Abstract
Declines in abalone populations throughout the world have led to conservation measures including fishery closures and captive breeding programs aimed at stock restoration. Restoration of endangered pinto abalone (Haliotis kamtschatkana) in the southern Salish Sea (Washington State, USA) began in the mid-2000s, and since 2009, nearly 40,000 hatchery-produced juvenile abalone have been outplanted at 21 restoration sites. We used genotyping by sequencing to evaluate the efficacy of this restoration program from a genomic standpoint. Over 49,000 SNPs and 8,000 haplotypes were evaluated across both wild and hatchery-produced abalone. Compared to wild abalone, hatchery-bred abalone had similar heterozygosity, lower allelic richness and effective population size, and higher relatedness. However, more recently bred hatchery progeny sampled prior to outplanting showed intermediate allelic richness, lower relatedness and genomic divergence, and higher effective population size compared to older hatchery outplants sampled after outplanting. We attribute these differences to genetic drift among older hatchery outplants due to mortality and emigration as well as larger numbers of broodstock families in more recent hatchery operations. This suggests that current hatchery practices that combine higher output with larger numbers of broodstock families may more effectively overcome the effects of genetic drift. Effective population size estimates among wild Salish Sea abalone had no upper limit, indicating that these abalone have sufficient genetic diversity to support the restoration program, but also highlighting the need for large broodstock sizes to overcome potential Ryman-Laikre effects. The greatest challenge for the future of the restoration program will be finding sufficient numbers of wild broodstock in the Salish Sea, where pinto abalone remain extremely rare.
Introduction
Abalone are large marine snails found on shallow temperate and tropical rocky reefs throughout the world. Prized as a food, many abalone species have undergone significant population declines largely due to overharvest (Cook and Gordon, 2010; Karpov et al., 2000). Because abalone are diecious broadcast spawners with very small home ranges, depleted populations often occur at densities too low for successful fertilization (Babcock and Keesing, 1999; Zhang, 2008), and reproductive failure is therefore the primary factor limiting natural rebound of abalone populations (Carson et al., 2019; Masuda and Tsukamoto, 1998; Roberts et al., 2007; Rogers-Bennett et al., 2016). Stock status reviews and extinction risk models have indicated that human intervention is necessary to facilitate species recovery (Carson and Ulrich, 2019; Catton et al., 2016; Hobday and Tegner, 2000; Stierhoff et al., 2012; VanBlaricom et al., 2009), which has resulted in widespread efforts to rebuild wild abalone stocks through restoration aquaculture (Carson et al., 2019; Masuda and Tsukamoto, 1998; Roberts et al., 2007; Rogers-Bennett et al., 2016). Restoration aquaculture programs aim to replenish depleted populations with little or no natural reproductive output, often due to an Allee effect, and typically use remnant populations as broodstock to breed large quantities of progeny for eventual release into the wild (Grant et al., 2017). This serves the dual purpose of bringing together sparse populations of adults to breed while also reducing the effects of predation on early life history stages.
For restoration aquaculture programs, maintenance of wild stock genetic integrity through the production of genetically diverse progeny is of paramount concern (Grant et al., 2017). The relationship between organismal genetic diversity and fitness is well established (DeWoody et al., 2021), and preservation of genetic diversity is essential to protect the adaptive potential of a population, particularly those that are small or have been depleted (Crow and Kimura, 1970; Willi et al., 2006). Depleted populations may exhibit reduced genetic variation and increased inbreeding (Willi et al., 2006), and outplants of large numbers of insufficiently diverse hatchery-produced individuals risks further undermining genetic diversity and effective population size (Grant et al., 2017).
Despite efforts to preserve genetic diversity in abalone captive breeding programs, several studies have reported lower diversity among hatchery produced abalone relative to wild populations, including reduced heterozygosity, effective population size, and allelic richness, (Evans et al., 2004; Gruenthal et al., 2014; Lemay and Boulding, 2009; Smith and Conroy, 1992). Moreover, the genetic signal of outplanted hatchery-produced abalone has been detected in areas experiencing stock enhancement, a further indication of differentiation between hatchery-produced and wild populations (Gaffney et al., 1996; Sekino et al., 2005). These studies have underscored the importance of large broodstocks, hatchery practices that maximize diverse progeny, and the monitoring of genetic diversity among hatchery-produced and wild populations (Gaffney et al., 1996; Rhode et al., 2014; Smith and Conroy, 1992).
Pinto abalone (Haliotis kamtschatkana Jonas, 1845), the widest ranging abalone species along the Pacific coast of North America, have become rare in many parts of their range as a result of overharvest (Neuman et al., 2018). In the southern Salish Sea, Washington, USA, pinto abalone have undergone a 97% decline since the early 1990s, despite a 1994 harvest ban (Carson and Ulrich, 2019). Pinto abalone are now considered threatened with local extinction, which led the State of Washington to list the species as a State Endangered Species in 2019. Federally, pinto abalone are listed as a Species of Concern (Neuman et al., 2018). As with other imperiled abalone species, low remaining populations of pinto abalone in Washington waters are thought to be below critical thresholds for successful reproduction (Rothaus et al., 2008). This is supported by observations of extremely low natural recruitment in the San Juan Archipelago, where a significant portion of Washington’s remnant populations are found (Bouma et al., 2012; Rogers-Bennett et al., 2011).
In an attempt to mitigate natural recruitment failure, pinto abalone recovery efforts began over 15 years ago as a collaborative effort between federal, state, tribal, local, and non-governmental organizations. Since 2009, 40,000 hatchery-raised juvenile abalone have been outplanted at 21 sites in the San Juan Archipelago. A recent study found that observed survival one year after outplant averaged approximately 10% and was highly site-dependent (Carson et al., 2019). Initially a relatively small-scale pilot program, the recovery effort has recently transitioned to a larger-scale production phase that includes satellite hatchery facilities.
As the program expands in scope, detailed genetic data on broodstocks, hatchery-produced outplants, and wild populations are essential to help inform future conservation aquaculture practices and restoration strategies. In this study, we used restriction site associated DNA sequencing (RADseq) to determine the efficacy of restoration practices to date with respect to the genetic diversity of broodstocks, progeny, surviving outplants, and wild populations. Using a robust set of single nucleotide polymorphisms (SNPs) and haplotypes, our goal was to determine if pre- and post-outplant hatchery progeny have low genomic divergence and comparable genomic diversity relative to wild abalone.
Methods
Broodstock Collection & Hatchery Production
Broodstock collection and hatchery production methods are described in detail by Carson et al. (2019). In brief, broodstock collection targeted reproductively isolated individuals occurring over a broad geographic area within the San Juan Archipelago; aggregations of abalone were generally left undisturbed, and only lone individuals were targeted. Each year of the program, new broodstock were collected, and only F1 progeny bred from wild broodstock were used for restoration. Broodstock collection began in 2003 followed by hatchery production in 2007, with the first outplants in 2009. The process has been repeated nearly every year since the first year of outplant (Table 1). Hatchery operations took place at the NOAA Mukilteo Research Station in Mukilteo, WA from 2009 to 2018, and transitioned to the Kenneth K. Chew Center for Shellfish Research and Restoration at NOAA’s Manchester Research Station in 2019. Single-parent crosses were produced from gametes obtained via induced spawning. Herein, the term ‘broodstock family’ refers to the offspring of each pairing of one female and one male broodstock. Some females were paired with more than one male, resulting in multiple families of half-siblings. Broodstock families were reared in separate aquaria through all life stages.
Table 1
| Outplant Year | Site | N | # Broodstock | # Families |
|---|---|---|---|---|
| 2009 | Omaha | 260 | 5f, 6m | 7 |
| 2009 | Gold | 257 | 5f, 6m | 7 |
| 2011 | Omaha | 330 | 9f, 13m | 17 |
| 2011 | Gold | 350 | 9f, 13m | 17 |
| 2014 | Omaha | 358 | 7f, 5m | 10 |
| 2014 | Gold | 358 | 7f, 5m | 10 |
| 2015 | Omaha | 218 | 2f, 4m | 5 |
| 2015 | Gold | 218 | 2f, 4m | 5 |
| 2015 | Baytown | 726 | 9f, 10m | 14 |
| 2016 | Baytown | 601 | 9f, 13m | 14 |
| 2016 | Dragoon | 619 | 9f, 13m | 14 |
| 2017 | Omaha | 491 | 6f, 8m | 9 |
| 2017 | Gold | 488 | 6f, 8m | 9 |
| 2017 | Baytown | 488 | 6f, 8m | 9 |
| 2017 | Dragoon | 592 | 8f, 14m | 17 |
Outplant history for each of the outplant sites represented in the manuscript.
