 Ludwig Triest
Ludwig Triest Jasper Dierick
Jasper Dierick Thi Thuy Hang Phan
Thi Thuy Hang Phan Quang Doc Luong
Quang Doc Luong Nguyen Quang Huy
Nguyen Quang Huy Tim Sierens
Tim Sierens
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Conserv. Sci. , 17 September 2021
Sec. Conservation Genetics and Genomics
Volume 2 - 2021 | https://doi.org/10.3389/fcosc.2021.723376
This article is part of the Research Topic Integrated Phylogeography and Population Genomics of Coastal Ecosystems View all 8 articles
Lagoonal environments exhibit high levels of instability depending on hydrological, climatic and ecological factors, thereby influencing the distribution and structure of submerged plant communities. Conditions typically fluctuate widely due to the interaction of freshwater from rivers with saltwater from the sea, as well as from aquaculture activities that together influence submerged hydrophyte community spatial and temporal variability depending on plant survival strategies. Ruppia species feature either underwater pollination mediated by an air bubble or by the release of pollen floating at the water surface, the former promoting self-pollination. Tropical Asian Ruppia brevipedunculata Yu and den Hartog was assumed to pollinate below the water surface and identified as a separate lineage among selfed Ruppia taxa. We used nine nuclear microsatellites to estimate inbreeding levels and connectivity of R. brevipedunculata within a large SE Asian lagoon complex. Ruppia brevipedunculata meadows were strongly inbred as could be derived from the many monomorphic or totally fixed loci for unique alleles in different parts of the lagoon, which appears consistent with selfing behavior. Those from aquaculture ponds were highly inbred (FIS = 0.620), though less than open lagoon sites that showed nearly total inbreeding (FIS = 0.942). Ruppia brevipedunculata from two major lagoon parts were highly differentiated with spatially structured gene pools and a strong barrier between parts of the lagoon over a 30 km distance. Migration-n analysis indicated unidirectional though limited gene flow and following potential hydrological connectivity. Overall, private alleles under homozygote conditions explained a stronger genetic differentiation of populations situated inside aquaculture ponds than of open lagoon populations. Kinship values were only relevant up to 5 km distance in the open lagoon. Within a confined area of aquaculture ponds featuring dense vegetation in stagnant water, there would be opportunity for mixed pollination, thereby explaining the higher diversity of unique multilocus genotypes of aquaculture pond habitats. Low connectivity prevents gene pools to homogenize however promoted sites with private alleles across the lagoon. Complex hydrodynamic systems and human-made habitats enclosed by physical structures impose barriers for propagule dispersal though may create refugia and contribute to conserving regional genetic diversity.
Physical alterations of the environment due to coastal development (Waycott et al., 2009; Grech et al., 2011; Unsworth et al., 2019), run-off from urban, agricultural and industrial areas (Waycott et al., 2009; Grech et al., 2011; Unsworth et al., 2019) together with ocean warming (Duarte et al., 2018; Kendrick et al., 2019) are the main stressors resulting in worldwide seagrass decline. Ruppia species appear to be more resistant toward environmental perturbations because of their high tolerance toward fluctuations in salinity, temperature and turbidity (Verhoeven, 1979; Mannino et al., 2015), their high colonization potential due the capability of self-pollination, large seed sets (Kendrick et al., 2012), fast rhizome growth rates (Marbà and Duarte, 1998), and the possibility of recovery after disturbance from seed banks (Strazisar et al., 2013). Consequently, Ruppia may replace more sensitive seagrass species and dominate in disturbed habitats (Cho et al., 2009). Nevertheless, with increasing coastal development, landform modifications and environmental perturbations, Ruppia populations may be particularly vulnerable in the future because of their proximity to human-induced land use and thus high exposure to direct and indirect effects of physical disturbance, sedimentation and eutrophication (Waycott et al., 2009; Quiros et al., 2017; Dierick et al., 2021).
The cosmopolitan genus Ruppia, found from northern to tropical regions, consists of submerged annual or perennial plants which occur in a broad array of coastal habitats ranging from brackish to hypersaline water (Short et al., 2007). Ruppia dominated seagrass meadows, which are often monospecific in saltwater lagoons of temperate regions or combined with other submerged macrophytes and seagrasses in brackish water lagoons in the tropics (Verhoeven, 1979), are important habitats for fish, invertebrates and birds and thus essential for the functioning of coastal ecosystems and related ecosystem services (Verhoeven, 1979; Triest et al., 2018a). Ruppia species are monoecious plants capable of both sexual reproduction by bisexual flowers, sometimes regarded as unisexual flowers which occur in pairs on a peduncle, as by asexual reproduction involving the formation of new shoots following rhizome growth (Verhoeven, 1979; den Hartog and Kuo, 2006). Pollination in Ruppia is characterized by hydrophily with either the release of pollen underwater in an air bubble or on the water surface (Cox and Knox, 1989; Kaul, 1993; Lacroix and Kemp, 1997; Yu and den Hartog, 2014). Flexibility in pollen release and capture location has been observed in R. maritima where the location depends on local environmental conditions and the time of pollen maturation (Lacroix and Kemp, 1997; Taylor et al., 2020). Ruppia fruits, which contain negatively buoyant seeds, sink almost immediately to the bottom when ripe and detached, and contribute to local seed banks which most likely have an important role in the yearly replenishment of Ruppia populations (Strazisar et al., 2013; Triest and Sierens, 2013; Mannino et al., 2015). The seed dispersal capacity of Ruppia species is therefore low with the most important dispersal vectors being currents, wave action and tidal flow, though birds can additionally play a role in propagule dispersal (Les et al., 2003; Ito et al., 2010; Triest and Sierens, 2013). In addition, the dispersal of seeds by rafting on detached floating reproductive shoots and fragments is considered to be an important dispersal mechanism especially for long-distance transport in Ruppia and may promote connectivity among distant populations (Orth et al., 1994; Källström et al., 2008; Triest et al., 2018b). Most population genetic studies focused on Ruppia species of Europe, more specifically in the Mediterranean, and recorded low levels of genetic diversity and strong inbreeding in R. maritima L. populations due to high levels of self-fertilization (Triest and Sierens, 2015). Consequently, selfing, which is promoted by bisexual flowers and the location of pollen release and capture, is considered to be the predominant reproduction strategy in those Ruppia species closely related to R. maritima (Triest et al., 2018a) though an incomplete protogyny that facilitates delayed selfing increases the potential for outcrossing before self-pollination can occur (Goodwillie and Weber, 2018; Taylor et al., 2020). Nevertheless, inter- and intraspecific variation in Ruppia has been shown in the contribution of self-fertilization, biparental inbreeding and outcrossing for population growth and sustainability (Triest and Sierens, 2015; Triest et al., 2018a; Beirinckx et al., 2020).
High phenotypic plasticity to cope with fluctuations in salinity, water level, temperature and irradiance (Verhoeven, 1979; Mannino et al., 2015), together with recurrent hybridizations (Beirinckx et al., 2020), historical misinterpretations and local, incomplete descriptions have complicated Ruppia genus taxonomy and phylogeny (Yu and den Hartog, 2014; den Hartog and Triest, 2020). As a result, Ruppia was thought to consist of only two cosmopolitan species (R. maritima and R. spiralis L. ex Dum.—the latter often cited under R. cirrhosa (Petagna) Grande) whose species range sizes were overestimated (Yu and den Hartog, 2014; den Hartog and Triest, 2020). Yu and den Hartog (2014) described two new (sub-)tropical species in Asia (China) using extensive morphological and molecular features (Yu et al., 2020) i.e., R. brevipedunculata Yu and den Hartog and R. sinensis Yu and den Hartog, which were previously considered as R. maritima and R. cirrhosa, respectively. The South East Asian R. brevipedunculata is found in shallow lagoons up to a depth of 1 m where it can grow as an annual together with other Ruppia, seagrass or aquatic plant species. It can be distinguished from related Ruppia species by its very short peduncle which restricts pollen release and capture under the water surface (Yu and den Hartog, 2014). Because of the sexual traits of R. brevipedunculata which promote self-pollination (i.e., bisexual flowers, short peduncle), selfing is expected to be an essential strategy for the maintenance of R. brevipedunculata meadows. We hypothesize that this strategy will result in reduced levels of genetic diversity and strong genetic differentiation among populations.
In this study, we determined the gene diversity and genetic structure within and among R. brevipedunculata meadows to assess the contribution of self-fertilization and inbreeding in a tropical aquaculture-dominated environment. We specifically tested for (1) evidence of selfing as mating system, and (2) connectivity levels and directionality between sites over various distances. Therefore, we developed nuclear microsatellites for the species and studied 12 sites in two distinct though hydrologically connected parts of a tropical lagoon complex (Hue, Central Viet Nam). In addition, we aim to test for a local spatial structure driven by enclosed aquaculture ponds. The consequence of a selfing plant mating system and the potential role of hydrodynamics in lagoons and aquaculture ponds is discussed in a conservation and coastal landscape context.
