
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Conserv. Sci. , 15 March 2023
Sec. Animal Conservation
Volume 4 - 2023 | https://doi.org/10.3389/fcosc.2023.1069490
 Sonia L. Ghose1,2,3
Sonia L. Ghose1,2,3 Tiffany A. Yap4,5,6
Tiffany A. Yap4,5,6 Allison Q. Byrne5,7
Allison Q. Byrne5,7 Hasan Sulaeman6
Hasan Sulaeman6 Erica Bree Rosenblum5,7
Erica Bree Rosenblum5,7 Alan Chan-Alvarado6
Alan Chan-Alvarado6 Shruti Chaukulkar6Eli Greenbaum8
Shruti Chaukulkar6Eli Greenbaum8 Michelle S. Koo5
Michelle S. Koo5 Marcel T. Kouete3,9Kurt Lutz6
Marcel T. Kouete3,9Kurt Lutz6 Denise McAloose10
Denise McAloose10 Alessandra J. Moyer6Eliseo Parra6Daniel M. Portik3,5
Alessandra J. Moyer6Eliseo Parra6Daniel M. Portik3,5 Heidi Rockney6
Heidi Rockney6 Andrew G. Zink6David C. Blackburn3,9
Andrew G. Zink6David C. Blackburn3,9 Vance T. Vredenburg3,5,6*
Vance T. Vredenburg3,5,6*Introduction: Emerging infectious diseases are increasingly recognized as a global threat to wildlife. Pandemics in amphibians, caused by the fungal pathogen Batrachochytrium dendrobatidis (Bd), have resulted in biodiversity loss at a global scale. Genomic data suggest a complex evolutionary history of Bd lineages that vary in pathogenicity. Africa harbors a significant proportion of global amphibian biodiversity, and multiple Bd lineages are known to occur there; yet, despite the decline of many host species, there are currently no described Bd-epizootics. Here, we describe the historical and recent biogeographical spread of Bd and assess its risk to amphibians across the continent of Africa.
Methods: We provide a 165-year view of host-pathogen interactions by (i) employing a Bd assay to test 4,623 specimens (collected 1908–2013); (ii) compiling 12,297 published Bd records (collected 1852–2017); (iii) comparing the frequency of Bd-infected amphibians through time by both country and region; (iv) genotyping Bd lineages; (v) histologically identifying evidence of chytridiomycosis, and (vi) using a habitat suitability model to assess future Bd risk.
Results: We found a pattern of Bd emergence beginning largely at the turn of the century. From 1852–1999, we found low Bd prevalence (3.2% overall) and limited geographic spread, but after 2000 we documented a sharp increase in prevalence (18.7% overall), wider geographic spread, and multiple Bd lineages that may be responsible for emergence in different regions. We found that Bd risk to amphibians was highest in much of eastern, central, and western Africa.
Discussion: Our study documents a largely overlooked yet significant increase in a fungal pathogen that could pose a threat to amphibians across an entire continent. We emphasize the need to bridge historical and contemporary datasets to better describe and predict host-pathogen dynamics over larger temporal scales.
Amphibians are one of the most vulnerable groups of vertebrates with 41% of all species currently threatened with extinction (Stuart et al., 2004; Wake & Vredenburg, 2008; The IUCN Red List of Threatened Species, 2021). An emerging infectious disease, chytridiomycosis, caused by the fungal pathogen Batrachochytrium dendrobatidis (Bd), has been a major driver of amphibian declines (Skerratt et al., 2007; Wake and Vredenburg, 2008; Fisher et al., 2009). Invasion and spread of the Bd pathogen are linked with declines and extinctions of hundreds of amphibian species worldwide and Bd has been documented on every continent where amphibians occur (Skerratt et al., 2007; Fisher et al., 2009; Olson et al., 2013; Scheele et al., 2019; Fisher and Garner, 2020). However, the pathogen’s specific historical distribution and geographic spread remain unclear, hampering efforts to predict which amphibian species are at greatest risk of disease outbreak (Berger et al., 2016).
While effects of Bd on vertebrates are greater than those of any other observed pathogen (Scheele et al., 2019; Fisher and Garner, 2020), there is still much that is not understood. For example, while Bd-caused epizootics resulting in amphibian species decline or extirpation have been documented in detail in various parts of the world (Lips et al., 2006; Fisher et al., 2009; Vredenburg et al., 2010; Walker et al., 2010), it is still not known why epizootics are found in some areas but not others (Berger et al., 2016). Delineating the geographic origin and spread of the pathogen, as well as its evolutionary history and population genetic structure, are necessary to describe the dynamics of this pathogen-host system. Recent studies reveal a complicated geographic and phylogenetic history for Bd, including multiple lineages that vary in pathogenicity and may have spread across and between continents over a period of hundreds or even thousands of years. The genetic structure of Bd populations suggests that competition and hybridization among and between Bd lineages has occurred (Farrer et al., 2011; Schloegel et al., 2012; Bataille et al., 2013; Rosenblum et al., 2013; O’Hanlon et al., 2018). The global panzootic lineage (Bd-GPL; Farrer et al., 2011) is regarded as the most virulent lineage, has the widest geographic distribution, and is associated with epizootic events (Schloegel et al., 2012; O’Hanlon et al., 2018). Although Bd is believed to have originated in Asia, it is unclear when, where, or how Bd-GPL diverged from older lineages (O’Hanlon et al., 2018). Other lineages thought to be less virulent are associated with areas that generally lack epizootics (Berger et al., 2016; O’Hanlon et al., 2018), including several identified in Asia (Bd-ASIA-1, believed to have originated in Korea, which includes Bd-CH, originally thought to have originated in Switzerland), South America (Bd-ASIA-2/Bd-BRAZIL) and southern Africa (Bd-CAPE, first described from Cape Province, South Africa) (Farrer et al., 2011; Bataille et al., 2013; Rodriguez et al., 2014; O’Hanlon et al., 2018).
Africa harbors nearly 16% of known living species of amphibians (AmphibiaWeb, 2021) and multiple lineages of Bd have been found to infect these hosts in the wild (including Bd-CAPE and Bd-GPL). There are few relevant Bd host studies in Africa and no described Bd-epizootics in the region (Berger et al., 2016; O’Hanlon et al., 2018; Doherty-Bone et al., 2020; Zimkus et al., 2020). However, this is likely due to a lower Bd sampling effort in Africa relative to other continents rather than a true absence of such events. Several studies have reported a small number of Bd-infected amphibians collected in the early 20th century in Africa (Weldon et al., 2004; Soto-Azat et al., 2010). More recent African amphibian studies have revealed much higher Bd infection prevalence that may be associated with host declines (Goldberg et al., 2007; Kielgast et al., 2010; Bell et al., 2011; Gower et al., 2012; Tarrant et al., 2013; Seimon et al., 2015; Hirschfeld et al., 2016; Jongsma et al., 2016). Although these Bd-host infection data vary in spatial and temporal resolution, they suggest a pattern of Bd emergence that may represent a threat to African amphibian species, warranting a comprehensive analysis encompassing the broadest possible temporal and geographic scales.
We set out to address an important prediction regarding continent-wide infection dynamics of Bd: that the emergence of Bd in Africa is not limited to isolated outbreaks but instead represents a broad pattern of synchronized emergence across the entire continent. We address this prediction by comparing the historical frequency of Bd occurrence (spanning over a century) to the recent frequency of Bd occurrence (roughly the last 20 years). These comparisons of the frequency of Bd occurrence (revealing epizootic dynamics) are made both within African countries and within broader geographic regions. In addition, we include comparisons of changes in overall Bd infection intensity (zoospore counts) for specific countries in central Africa (Cameroon) and eastern Africa (Kenya) that are known to harbor widespread Bd in recent years (Hirschfeld et al., 2016; Kielgast et al., 2010). For our analyses, we have gathered 16,920 original and previously published museum- and field-collected records between 1852 and 2017 from a total of 36 countries. Importantly, our survey of Africa is unique in that it includes both positive and negative Bd samples as opposed to a more restricted reporting of only the number of Bd positives over time (Zimkus et al., 2020).
Additionally, if there is a continent-wide synchronous emergence of Bd, there could be regional differences in the magnitude of Bd prevalence. Such differences are likely associated with the distributions of the two described Bd lineages detected on the African continent (Bd-CAPE and Bd-GPL), depending on the presence of one or a combination of both in certain areas. Bd-CAPE has been detected in Cameroon and was found to be widely present in South Africa, while the Bd-GPL lineage has invaded almost every region of the continent (Rosenblum et al., 2013; O’Hanlon et al., 2018; Byrne et al., 2019). While it is speculated that the hypervirulent Bd-GPL is the more virulent of these two lineages based on ex situ experiments, less is known about the impacts of Bd-CAPE in nature, and there is evidence that Bd-CAPE does exhibit high virulence under certain conditions (Doddington et al., 2013; O’Hanlon et al., 2018; Fisher et al., 2021). Competition among invasive Bd strains, and the potential for hybridization has been documented on other continents (Jenkinson et al., 2016) and genotyping has revealed lineages in Africa that are intermediary to the more derived Bd-GPL complex and the more early diverging Bd-CAPE clade (Rosenblum et al., 2013; O’Hanlon et al., 2018; Byrne et al., 2019). These intermediaries are likely to be hybrids of the two described lineages and could be impacting disease dynamics (Farrer et al., 2011; Greenspan et al., 2018).
To understand how the dynamics of Bd lineage competition and/or hybridization may lead to future epizootics in some areas and not in others, we utilize our large database to document changes in Bd prevalence in regions that have experienced invasion by different lineages. To predict the risk to future populations of amphibians across the continent, we develop a spatially explicit model of Bd risk across regions of Africa, based on our current understanding of the habitat suitability of these Bd lineages and amphibian species richness. Combined, this work provides novel predictions for the regions in Africa that are at high risk of Bd invasion and is complementary to a recent spatially explicit suitability model for the African continent (Zimkus et al., 2020).
We sought to characterize historical and recent Bd prevalence in African amphibians. To do this we first compiled a dataset including (1) original records we generated (referred to as “our data”) from amphibian skin swabs assayed for Bd and (2) published Bd records (referred to as “published data”) for African amphibians.
For records we generated (our data), we collected skin swabs from 2,972 amphibian museum specimens collected from 1908–2009 in Cameroon, Ethiopia, Kenya, Lesotho, Tanzania and Uganda, and from 1,651 live field animals collected by coauthors and collaborators from Burundi and Equatorial Guinea (in 2011), and from Cameroon and the Democratic Republic of Congo (in 2011 and 2013) for a combined total of 4,623 samples. We used a standard Taqman Bd-qPCR assay to determine the frequency of Bd occurrence and Bd-infection intensity (Boyle et al., 2004; Hyatt et al., 2007). This assay was designed to detect Bd-GPL, and therefore a limitation is that divergent lineages with mutations in the Taqman binding site may not amplify (Mutnale et al., 2018). Other retrospective studies utilized historical specimens (collected during a similar time-frame) and using the same Bd-qPCR assay, found evidence for Bd-infection even in their oldest specimens (Cheng et al., 2011; Rodriguez et al., 2014; Talley et al., 2015). This technique was shown to generate a recovery rate of 82% from museum specimens (Cheng et al., 2011), and has robust power of detection, especially with large datasets. We focused historical sampling on areas with putative Bd-related declines [Lesotho (Minter et al., 2004), Ethiopia (Gower et al., 2012; Gower et al., 2013), Tanzania (Channing et al., 2006; Weldon et al., 2020)], areas with many available samples (Uganda and Kenya), and those with the most extensive recent sampling [Cameroon (Blackburn et al., 2010; Hirschfeld et al., 2016) and Kenya (Kielgast et al., 2010)]. Museum specimens are housed in the permanent collections of the California Academy of Sciences (CAS), the Museum of Vertebrate Zoology (MVZ) at the University of California, Berkeley, and Harvard University’s Museum of Comparative Zoology (MCZ). Field sampling locations (live animals) were selected opportunistically based on field trips of coauthors and collaborators. As our goal was to assess Bd prevalence across all amphibians, we sampled amphibians from all taxonomic classes that were available in collections or captured at field sites (these included Anura and Gymnophiona).
