan-producing engineered microorganisms, reducing the number of glycans used would simplify the task both in the immediate and long-term future. A potential solution would be to streamline existing structure-activity relationship for specific glycan features into a small set of defined structures which afford improvement of the targeted properties for a therapeutic class (anti-inflammatory, cytotoxic, etc.), allowing batch glycan synthesis. The term “universal glycan” was coined by Wong et al. to describe the non-fucosylated, bi-antennary sialylated complex type glycan which, upon homogenous addition to anti-cancer mAbs, proved optimal for enhancing effector functions (Lin et al., 2015). Homogenization of rituximab with this universal glycan stimulated activity on generic rituximab-resistant cell lines, and both remodeled rituximab and trastuzumab demonstrated improved binding to FcγIIIaR and enhanced ADCC, primarily due to the removal of core-fucose (Shields et al., 2002; Nakamura et al., 2003). Furthermore, the incorporated terminal α-(2,6)-sialylation confers anti-inflammatory activity to mAbs (Anthony et al., 2008).
Three additional mAbs of the IgG type, which target the surface glycoprotein of Ebola virus, were engineered with such homogenous, non-fucosylated, sialylated complex-type-Fc glycoprofile (Chen et al., 2017). When assessing their ADCC activity in comparison to the parent, heterogenous mAbs, the authors observed “gain-of-function” for two of the engineered variants. The parent counterparts showed no measurable ADCC activity even at concentrations as high as 40 μg/ml, which suggests that they exert their therapeutic function via neutralizing the virus itself. However, the according engineered variants demonstrated significant ADCC, with EC50 values of 0.219 μg/ml and 0.387 μg/ml. These striking results confirm that careful control over a therapeutic’s glycoprofile can introduce and enhance functionality, with the non-fucosylated bi-antennary sialylated complex type glycoform appearing as the optimal choice for Fc-mediated effector function improvement.
Chemoenzymatic glycoengineering is a readily applicable technique for accessing glycovariants that may be challenging to obtain through traditional chemical synthesis. These glycoforms can have very dramatic impact on the pharmacokinetics, bioavailability, and efficacy of the therapeutic protein. For-example, Endo-A N171A can successfully glycosylate therapeutics with high-mannose type oxazolines containing terminal mannose-6-phosphate moieties, thus enabling selective drug targeting to the lysosome, which is essential in enzyme replacement therapies for lysosomal storage disorders (Priyanka et al., 2016; Yamaguchi et al., 2016). Furthermore, through careful control and homogenization of glycosylation patterns, one can access improved therapeutic constructs with novel and enticing activities, capable of addressing efficacy challenges in minor patient populations.
There is now prevailing evidence that Fc-glycosylation patterns have notable impact on mAb interactions with immune cells, affecting ADCC and antibody-mediated inflammation. Removal of core fucose has been shown to significantly improve ADCC through enhancing mAb affinity for the FcγIIIa receptor (Shields et al., 2002; Nakamura et al., 2003). This is of particular significance for patients with the low-affinity FcγRIIIa-F158 allelic polymorphism and provides a sought-after improvement of anti-cancer activity in these cases (Huang et al., 2012). Homogenous afucosylated glycoforms of rituximab, accessed through chemoenzymatic glycoengineering, demonstrated much improved binding to both forms of the FcγIIIa receptor when compared to their commercially available heterogeneous counterpart, that includes primarily fucosylated motifs (Huang et al., 2012; Li et al., 2017). Similarly, the terminal α-(2,6)-sialylated glycoforms are known to confer the anti-inflammatory effect of the widely used intravenous immunoglobulin G (IVIG); it is proposed that one of the reasons for the high doses required to elicit therapeutic effect is that the active glycoform is only a minor component of the glycoprofile in commercially available preparations (Kaneko et al., 2006; Anthony et al., 2008).
Chemoenzymatic glycoengineering provides a viable strategy for accessing mAb constructs with consistent, enhanced targeted activity. Up to date, only three mAb therapeutics approved by the U.S. Food and Drug Administration (FDA), have been subjected to some form of glycoengineering: obinutuzumab (GA101 or Gazyva®) was first in 2013, followed by mogamulizumab (POTELIGEO®) and benralizumab (MEDI-563, Fasenra™) in 2017 (Lin et al., 2015; Pereira et al., 2018). All three drugs are produced in glyco-engineered CHO cell lines, which leads to enrichment of specific glycoforms without full homogeneity. CHO Pharma Inc. is the first pharmaceutical company to develop fully homogenous “biobetter” versions of commercially available antibodies through their propriety glycan engineering platform (Lin et al., 2018). The company patented the term “glycoantibodies” to describe antibody therapeutics with homogenous Fc-region glycoprofile, constructed through chemoenzymatic glycoengineering of the Fc region (Wong et al., 2015). In March 2018, its first ever anti-CD20 glycoantibody entered Phase I clinical trial (Trial ID: NCT03221348) for refractory or relapsed follicular lymphoma, evidencing the applicability of glycoprofile homogenization to industrial formulations (2017).
Through the enzymatic addition of unnatural glycans with a precise number of chemical handles for cargo attachment, one can access ADCs with defined and tunable drug to antibody ratio (DAR) (OY, 2018). Conjugation of payloads to the Fc N-glycan of the antibody component provides an optimal strategy for ADC creation, as the glycan position is far away from the antigen-binding region, ensuring that the cargo does not interfere with antigen recognition (Manabe et al., 2019). Primarily, the glycosynthase mutants of Endo-S have catalyzed the transfer of chemically modified glycans to deglycosylated mAbs, facilitating further payload conjugation through click-chemistry. Transfer of an azide-tagged glycan onto trastuzumab, catalyzed by Endo-S D233Q, allowed for subsequent biorthogonal conjugation to introduce the cytotoxic agent monomethyl auristatin E (MMAE), creating a glycosite-specific antibody-drug conjugate with doubly enhanced therapeutic activity (Manabe et al., 2019). Each step of the ADC preparation produced a single UPLC product peak, in contrast to the traditionally employed chemical conjugation between the maleimide and sulfhydryl group which results in multiple impurity peaks in UPLC analysis.