Ruppia brevipedunculata plants were collected in 12 sites of the Hue lagoon complex, of which four sites in Tam Giang (TG) and eight sites of the Cau Hai (CH) lagoon (Table 1, Figure 1A). In each site, depending on the number of clumps we collected up to 24 individual shoots along a transect at distance intervals of more than 2 m, thereby putatively avoiding duplicates of a same individual. Individual clumps develop seasonally and were usually smaller than 1 m diameter, sometimes intermingled with other species, namely Najas malesiana Wild (often reported as Najas indica (Willd.) Cham., occasionally with Halophila beccarii Aschers. or Halophila ovalis (R.Br.) Hook.f. We sampled 230 Ruppia individual shoots that were dried on silica gel. The identity of this collection was previously checked on a single sample with cpDNA haplotypes (intron trnH-psbA) (Triest et al., 2018a), ITS sequences corresponding to the ITS-A variant (Triest and Sierens, 2014) and with nuclear microsatellites (Triest et al., 2018a). In the largest lagoon of South East Asia situated near Hue in Central Viet Nam, two main and distant parts of the Hue lagoon were sampled (Table 1, Figure 1A). The maximum geographical distance between sites and among both lagoons was 45 km whereas the distance between sites within a lagoon ranged from 0.94 to 12.6 km for TG and from 0.71 to 6.9 km for CH. All sites of the Hue lagoon were brackish to saltwater (Conductivity from 21.6 to 38.8 mS/cm; Salinity from 13.1 to 24.8 mg/l). The TG lagoon was most impacted by intensive aquaculture activities along shallowest borders (otherwise deep and less suitable habitats for submerged macrophytes) and comprised monospecific R. brevipedunculata vegetations inside the shrimp ponds with turbid water (Table 1). The so-called high-tide aquaculture ponds were surrounded by an elevated earth wall and a narrow and temporary inlet/outlet. The CH lagoon is a more open system of very shallow (<2 m deep) clear water connected to the sea though influenced by freshwater river input and a brackish water connection (Thuy Tu) from major aquaculture areas of the TG lagoon. Total phosphorus ranged from (0.02-) 0.24 to 0.96 mg/l in TG and from 0.03 to 0.49 mg/l in CH (Table 1). Using QGIS (v.3.10.3) (QGIS Development Team, 2021) we constructed a digital elevation model (DEM) map of the sampling area (Figures 2A,B). The topography of the surrounding area (<5 km) around both lagoons is characterized by lowlands never exceeding 50 m height except for Bạch Mã national park which borders the CH lagoon in the South and South-East. We mapped the water flow direction in both the high tide (Figure 2A) and low tide (Figure 2B) during the dry season in the lagoon which was adapted from Tran (2005) who determined water flow directionality and velocity by the combination of direct measurements and modeling based on topographic maps. During the dry season at high tide, seawater enters the lagoon complex from the inlet in CH (Tu Hien) and in TG (Thuan An) with the latter generating a North-West current toward TG and a South-East current toward the narrow zone which connects TG and CH (Thuy Tu) (Figure 2A) (Nguyen Tien Lam, 2002; Tran, 2005). In contrast, the Perfume river and other small rivers discharges water in the Tam Giang-Cau Hai lagoon complex during low tide in the dry season and during the rainy season where water travels through the narrow connection from TG to CH at higher velocities (>0.200 m/s) and exits the lagoon complex at Tu Hien in TG and Thuan An in CH (Figure 2B) (Nguyen Tien Lam, 2002; Tran, 2005).
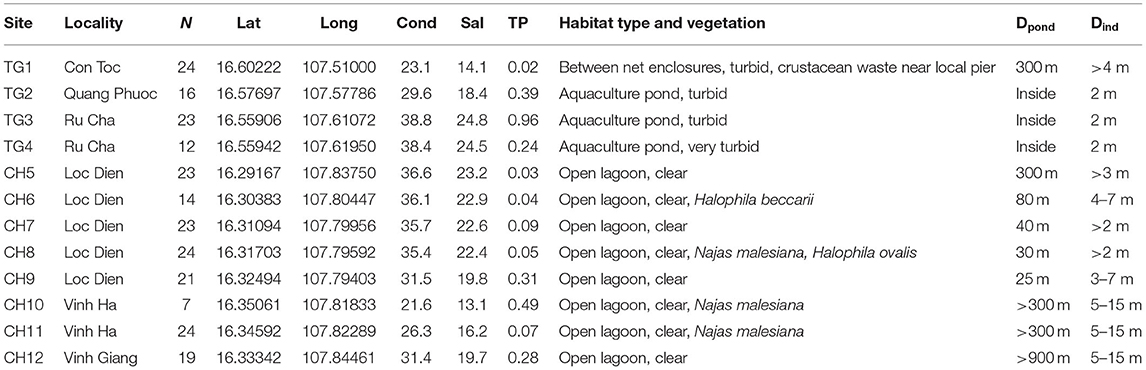
Table 1. Characteristics of 12 sites in the Tam Giang (TG)-Cau Hai (CH) lagoon system (Viet Nam) with N: number of samples; decimal coordinates; conductivity (mS/cm); salinity (‰); total phosphorus (TP in mg/l); habitat features; Dpond: distance to nearest aquaculture pond (m) and Dind: distance interval between individual samples (m).
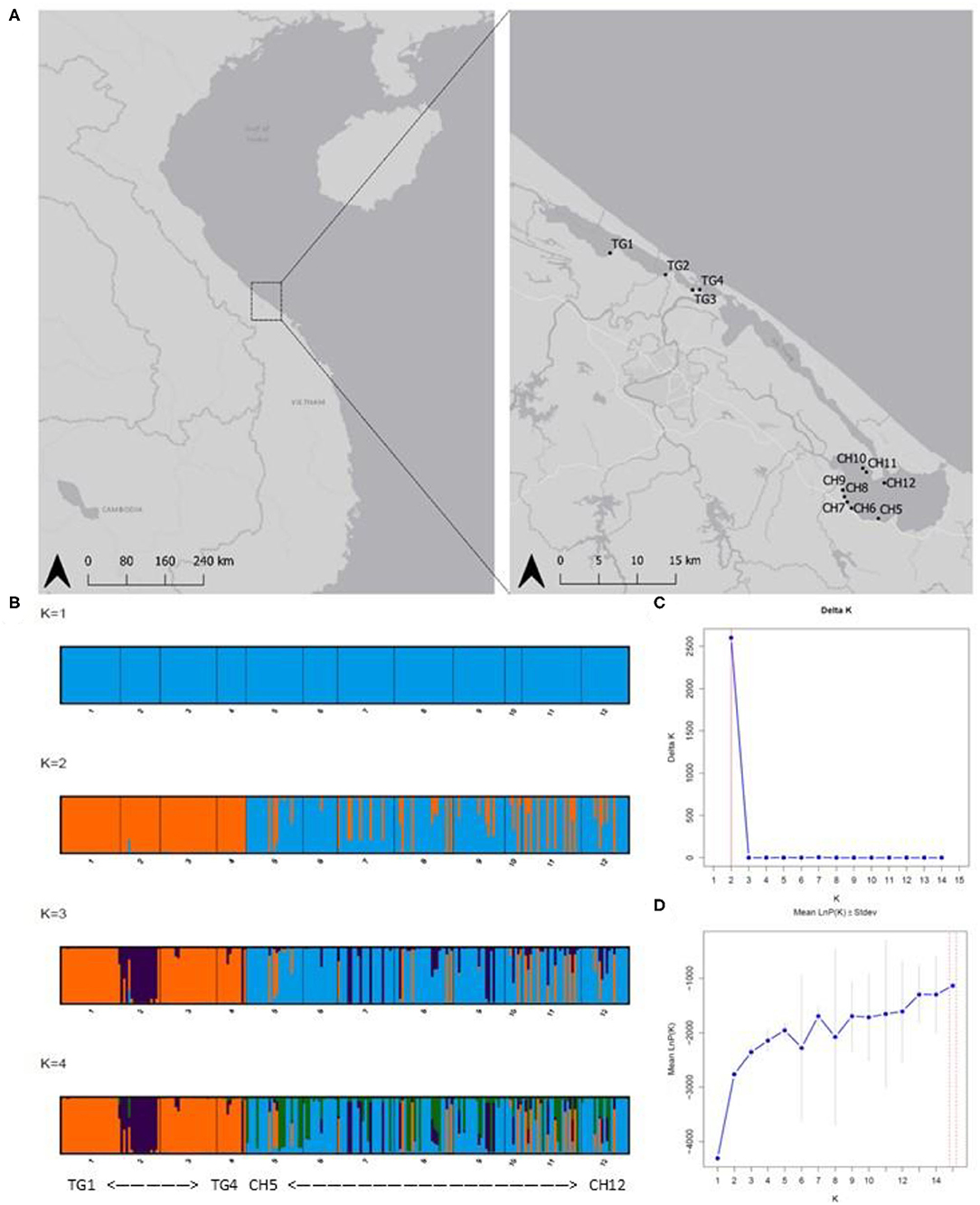
Figure 1. (A) Sampled Ruppia brevipedunculata sites in the Tam Giang (TG)-Cau Hai (CH) lagoon system in Central Viet Nam. Map generated using the Quantum Geographical Information System, version 3.10.3 (QGIS). Basemap source: QGIS. (B) Barplots of Baysian clustering analysis (STRUCTURE from K = 1 to K = 4); (C) Delta K indicating K = 2 clusters; (D) LnP(K) graphs. Abbreviations of sample locations are denoted in Table 1.