Gloves were worn during swab sampling and changed between animals to prevent cross contamination. In addition, archived museum specimens were rinsed with 70% ethanol before swabbing to reduce cross contamination between animals housed in the same jar. We used standard skin swabbing procedures (which include 30 strokes of ventral surfaces) for Bd qPCR assays on live animals and museum specimens using Bd-specific ITS1 primers (Boyle et al., 2004; Hyatt et al., 2007; Cheng et al., 2011). Swabs were dried and stored in 1.5 ml microcentrifuge tubes at 4°C until DNA extraction using Prepman Ultra DNA Extraction Kit following the manufacturer’s protocol (using 40 μl Prepman Ultra per sample). Standard real-time PCR quantification techniques were used: one replicate per sample was run with standard controls of 0, 0.1, 1, 10, and 100 zoospore equivalents (ZE) (Boyle et al., 2004; Hyatt et al., 2007; Cheng et al., 2011). Bd standards were created using Bd isolate CJB7 collected in 2009 from host Rana muscosa during an epizootic at Sixty Lake Basin in the southern Sierra Nevada, California (Vredenburg et al., 2010), identified as part of the highly virulent Bd Global Panzootic Lineage (Bd-GPL1; James et al., 2015; Piovia-Scott et al., 2014; Rosenblum et al., 2013). We multiplied the qPCR genomic equivalents (GE) by a PCR dilution factor of 80 in order to quantify the number of zoospore equivalents on each swab (Zswab; Briggs et al., 2010; Vredenburg et al., 2010). Any sample with a Zswab >0 was considered Bd-infected or Bd-positive. Because some animals had very high Zswab values, we report infection intensity on a base-10 logarithmic scale, represented with units of logZE.
For our compilation of published data, we obtained records from Bd surveys conducted in continental Africa (excluding Madagascar but including the islands of the Gulf of Guinea) from scientific literature and from the Bd-maps Legacy Project (Olson et al., 2021; dataset available at: https://geome-db.org/workbench/project-overview?projectId=300). We included records from Anura, Caudata, and Gymnophiona from all life-stages with location data (i.e., country, latitude and longitude when available), collection year, and Bd infection status (i.e., presence or absence), and excluded food markets and the pet trade. While many published records used methods for swab sampling and Bd qPCR described above, we also included records using different sample materials (including skin scrapes/brush samples, toe clips, skin/mouthpart tissue, Bd isolates) and/or Bd screening methods (including conventional PCR, histology, microscopic visualization, Bd genome sequencing or genotyping). These published data include 12,297 Bd records (collected 1852–2017). When combined with our data (museum and field records we generated; N=4,623), we include a total of 16,920 Bd records from 36 African countries collected between 1852 to 2017 for this study. For more information on the dataset, see the Supplementary Material.
To characterize the temporal pattern of Bd throughout Africa and estimate the timing of Bd emergence, we examined the frequency of Bd occurrence over time for all of Africa, for individual countries, and for regions (as defined by the United Nations). For frequency of Bd occurrence across all of Africa, we analyzed 16,857 records from our compiled dataset (summarized in Table 1). These included our data (all new records generated for this study; N=4,623) and 12,234 records from published data (we excluded 63 of the 12,297 published records in analyses of frequency of Bd occurrence because the associated studies did not report both Bd-negative and Bd-positive samples for Bd detection in amphibian hosts; these 63 Bd-positive records were included, however, in our Bd risk analysis described below that only required Bd-positive samples with latitude and longitude data). To examine temporal changes in Bd occurrence throughout Africa, we binned records by decade (excluding an additional 171 records from published data for which collection year did not fall into a single decade; N=16,686; see Supplementary Material; Table S1) and calculated the frequency of Bd occurrence by dividing the number of Bd-positive records by the total number of animals tested. We calculated a 95% binomial confidence interval (95% CI) for each decade using the R function binom.test (R Core Team, 2022; Phillips and Puschendorf, 2013; De León et al., 2017). We assessed our power to detect a prevalence <11%, a conservative estimate of Bd endemism based on a previous study that spanned over a 100 year period (Talley et al., 2015), by calculating the probability of detecting zero positives for each decade (defined as “Pr(no Bd)”). To do so, we used the R function dbinom (R Core Team, 2022; Doraj-Raj, 2014), which is based on the binomial distribution; we used 11% as the “true” probability that each sample was positive and the total number of samples evaluated in a particular time period as the number of trials.
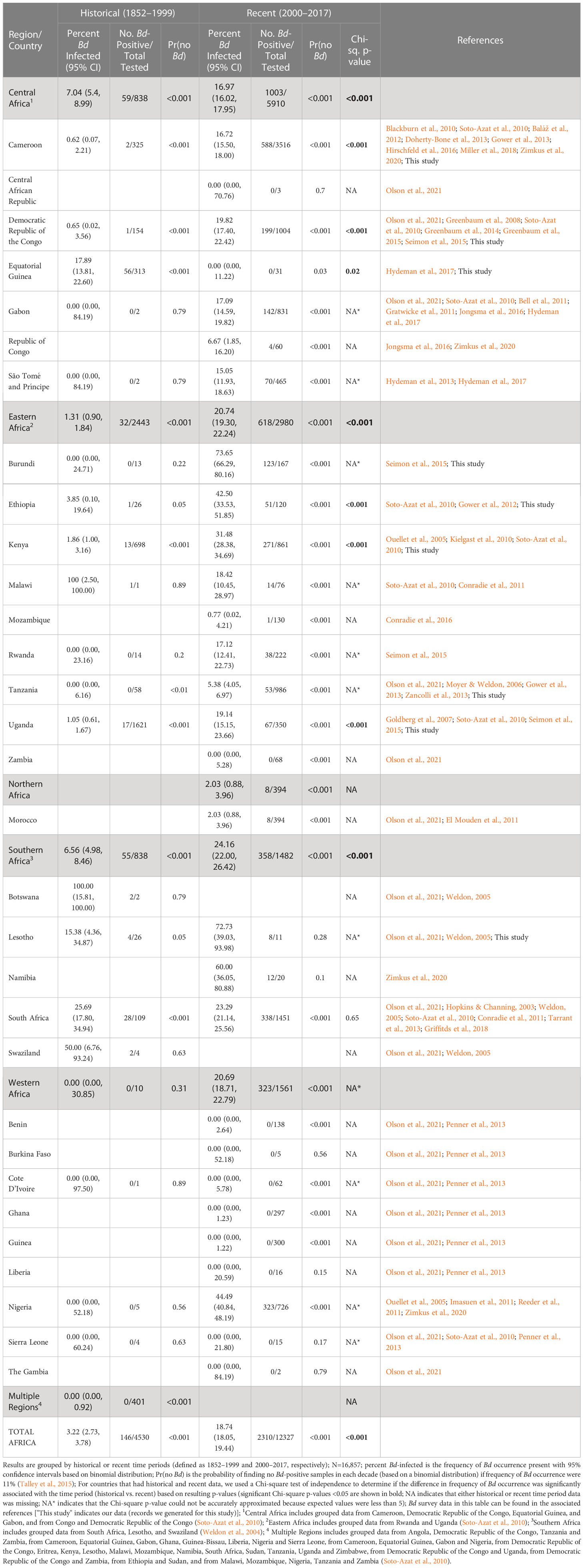
Table 1 Compiled historical and recent frequency of Batrachochytrium dendrobatidis (Bd) occurrence by country and region.
Next, we examined the frequency of Bd occurrence and Bd-infection intensity by decade in countries for which we had the most comprehensive data: Cameroon and Kenya. For these countries, we analyzed a subset of records for which we had data on Bd infection intensity in addition to infection status (this included our data generated for this study for Cameroon and Kenya, data from Hirschfeld et al. (2016) and Zimkus et al. (2020) for Cameroon, and data from Kielgast et al. (2010) for Kenya; see Supplementary Material; Tables S2, S3). In addition to calculating frequency of Bd occurrence with 95% CI and Pr(no Bd) (as described above), mean and maximum Bd-infection intensity for Bd-positive samples were calculated and visualized by decade. We emphasize, however, that estimates of infection intensity from museum specimens are unreliable due to DNA degradation that occurs over time, so our historical infection intensities may be inaccurate estimates.
Finally, we defined a time of emergence based on the increase in frequency of Bd occurrence we observed across decades for all of Africa, Cameroon, and Kenya (year = 2000). We binned Bd records by whether they were pre-emergence (“historical,” which we define as “pre-2000”) and post-emergence (“recent,” defined as “2000-present”) and calculated frequency of Bd occurrence with 95% CI and Pr(no Bd) (as described above) by country and region. To assess whether data across Africa support our proposed timing of emergence statistically, we tested whether the frequency of samples with Bd-infection was dependent on time period for countries and regions that had both pre-emergence and post-emergence records (Chi-square tests using the chisq.test function in R; R Core Team, 2022). For cases where we rejected the null (p<0.05), we inferred the direction of change based on the values for frequency of Bd occurrence in historical and recent time periods. Statistical analyses were conducted in R (version 4.0.4) in RStudio (version 1.4.1106) using ‘pwr,’ ‘binom,’ and ‘Hmisc’ packages (R Core Team, 2022; Harrell, 2012; Doraj-Raj, 2014; RStudio Team, 2016; Champely, 2018).
To determine which lineages of Bd were present, we genotyped Bd from a subset of our swab samples taken from 32 frogs collected in Cameroon in 2013 (N=25) and Burundi in 2011 (N=7) using a custom genotyping assay (Table S4) (Byrne et al., 2017). Briefly, this genotyping assay uses the Fluidigm Access Array platform to perform microfluidic multiplex PCR on 192 target loci of which 188 are on the Bd nuclear genome, 3 are on the Bd mitochondrial genome, and one is designed to target the internal transcribed spacer (ITS) region of the related pathogenic fungus Batrachochytrium salamandrivorans. Each target locus is 150–200 base pairs (bp) long. First, extracted DNA from swab samples was cleaned and concentrated from 30µl to 10µl using an isopropanol precipitation protocol. All samples were preamplified in two separate PCR reactions, each containing 96 primer pairs at a final concentration of 500nM. For each preamplification PCR reaction, we used the FastStart High Fidelity PCR System (Roche) at the following concentrations: 1x FastStart High Fidelity Reaction Buffer with MgCl2, 4.5mM MgCl2, 5% DMSO, 200µM PCR Grade Nucleotide Mix, 0.1 U/µl FastStart High Fidelity Enzyme Blend. We added 1µl of DNA to each preamplification reaction and used the following thermocycling profile: 95˚C for 10min, 15 cycles of 95˚C for 15sec and 60˚C for 4min. Preamplified products were treated with 4µl ExoSAP-it (Affymetrix Inc.) and incubated for 15min at 30˚C, then 15min at 80˚C. Treated products were diluted 1:5 in PCR-grade water. The diluted products from each of the two preamplification reactions were combined in equal proportions and used for downstream amplification and sequencing.
Each preamplified sample was loaded into the Fluidigm Access Array IFC (Fluidigm, Inc.) for amplification. All samples were barcoded and pooled for sequencing on ¼ of an Illumina MiSeq lane using the 300bp paired-end kits at the University of Idaho IBEST Genomics Resources Core. We pre-processed all sequencing data as described in Byrne et al. (2017) and generated consensus sequences for all variants present for each sample at each locus. We filtered reads by selecting sequence variants that were present in at least five reads and represented at least 5% of the total number of reads for that sample/locus using dbcamplicons (https://github.com/msettles/dbcAmplicons).
We constructed a phylogeny to explore the relationship of the Bd sequences collected for this study (N=32) to those from previously-published Bd isolates (N=31; Rosenblum et al., 2013; O’Hanlon et al., 2018). The isolates from Rosenblum et al. (2013) (N=28) were sequenced and processed exactly as described for the swab samples. The consensus amplicon sequences for the isolates from O’Hanlon et al. (2018) (N=3) were calculated from raw sequencing reads. Briefly, we downloaded raw reads from NCBI’s Sequence Read Archive (accession numbers SRS2757071, SRS2757072, SRS2757205) and cleaned the reads using seqyclean v 1.9.9 (https://github.com/ibest/seqyclean). We aligned the paired reads to the reference genome JEL423 (Broad Institute v. 17-Jan-2007) using BWA MEM (Li, 2013). Aligned reads were sorted using picard v.2.9.0 (http://broadinstitute.github.io/picard) and variants were called using samtools mpileup v.1.6 and bcftools call v.1.6 (Li, 2011). We produced a consensus genome for each isolate using the bcftools consensus. After producing a consensus genome for each of the three isolates, we used BLAST v.2.7.1 to match our reference amplicon sequences to the amplicon sequences in each genome. We then extracted the best-scoring BLAST match for each isolate to produce a single FASTA containing a consensus sequence for each target locus.