Huang et al. further applied their one-pot, in-situ oxazoline formation technology to obtain homogenously glycosylated variants of trastuzumab with azide or alkyne chemical handles (Tang et al., 2016). The chemical handles on the homogenized mAb enabled ADC formation via strain-promoted copper-free click chemistry with a dibenzocyclooctyne (DBCO)-conjugated payload (cytotoxic or reporter) like DM1, MMAE or the fluorescent tag Cy5. The advantage of site-specific payload conjugation to the glycan component was evidenced through the antigen-binding activity (KD values) of commercial trastuzumab, glycoside-labelled Cy5-trastuzumab and random-labelled Cy5-trastuzumab, which were measured as 1.2, 3.5, and 24 μg ml−1 respectively. Glycoside-specific Cy5 conjugation resulted in significantly higher antigen-binding activity, demonstrating that ADC formation through glycan homogenization avoids unwanted influences on the antigen-binding domain, which are common in random conjugation to Lys mAb residues.
Implementing chemoenzymatic glycoengineering to the large-scale glycan homogenization of commercially available glycotherapeutics is an exciting strategy towards the production of safer and improved therapies with enhanced targeted activity. However, there are several hurdles preventing the current application of such in vitro glycoengineering to large-scale therapeutic production. While there is clear evidence that single glycoforms of therapeutic proteins have not only higher batch-to-batch reproducibility, but also enhanced therapeutic properties from targeting to efficacy, their posttranslational engineering needs to meet the strict regulatory challenges of the industry. Therefore, the current industrial perspective is that it is still more costly to produce single glycoforms than it is to provide a defined, albeit heterogeneous, population of glycoforms which confers a sub-optimal but reproducible therapeutic profile. The latter can be usually achieved by cell line engineering with contained investment and no impact on the mAb production process.
Further development is still necessary to make chemoenzymatic glycoengineering economically attractive: given that the glycoengineered mAb is the final drug substance, it is safe to assume that the glycoengineering step would need to be performed under GMP, which has a higher cost associated with manufacturing plant occupancy and also very strict characterization and quantification of impurities; to address the latter, both enzymes used in the glycoengineering cascades would have to be purified, which in itself is a high cost contributor. The significant portion of unproductive conversions during chemoenzymatic glycoengineering leads to poor atom economy and further decreases process sustainability. Furthermore, we require improved, sustainable and cost-effective methods for production of complex glycan donors on demand, ideally from agricultural waste.
Throughout this review, we have highlighted potential solutions to all of the outstanding barriers to the wider implementation of chemoenzymatic glycoengineering, which build on the tremendous scientific advances in enzyme engineering, synthetic biology, and process chemistry. In the first instance, a viable process for the homogenization of mAb N-glycoprofiles would likely focus on optimization of only one enzyme (probably Endo-S2 due to its relaxed substrate specificity) acting on a single glycan donor set (the universal glycan), to alleviate the aforementioned difference in substrate scope and acceptance between enzymes (Heidecke et al., 2009). It would implement process solutions (such as enzyme immobilization and packed bed reactors) to comply with the regulatory framework in a cost-efficient manner, and it would address the issue of starting material (glycans) sustainable supply.
Ultimately, it will be the change in the regulatory landscape, spearheaded by the enhanced therapeutic activity and functionality of engineered glycoform, that will make the production of single glycoforms the norm in the industry. In this context, chemoenzymatic glycan remodeling, with all the improvements discussed and suggested within this review, will position itself as a strong enabling technology.
AI conducted the majority of reviewing of the literature. AI and FF prepared and wrote the manuscript. FF provided key opinions on the discussed industrial challenges. All authors reviewed the manuscript.
The publication of this work is part of a joint research project between the University of Edinburgh and AstraZeneca.