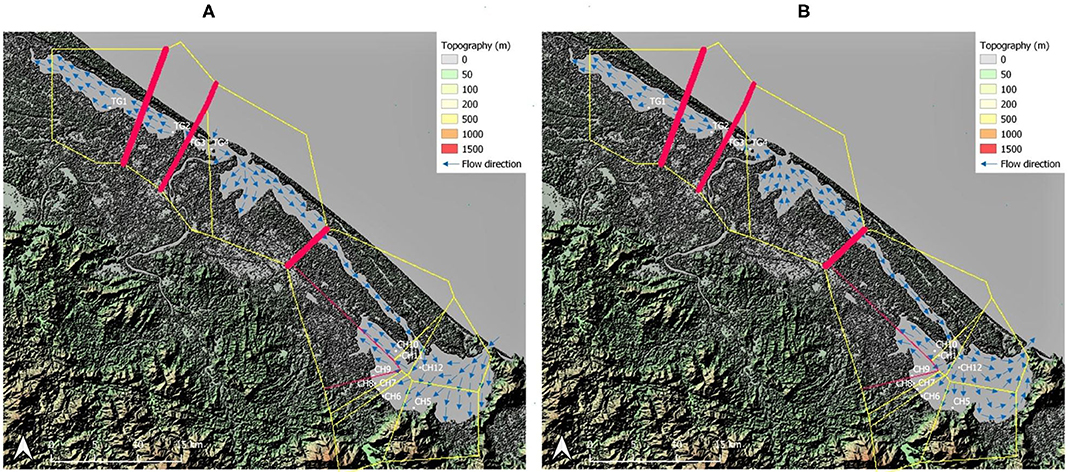
Figure 2. Barrier analysis with thickest red lines indicating strongest differentiation between neighboring sampled sites. Topographic map of the sampling area including water flow direction adapted from Tran (2005) within the Tam Giang (TG)-Cau Hai (CH) lagoon system. (A) Flow direction during high tide in dry season; (B) Flow direction during low tide in dry season and during the rainy season. Map generated using the Quantum Geographical Information System, version 3.10.3 (QGIS).
Genomic DNA was extracted from leaf tissue using the E.Z.N.A SP plant DNA Mini Kit (Omega bio-tek, Norcross, GA, USA) as in Triest and Sierens (2010). Microsatellite loci were developed and isolated from a microsatellite-enriched genomic library following an enrichment procedure with Dynabeads. We used RsaI and SspI as restriction enzymes and motifs AG, TG, GATA and GACA. Out of 96 sequences, 39 were identified as potential for designing primers with Primer3 (http://primer3.wi.mit.edu/). From these 39 primer pairs, the purity and quantity of the DNA were determined using a Nanodrop one Spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). An Illumina paired-end library was constructed and sequenced using the Illumina HiSeq platform at Macrogen (Seoul, Republic of Korea). SSR_pipeline (Miller et al., 2013) was used to identify microsatellites and design primers. Out of 18.2 million 100 bp paired-end reads, 2.3 million pairs were successfully joined by the module joinseqs. The module SSR_search found 1547 dinucleotide SSRs with at least 14 repeats, 4,602 trinculeotide SSRs with at least 12 repeats and 3,776 tetranucleotide SSRs with at least 10 repeats. Batchprimer 3 (You et al., 2008) was used for designing primers and 48 primer-pairs were selected for synthesis based on fragment length and number of repeats. We selected nine polymorphic loci presenting di- and trinucleotide repeats flanked by more than 10 nucleotides between primers region and motifs (Supplementary Table 1). Motifs were repeated 9–23 times within allele lengths of 103–189 bp. Using Multiplex Manager (Holleley and Geerts, 2009), one single multiplex reaction of nine amplifiable primer pairs with four different dye-labeled primers (6FAM/VIC/NED/PET) was designed.
Microsatellite amplification of all individuals was done using the abovementioned 9 loci. Multiplex polymerase chain reaction (PCR) conditions for each PCR reaction were: 6.25 μl master mix (Qiagen multiplex PCR kit master mix), 1.25 μl primer mix, 2.5μl H2O and 2.5μl of genomic DNA, making a total volume of 12.5 μl. PCR was performed in a thermal cycler (MJ research PTC-200 and Bio-Rad MyCycler) with the following conditions: an initial denaturation of 95°C for 15 min followed by 35 cycles of 30 s denaturation at 95°C, 90 s annealing at 57°C and 80 s elongation at 72°C followed by a final extension of 30 min at 60°C. PCR products were run on an ABI3730XL sequencer (Macrogen, Seoul, Korea) and fragments were analyzed with GeneMarker (v.2.60) (Holland and Parson, 2011).
A linkage test between all pairs of loci (1,000 permutations) was done across all populations to verify any genotypic disequilibrium at the 0.001 level using FSTAT (v.2.9.3) (Goudet, 2001) MICRO-CHECKER (Van Oosterhout et al., 2004) was used to verify data quality, potential null alleles, scoring errors or large allele dropout. This test, however, does not consider the effect of inbreeding situations and therefore INEST 2.2 (Chybicki and Burczyk, 2009) was applied as a Bayesian approach for estimating both null alleles and inbreeding simultaneously. Three parameters were used for the model comparison: n, null alleles; f, inbreeding; b, genotyping failure. With these three parameters, seven models are composed: f, n, b, nf, nb, bf, nfb (e.g., nf means the model includes null alleles and inbreeding but does not include genotyping failures). The models were run with 10,000 burn-ins and 50,000 cycles for each site. Summary values of the Deviance Information Criterion (DIC) were obtained for comparison of seven models to (1) verify whether several zero values scored could be explained from the presence of null alleles and (2) explain the large homogeneity of multilocus genotypes encountered. Post-processing was done for each sample on the full model “nbf” from the marginal posterior distribution of hyper-parameters, namely the empirical average inbreeding coefficient Avg(Fi) with 95% highest posterior density interval (HPD low—HPD high).
We calculated the number of multilocus genotypes (MLG's) which are shared among individuals using GenAlEx (v.6.5) (Peakall and Smouse, 2012). In addition, we determined the number and identity of shared MLG's among sites and visually represented the shared MLG's within sites by bar-charts using Poppr (Kamvar et al., 2014). To assess if shared MLG's are the result of an asexual reproduction event in the past or due to high levels of inbreeding, we calculated the probability of occurrence for each MLG (Pgen_FIS) and the probability for each identical pair within a population that they have arisen from distinct sexual reproduction events (psex_FIS) taking account of departures from H-W equilibrium using RClone (Bailleul et al., 2016). Moreover, we calculated and tested the significance of the aggregation index (Ac), the probability that closest sampled individuals have identical MLG's relative to randomly drawn pairs in a population.
The probability of identity for a combination of 9 loci was estimated for the full data set of both lagoons using GenAlex. Deviations from Hardy-Weinberg equilibrium per locus and per population were tested using a Chi-Square test (GenAlex). At site and lagoon level, we estimated the mean number of alleles (NA), effective number of alleles (Ae), observed heterozygosity (Ho), unbiased expected heterozygosity (uHe) corrected as 2N/(2N-1), number of monomorphic and fixed loci (i.e., loci with 1 or more alleles, but lacking any heterozygote conditions) using GenAlex. Allelic richness (Ar) for site and lagoon level, overall FST and population inbreeding coefficient (FIS) with p-values after randomization tests (1/1,000) were obtained from FSTAT. We tested for recent bottlenecks in each site under the two-phase model (TPM) with 95% single-step mutations and 5% multiple-step mutations (Wilcoxon's test 1-tailed) using BOTTLENECK 1.2.02 (Piry et al., 1999). Selfing rate (S) in TG and CH were estimated assuming a mixed mating model using SPAGeDi (v.1.5a) (Hardy and Vekemans, 2002). We explored the correlation between environmental features (conductivity, salinity, TP, distance to nearest aquaculture ponds) of 12 sites and their genetic variables (allelic richness, gene diversity, number of monomorphic or fixed loci, FIS).