Outplant Year is the year of outplanting, Site is the name of the outplant site, N is the number of juvenile abalone outplanted, # Broodstock is the number of female (f) and male (m) broodsock used, and # Families is the total number of unique broodstock families produced.
Juvenile abalone were outplanted at approximately 20 months post-fertilization. Tagging of individuals with bee tags glued on the shell surface prior to outplanting allowed tracking of broodstock families in the field for up to five years, by which time nearly all tags had worn off (Carson et al., 2019).
Hatchery Outplants
The restoration program has outplanted juvenile abalone at 21 sites in the San Juan Archipelago, but the data reported herein reflect abalone from four of the oldest and most successful sites with the highest abalone survival (Table 1; Table 2). Details on site selection criteria are provided in Carson et al. (2019). As in Carson et al. (2019), restoration site geographic names and coordinates are not reported to protect abalone from potential illegal harvest. Instead, the site naming convention used in Carson et al. (2019) is also used here. Abalone samples from sites Omaha, Gold, Baytown, and Dragoon were collected for this study during annual surveys in 2019. Omaha and Gold were established in 2009, and are located on separate islands approximately 1.5 km apart. These two sites received outplants in 2009, 2011, 2014, 2015, and 2017. Baytown was established in 2015 on another island 7.5 km from Omaha and Gold, and received outplants in 2015, 2016, and 2017. Dragoon was established in 2016 on an offshore reef 20 km from Omaha and Gold and received outplants in 2016 and 2017. Due to extremely low natural recruitment, all tagged or untagged abalone were assumed to be a hatchery outplant (Rogers-Bennett et al., 2011).
Hatchery production has increased significantly in recent years, and to provide a picture of genetic diversity among hatchery-bred abalone from more recent hatchery operations, hatchery progeny bred between 2017 and 2020 (Progeny ‘17-’20 H) were randomly sampled prior to outplanting (Table 3; Table 2). Starting in 2019, the restoration program began outplanting a combination of 10-month and 20-month-old juveniles. Therefore, two production years are listed separately for each outplant year in Table 3. For the sake of clarity, and because it bears critically on the interpretation of results, it is worth reiterating that Progeny ‘17-’20 H were sampled prior to outplanting, whereas Omaha H, Gold H, Baytown H, and Dragoon H were sampled after abalone of multiple cohorts had been living at outplant sites for 2-10 years.
Table 3
| Year Produced | Outplant Year | Sites | N | # Broodstock | # Families |
|---|---|---|---|---|---|
| 2017 | 2019 | 6 | 1721 | 7f, 13m | 12 |
| 2018 | 2019 | 6 | 4838 | 5f, 13m | 14 |
| 2018 | 2020 | 6 | 370 | 5f, 7m | 7 |
| 2019 | 2020 | 6 | 3246 | 12f, 15m | 24 |
| 2019 | 2021 | 10 | 2548 | 9f, 13m | 13 |
| 2020 | 2021 | 10 | 8145 | 7f, 19m | 24 |
Recent (2017-2020) hatchery production.
Data from a random subsample of these juveniles are included in this study (Progeny 2017-2020). Year Produced is the year the abalone were bred, Outplant Year is the year of outplanting, Sites is the number of outplant sites to which each cohort was outplanted, N is the number of juvenile abalone outplanted, # Broodstock is the number of female (f) and male (m) broodsock used, and # Families is the total number of unique broodstock families produced.
Table 2
| Sample | N | Shell length(mm, mean ± SD) | Sex ratio M:F | Ho | He | FIS |
|---|---|---|---|---|---|---|
| Gold H | 37 | 88.7 ± 27.4 | 0.625 | 0.0629 | 0.0685 | 0.0616 |
| Omaha H | 96 | 99.3 ± 24.4 | 0.529 | 0.0623 | 0.0681 | 0.0662 |
| Baytown H | 72 | 81.4 ± 17.4 | 0.5 | 0.0628 | 0.0685 | 0.0655 |
| Dragoon H | 72 | 66.5 ± 18.7 | 0.666 | 0.0632 | 0.0676 | 0.0526 |
| Progeny ‘17-’20 H | 45 | 9.8 ± 3.3 | NA | 0.0635 | 0.0711 | 0.0800 |
| Broodstock W | 59 | ND | 0.356 | 0.0646 | 0.0715 | 0.0717 |
| San Juan W | 36 | 120.6 ± 14.4 | 0.583 | 0.0642 | 0.0721 | 0.0833 |
| Ketchikan W | 40 | ND | ND | 0.0638 | 0.0710 | 0.0796 |
Sample sizes, shell lengths, sex ratios (M/F; even = 0.5), and genetic diversity statistics for each group of samples included in the final assembly.
Wild versus hatchery-raised abalone are denoted by ‘W’ and ‘H’ respectively. NA, not applicable; ND, not determined.
Wild Abalone Samples
Three wild abalone groups are included in this analysis to provide a baseline of natural genetic diversity within the Salish Sea region and beyond with which to compare hatchery-produced individuals (Table 2). Older broodstock collected in the San Juan Islands between 2007 and 2015 (Broodstock W) represent parents of the abalone outplanted to the sites reported herein between 2009 and 2017. In 2019, additional wild abalone were sampled in the San Juan Islands (San Juan W), and some of these individuals served as broodstock for the Progeny ‘17-’20 H group. To provide additional perspective on wild abalone genomic diversity, we also included a sample of individuals from Ketchikan, Alaska (Ketchikan W).
Sample Collection, DNA Extraction, Library Preparation, and Sequencing
Non-lethal tissue samples from abalone were taken by clipping epipodial tentacles and preserving them in 95% ethanol. DNA was extracted from samples using the Qiagen DNeasy Blood and Tissue kit, with overnight lysis in proteinase k. An Agilent Technologies TapeStation was used to quantify DNA quality and concentration. RADseq library preparation followed the BestRAD technique (Ali et al., 2016) with the restriction enzyme SbfI (5’-CCTGCAGG-3’ cut site). Paired-end, 100 bp sequencing was carried out by Novogene Corp. on an Illumina NovaSeq 6000. Five libraries with 96 samples each were each sequenced on an individual NovaSeq lane (5 lanes total). Samples from different populations were randomly allocated across each library, along with a total of 25 technical replicates represented by samples sequenced in duplicate or triplicate.
Bioinformatic Processing
Raw reads were filtered and demultiplexed with the Stacks v.2 process_radtags program (Rochette et al., 2019) using default settings. Demultiplexed reads were then mapped to the red abalone (Haliotis rufescens) genome (Masonbrink et al., 2019) with the BWA mem program (Li and Durbin, 2010) using default settings. Phylogenetic analysis of northeastern Pacific abalone species indicates relatively shallow divergence between pinto and red abalone (Masonbrink et al., 2019), justifying the use of this genome for read mapping. High-quality (mapQ 20+) and properly-paired mapped reads were sorted and converted to BAM format with samtools v.1.9 (Li et al., 2009) and assembled with the Stacks v.2 gstacks program with the PCR duplicates removal option, followed by SNP and haplotype processing with the Stacks v.2 populations program (Rochette et al., 2019).
SNP Analysis
To generate the SNP dataset, the following settings in Stacks v.2 populations were used: minimum percentage of individuals in a population required to process a locus for that population (0.7), minimum minor allele count required to process a SNP (3). Further SNP filtering of the resulting VCF file was undertaken with the R package vcfR (Knaus and Grünwald, 2017). Variants with sequencing depth outside of 95% confidence intervals were excluded, as were those with quality scores below 20 and with greater than 20% missing data. Lastly, to exclude variants according to linkage disequilibrium (LD), LD-based SNP pruning was performed with the R package SNPRelate (Zheng et al., 2012) using an LD threshold of r2 = 0.2.
Population statistics, including gene diversity, observed heterozygosity, inbreeding coefficients, and genetic differentiation were computed with the R package hierfstat (Goudet, 2005). Pairwise fixation index (FST) was computed according to the method of (Weir and Cockerham, 1984) as implemented in hierfstat and StAMPP (Pembleton et al., 2013), with 95% confidence intervals and p values computed with StAMPP. Visualization of population differentiation in two-dimensions was facilitated with discriminant analysis of principal components (DAPC) implemented in the R package adegenet (Jombart, 2008). The xvalDAPC cross-validation procedure was used in adegenet to select the appropriate number of principal components (100) for the DAPC (Figure S1).