AI was employed by AstraZeneca when this review was being prepared. FF is currently employed by AstraZeneca. This study received funding from AstraZeneca. The funder had the following involvement with the study: supporting the publication of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to acknowledge Prof Dominic Campopiano for his valuable inputs and Dr Nathaniel Gordon for his help with the lab work on the glycosidase and glycosynthase expression and his feedback during manuscript production.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2021.810779/full#supplementary-material
Anthony, R. M., Nimmerjahn, F., Ashline, D. J., Reinhold, V. N., Paulson, J. C., and Ravetch, J. V. (2008). Recapitulation of IVIG Anti-inflammatory Activity with a Recombinant IgG Fc. Science 320 (5874), 373–376. doi:10.1126/science.1154315
Arico, C., Bonnet, C., and Javaud, C. (2013). N-glycosylation Humanization for Production of Therapeutic Recombinant Glycoproteins in Saccharomyces cerevisiae. Methods Mol. Biol. 988, 45–57. doi:10.1007/978-1-62703-327-5_4
Baeshen, N. A., Baeshen, M. N., Sheikh, A., Bora, R. S., Ahmed, M. M. M., Ramadan, H. A. I., et al. (2014). Cell Factories for Insulin Production. Microb. Cel Fact 13, 141. doi:10.1186/s12934-014-0141-0
Balogh, T., Boross, L., and Kosáry, J. (2004). Novel Reaction Systems for the Synthesis of O-Glucosides by Enzymatic Reverse Hydrolysis. Tetrahedron 60, 679–682. doi:10.1016/j.tet.2003.10.098
Batra, J., and Rathore, A. S. (2016). Glycosylation of Monoclonal Antibody Products: Current Status and Future Prospects. Biotechnol. Prog. 32, 1091–1102. doi:10.1002/btpr.2366
Baumeister, H., and Härter, M. (2010). Psychische Komorbidität bei muskuloskelettalen Erkrankungen. Bundesgesundheitsbl. 54, 52–58. doi:10.1007/s00103-010-1185-x
Beck, A., Cochet, O., and Wurch, T. (2010). GlycoFi's Technology to Control the Glycosylation of Recombinant Therapeutic Proteins. Expert Opin. Drug Discov. 5 (1), 95–111. doi:10.1517/17460440903413504
Bennett, L., Yang, Q., Berquist, B., Giddens, J., Ren, Z., Kommineni, V., et al. (2018). Implementation of Glycan Remodeling to Plant-Made Therapeutic Antibodies. Int. J. Mol. Sci. 19 (2), 421. doi:10.3390/ijms19020421
Bonnet, C., Arico, C., Rigaud, C., Blandais, C., Chanteclaire, E., Tassy-Freches, E., et al. (2012). “GlycodExpressTM and YAC-ExpressTM: Two Innovative Technologies Developed for N-Glycosylation Humanization and Production of Therapeutic Recombinant Glycoproteins in Saccharomyces cerevisiae,” in IUBMB-FEBS congress (Sevilla, Spain: GlycodExpress).
Brik, A., Yang, Y.-Y., Ficht, S., and Wong, C.-H. (2006). Sugar-assisted Glycopeptide Ligation. J. Am. Chem. Soc. 128 (17), 5626–5627. doi:10.1021/ja061165w
Britton, J., Majumdar, S., and Weiss, G. A. (2018). Continuous Flow Biocatalysis. Chem. Soc. Rev. 47, 5891–5918. doi:10.1039/c7cs00906b
Chao, Q., Ding, Y., Chen, Z.-H., Xiang, M.-H., Wang, N., and Gao, X.-D. (2020). Recent Progress in Chemo-Enzymatic Methods for the Synthesis of N-Glycans. Front. Chem. 8, 513. doi:10.3389/fchem.2020.00513
Chen, C.-L., Hsu, J.-C., Lin, C.-W., Wang, C.-H., Tsai, M.-H., Wu, C.-Y., et al. (2017). Crystal Structure of a Homogeneous IgG-Fc Glycoform with the N-Glycan Designed to Maximize the Antibody Dependent Cellular Cytotoxicity. ACS Chem. Biol. 12 (5), 1335–1345. doi:10.1021/acschembio.7b00140
Collin, M., and Olsén, A. (2001). Effect of SpeB and EndoS from Streptococcus Pyogenes on Human Immunoglobulins. Infect. Immun. 69 (11), 7187–7189. doi:10.1128/IAI.69.11.7187-7189.2001
Dawson, P. E., and Kent, S. B. H. (2000). Synthesis of Native Proteins by Chemical Ligation. Annu. Rev. Biochem. 69 (1), 923–960. doi:10.1146/annurev.biochem.69.1.923
Dawson, P. E., Muir, T. W., Clark-Lewis, I., and Kent, S. B. H. (1994). Synthesis of Proteins by Native Chemical Ligation. Science 266 (5186), 776–779. doi:10.1126/science.7973629
Deng, S., Ma, X., Su, E., and Wei, D. (2016). Efficient cascade Synthesis of Ampicillin from Penicillin G Potassium Salt Using Wild and Mutant Penicillin G Acylase from Alcaligenes Faecalis. J. Biotechnol. 219, 142–148. doi:10.1016/j.jbiotec.2015.12.034
Du, J. J., Klontz, E. H., Guerin, M. E., Trastoy, B., and Sundberg, E. J. (2020). Structural Insights into the Mechanisms and Specificities of IgG-Active Endoglycosidases. Glycobiology 30 (4), 268–279. doi:10.1093/glycob/cwz042
Fairbanks, A. J. (2021). Applications of Shoda's Reagent (DMC) and Analogues for Activation of the Anomeric centre of Unprotected Carbohydrates. Carbohydr. Res. 499, 108197. doi:10.1016/j.carres.2020.108197
Fairbanks, A. J. (2019). Chemoenzymatic Synthesis of Glycoproteins. Curr. Opin. Chem. Biol. 53, 9–15. doi:10.1016/j.cbpa.2019.05.015
Fairbanks, A. J. (2018). Synthetic and Semi-synthetic Approaches to Unprotected N-Glycan Oxazolines. Beilstein J. Org. Chem. 14, 416–429. doi:10.3762/bjoc.14.30
Fairbanks, A. J. (2017). The ENGases: Versatile Biocatalysts for the Production of Homogeneous N-Linked Glycopeptides and Glycoproteins. Chem. Soc. Rev. 46, 5128–5146. doi:10.1039/c6cs00897f
Fan, S.-Q., Huang, W., and Wang, L.-X. (2012). Remarkable Transglycosylation Activity of Glycosynthase Mutants of Endo-D, an Endo-β-N-Acetylglucosaminidase from Streptococcus Pneumoniae. J. Biol. Chem. 287, 11272–11281. doi:10.1074/jbc.m112.340497
Fernández-Tejada, A., Brailsford, J., Zhang, Q., Shieh, J. H., Moore, M. A., and Danishefsky, S. J. (2015). Total Synthesis of Glycosylated Proteins. Top. Curr. Chem. 362, 1–26. doi:10.1007/128_2014_622
Fischer, R., Holland, T., Sack, M., Schillberg, S., Stoger, E., Twyman, R. M., et al. (2018). “Glyco-engineering of Plant-Based Expression Systems,” in Advances in Glycobiotechnology. Editors E. Rapp, and U. Reichl (Cham: Springer), 137–166. doi:10.1007/10_2018_76
Fujita, M., Shoda, S.-i., Haneda, K., Inazu, T., Takegawa, K., and Yamamoto, K. (2001). A Novel Disaccharide Substrate Having 1,2-oxazoline Moiety for Detection of Transglycosylating Activity of Endoglycosidases. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1528, 9–14. doi:10.1016/s0304-4165(01)00164-7
FyoniBio (2021). GlycoExpress®(GEX®) [Online]. Available at: https://www.fyonibio.com/wp-content/uploads/2021/10/Flyer-FyoniBio-GlycoExpress_10-2021_web.pdf (Accessed October, , 2021).
