 Anna K. Schweiger
Anna K. Schweiger Kenji Miyamoto2
Kenji Miyamoto2 Robert Kourist
Robert Kourist- 1Institute of Molecular Biotechnology, Graz University of Technology, Graz, Austria
- 2Department of Biosciences and Informatics, Keio University, Yokohama, Japan
Bacterial arylmalonate decarboxylase (AMDase) is an intriguing cofactor-independent enzyme with a broad substrate spectrum. Particularly, the highly stereoselective transformation of diverse arylmalonic acids into the corresponding chiral α-arylpropionates has contributed to the broad recognition of this biocatalyst. While, more than 30 years after its discovery, the native substrate and function of AMDase still remain undiscovered, contributions from multiple fields have ever since brought forth a powerful collection of AMDase variants to access a wide variety of optically pure α-substituted propionates. This review aims at providing a comprehensive overview of the development of AMDase from an enzyme with unknown function up to a powerful tailored biocatalyst for the synthesis of industrially relevant optically pure α-arylpropionates. Historical perspectives as well as recent achievements in the field will be covered within this work.
Introduction
Arylmalonate decarboxylase (AMDase, EC 4.1.1.76), originally isolated from the soil bacterium Bordetella bronchiseptica, catalyzes the decarboxylation of α-aromatic- or α-alkenylmalonic acids to yield the corresponding optically pure mono-acid without the aid of a cofactor. While the wildtype enzyme exhibits strict (R)-selectivity, enzyme engineering afforded efficient (S)-selective or racemizing enzyme variants (Figure 1A). The native function or the natural substrate of AMDase, however, both remain unknown to date. The unique reactivity and broad substrate tolerance allows for the production of diverse aryl- or alkenylaliphatic carboxylic acids in outstanding optical purity, amongst them several α-arylpropionates with non-steroidal anti-inflammatory activity, the so-called profens (Miyamoto and Kourist, 2016).
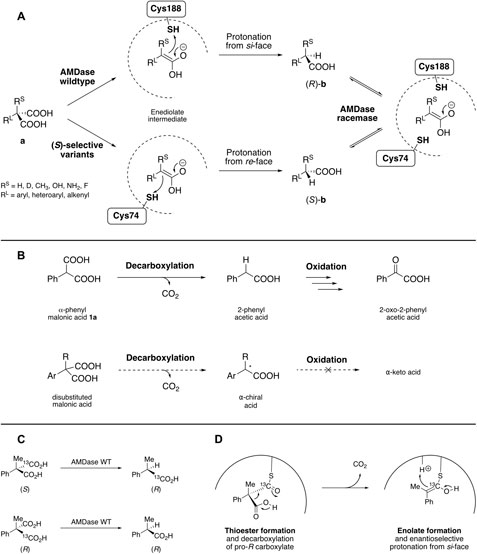
FIGURE 1. (A) Overview on AMDase catalyzed asymmetric decarboxylation by wildtype (variants with Cys188) and (S)-selective variants (Cys74) and AMDase catalyzed racemization (variants with Cys188/Cys74). (B) Assumed reaction pathway of α-phenylmalonic acid-converting microorganisms (top) and proposed route towards α-chiral acids via the same decarboxylative activity (bottom). (C) Detected reaction products after decarboxylation of 13C-labelled chiral malonic acid substrates by wildtype AMDase indicates, that the pro-(R) carboxylate is eliminated during the reaction in any case. (D) First postulated mechanism based on the assumption, that AMDase acts as a thiol decarboxylase. After thioester formation between the substrate and a cysteine in the active site, the pro-(R) carboxylate is cleaved and the resulting enolate is enantioselectively protonated from the si-face.
Discovery of Arylmalonate Decarboxylase
Miyamoto et al. discovered arylmalonate decarboxylase (AMDase) in the 1990’s in a screening to identify enzymes for the generation of chiral molecules from prochiral malonates by enzymatic decarboxylation (Miyamoto Ohta and, Hiromichi, 1990; Miyamoto and Ohta, 1992a) As the decarboxylation of malonyl-ACP is a key step in metabolism, it appeared likely that the bacterial catabolism might possess malonate decarboxylases. The screening followed the assumption that decarboxylation of α-phenylmalonic acid 1a could represent the initial step in the metabolism, followed by oxidation to yield 2-oxo-2-phenylacetic acid. Accordingly, it was proposed that analogous decarboxylation of disubstituted malonic acids would lead to α-chiral acids, as further oxidation would not be possible (Figure 1B).
An assay for microorganisms with the ability to grow on phenylmalonic acid 1a as the sole carbon source led to the identification of the soil bacterium Alcaligenes bronchisepticus KU 1201 (now: Bordetella bronchiseptica). It could be shown, that whole cells of this bacterium also converted α-methyl-α-phenylmalonic acid 2a and analogous substrates with various aromatic residues (Miyamoto Ohta and, Hiromichi, 1990; Miyamoto and Ohta, 1992b). The decarboxylase was purified from the microorganism and named arylmalonate decarboxylase (AMDase), due to its preference towards α-arylmalonates. Characterization of the purified enzyme revealed that it acts without a cofactor and is biotin-independent. Yet, the native role and substrate of AMDase remained unclear, as aryl malonates are not naturally abundant (Miyamoto and Ohta, 1992b).
Elucidation of Enzyme Mechanism and Selectivity
Shortly after the enzyme was discovered, Miyamoto et al. aimed at revealing the enzyme stereoselectivity by means of isotope labelling studies (Miyamoto et al., 1992b). For that, both enantiomers of 13C-labelled α-methyl-α-phenylmalonic acid 2a where synthesized. Products of the enzymatic reaction were analyzed via 13C NMR spectroscopy, which disclosed that if the (S)-substrate was used, the (R)-configured product still contained the 13C-labelled carboxylate, whereas in case of the (R)-substrate, enrichment of 13C was not detected in the remaining carboxylate of the (R)-configured product (Figure 1C). These results indicated that, in both cases, exclusively the pro-(R) carboxylate is cleaved, yielding the final product via inversion of configuration. Later on, this finding was confirmed by Okrasa et al. using 18O labeling of the enantiotopic carboxylate groups (Okrasa et al., 2009).
The observation that AMDase was inhibited by sulfhydryl reagents indicated that AMDase is likely to be a thiol decarboxylase (Miyamoto and Ohta, 1992b). Thus, the initial step of the reaction was believed to be the formation of a thioester between the pro-(S) carboxylate of the substrate and a cysteine residue in the active site (cf. activation by coenzyme A in fatty acid biosynthesis) (Figure 1D). The observed inversion of configuration, however, was in discrepancy to other known decarboxylases, where configuration was strictly retained (Kim and Kolattukudy, 1980). This prompted the authors to suggest either a SE2-type concerted mechanism with complete inversion, or the formation of an intermediary enolate, which further gets protonated in an enantioselective fashion from the si-face (Figure 1D) (Miyamoto et al., 1992b). Formation of a charged intermediate was also supported by the finding that substrates bearing electron-withdrawing groups (EWGs) were converted at a higher rate, due to their ability to stabilize the supposedly formed carbanion intermediate (Miyamoto and Ohta, 1992b).
Identification of the AMDase gene and sequence analysis thereof disclosed that the enzyme contains four cysteine residues (C101, C148, C171, C188) (Miyamoto and Ohta, 1992a). All were present in a reduced state and only mutation of C188 to serine (C188S) proved detrimental to enzyme activity (kcat), indicating its critical role in the reaction mechanism (Miyazaki et al., 1997). Soon thereafter, however, it became apparent that thioester formation does not play a role in the mechanism of AMDase. Instead, it was suggested that Cys188 rather acts as a proton donor. This became obvious after the wildtype enzyme, having the more acidic cysteine residue in the active center, was inactivated at a pH > 9, whereas the corresponding C188S variant was not. Interesting evidence was also found among quite remotely related (30% homology) enzymes from the racemase and isomerase family (Matoishi et al., 2004). Those enzymes, which were well-studied with regards to their mechanism, also exhibit a highly conserved cysteine residue in this region, which functions as a proton donating residue (Glavas and Tanner, 1999).
Inversion Of Enantioselectivity And Introduction Of Racemase Activity
Some enzymes with about 30% homology to AMDase were identified via PSI-BLAST (position-specific iterative basic local alignment search tool), all belonging to the class of isomerases (EC 5). Glutamate racemase (Lactobacillus fermenti), aspartate racemase (Streptococcus thermophilus), hydantoine racemase (Pseudomonas sp. strain NS671) and maleate isomerase (Alcaligenes faecalis) all share the conserved cysteine at position 188 with AMDase, while enzymes from the isomerase class have an additional cysteine at around residue 74 (Figure 2A) (Ijima et al., 2005). As the reaction mechanism and crystal structure of glutamate racemase were already well-studied, two cysteine residues on both sides of the substrate were proposed to be essential for the racemizing activity, by either abstracting or donating protons from opposite sites in a so-called two-base mechanism (Glavas and Tanner, 1999).
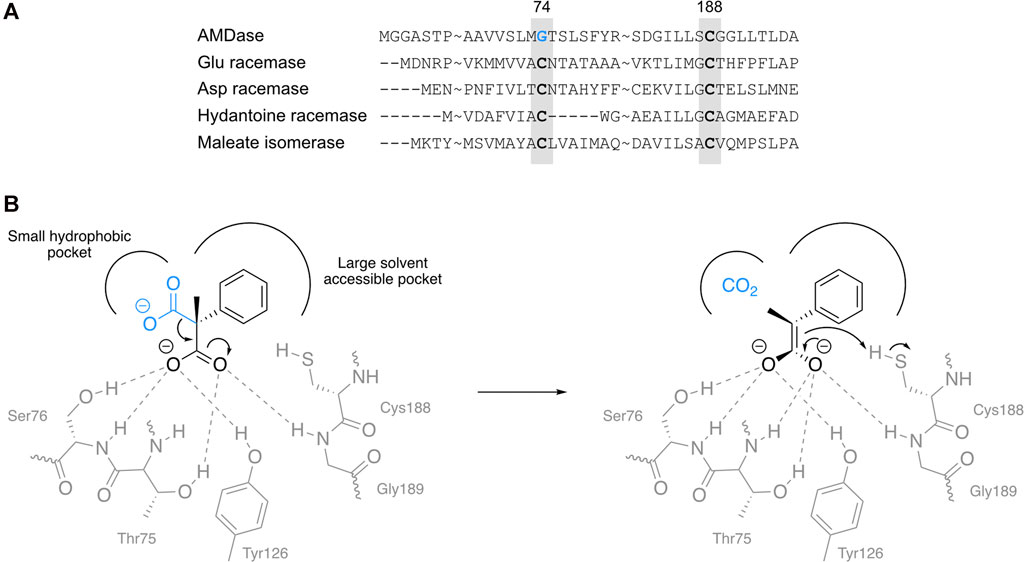
FIGURE 2. (A) Amino acid sequence homology between AMDase and selected racemases identified via PSI-BLAST. Glutamate racemase from Lactobacillus fermenti, aspartate racemase from Streptococcus thermophilus, hydantoine racemase from Pseudomonas sp. strain NS671, maleate isomerase from Alcaligenes faecalis and AMDase from Bordetella bronchiseptica. (B) Proposed binding mode and interaction network of α-methyl-α-phenylmalonic acid 2a (left) and the resulting enediolate intermediate (right) in the active site of AMDase WT. The pro-(R) carboxylate is shown in blue and the dioxyanion hole in gray.
Inspired by these findings, Ijima et al. decided to introduce a cysteine at position 74 instead of the glycine residue present in AMDase (Ijima et al., 2005). Additionally, previous studies have shown that the mutation G188S led to a drastic decrease of native AMDase activity (Miyazaki et al., 1997). While the proton-donating ability of serine is already low compared to cysteine, the authors proposed that an amino acid without any acidic proton would be beneficial for the enantiomeric excess of the obtained product. Surprisingly, the mutant G188A proved to be completely inactive. Hence, the double-variant G74C/C188S was prepared, and was found to produce the opposite enantiomers in 94–96 %ee as compared to wildtype AMDase. Yet, activity of the G74C/C188S variant even fell below the tremendously reduced activity of C188S (Ijima et al., 2005). Interestingly, also for the (S)-selective variant S36N/G74C/C188S, preference for cleavage of the pro-(R) carboxylate was observed by isotope labeling studies (Terao et al., 2006a), thus confirming that enantioselective reprotonation of the enolate intermediate is decisive for the final configuration of the product (Miyamoto et al., 2007a). As already observed by Ijima et al., the single amino acid exchange G74C rendered arylmalonate decarboxylase a racemase (Terao et al., 2006b). Notably, the decarboxylase activity of AMDase was preserved in the G74C variant, yielding racemic arylpropionates from arylmalonates. Kinetic analysis disclosed that, in terms of catalytic efficiency (kcat/KM), decarboxylation (0.96 s−1mM−1) exceeded racemization (0.56 s−1mM−1), which was also reflected by the racemic product already detected at an early stage of the reaction (Terao et al., 2006b).