Relatedness was evaluated using a genomic relatedness matrix calculated by StAMPP (Pembleton et al., 2013) as well as by the R package related (Pew et al., 2015). To evaluate and choose among the different relatedness estimators used by related, allele frequencies from the dataset were used to simulate individuals of known relatedness (parent-offspring, full siblings, half siblings, or unrelated). Relationships were computed using four different estimators, and the optimal estimator was chosen based on the correlation between observed and expected relatedness values (Pew et al., 2015). After using the optimal estimator to calculate relatedness in the real dataset, we also tested its classification accuracy by using the simulated dataset to train a k-nearest neighbor model with 10-fold repeat cross validation implemented in the caret R package (Kuhn, 2008). Pairwise relationships computed in related were used as a proxy for hypothetical mate pairings, and they were also used to cross-check broodstock family structure based on hierarchical clustering of the relatedness matrix calculated by StAMPP.
Haplotype Analysis
Because SNPs are biallelic, they are not ideal for providing information on allelic richness. Instead, multi-allelic haplotypes provide much richer datasets with which to evaluate allelic diversity and rare alleles. Haplotypes output by Stacks v.2 were used to assess haplotype richness. We ran the populations program on multiple sample sizes from N = 4-36 in increments of 4 to evaluate allelic richness as a function of sample size. Samples were randomly selected and added for each increment, and only loci that were present across all increments were used for comparison. This resulted in ten datasets with no missing data. The following settings in Stacks v.2 populations were used: minimum percentage of individuals in a population required to process a locus for that population (1.0), minimum minor allele count required to process a SNP (3).
Haplotypes from the N = 36 increment were also used to calculate effective population size (Ne) using the LD method in NeEstimator v2.1 (Do et al., 2014). Since estimates of census population size (N) were available for each outplant site (Table S1), we also computed Ne/N.
Results
Data Yield and Error Rates
The final SNP dataset consisted of 49,289 SNPs across 1,131 scaffolds and 477 samples, with 6.5% missing data. Genotyping error, as assessed by SNP mismatches between the 25 technical replicates, was 0.03%, meaning that SNPs in the final assembly were called with 99.97% accuracy.
The final haplotype dataset for N = 36 across all groups (N = 288 total samples) with no missing data included 8,345 loci across 929 scaffolds.
Genetic Diversity
Based on the SNP dataset, slight heterozygote deficiencies were observed in all groups (Table 2), with inbreeding coefficients (FIS) ranging from 0.0526 (Dragoon H) to 0.0833 (San Juan W). Although wild abalone generally had higher FIS, Progeny ‘17-’20 H had FIS comparable to wild individuals. Analysis of allelic richness in the haplotype data series showed an increase in allelic richness with sample size in all groups (Figure 1). At N > 20, clear patterns were observed with wild individuals having higher allelic richness than hatchery-raised individuals. Progeny ‘17-’20 H had intermediate levels of allelic richness between the three wild groups and the four older outplant sites.
Figure 1
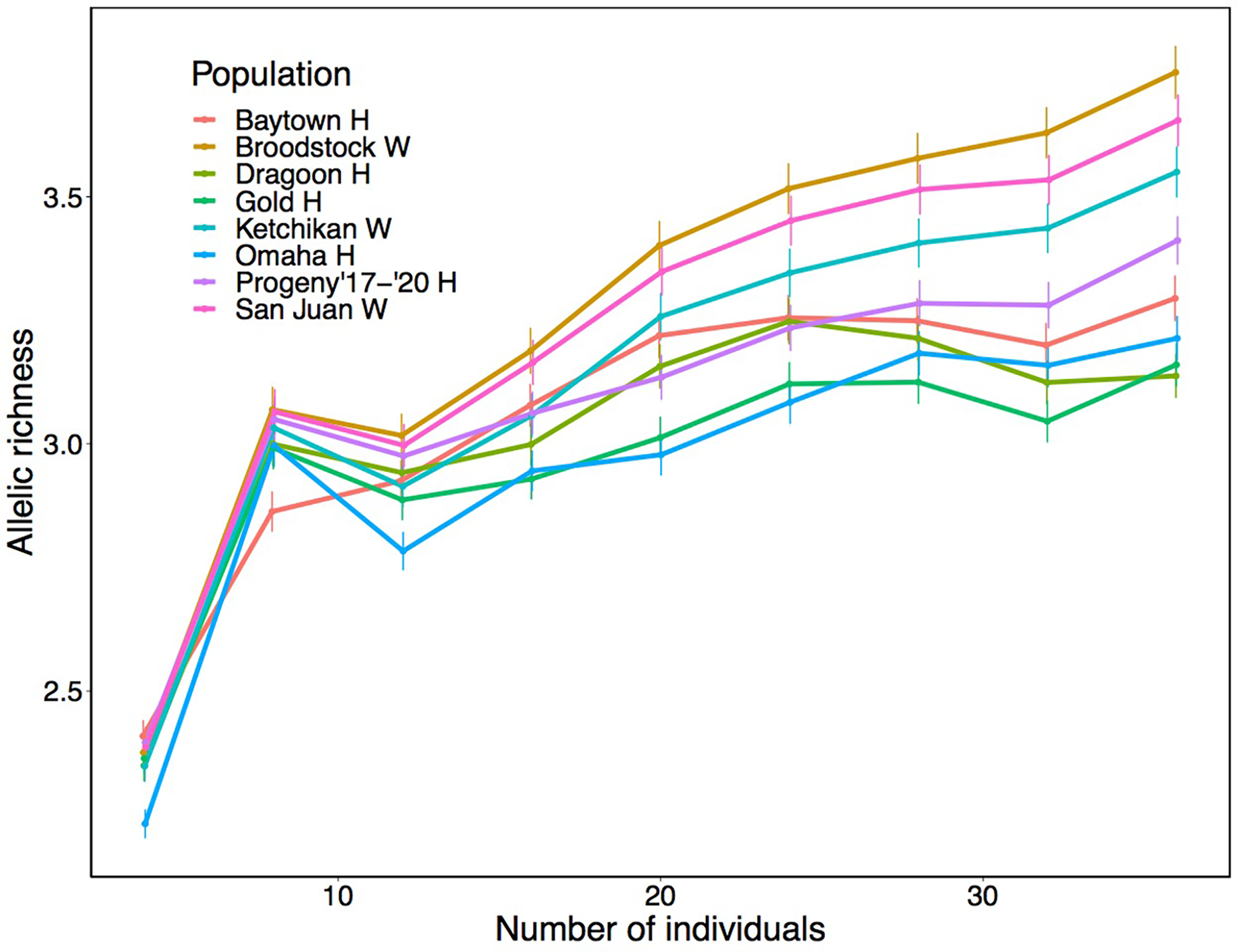
Allelic richness as a function of sample size for haplotype-based assemblies ranging from 4–36 individuals per group. Error bars represent 95% confidence intervals.
Genetic Differentiation Among Groups
Pairwise FST values ranged from 0.001 - 0.019, with the lowest values between wild abalone groups (Figure 2). All FST values were significantly different from zero (p < 0.001). FST values were also low between Progeny ‘17-’20 H and wild abalone, as well as between Gold H and Omaha H. All outplant sites had intermediate FST values from their parent group, Broodstock W. The highest FST values were between outplant sites and Ketchikan W, San Juan W, and Progeny ‘17-’20 H. Additionally, Gold H and Omaha H were relatively strongly differentiated from Dragoon H and Baytown H. Hierarchical clustering of FST values showed two primary clusters; one cluster including all outplant sites, and another cluster including all wild groups plus Progeny ‘17-’20 H.