Giddens, J. P., Lomino, J. V., Amin, M. N., and Wang, L.-X. (2016). Endo-F3 Glycosynthase Mutants Enable Chemoenzymatic Synthesis of Core-Fucosylated Triantennary Complex Type Glycopeptides and Glycoproteins. J. Biol. Chem. 291 (17), 9356–9370. doi:10.1074/jbc.m116.721597
Giddens, J. P., Lomino, J. V., DiLillo, D. J., Ravetch, J. V., and Wang, L.-X. (2018). Site-selective Chemoenzymatic Glycoengineering of Fab and Fc Glycans of a Therapeutic Antibody. Proc. Natl. Acad. Sci. USA 115 (47), 12023–12027. doi:10.1073/pnas.1812833115
Goetze, A. M., Liu, Y. D., Zhang, Z., Shah, B., Lee, E., Bondarenko, P. V., et al. (2011). High-mannose Glycans on the Fc Region of Therapeutic IgG Antibodies Increase Serum Clearance in Humans. Glycobiology 21 (7), 949–959. doi:10.1093/glycob/cwr027
Heidecke, C. D., Ling, Z., Bruce, N. C., Moir, J. W. B., Parsons, T. B., and Fairbanks, A. J. (2008). Enhanced Glycosylation with Mutants of Endohexosaminidase A (Endo A). ChemBioChem 9 (13), 2045–2051. doi:10.1002/cbic.200800214
Heidecke, C. D., Parsons, T. B., and Fairbanks, A. J. (2009). Endohexosaminidase-catalysed Glycosylation with Oxazoline Donors: Effects of Organic Co-solvent and pH on Reactions Catalysed by Endo A and Endo M. Carbohydr. Res. 344 (18), 2433–2438. doi:10.1016/j.carres.2009.09.013
Hober, S., Nord, K., and Linhult, M. (2007). Protein A Chromatography for Antibody Purification. J. Chromatogr. B 848 (1), 40–47. doi:10.1016/j.jchromb.2006.09.030
Huang, W., Giddens, J., Fan, S.-Q., Toonstra, C., and Wang, L.-X. (2012). Chemoenzymatic Glycoengineering of Intact IgG Antibodies for Gain of Functions. J. Am. Chem. Soc. 134 (29), 12308–12318. doi:10.1021/ja3051266
Huang, W., Li, C., Li, B., Umekawa, M., Yamamoto, K., Zhang, X., et al. (2009). Glycosynthases Enable a Highly Efficient Chemoenzymatic Synthesis of N-Glycoproteins Carrying Intact Natural N-Glycans. J. Am. Chem. Soc. 131 (6), 2214–2223. doi:10.1021/ja8074677
Iwamoto, M., Sekiguchi, Y., Nakamura, K., Kawaguchi, Y., Honda, T., and Hasegawa, J. (2018). Generation of Efficient Mutants of Endoglycosidase from Streptococcus Pyogenes and Their Application in a Novel One-Pot Transglycosylation Reaction for Antibody Modification. PLoS One 13 (2), e0193534. doi:10.1371/journal.pone.0193534
Jacobs, P. P., Geysens, S., Vervecken, W., Contreras, R., and Callewaert, N. (2009). Engineering Complex-type N-Glycosylation in Pichia pastoris Using GlycoSwitch Technology. Nat. Protoc. 4 (58), 58–70. doi:10.1038/nprot.2008.213
Jefferis, R. (2009). Glycosylation as a Strategy to Improve Antibody-Based Therapeutics. Nat. Rev. Drug Discov. 8 (3), 226–234. doi:10.1038/nrd2804
Kaneko, Y., Nimmerjahn, F., and Ravetch, J. V. (2006). Anti-inflammatory Activity of Immunoglobulin G Resulting from Fc Sialylation. Science 313 (5787), 670–673. doi:10.1126/science.1129594
Karamanos, Y., Bourgerie, S., Barreaud, J.-P., and Julien, R. (1995). Are There Biological Functions for Bacterial Endo-N-Acetyl-β-D-Glucosaminidases? Res. Microbiol. 146 (6), 437–443. doi:10.1016/0923-2508(96)80289-0
Laukens, B., De Wachter, C., and Callewaert, N. (2015). Engineering the Pichia pastoris N-Glycosylation Pathway Using the GlycoSwitch Technology. Methods Mol. Biol. 1321, 103–122. doi:10.1007/978-1-4939-2760-9_8
Li, C., and Wang, L.-X. (2018). Chemoenzymatic Methods for the Synthesis of Glycoproteins. Chem. Rev. 118 (17), 8359–8413. doi:10.1021/acs.chemrev.8b00238
Li, T., DiLillo, D. J., Bournazos, S., Giddens, J. P., Ravetch, J. V., and Wang, L.-X. (2017). Modulating IgG Effector Function by Fc Glycan Engineering. Proc. Natl. Acad. Sci. USA 114 (13), 3485–3490. doi:10.1073/pnas.1702173114
Li, T., Li, C., Quan, D. N., Bentley, W. E., and Wang, L.-X. (2018). Site-specific Immobilization of Endoglycosidases for Streamlined Chemoenzymatic Glycan Remodeling of Antibodies. Carbohydr. Res. 458-459, 77–84. doi:10.1016/j.carres.2018.02.007
Li, T., Tong, X., Yang, Q., Giddens, J. P., and Wang, L.-X. (2016). Glycosynthase Mutants of Endoglycosidase S2 Show Potent Transglycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling. J. Biol. Chem. 291, 16508–16518. doi:10.1074/jbc.m116.738765
Lin, C.-W., Tsai, M.-H., Li, S.-T., Tsai, T.-I., Chu, K.-C., Liu, Y.-C., et al. (2015). A Common Glycan Structure on Immunoglobulin G for Enhancement of Effector Functions. Proc. Natl. Acad. Sci. USA 112, 10611–10616. doi:10.1073/pnas.1513456112
Lin, N.-H., Huang, L.-Y., Shivatare, S. S., Chrn, L.-T., Wong, C.-H., Wu, C.-Y., et al. (2018). Endoglycosidase Mutants for Glycoprotein Remodelling and Methods of Using it. USA patent Appl. 15/684, 897.