Crystal Structure and Mechanism
Despite the crystal structure of AMDase remained unsolved until 2008, considerable insights were already acquired by that time, including suggestions on the enzyme mechanism (Miyamoto et al., 1992b; Miyamoto et al., 2007a; Matoishi et al., 2004), identification of key residues (Matoishi et al., 2004; Terao et al., 2007), switch in enantioselectivity (Ijima et al., 2005; Terao et al., 2006a) and introduction of a racemase activity (Terao et al., 2006b). Yet, the exact mechanism for stabilization of the highly unstable enediolate intermediate remained elusive. While other enzymes with related enolate intermediates use Mg2+ for stabilization (Gerlt et al., 2005), activity of AMDase is not dependent on metal ions or other cofactors (Miyamoto and Ohta, 1992b). Further, delocalization of electron density into the aromatic ring system might partly account for stabilization of the enediolate but would be insufficient to explain the observed highly efficient enzymatic decarboxylation (Okrasa et al., 2008).
By solving the first crystal structure of wildtype AMDase (PDB: 3DG9), Okrasa et al. were able to unravel key features of enzyme activity and selectivity in more detail. Interestingly, in the core structure consisting of two four-stranded parallel β-sheets surrounded by several α-helices, a tightly bound phosphate ion was found near the active site cysteine 188. A total of six hydrogen bonds were established by side-chain and backbone interactions to Thr75, Ser76, Tyr126, Cys188 and Gly189, belonging to two adjacent oxyanion holes (Okrasa et al., 2008). The authors suggested that the phosphate might resemble the position of the enediolate intermediate. In retrospective, this structural motif termed “dioxyanion hole” was also found in related enzymes of the isomerase family and mutational studies on Thr75 and Ser76 or Tyr126 of AMDase have already demonstrated the essential role of this region for enzyme activity (Terao et al., 2007; Okrasa et al., 2008).
After reconfirming loss of the pro-(R) carboxylate by a 18O-labelling strategy, the enediolate intermediate resulting from decarboxylation of α-methyl-α-phenylmalonic acid 2a was placed in the active site. It became obvious, that the phenyl moiety occupies a large solvent accessible pocket, whereas the small methyl substituent is left at an orientation, where only little space is available. Considering these restrictions and the fixed position of the pro-(S) carboxylate entrapped in the dioxyanion hole, it was suggested that the pro-(R) carboxylate must point towards a small hydrophobic pocket (Leu40, Val43, Val156, Tyr48), which could act as a driving force for decarboxylation by hydrophobic destabilization (Figure 2B). Thus, the key for cofactor-free decarboxylation by AMDase was proposed to be the extensive stabilization of one carboxylate, while the other one is destabilized by unfavorable electrostatic interactions (Okrasa et al., 2008).
This proposed binding mode was confirmed when AMDase was co-crystallized with the mechanism-based inhibitor benzylphosphonate (Ki = 5.2 mM) (PDB: 3IP8) (Okrasa et al., 2009). Similarly, the phosphonate dianion was tightly engaged by six hydrogen bonds to the residues of the dioxyanion hole and the phenyl residue was positioned in the large pocket stacked within the Gly189-Gly190 amide bond and Pro14 through van der Waals interactions.
Obata et al. solved the crystal structure of the G74C/C188S variant containing the ligand 2-phenyl acetate (PDB: 3IXL, Figure 3A) and of the G74C variant (PDB: 3IXM, Figure 3C) and WT enzyme (PDB: 3DTV, Figure 3D) in a sulfate associated form (Obata and Nakasako, 2010). While the overall structure of AMDase G74C/C188S was mostly preserved when compared to wildtype structures either containing BnzPO (PDB: 3IP8, Figure 3A) or PO43− (PDB: 3DG9) in the active site (RMSD of Cα atoms approx. 0.25 Å), considerable conformational differences became obvious by comparing to the SO42−-associated structures of the wildtype and G74C variant (PDB: 3DTV and 3IXM). It was suggested that a positional shift of Cys188-Gly189-Gly190 towards Gly74-Thr75 was induced upon ligand binding, thereby triggering the formation of a hydrophobic network covering the active site (Val43, Thr154, Val156). In the unliganded (SO42−-associated) crystal structures, such cluster was not observed, as the corresponding loop regions are not contacting each other. Additionally, in this conformation, the key residue 188 is rotated away from the ligand towards the hydrophobic pocket in a rather “non-productive” orientation (Figures 3C,D). These observations indicated the presence of either an “open” or “closed” conformation, regulated by ligand binding (Figure 3) (Obata and Nakasako, 2010). A similar behavior between structures of the empty and the ligand-bound enzyme was found for the related glutamate racemase (Puig et al., 2009).
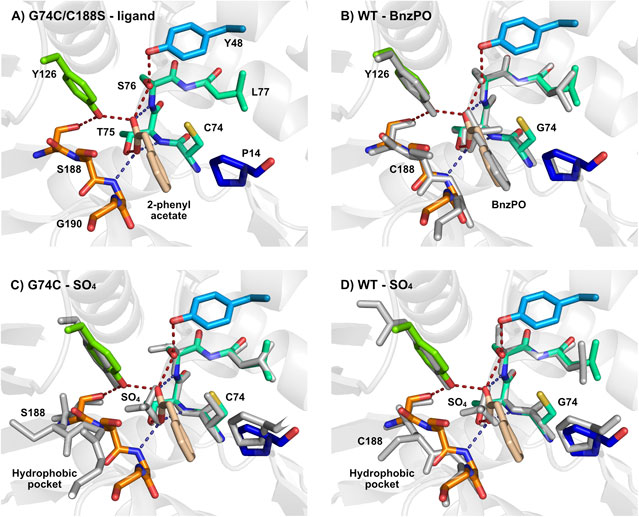
FIGURE 3. Representation of the active site of (A) AMDase G74C/C188S in association with 2-phenyl acetate (PDB: 3IXL) and superimposed with (B) AMDase WT in complex with benzylphosphonate (BnzPO) (PDB: 3IP8), (C) AMDase G74C in complex with SO42- (PDB: 3IXM) and (D) AMDase WT in complex with SO42- (PDB: 3DTV). Active site residues of AMDase G74C/C188S are shown in color and the respective superimposed structure in gray. Dotted lines represent H-bonds formed between the carboxylate and residue side-chains (red) or backbone NH groups (blue). Figure created with PyMOL.
Most important, however, was the observation that the position of Cys74 of the G74C/C188S variant was indeed in mirror symmetry to C188 of AMDase WT with regards to the Cα atom of the enediolate intermediate, which gets protonated during the reaction, thereby confirming the early proposed rationale for inversion of enantioselectivity (Ijima et al., 2005; Terao et al., 2007). In this binding mode, configuration of the final product is only dependent on the enantioface-selective protonation, which occurs from si-face in case of Cys188 or from re-face in case of Cys74, resulting in (R)- or (S)-2-phenylpropionate, respectively in accordance to experimental data (Miyauchi et al., 2011).
Computational Modelling of the Enzyme Mechanism
The reaction mechanism of AMDase was also subject of computational studies. Lind and Himo studied the mechanism by means of density functional theory (DFT) calculations (Lind and Himo, 2014). By employing the quantum chemical cluster approach, the mechanism, stationary points thereof and enantioselectivity were investigated with the aid of two different truncated active site models. The smaller model only partly contained the residues of the dioxyanion hole and the catalytic Cys188 (81 atoms), whereas the second model additionally included residues from the small and large binding pockets (223 atoms). Results obtained from using the small model indicated that a two-step mechanism proceeding via a planar enediolate intermediate is plausible, with the decarboxylation step being rate-limiting. However, binding modes leading either to cleavage of the pro-(R) or pro-(S) carboxylate were very similar in energy (1.9 kcal mol−1 for initial binding pose; 1.5 kcal mol−1 for transition state) probably due to the absence of the binding pockets determining the orientation of the remaining substituents, therefore being unable to explain the observed enantioselectivity with this small model (>99 %ee corresponds to at least 3 kcal mol−1). By using the larger model, the calculated energies for binding and decarboxylation were 14.1 and 18.3 kcal mol−1 higher for the formation of the (S)-enantiomer than for the (R)-product, mostly caused by unfavorable steric clashes of the large aryl substituent and the residues of the small binding pocket. When the much smaller α-methyl-α-vinylmalonic acid was studied, ambiguous results were obtained concerning absolute transition state energies, probably being a result of the too small and rigid models of the binding pockets. Both binding modes and transition states, however, were much closer in energy as compared to α-methyl-α-phenylmalonic acid 2a. This would indicate that stereoselectivity is exclusively determined by substrate binding in case of bulky aromatic compounds, whereas in case of smaller substrates, the decarboxylation transition state could also contribute to enantiodiscrimination (Lind and Himo, 2014). Just recently, Dasgupta et al. emphasized that geometric constraints such as introduced in quantum-chemical studies can lead to artifacts like imaginary vibrational frequencies and impaired efficiency of the overall optimization process (Dasgupta and Herbert, 2020). The authors thus introduced soft harmonic confining potentials to the terminal atoms of the model, thereby avoiding the artificial strain and rigidity of fixed-atom truncated active site models. By employing this system, they were able to reproduce the results from previous studies (Lind and Himo, 2014) and further claimed that this methodology is easy to implement and can dramatically reduce optimization efforts (Dasgupta and Herbert, 2020).
In the previously described modeling approaches, results were substantially dependent on the size of the truncated enzyme models used (Lind and Himo, 2014; Dasgupta and Herbert, 2020). In order to obtain a more comprehensive overview of the molecular level origin of AMDase selectivity, a full enzyme model would be highly preferable. Particularly, counterintuitive effects observed after iterative saturation mutagenesis within the AMDase active site cavity (Okrasa et al., 2009; Miyauchi et al., 2011; Yoshida et al., 2015) require consideration of the entire first coordination sphere. Thus, the empirical valence bond (EVB) approach was the methodology of our choice, which was further complemented by metadynamics simulations (Warshel and Weiss, 1980; Barducci et al., 2008; Biler et al., 2020). EVB simulations were able to reproduce experimentally observed activation energy barriers (within 3 kcal mol−1) and the preferential cleavage of the pro-(R) carboxylate group which was already determined during substrate positioning in the Michaelis complex. Yet curiously, the (S)-selective CLGIPL variant showed preferential cleavage of the pro-(S) group which still led to the expected formation of (S)-enantiomers due to reacting via a different substrate binding pose. For compounds, which were not or only poorly converted by AMDase and variants thereof, an increased flexibility and motility within the active site was observed during simulations. The resulting inadequate substrate positioning furthermore allowed water to enter the active center, which is likely to destroy the vulnerable interaction network vital for catalysis (Biler et al., 2020). By consulting the grid inhomogeneous solvation theory (GIST) (Nguyen et al., 2012; Nguyen et al., 2014) to analyze the local hydrophobicity within the AMDase active site, we found clear evidence for a mechanism driven by ground-state destabilization by entrapment of the carboxylate to-be-cleaved in a hydrophobic environment. These analyses also revealed the emergence of an additional hydrophobic cavity in the CLGIPL variant, which allows this variant to react via an alternative binding pose and unexpected cleavage of the pro-(S) carboxylate. While the alteration of hydrophobic pockets in the CLGIPL variant was not by design, the targeted engineering of the active site hydrophobicity presents a seminal approach for future engineering of AMDase. WT-MetaD simulations further confirmed that the reactive binding pose (according to EVB simulations) also coincides with the most populated binding pose observed at the Michaelis complex in most of the studied cases. Thus, AMDase selectivity is partly already determined at the stage of substrate binding, but selective destabilization of a distinct carboxylate group (leading to ground-state destabilization) seems to be the true determinant of the eventual reactive transition state (Biler et al., 2020).