The isolates represent three of the major Bd clades: Bd-GPL (Global Panzootic Lineage), Bd-BRAZIL/Bd-ASIA-2, and Bd-CAPE (Farrer et al., 2011; O’Hanlon et al., 2018). First, we trimmed our consensus sequence dataset to eliminate loci that had more than 50% missing data. The remaining dataset contained 186 loci (183 nuclear, 3 mitochondrial). To ensure proper alignment, we filled in missing sequences with a string of Ns (length = maximum sequence length of that locus) using custom R scripts. Next, we individually aligned all loci using the MUSCLE package v.3.18.0 (Edgar, 2004) in R (v.3.4.3) (R Core Team, 2022) and concatenated all aligned loci for each sample for a total length of 25,617bp. We visually checked the concatenated alignment for errors in Geneious v.10.2.3 (Kearse et al., 2012) and used the RAxML plugin (Stamatakis, 2014) in Geneious to search for the best scoring maximum likelihood tree using rapid bootstrapping (1000 replicates).
We integrated abiotic and biotic factors to predict the areas in Africa with the greatest risk of Bd establishment, following previous studies (Murray et al., 2011; Liu et al., 2013; Yap et al., 2018). We first created a presence-only habitat suitability model (HSM) driven by climate and land use factors using Maxent v.3.4.1 (Phillips et al., 2017). We used 1,914 georeferenced Bd occurrence records for continental Africa and the islands in the Gulf of Guinea (Bioko, São Tomé, Príncipe); north of the Sahara, recent data were only available from Morocco. Records included all Bd-positives with latitude and longitude from our data and from published data. To reduce sampling bias, we applied a 10 arc-minute spatial filter by randomly choosing one Bd occurrence site from every 10 arc-minute (~12 km2) area (Boria et al., 2014) using R packages 'dismo' (Hijmans et al., 2013) and 'maptools' (Bivand and Lewin-Koh, 2014). This resulted in 202 Bd-positive sites; 195 sites had environmental data and were used for model training. We further minimized sampling bias by restricting background sampling areas to a minimum convex hull polygon around large clusters of occurrence points (Phillips et al., 2009; Rödder et al., 2009; Thorne et al., 2012; Merow et al., 2013; Phillips and Elith, 2013; Mainali et al., 2015; Yap et al., 2018).
Similar to Yap et al. (2018), we used 19 bioclimatic variables from the Worldclim database (http://www.worldclim.org/bioclim) (Hijmans et al., 2005) and the global human footprint (HF) (Venter et al., 2016) as predictor variables. To reduce the chances of overfitting the model due to multicollinearity, we calculated Spearman’s rank correlations (r) among all the variables to determine which variables were highly correlated with each other (James et al., 2015). We removed one variable from each pair that had an r2 > 0.7. We used the resulting subset of 11 environmental variables to create the Bd HSM: mean diurnal temperature range (Bio2), isothermality (Bio3), maximum temperature of the warmest month (Bio5), minimum temperature of the coldest month (Bio6), mean temperature of driest quarter (Bio9), precipitation of the wettest month (Bio13), precipitation of the driest month (Bio14), precipitation seasonality (Bio15), precipitation of the warmest quarter (Bio18), precipitation of the coldest quarter (Bio19), and human footprint (HF). These variables are biologically relevant to Bd and performed well in previous Bd HSMs (Ron, 2005; Rödder et al., 2009; Murray et al., 2011; Liu et al., 2013; James et al., 2015; Yap et al., 2018). We ran 20 replicates using cross-validation: occurrence data were divided into 20 equal-sized folds, or groups, and for each replicate 19 folds were used for model training and one fold was used for model testing, and the results were then averaged by Maxent to produce a final occurrence probability (Phillips et al., 2017).
In addition to abiotic factors, biotic factors such as host availability are critical for disease maintenance and spread (Lloyd-Smith et al., 2005). Although host abundance data would be ideal, such data at the community level for a continental assessment are not available for Africa. However, several studies have shown species richness can be an important factor in Bd spread (Rödder et al., 2009; Liu et al., 2013; Boria et al., 2014; Yap et al., 2016; Hydeman et al., 2017; Yap et al., 2018). Thus, in lieu of abundance data, we incorporated species richness to refine our predictions of Bd risk. We obtained amphibian richness data from AmphibiaWeb (amphibiaweb.org; AmphibiaWeb, 2021), which included range maps from the IUCN Red List as well as expert-based range maps supplied by AmphibiaWeb to calculate richness.
We calculated the product of the Bd HSM and amphibian richness to determine Bd risk (i.e., areas of overlap between suitable Bd habitat and potential hosts) (Yap et al., 2015; Yap et al., 2016; Yap et al., 2018). We show Bd risk on a scale of low to high using the Jenks optimization method, which identifies natural clusters, or classes, of data and reduces variance within the classes while maximizing the differences between classes (Jenks, 1967). This method is more informative than the binary threshold options provided by Maxent when predicting species range expansions (Creley et al., 2019).
Our Bd-assay data for 4,623 amphibian specimens (collected 1908–2013) compiled with 12,234 Bd records from published data (collected 1852–2017) over a 165-year period show a pattern of rapid Bd emergence beginning largely at the turn of the century (Figure 1A). From 1852–1999, we found low overall Bd prevalence (3.2%, 2.7–3.7% CI) and limited geographic spread, but after the year 2000 (from 2000-2017), we found a sharp increase in continent-wide prevalence (18.7%, 18.1–19.4% CI) and wider geographic spread. When we examined Bd prevalence by decade, we found that it remained below 5% for every decade until the 2000s when it increased to 17.2% (16.6–18.0% CI; Figure 1B; Table S1). In the second decade of the new century (2010s), overall frequency continued to increase to 21.6% (20.4–22.9% CI). In several countries where we had more complete data, the pattern was more pronounced. For example, in Cameroon, we found only one Bd positive in samples we collected up to 1999, but in the next decade, the 2000s, the frequency of Bd occurrence rose to 11.0% (8.9–13.3% CI), followed in the 2010s, by an increase to 36.2% (33.0–39.5% CI; Figure 1C; Table S2). In Cameroon, the average Bd infection intensity also increased over time and in the most recent samples, maximum values (2000–2009, 5.5 logZE; 2010–present, 6.4 logZE) surpassed a threshold infection intensity associated with chytridiomycosis-caused mortality (4.0 logZE; Vredenburg’s “10,000 zoospore rule”; Vredenburg et al., 2010; Kinney et al., 2011). In Kenya, frequency of Bd occurrence was zero or very low in the first four decades of data (the highest frequency in these early decades was in the 1970’s at 5.2%, 2.5–9.3% CI; Figure 1D; Table S3) and increased approximately six-fold in the 2000s (31.5%, 28.4–34.7% CI; Figure 1D; Table S3). The average Bd infection intensity in Kenya also increased during this same time period, with maximum Bd-infection intensity surpassing the mortality threshold in the 2000s by several orders of magnitude (7.9 logZE; Figure 1D; Table S3).
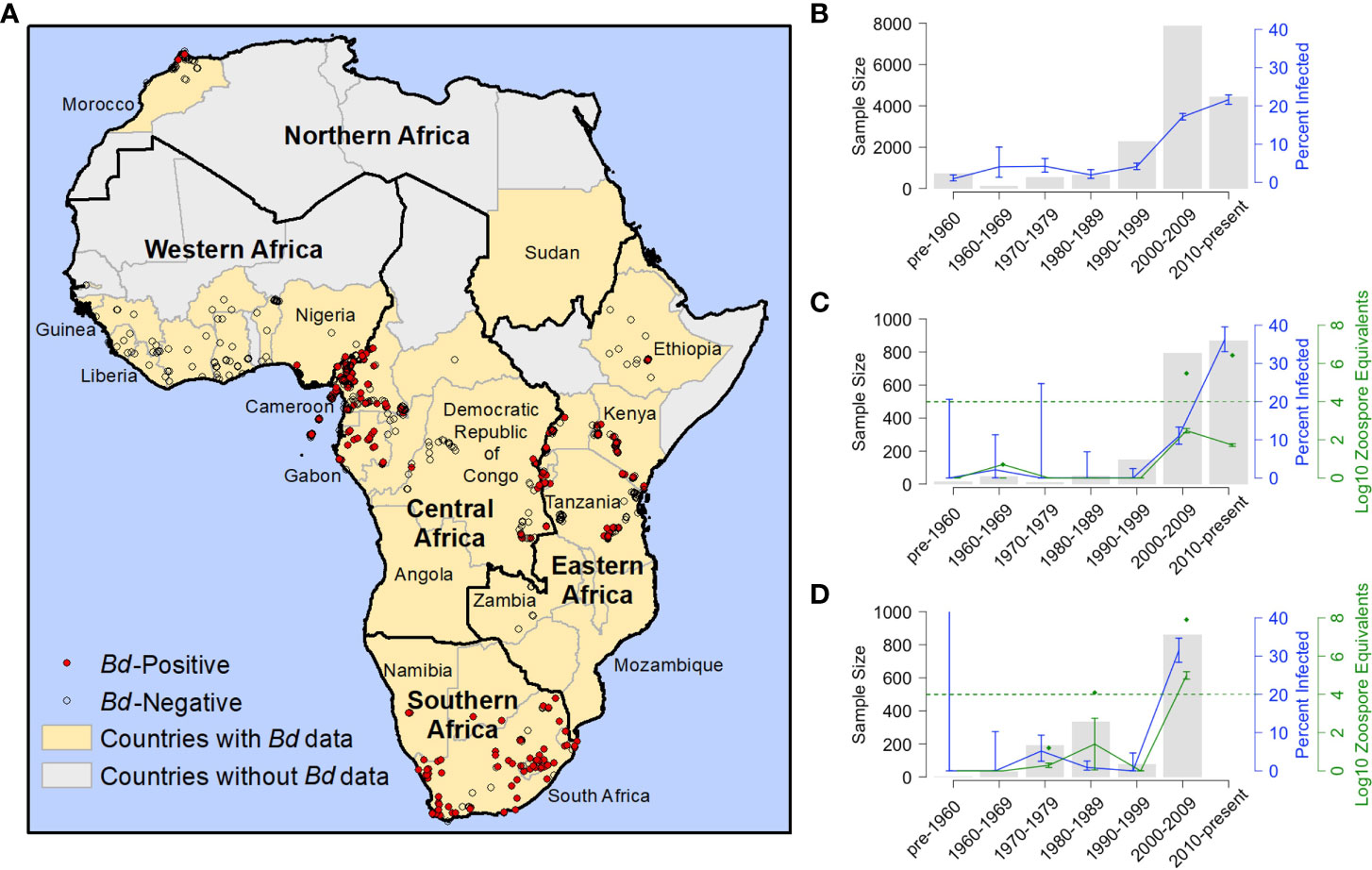
Figure 1 Distribution and frequency Batrachochytrium dendrobatidis (Bd) occurrence in Africa 1852-2017. (A) Georeferenced Bd survey data for Africa (N=11,472; includes records generated for this study and previously published records for which we had latitude and longitude); red dots indicate Bd-positive records (N=1,914); open circles indicate Bd-negative records (N=9,558); yellow fill indicates countries with Bd data (including georeferenced and non-georeferenced locations); black outlines indicate regions, as defined by the United Nations; (B) frequency of Bd occurrence for all of Africa by decade; N=16,686 (171 samples included in Table 1 not included here because collection years did not fall into a single decade); (C) frequency of Bd occurrence and infection intensity results from Cameroon; N=1,940 [includes our data records generated for this study, records from Hirschfeld et al. (2016) and from Zimkus et al. (2020)]; (D) frequency of Bd occurrence and infection intensity results from Kenya; N=1,503 [records through 1999 generated for this study; 2006 records from Kielgast et al. (2010)]; (B–D) grey bars represent sample size (N); blue line represents frequency of occurrence (percent infected) and blue bars represent the corresponding 95% binomial confidence intervals (95% CI); (C, D) green line represents the average infection intensity (the logarithm of zoospore equivalents or logZE) and green bars represent the corresponding standard error; green diamonds represent maximum logZE for each time period; green hashed horizontal line indicates a logZE value of 4, the threshold for deadly infection intensity (Vredenburg et al., 2010; Kinney et al., 2011).