Figure 2
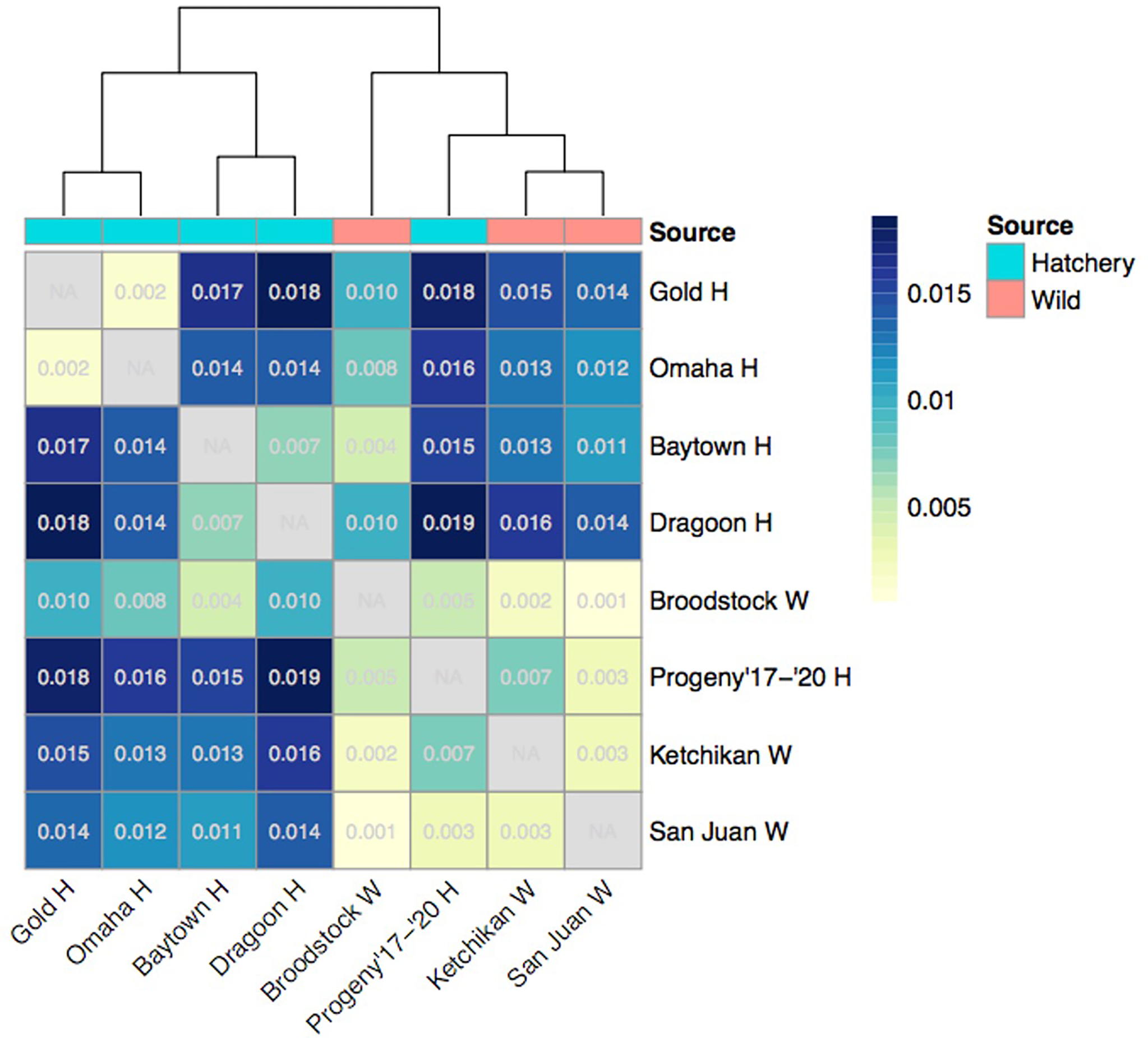
Heatmap and hierarchical clustering of pairwise FST values between groups. All FST values were significantly different from zero (p < 0.001).
These patterns were generally corroborated by discriminant analysis of principal components (DAPC) analysis (Figure 3). As expected, outplant sites clustered closely with Broodstock W. In turn, Broodstock W overlapped with San Juan W, which overlapped with Progeny ‘17-’20 H. Ketchikan W formed a separated cluster that was largely spread along the secondary axis.
Figure 3
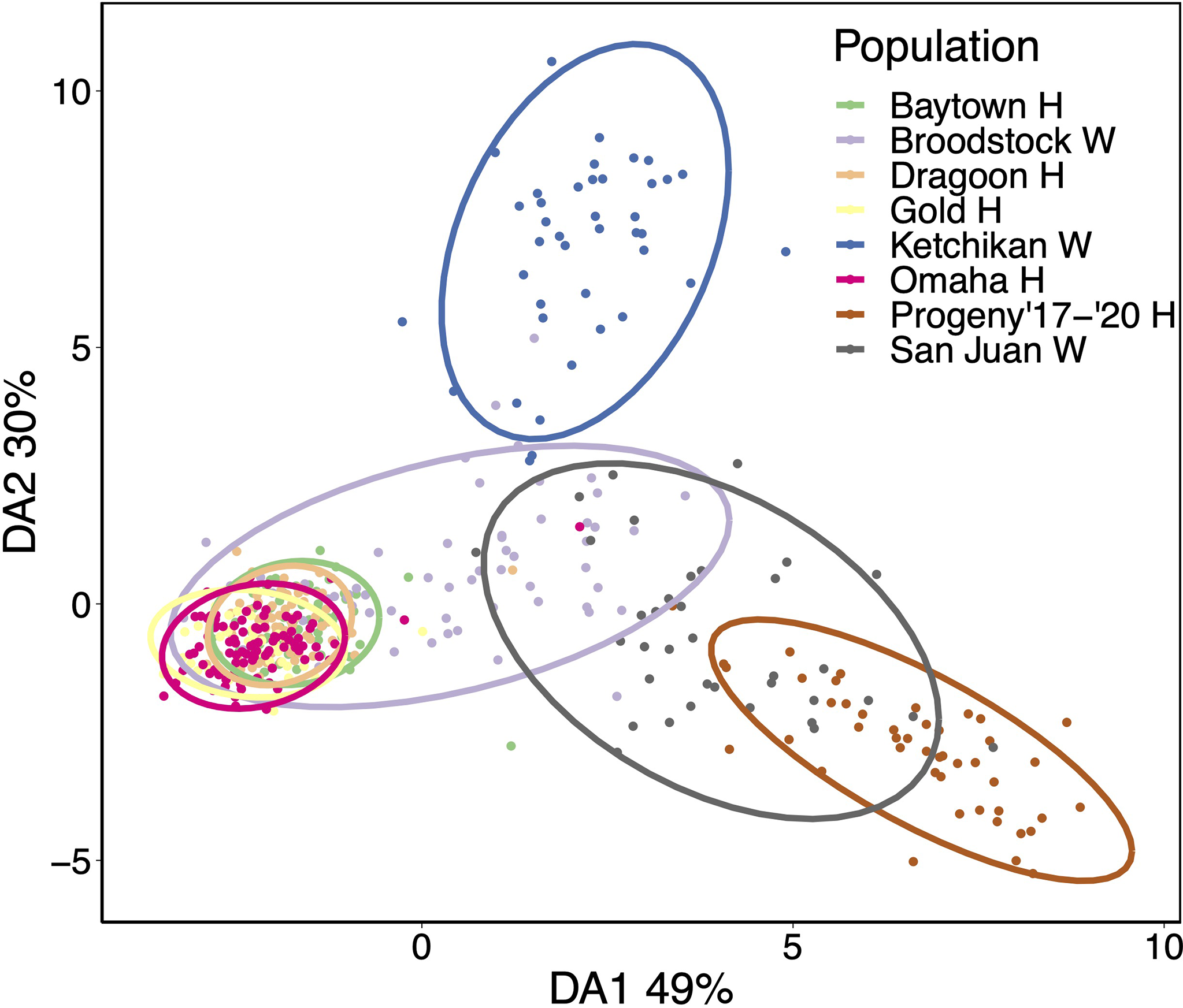
Ordination of samples and groups according to the first two discriminant functions of the discriminant analysis of principal components (DAPC). The percentage of DAPC variance captured by each discriminant axis (DA) is shown in axis labels.
Relatedness
Based on simulated broodstock family relationships from allele frequencies in the SNP dataset, the method of Queller and Goodnight (1989) had the highest correlation coefficient with expected values (0.72) and was chosen for further analyses (Figure S2). The overall classification accuracy of this estimator was 0.59 based on cross-validation of simulated data; overlap of relatedness values for each relationship limits classification accuracy (Figure S3). Parent-offspring relationships were excluded from further analyses because we were primarily interested in relationships among hatchery-bred animals, which were not expected to have parent-offspring relationships because all animals were F1 offspring bred from wild broodstock.
At the four outplant sites, we detected a total of 25 unique broodstock families. Baytown, Dragoon, and Omaha each had 14 broodstock families, while Gold had 10, probably due in part to the much smaller sample size for Gold than for the other three sites (Figure 4). In fact, the ratio of number of broodstock families to number of samples was highest for Gold (0.23), intermediate for Dragoon and Baytown (0.19), and lowest for Omaha (0.15). There was considerable variability in broodstock family size, with one broodstock family represented by 42 individuals across all four sites, while several broodstock families were represented by just one or two individuals.