Busch et al. used semiempirical QM/MM calculations to study the mechanism of the AMDase racemase variant G74C (Busch et al., 2016). MD simulations revealed that both catalytic cysteines need to be in their deprotonated state for successful catalysis. In contrast to glutamate racemase, where co-catalytic residues activate the cysteines (Glavas and Tanner, 2001), water is suggested to deprotonate the corresponding residues of AMDase G74C prior to substrate binding, which are then further stabilized by thiolate holes. This might also explain the pronounced pH-dependency of the reaction. Further semiempirical calculations indicated a stepwise mechanism similar to decarboxylation, where a delocalized π-electron system within the substrate is necessary to stabilize the enediolate intermediate (Busch et al., 2016). Likewise to previous studies (Lind and Himo, 2014), there was no indication for a concerted mechanism as described for glutamate racemase (Puig et al., 2009).
Another study conducted by Karmakar et al. aimed at the simulation of CO2 and product release from the active site (Karmakar and Balasubramanian, 2016). By performing (steered) MD simulations it was demonstrated that release of the decarboxylated product is energetically unfavorable and might even surpass the decarboxylation step, thus potentially rendering product release the true rate-limiting step (WT: ∼14 kcal mol−1 for decarboxylation vs. ∼23 kcal mol−1 for product release). The authors claimed that this might also partly account for the reduced activity of the G188S/G74C variant (20,000-fold) (Miyauchi et al., 2011), as the calculated energy barrier for product release was even higher in this case (∼20 kcal mol−1 for decarboxylation vs. ∼37 kcal mol−1 for product release) (Karmakar and Balasubramanian, 2016).
Substrate Scope
Already after the discovery of AMDase, Miyamoto and Ohta thoroughly characterized the enzyme and its substrate scope (Miyamoto and Ohta, 1992b). These fundamental findings, obtained in absence of any structural and mechanistic knowledge on the enzyme, provided the basis for studies up to the present by demonstrating the essential limitations of AMDase substrates. 1) Steric effects play a crucial role for the small substituent, as mono-substituted malonates were converted faster than the corresponding methylated substrates. Further, the ethyl analogue was not converted. 2) The aromatic substituent is essential for activity, as substrates with a benzyl-, phenoxy- or phenylthio- instead of an phenyl-group were inert (Miyamoto Ohta and, Hiromichi, 1990). This was also true for malonic acid (RL = H) or methylmalonic acid (RL = CH3). 3) Only free malonic acids can act as a substrate, as the corresponding mono- and diesters were not accepted. 4) Electron-withdrawing substituents on the aromatic moiety enhance the reaction rate by better stabilizing the proposed carbanion intermediate. Table 1 summarizes substrates studied in AMDase catalyzed decarboxylation and racemization.
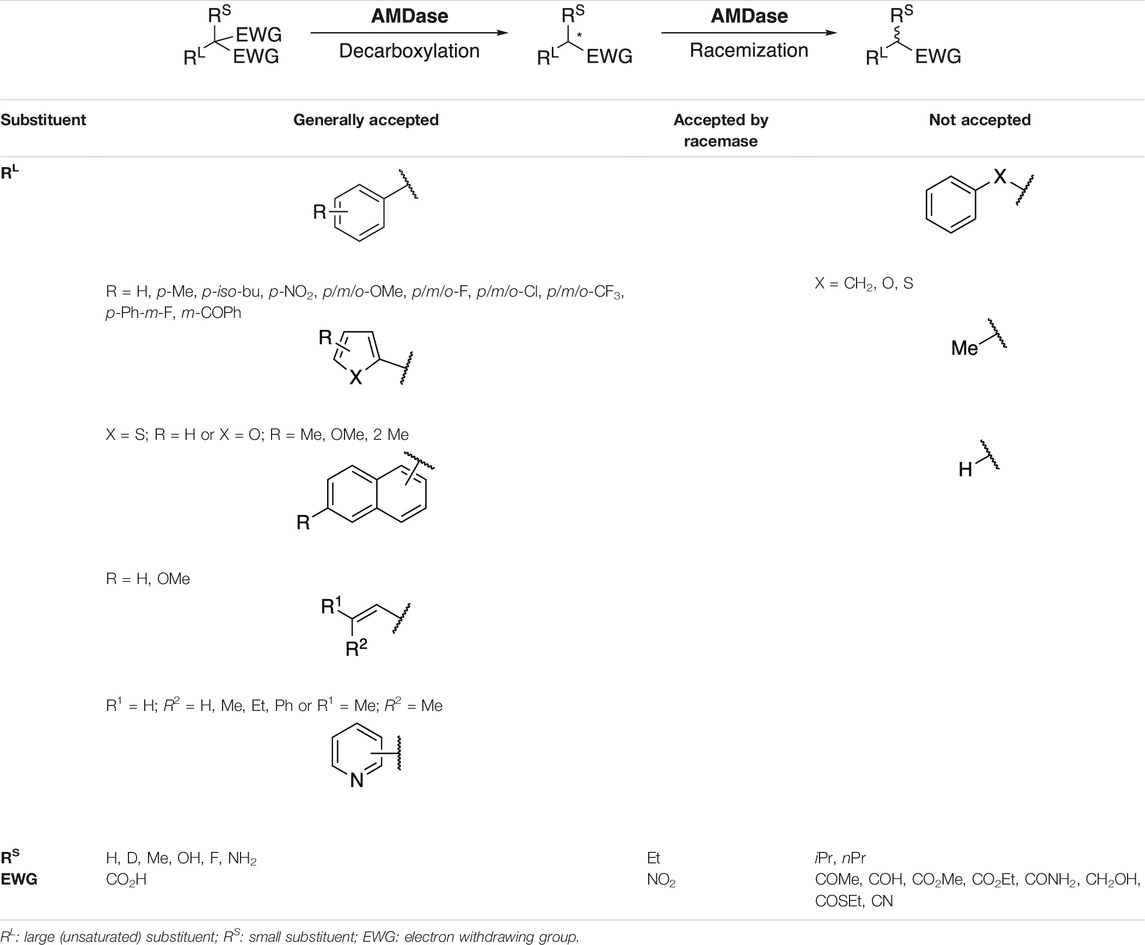
TABLE 1. Overview on the AMDase substrate scope.
The large substituent RL offers major variability within the substrate scaffold. Phenyl- (Miyamoto Ohta and, Hiromichi, 1990; Miyamoto and Ohta, 1992b; Miyamoto et al., 1992a; Miyamoto et al., 1994; Miyazaki et al., 1997; Fukuyama et al., 1999; Matoishi et al., 2000; Terao et al., 2006b; Terao et al., 2007; Okrasa et al., 2008; Okrasa et al., 2009; Tamura et al., 2008; Kourist et al., 2011b; Miyauchi et al., 2011; Yoshida et al., 2015; Lewin et al., 2015), 2-naphthyl- (Miyamoto and Ohta, 1992b; Ijima et al., 2005; Terao et al., 2006a; Terao et al., 2006b; Terao et al., 2007; Miyauchi et al., 2011) and thienyl- (Miyamoto Ohta and, Hiromichi, 1990; Miyamoto and Ohta, 1992b; Ijima et al., 2005; Terao et al., 2006a; Terao et al., 2006b; Terao et al., 2007; Lewin et al., 2015) malonic acids are typical substrates of AMDase and omnipresent in literature. Over time, the substrate scope was extended to a vast number of variously substituted phenyl derivatives (o-Me (Miyamoto Ohta and, Hiromichi, 1990), p-Me (Miyamoto and Ohta, 1992b; Kourist et al., 2011b), p-iso-butyl (Yoshida et al., 2015), p-NO2 (Kourist et al., 2011b), OMe (Miyamoto Ohta and, Hiromichi, 1990; Miyamoto and Ohta, 1992b; Kourist et al., 2011b), F (Miyamoto et al., 1992a; Miyamoto and Ohta, 1992b), Cl (Miyamoto Ohta and, Hiromichi, 1990; Miyamoto and Ohta, 1992b; Miyamoto et al., 1994), CF3 (Miyamoto et al., 1992a), p-Ph-m-F (Kourist et al., 2011b; Gaßmeyer et al., 2016), m-COPh (Kourist et al., 2011b)), naphthyl derivatives (1-naphthyl (Miyamoto and Ohta, 1992b), 6-methoxynaphthalen-2-yl (Miyamoto Ohta and, Hiromichi, 1990; Kourist et al., 2011b; Miyauchi et al., 2011; Yoshida et al., 2015; Gaßmeyer et al., 2016)) heterocyclic (pyridinyl (Kourist et al., 2011b; Lewin et al., 2015), furanyl and bicyclic systems (Lewin et al., 2015)) and also alkenyl (Okrasa et al., 2009; Kourist et al., 2011b) substrates.
With regards to the small substituent RS, it was soon recognized that, besides H and CH3, other small groups like D (Matoishi et al., 2000), F (Miyamoto et al., 1992a), OH or NH2 (Tamura et al., 2008) were generally tolerated, while larger alkyl groups (ethyl (Miyamoto and Ohta, 1992b) or propyl (Terao et al., 2006b; Kourist et al., 2011b)) were not or only converted at a very low rate (Terao et al., 2006b; Kourist et al., 2011b). This was later on explained by structural analysis, as only limited space is available at the position left for this group (Okrasa et al., 2008; Obata and Nakasako, 2010).
As already exposed by Miyamoto et al. in 1992, presence of the free di-acid is indispensable for decarboxylation to proceed (Miyamoto and Ohta, 1992b). This was also proven valid for the racemization activity of AMDase, as carboxylate derivatives such as alcohols, amides, nitriles (Terao et al., 2006b), esters (Terao et al., 2006b; Kourist et al., 2011b), thioesters, ketones and aldehydes (Kourist et al., 2011b) were completely inactive. Thus, stabilization of the free carboxylate via the complex network within the dioxyanion hole seems crucial, regardless of decarboxylation or racemization. The only exceptional case known so far is (nitromethyl)benzene, which was accepted as a substrate by AMDase racemase G74C and G74C/V43A (Kourist et al., 2011b). It was generally concluded that the substrate structure seems more decisive for successful conversion, than α-proton acidity of the α-aryl propionates (Kourist et al., 2011b).
In terms of spatial arrangement, Miyamoto et al. proposed that a co-planar alignment of the phenyl group and α-substituent would allow optimal overlap of the π-orbitals in order to stabilize the formed charge on the enolate intermediate (Miyamoto et al., 1994). In case of ortho-substituted substrates like α-(o-chlorophenyl)malonic acid, two such conformations come into consideration, namely a syn- and anti-periplanar arrangement (Figures 4A,B).
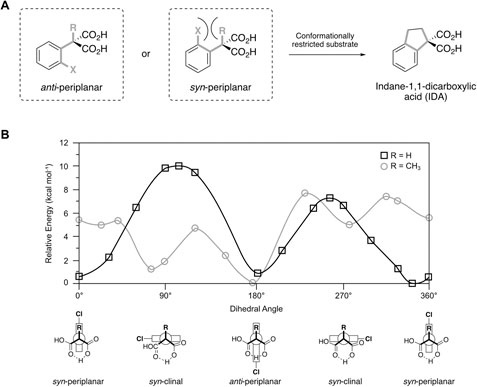
FIGURE 4. (A) Coplanar arrangement of aromatic ring and α-substituent R in a syn- or anti-periplanar conformation with respect to the ortho-substituent X (bold gray bonds). Indane-1,1-dicarboxylic acid (IDA) serves as conformationally restricted model substrate to study entropic contributions for substrate acceptance. (B) Potential energy diagram for the C-C bond rotation of α-(o-chlorophenylmalonic) acid (black squares) and α-(o-chlorophenyl)-α-methylmalonic acid (gray circles) calculated with HF/3-21G*. The phenyl ring is represented as rectangle in the Newman projection of the respective rotamers. Figure created based on data presented in Miyamoto et al. (1994).