Genetic structure was assessed with a three-level analysis of molecular variance (AMOVA) (Michalakis and Excoffier, 1996) using hierarchical fixation indices for two regions, 12 sites and 230 individuals (GenAlex) giving FRT and the standardized F'RT between two lagoons. Pairwise FST-values and standardized F'ST-values were calculated between all pairs of sites and FST-values were tested for significant differentiation using 999 permutations. Isolation-by-distance was tested for pairwise FST of sites and their geographical distances considering straight flight distances between sites in a Mantel test using 1,000 randomizations (Mantel, 1967) with GenAlex. Additionally, pairwise FST values were tested for five distance classes (1,000 permutations) and regressed over full range (slope b tested with 1,000 permutations) using SPAGeDi. At the Hue lagoon scale, relative to the whole sample, we estimated Kinship values (FIJ according to (Loiselle et al., 1995) of individuals between sites and their spatial autocorrelation. Five distance classes (up to 2, 5, 20, 35, 45 km) were considered and represented an equal number of individual comparisons for each class, using 1,000 randomizations (SPAGeDi).
With the R-package Adegenet (Jombart, 2008; R Core Team, 2021) we performed a Discriminant Analysis of Principal Components (DAPC). We used the function “find.clusters” as a clustering algorithm to infer the genetic structure on both lagoon (with K = 2 clusters) as site level (with K = 4 clusters). In addition, we assigned individuals to 12 clusters and compared the membership of the newly assigned clusters to our 12 predefined sites using the function “assignplot.” A Bayesian clustering method (Pritchard et al., 2000) was carried out using STRUCTURE version 2.3. We tested K in 20 independent runs from 1 to 15 (50,000 burn-in and 500,000 Markov chain Monte Carlo replicates in each run), without using sampling location as a prior. Runs were carried out assuming admixture and an independent model of allele frequencies. The optimal K was inferred with the ΔK statistic (Evanno et al., 2005) and LnPK using Structure Harvester (Earl and vonHoldt, 2012) calculated with StructureSelector (Li and Liu, 2018). To determine zones of sharp genetic changes across the lagoon, the geographic coordinates and nine FST data matrices of the individual loci were analyzed with the Monmonier's algorithm, implemented in BARRIER 2.2 software (Manni et al., 2004) and allowed for two barriers per locus. Three hypotheses to estimate gene flow were tested with Migrate-n (Beerli, 2006; Beerli and Palczewski, 2010) from the mutation-scaled population sizes (Theta) and immigration rates (M) assuming a constant population size through time. The Brownian model was tested locus by locus along with the product of all distributions of all loci. Uniform prior distribution settings (min, max, delta) were as follows for Theta = 0, 20, 2 and for M = 0, 20, 2. The number of recorded steps was 106 at a sampling frequency of 104 after an initial burn-in. The effective number of immigrants per generation (Nem) was calculated as [Theta × M]/4. More precisely, we considered two pooled R. brevipedunculata populations of Tam Giang and Cau Hai lagoons to test for uni- and bidirectional migration models. The Brownian motion mutation model within each case was adopted for a subsample of 35 individuals of each lagoon part, following the abovementioned settings, computing two replicate chains (with different seed), and using the Bezier thermodynamic integration (Beerli and Palczewski, 2010) for calculation of the Bayes factors from marginal likelihoods giving model probabilities. To calculate the relative migration matrix between the TG and CH lagoon and between the 12 sites, an alternative method divMigrate-online (Sundqvist et al., 2016) was used for the parameter Nm (eqn. 12 from Alcala et al., 2014) considering 1,000 bootstraps (alpha = 0.5).
A linkage test between all pairs of loci (1,000 permutations) was done across all populations to verify any genotypic disequilibrium (Supplementary Table 2) and gave a putative disequilibrium for RMV22 × RV32 (at p < 0.001) and for RV32 × RV40 (at p < 0.05). Both loci contain many homozygotes, though could not be considered as effectively linked. Data quality was verified with MICRO-CHECKER and gave neither scoring errors, nor evidence of large allele dropout. Potential presence of null alleles was suggested in eight out of nine loci. This can be better explained from a consistent very low heterozygosity due to strong inbreeding instead of null alleles because no single missing value that would refer to a homozygous null allele was recorded. Summary values of the Deviance Information Criterion (DIC) as obtained from INEST further allowed for comparison of seven models to explain the large homogeneity encountered or potential null alleles considering “f” the inbreeding; “n” the null alleles; and “b” genotyping failures and all combinations thereof. All models that included inbreeding (f, nf, fb, nbf) gave a comparable lowest DIC value within each of the 12 populations (Supplementary Table 3). For most sites, the “f” inbreeding model could not be calculated because of one or a few samples with zero score for alleles in a locus. Nonetheless, a comparison of those models with the lowest Deviance Information Criterion (DIC) selected nf, bf and nbf (in all 12 populations) as best fit, thereby outperforming the other models and evidencing an effect of inbreeding, hence putting inbreeding instead of null alleles as a most likely explanation for the lack of heterozygotes. The FIS of each site was corrected for putative null alleles and ranged from 0.473 to 0.999 (Table 2), though 10 out of 12 sites reached values exceeding 0.7.
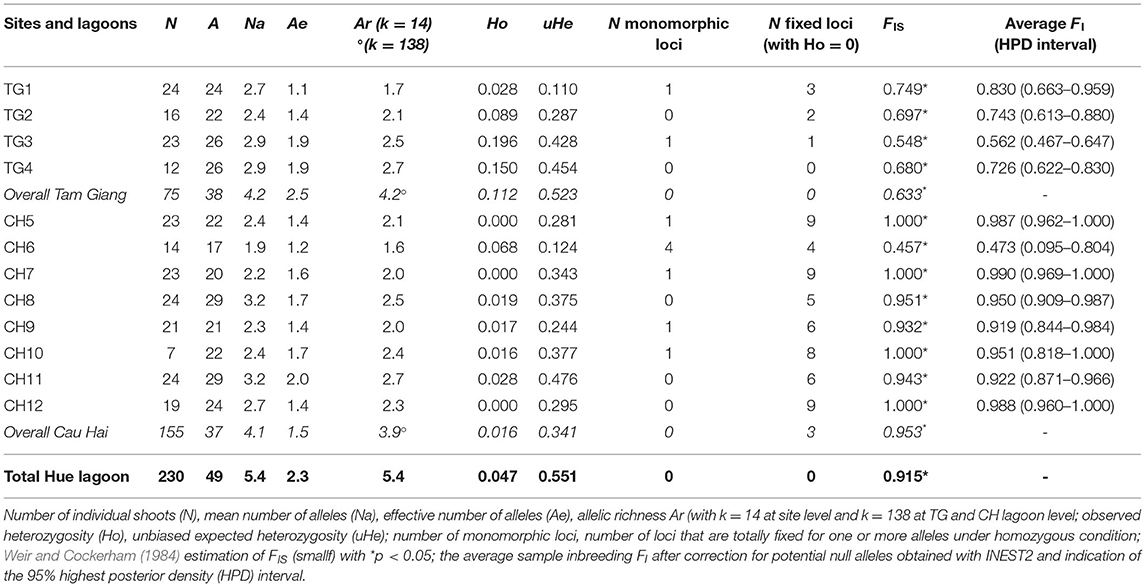
Table 2. Gene diversity measures of 12 Ruppia brevipedunculata sites and over 2 lagoons (TG and CH).
Over the total dataset, 152 individuals shared identical MLG's with sites TG1, CH5, CH7, CH8, and CH12 containing an MLG being present in more than 10 individuals (Supplementary Figure 1). Identical genotypes were identified among sites in CH but none were found among sites in TG or between lagoons TG and CH (Supplementary Table 4). One dominant, fully homozygous multilocus genotype (MLG45) was identified within 71 individuals spanning all sites in CH and was composed of alleles with the highest frequencies at each locus when considering all individuals though only one individual within each site for shared MLG (Supplementary Table 4). Ac ranged from 0.41 to 0.54 (mean = 0.47) and was not significant for all populations.
Nine polymorphic microsatellite loci gave interpretable patterns for a total of 49 alleles in the lagoon (Supplementary Table 1). We found a low probability of identity for 9 loci when considering both lagoons (PI = 0.000005 in TG; PI = 0.001 in CH). Regarding the total population, the allelic richness reached Ar = 5.4 though a low effective size (Ae = 2.3) and very low heterozygosity levels were observed (Ho = 0.047; He = 0.551) (Table 2). A very high level of inbreeding with FIS = 0.991 might be subject to a Wahlund effect and therefore was reconsidered at smaller geographic scales.