In our test of whether frequency of samples with Bd-infection was dependent on time period (historical vs recent), we found that the overall frequency of Bd occurrence across Africa in the historical time period (pre-2000; 3.2%, 2.7–3.8% CI), was significantly different from the recent time period (2000–2017; 18.7%, 18.1–19.4% CI; Chi-sq., p<0.001; Table 1). We also found that the frequency of Bd occurrence was significantly different regionally (see regions; Figure 1A) across central, eastern, and southern Africa, between historical (<7%) and recent (>17%) time periods (Chi-sq., p<0.001, p<0.001, p<0.001, respectively; Table 1). One country in central Africa, Equatorial Guinea, showed a significant decrease in the frequency of Bd occurrence between historical and recent time periods (all samples from Bioko Island, Equatorial Guinea; Chi-sq., p=0.02), though all the Bd-positives from the historical time-period were collected close to the year 2000 (in 1998). Burundi, in eastern Africa, had the highest frequency of Bd infections after 2000 (73.7%, 66.3–80.2% CI) with no Bd positives detected historically; however, a Chi-square p-value could not be accurately estimated for this country because of the small historical sample size (N=13). We did not detect a recent surge of Bd infections in the country of South Africa (25.7%, 17.8–34.9% CI, historic vs. 23.3%, 21.1–25.6% CI, recent; Chi-sq, p=0.65), though we did in the region overall (6.6%, 5.0–8.5% CI, historic vs. 24.2%, 22.0–26.4% CI, recent; Chi-sq, p<0.001). There were no historical data for northern Africa, which only included data from Morocco, and the recent frequency of Bd occurrence there was low (2.0%, 0.9–4.0% CI). In western Africa, although the frequency of Bd occurrence also showed an increase from 0% historically to >20% recently, low historical sample size (N=10) prevented accurate estimation of a Chi-square p-value. Bd was almost entirely absent from western Africa (Table 1) except in Nigeria, where the recent frequency of Bd occurrence was 44.5% (40.8–48.2% CI).
We found multiple lineages of Bd in Burundi and Cameroon (Figures 2A, B; Table S4; Byrne et al., 2019). In Cameroon, all genotyped samples formed a well-supported clade that includes the previously-published Bd-CAPE isolate CCB1 (Rosenblum et al., 2013), indicating that amphibians in Cameroon are widely infected by the Bd-CAPE lineage (Figure 2B). In contrast, Bd samples collected in Burundi were more variable, with some samples grouping with previously-published Bd-GPL isolates (CAS250667, CAS250764, CAS250826, CAS250829) while others were not clearly members of either the Bd-GPL or Bd-CAPE lineages (CAS250662, CAS250742, CAS250744) (Figure 2B). For one of these ambiguous samples, CAS250742, the average number of alleles found at each locus across all amplified loci was 2, which was higher than for all other sequenced samples (Figure S1). Bootstrap support in this group of samples is low, thus no firm conclusions can be made from data for these three samples. However, the ambiguous Burundi samples are distinct from both from Bd-CAPE samples as well as other previously published Bd-CAPE/Bd-GPL hybrids (Figure 2B; O’Hanlon et al., 2018).
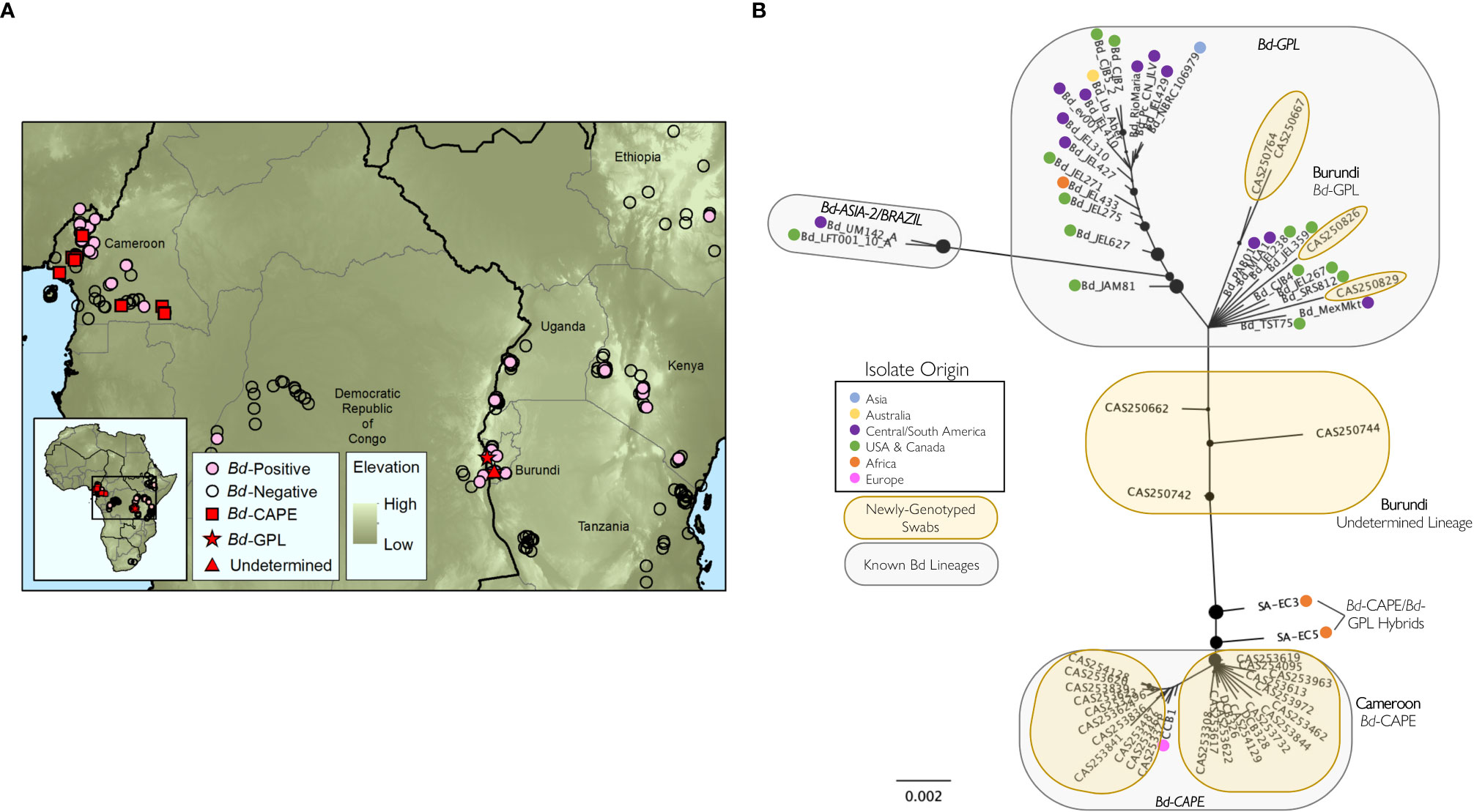
Figure 2 Genotyping of Batrachochytrium dendrobatidis (Bd) lineages from samples processed in this study. (A) Locations and infection status of georeferenced samples processed by the authors of this study (N=4,349) showing Bd-positive (pink circles) and Bd-negative (open circles) locations; locations of Bd samples genotyped in this study (N=32) are indicated by red squares (Bd-GPL), red stars (Bd-CAPE), and red triangles (Undetermined lineage); (B) best-scoring unrooted maximum likelihood tree for 61 Bd samples (32 sequenced for this study, 31 previously-published Bd isolates). Tree created from concatenated, aligned consensus sequences from 186 Bd loci (25,617 bp). Nodes with less than 10% bootstrap support have been collapsed and the size of the black node markers are scaled based on bootstrap support. Geographic origin for each previously published isolate is indicated with a colored dot. Known Bd lineages are inside grey circles and samples genotyped for this study are in yellow circles. See Table S4 for sample information associated with the newly genotyped swabs.
The Bd risk model predicted moderate to high Bd vulnerability throughout much of western, central, and eastern Africa, including within the Ethiopian Plateau (Figure 3). Some portions of Mediterranean northern Africa and the southeastern tip of Africa were also predicted to have some Bd risk. Our model predicted the highest Bd risk in the tropical rainforests of western and central Africa and the montane forests surrounding the Great Rift Valley, while less vulnerable areas included the arid Sahara Desert throughout most of northern Africa and the semi-arid Kalahari Desert in the western region of southern Africa.
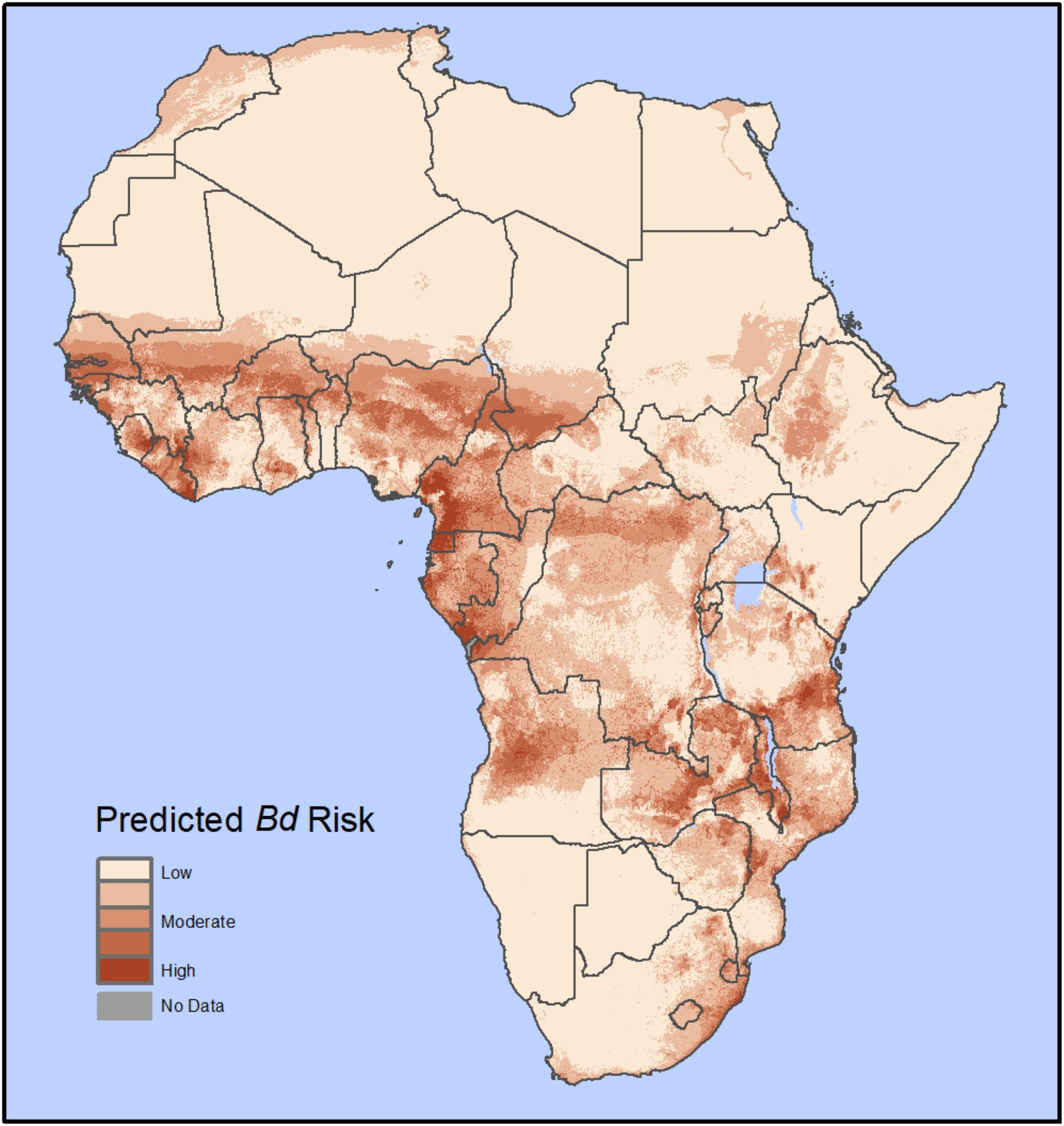
Figure 3 Predicted Bd disease risk in Africa from our habitat suitability model (HSM); Based on 1,914 georeferenced Bd occurrence records with 195 unique Bd-positive localities in the African mainland and islands in the Gulf of Guinea. The Bd risk model is a product of a Bd HSM model (similar to Yap et al. (2018); here using 10 bioclimatic variables and the global human footprint as predictor variables) and host richness maps (AmphibiaWeb) for the entire African continent. The results are shown on a color scale from low risk (tan color) to high risk (red color).