Kinetic studies revealed that, while the activity of AMDase towards α-(o-chlorophenyl)malonic acid even surpassed α-phenylmalonic acid 1a, the corresponding α-methyl-substituted compound was not accepted at all. This led to the assumption, that a syn-periplanar arrangement is necessary for AMDase-mediated decarboxylation, which might in turn disfavor substrates with an additional α-substituent, due to steric clashes with the ortho-substituent (Miyamoto et al., 1994). Ab initio calculations were thus performed to assess the energy barriers encountered during rotation along the Cα-CAr bond. Results obtained for α-(o-chlorophenyl)malonic acid indicated two stable conformations corresponding to a syn- or anti-periplanar orientation (approx. 0° or 360° and 180° respectively). The small energy difference between both structures (<1 kcal mol−1) implies, that steric repulsion between o-Cl and α-H is negligible. Repulsive contributions were mainly caused by interaction of the chlorine residue with the carboxylates (Figure 4B). In case of α-(o-chlorophenyl)-α-methylmalonic acid, an anti-periplanar or perpendicular arrangement of substituents was energetically favored, whereas syn-periplanar-like conformations were found to be unstable (Figure 4B).
It was further proposed that by using the conformationally restricted indane-1,1-dicarboxylic acid (IDA), a syn-periplanar conformation would be mimicked, thereby lowering the activation entropy ΔS‡ (Figure 4A). Indeed, the model substrate IDA was accepted by AMDase in contrast to the corresponding α-methyl-α-(o-tolyl)malonic acid, thus underlining the inability of the latter to overcome rotational energy barriers towards a suitable syn-periplanar arrangement for conversion (Miyamoto et al., 1994). The remarkably low KM value observed for IDA in comparison to other studied substrates further affirmed the suitability of such spatial arrangement for AMDase-catalyzed decarboxylation (Miyamoto et al., 1994; Kawasaki et al., 1996). In this context it was also proposed that a hydrophobic pocket within the active site might be responsible for correct positioning of unrestricted substrates via CH-π interactions (Nishio et al., 1995), thereby reducing the activation entropy in a similar manner (Kawasaki et al., 1996).
Enzyme Engineering
In view of the dramatically reduced efficiency of the initially created variants with inversed (Ijima et al., 2005) or racemizing activity (Terao et al., 2006b) in comparison to the native enzyme, considerable efforts to recover or even to increase the activity by enzyme engineering have been made until today.
Enzyme Engineering of (S)-Selective C188X/G74C-Based Variants
At the time, when Ijima et al. first described the AMDase G74C/C188S variant, the obtained inversion of enantioselectivity by merely employing the yet uncovered spatial mirror symmetry of residues 188 and 74 was intriguing. Yet, the variant suffered from an incisive loss of activity due to diminished kcat (see Table 2) (Ijima et al., 2005). Early attempts on recovering the activity by random mutagenesis led to a variant (S36N/G74C/C188S) with increased (10-fold) activity among a total of 50,000 screened clones (Terao et al., 2006a). This reflected the difficulty of enzyme evolution purely relying on randomization in the absence of rational guidance.
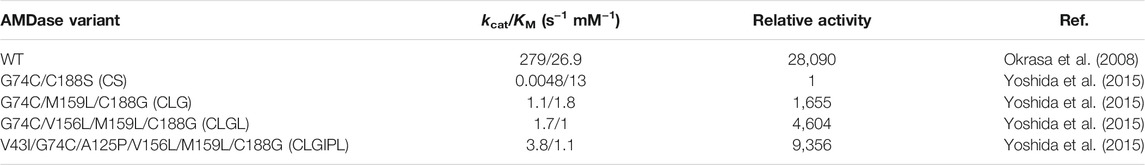
TABLE 2. Kinetic parameters of AMDase WT and (S)-selective variants towards α-methyl-α-phenylmalonic acid 2a according to Yoshida et al. (2015). Relative activity was calculated by dividing the catalytic efficiency (kcat/KM) of each variant by the catalytic efficiency of G74C/C188S variant.
After structural data of AMDase became available and, inspired by previous findings, that variations of the hydrophobic pocket residues can strongly enhance enzyme activity (Okrasa et al., 2009) Miyauchi et al. also attempted directed evolution of the G74C/C188S variant. This time, structural information provided the basis for three rounds of directed evolution via iterative saturation mutagenesis (ISM) (Reetz et al., 2008) of Ser188 in the first place, and residues of the hydrophobic pocket (Leu40, Val43, Tyr48, Leu77, Val156, Met159) in the following rounds (NNK codon for introduction of all 20 amino acids) (Figure 5) (Miyauchi et al., 2011). The critical role of the Cys188-substituting residue on enzyme activity was demonstrated earlier by the G74C/C188A variant being completely inactive (Ijima et al., 2005). From the first round of screening, variant G74C/C188G was identified to exhibit 5.6-fold increased activity towards α-phenylmalonic acid 1a. By using this variant as a template for the second generation, a triple mutant (G74C/M159L/C188G) with 210-fold higher activity evolved. Interestingly, the mutant selected from the last screening round (Y48F/G74C/M159L/C188G) with 920-fold increased activity carried the mutation Y48F, which was already found beneficial in the second-generation mutants. All identified beneficial mutations within the hydrophobic pocket were due to hydrophobic substitutions, thus underlining the high importance of this destabilizing environment for decarboxylation.
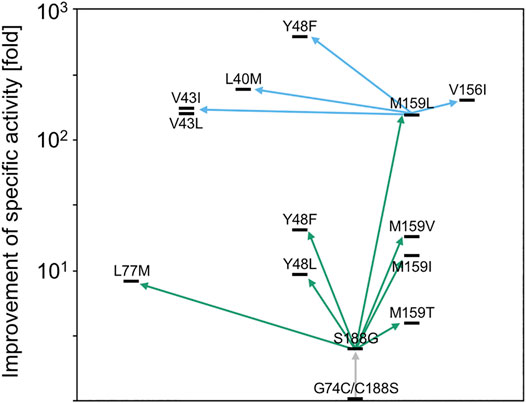
FIGURE 5. Enzyme variants evolved from AMDase G74C/C188S in three rounds of directed evolution via iterative saturation mutagenesis (ISM) and their improvement of specific activity towards α-phenylmalonic acid 1a. First (gray), second (green) and third (blue) screening are shown as arrows. Reproduced from Miyauchi et al. Miyauchi et al. (2011) with permission from the Royal Society of Chemistry.
As selection was based on conversion of α-phenylmalonic acid 1a, activity towards α-aryl-α-methylmalonic acids differed considerably. While the G74C/M159L/C188G variant showed a comparably increased activity in the synthesis of Naproxen 8b from its corresponding malonate (220-fold higher specific activity), the quadruple variant Y48F/G74C/M159L/C188G, superior for conversion of α-phenylmalonic acid 1a, exhibited only reduced activity. Additionally, the slightly impaired enantioselectivity of the G74C/C188S variant was completely abolished in the C188G-based variants (>99%ee), indicating potential disadvantageous interactions of the serine moiety with the enolate intermediate (Miyauchi et al., 2011).
Due to previously created variants, synergistic effects through multiple amino acid exchanges within the hydrophobic pocket were strongly indicated. Yoshida et al. conducted simultaneous saturation mutagenesis (SSM) on the previously evolved G74C/M159L/C188G (CLG) variant (Miyauchi et al., 2011), which allowed for simultaneous variation of multiple sites (Yoshida et al., 2015). Further, the authors argued, that evolution towards methyl substituted α-arylmalonic acids would be advantageous compared to simple α-phenylmalonic acid 1a, as the corresponding optical pure products are of major interest (Kourist et al., 2011a). Again, residues from the hydrophobic pocket were chosen as mutagenesis targets, due to their vicinity to the aryl residue (Leu40, Val156) or the proton-donating Cys74 (Met73). AMDase CLG was used as the starting variant, due to its superior activity in the synthesis of Naproxen 8b (Miyauchi et al., 2011) and a limited set of amino acids was introduced at the selected sites via the VTK codon (Leu, Ile, Val and Met). Surprisingly, only variants with mutations at V156 were selected from the first-generation library, thus indicating its crucial role in the decarboxylation mechanism. The best variant CLG-L (G74C/V156L/M159L/C188G) was chosen for the second round of SSM (see Table 2), where the region around the important residues Val156 and Met159 was targeted (Val43 and Ala125). Interestingly, from the three selected variants with improved activity, two included the mutation A125P, which seemed surprising, as A125 was part of an α-helix. The most active variant identified in this screening was CLGL-IP (V43I/G74C/A125P/V156L/M159L/C188G), where the catalytic efficiency (kcat/KM) was improved 2.1-fold compared to CLG-L and over 9,000-fold compared to the initial G74C/C188S variant (see Table 2).
The CLGIPL variant turned out to be highly (S)-selective and most active amongst the (S)-selective enzyme variants towards all tested substrates. Notably, malonate 7a was converted by AMDase CLGIPL with 1.3 U mg−1 (not converted by G74C/C188S) which was also more than twofold faster than the wildtype enzyme (0.53 U mg−1). Also Naproxen 8b was formed by CLGIPL (17 U mg−1) with high activity, which was yet surpassed by the (R)-selective wildtype (88 U mg−1) (Yoshida et al., 2015). The AMDase CLGIPL variant also turned out highly efficient in the conversion of malonate 6a (55 U mg−1) when compared to the wildtype (33.1 U mg−1) or the (S)-selective CLG variant (15.8 U mg−1) (Gaßmeyer et al., 2016). Interestingly, AMDase wildtype possesses higher activity towards the formation of Naproxen 8b compared to Flurbiprofen 6b, while AMDase CLGIPL shows an inversed preference. This emphasizes once more that the inherent reactivity of a substrate is less decisive for AMDase activity compared to the ability of productive binding in the active site and the sterical and electronical interactions resulting thereof.
Enzyme Engineering of (R)-Selective WT-Based Variants
By solving the AMDase WT crystal structure in complex with the mechanism-based inhibitor benzyl phosphonate, Okrasa et al. were able to further rationalize interactions of the enzyme active site residues and the substrate scaffold (Okrasa et al., 2009). They further proposed that alkenyl moieties might also afford suitable delocalization of electron density during the reaction, similarly to the previously studied substrates with aryl residues. Indeed, the tested alkenyl substrates proved to be suitable for AMDase-catalyzed decarboxylation, yet at lower efficiencies as their aromatic counterparts. Despite their relatively low turnover numbers (kcat), most of the alkenyl substrates often showed a higher affinity (KM) for AMDase than the respective phenyl derivatives. To prove the hypothesized interaction patterns in the active center, two sets of residues (Pro14, Pro15 and Gly190 from the large binding pocket and Val43 and Met159 from the hydrophobic pocket) were selected for three rounds of iterative saturation mutagenesis (NNK coding for all 20 amino acids). Due to the established setup, mutants were screened for α-phenylmalonic acid 1a, but were later on also tested towards the newly introduced alkenyl substrates (Okrasa et al., 2009).
By introducing the single point mutation M159V, the relative activity towards α-phenylmalonic acid 1a was raised 51-fold, thus implying at the same time, that it is not the native substrate of AMDase (Table 3, entry 1). The double mutant P14V/P15G exhibited enhanced activity towards several different substrates (Table 3, entry 2–4), which could be attributed to a generally increased flexibility within the larger binding pocket. The applicability of the M159V and P14V/P15G variants was later on also exemplified with a series of α-heterocyclic prochiral malonic acids (Lewin et al., 2015). The importance of substrate-fit was also demonstrated by the G190A variant, which converted α-methyl-α-vinylmalonic acid 4a, the smallest of all tested substrates, with 4.6-fold increased activity (Table 3, entry 5). The authors proposed that the slightly decreased space within the pocket might be beneficial for binding of the small vinyl group, which was also reflected in the lowered KM value (0.8 mM) as compared to the wildtype (7.8 mM) (Okrasa et al., 2009).