Figure 4
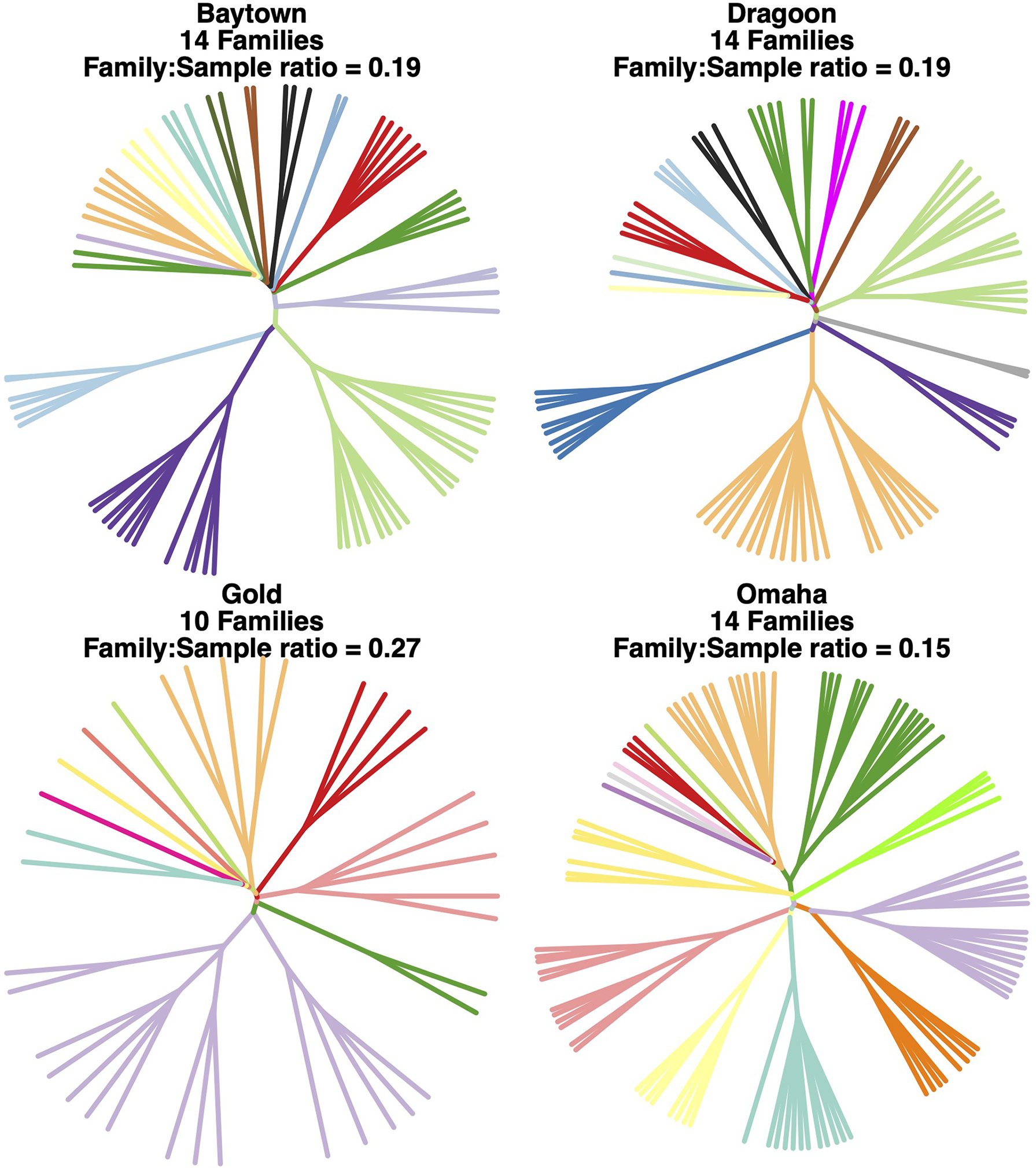
Family trees of hatchery-bred abalone at each of the outplant sites, based on hierarchical clustering of the StAMPP relatedness matrix. Unique broodstock families are represented by unique colors, and each branch represents an individual.
Given that abalone are dioecious broadcast spawners, pairwise relatedness estimates for each group can also be thought of in terms of relationships between potential sperm and egg mate pairs (Figure 5; Table 4). This analysis showed that 86-89% of potential matings at abalone outplant sites would occur between unrelated individuals, followed by 7-9% between half sibs, and 4-6% between full sibs. This is compared to a 25% chance of half-sibling matings and 11% chance of full-sibling matings estimated for an isolated cohort at these four sites based on tag recaptures (Carson et al., 2019). By contrast, 94-95% of matings among Progeny ‘17-’20 H and Ketchikan W would occur between unrelated individuals, with 5% between half sibs, and 100% of matings among Broodstock W and San Juan W would be between unrelated individuals. The presence of half sibs among the Ketchikan W abalone could be due to group synchronous spawning behavior, which has been observed in H. kamtschatkana (Breen and Adkins, 1980).
Figure 5
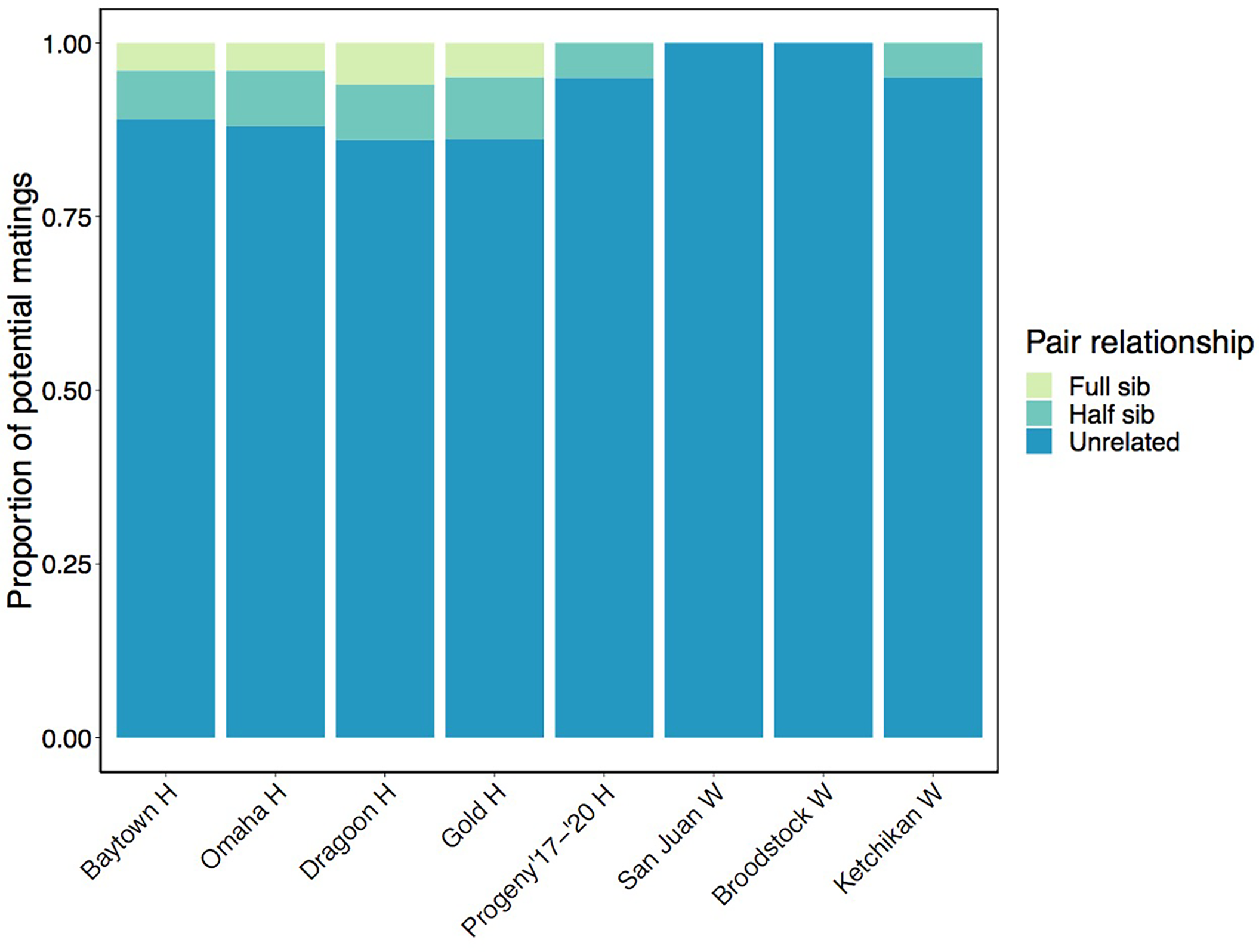
Pairwise relatedness among each group based on the method of Queller and Goodnight (1989), expressed as the proportion of potential matings in each group between full sibs, half sibs, or unrelated individuals.
Table 4
| Site | Baytown H | Omaha H | Dragoon H | Gold H | Progeny ‘17-’20 H | San Juan W | Broodstock W | Ketchikan W |
|---|---|---|---|---|---|---|---|---|
| Baytown H | 0.406 | 0.008 | 0.176 | <0.001 | <0.001 | <0.001 | <0.001 | |
| Omaha H | 0.001 | 0.525 | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Dragoon H | 0.098 | <0.001 | <0.001 | <0.001 | <0.001 | |||
| Gold H | <0.001 | <0.001 | <0.001 | <0.001 | ||||
| Progeny ‘17-’20 H | <0.001 | <0.001 | 0.243 | |||||
| San Juan W | 1 | <0.001 | ||||||
| Broodstock W | <0.001 | |||||||
| Ketchikan W |
Results of Pearson’s Chi-squared tests with Yates’ continuity correction comparing relatedness proportions for each of the populations.