Liu, H., Nowak, C., Shao, M., Ponniah, G., and Neill, A. (2016). Impact of Cell Culture on Recombinant Monoclonal Antibody Product Heterogeneity. Biotechnol. Prog. 32, 1103–1112. doi:10.1002/btpr.2327
Liu, L. (2015). Antibody Glycosylation and its Impact on the Pharmacokinetics and Pharmacodynamics of Monoclonal Antibodies and Fc-Fusion Proteins. J. Pharm. Sci. 104 (6), 1866–1884. doi:10.1002/jps.24444
Liu, L., Gomathinayagam, S., Hamuro, L., Prueksaritanont, T., Wang, W., Stadheim, T. A., et al. (2013). The Impact of Glycosylation on the Pharmacokinetics of a TNFR2:Fc Fusion Protein Expressed in Glycoengineered Pichia Pastoris. Pharm. Res. 30 (3), 803–812. doi:10.1007/s11095-012-0921-3
Liu, L., Hong, Z.-Y., and Wong, C.-H. (2006). Convergent Glycopeptide Synthesis by Traceless Staudinger Ligation and Enzymatic Coupling. ChemBioChem 7 (3), 429–432. doi:10.1002/cbic.200500437
Liu, L., Prudden, A. R., Bosman, G. P., and Boons, G.-J. (2017). Improved Isolation and Characterization Procedure of Sialylglycopeptide from Egg Yolk Powder. Carbohydr. Res. 452, 122–128. doi:10.1016/j.carres.2017.10.001
Liu, L., Prudden, A. R., Capicciotti, C. J., Bosman, G. P., Yang, J.-Y., Chapla, D. G., et al. (2019). Streamlining the Chemoenzymatic Synthesis of Complex N-Glycans by a Stop and Go Strategy. Nat. Chem 11, 161–169. doi:10.1038/s41557-018-0188-3
Lu, R. M., Hwang, Y. C., Liu, I. J., Lee, C. C., Tsai, H. Z., Li, H. J., et al. (2020). Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 27, 1. doi:10.1186/s12929-019-0592-z
Lugovskoy, A. A., Reichert, J. M., and Beck, A. (2012). 7th Annual European Antibody Congress 2011. mAbs 4 (2), 134–152. doi:10.4161/mabs.4.2.19426
Mackenzie, L. F., Wang, Q., Warren, R. A. J., and Withers, S. G. (1998). Glycosynthases: Mutant Glycosidases for Oligosaccharide Synthesis. J. Am. Chem. Soc. 120, 5583–5584. doi:10.1021/ja980833d
Maki, Y., Okamoto, R., Izumi, M., Murase, T., and Kajihara, Y. (2016). Semisynthesis of Intact Complex-type Triantennary Oligosaccharides from a Biantennary Oligosaccharide Isolated from a Natural Source by Selective Chemical and Enzymatic Glycosylation. J. Am. Chem. Soc. 138 (10), 3461–3468. doi:10.1021/jacs.5b13098
Maley, F., Trimble, R. B., Tarentino, A. L., and Plummer, T. H. (1989). Characterization of Glycoproteins and Their Associated Oligosaccharides through the Use of Endoglycosidases. Anal. Biochem. 180 (2), 195–204. doi:10.1016/0003-2697(89)90115-2
Manabe, S., Yamaguchi, Y., Abe, J., Matsumoto, K., and Ito, Y. (2018). Acceptor Range of Endo-β- N -acetylglucosaminidase Mutant Endo-CC N180H: from Monosaccharide to Antibody. R. Soc. Open Sci. 5 (5), 171521–171533. doi:10.1098/rsos.171521
Manabe, S., Yamaguchi, Y., Matsumoto, K., Fuchigami, H., Kawase, T., Hirose, K., et al. (2019). Characterization of Antibody Products Obtained through Enzymatic and Nonenzymatic Glycosylation Reactions with a Glycan Oxazoline and Preparation of a Homogeneous Antibody-Drug Conjugate via Fc N-Glycan. Bioconjug. Chem. 30 (5), 1343–1355. doi:10.1021/acs.bioconjchem.9b00132
Mastrangeli, R., Palinsky, W., and Bierau, H. (2019). Glycoengineered Antibodies: towards the Next-Generation of Immunotherapeutics. Glycobilogy 29, 199–210. doi:10.1093/glycob/cwy092
Meuris, L., Santens, F., Elson, G., Festjens, N., Boone, M., Dos Santos, A., et al. (2014). GlycoDelete Engineering of Mammalian Cells Simplifies N-Glycosylation of Recombinant Proteins. Nat. Biotechnol. 32, 485–489. doi:10.1038/nbt.2885