Both major lagoon areas, Tam Giang (TG) and Cau Hai (CH), contained a nearly similar number of alleles (A = 38 and 37, respectively) and allelic richness (Ar = 4.2 and 3.9, respectively) though differed in observed gene diversity (Ho = 0.112 and 0.016 respectively; p = 0.012, one-sided test) (Table 2). CH lagoon showed more cases of monomorph loci, of fixed loci under homozygous condition and higher levels of inbreeding than TG lagoon (Table 2), with FIS = 0.953 (in CH) and FIS = 0.633 (in TG) at p = 0.014 (one-sided test). Selfing rate estimations were very high within both TG (S = 0.90, SE = 0.02; p < 0.001) and CH (S = 0.94, SE = 0.07; p < 0.001) lagoons. Significant recent bottlenecks could be detected for sites TG3, CH6, CH7, and CH11. Out of the 49 alleles, 23 were unique to either part of the Hue lagoon complex. In the TG lagoon 12 private alleles at q = 0.007–0.620 (mean q = 0.165) and in CH lagoon 11 private alleles at q = 0.003–0.884 (mean q = 0.253) could be found.
At site (transect) level, R. brevipedunculata harbored 17–29 alleles out of the 49 for the whole lagoon and showed Ae ranging from 1.1 to 2.0 and Ar from 1.6 to 2.7 (Table 2). R. brevipedunculata sites showed very low to low gene diversities with HO ranging from 0 to 0.196 and unbiased He from 0.110 to 0.476 (Table 2), thereby giving high inbreeding values with FIS ranging from 0.457 to 1 (i.e., total fixation). Within-site inbreeding values consistently were significantly high, especially in CH sites and can be explained from the occurrence of several polymorphic loci in which alleles were only present in homozygous conditions (Table 2). A comparison between aquaculture pond habitats and open lagoon sites revealed lower levels of inbreeding (FIS = 0.620 and FIS = 0.942, respectively at p = 0.014) and consequently higher levels of observed heterozygosity (HO = 0.151 and HO = 0.018, respectively at p = 0.004) in aquaculture ponds. No correlation was found between environmental features (conductivity, salinity, TP, distance to nearest aquaculture ponds) of 12 sites and the genetic variables (allelic richness, gene diversity, number of monomorphic or fixed loci, FIS).
A three-level AMOVA-FST for 2 regions (TG and CH lagoon) and 12 sites resulted in strong differentiation at all hierarchical levels with 43% of the variance explained among lagoons and FRT = 0.472, FSR = 0.211, FST = 0.548, FIS = 0.864, FIT = 0.938 (all at p < 0.001) (Table 3). A maximum FRT of 0.553 gave a standardized F'RT = 0.772 between both lagoons (Table 3). Pairwise FST and F'ST values among sites between TG and CH, and within TG (except among TG3 and TG4 with FST = 0.148 and F'ST = 0.341) were high and always significant ranging from 0.438 to 0.802 (FST) corresponding to 0.660 – 0.837 when considering standardized F'ST (Supplementary Table 5). In contrast, sites within CH showed much less differentiation with relatively low pairwise FST values ranging from 0 to 0.379 (F'ST from 0 to 0.223) which were mostly not significant (Supplementary Table 2). A comparison test showed significant (p = 0.003) higher differentiation of the aquaculture sites within TG (FST = 0.497) than for sites within the CH lagoon (FST = 0.0035) due to many private alleles under homozygote conditions.
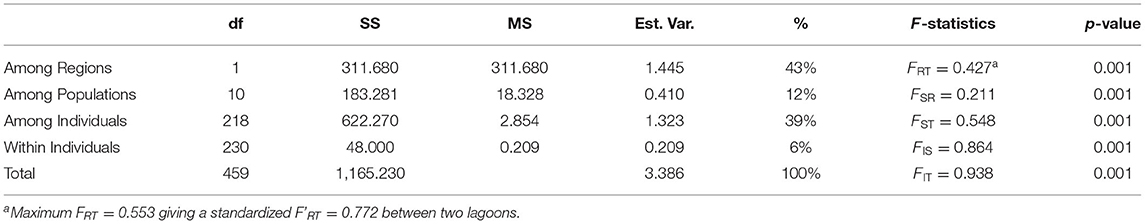
Table 3. Summary of AMOVA and hierarchical F-statistics of Ruppia brevipedunculata (df, degrees of freedom; SS, sum of squares; MS, mean of squares; % Est.Var., estimated variance).
Isolation-by-distance was obtained (Mantel test, p = 0.001), with high predictive value (y = 0.014x+0.034; R2 = 0.80) (Supplementary Figure 2). Pairwise FST testing showed significantly lower values than the average (FST = 0.353) at shortest distance classes up to 3.8 km (FST = 0.064; p = 0.002) and 5.4 km (FST = 0.109; p = 0.004) though significantly higher values for large distance classes above 32.5 km (FST = 0.348 p > 0.05) and up to 37 km (FST = 0.543; p = 0.013) and 49 km (FST = 0.677; p < 0.001). Regression with linear distance at locus level gave a significant positive slope for 8 out of 9 loci which strengthens the evidence of IBD (overall slope b = 0.015 with R2 = 0.78; p < 0.001). Kinship values (FIJ) of individuals among sites (thus omitting within-site comparisons, to avoid adverse effects from local inbreeding) were significantly higher than the average kinship value, relative to the whole sample, over shortest distances of 2 km (FIJ = 0.17; p = 0.002) and 5 km (FIJ = 0.14; p = 0.006) (Supplementary Figure 3).
DAPC results at lagoon level (K = 2) showed a strong genetic structure characterized by two genetically distinct clusters (Figure 3A) whose membership was consistent with those sampled in both main parts in the lagoon (126 individuals (81.3%) of CH in cluster1, all 75 individuals of TG in cluster2), except for 29 individuals (18.70%) of CH which were assigned to cluster2 (Figure 3B). At site level (K = 4), a similar pattern was identified with cluster3, including only individuals of sites in CH, being genetically distinct from the other clusters containing mostly TG individuals (Figures 3C,D). These results verify the low level of connectivity between TG and CH although a low number of individuals in all CH populations (except CH6) were assigned to TG clusters potentially reflecting migrants from TG but not vice versa (Figures 3B,D). Genetically distinct clusters, nearly consistent with individuals in TG1 (cluster1), TG2 (cluster4), TG3 and TG4 (cluster2), show strong population structure within TG and consequently, sites inside aquaculture ponds appear to be genetically isolated (Figures 3C,D). With 12 new clusters, most individuals of TG were reassigned to their predefined sites (Figure 3E). In contrast, individuals of CH were assigned over CH sites without distinct patterns and with only a few individuals being fully reassigned to their predefined site. Low genetic differentiation of CH sites and the absence of a population structure across CH, together with a high proportion of shared and fully homozygous MLG's among CH sites indicate high levels of genetic connectivity among these open lagoon sites.