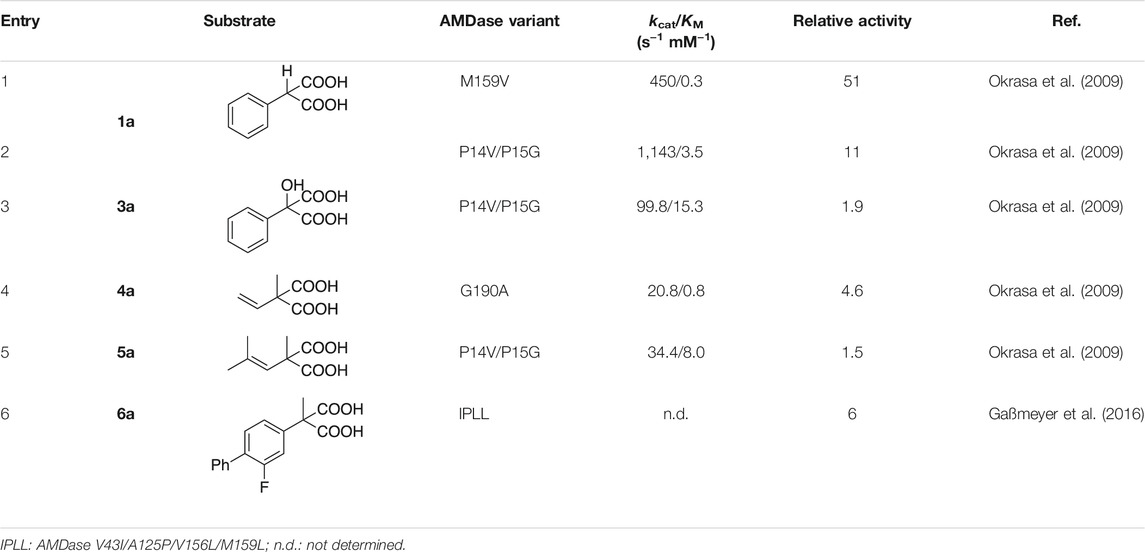
TABLE 3. Kinetic parameters of AMDase wildtype-derived variants towards different substrates. Relative activities were taken from literature and were either calculated by dividing the catalytic efficiency (kcat/KM) of each variant by the catalytic efficiency of wildtype AMDase (entry 1–5) or the corresponding specific activities (entry 6).
Gaßmeyer et al. showed that by transferring the set of amino acid substitutions previously found beneficial in the (S)-selective CLGIPL variant (V43I/A125P/ V156L/M159L excluding C188G/G74C), a potent (R)-selective variant (IPLL) for production of (R)-Flurbiprofen 6b is created. The quadruple variant converted the respective malonate with 209 U mg−1 to yield (R)-Flurbiprofen 6b in 98%ee, which represents a six-fold increase of specific activity compared to the native AMDase (Table 3, entry 6) (Gaßmeyer et al., 2016).
Enzyme Engineering of G74C-Based Variants With Racemising Activity
The limited activity of the AMDase G74C racemase towards bulkier and industrially relevant α-aryl propionic acid derivatives (also referred to as profens) prompted Kourist et al. to create more suitable racemase variants by rational enzyme engineering (Kourist et al., 2011b). While attempts to introduce co-catalytic residues (Asp, Glu, His) as present in the related glutamate racemase GluR (Glavas and Tanner, 2001) at the corresponding positions of AMDase (Val13, Pro14, Gly190) only led to inactive mutants, variation of the hydrophobic pocket residues proved more successful. Destabilization within the hydrophobic pocket is crucial for decarboxylation, however, only plays a minor role in racemization. Yet, most of its constituting residues are in close proximity of the reaction center, thereby indirectly affecting the activity of the racemase. In the case of AMDase G74C, MD simulations revealed, that Val43 and Met159 are potent engineering targets, due to their vicinity to the small substituent. Two of the designed variants, AMDase G74C/V43A and G74C/M159L, were found to exhibit enhanced racemization activity, while efficiency towards decarboxylation was decreased at the same time, thus underlining the different determinants for both promiscuous activities. In total, activity of the G74C/V43A variant was 20-fold shifted from decarboxylation towards racemization. Remarkably, activity towards racemization of ketoprofen 9b was also enhanced 30-fold (Kourist et al., 2011b). In a later study, structure-guided protein engineering of the racemase was performed based on STD-NMR (saturation-transfer-difference NMR) analysis, which eventually led to the quadruple variant V43A/G74C/A125P/V156L with generally increased activity towards all tested profen derivatives and a maximum of 40-fold activity-increase towards Naproxen 8b (Gaßmeyer et al., 2015).
Recent Applications
Production of Non-Steroidal Anti-Inflammatory Drugs
Chiral α-aryl propionates are members of the group of non-steroidal anti-inflammatory drugs (NSAIDs) and possess anti-inflammatory as well as analgesic properties (Brogden, 1986). Members of this drug class belong to top-selling over-the-counter drugs and are used for the treatment of rheumatic disease (muscoskeletal pain) and acute or chronic pain (Brooks, 1998). NSAIDs inhibit prostaglandin biosynthesis by acting on the cyclo-oxygenase (COX) enzyme system, which at the same time causes common adverse effects on gastrointestinal functions by non-specific inhibition of a different COX isoform (Brogden, 1986; Brooks, 1998). Amongst the commonly known profens, only Naproxen 8b is marketed as pure (S)-enantiomer, whereas Ibuprofen 7b, Ketoprofen 9b or Flurbiprofen 6b are administered in a mixture together with their inactive (R)-enantiomers (Brooks, 1998). However, unidirectional in vivo interconversion of the inactive (R)- to the pharmacologically active (S)-enantiomer can occur to a varying extent (Tracy and Hall, 1992; Brooks, 1998). Even though Flurbiprofen 6b is mostly sold as a racemate, the (S)-enantiomer primarily accounts for the anti-inflammatory effect (Abdel-Aziz et al., 2012). Yet, other biological effects of (R)-Flurbiprofen 6b had been described (Geerts, 2007; Jin et al., 2010; Liu et al., 2012), thus making the accessibility of both enantiomers in pure form a desirable goal. The ability of AMDase to convert prochiral malonates into optically pure arylpropionic acids was early recognized in the context of profen synthesis, yet hampered by the availability of efficient enzyme variants or limited stability of the malonate precursors (Terao et al., 2003). Table 4 should give an overview on NSAID precursors studied in AMDase catalyzed decarboxylation and the corresponding best-performing enzyme variants to date.

TABLE 4. Overview on AMDase substrates with relevance for research and industry.
Interestingly, several industrially applied routes for profen synthesis employ chemical malonate decarboxylation (Hylton and Walker, 1981; Mizushima et al., 2014). Due to the inherent instability of the intermediately formed malonic acid, ester hydrolysis and decarboxylation are often carried out simultaneously, thus yielding racemic product mixtures (Hylton and Walker, 1981; Lu et al., 2006; Mizushima et al., 2014). Yet, efforts to obtain pure enantiomers, like recrystallization of diastereomeric mixtures or enzymatic kinetic resolution, are often tedious and suffer from low yields or limited enantiomeric excess (Terao et al., 2003). In this respect, also enzymatic racemization under mild conditions could contribute to higher overall yields by recycling the undesired enantiomer for resolution processes (Kourist et al., 2011b).
In the synthesis of Flurbiprofen 6b, the low stability of the corresponding malonic acid substrate with its electron-withdrawing fluorine substituent represents a serious problem for its work-up and isolation after the saponification of α-arylmalonic acid esters. Inspired by a protection group strategy used by Miyamoto and Ohta for the preparation of isotope-labelled pseudochiral malonates (Miyamoto et al., 1992b), we implemented hydrogenolysis of benzyl esters instead of alkyl ester hydrolysis to avoid problems associated to spontaneous decarboxylation (Gaßmeyer et al., 2016). This strategy is particularly suitable for all malonates with electron-poor larger substituents.
We then developed an integrated chemo-enzymatic pathway towards Flurbiprofen 6b including AMDase-mediated decarboxylation and the previously introduced protecting group strategy (Gaßmeyer et al., 2016; Enoki et al., 2019a). We envisioned two potential chemo-enzymatic routes similarly to previously described chemical synthesis strategies, yet adapted to the corresponding dibenzyl malonic acid esters (Figure 6A) (Terao et al., 2003; Lu et al., 2006). Interestingly, the AMDase-catalyzed reaction step (Figure 6A, step (viii)) was planned at two different stages of the process. Either after Suzuki coupling, thereby using the established malonate 6a as substrate (Figure 6A, step (viii-a)), of before Suzuki coupling, which would require that AMDase exhibits activity towards α-(4-bromo-3-fluorophenyl)-α-methylmalonic acid (Figure 6A, step (viii-b)) (Enoki et al., 2019a).
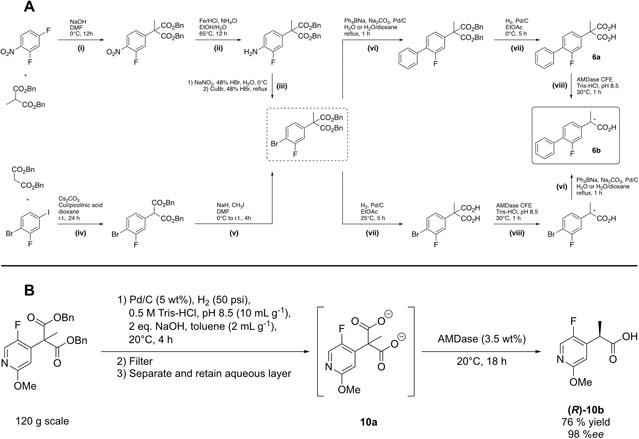
FIGURE 6. (A) Chemo-enzymatic synthesis routes towards optically pure Flurbiprofen 6b. The upper route was inspired and adapted from an industrial chemical synthesis and the lower route was adapted to the aspired conditions of the chemo-enzymatic process. (B) Chemo-enzymatic synthesis of electron-deficient N-heteroaromatic propionic acids.
After necessary adaptions were established for the initial chemical steps towards the central intermediate dibenzyl α-(4-bromo-3-fluorophenyl)-α-methylmalonic acid ester (Figure 6A, steps (i)-(v)), the authors focused on the second half of the cascade, including Pd-catalyzed Suzuki coupling (step (vi)), deprotection of the dibenzyl-protected malonates (step (vii)) and eventually AMDase catalyzed decarboxylation (step (viii)). It was suggested that palladium on charcoal (Pd/C) should be a suitable catalyst for both Suzuki coupling and cleavage of benzyl esters. Notably, during hydrogenolysis, the central intermediate was less reactive (step (vii-b)) compared to dibenzyl flurbiprofen malonic acid ester (step (vii-a)), yet more stable towards spontaneous decarboxylation, which proved advantageous for obtaining a high final ee of the product (Enoki et al., 2019a). While Suzuki coupling under mild conditions in water, using Pd/C as a catalyst and Ph4BNa as phenyl group donor, was reported (step (vi-b)) (Lu et al., 2006), those particular conditions proved impractical for the corresponding reaction of the alternative route (step (vi-a)), due to solvent incompatibilities of the reagents. Alternative Suzuki coupling strategies in organic solvents were not considered at this point, by reasons of the aspired environmentally benign reaction conditions. Eventually, AMDase variants also proved active towards the yet uncharacterized substrate in step (viii-b) with 114.3 U mg−1 (WT), 480.3 U mg−1 (IPLL), 41.7 U mg−1 (CLG) or 40.7 U mg−1 (CLGIPL), which was comparable to the activities towards malonate 6a. Both enantiomers of Flurbiprofen 6b could thus be produced in high yields and >99%ee (Enoki et al., 2019a).
Synthesis of Decarboxylation of Heteroaromatic Malonic Acids
The deprotection strategy was also successfully applied for the preparation of electron-deficient N-heteroaromatic malonic acids (Blakemore et al., 2020). The inherent instability of this compound class even surpasses the one of their common aromatic counterparts, thus fully preventing successful isolation without undesired spontaneous decarboxylation even under the typically mild hydrogenolysis conditions. After optimization, hydrogenolysis was performed at carefully balanced basic conditions in a biphasic system (5 wt% Pd/C; 10 ml g−1 0.5 M Tris-HCl, pH 8.5; 2 eq. NaOH; 2 ml g−1 toluene, 20°C, 4 h), which furnished the respective malonate dianions in the buffered aqueous phase. This layer was separated and directly used for enzymatic decarboxylation (3.5 wt% lyophilized CFE, 20°C, 18 h), thus circumventing intermediary isolation of the unstable malonic acid derivatives. The usability of this telescoped hydrogenation-AMDase process was demonstrated on a 120 g scale, producing optically pure 10b in 76% yield and 98%ee (R) (Figure 6B). To prevent undesired spontaneous decarboxylation of the AMDase substrates, the importance of careful pH and temperature control throughout the process was highlighted.
Synthesis of Optically Pure Alkanoic Acids
AMDase requires a substituent with a π-electron system on the substrate and does not convert aliphatic malonic acids. Nevertheless, enantiopure alkanoic acids could be accessed in two steps by combining AMDase-mediated decarboxylation and reduction of the non-activated double bond (see Table 5). With other catalytic methods, such molecules are not easily accessible, particularly due to the difficulty of chemical catalysts to distinguish the structurally similar substituents (Enoki et al., 2019b). While the activity of wildtype AMDase towards alkenyl malonic acids was already reported (Okrasa et al., 2009), reactivity of other (R)- and (S)-selective enzyme variants was not yet studied with this class of substrates.