Mimura, Y., Katoh, T., Saldova, R., O’Flaherty, R., Izumi, T., Mimura-Kimura, Y., et al. (2018). Glycosylation Engineering of Therapeutic IgG Antibodies: Challenges for the Safety, Functionality and Efficacy. Protein Cell 9, 47–62. doi:10.1007/s13238-017-0433-3
Montero-Morales, L., and Steinkellner, H. (2018). Advanced Plant-Based Glycan Engineering. Front. Bioeng. Biotechnol. 6, 81. doi:10.3389/fbioe.2018.00081
Muir, T. W. (2003). Semisynthesis of Proteins by Expressed Protein Ligation. Annu. Rev. Biochem. 72 (1), 249–289. doi:10.1146/annurev.biochem.72.121801.161900
Noguchi, M., Fujieda, T., Huang, W. C., Ishihara, M., Kobayashi, A., and Shoda, S.-i. (2012). A Practical One-step Synthesis of 1,2-Oxazoline Derivatives from Unprotected Sugars and its Application to Chemoenzymaticβ-N-Acetylglucosaminidation of Disialo-Oligosaccharide. Hca 95 (10), 1928–1936. doi:10.1002/hlca.201200414
Noguchi, M., Tanaka, T., Gyakushi, H., Kobayashi, A., and Shoda, S.-i. (2009). Efficient Synthesis of Sugar Oxazolines from Unprotected N-Acetyl-2-Amino Sugars by Using Chloroformamidinium Reagent in Water. J. Org. Chem. 74 (5), 2210–2212. doi:10.1021/jo8024708
Parsons, T. B., Moir, J. W. B., and Fairbanks, A. J. (2009). Synthesis of a Truncated Bi-antennary Complex-type N-Glycan Oxazoline; Glycosylation Catalysed by the Endohexosaminidases Endo A and Endo M. Org. Biomol. Chem. 7 (15), 3128–3140. doi:10.1039/b907273j
Parsons, T. B., Struwe, W. B., Gault, J., Yamamoto, K., Taylor, T. A., Raj, R., et al. (2016). Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem. Int. Ed. 55 (7), 2361–2367. doi:10.1002/anie.201508723
Pereira, N. A., Chan, K. F., Lin, P. C., and Song, Z. (2018). The "Less-Is-More" in Therapeutic Antibodies: Afucosylated Anti-cancer Antibodies with Enhanced Antibody-dependent Cellular Cytotoxicity. mAbs 10 (5), 693–711. doi:10.1080/19420862.2018.1466767
Pichia GlycoSwitch (2021). Pichia GlycoSwitch® [Online]. Available at: https://pichia.com/glycoswitch/(Accessed 09 25, 2021).
Priyanka, P., Parsons, T. B., Miller, A., Platt, F. M., and Fairbanks, A. J. (2016). Chemoenzymatic Synthesis of a Phosphorylated Glycoprotein. Angew. Chem. Int. Ed. 55 (16), 5058–5061. doi:10.1002/anie.201600817
Reason, A. J., Weiskopf, A., and Rathore, A. S. (2014). Defining Critical Quality Attributes for Monoclonal Antibody Therapeutics. Biopharm. Int. 27 (7), 34–43.
Reusch, D., and Tejada, M. L. (2015). Fc Glycans of Therapeutic Antibodies as Critical Quality Attributes. Glycobiology 25, 1325–1334. doi:10.1093/glycob/cwv065
Rising, T. W. D. F., Claridge, T. D. W., Davies, N., Gamblin, D. P., Moir, J. W. B., and Fairbanks, A. J. (2006). Synthesis of N-Glycan Oxazolines: Donors for Endohexosaminidase Catalysed Glycosylation. Carbohydr. Res. 341 (10), 1574–1596. doi:10.1016/j.carres.2006.03.007
Rising, T. W. D. F., Heidecke, C. D., Moir, J. W. B., Ling, Z., and Fairbanks, A. J. (2008). Endohexosaminidase-Catalysed Glycosylation with Oxazoline Donors: Fine Tuning of Catalytic Efficiency and Reversibility. Chem. Eur. J. 14, 6444–6464. doi:10.1002/chem.200800365
Rosati, S., van den Bremer, E. T., Schuurman, J., Parren, P. W., Kamerling, J. P., and Heck, A. J. (2013). In-depth Qualitative and Quantitative Analysis of Composite Glycosylation Profiles and Other Micro-heterogeneity on Intact Monoclonal Antibodies by High-Resolution Native Mass Spectrometry Using a Modified Orbitrap. MAbs 5 (6), 917–924. doi:10.4161/mabs.26282
Seeberger, P. H., and Cummings, R. D. (2017). “Glycans in Biotechnology and the Pharmaceutical Industry,” in Essentials in Glycobiology. Editors R. D. C. A. Varki, and J. D. Esko 3 ed (New York: Cold Spring Harbor Laboratory Press).