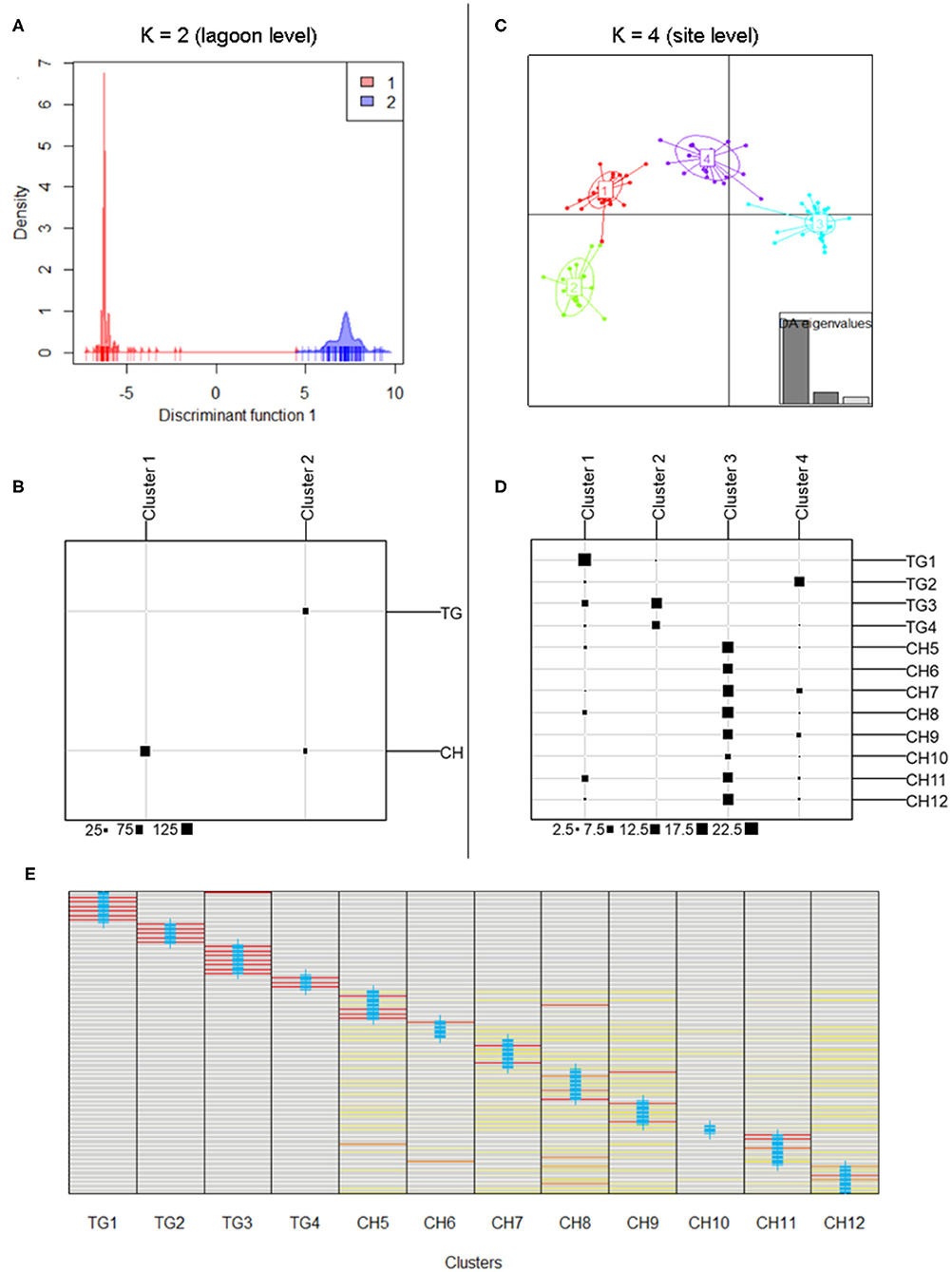
Figure 3. Discriminant Analysis of Principal Components (DAPC) indicating separate clusters at lagoon level and site level. (A,B) Discriminant function of DAPC results at lagoon level with K = 2; membership of inferred clusters (columns) compared to actual groups (rows). (C,D) Scatterplot of DAPC results at site level with K = 4; membership of inferred clusters (columns) compared to actual groups (rows). (E) Comparison of individual membership between 12 newly assigned clusters and original clusters (blue crosses) (red = fully assigned to one cluster, yellow = admixed between clusters).
The model-based clustering method as implemented in STRUCTURE showed K = 2 [LnP(K) = −2,762] because the ΔK was highest and standard deviation was lowest. These two clusters corresponded largely to the TG and the CH lagoon, indicating their different gene pools (Figure 1B). We further implicitly considered the inbred nature of the species. The LnP(K) values increased for K = 3 [LnP(K) = −2,353] and K = 4 [LnP(K) = −2,143] after which the larger K values converged to a plateau (Figures 1C,D). We also presented K = 3 and K = 4 because additional clusters distinguished isolated populations inside aquaculture ponds. The scenarios of K = 2, K = 3, and K = 4 allowed to indicate several individuals of another gene pool within the CH cluster, potentially reflecting migrants coming from TG which hereafter was tested in a migration analysis.
The strongest barriers for most loci and their highest pairwise FST were indicated between TG and CH lagoon and between neighboring TG populations (TG1, TG2, and TG3) located inside aquaculture ponds (Figures 2A,B). The specific testing with migrate-n on directionality for R. brevipedunculata across both lagoon parts, indicated that a unidirectional stepping stone model from TG to CH (Bezier value = −1,345,084) appeared more likely compared to a unidirectional model from CH to TG (Bezier value = −1,385,917) and to the bidirectional model (Bezier value = −1,452,903) (Supplementary Table 6), although the estimated gene flow value from TG toward CH was low (Nem = 0.6). The migration between the TG and CH lagoon using divMigrate gave a significant direction from TG to CH with a relative value 1. The migration between all 12 sites of the area gave only a significant direction from CH10 to CH11 and with a relative value of 0.23 Nm.
The very high levels of inbreeding in all sites with selfing rate estimations exceeding 0.90 are presumed to be the result of the self-pollination mating system of R. brevipedunculata. These findings are consistent with a broad-scale study on R. maritima populations in the Mediterranean where low genetic diversity, a high number of monomorphic and fixed loci and significant though lower levels of inbreeding were attributed to self-fertilization (Triest and Sierens, 2015). R. cirrhosa populations in the Mediterranean were also significantly inbred but lacking fixed loci when compared to R. maritima which suggests that outcrossing, though with biparental inbreeding, is favored over self-pollination in R. cirrhosa (Triest et al., 2018b). In contrast, populations of Halophila beccarii studied in the same lagoon complex (Cau Hai) did not show evidence of inbreeding or selfing and were maintained by local seed recruitment and clonal extension (Phan et al., 2017). The strong ability of self-fertilization in Ruppia is strongly determined by the location of pollen release, with hypohydrophily hypothesized to be less efficient for outcrossing than pollen transfer along the water surface (Taylor et al., 2020). Due to the very short peduncle, R. brevipedunculata may be restricted to pollen release and pollen capture under the water surface which most likely explains self-fertilization as the most prevalent pollination strategy for R. brevipedunculata in our study. The ability of selfing in Ruppia may increase seed-set (Les, 1988; Taylor et al., 2020) and provides reproductive assurance when mates are scarce or vectors for pollen flow are absent or inadequate (Wright et al., 2013; Razanajatovo et al., 2016). Autogamy could therefore promote the establishment and sustainability of R. brevipedunculata populations in enclosed and isolated habitats.
Somewhat less elevated inbreeding values within high tide ponds in aquaculture sites when compared to open lagoon populations indicate that pollen flow among distinct but potentially related individuals appears to be promoted within confined aquaculture areas. Dense vegetation in stagnant water may explain the elevated levels of pollen flow between individuals in aquaculture ponds whereas pollen in open lagoon sites could be lost when not immediately used upon release for self-fertilization (Van Vierssen et al., 1984; Reusch, 2003). Geitonogamy could be an additional and important form of self-fertilization alongside autogamy (Reusch, 2001; Taylor et al., 2020), especially when pollen are released between entangled branches of a same plant. Due to the physical enclosure and dense patches within high-tide ponds, both local outcrossing, evidenced by the lower inbreeding values and the higher number of multilocus genotypes in these particular man-made habitats, and geitonogamy can be hypothesized. If outcrossing is possible, cases of geitonogamy may also occur.
Thus far, we have interpreted the obtained levels and patterns of genetic diversity and structure of R. brevipedunculata as the result of an overarching effect of selfing and inbreeding. Nevertheless, this species showed a high proportion of shared multilocus genotypes (MLGs) within sites as well as across open lagoon sites, a phenomenon usually indicative of asexual reproduction and vegetative dispersal (Arnaud-Haond et al., 2007; McMahon et al., 2017; Dierick et al., 2021). Due to high Pgen_FIS and Psex_FIS values, non-significant aggregation of identical MLGs and the presence of several repeated genotypes that are completely homozygous, although they are widely shared among individuals, we assume that multilocus genotypes similarities in our study are the result from inbreeding events. This assumption is further reinforced by the knowledge about the mating system of R. brevipedunculata, the description of R. brevipedunculata being a species that grows annually from seeds (Yu and den Hartog, 2014) and our sampling strategy that avoided clumps of shoots. The possibility of inadvertent re-sampling or the existence of larger individuals cannot be completely ruled out, but after careful consideration, we maintain the view that repetitive MLGs are the result of selfing/inbreeding events based on the observations and interpretations discussed below.