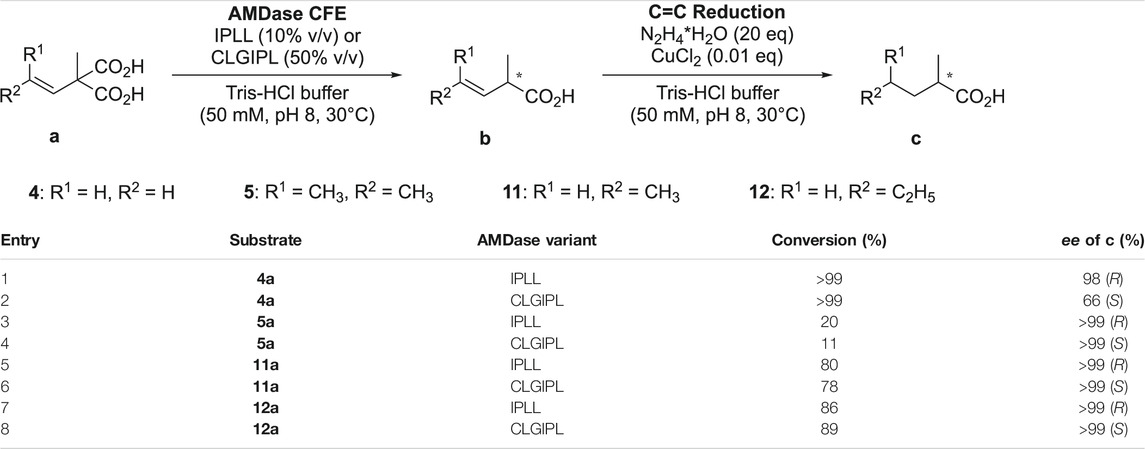
TABLE 5. Sequential chemo-enzymatic cascade of AMDase catalyzed decarboxylation of α-alkenyl-α-methylmalonic acids followed by C=C bond reduction with in situ generated diimide. After completion of the biocatalytic reaction step, hydrazine (20 eq) and CuCl2 (0.01 eq) were added directly to the reaction mixture. The stated conversion refers to the final reaction step and was detected after 23 h.
As already demonstrated with other substrate types, AMDase IPLL and CLGIPL outperformed the wildtype enzyme and other (S)-selective variants, yet with a 19–170 and 190–2,200 times reduced activity, respectively, as compared to aromatic substrates. This reflected the lower capacity of an alkene to stabilize the enediolate intermediate (Enoki et al., 2019b).
Interestingly, all tested enzyme variants proved to be highly stereoselective, with the conversion of α-methyl-α-vinylmalonic acid 4a by AMDase CLGIPL as only exception. This reaction produced (S)-2-methylbut-3-enoic acid with surprisingly low 66%ee (S) (Table 5, entry 2). In this case, either an alternative proton donor (e.g. water) or binding mode was discussed to cause such impaired enantioselectivity, as control reactions with other, highly selective AMDase variants ruled out a racemizing side-reaction as source of this low enantiomeric excess (Lind and Himo, 2014). Moreover, AMDase CLGIPL decarboxylated α-vinyl malonates with additional substituents present on the alkene (11–12a) with outstanding enantioselectivity, thus reflecting the fragile interaction network responsible for ligand recognition and binding.
Regarding chemical C=C bond reduction, the potential risk of double bond isomerization during transition-metal catalyzed reduction, which would cause product racemization, could be eventually eliminated by using in situ generated diimide as reductant. The high enantiomeric excess observed after AMDase-catalyzed decarboxylation could be thus retained until after the final chemical reduction step (Table 5) (Enoki et al., 2019b).
Immobilization of Arylmalonate Decarboxylase
Enzyme immobilization can significantly contribute to an economical use of a biocatalyst (e.g.: stability, reuse) and facilitate downstream processing (Cantone et al., 2013). Considering the limited stability of purified AMDase under process conditions (t1/2 = 1.2 h) (Aßmann et al., 2017a), differing immobilization strategies were tested recently, including site-specific (Wong et al., 2010) or conservative (Gaßmeyer et al., 2016; Aßmann et al., 2017b; Aßmann et al., 2017a) covalent immobilization, adsorption, complexation (Aßmann et al., 2017a), entrapment or combined approaches (Table 6) (Markošová et al., 2018). Wong et al. studied the phosphopantetheinyl transferase (Sfp)-catalyzed immobilization of ybbR-tagged proteins (12-mer N-terminal tag) on CoA-functionalized polystyrene nanoparticles (Table 6, entry 1) (Wong et al., 2010). The covalent and site-specific linkage was efficiently achieved under mild conditions, and the obtained biocatalyst revealed high operational stability over four cycles (approx. 7% loss of activity). Yet, the observed KM values were approximately three times higher compared to the free enzyme, whereas KM values for AMDase and ybbR-AMDase were similar, thus indicating that unfavorable interactions between the nanoparticle and substrate are causing this observed loss in efficiency rather than the immobilization itself (Wong et al., 2010).
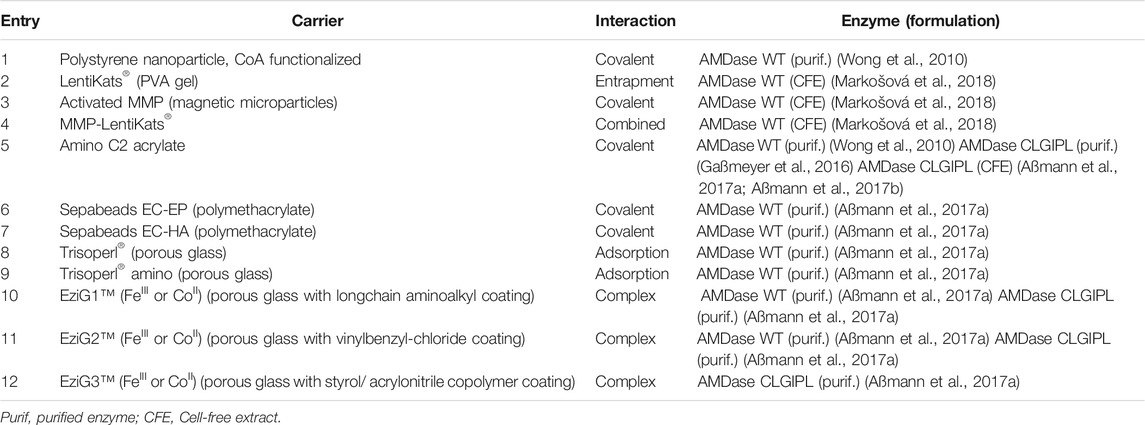
TABLE 6. Overview on immobilization techniques studied in AMDase catalyzed decarboxylation.
Recently, Markošová et al. attempted entrapment of AMDase in polyvinyl alcohol (PVA) gel (Table 6, entry 2) (Markošová et al., 2018). While AMDase showed enhanced stability, the formulation suffered from severe activity loss after repeated use, probably due to leaching of the enzyme. In a combined approach (Table 6, entry 4), the combination of the operational stability of covalently bound AMDase on magnetic microparticles (MMP) and easy handling of PVA gel beads afforded a biocatalyst, which was stable over eight biocatalytic reactions and upon storage at 4°C (64% retained activity after 3 months) (Markošová et al., 2018).
Covalent immobilization on well-established amino C2 acrylate carrier proved practical to obtain a stable biocatalyst (approx. 20% activity yield) with a half-life of 16.5 h and a total turnover number (TTN) of over 20,000 (Table 6, entry 5) (Gaßmeyer et al., 2016). The same strategy was further studied and applied by Aßmann et al. in an upscaled process for (S)-naproxen 8b synthesis via decarboxylation of 8a (Aßmann et al., 2017a; Aßmann et al., 2017b). This example demonstrates the complementarity of enzyme engineering and process engineering. While enzyme engineering could recover the initially reduced activity of the (S)-selective variants by several hundred folds to levels comparable to the wildtype, enzyme immobilization achieved a substantial improvement of the stability. Compared to the (S)-selective variant G74C/C188S in solution, both protein and reaction engineering achieved a tremendously improved productivity of the enzyme variants with TTNs of 83,000–107,000 over five batches (Aßmann et al., 2017a; Aßmann et al., 2017b). Also in this case, immobilization increased the KM-value towards 8a from 0.08 to 22 mM, underlining the strong effect of mass-transfer limitation. At the same time, different other support materials and binding strategies for immobilization were evaluated (Table 6, entry 5–12). Overall, covalently attached enzyme preparations were clearly preferred in terms of enzyme loading and long-term stability in repeated batches. For example, activity of AMDase immobilized on EziG1 was formidable, yet the coordinative binding between the enzyme’s 6xHis-tag and Fe(III) or Co(II) on the carrier is considerably weaker and thus led to extensive leaching during repeated experiments. Immobilization on amino C2 acrylate was further optimized by using CFE instead of purified protein, which certainly led to a decreased catalytic activity (30% compared to purified AMDase), yet to an enhanced long-term operational stability (t1/2 = 8.6 d) (Aßmann et al., 2017a).
Immobilized AMDase was used by Aßmann et al. for an intensification and up-scale of the synthesis of (S)-Naproxen 8b. A careful analysis of kinetic parameters was greatly facilitated by an in-line reaction monitoring via Raman spectroscopy. Importantly, AMDase CLGIPL was inhibited by the product, but only to an extent, where in situ product removal was not necessary. A productivity of 140 kgproduct kgenzyme−1 was calculated for the implemented process, which exceeds the minimum specified value for pharmaceutical processes of 50–100 kgproduct kgenzyme−1. Further, product isolation via precipitation was optimized and (S)-naproxen 8b was isolated in 92% yield and 99%ee (Aßmann et al., 2017b).
Arylmalonate Decarboxylases From Other Organisms
In search of organisms with similar reactivities and characteristics as AMDase from Alcaligenes bronchisepticus (now: Bordetella bronchiseptica) KU1201, Ohta and co-workers were able to identify two further strains from soil samples by their ability to degrade α-phenylmalonic acid 1a (Miyamoto et al., 2007b; Yatake et al., 2008). The strains KU1311 (Miyamoto et al., 2007b) and KU1313 (Yatake et al., 2008) were the most active ones and identified as Achromobacter sp. and Enterobacter cloacae, respectively. Respective genes were amplified from the genomic DNA and heterologously produced in E. coli for characterization. Both genes encoded a protein of 240 amino acids length, consistent with the originally identified AMDase from strain KU1201 (Miyamoto and Ohta, 1992a) and both shared a high sequence homology of 94% (KU1311) and 85% (KU1313). Accordingly, the observed substrate selectivity and activity were comparable to the original AMDase and all shared the same strict enantiopreference (Miyamoto and Ohta, 1992a, 1992b; Miyamoto et al., 2007b; Yatake et al., 2008).
While those three AMDases show considerable similarity, the shifted pH optimum (pH 5.5) towards acidic conditions of AMDase from strain KU1313 was intriguing. It was the only microorganism, which showed AMDase activity when soil samples were screened at acidic pH, others were rather active at neutral to slight basic pH (pH 7–8.5) (Table 7).
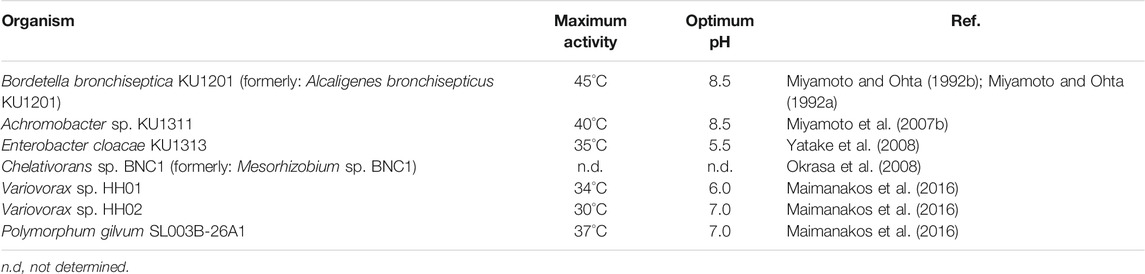
TABLE 7. Overview on AMDases identified from different organisms.