The 165 years of amphibian sample collections show a distinct signal of Bd emergence and geographic spread across the African continent that began in the first decade of the 21st century. In general, in the 20th century (1852-1999), we found few, geographically scattered Bd-positives, showing no signal of epizootic spread (Figures 1A, B; earliest Bd infected host was 1933 in Cameroon; Soto-Azat et al., 2010). However, beginning in the 2000–2009 sample period (Figures 1A, B) we saw a large increase in prevalence and evidence of widespread infection across the continent, which are signals consistent with epizootics. Similarly, in Central America and western North America, studies found few and scattered Bd-infected specimens historically in areas that decades later experienced rapid Bd expansion, epizootics and host population collapse (Vredenburg et al., 2010; Cheng et al., 2011; Sette et al., 2015; Yap et al., 2016; Adams et al., 2017; De León et al., 2017; Yap et al., 2018; Vredenburg et al., 2019; Sette et al., 2020). In Africa, we saw only one major exception to this general pattern of emergence: in South Africa, infections were widespread even in our oldest samples (Table 1). Thus, generally we found strong evidence indicating a recent surge (beginning in 2000) in the frequency of Bd occurrence in amphibian specimens collected across a large portion of the continent (Figures 1A, B; Tables 1, S1). Disturbingly, we found this broad pattern of Bd emergence in areas of Africa where many amphibian species are either suspected or known to have collapsed (Channing et al., 2006; Gower et al., 2012; Gower et al., 2013; Hirschfeld et al., 2016; Weldon et al., 2020). Considering that Bd invasion was followed by Bd-epizootics resulting in extinctions of amphibian populations in western North America (Briggs et al., 2010; Vredenburg et al., 2010), Central America (Berger et al., 1998; Lips et al., 2006), South America (Catenazzi et al., 2011; Carvalho et al., 2017), Europe (Walker et al., 2010; Bates et al., 2018), and Australia (Laurance et al., 1996; Berger et al., 1998), amphibian declines in Africa that coincide with widespread expansions of Bd may also be linked.
Recent Bd emergence was most pronounced in eastern and central Africa where many species are threatened amid reports of declines and die-offs. In fact, most reports place the declines and extinctions occurring near or after the year 2000 (e.g. in Tanzania, Ethiopia, and Cameroon; Channing et al., 2006; Gower et al., 2012; Gower et al., 2013; Hirschfeld et al., 2016; Weldon et al., 2020), which coincides with our Bd emergence data. The historical and recent Bd occurrence data from Cameroon and Kenya are the most complete. In these countries, we saw a large and significant increase in the number and proportion of Bd infected hosts after 2000 (Figures 1C, D; Tables S2, S3). Studies have shown that high levels of infection (i.e. Bd infection intensity or Bd load) are associated with severe chytridiomycosis, death, and population collapse (e.g., mortality threshold, Vredenburg’s “10,000 zoospore rule”; Vredenburg et al., 2010; Kinney et al., 2011); and, low Bd infection intensity is associated with host survival and coexistence with Bd suggestive of enzootic dynamics (Briggs et al., 2010; Knapp et al., 2016; Catenazzi et al., 2017). In both Cameroon and Kenya, we detected a recent increase in the average Bd-infection intensity of hosts. We note, however, that Bd infection intensities from historical Bd occurrences may be unreliable (see Methods). Regardless, the maximum values for infection intensity that we documented from live animals in the most recent decade for Cameroon and Kenya surpass the mortality threshold (Figures 1C, D; Tables S2, S3). Thus, we conclude that our evidence of recent and rapid emergence of Bd and presence of highly infected animals signal that Bd epizootics may be occurring in these regions. By contrast, in South Africa, we found no evidence of a recent Bd emergence (Table 1). There have been a few cases of observed mortality and population bottlenecks associated with Bd in some regions of South Africa, however no widespread declines or extinctions have been linked to Bd despite comprehensive population survey data (Hopkins & Channing, 2003; Tarrant et al., 2013; Griffiths et al., 2018; Doherty-Bone et al., 2020). This suggests that either Bd may be endemic to South Africa, that there may be Bd genetic lineages with lower virulence there, or that sampling remains incomplete.
Global studies on Bd genomics suggest that Bd has a complex history with many lineages that appear to have varied effects on amphibian populations (Rosenblum et al., 2013; Fisher and Garner, 2020). Generally, lineages identified as endemic to a single part of the world are thought to have a long evolutionary history of co-occurrence with hosts, and thus have evolved lower virulence than invasive (or novel) lineages that cause epizootics and host die-offs. In some places, endemic and invasive lineages are hypothesized to be co-occurring and hybridizing to form new lineage variants (O’Hanlon et al., 2018), and the effects of these hybrid lineages on hosts is unknown. Bd-epizootics can also be triggered by enigmatic abiotic or biotic factors that have not been deeply explored (Fisher and Garner, 2020; Fisher et al., 2021). Previous work has reported multiple genetic lineages of Bd in Africa (O’Hanlon et al., 2018; Byrne et al., 2019), suggesting the potential for complex hybridization and epizootic dynamics.
In Cameroon, we found evidence of Bd-CAPE, a lineage generally considered to be endemic to Africa and less virulent than Bd-GPL; however, our emergence data and results of histology (see Supplementary Material) suggest this lineage may in fact be virulent in Africa. All genotyped samples from Cameroon clustered genetically with Bd-CAPE (Figure 2B), which supports previous work (O’Hanlon et al., 2018). Histological examination of animals from Cameroon show evidence of chytridiomycosis (see Supplementary Material; Table S5; Figure S2), and we found Bd prevalence in the area has increased and spread geographically. The reported amphibian declines from Cameroon (Hirschfeld et al., 2016) coincide with the recent increase in frequency of Bd occurrence that we report here. We concur with previous work that states that pathogen lineage alone cannot fully predict disease outcomes since lineages that appear less pathogenic in native hosts may cause negative impacts on new or naïve host populations, especially when abiotic factors promote infection and disease (O’Hanlon et al., 2018; Fisher et al., 2021). We suggest this may be the case with Bd-CAPE in parts of Africa. It is possible that Bd-CAPE was present in Cameroon historically and has only recently increased in frequency due to shifts in some environmental, human, or other unknown factor, but it is also possible that the recent increase in prevalence represents a new invasion and spread of Bd-CAPE in Cameroon, perhaps from elsewhere on the continent. Bd-CAPE has been widely detected in South Africa (O’Hanlon et al., 2018; Byrne et al., 2019) and may be endemic there. In South Africa, we found a relatively constant proportional occurrence of Bd in hosts through a long time-period, and while there have been cases of Bd-associated mortality there, these could be associated with different Bd lineages (Bd-GPL is also present) or result from cofactors that are not yet known. Previous work has proposed that Bd-CAPE spread from South Africa to Europe, where it has thus far been associated with host declines (Doddington et al., 2013; O’Hanlon et al., 2018). Thus, we propose that Bd-CAPE may have recently spread within continental Africa and may also be associated with Bd-epizootics in some locations where conditions are suitable. Studies have reported recent evidence of mass amphibian die-offs in Cameroon (Blackburn et al., 2010; Hirschfeld et al., 2016) and Lesotho (Minter et al., 2004), areas where more detailed studies are needed to discern whether Bd-CAPE is associated with Bd-epizootics.
Our genomic data also identified Bd-GPL (Global Panzootic Lineage) in samples from Burundi. The Bd-GPL lineage is considered the most virulent Bd lineage (Farrer et al., 2011; Rosenblum et al., 2013; O’Hanlon et al., 2018; Byrne et al., 2019). Consistent with this, histological examination of specimens from Burundi showed signs of chytridiomycosis (see Supplementary Material; Table S5). In fact, our samples from Burundi had the highest proportion of Bd infected hosts in our recent time frame (73.7%; 66.3–80.2% CI), and this could suggest Bd-epizootics are occurring there. Furthermore, we found no historical Bd positive data in Burundi (Table 1) or in adjacent Tanzania (samples collected 1926–2003). In 2003, the Tanzanian Kihansi spray toad (Nectophrynoides asperginis) went extinct in the wild purportedly due to a Bd epizootic (Weldon et al., 2020). While there was no statistical support that frequency of Bd occurrence increased in the recent timeframe in Burundi and Tanzania (due to low availability of historical samples), we did find that frequency of occurrence was significantly higher statistically after 2000 in eastern Africa overall (Table 1). Therefore, the recent presence of Bd-GPL detected after a dramatic increase in Bd occurrence in eastern Africa along with histological evidence of chytridiomycosis, may indicate that Bd-GPL driven epizootics are occurring there.
Hybridization between distinct Bd lineages has been proposed as a way that virulent lineages have emerged, and these hybrid lineages may be more virulent than either parent lineage (Farrer et al., 2011; Greenspan et al., 2018). Three of our genotyped samples from Burundi did not clearly cluster with previously characterized Bd lineages on the phylogenetic tree, but rather were positioned between Bd-GPL and Bd-CAPE (Figure 2B). We see three possibilities for the identity of Bd lineages from these samples: (i) Bd-GPL and Bd-CAPE are able to hybridize, and while our ambiguous samples did not cluster with known hybrids, they could be novel hybrids of these two lineages (Figure 2B; O’Hanlon et al., 2018); (ii) these samples could each contain multiple lineages of Bd, as evidenced by a higher average number of alleles found in one of these samples than any other (Figure S1), and this supports the possibility that more than one lineage was present in that sample; or (iii) Bd detected in these samples could represent novel genetic variation that has not previously been characterized. Further work on ambiguous lineages in Burundi could resolve this issue and help in our general understanding of this complex pathogen. Further studies may identify lethal and sub-lethal infections (potentially from different Bd lineages) that could help us understand factors that could affect pathogen success (and how it may vary across lineages) such as differences in pathogen dispersal ability, reproductive ability, and vulnerability to predators. Additionally, we emphasize that future studies should prioritize genotyping Bd throughout Africa. The genotyping work presented here is limited, and further Bd genotyping is needed to better characterize the distribution of lineages across the continent.
Habitat suitability models combined with potential host availability provide an opportunity to understand the potential distribution of Bd risk over large geographic areas (Murray et al., 2011; Liu et al., 2013; Yap et al., 2018). The results of our HSM generally align with previous potential distribution models (Penner et al., 2013; Zimkus et al., 2020); however, two key distinctions in our model are that (1) we incorporated potential host availability to determine risk of disease spread (Yap et al., 2015; Yap et al., 2016), and (2) we included the global human footprint (Venter et al., 2016). Although host abundance data at the community and continental scale would be ideal to identify host availability, such data are not available. Therefore, as a proxy, we used species richness, which has been shown to be an important factor in Bd spread (Rödder et al., 2009; Liu et al., 2013; Boria et al., 2014; Yap et al., 2016; Hydeman et al., 2017; Yap et al., 2018). We included the global human footprint (Venter et al., 2016) in our model because the presence of humans or human activities can play a role in disease spread (Liu et al., 2013; James et al., 2015). We also took additional steps to reduce sampling bias and minimize the chances of overfitting due to multicollinearity of the abiotic variables. Thus, our model is complementary to previous potential distribution models (Penner et al., 2013; Zimkus et al., 2020). Our model identifies geographic areas with moderate to high risk of Bd spread as well as further highlighting African regions with high Bd suitability and supporting the assertion that countries in these regions are potential hotspots for the negative impacts of Bd (Figure 3).
In central Africa, the HSM predicted high Bd risk in Angola, Cameroon, Democratic Republic of Congo, Equatorial Guinea, Gabon, and Republic of Congo. The observed Bd dynamics in Cameroon and Democratic Republic of Congo, coupled with population declines in Cameroon (Hirschfeld et al., 2016) suggest a recent emergence, yet more information and monitoring is also needed in Angola, Equatorial Guinea, Gabon, and Republic of Congo to determine their Bd status (Table 1). We found a high proportion of Bd positives from Bioko Island (Equatorial Guinea) from 1998, and Bd-GPL has also been detected there (O’Hanlon et al., 2018; Byrne et al., 2019; Zimkus et al., 2020), but the lack of population survey data across a longer timescale prevents a proper understanding of Bd-host dynamics there.
In eastern Africa, the HSM model predicted high Bd risk in Kenya, Malawi, Mozambique, Tanzania, Zambia, and Zimbabwe. Mozambique and Zimbabwe border South Africa, where Bd-CAPE may be endemic and there is evidence suggesting spread to Lesotho could be associated with amphibian declines (Minter et al., 2004). Tanzania experienced a documented Bd-driven species extinction in the wild in 2003 (Weldon et al., 2020). Additionally, Tanzania borders Kenya, where we found evidence of Bd emergence and high Bd risk, and Burundi, where we report high Bd prevalence and ambiguous Bd lineages (Seimon et al., 2015). Because eastern Africa is identified as having widespread high Bd risk and multiple locations have high recent Bd prevalence, Bd-host dynamics in these countries should be closely monitored and studied.