Significant p-values are shown in bold.
Effective Population Size
Each of the abalone outplant sites showed similar estimates of effective population size (Ne), ranging from 26.2 (Dragoon) to 40.4 (Baytown; Table 5). The ratio of Neto estimates of census size (Ne/N) was also comparable at each outplant site, with a low of 0.11 (Dragoon) and a high of 0.18 (Gold). Among the Progeny ‘17-’20 H group, Newas 99.9, more than double that of any of the outplant sites. Meanwhile, Ne was considerably larger in all of the wild abalone groups. The Broodstock W and San Juan W groups both had infinite Ne, which should be interpreted as inconclusive but potentially very large, while Ketchikan W had an Ne of 2306.5.
Table 5
| Population | Ne (95% CI) | N (range) | Ne/N |
|---|---|---|---|
| Baytown H | 40.4 (40.4 - 40.4) | 232 (231-234) | 0.17 |
| Dragoon H | 26.2 (26.2 -26.2) | 235 (233-236) | 0.11 |
| Gold H | 27.9 (27.9 -27.9) | 154 (147-167) | 0.18 |
| Omaha H | 35.0 (34.9 - 35.0) | 289 (280-301) | 0.12 |
| Progeny ‘17-’20 H | 99.9 (99.7 - 100.1) | ||
| Broodstock W | ∞ (∞ - ∞) | ||
| San Juan W | ∞ (∞ - ∞) | ||
| Ketchikan W | 2306.5 (2220.4 - 2399.6) |
Effective population size estimates (Ne) based on allele frequencies of 0.01, as well as census population size estimates (N) and the Ne/N ratio for each population.
Discussion
Our analysis of the H. kamtschatkana restoration program revealed several findings that mirror other studies of abalone restoration and aquaculture in other parts of the world. First, we found that expected heterozygosity among hatchery-bred F1 abalone was only slightly lower relative to wild broodstock. Similarly, no significant loss of heterozygosity among F1 offspring bred from wild broodstock was observed among Australian H. rubra (Evans et al., 2004) or South African H. midae (Evans et al., 2004; Rhode et al., 2012; Rhode et al., 2014; Slabbert et al., 2009). In fact, as in Rhode et al. (2014), we found slightly lower FIS, indicative of a relative heterozygote excess, among hatchery-bred abalone. Although counterintuitive, this is commonly observed among population bottlenecks (in this case, hatchery breeding), where a decrease in the number of alleles is not initially accompanied by a loss of heterozygosity Luikart and Cornuet, 2008. Second, corroborating this effect, we found significantly lower allelic richness among hatchery-bred abalone relative to all wild populations. Loss of allelic richness and rare alleles in cultured abalone is a consequence of a relatively small number of progenitors in hatchery settings and has been widely reported in the literature (Evans et al., 2004; Gaffney et al., 1996; Slabbert et al., 2009; Smith and Conroy, 1992). Third, there was significant genomic divergence of hatchery-bred abalone from wild abalone, although this divergence was less pronounced when comparing hatchery-bred abalone directly to their source broodstock. Genetic divergence between wild and hatchery-raised abalone has also been reported by a number of studies and is likely a consequence of small numbers of breeders, genetic drift, and potentially, artificial selection (Evans et al., 2004; Gaffney et al., 1996; Gruenthal et al., 2014; Rhode et al., 2012; Rhode et al., 2014). Lastly, Ne among hatchery-bred H. kamtschatkana was substantially lower than that of wild abalone. Again, this is a consequence of limited broodstock and broodstock family sizes and is comparable to data reported in other abalone breeding studies Rhode et al., 2012; Rhode et al., 2014).
A key finding, however, is that the most recently produced pre-outplant hatchery progeny (Progeny ‘17-’20 H) had genomic profiles much more similar to wild abalone. This suggests that larger broodstock sizes and resultant broodstock family sizes in recent years may have been effective in increasing allelic richness and Ne and reducing genomic divergence from wild abalone. For example, during outplant years 2009-2017, broodstock family sizes ranged from 5-17 per year, compared to 7-26 broodstock families per year from 2019-2021. However, abalone in the more recent group were sampled prior to outplant, so it is not a direct comparison to the older cohorts that were sampled after several years in the wild. Indeed, it is likely that some of the genomic divergence, reduced allelic richness, and lower Ne among the older outplants is the result of genetic drift due to emigration or mortality in the wild over time. Mark-recapture studies of pinto abalone at these outplant sites indicate that the majority of individuals have little net movement over annual time scales, but some emigration is expected (Carson et al., 2019). Meanwhile, predation may be significant, particularly on small, newly outplanted pinto abalone (Hansen and Gosselin, 2013). Observed 1-year survival rates for abalone at the outplant sites reported herein ranged from 11.4% (Baytown) to 23% (Omaha; Carson et al. (2019), and in the present study, the percentage of original broodstock families observed in 2019 ranged from 21% (Gold) to 45% (Omaha). Taken together, these data suggest that a significant amount of the original genetic diversity at outplant sites has been lost through emigration or mortality. It is encouraging that even with this loss of diversity, however, relatedness analyses indicate that the probability of inbreeding between siblings and half-siblings at outplant sites remains relatively low.
While these results suggest that current hatchery practices alone cannot account for the more favorable genomic profiles among recent pre-outplant progeny compared to older post-outplant progeny, they also provide evidence that current practices are capable of producing genetically diverse progeny in the absence of post-outplant genetic drift. Furthermore, lack of strong differentiation between recent pre-outplant progeny and wild abalone also provides some evidence against significant artificial selection in the hatchery, and suggests that the strong divergence between older post-outplant progeny and wild abalone is more likely due to genetic drift and small broodstock family sizes than to artificial selection. With larger total numbers of abalone now being produced and outplanted in addition to larger broodstock family sizes, current practices may now be better able to overcome the effects of genetic drift. Hatchery operations now include multiple rearing facilities, and since 2009, the number of outplants has increased approximately eightfold.
With this increase in production, however, comes an increased risk of a Ryman-Laikre effect, defined as a reduction of the combined wild/captive-bred Ne by a low captive-bred Ne (Ryman and Laikre, 1991). To avoid this, hatchery practices should aim for the highest possible broodstock sizes, limit variance in broodstock family size, and continually outplant new individuals at existing outplants sites (Ryman and Laikre, 1991; Wang and Ryman, 2001; Waples et al., 2016). Finding sufficient wild abalone to serve as broodstock continues to be among the greatest challenges for pinto abalone restoration in Washington. In 2021, a single pinto abalone was counted in 57 subtidal sea urchin surveys across the San Juan Islands; a basic extrapolation of abalone numbers from these surveys gives a current population on the order of 5,000-10,000 individuals (WA Dept. of Fish and Wildlife, unpublished). With Ne among these wild abalone potentially several times higher than this rough census estimate, wild Ne/N may be very high, providing evidence against a genetic bottleneck despite the severe population decline. This is consistent with earlier results from remnant populations of this species in British Columbia, where Ne was estimated at >350,000 (Withler et al., 2003). Furthermore, we found that Salish Sea pinto abalone appear to have higher Ne than those in Ketchikan, Alaska, where abalone population density is higher and natural recruitment has been observed (Donnellan and Hebert, 2017). This indicates that Washington abalone have the necessary genetic diversity to continue supporting the breeding program. However, perhaps counterintuitively, this situation also poses some challenges. It means that broodstock sizes need to be large in order to avoid a Ryman-Laikre effect, particularly as the captive-bred proportion of the total population increases (Ryman and Laikre, 1991; Hare et al., 2011). The ratio of captive-bred Ne/N to wild Ne/N [β Waples et al. (2016)] can be used to assess risk of a Ryman-Laikre effect, with values below 1 leading to reduced overall Ne (Waples et al., 2016). In this case, with current captive-bred Ne/N ranging from 0.11 to 0.18 and wild Ne/N perhaps much higher, β is likely to be below 1, emphasizing the importance of large broodstocks.