Sehn, L. H., Assouline, S. E., Stewart, D. A., Mangel, J., Gascoyne, R. D., Fine, G., et al. (2012). A Phase 1 Study of Obinutuzumab Induction Followed by 2 Years of Maintenance in Patients with Relapsed CD20-Positive B-Cell Malignancies. Blood 119 (22), 5118–5125. doi:10.1182/blood-2012-02-408773
Shields, R. L., Lai, J., Keck, R., O'Connell, L. Y., Hong, K., Meng, Y. G., et al. (2002). Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human FcγRIII and Antibody-dependent Cellular Toxicity. J. Biol. Chem. 277 (30), 26733–26740. doi:10.1074/jbc.m202069200
Shinkawa, T., Nakamura, K., Yamane, N., Shoji-Hosaka, E., Kanda, Y., Sakurada, M., et al. (2003). The Absence of Fucose but Not the Presence of Galactose or Bisecting N-Acetylglucosamine of Human IgG1 Complex-type Oligosaccharides Shows the Critical Role of Enhancing Antibody-dependent Cellular Cytotoxicity. J. Biol. Chem. 278 (5), 3466–3473. doi:10.1074/jbc.M210665200
Shivatare, S. S., Huang, L.-Y., Zeng, Y.-F., Liao, J.-Y., You, T.-H., Wang, S.-Y., et al. (2018). Development of Glycosynthases with Broad Glycan Specificity for the Efficient Glyco-Remodeling of Antibodies. Chem. Commun. 54, 6161–6164. doi:10.1039/c8cc03384f
Sjögren, J., Struwe, W. B., Cosgrave, E. F., Rudd, P. M., Stervander, M., Allhorn, M., et al. (2013). EndoS2 Is a Unique and Conserved Enzyme of Serotype M49 Group A Streptococcus that Hydrolyses N-Linked Glycans on IgG and α1-acid Glycoprotein. Biochem. J. 455 (1), 107–118. doi:10.1042/BJ20130126
Sjögren, J., Cosgrave, E. F. J., Allhorn, M., Nordgren, M., Björk, S., Olsson, F., et al. (2015). EndoS and EndoS2 Hydrolyze Fc-Glycans on Therapeutic Antibodies with Different Glycoform Selectivity and Can Be Used for Rapid Quantification of High-Mannose Glycans. Glycobiology 25, 1053–1063. doi:10.1093/glycob/cwv047
Song, T., Ozcan, S., Becker, A., and Lebrilla, C. B. (2014). In-depth Method for the Characterization of Glycosylation in Manufactured Recombinant Monoclonal Antibody Drugs. Anal. Chem. 86 (12), 5661–5666. doi:10.1021/ac501102t
Sun, B., Bao, W., Tian, X., Li, M., Liu, H., Dong, J., et al. (2014). A Simplified Procedure for Gram-Scale Production of Sialylglycopeptide (SGP) from Egg Yolks and Subsequent Semi-synthesis of Man3GlcNAc Oxazoline. Carbohydr. Res. 396, 62–69. doi:10.1016/j.carres.2014.07.013
Tang, F., Wang, L.-X., and Huang, W. (2017). Chemoenzymatic Synthesis of Glycoengineered IgG Antibodies and Glycosite-specific Antibody-Drug Conjugates. Nat. Protoc. 12 (8), 1702–1721. doi:10.1038/nprot.2017.058
Tang, F., Yang, Y., Tang, Y., Tang, S., Yang, L., Sun, B., et al. (2016). One-pot N-Glycosylation Remodeling of IgG with Non-natural Sialylglycopeptides Enables Glycosite-specific and Dual-Payload Antibody-Drug Conjugates. Org. Biomol. Chem. 14, 9501–9518. doi:10.1039/c6ob01751g
Tischer, W., and Kasche, V. (1999). Immobilized Enzymes: Crystals or Carriers? Trends Biotechnol. 17 (8), 326–335. doi:10.1016/s0167-7799(99)01322-0
Tomabechi, Y., Krippner, G., Rendle, P. M., Squire, M. A., and Fairbanks, A. J. (2013). Glycosylation of Pramlintide: Synthetic Glycopeptides that Display In Vitro and In Vivo Activities as Amylin Receptor Agonists. Chem. Eur. J. 19 (45), 15084–15088. doi:10.1002/chem.201303303
Trincone, A., and Giordano, A. (2006). Glycosyl Hydrolases and Glycosyltransferases in the Synthesis of Oligosaccharides. Curr. Org. Chem. 10, 1163–1193. doi:10.2174/138527206777698075
Umekawa, M., Huang, W., Li, B., Fujita, K., Ashida, H., Wang, L.-X., et al. (2008). Mutants of Mucor Hiemalis Endo-β-N-Acetylglucosaminidase Show Enhanced Transglycosylation and Glycosynthase-like Activities. J. Biol. Chem. 283, 4469–4479. doi:10.1074/jbc.m707137200
Umekawa, M., Li, C., Higashiyama, T., Huang, W., Ashida, H., Yamamoto, K., et al. (2010). Efficient Glycosynthase Mutant Derived from Mucor Hiemalis Endo-β-N-Acetylglucosaminidase Capable of Transferring Oligosaccharide from Both Sugar Oxazoline and Natural N-Glycan. J. Biol. Chem. 285, 511–521. doi:10.1074/jbc.m109.059832
Unverzagt, C., and Kajihara, Y. (2013). Chemical Assembly of N-Glycoproteins: a Refined Toolbox to Address a Ubiquitous Posttranslational Modification. Chem. Soc. Rev. 42, 4408–4420. doi:10.1039/c3cs35485g
Urquhart, L. (2020). Top Companies and Drugs by Sales in 2019. Nat. Rev. Drug Discov. 19, 228. doi:10.1038/d41573-020-00047-7
U.S. National Library of Medicine (2017). A Phase I Study of Intravenous CHO-H01 in Patients with Refractory or Relapsed Follicular Lymphoma [Online]. Bethesda, MD, USA: U.S. National Library of Medicine. Available at: https://clinicaltrials.gov/ct2/show/NCT03221348 (Accessed 05 29, 2020).