In western Africa, our HSM predicted high Bd risk in Ghana, Côte d’Ivoire, Liberia, Guinea, and Sierra Leone. However, there are no records of Bd positives there, and it has been hypothesized that the Dahomey Gap (a non-forested area between western Nigeria and eastern Ghana) may act as a natural barrier creating a safe haven from chytridiomycosis for amphibians (Penner et al., 2013). However, it is worrisome that in nearby Nigeria (a country where we identified moderate Bd risk) the frequency of Bd occurrence is very high (44.5%; 40.1–48.2% CI), and our model predicted highly suitable habitat just west of the Dahomey Gap, similar to previous studies (Penner et al., 2013; Zimkus et al., 2020). Also, other studies have shown that once Bd invades an area, it spreads over large distances and into remote areas rapidly (Lips et al., 2006; Berger et al., 2016; Yap et al., 2016; Vredenburg et al., 2019; Fisher and Garner, 2020). We suggest that future research should focus on Bd-host relationships in western Africa, including monitoring disease presence and spread to determine whether species are threatened by future Bd invasion and surveillance of amphibians in Nigeria to determine whether population declines have occurred.
In northern Africa, our HSM predicted moderate risk in Morocco and in northern Algeria, Tunisia, and Egypt. However, this risk may be underestimated as our analysis did not include recent studies showing an expansion of Bd positives in Morocco (El Cadi et al., 2019; Thumsová et al., 2022). Despite these newer records for Morocco, data for northern Africa are limited; in fact, we are not aware of any published records for other countries in the region. Thus, northern Africa represents another understudied region where monitoring of amphibian populations and Bd are crucial.
New studies of Bd-host dynamics and interactions between Bd lineages in African amphibians could greatly advance our knowledge of chytridiomycosis. By bringing together new and published data spanning 165 years, our study demonstrates recent increases in the frequency of Bd occurrence in much of Africa; yet, we also find areas where frequency of Bd occurrence has not changed over time. In some areas, increases in frequency of Bd occurrence are coincident with amphibian declines (e.g., Cameroon Tanzania, and Ethiopia;Hirschfeld et al., 2016; Channing et al., 2006; Weldon et al., 2020; Gower et al., 2012; Gower et al., 2013), whereas in regions where Bd has not changed in frequency over long periods of time, evidence of widespread declines has not been detected (e.g., South Africa; Hopkins and Channing, 2003; Tarrant et al., 2013). Combined, these results indicate that Africa may harbor regions of Bd endemism (where host species may not be susceptible to epizootics) as well as regions where Bd is invading (posing large risks to host species). We find that multiple genetic lineages of Bd occur in Africa and suggest that (1) lineages generally considered to be low in virulence may actually be responsible for epizootic dynamics in some areas (e.g. Bd-CAPE), and that (2) co-occurring lineages may be hybridizing. Our results provide an opportunity to further investigate virulence of Bd lineages in nature and to determine how Bd hybridization may affect virulence, pathogenicity, and health outcomes in hosts. Finally, our initial data and model predictions help expand our study to enable risk prediction in areas where no Bd data are presently available. Our study highlights the importance of determining the temporal context of host-pathogen dynamics and emphasizes the need to bridge historical and contemporary datasets to better describe host-pathogen dynamics over larger temporal scales and predict future scenarios.
The datasets presented in this study are deposited in the following repositories: Zenodo, doi: https://zenodo.org/record/5514129#.Y_Th5XbP02w; Amphibian Disease Project hosted on the Genomic Observatories MetaDatabase, doi: https://n2t.net/ark:/21547/Dvp2; National Center for Biotechnology Information Sequence Read Archive, bioproject PRJNA555719.
The animal study was reviewed and approved by San Francisco State University Institutional Animal Care and Use Committee (#A20-05-SFSU).
VV and DB conceived and designed the study. DB, EG, MTK, and DP collected field samples. DB and SG collected museum samples. SG and AB performed laboratory work. DM performed histology. SG, TY, HS, AC-A, SC, KL, AM, EP, MSK, and HR compiled and organized data from the literature. SG, TY, AB, ER, VV, and HS contributed to the analysis of the data. SG, VV, TY, AB, HS, DM, and AZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the National Science Foundation (Belmont Forum project NSF ICER-1633948 to VV, NSF DEB-1202609 to DB, NSF Graduate Research Fellowship Program to AB, and NSF IOS-1354241 to ER), the California Academy of Sciences (Hagey Venture Research Fund to DB and VV), and the National Institutes of Health (Animal Models of Infectious Diseases Training Program T32 AI060555 Ruth L. Kirschstein National Research Service Award to SG). DNA sequencing, conducted at the University of Idaho IBEST Genomics Resources Core, was supported in part by NIH COBRE Phase III grant P30GM103324. VV gratefully acknowledges financial support for this research by the Fulbright U.S. Morocco-Spain Joint Teaching/Research Award, which is sponsored by the U.S. Department of State and the Moroccan-American Commission for Educational and Cultural Exchange and the Commission for Cultural, Educational, and Scientific Exchange between the United States of America and Spain. Its contents are solely the responsibility of the author and do not necessarily represent the official views of the Fulbright Program, the Government of the United States, or the U.S. Fulbright Commission’s in Morocco or Spain.
We thank the following natural history museums for access to their specimens: California Academy of Sciences, Museum of Vertebrate Zoology (University of California Berkeley), and Museum of Comparative Zoology (Harvard University). We also thank Marvalee Wake at UC Berkeley for allowing us to sample museum preserved specimens collected in Tanzania. We thank Ben J. Evans for contributing field collected samples for processing and analysis in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The content is solely the responsibility of the authors and does not necessarily represent the official views of funding agencies.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcosc.2023.1069490/full#supplementary-material
Adams A. J., Pessier A. P., Briggs C. J. (2017). Rapid extirpation of a North American frog coincides with an increase in fungal pathogen prevalence: Historical analysis and implications for reintroduction. 10216 | Ecol. Evol. 7, 10216–10232. doi: 10.1002/ece3.3468
AmphibiaWeb. (2021). (Berkeley, CA, USA: University of California). Available at: https://amphibiaweb.org.
Baláž V., Kopecký O., Gvoždík V. (2012). Presence of the amphibian chytrid pathogen confirmed in Cameroon. Herpetological J. 22 (3), 191–194.
Bataille A., Fong J. J., Cha M., Wogan G. O. U., Baek H. J., Lee H., et al. (2013). Genetic evidence for a high diversity and wide distribution of endemic strains of the pathogenic chytrid fungus Batrachochytrium dendrobatidis in wild Asian amphibians. Mol. Ecol. 22 (16), 4196–4209. doi: 10.1111/mec.12385
Bates K. A., Clare F. C., O'Hanlon S., Bosch J., Brookes L., Hopkins K., et al. (2018). Amphibian chytridiomycosis outbreak dynamics are linked with host skin bacterial community structure. Nature communications 9:693. doi: 10.1038/s41467-018-02967-w
Bell R. C., Garcia A. V. G., Stuart B. L., Zamudio K. R. (2011). High prevalence of the amphibian chytrid pathogen in Gabon. EcoHealth 8 (1), 116–120. doi: 10.1007/s10393-010-0364-4
Berger L., Speare R., Daszak P., Green D. E., Cunningham A. A., Goggin C. L., et al. (1998). Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc. Natl. Acad. Sci. U.S.A 95 (15), 9031–9036. doi: 10.1073/pnas.95.15.9031
Berger L., Roberts A. A., Voyles J., Longcore J. E., Murray K. A., Skerratt L. F., et al. (2016). History and recent progress on chytridiomycosis in amphibians. Fungal ecology 19, 89–99. doi: 10.1016/j.funeco.2015.09.007
Bivand R., Lewin-Koh N. (2014). Maptools: Tools for reading and handling spatial objects. R package version 0 8–30. Available at: http://cran.r-project.org/package=maptools.
Blackburn D. C., et al. (2010). An enigmatic mortality event in the only population of the critically endangered cameroonian frog Xenopus longipes. Afr. J. Herpetology 59 (2), 111–122. doi: 10.1080/04416651.2010.495674
Boria R. A., Evans B. J., Pessier A. P., Vredenburg V. T. (2014). Spatial filtering to reduce sampling bias can improve the performance of ecological niche models. Ecological modelling 275, 73–77. doi: 10.1016/j.ecolmodel.2013.12.012
Boyle D. G., Olson L. E., Goodman S. M., Anderson R. P. (2004). Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time taqman PCR assay. Dis. Aquat. Organisms 60 (2), 141–148. doi: 10.3354/dao060141
Briggs C. J., Knapp R. A., Vredenburg V. T. (2010). Enzootic and epizootic dynamics of the chytrid fungal pathogen of amphibians. Proc. Natl. Acad. Sci. U.S.A 107 (21), 9695–9700. doi: 10.1073/pnas.0912886107
Byrne A. Q., Rothstein A. P., Poorten T. J., Erens J., Settles M. L., Rosenblum E. B., et al. (2017). Unlocking the story in the swab: A new genotyping assay for the amphibian chytrid fungus Batrachochytrium dendrobatidis. Mol. Ecol. Resour 17 (6), 1283–1292. doi: 10.1111/1755-0998.12675
Byrne A. Q., Vredenburg V. T., Martel A., Pasmans F., Bell R. C., Blackburn D. C., et al. (2019). Cryptic diversity of a widespread global pathogen reveals expanded threats to amphibian conservation. Proc. Natl. Acad. Sci. U.S.A 116 (41), 20382–20387. doi: 10.1073/pnas.1908289116
Carvalho T., Guilherme Becker C., Toledo L. F. (2017). Historical amphibian declines and extinctions in Brazil linked to chytridiomycosis. Proc. R. Soc. B. 284:20162254. doi: 10.1098/rspb.2016.2254
Catenazzi A., Lehr E., Rodriguez L. O., Vredenburg V. T. (2011). Batrachochytrium dendrobatidis and the collapse of anuran species richness and abundance in the Upper Manu National Park, Southeastern Peru. Conservation biology 25 (2), 382–391. doi: 10.1111/j.1523-1739.2010.01604.x
Catenazzi A., Swei A., Finkle J., Foreyt E., Wyman L, Vredenburg V. T., et al. (2017). Epizootic to enzootic transition of a fungal disease in tropical Andean frogs: Are surviving species still susceptible?. PLoS One 12 (10), e0186478. doi: 10.1371/journal.pone.0186478
Champely S. (2018) Pwr: Basic functions for power analysis. Available at: https://cran.r-project.org/package=pwr.
Channing A., Finlow-Bates K. S., Haarklau Erik S., Hawkesc P. G. (2006). The biology and recent history of the critically endangered Kihansi spray toad Nectophrynoides asperginis in Tanzania. J. East Afr. Natural History 95 (2), 117–138. doi: 10.2982/0012-8317(2006)95[117:tbarho]2.0.co;2
Cheng T. L., Rovito S. M., Wake D. B., Vredenburg V. T. (2011). Coincident mass extirpation of neotropical amphibians with the emergence of the infectious fungal pathogen Batrachochytrium dendrobatidis. Proc. Natl. Acad. Sci. U.S.A 108 (23), 1–6. doi: 10.1073/pnas.1105538108
Conradie W., Bittencourt-Silva G. B., Loader S. P., Menegon M., Nanvonamuquitxo C., Kotzé A., et al. (2016). Batrachochytrium dendrobatidis survey of amphibians in the Northern Mozambique “Sky islands” and low-lying areas. Herpetological Rev. 47 (1), 42–46.
Conradie W., Harvey J., Kotzé A., Dalton D. L., Cunningham M. J. (2011). Confirmed amphibian chytrid in Mount Mulanje area, Malawi. Herpetological Rev. 42 (3), 369–371.
Conradie W., Weldon C., Smith K. G., Preez Du L. K. (2011). Seasonal pattern of chytridiomycosis in common river frog (Amietia angolensis) tadpoles in the South African grassland biome. Afr. Zoology 46 (1), 95–102. doi: 10.1080/15627020.2011.11407483
Creley C. M., Shilling F. M., Muchlinski A. E. (2019). An ecological niche model to predict range expansion of the eastern gray squirrel in California. Bull. South. California Acad. Sci. 118 (1), 58–70. doi: 10.3160/0038-3872-118.1.58
De León M. E., Vredenburg V. T., Piovia-Scott J. (2017). Recent emergence of a chytrid fungal pathogen in California cascades frogs (Rana cascadae). EcoHealth 14, 155–161. doi: 10.1007/s10393-016-1201-1
Doddington B. J., Bosch J., Oliver J. A., Grassly N. C., Garcia G., Schmidt B. R., et al. (2013). Context-dependent amphibian host population response to an invading pathogen. Ecology 94 (8), 1795–1804. doi: 10.1890/12-1270.1
Doherty-Bone T. M., Gonwouo N. L., Hirschfeld M., Ohst T., Weldon C., Perkins M., et al. (2013). Batrachochytrium dendrobatidis in amphibians of Cameroon, including first records for caecilians. Dis. Aquat. Organisms 102 (3), 187–194. doi: 10.3354/dao02557
Doherty-Bone T. M., Cunningham A. A., Fisher M. C., Garner T. W. J., Ghosh P., Gower D. J., et al. (2020). Amphibian chytrid fungus in Africa - realigning hypotheses and the research paradigm. Anim. Conserv. 23 (3), 239–244. doi: 10.1111/acv.12538
Doraj-Raj S. (2014). Binom: Binomial confidence intervals for several parameterizations. R package version 1.1-1.1. Available at: https://CRAN.R-project.org/package=binom.