Okrasa et al. tried to identify novel AMDase enzymes from protein sequences with a similarity of 30-52% and a requisite cysteine residue at the same relative position as in the enzyme from B. bronchiseptica. The putative AMDases were characterized, however only the enzyme from Mesorhizobium sp. (now: Chelativorans sp.) BNC1, exhibiting the highest similarity, showed satisfactory decarboxylase activity, while the other candidates rather acted as racemases (Okrasa et al., 2008).
The difficulty of identifying novel AMDases accompanied by the issue of inappropriate gene annotation was addressed by the work of Maimanakos et al., who developed a sequence-based search algorithm for a more reliable prediction of enzymes possessing AMDase activity (Maimanakos et al., 2016). Sequence information of confirmed AMDases was used to specify 12 conserved sequence patterns (e.g. residues of the hydrophobic pocket, dioxyanion hole, aryl binding pocket and residues C188 and G74 including their surroundings), which, in turn, were used for concise database screening. In this way, 58 additional ORFs encoding putative AMDase-like enzymes were found and used for generating a Hidden Markov Model (HMM), which furnished six specific motifs (Table 8) and was implemented in the applied search algorithm. To prove the reliability and applicability of these search criteria, the enzyme from Polymorphum gilvum SL003B-26A1 identified via data base search was characterized together with two enzymes from Variovorax sp. HH01 and HH02 identified via screening of soil samples. All three enzymes showed the desired AMDase activity within common mesophilic temperature and around neutral to slightly acidic pH (Table 7) (Maimanakos et al., 2016).

TABLE 8. Identified conserved sequence motifs and residues involved in the catalytic mechanism of AMDase (in bold). Unconserved residues are denoted with x. Residue numbering according to B. bronchiseptica AMDase (Maimanakos et al., 2016).
Interestingly, all putative AMDase-encoding ORFs identified by Maimanakos et al. belonged to the class of α- β- and γ-proteobacteria. Interestingly, when AMDase flanking regions were analyzed, genes encoding mandelate racemase/muconate lactonizing enzyme or several transporters (e.g. tripartite tricarboxylate transporter TTT, TRAP or ABC transporters) were commonly found. According to these flanking regions and sequence similarities, AMDases were classified into eight enzyme clusters. While the natural role of this enzyme still remained elusive, the fact, that close relatives of AMDase-producing bacteria often do not encode this enzyme, strongly indicated, that this gene was frequently lost in evolution and might be therefore only temporarily advantageous for organisms under certain physiologic conditions (Maimanakos et al., 2016).
Conclusion
30 years after its discovery, AMDase has arrived at the stage of industrial applications. The history of research on this unique decarboxylase is exemplary for the technological progress in biocatalysis within the last decades. After AMDase discovery, based on a purely functional screen, the first important milestone achieved by the Ohta group was the identification of the corresponding gene, which allowed recombinant production of the enzyme. Enzymological characterization and studies with isotope-labelled pseudochiral malonic acids allowed initial mechanistic insights: During the reaction, only one carboxylate group is cleaved, and this step is not directly determining the stereooutcome of the reaction. Based on homology models, the enantioselectivity was completely inverted, and a promiscuous, unique profen racemase was generated. Yet, elucidation of the crystal structure of AMDase in its liganded form in the early 2010s was groundbreaking for the formulation of a hypothetical decarboxylation mechanism, which was confirmed by several computational studies in recent years. Despite the availability of several crystal structures, it remains exceedingly difficult to accurately predict the influence of specific amino acid substitutions in the hydrophobic pocket on enzyme activity. In lieu of predictability, site-saturation mutagenesis and simultaneous saturation mutagenesis proved to be efficient methods to increase the activity of enzyme variants in the synthesis of both enantiomers of α-aryl propionic acids, and to identify optimal variants for different substrates.
The tendency of α-aryl α-methylmalonic acids to undergo spontaneous decarboxylation proved to be an obstacle for industrial application, particularly regarding the decarboxylation of substrates with large electron-poor substituents. By developing a carefully balanced deprotection strategy for the corresponding malonic acid esters, conditions leading to spontaneous decarboxylation could be avoided. A telescoped deprotection/decarboxylation approach resulted in a robust, practicable reaction that allows to obtain α-aryl propionates in either their (S)- or (R)-form in outstanding optical purity. While the activity of the enzyme is generally high, immobilization greatly increased the stability, resulting in an excellent productivity.
Starting with a completely unknown enzyme with unknown mechanism that produced the “wrong” enantiomer of profens, a combination of biocatalysis, enzymology, structure elucidation, molecular modeling, protein engineering, organic chemistry and process intensification succeeded to create a powerful toolbox of AMDase variants that provide access to a large diversity of α-substituted propionates in their enantiopure form.
Author Contributions
AS devised the article and prepared figures, RK and KM participated in planning of the article and contributed to the text.
Funding
All sources of funding have been submitted. RK is funded by the Austrian Science Fund (FWF) P34820.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors RK
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
RK would like to thank the Austrian Science Funds (FWF, P34280) for financial support.
References
Abdel-Aziz, A. A.-M., Al-Badr, A. A., and Hafez, G. A. (2012). “Flurbiprofen,” in Profiles Of Drug Substances, Excipients And Related Methodology. Editor H. G. Brittain (Elsevier), 113–181. doi:10.1016/B978-0-12-397220-0.00004-0
Aßmann, M., Mügge, C., Gaßmeyer, S. K., Enoki, J., Hilterhaus, L., Kourist, R., et al. (2017a). Improvement of the Process Stability of Arylmalonate Decarboxylase by Immobilization for Biocatalytic Profen Synthesis. Front. Microbiol. 8, 448. doi:10.3389/fmicb.2017.00448
Aßmann, M., Stöbener, A., Mügge, C., Gaßmeyer, S. K., Hilterhaus, L., Kourist, R., et al. (2017b). Reaction Engineering of Biocatalytic (S)-Naproxen Synthesis Integrating In-Line Process Monitoring by Raman Spectroscopy. React. Chem. Eng. 2, 531–540. doi:10.1039/C7RE00043J
Barducci, A., Bussi, G., and Parrinello, M. (2008). Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 100, 020603. doi:10.1103/PhysRevLett.100.020603
Biler, M., Crean, R. M., Schweiger, A. K., Kourist, R., and Kamerlin, S. C. L. (2020). Ground-State Destabilization by Active-Site Hydrophobicity Controls the Selectivity of a Cofactor-free Decarboxylase. J. Am. Chem. Soc. 142, 20216–20231. doi:10.1021/jacs.0c10701
Blakemore, C. A., France, S. P., Samp, L., Nason, D. M., Yang, E., Howard, R. M., et al. (2020). Scalable, Telescoped Hydrogenolysis-Enzymatic Decarboxylation Process for the Asymmetric Synthesis of (R)-α-Heteroaryl Propionic Acids. Org. Process. Res. Dev. 25, 421–426. doi:10.1021/acs.oprd.0c00397
Brogden, R. N. (1986). Non-Steroidal Anti-inflammatory Analgesics Other Than Salicylates. Drugs 32, 27–45. doi:10.2165/00003495-198600324-00004
Brooks, P. (1998). Use and Benefits of Nonsteroidal Anti-inflammatory Drugs. Am. J. Med. 104, 9S–13S. doi:10.1016/S0002-9343(97)00204-0
Busch, F., Enoki, J., Hülsemann, N., Miyamoto, K., Bocola, M., and Kourist, R. (2016). Semiempirical QM/MM Calculations Reveal a Step-wise Proton Transfer and an Unusual Thiolate Pocket in the Mechanism of the Unique Arylpropionate Racemase AMDase G74C. Catal. Sci. Technol. 6, 4937–4944. doi:10.1039/c5cy01964h
Cantone, S., Ferrario, V., Corici, L., Ebert, C., Fattor, D., Spizzo, P., et al. (2013). Efficient Immobilisation of Industrial Biocatalysts: Criteria and Constraints for the Selection of Organic Polymeric Carriers and Immobilisation Methods. Chem. Soc. Rev. 42, 6262–6276. doi:10.1039/c3cs35464d
Dasgupta, S., and Herbert, J. M. (2020). Using Atomic Confining Potentials for Geometry Optimization and Vibrational Frequency Calculations in Quantum-Chemical Models of Enzyme Active Sites. J. Phys. Chem. B. 124, 1137–1147. doi:10.1021/acs.jpcb.9b11060
Enoki, J., Linhorst, M., Busch, F., Baraibar, Á. G., Miyamoto, K., Kourist, R., et al. (2019a). Preparation of Optically Pure Flurbiprofen via an Integrated Chemo-Enzymatic Synthesis Pathway. Mol. Catal. 467, 135–142. doi:10.1016/j.mcat.2019.01.024
Enoki, J., Mügge, C., Tischler, D., Miyamoto, K., and Kourist, R. (2019b). Chemoenzymatic Cascade Synthesis of Optically Pure Alkanoic Acids by Using Engineered Arylmalonate Decarboxylase Variants. Chem. Eur. J. 25, 5071–5076. doi:10.1002/chem.201806339
Fukuyama, Y., Matoishi, K., Iwasaki, M., Takizawa, E., Miyazaki, M., Ohta, H., et al. (1999). Preparative-scale Enzyme-Catalyzed Synthesis of (R)-α-Fluorophenylacetic Acid. Biosci. Biotechnol. Biochem. 63, 1664–1666. doi:10.1271/bbb.63.1664
Gaßmeyer, S. K., Wetzig, J., Mügge, C., Assmann, M., Enoki, J., Hilterhaus, L., et al. (2016). Arylmalonate Decarboxylase-Catalyzed Asymmetric Synthesis of Both Enantiomers of Optically Pure Flurbiprofen. ChemCatChem. 8, 916–921. doi:10.1002/cctc.201501205
Gaßmeyer, S. K., Yoshikawa, H., Enoki, J., Hülsemann, N., Stoll, R., Miyamoto, K., et al. (2015). STD-NMR-Based Protein Engineering of the Unique Arylpropionate-Racemase AMDase G74C. ChemBioChem. 16, 1943–1949. doi:10.1002/cbic.201500253
Geerts, H. (2007). Drug Evaluation: (R)-flurbiprofen--an Enantiomer of Flurbiprofen for the Treatment of Alzheimer's Disease. IDrugs 10, 121–133.
Gerlt, J. A., Babbitt, P. C., and Rayment, I. (2005). Divergent Evolution in the Enolase Superfamily: the Interplay of Mechanism and Specificity. Arch. Biochem. Biophys. 433, 59–70. doi:10.1016/j.abb.2004.07.034
Glavas, S., and Tanner, M. E. (2001). Active Site Residues of Glutamate Racemase†. Biochemistry 40, 6199–6204. doi:10.1021/bi002703z
Glavas, S., and Tanner, M. E. (1999). Catalytic Acid/Base Residues of Glutamate Racemase†. Biochemistry 38, 4106–4113. doi:10.1021/bi982663n
Hylton, T. A., and Walker, J. A. (1981). Process for Preparing Arylmethylmaonate Esters, Novel Products Thereof, and Processes for Converting the Products to Therapeutic 2-arylpropionic Aicds and Esters.