Nearly all MLGs were found in a fully homozygous state for all 9 loci, despite an intrinsic allelic diversity at site and lagoon level. Only twice, within the same site, did a repeated genotype (three shoots) contain a single heterozygous locus. Exceptional to observe was that one MLG occurred 71 times in all eight sampled meadows of the open lagoon, although it showed complete homozygous conditions for the most common allele from each locus (when considering the allele frequencies of a subset omitting the repeated MLGs, this to avoid bias when including so many repeats). On the contrary, in case of clonal growth and vegetative propagation, one should expect a much higher number of heterozygous clones (i.e., heterozygous in at least one locus but rather any combination of up to 9 loci) than from fully homozygous clones. Thus, shared MLGs were extremely skewed toward a homozygous state, despite the presence of numerous unique MLGs harboring one to multiple polymorphic loci. It seems highly unlikely that the shared MLGs were so disproportionate by chance or selection. Such homogeneity of the common alleles as we observed in R. brevipedunculata is therefore best explained by selfing behavior and inbreeding over several generations rather than clonal growth and asexual spread. Thus, in addition to a lack of heterozygosity, R. brevipedunculata meadows were also characterized by completely fixed and monomorphic loci, a combination of characteristics as might be expected from an inbreeding behavior of different strengths and historical origins. This situation is very similar to the inbred R. maritima from Europe (Triest and Sierens, 2015) but completely different from the outcrossed R. spiralis (Triest et al., 2018b). An alternative explanation, such as bias in the sampling design performed (e.g., spatially clustered patches) is not expected to hold here, as individual shoots were taken at a distance interval of 2 m which is sufficient to avoid repeated sampling of the same individual. Meadows of Ruppia were spatially monitored over a gradient of distance classes. The 12 sites were at least 0.7 km but usually several km or tens of km apart and in fact represented replica sites within two lagoons, all sites of which exhibited nearly similar genetic diversity levels, similar patterns, and consistently elevated levels of inbreeding levels, giving some widespread occurrence of the homozygous state of shared MLGs. Occasionally, population genetic studies may suffer from an unfortunate choice of uninformative microsatellite markers yielding large amounts of homozygous genotypes, e.g., a microsatellite locus with a very common allele at near maximum frequency and one or a few alleles at very low frequencies. However, we used nine microsatellite loci each containing 2–8 alleles in a lagoon and were recovered under different heterozygous conditions and in all locations. Allele lengths from 9 to 23 motif repeats were appropriately sized for consistent PCR amplification. Scoring failures or null alleles was not a problem in this study as shown by the INEST analysis. Cross-amplification of 24 primers for microsatellite loci of other Ruppia species on R. brevipedunculata was possible but gave only a single amplification product (Triest et al., 2018a), hence an indication of a reorganized nuclear genome and monomorphism of R. brevipedunculata.
Therefore, in view of the above discourse, we hold the view that R. brevipedunculata in the studied lagoon system was subject to selfing and inbreeding events explaining homozygosity and homogeneity of MLGs. Alternative explanations such as clonal growth and dispersal, severe bias in sampling design and uninformative microsatellite loci are considered less likely or even unlikely to explain the patterns obtained. We further hypothesize that subsequent self-pollination could lead to a range of inbred lines, some even spanning an entire genome, and this should be explored in future studies using nuclear single nucleotide polymorphic markers covering a wide range of the genome.
Despite being physically connected by water, the main lagoon parts of the Tam Giang-Cau Hai complex are highly differentiated which is confirmed by an overall high fixation index among these regions, high values of pairwise genetic differentiation among sites between TG and CH, nearly half of the total alleles being private for one of both lagoon parts and a strong spatial structure based on the DAPC (K = 2) and STRUCTURE results. In both TG and CH the mean number of alleles is higher than 4 with the allele number for each locus exceeding 2. Consequently, we hypothesize that the presence of viable, but highly inbred and differentiated populations within the lagoon and aquaculture ponds are most likely the result of various founders who colonized empty patches and established populations predominantly by self-fertilization as a result of a small number of independent historical introductions in CH and TG or multiple historical recurrent introductions in the total Tam Giang-Cau Hai lagoon complex followed by subsequent inbreeding which led to a different subset of alleles in both parts. The subsequent yearly replenishment and recruitment may then originate from local seed banks (Strazisar et al., 2013). A pattern of isolation-by-distance was detected with lowest FST - values and highest relative migration estimates among closely sampled sites in CH, thereby largely explaining the lower levels of genetic differentiation over small distances. Four sites at close vicinity within CH (CH8, CH10, CH11, and CH12) were not differentiated from each other, being genetically identical or nearly identical inbred exhibiting homozygous loci. Consequently, highly inbred seeds are dispersed and mixed among open lagoon sites in CH leading to the absence of population structure and an overall higher relatedness between individuals up to 5 km compared to the total Tam Giang-Cau Hai system. In contrast, sites within aquaculture ponds in TG were strongly structured and highly differentiated from each other except for closely sampled sites TG3 and TG4. Hence, aquaculture ponds such as these high-tide systems surrounded by an elevated earth wall, act as strong barriers for propagule dispersal of R. brevipedunculata.
The low genetic connectivity between both main lagoon parts over a distance exceeding 30 km's for R. brevipedunculata is most likely explained by the low seed dispersal capacity of the Ruppia genus which fully ripened fruits almost directly sink to the bottom when detached (Triest and Sierens, 2013), however, seedlings may become detached and get distributed throughout a water column (Koch et al., 2010). Besides the relatively large distance between TG and CH, outlets to the open sea and water discharge from the Perfume river in TG during high tide and the rainy season may impose physical barriers for the dispersal of seedlings or ripening fruit-bearing shoots and most likely contributes to the low observed genetic connectivity between both parts. Nevertheless, migration was proposed from TG to CH but the estimated number of migrants per generation appeared low (Nem = 0.6). Such one-way migration of dispersal propagules aligns with the pattern of unidirectional water flow (from TG to CH) found in the narrow connection during both low and high tide which makes it nearly impossible for propagules to disperse from CH to TG. An estimated dispersal distance of 20 km for R. cirrhosa in the Mediterranean Sea was attributed to detached seedlings, rooted shoot fragments containing ripening fruits that connected sites over these larger distances but only within hydrologically connected lagoon areas (Triest et al., 2018b). Likewise, detached fragments may promote gene flow for R. brevipedunculata among distant lagoon parts though no direct evidence is available to support this. Next to currents and tidal waves, birds are reported to be important or occasional dispersal vectors for Ruppia (Ito et al., 2010; Martínez-Garrido et al., 2017). A high number of bird species (S = 73), with a large proportion being migratory (nearly 50%), have been recorded in the estuaries in the North-West of TG (mainly Anas sp. and Egretta sp.) (Tuan 2012). We suggest that the dispersal of seeds by birds cannot fully explain the pattern of unidirectional gene flow between TG and CH. Still, we do not rule out birds as potential dispersal vectors, though their prevalence and effect on population structure is probably low and difficult to detect compared to the hydrodynamics within the lagoon (Triest and Sierens, 2013). In addition, a large proportion of people that live in and around the lagoon depends on fishing activities which may promote connectivity within and between lagoon parts, though more information is needed on the adherence and transport capacity of Ruppia fragments by boats, nets and other fishing gear.
From a phylogeographical perspective, it is important to consider coastal landform changes in recent historical times as they might shed light on the dynamics that populations have experienced. For a long time, Tu Hien (CH) was the only direct connection to the open sea in the lagoon complex and water was mainly transported unidirectionally from the estuaries in TG to the outlet in CH (Nguyen Tien Lam, 2002). Since the early fifteenth century, a new inlet in TG (Thuan An) was created which changed the hydrodynamics in the lagoon due to the presence of an additional exit for water discharge during low tide and floods and the formation of two reverse currents during high tide, one toward the estuaries in TG and another toward CH (Nguyen Tien Lam, 2002). Our study indicates that this new man-made connection along with the creation of high-tide aquaculture ponds affect the population structure and genetic diversity within and among lagoon parts by decreasing genetic connectivity, reducing population viability and adaption potential toward environmental perturbations (Takebayashi and Morrell, 2001; Wright et al., 2013). However, a higher number of unique multilocus genotypes found in Tam Giang compared to the open lagoon system in Cau Hai may indicate that man-made habitats enclosed by physical structures can provide refuge for rare alleles and genotypes and thus enhance genotypic diversity relative to highly inbred populations. Nevertheless, we assume that the positive effects of preserving rare alleles at isolated sites can be neglected compared to the negative effects of reduced gene flow.
The wide variety in survival strategies (e.g., selfing, outcrossing, clonal persistent, etc.) and their effect on population dynamics and diversification have been overlooked for the genus Ruppia due to largely focusing on two wrongly assumed cosmopolitan Ruppia species in the past (Mannino et al., 2015; Triest et al., 2018a). Correct taxon identification and comprehensive molecular studies at different spatial scales focusing on a variety of Ruppia lineages are therefore needed to understand how Ruppia populations are maintained and persist, which is essential information for the design and application of effective conservation measures to mitigate the effects of habitat deterioration and climate change. Our study highlights the importance of self-fertilization and local recruitment for population maintenance and sustainability for R. brevipedunculata, as recorded in other Ruppia species, though the potential for connectivity beyond the genetic neighborhood should be explored in future studies (Triest and Sierens, 2015; Triest et al., 2018a). Recurrent selfing and mating among relatives may result in inbreeding depression and the loss of populations in the long term due to low effective population sizes and the accumulation of deleterious mutations (Takebayashi and Morrell, 2001; Wright et al., 2013). Therefore, information on the genetic and demographic consequences of long-term inbreeding for R. brevipedunculata is essential to assess if self-fertilization is an advantageous strategy both in the short and long term.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
LT: conceptualization (lead), investigation (lead), methodology (lead), collecting field data (supporting), data curation (lead), formal analysis (lead), visualization (lead), writing-original draft (lead), writing-review and editing (lead), funding acquisition (lead), and resources (supporting). JD: conceptualization (lead), investigation (lead), methodology (lead), collecting field data (supporting), data curation (lead), formal analysis (lead), visualization (lead), writing-original draft (lead), writing-review and editing (lead), funding acquisition (supporting), and resources (supporting). TP and QL: investigation (supporting), methodology (supporting), collecting field data (supporting), and writing-original draft (supporting). NH: investigation (supporting), methodology (supporting), and collecting field data (lead). TS: methodology (lead). All authors contributed to the article and approved the submitted version.