Edgar R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
El Cadi R. A., Laghzaoui E.-M., Crottini A., Slimani T., Bosch J., Mouden El E. H., et al. (2019). Occurrence of Batrachochytrium dendrobatidis in the tensift region, with comments on its spreading in Morocco. Acta Herpetologica. Firenze Univ. Press 14 (2), 109–115. doi: 10.13128/A_H-7748
El Mouden E. H., Slimani T., Donaire D., Fernández-Beaskoetxea S., Fisher M. C., Bosch J., et al. (2011). First record of the chytrid fungus Batrachochytrium dendrobatidis in North Africa. Herpetological Rev. 42 (1), 71–75.
Farrer R. A., Weinert L. A., Bielby J., Garner T. W. J., Balloux F., Clare F., et al. (2011). Multiple emergences of genetically diverse amphibian-infecting chytrids include a globalized hypervirulent recombinant lineage. Proc. Natl. Acad. Sci. U.S.A 108 (46), 18732–18736. doi: 10.1073/pnas-1111915108
Fisher M. C., Garner T. W. J. (2020). Chytrid fungi and global amphibian declines. Nature Reviews Microbiology 18 (6), 332–343. doi: 10.1038/s41579-020-0335-x
Fisher M. C., Garner T. W. J., Walker S. F. (2009). Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu. Rev. Microbiol. 63 (1), 291–310. doi: 10.1146/annurev.micro.091208.073435
Fisher M. C., Pasmans F., Martel A. (2021). Virulence and pathogenicity of chytrid fungi causing amphibian extinctions. Annu. Rev. Microbiol. Annu. Rev. Microbiol. 75, 673–693. doi: 10.1146/ANNUREV-MICRO-052621-124212
Goldberg T. L., Readel A. M., Lee M. H. (2007). Chytrid fungus in frogs from an equatorial African montane forest in western Uganda. J. Wildlife Dis. 43 (3), 521–524. doi: 10.7589/0090-3558-43.3.521
Gower D. J., Aberra R. K., Schwaller S., Largen M. J., Collen B., Spawls S., et al. (2013). Long-term data for endemic frog genera reveal potential conservation crisis in the Bale Mountains, Ethiopia. Oryx 47 (1), 59–69. doi: 10.1017/S0030605311001426
Gower D. J., Doherty-Bone T., Loader S. P., Wilkinson M., Kouete M. T., Tapley B., et al. (2013). Batrachochytrium dendrobatidis infection and lethal chytridiomycosis in caecilian amphibians (Gymnophiona). EcoHealth 10 (2), 173–183. doi: 10.1007/s10393-013-0831-9
Gower D. J., Doherty-Bone T. M., Aberra R. K., Mengistu A., Schwaller S., Menegon M., et al. (2012). High prevalence of the amphibian chytrid fungus (Batrachochytrium dendrobatidis) across multiple taxa and localities in the highlands of Ethiopia. Herpetological J. 22 (4), 225–233.
Gratwicke B., Alonso A., Elie T., Kolowski J., Lock J., Rotzel N., et al. (2011). Batrachochytrium dendrobatidis not detected on amphibians from two lowland sites in Gabon, Africa. Herpetological Rev. 42 (1), 69–71.
Greenbaum E., Kusamba C., Aristote M. M., Reed K. (2008). Amphibian chytrid fungus infections in Hyperolius (Anura: Hyperoliidae) from eastern Democratic Republic of Congo. Herpetological Rev. 39 (1), 70–73.
Greenbaum E., Meece J., Reed K. D., Kusamba C. (2014). Amphibian chytrid infections in non-forested habitats of Katanga, Democratic Republic of the Congo. Herpetological Rev. 45 (4), 610–614.
Greenbaum E., Meece J., Reed K. D., Kusamba C. (2015). Extensive occurrence of the amphibian chytrid fungus in the Albertine Rift, a Central African amphibian hotspot. Herpetological J. 25, 91–100.
Greenspan S. E., Lambertini C., Carvalho T., James T. Y., Toledo L. F., Haddad C. F. B., et al. (2018). Hybrids of amphibian chytrid show high virulence in native hosts. Sci. Rep. Nat. Publishing Group 8 (1), 9600. doi: 10.1038/s41598-018-27828-w
Griffiths S. M., Harrison X. A., Weldon C., Wood M. D., Pretorius A., Hopkins K., et al. (2018). Genetic variability and ontogeny predict microbiome structure in a disease-challenged montane amphibian. ISME J. 12 (10), 2506–2517. doi: 10.1038/s41396-018-0167-0
Harrell F. E. (2012). ‘Hmisc: Harrell miscellaneous. R package version 4.7-1. Available at: https://CRAN.R-project.org/package=Hmisc.
Hijmans R. J., Cameron S. E., Parra J. L., Jones P. G., Jarvis A. (2005). Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatology 25 (15), 1965–1978. doi: 10.1002/joc.1276
Hijmans R. J., Phillips S., Leathwick J., Elith J. (2013). dismo: Species distribution modeling [Internet]. R package version 0.9-3 9–3. Available at: http://cran.r-project.org/package=dismo.
Hirschfeld M., Blackburn D. C., Doherty-Bone T. M., Gonwouo Nono L., Ghose S., Rödel M.-O., et al. (2016). Dramatic declines of montane frogs in a Central African biodiversity hotspot. PloS One 11 (5), e0155129. doi: 10.1371/journal.pone.0155129
Hopkins S., Channing A. (2003). Chytrid fungus in Northern and Western cape frog populations, South Africa. Herpetological Rev. 34 (4), 334–336.
Hyatt A. D., Boyle D. G., Olsen V., Boyle D. B., Berger L., Obendorf D., et al. (2007). Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis. Aquat. Organisms 73 (3), 175–192. doi: 10.3354/dao073175
Hydeman M. E., Bell R. C., Drewes R. C., Zamudio K. R. (2013). Amphibian chytrid fungus confirmed in endemic frogs and caecilians on the island of São Tomé, Africa. Herpetological Rev. 44 (2), 254–257.
Hydeman M. E., Longo A. V., Velo-Antón G., Rodriguez D., Zamudio K. R., Bell R. C., et al. (2017). Prevalence and genetic diversity of Batrachochytrium dendrobatidis in Central African island and continental amphibian communities. Ecol. Evol 7, 7729–7738. doi: 10.1002/ece3.3309
Imasuen A. A., Aisien M. S. O., Weldon C., Dalton D. L., Kotze A., Preez Du L. H., et al. (2011). Occurrence of Batrachochytrium dendrobatidis in amphibian populations of Okomu National Park, Nigeria. Herpetological Rev. 42 (3), 379–382.
James T. Y., Toledo L. F., Rödder D., Leite Silva D., Belasen A. M., Betancourt-Román C. M., et al. (2015). Disentangling host, pathogen, and environmental determinants of a recently emerged wildlife disease: lessons from the first 15 years of amphibian chytridiomycosis research. Ecol. Evol. 5 (18), 4079–4097. doi: 10.1002/ece3.1672
Jenkinson T. S., Román Betancourt C. M., Lambertini C., Valencia-Aguilar A., Rodriguez D., Nunes-De-Almeida C. H. L., et al. (2016). Amphibian-killing chytrid in Brazil comprises both locally endemic and globally expanding populations. Mol. Ecol. 25 (13), 2978–2996. doi: 10.1111/mec.13599
Jenks G. F. (1967). The data model concept in statistical mapping. Int. Yearbook Cartography 7, 186–190.
Jongsma G. F. M., Kaya A. B., Yoga J. A., Mbega J. D., Beh Mve J. H., Tobi E., et al. (2016). Widespread presence and high prevalence of Batrachochytrium dendrobatidis in Gabon. Herpetological Rev. 47 (2), 227–230.
Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 (12), 1647–1649. doi: 10.1093/bioinformatics/bts199
Kielgast J., Röder D., Veith M., Lötters S. (2010). Widespread occurrence of the amphibian chytrid fungus in Kenya. Anim. Conserv. 13, 36–43. doi: 10.1111/j.1469-1795.2009.00297.x
Kinney V. C., Heemeyer J. L., Pessier A. P., Lannoo M. J. (2011). Seasonal pattern of Batrachochytrium dendrobatidis infection and mortality in Lithobates areolatus: Affirmation of Vredenburg’s “10,000 zoospore rule”. PloS One 6 (3), e16708. doi: 10.1371/journal.pone.0016708
Knapp R. A., Fellers G. M., Kleeman P. M., Miller D. A. W., Vredenburg V. T., Rosenblum E. B., et al. (2016). Large-Scale recovery of an endangered amphibian despite ongoing exposure to multiple stressors. PNAS 113 (42), 11889–11894. doi: 10.1073/pnas.1600983113
Laurance W. F., Mcdonald K. R., Speare R. (1996). Epidemic disease and the catastrophic decline of Australian rain forest frogs. Conserv. Biol. 10 (2), 406–413. doi: 10.1046/j.1523-1739.1996.10020406.x
Li H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27 (21), 2987–2993. doi: 10.1093/bioinformatics/btr509
Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303–3997. doi: 10.48550/arXiv.1303.3997
Lips K. R., Brem F., Brenes R., Reeve J. D., Alford R. A., Voyles J., et al. (2006). Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proc. Natl. Acad. Sci. 103 (9), 3165–3170. doi: 10.1073/pnas.0506889103
Liu X., Rohr J. R., Li Y. (2013). Climate, vegetation, introduced hosts and trade shape a global wildlife pandemic. Proc. R. Soc. B: Biol. Sci. 280 (1753), 20122506. doi: 10.1098/rspb.2012.2506
Lloyd-Smith J. O., Schreiber S. J., Kopp P. E., Getz W. M. (2005). Superspreading and the effect of individual variation on disease emergence. Nature 438 (7066), 355–359. doi: 10.1038/nature04153
Mainali K. P., Warren D. L., Dhileepan K., McConnachie A., Strathie L., Hassan G., et al. (2015). Projecting future expansion of invasive species: Comparing and improving methodologies for species distribution modeling. Global Change Biol. 21, 4464–4480. doi: 10.1111/gcb.13038
Merow C., Smith M. J., Silander J. A. (2013). A practical guide to MaxEnt for modeling species’ distributions: What it does, and why inputs and settings matter. Ecography 36 (10), 1058–1069. doi: 10.1111/j.1600-0587.2013.07872.x
Miller C. A., Taboue G. C. T., Ekane M. M. P., Robak M., Clee P. R. S., Richards-Zawacki C., et al. (2018). Distribution modeling and lineage diversity of the chytrid fungus Batrachochytrium dendrobatidis (Bd) in a Central African amphibian hotspot. PloS One 13 (6), 1–16. doi: 10.1371/journal.pone.0199288
Minter L. R., Burger M., Harrison J. A., Braack H. H., Bishop P. J., Kloepfer D., et al (Eds). (2004). (Eds) Atlas and red data book of the frogs of South Africa, Lesotho and Swaziland. (Washington, DC: SI/MAB Biodiversity Program, Smithsonian Institution,) 9 series.
Moyer D. C., Weldon C. (2006). Chytrid distribution and pathogenicity among frogs of the Udzungwa Mountains, Tanzania (Iringa: The Critical Ecosystem Partnership Fund (CEPF) Small Grant Final Project Completion Report).