For the future, identifying new broodstock sources will clearly be a priority for the restoration program. Remnant populations exist outside the San Juan Islands in the Strait of Juan de Fuca, and may act as an additional broodstock source within the state. The Strait will also likely receive restoration outplants in the future, so care must be taken in siting those efforts. Currently, the restoration program does not outplant hatchery juveniles near where the remnant wild populations are most prevalent in the San Juan Islands, to avoid the possibility of collecting a hatchery-produced animal as broodstock. Subtidal diver surveys continue to be the gold standard for locating wild abalone, but environmental DNA survey methods are currently being tested and may aid the detection effort (Dimond, unpublished), especially in the Strait of Juan de Fuca where fewer surveys have been conducted. Meanwhile, bringing in broodstock from outside the region may become necessary as wild abalone continue to age out of the population and natural recruitment remains low. Studies are underway to determine range-wide pinto abalone population genetics to help identify optimal populations for assisted gene flow if sufficient wild abalone can longer be found in Washington.
Funding
Pinto abalone restoration in Washington has benefitted from numerous funding sources over the years, including the Benjamin & Margaret Hall Foundation, the Vincent J. Coates Foundation, the Skagit County Marine Resources Committee, Washington Department of Fish and Wildlife, Washington Department of Natural Resources, NOAA Species of Concern Grants, and the SeaDoc Society through the Karen C. Drayer Wildlife Health Center of the School of Veterinary Medicine at the University of California, Davis.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA816092.
Author contributions
JD contributed to study design, analyzed data, and wrote the manuscript. JB conceived the study, contributed to study design, managed hatchery operations and sample collection, and edited the manuscript. HC contributed to study design, analyzed data, and edited the manuscript. MG contributed to study design, analyzed data, and edited the manuscript. CO’B contributed to study design, hatchery operations and sample collection, and edited the manuscript. CS prepared RADseq libraries and managed sequencing. KS contributed to study design, sample collection, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We are grateful for support from students, staff, and volunteers affiliated with the NOAA Mukilteo Research Station, the Kenneth K. Chew Center for Shellfish Research and Restoration, Puget Sound Restoration Fund, SeaDoc Society, Washington Department of Fish and Wildlife, Western Washington University, the University of Washington, Shikat Bay Oysters and the University of Alaska, Fairbanks. These include M. Adkisson, B. Allen, A. Benolkin, J. Davis, P. Dinnell, R. Downes, G. Eckert, C. Friedman, T. Frierson, J. Gaydos, R. Goetz, J. Hester, L. Hillier, E. Jinéte, T. Kimball, G. Parsley, B. Peabody, P. Plesha, P. Pratt, C. Rice, S. Ryan, K. Rosenberger, D. Rothaus, N. Schwarck, B. Sizemore, B. Stevick, A. Thomson, M. Ulrich, B. Vadopalas, J. Walker, J. Watson, and O. Working.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcosc.2022.911218/full#supplementary-material
References
1
AliO. A.O’RourkeS. M.AmishS. J.MeekM. H.LuikartG.JeffresC.et al. (2016). RAD Capture (Rapture): Flexible and Efficient Sequence-Based Genotyping. Genetics202, 389–400. doi: 10.1534/genetics.115.183665
2
BabcockR.KeesingJ. (1999). Fertilization Biology of the Abalone Haliotis Laevigata: Laboratory and Field Studies. Can. J. Fish. Aquat. Sci.56, 1668–1678. doi: 10.1139/f99-106
3
BoumaJ. V.RothausD. P.StrausK. M.VadopalasB.FriedmanC. S. (2012). Low Juvenile Pinto Abalone Haliotis Kamtschatkana Kamtschatkana Abundance in the San Juan Archipelago, Washington State. Trans. Am. Fish. Soc.141, 76–83. doi: 10.1080/00028487.2011.651551
4
BreenP. A.AdkinsB. E. (1980). Spawning in a British Columbia Population of Northern Abalone. Haliotis. kamtschatkana. Veliger.23, 177–179.
5
CarsonH. S.MorinD. J.BoumaJ. V.UlrichM.SizemoreR. (2019). The Survival of Hatchery-Origin Pinto Abalone Haliotis Kamtschatkana Released Into Washington Waters. Aquat. Conserv.: Mar. Freshw. Ecosyst. 29 (3), 424–441. doi: 10.1002/aqc.3004
6
CarsonH. S.UlrichM. (2019). Status Report for the Pinto Abalone in Washington (Olympia: Washington Department of Fish and Wildlife).
7
CattonC. A.StierhoffK. L.Rogers-BennettL. (2016). Population Status Assessment and Restoration Modeling of White Abalone Haliotis Sorenseni in California. J. Shellfish Res.35, 593–599. doi: 10.2983/035.035.0304
8
CookP. A.GordonH. R. (2010). World Abalone Supply, Markets, and Pricing. J. Shellfish Res.29, 569–571. doi: 10.2983/035.029.0303
9
CrowJ. F.KimuraM. (1970). An Introduction to Population Genetics Theory. (eds HarperRow), p. 591. New-York, USA.
10
DeWoodyJ. A.HarderA. M.MathurS.WilloughbyJ. R. (2021). The Long-Standing Significance of Genetic Diversity in Conservation. Mol. Ecol.30, 4147–4154. doi: 10.1111/mec.16051
11
DonnellanM.HebertK. (2017). Pinto Abalone (Haliotis Kamtschatkana Jonas 1845) Surveys in Southern Southeast Alaska 2016 (Alaska Department of Fish and Game, Fishery Data Series No. 17-40, Anchorage).
12
DoC.WaplesR. S.PeelD.MacbethG. M.TillettB. J.OvendenJ. R. (2014). NeEstimator V2: Re-Implementation of Software for the Estimation of Contemporary Effective Population Size (Ne ) From Genetic Data. Mol. Ecol. Resour.14, 209–214. doi: 10.1111/1755-0998.12157
13
EvansB.BartlettJ.SweijdN.CookP.ElliottN. G. (2004). Loss of Genetic Variation at Microsatellite Loci in Hatchery Produced Abalone in Australia (Haliotis Rubra) and South Africa (Haliotis Midae). Aquaculture233, 109–127. doi: 10.1016/j.aquaculture.2003.09.037
14
GaffneyP. M.RubinV. P.HedgecockD.PowersD. A.MorrisG.HerefordL. (1996). Genetic Effects of Artificial Propagation: Signals From Wild and Hatchery Populations of Red Abalone in California. Aquaculture143, 257–266. doi: 10.1016/0044-8486(96)01278-1
15
GoudetJ. (2005). Hierfstat, a Package for R to Compute and Test Hierarchical F-Statistics. Mol. Ecol. Notes5, 184–186. doi: 10.1111/j.1471-8286.2004.00828.x
16
GrantW. S.JasperJ.BekkevoldD.AdkisonM. (2017). Responsible Genetic Approach to Stock Restoration, Sea Ranching and Stock Enhancement of Marine Fishes and Invertebrates. Rev. Fish. Biol. Fish.27, 615–649. doi: 10.1007/s11160-017-9489-7
17
GruenthalK. M.WittingD. A.FordT.NeumanM. J.WilliamsJ. P.PondellaD. J.et al. (2014). Development and Application of Genomic Tools to the Restoration of Green Abalone in Southern California. Conserv. Genet.15, 109–121. doi: 10.1007/s10592-013-0524-5
18
HansenS. C.GosselinL. A. (2013). Do Predators, Handling Stress or Field Acclimation Periods Influence the Survivorship of Hatchery-Reared Abalone Haliotis Kamtschatkana Outplanted Into Natural Habitats? Aquat. Conserv.: Mar. Freshw. Ecosyst.23, 246–253. doi: 10.1002/aqc.2315
19
HareM. P.NunneyL.SchwartzM. K.RuzzanteD. E.BurfordM.WaplesR. S.et al. (2011). Understanding and Estimating Effective Population Size for Practical Application in Marine Species Management. Conserv. Biol.25, 438–449. doi: 10.1111/j.1523-1739.2010.01637.x
20
HobdayA. J.TegnerM. J. (2000). Status Review of White Abalone (Haliotis Sorenseni) Throughout its Range in California and Mexico. (Washington, D.C.: U.S. Department of Commerce; National Oceanic and Atmospheric Administration).
21
JombartT. (2008). Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics24, 1403–1405. doi: 10.1093/bioinformatics/btn129
22
KarpovK.HaakerP.TaniguchiI. (2000). Serial depletion and the collapse of the California abalone (Haliotis spp.) fishery. Workshop on Rebuilding Abalone Stocks in British Columbia.
23
KnausB.J.GrünwaldN.J. (2017). vcfr: A Package to Manipulate and Visualize Variant Call Format Data in R. Mol. Eco. Res., 17 (1), 44–53.