Ijima, Y., Matoishi, K., Terao, Y., Doi, N., Yanagawa, H., and Ohta, H. (2005). Inversion of Enantioselectivity of Asymmetric Biocatalytic Decarboxylation by Site-Directed Mutagenesis Based on the Reaction Mechanism. Chem. Commun., 877–879. doi:10.1039/b416398b
Jin, H., Wang, Z., Liu, L., Gao, L., Sun, L., Li, X., et al. (2010). R-flurbiprofen Reverses Multidrug Resistance, Proliferation and Metastasis in Gastric Cancer Cells by p75NTR Induction. Mol. Pharmaceutics 7, 156–168. doi:10.1021/mp900189x
Karmakar, T., and Balasubramanian, S. (2016). Molecular Dynamics and Free Energy Simulations of Phenylacetate and CO2 Release from AMDase and its G74C/C188S Mutant: A Possible Rationale for the Reduced Activity of the Latter. J. Phys. Chem. B. 120, 11644–11653. doi:10.1021/acs.jpcb.6b07034
Kawasaki, T., Horimai, E., and Ohta, H. (1996). On the Conformation of the Substrate Binding to the Active Site during the Course of Enzymatic Decarboxylation. Bcsj 69, 3591–3594. doi:10.1246/bcsj.69.3591
Kim, Y. S., and Kolattukudy, P. E. (1980). Stereospecificity of Malonyl-CoA Decarboxylase, Acetyl-CoA Carboxylase, and Fatty Acid Synthetase from the Uropygial Gland of Goose. J. Biol. Chem. 255, 686–689. doi:10.1016/s0021-9258(19)86232-4
Kourist, R., Domínguez de María, P., and Miyamoto, K. (2011a). Biocatalytic Strategies for the Asymmetric Synthesis of Profens - Recent Trends and Developments. Green. Chem. 13, 2607. doi:10.1039/c1gc15162b
Kourist, R., Miyauchi, Y., Uemura, D., and Miyamoto, K. (2011b). Engineering the Promiscuous Racemase Activity of an Arylmalonate Decarboxylase. Chem. Eur. J. 17, 557–563. doi:10.1002/chem.201001924
Lewin, R., Goodall, M., Thompson, M. L., Leigh, J., Breuer, M., Baldenius, K., et al. (2015). Enzymatic Enantioselective Decarboxylative Protonation of Heteroaryl Malonates. Chem. Eur. J. 21, 6557–6563. doi:10.1002/chem.201406014
Lind, M. E. S., and Himo, F. (2014). Theoretical Study of Reaction Mechanism and Stereoselectivity of Arylmalonate Decarboxylase. ACS Catal. 4, 4153–4160. doi:10.1021/cs5009738
Liu, J. K., Patel, S. K., Gillespie, D. L., Whang, K., and Couldwell, W. T. (2012). R-flurbiprofen, a Novel Nonsteroidal Anti-inflammatory Drug, Decreases Cell Proliferation and Induces Apoptosis in Pituitary Adenoma Cells In Vitro. J. Neurooncol. 106, 561–569. doi:10.1007/s11060-011-0712-4
Lu, G., Franzén, R., Yu, X. J., and Xu, Y. J. (2006). Synthesis of Flurbiprofen via Suzuki Reaction Catalyzed by Palladium Charcoal in Water. Chin. Chem. Lett. 17, 461–464.
Maimanakos, J., Chow, J., Gaßmeyer, S. K., Güllert, S., Busch, F., Kourist, R., et al. (2016). Sequence-Based Screening for Rare Enzymes: New Insights into the World of AMDases Reveal a Conserved Motif and 58 Novel Enzymes Clustering in Eight Distinct Families. Front. Microbiol. 7, 1332. doi:10.3389/fmicb.2016.01332
Markošová, K., Husarčíková, J., Halásová, M., Kourist, R., Rosenberg, M., Stloukal, R., et al. (2018). Immobilization of Arylmalonate Decarboxylase. Catalysts 8, 603. Available at: http://www.mdpi.com/2073-4344/8/12/603.
Matoishi, K., Kakidani, H., Suzuki, M., Sugai, T., Ohta, H., and Hanzawa, S. (2000). The First Synthesis of Both Enantiomers of [α-2H]phenylacetic Acid in High Enantiomeric Excess. Chem. Commun., 1519–1520. doi:10.1039/b003941l
Matoishi, K., Ueda, M., Miyamoto, K., and Ohta, H. (2004). Mechanism of Asymmetric Decarboxylation of α-aryl-α-methylmalonate Catalyzed by Arylmalonate Decarboxylase Originated from Alcaligenes Bronchisepticus. J. Mol. Catal. B: Enzymatic 27, 161–168. doi:10.1016/j.molcatb.2003.11.005
Miyamoto, K., and Kourist, R. (2016). Arylmalonate Decarboxylase-A Highly Selective Bacterial Biocatalyst with Unknown Function. Appl. Microbiol. Biotechnol. 100, 8621–8631. doi:10.1007/s00253-016-7778-z
Miyamoto, K., and Ohta, H. (1992a). Cloning and Heterologous Expression of a Novel Arylmalonate Decarboxylase Gene from Alcaligenes Bronchisepticus KU 1201. Appl. Microbiol. Biotechnol. 38, 234–238. doi:10.1007/BF00174474
Miyamoto, K., Ohta, H., and Osamura, Y. (1994). Effect of Conformation of the Substrate on Enzymatic Eecarboxylation of α-Arylmalonic Acid. Bioorg. Med. Chem. 2, 469–475. doi:10.1016/0968-0896(94)80016-2
Miyamoto, K., and Ohta, H. (1992b). Purification and Properties of a Novel Arylmalonate Decarboxylase from Alcaligenes Bronchisepticus KU 1201. Eur. J. Biochem. 210, 475–481. doi:10.1111/j.1432-1033.1992.tb17445.x
Miyamoto, K., Tsuchiya, S., and Ohta, H. (1992a). Microbial Asymmetric Decarboxylation of Fluorine-Containing Arylmalonic Acid Derivatives. J. Fluorine Chem. 59, 225–232. doi:10.1016/S0022-1139(00)82414-8
Miyamoto, K., Tsuchiya, S., and Ohta, H. (1992b). Stereochemistry of Enzyme-Catalyzed Decarboxylation of .alpha.-methyl-.alpha.-phenylmalonic Acid. J. Am. Chem. Soc. 114, 6256–6257. doi:10.1021/ja00041a060
Miyamoto, K., Tsutsumi, T., Terao, Y., and Ohta, H. (2007a). Stereochemistry of Decarboxylation of Arylmalonate Catalyzed by Mutant Enzymes. Chem. Lett. 36, 656–657. doi:10.1246/cl.2007.656
Miyamoto, K., Yatake, Y., Tamura, K., Terao, Y., and Ohta, H. (2007b). Purification and Characterization of Arylmalonate Decarboxylase from Achromobacter Sp. KU1311. J. Biosci. Bioeng. 104, 263–267. doi:10.1263/jbb.104.263
Miyauchi, Y., Kourist, R., Uemura, D., and Miyamoto, K. (2011). Dramatically Improved Catalytic Activity of an Artificial (S)-selective Arylmalonate Decarboxylase by Structure-Guided Directed Evolution. Chem. Commun. 47, 7503. doi:10.1039/c1cc11953b
Miyazaki, M., Kakidani, H., Hanzawa, S., and Ohta, H. (1997). Cysteine188 Revealed as Being Critical for the Enzyme Activity of Arylmalonate Decarboxylase by Site-Directed Mutagenesis. Bcsj 70, 2765–2769. doi:10.1246/bcsj.70.2765
Mizushima, T., Otsuka, M., Okamoto, Y., and Yamakawa, N. (2014). Fluorophenylpropionic Acid Derivative, 2.
Nguyen, C. N., Cruz, A., Gilson, M. K., and Kurtzman, T. (2014). Thermodynamics of Water in an Enzyme Active Site: Grid-Based Hydration Analysis of Coagulation Factor Xa. J. Chem. Theor. Comput. 10, 2769–2780. doi:10.1021/ct401110x
Nguyen, C. N., Kurtzman Young, T., and Gilson, M. K. (2012). Grid Inhomogeneous Solvation Theory: Hydration Structure and Thermodynamics of the Miniature Receptor Cucurbit[7]uril. J. Chem. Phys. 137, 044101. doi:10.1063/1.4733951
Nishio, M., Umezawa, Y., Hirota, M., and Takeuchi, Y. (1995). The CH/π Interaction: Significance in Molecular Recognition. Tetrahedron 51, 8665–8701. doi:10.1016/0040-4020(94)01066-9
Obata, R., and Nakasako, M. (2010). Structural Basis for Inverting the Enantioselectivity of Arylmalonate Decarboxylase Revealed by the Structural Analysis of the Gly74Cys/Cys188Ser Mutant in the Liganded Form. Biochemistry 49, 1963–1969. doi:10.1021/bi9015605
Ohta, Miyamoto., and Hiromichi, K. (1990). Enzyme-mediated Asymmetric Decarboxylation of Disubstituted Malonic Acids. J. Am. Chem. Soc. 112, 4077–4078.
Okrasa, K., Levy, C., Hauer, B., Baudendistel, N., Leys, D., and Micklefield, J. (2008). Structure and Mechanism of an Unusual Malonate Decarboxylase and Related Racemases. Chem. Eur. J. 14, 6609–6613. doi:10.1002/chem.200800918
Okrasa, K., Levy, C., Wilding, M., Goodall, M., Baudendistel, N., Hauer, B., et al. (2009). Structure-Guided Directed Evolution of Alkenyl and Arylmalonate Decarboxylases. Angew. Chem. Int. Ed. 48, 7691–7694. doi:10.1002/anie.200904112
Puig, E., Mixcoha, E., Garcia-Viloca, M., González-Lafont, À., and Lluch, J. M. (2009). How the Substrate D-Glutamate Drives the Catalytic Action of Bacillus Subtilis Glutamate Racemase. J. Am. Chem. Soc. 131, 3509–3521. doi:10.1021/ja806012h
Reetz, M. T., Kahakeaw, D., and Lohmer, R. (2008). Addressing the Numbers Problem in Directed Evolution. ChemBioChem. 9, 1797–1804. doi:10.1002/cbic.200800298
Tamura, K., Terao, Y., Miyamoto, K., and Ohta, H. (2008). Asymmetric Decarboxylation of α-hydroxy- and α-amino-α-phenylmalonate Catalyzed by Arylmalonate Decarboxylase fromAlcaligenes Bronchisepticus. Biocatal. Biotransformation 26, 253–257. doi:10.1080/10242420701685668
Terao, Y., Ijima, Y., Kakidani, H., and Ohta, H. (2003). Enzymatic Synthesis of (R)-Flurbiprofen. Bcsj 76, 2395–2397. doi:10.1246/bcsj.76.2395
Terao, Y., Ijima, Y., Miyamoto, K., and Ohta, H. (2007). Inversion of Enantioselectivity of Arylmalonate Decarboxylase via Site-Directed Mutation Based on the Proposed Reaction Mechanism. J. Mol. Catal. B: Enzymatic 45, 15–20. doi:10.1016/j.molcatb.2006.11.002
Terao, Y., Miyamoto, K., and Ohta, H. (2006a). Improvement of the Activity of Arylmalonate Decarboxylase by Random Mutagenesis. Appl. Microbiol. Biotechnol. 73, 647–653. doi:10.1007/s00253-006-0518-z
Terao, Y., Miyamoto, K., and Ohta, H. (2006b). Introduction of Single Mutation Changes Arylmalonate Decarboxylase to Racemase. Chem. Commun., 3600–3602. doi:10.1039/b607211a
Tracy, T. S., and Hall, S. D. (1992). Metabolic Inversion of (R)-ibuprofen. Epimerization and Hydrolysis of Ibuprofenyl-Coenzyme A. Drug Metab. Dispos. 20, 322–327.
Warshel, A., and Weiss, R. M. (1980). An Empirical Valence Bond Approach for Comparing Reactions in Solutions and in Enzymes. J. Am. Chem. Soc. 102, 6218–6226. doi:10.1021/ja00540a008
Wong, L. S., Okrasa, K., and Micklefield, J. (2010). Site-selective Immobilisation of Functional Enzymes on to Polystyrene Nanoparticles. Org. Biomol. Chem. 8, 782–787. doi:10.1039/B916773K
Yatake, Y., Miyamoto, K., and Ohta, H. (2008). Screening, Cloning, Expression, and Purification of an Acidic Arylmalonate Decarboxylase from Enterobacter cloacae KU1313. Appl. Microbiol. Biotechnol. 78, 793–799. doi:10.1007/s00253-008-1375-8
Keywords: decarboxylase, enantioselectivity, asymmetric synthesis, biocatalysis, optically pure carboxylic acids
Citation: Schweiger AK, Miyamoto K and Kourist R (2021) Arylmalonate Decarboxylase—A Versatile Biocatalyst for the Synthesis of Optically Pure Carboxylic Acids. Front. Catal. 1:742024. doi: 10.3389/fctls.2021.742024
Received: 15 July 2021; Accepted: 13 September 2021;
Published: 12 October 2021.
Edited by:
Frank Hollmann, Delft University of Technology, NetherlandsReviewed by:
Vicente Gotor-Fernández, University of Oviedo, SpainDunming Zhu, Tianjin Institute of Industrial Biotechnology (CAS), China
Copyright © 2021 Schweiger, Miyamoto and Kourist. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Kourist, a291cmlzdEB0dWdyYXouYXQ=