Murray K., Retallick R. W. R., Puschendorf R., Skerratt L. F., Rosauer D., McCallum H. I., et al. (2011). Assessing spatial patterns of disease risk to biodiversity: implications for the management of the amphibian pathogen, Batrachochytrium dendrobatidis. J. Appl. Ecol. 48 (1), 163–173. doi: 10.1111/j.1365-2664.2010.01890.x
Mutnale M. C., Anand S., Eluvathingal L. M., Roy J. K., Reddy G. S., Vasudevan K., et al. (2018). Enzootic frog pathogen Batrachochytrium dendrobatidis in Asian tropics reveals high ITS haplotype diversity and low prevalence OPEN. Sci. Rep. 8, 10125. doi: 10.1038/s41598-018-28304-1
O’Hanlon S. J., Rieux A., Farrer R. A., Rosa G. M., Waldman B., Bataille A., et al. (2018). Recent Asian origin of chytrid fungi causing global amphibian declines. Science 360 (6389), 621–627. doi: 10.1126/science.aar1965
Olson D. H., Aanensen D. M., Ronnenberg K. L., Powell C. I., Walker S. F., Bielby J., et al. (2013). Mapping the global emergence of Batrachochytrium dendrobatidis, the amphibian chytrid fungus. PLoS One 8 (2), e56802. doi: 10.1371/journal.pone.0056802
Olson D. H., Ronnenberg K. L., Glidden C. K., Christiansen K. R., Blaustein A. R. (2011). Global patterns of the fungal pathogen Batrachochytrium dendrobatidis support conservation urgency. Front. Vet. Sci. 8, 685877. doi: 10.3389/fvets.2021.685877
Ouellet M., Mikaelian I., Pauli B. D., Rodrigue J., Green D. M. (2005). Historical evidence of widespread chytrid infection in North American amphibian populations. Conserv. Biol. 19 (5), 1431–1440. doi: 10.1111/j
Penner J., Adum G. B., McElroy M. T., Doherty-Bone T., Hirschfeld M., Sandberger L., et al. (2013). West Africa - a safe haven for frogs? a sub-continental assessment of the chytrid fungus (Batrachochytrium dendrobatidis). PLoS One 8 (2), e56236. doi: 10.1371/journal.pone.0056236
Phillips S. J., Elith J. (2013). On estimating probability of presence from use – availability or presence – background data. Ecology 94 (6), 1409–1419. doi: 10.1890/12-1520.1
Phillips S. J., Dudík M., Elith J., Graham C. H., Lehmann A., Leathwick J., et al. (2009). Sample selection bias and presence-only distribution models: Implications for background and pseudo-absence data. Ecol. Appl. 19 (1), 181–197. doi: 10.1890/07-2153.1
Phillips S. J., Anderson R. P., Dudík M., Schapire R. E., Blair M. E. (2017). Opening the black box: an open-source release of maxent. Ecography 40 (7), 887–893. doi: 10.1111/ecog.03049
Phillips B. L., Puschendorf R. (2013). Do pathogens become more virulent as they spread? evidence from the amphibian declines in Central America. Proc. R. Soc. B (Vienna, Austria) 280 (1766), 20131290. doi: 10.1098/rspb.2013.1290
Piovia-Scott J., Pope K., Worth S. J., Rosenblum E. B., Poorten T., Refsnider J., et al. (2015). Correlates of virulence in a frog-killing fungal pathogen: Evidence from a California amphibian decline. ISME J. 9, 1570–1578. doi: 10.1038/ismej.2014.241
R Core Team. (2022). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available at: https://www.R-project.org/.
Reeder N. M. M., Cheng T. L., Vredenburg V. T., Blackburn D. C. (2011). Survey of the chytrid fungus Batrachochytrium dendrobatidis from montane and lowland frogs in eastern Nigeria. Herpetology Notes 4 (1), 83–86.
Rödder D., Kielgast J., Bielby J., Schmidtlein S., Bosch J., Garner T. W. J., et al. (2009). Global amphibian extinction risk assessment for the panzootic chytrid fungus. Diversity 1 (1), 52–66. doi: 10.3390/d1010052
Rodriguez D., Becker C. G., Pupin N. C., Haddad C. F. B., Zamudio K. R. (2014). Long-term endemism of two highly divergent lineages of the amphibian-killing fungus in the Atlantic forest of Brazil. Mol. Ecol. 23 (4), 774–787. doi: 10.1111/mec.12615
Ron S. R. (2005). Predicting the distribution of the amphibian pathogen Batrachochytrium dendrobatidis in the new world. Biotropica 37 (2), 209–221. doi: 10.2307/30043173
Rosenblum E. B., James T. Y., Zamudio K. R., Poorten T. J., Ilut D., Rodriguez D., et al. (2013). Complex history of the amphibian-killing chytrid fungus revealed with genome resequencing data. Proc. Natl. Acad. Sci. 110 (23), 9385–9390. doi: 10.1073/pnas.1300130110
RStudio Team. (2016). RStudio: Integrated development for r (Boston, MA: RStudio, Inc). Available at: http://www.rstudio.com/.
Scheele B. C., Pasmans F., Skerratt L. F., Berger L., Martel A., Beukema W., et al. (2019). Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 363 (6434), 1459–1463. doi: 10.1126/science.aav0379
Schloegel L. M., Toledo L. F., Longcore J. E., Greenspan S. E., Vieira C. A., Lee M., et al. (2012). Novel, panzootic and hybrid genotypes of amphibian chytridiomycosis associated with the bullfrog trade. Mol. Ecol. 21 (21), 5162–5177. doi: 10.1111/j.1365-294X.2012.05710.x
Seimon T. A., Ayebare S., Sekisambu R., Muhindo E., Mitamba G., Greenbaum E., et al. (2015). Assessing the threat of amphibian chytrid fungus in the Albertine Rift: Past, present and future. PloS One 10 (12), 1–24. doi: 10.1371/journal.pone.0145841
Sette C. M., Vredenburg V. T., Zink A. G. (2015). Reconstructing historical and contemporary disease dynamics: A case study using the California slender salamander. Biol. Conserv. 192, 20–29. doi: 10.1016/j.biocon.2015.08.039
Sette C. M., Vredenburg V. T., Zink A. G. (2020). Differences in fungal disease dynamics in co-occurring terrestrial and aquatic amphibians. EcoHealth 17, 302–314. doi: 10.1007/s10393-020-01501-z
Skerratt L. F., Berger L., Speare R., Cashins S., McDonald K. R., Phillott A. D., et al. (2007). Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. EcoHealth 4 (2), 125–134. doi: 10.1007/s10393-007-0093-5
Soto-Azat C., Clarke B. T., Poynton J. C., Cunningham A. A. (2010). Widespread historical presence of Batrachochytrium dendrobatidis in African pipid frogs. Diversity Distributions 16 (1), 126–131. doi: 10.1111/j.1472-4642.2009.00618.x
Stamatakis A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stuart S. N., Chanson J. S., Cox N. A., Young B. E., Rodrigues A. S. L., Fischman D. L., et al. (2004). Status and trends of amphibian declines and extinctions worldwide. Sci. (New York N.Y.) 306 (2002), 1783–1786. doi: 10.1126/science.1103538
Talley B. L., Muletz C. R., Vredenburg V. T., Fleischer R. C., Lips K. R. (2015). A century of Batrachochytrium dendrobatidis in Illinois amphibians, 1888-1989. Biol. Conserv. 182, 254–261. doi: 10.1016/j.biocon.2014.12.007
Tarrant J., Cilliers D., Preez L. H., Weldon C. (2013). Spatial assessment of amphibian chytrid fungus (Batrachochytrium dendrobatidis) in South Africa confirms endemic and widespread infection. PloS One 8 (7), e69591. doi: 10.1371/journal.pone.0069591
The IUCN Red List of Threatened Species. (2021). (IUCN). Available at: https://www.iucnredlist.org (Accessed 2 September 2021).
Thorne L. H., Johnston D. W., Urban D. L., Tyne J., Bejder L., Baird R. W., et al. (2012). Predictive modeling of spinner dolphin (Stenella longirostris) resting habitat in the main Hawaiian islands. PloS One 7 (8), e43167. doi: 10.1371/journal.pone.0043167
Thumsová B., Donaire-Barroso D., Mouden El E. H., Bosch J. (2022). Fatal chytridiomycosis in the Moroccan midwife toad alytes maurus and potential distribution of batrachochytrium dendrobatidis across Morocco. African Journal of Herpetology 71, 1–11. doi: 10.1080/21564574.2021.1998235
Venter O., Sanderson E. W., Magrach A., Allan J. R., Beher J., Jones K. R., et al. (2016). Sixteen years of change in the global terrestrial human footprint and implications for biodiversity conservation. Nat. Commun. 7, 1–11. doi: 10.1038/ncomms12558
Vredenburg V. T., Knapp R. A., Tunstall T. S., Briggs C. J. (2010). Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proc. Natl. Acad. Sci. U.S.A 107 (21), 9689–9694. doi: 10.1073/pnas.0914111107
Vredenburg V. T., McNally S. V. G., Sulaeman H., Butler H. M., Yap T., Koo M. S., et al. (2019). Pathogen invasion history elucidates contemporary host pathogen dynamics. PloS One 14 (9), e0219981. doi: 10.1371/journal.pone.0219981
Wake D. B., Vredenburg V. T. (2008). Are we in the midst of the sixth mass extinction? a view from the world of amphibians. PNAS 105 (1), 11466–11473. doi: 10.1073/pnas.0801921105
Walker S. F., Bosch J., Gomez V., Garner T. W. J., Cunningham A. A., Schmeller D. S., et al. (2010). Factors driving pathogenicity vs. prevalence of amphibian panzootic chytridiomycosis in Iberia. Ecol. Lett. 13 (3), 372–382. doi: 10.1111/j.1461-0248.2009.01434.x
Weldon C. (2005). Chytridiomycosis, an emerging infectious disease of amphibians in South Africa [dissertation thesis] (Potchefstroom, South Africa: North-West University).
Weldon C., Preez L. H., Hyatt A. D., Muller R., Speare R. (2004). Origin of the amphibian chytrid fungus. Emerging Infect. Dis. 10 (12), 2100–2105. doi: 10.3201/eid1012.030804
Weldon C., Channing A., Misinzo G., Cunningham A. A. (2020). Disease driven extinction in the wild of the kihansi spray toad, nectophrynoides asperginis. African Journal of herpetology 69, 151–164. doi: 10.1080/21564574.2020.1752313
Yap T. A., Koo M. S., Ambrose R. F., Wake D. B., Vredenburg V. T. (2015). Averting a North American biodiversity crisis: A newly described pathogen poses a major threat to salamanders via trade. Science. Am. Assoc. Advancement Sci. 349 (6247), 481–482. doi: 10.1126/SCIENCE.AAB1052/SUPPL_FILE/TABLES3.XLSX
Yap T. A., Gillespie L., Ellison S., Flechas S. V., Koo M. S., Martinez A. E., et al. (2016). Invasion of the fungal pathogen batrachochytrium dendrobatidis on California islands. EcoHealth 13, 145–150. doi: 10.1007/s10393-015-1071-y
Yap T. A., Koo M. S., Ambrose R. F., Vredenburg V. T. (2018). Introduced bullfrog facilitates pathogen invasion in the western United States. PLoS One 13 (4), e0188384. doi: 10.1371/journal.pone.0188384
Zancolli G., Storfer A., Rödel M. O. (2013). Detection of Batrachochytrium dendrobatidis in river frogs (genus Amietia) on mount Kilimanjaro, Tanzania. Herpetological Rev. 44 (4), 611–614.
Zimkus B. M., Baláz V., Belasen A. M., Bell R. C., Channing A., Doumbia J., et al. (2020). Chytrid pathogen (Batrachochytrium dendrobatidis) in African amphibians: A continental analysis of occurrences and modeling of its potential distribution. Herpetologica 76 (2), 201–215. doi: 10.1655/0018-0831-76.2.201
Keywords: Batrachochytrium dendrobatidis, chytridiomycosis, epizootic, Africa, amphibians, emerging infectious disease, fungal pathogen, historical survey
Citation: Ghose SL, Yap TA, Byrne AQ, Sulaeman H, Rosenblum EB, Chan-Alvarado A, Chaukulkar S, Greenbaum E, Koo MS, Kouete MT, Lutz K, McAloose D, Moyer AJ, Parra E, Portik DM, Rockney H, Zink AG, Blackburn DC and Vredenburg VT (2023) Continent-wide recent emergence of a global pathogen in African amphibians. Front. Conserv. Sci. 4:1069490. doi: 10.3389/fcosc.2023.1069490
Received: 13 October 2022; Accepted: 31 January 2023;
Published: 15 March 2023.
Edited by:
Ricardo Lourenço-de-Moraes, Federal University of Paraíba, BrazilReviewed by:
Paul M. Severns, University of Georgia, United StatesCopyright © 2023 Ghose, Yap, Byrne, Sulaeman, Rosenblum, Chan-Alvarado, Chaukulkar, Greenbaum, Koo, Kouete, Lutz, McAloose, Moyer, Parra, Portik, Rockney, Zink, Blackburn and Vredenburg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vance T. Vredenburg, dmFuY2V2QHNmc3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.