24
KuhnM. (2008). Building Predictive Models in R Using the Caret Package. J. Stat. Softw.28, 1–26. doi: 10.18637/jss.v028.i05
25
LemayM. A.BouldingE. G. (2009). Microsatellite Pedigree Analysis Reveals High Variance in Reproductive Success and Reduced Genetic Diversity in Hatchery-Spawned Northern Abalone. Aquaculture295, 22–29. doi: 10.1016/j.aquaculture.2009.06.029
26
LiH.DurbinR. (2010). Fast and Accurate Long-Read Alignment With Burrows-Wheeler Transform. Bioinformatics26, 589–595. doi: 10.1093/bioinformatics/btp698
27
LiH.HandsakerB.WysokerA.FennellT.RuanJ.HomerN.et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics25, 2078–2079. doi: 10.1093/bioinformatics/btp352
28
LuikartG.CornuetJ.-M. (2008). Empirical Evaluation of a Test for Identifying Recently Bottlenecked Populations From Allele Frequency Data. Conserv. Biol.12, 228–237. doi: 10.1111/j.1523-1739.1998.96388.x
29
MasonbrinkR. E.PurcellC. M.BolesS. E.WhiteheadA.HydeJ. R.SeetharamA. S.et al. (2019). An Annotated Genome for Haliotis Rufescens (Red Abalone) and Resequenced Green, Pink, Pinto, Black, and White Abalone Species. Genome Biol. Evol.11, 431–438. doi: 10.1093/gbe/evz006
30
MasudaR.TsukamotoK. (1998). Stock Enhancement in Japan: Review and Perspective. Bull. Mar. Sci.62 (2), 337–358.
31
NeumanM. J.WangS.BuschS.FriedmanC.GruenthalK.GustafsonR.et al. (2018). A Status Review of Pinto Abalone (Haliotis Kamtschatkana ) Along the West Coastof North America: Interpreting Trends, Addressing Uncertainty, and Assessing Risk for a Wide-Ranging Marine Invertebrate. J. Shellfish Res.37, 869–910. doi: 10.2983/035.037.0415
32
PembletonL. W.CoganN. O. I.ForsterJ. W. (2013). StAMPP: An R Package for Calculation of Genetic Differentiation and Structure of Mixed-Ploidy Level Populations. Mol. Ecol. Resour.13, 946–952. doi: 10.1111/1755-0998.12129
33
PewJ.MuirP. H.WangJ.FrasierT. R. (2015). Related: An R Package for Analysing Pairwise Relatedness From Codominant Molecular Markers. Mol. Ecol. Resour.15, 557–561. doi: 10.1111/1755-0998.12323
34
QuellerD. C.GoodnightK. F. (1989). Estimating Relatedness Using Genetic Markers. Evolution43, 258–275. doi: 10.1111/j.1558-5646.1989.tb04226.x
35
RhodeC.HeppleJ.JansenS.DavisT.VervalleJ.Bester-van der MerweA. E.et al. (2012). A Population Genetic Analysis of Abalone Domestication Events in South Africa: Implications for the Management of the Abalone Resource. Aquaculture356–357, 235–242. doi: 10.1016/j.aquaculture.2012.05.012
36
RhodeC.MadunaS. N.Roodt-WildingR.Bester-van der MerweA. E. (2014). Comparison of Population Genetic Estimates Amongst Wild, F1 and F2 Cultured Abalone (Haliotis Midae). Anim. Genet.45, 456–459. doi: 10.1111/age.12142
37
RobertsR. D.KeysE. F.PrendevilleG.PilditchC. A. (2007). Viability of Abalone (Haliotis Iris) Stock Enhancement by Release of Hatchery-Reared Seed in Marlborough, New Zealand. J. Shellfish Res.26, 697–703. doi: 10.2983/0730-8000(2007)26[697:VOAHIS]2.0.CO;2
38
RochetteN. C.Rivera-ColónA. G.CatchenJ. M. (2019). Stacks 2: Analytical Methods for Paired-End Sequencing Improve RADseq-Based Population Genomics. Mol. Ecol.28, 4737–4754. doi: 10.1111/mec.15253
39
Rogers-BennettL.AllenB. L.RothausD. P. (2011). Status and Habitat Associations of the Threatened Northern Abalone: Importance of Kelp and Coralline Algae. Aquat. Conserv.: Mar. Freshw. Ecosyst.21, 573–581. doi: 10.1002/aqc.1218
40
Rogers-BennettL.AquilinoK. M.CattonC. A.KawanaS. K.WalkerB. J.AshlockL. W.et al. (2016). Implementing a Restoration Program for the Endangered White Abalone (Haliotis Sorenseni ) in California. J. Shellfish Res.35, 611–618. doi: 10.2983/035.035.0306
41
RothausD. P.VadopalasB.FriedmanC. S. (2008). Precipitous Declines in Pinto Abalone (Haliotis Kamtschatkana Kamtschatkana) Abundance in the San Juan Archipelago, Washington, USA, Despite Statewide Fishery Closure. Can. J. Fish. Aquat. Sci.65, 2703–2711. doi: 10.1139/F08-168
42
RymanN.LaikreL. (1991). Effects of Supportive Breeding on the Genetically Effective Population Size. Conserv. Biol.5, 325–329. doi: 10.1111/j.1523-1739.1991.tb00144.x
43
SekinoM.SaidoT.FujitaT.KobayashiT.TakamiH. (2005). Microsatellite DNA Markers of Ezo Abalone (Haliotis Discus Hannai): A Preliminary Assessment of Natural Populations Sampled From Heavily Stocked Areas. Aquaculture243, 33–47. doi: 10.1016/j.aquaculture.2004.10.013
44
SlabbertR.BesterA. E.D’AmatoM. E. (2009). Analyses of Genetic Diversity and Parentage Within a South African Hatchery of the Abalone Haliotis Midae Linnaeus Using Microsatellite Markers. J. Shellfish Res.28, 369–375. doi: 10.2983/035.028.0220
45
SmithP. J.ConroyA. M. (1992). Loss of Genetic Variation in Hatchery-Produced Abalone, Haliotis Iris. N. Z. J. Mar. Freshw. Res.26, 81–85. doi: 10.1080/00288330.1992.9516503
46
StierhoffK. L.NeumanM.ButlerJ. L. (2012). On the Road to Extinction? Population Declines of the Endangered White Abalone, Haliotis Sorenseni. Biol. Conserv.152, 46–52. doi: 10.1016/j.biocon.2012.03.013
47
VanBlaricomGNeumanMButlerJ. L.De VogelaereA.GustafsonR. G.MobleyC. (2009). Status Review Report for Black Abalone (National Marine Fisheries Service, Southwest Region. Long Beach, CA). 1–135
48
WangJ.RymanN. (2001). Genetic Effects of Multiple Generations of Supportive Breeding. Conserv. Biol.15, 1619–1631. doi: 10.1046/j.1523-1739.2001.00173.x
49
WaplesR. S.HindarK.KarlssonS.HardJ. J. (2016). Evaluating the Ryman-Laikre Effect for Marine Stock Enhancement and Aquaculture. Curr. Zool.62, 617–627. doi: 10.1093/cz/zow060
50
WeirB. S.CockerhamC. C. (1984). Estimating F-Statistics for the Analysis of Population Structure. Evolution381358 . doi: 10.2307/2408641
51
WilliY.Van BuskirkJ.HoffmannA. A. (2006). Limits to the Adaptive Potential of Small Populations. Annu. Rev. Ecol. Evol. Syst.37, 433–458. doi: 10.1146/annurev.ecolsys.37.091305.110145
52
WithlerR. E.CampbellA.LiS.BrouwerD.SupernaultK. J.MillerK. (2003). Implications of High Levels of Genetic Diversity and Weak Population Structure for the Rebuilding of Northern Abalone in British Columbia, Canada. J. Shellfish Res.22, 839–847.
53
ZhangZ. (2008). A Simulation Study of Abalone Fertilization. J. Shellfish Res.27, 857–864. doi: 10.2983/0730-8000(2008)27[857:ASSOAF]2.0.CO;2
Summary
Keywords
abalone, RADseq, Haliotis kamtschatkana, restoration aquaculture, captive breeding, effective population size
Citation
Dimond JL, Bouma JV, Carson HS, Gavery MR, O’Brien C, Simchick C and Sowul K (2022) Efficacy of Endangered Pinto Abalone (Haliotis kamtschatkana) Stock Restoration in the Southern Salish Sea From a Genomic Perspective. Front. Conserv. Sci. 3:911218. doi: 10.3389/fcosc.2022.911218
Received
02 April 2022
Accepted
10 May 2022
Published
09 June 2022
Volume
3 - 2022
Edited by
Om Rajora, University of New Brunswick Fredericton, Canada
Reviewed by
Laura Rogers-Bennett, University of California System, United States; Hua Wu, Central China Normal University, China
Updates
Copyright
© 2022 Dimond, Bouma, Carson, Gavery, O’Brien, Simchick and Sowul.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James L. Dimond, dimondj2@wwu.edu
This article was submitted to Conservation Genomics, a section of the journal Frontiers in Conservation Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.