Varki, A., Cummings, R. D., Aebi, M., Packer, N. H., Seeberger, P. H., Esko, J. D., et al. (2015). Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 25 (12), 1323–1324. doi:10.1093/glycob/cwv091
Vetere, A., Gamini, A., Campa, C., and Paoletti, S. (2000). Regiospecific Transglycolytic Synthesis and Structural Characterization of 6-O-α-Glucopyranosyl- Glucopyranose (Isomaltose). Biochem. Biophysical Res. Commun. 274 (1), 99–104. doi:10.1006/bbrc.2000.3068
Vic, G., Hastings, J. J., and Crout, D. H. G. (1996). Glycosidase-catalysed Synthesis of Glycosides by an Improved Procedure for Reverse Hydrolysis: Application to the Chemoenzymatic Synthesis of Galactopyranosyl-(1→4)-O-α-Galactopyranoside Derivatives. Tetrahedron: Asymmetry 7 (7), 1973–1984. doi:10.1016/0957-4166(96)00238-8
Walsh, G. (2005). Therapeutic Insulins and Their Large-Scale Manufacture. Appl. Microbiol. Biotechnol. 67, 151–159. doi:10.1007/s00253-004-1809-x
Wang, L.-X., and Amin, M. N. (2014). Chemical and Chemoenzymatic Synthesis of Glycoproteins for Deciphering Functions. Chem. Biol. 21 (1), 51–66. doi:10.1016/j.chembiol.2014.01.001
Wang, L.-X., Ni, J., Singh, S., and Li, H. (2004). Binding of High-mannose-type Oligosaccharides and Synthetic Oligomannose Clusters to Human Antibody 2G12. Chem. Biol. 11 (1), 127–134. doi:10.1016/j.chembiol.2003.12.020
Wang, L.-X., Tong, X., Li, C., Giddens, J. P., and Li, T. (2019). Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem. 88, 433–459. doi:10.1146/annurev-biochem-062917-012911
Wang, N., Seko, A., Daikoku, S., Kanie, O., Takeda, Y., and Ito, Y. (2016). Non-enzymatic Reaction of Glycosyl Oxazoline with Peptides. Carbohydr. Res. 436, 31–35. doi:10.1016/j.carres.2016.11.002
Wang, X., Ashhurst, A. S., Dowman, L. J., Watson, E. E., Li, H. Y., Fairbanks, A. J., et al. (2020). Total Synthesis of Glycosylated Human Interferon-γ. Org. Lett. 22 (17), 6863–6867. doi:10.1021/acs.orglett.0c02401
Wei, Y., Li, C., Huang, W., Li, B., Strome, S., and Wang, L.-X. (2008). Glycoengineering of Human IgG1-Fc through Combined Yeast Expression and In Vitro Chemoenzymatic Glycosylation. Biochemistry 47 (39), 10294–10304. doi:10.1021/bi800874y
Witte, K., Sears, P., Martin, R., and Wong, C.-H. (1997). Enzymatic Glycoprotein Synthesis: Preparation of Ribonuclease Glycoforms via Enzymatic Glycopeptide Condensation and Glycosylation. J. Am. Chem. Soc. 119 (9), 2114–2118. doi:10.1021/ja961846z
Wong, C.-H., Wu, C.-Y., and Tsai, M.-H. (2015). Anti-CD20 Glycoantibodies and Uses of Thereof. USA patent application.
Yamaguchi, T., Amin, M. N., Toonstra, C., and Wang, L.-X. (2016). Chemoenzymatic Synthesis and Receptor Binding of Mannose-6-Phosphate (M6P)-Containing Glycoprotein Ligands Reveal Unusual Structural Requirements for M6P Receptor Recognition. J. Am. Chem. Soc. 138 (38), 12472–12485. doi:10.1021/jacs.6b05762
Yamane-Ohnuki, N., Kinoshita, S., Inoue-Urakubo, M., Kusunoki, M., Iida, S., Nakano, R., et al. (2004). Establishment ofFUT8 Knockout Chinese Hamster Ovary Cells: An Ideal Host Cell Line for Producing Completely Defucosylated Antibodies with Enhanced Antibody-dependent Cellular Cytotoxicity. Biotechnol. Bioeng. 87 (5), 614–622. doi:10.1002/bit.20151
Zhao, K., Tang, F., Shi, W., Hong, H., Zhou, Z., Huang, W., et al. (2019). One-step Immobilization and Purification of Genetic Engineering CBD Fusion EndoS on Cellulose for Antibodies Fc-Glycan Remodeling. Bioorg. Chem. 91, 103114. doi:10.1016/j.bioorg.2019.103114
Keywords: biocatalysis, glycan, glycosylation, remodeling, glycosynthase, glycoform
Citation: Ivanova A and Falcioni F (2022) Challenges and Opportunities for the Large-Scale Chemoenzymatic Glycoengineering of Therapeutic N-Glycosylated Monoclonal Antibodies. Front. Catal. 1:810779. doi: 10.3389/fctls.2021.810779
Received: 07 November 2021; Accepted: 31 December 2021;
Published: 03 February 2022.
Edited by:
Sandy Schmidt, University of Groningen, NetherlandsCopyright © 2022 Ivanova and Falcioni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adelina Ivanova, YWFpMjdAY2FtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.