This research was supported by the Vrije Universiteit Brussel (BAS42), the VLIR-International University Cooperation program 2013-2022 with Hue University and the Erasmus Mundus Lotus Action II Program (NQH research fellowship at Vrije Universiteit Brussel.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We gratefully thank VNU-University of Science for administration support. We also thank Mr. Nguyen Huu Tien for his fieldwork assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcosc.2021.723376/full#supplementary-material
Alcala, N., Goudet, J., and Vuilleumier, S. (2014). On the transition of genetic differentiation from isolation to panmixia: what we can learn from G S T and D. Theor. Popul. Biol. 93, 75–84. doi: 10.1016/j.tpb.2014.02.003
Arnaud-Haond, S., Duarte, C. M., Alberto, F., and Serrão, E. A. (2007). Standardizing methods to address clonality in population studies. Mol. Ecol. 16, 5115–5139. doi: 10.1111/j.1365-294X.2007.03535.x
Bailleul, D., Stoeckel, S., and Arnaud-Haond, S. (2016). RClone: a package to identify MultiLocus Clonal Lineages and handle clonal data sets in r. Methods Ecol. Evol. 7, 966–970. doi: 10.1111/2041-210X.12550
Beerli, P. (2006). Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics. 22, 341–345. doi: 10.1093/bioinformatics/bti803
Beerli, P., and Palczewski, M. (2010). Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics. 185, 313–326. doi: 10.1534/genetics.109.112532
Beirinckx, L., Vanschoenwinkel, B., and Triest, L. (2020). Hidden hybridization and habitat differentiation in a mediterranean macrophyte, the euryhaline genus ruppia. Front. Plant Sci. 11:830. doi: 10.3389/fpls.2020.00830
Cho, H. J., Biber, P., and Nica, C. (2009). “The rise of Ruppia in seagrass beds: changes in coastal environment and research needs,” in Seagrass: Ecology, Uses and Threats, eds E. K. Drury and T. S. Pridgen (NOVA Science Publisher), 209–223.
Chybicki, I. J., and Burczyk, J. (2009). Simultaneous estimation of null alleles and inbreeding coefficients. J. Hered. 100, 106–113. doi: 10.1093/jhered/esn088
Cox, P. A., and Knox, R. B. (1989). Two-dimensional pollination in hydrophilous plants: convergent evolution in the genera Halodule (Cymodoceaceae), Halophila (Hydrocharitaceae), Ruppia (Ruppiaceae), and Lepilaena (Zannichelliaceae). Am. J. Bot. 76, 164–175. doi: 10.1002/j.1537-2197.1989.tb11299.x
den Hartog, C., and Kuo, J. (2006). “Taxonomy and biogeography of seagrasses,” in Seagrasses: Biology, Ecology and Conservation, eds A. W. D. Larkum, C. Orth, and C. Duarte (Dordrecht: Springer), 1–23.
den Hartog, C., and Triest, L. (2020). A profound view and discourse on the typification and status of three confused taxa: Ruppia maritima, R. spiralis and R. cirrhosa. Bot. Mar. 63, 229–239. doi: 10.1515/bot-2019-0045
Dierick, J., Phan, T. T. H., Luong, Q. D., and Triest, L. (2021). Persistent clones and local seed recruitment contribute to the resilience of Enhalus acoroides populations under disturbance. Front. Plant Sci. 12:1048. doi: 10.3389/fpls.2021.658213
Duarte, B., Martins, I., Rosa, R., Matos, A. R., Roleda, M. Y., Reusch, T. B. H., et al. (2018). Climate change impacts on seagrass meadows and macroalgal forests: an integrative perspective on acclimation and adaptation potential. Front. Mar. Sci. 2018:190. doi: 10.3389/fmars.2018.00190
Earl, D. A., and vonHoldt, B. M. (2012). Structure Harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Goodwillie, C., and Weber, J. J. (2018). The best of both worlds? A review of delayed selfing in flowering plants. Am. J. Bot. 105, 641–655. doi: 10.1002/ajb2.1045
Goudet, J. (2001). FSTAT. A Program to Estimate and Test Gene Diversities and Fixation Indices. Available online at: https://www.scienceopen.com/document?vid=79097bb4-ec3c-47c3-94a1-47085d721e6b (accessed February 3, 2021).
Grech, A., Coles, R., and Marsh, H. (2011). A broad-scale assessment of the risk to coastal seagrasses from cumulative threats. Mar. Policy 35, 560–567. doi: 10.1016/j.marpol.2011.03.003
Hardy, O. J., and Vekemans, X. (2002). SPAGeDI: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2, 618–620. doi: 10.1046/j.1471-8286.2002.00305.x
Holland, M. M., and Parson, W. (2011). GeneMarker® HID: a reliable software tool for the analysis of forensic STR data. J. Forensic Sci. 56, 29–35. doi: 10.1111/j.1556-4029.2010.01565.x
Holleley, C. E., and Geerts, P. G. (2009). Multiplex Manager 1.0: a cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques 46, 511–517. doi: 10.2144/000113156
Ito, Y., Ohi-Toma, T., Murata, J., and Tanaka, N. (2010). Hybridization and polyploidy of an aquatic plant, Ruppia (Ruppiaceae), inferred from plastid and nuclear DNA phylogenies. Am. J. Bot. 97, 1156–1167. doi: 10.3732/ajb.0900168
Jombart, T. (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Källström, B., Nyqvist, A., Åberg, P., Bodin, M., and André, C. (2008). Seed rafting as a dispersal strategy for eelgrass (Zostera marina). Aquat. Bot. 88, 148–153. doi: 10.1016/j.aquabot.2007.09.005
Kamvar, Z. N., Tabima, J. F., and Gr¨unwald, N. J. (2014). Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. 2014, 1–14. doi: 10.7717/PEERJ.281
Kaul, R. B. (1993). Meristic and organogenetic variation in Ruppia occidentalis and R. maritima. Int. J. Plant Sci. 154, 416–424. doi: 10.1086/297124
Kendrick, G. A., Nowicki, R., Olsen, Y. S., Strydom, S., Fraser, M. W., Sinclair, E. A., et al. (2019). A systematic review of how multiple stressors from an extreme event drove ecosystem-wide loss of resilience in an iconic seagrass communit. Front. Mar. Sci. 6:455. doi: 10.3389/fmars.2019.00455
Kendrick, G. A., Waycott, M., Carruthers, T. J. B., Cambridge, M. L., Hovey, R., Krauss, S. L., et al. (2012). The central role of dispersal in the maintenance and persistence of seagrass populations. Bioscience 62, 56–65. doi: 10.1525/bio.2012.62.1.10
Koch, E. W., Ailstock, M. S., Booth, D. M., Shafer, D. J., and Magoun, A. D. (2010). The role of currents and waves in the dispersal of submersed angiosperm seeds and seedlings. Restorat. Ecol. 18, 584–595. doi: 10.1111/j.1526-100X.2010.00698.x
Lacroix, C. R., and Kemp, J. R. (1997). Developmental morphology of the androecium and gynoecium in Ruppia maritima L.: considerations for pollination. Aquatic Bot. 59, 253–262. doi: 10.1016/S0304-3770(97)00074-0
Les, D. H. (1988). Breeding systems, population structure, and evolution in hydrophilous angiosperms. Ann. Missouri Bot. Gard. 75:819. doi: 10.2307/2399370
Les, D. H., Crawford, D. J., Kimball, R. T., Moody, M. L., and Landolt, E. (2003). Biogeography of discontinuously distributed hydrophytes: a molecular appraisal of intercontinental disjunctions. Int. J. Plant Sci. 164, 917–932. doi: 10.1086/378650
Li, Y. L., and Liu, J. X. (2018). StructureSelector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 18, 176–177. doi: 10.1111/1755-0998.12719
Loiselle, B. A., Sork, V. L., Nason, J., and Graham, C. (1995). Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am. J. Bot. 82, 1420–1425. doi: 10.1002/j.1537-2197.1995.tb12679.x
Manni, F., Guérard, E., and Heyer, E. (2004). Geographic patterns of (genetic, morphologic, linguistic) variation: How barriers can be detected by using Monmonier's algorithm. Hum. Biol. 76, 173–190. doi: 10.1353/hub.2004.0034
Mannino, A. M., Menéndez, M., Obrador, B., Sfriso, A., and Triest, L. (2015). The genus Ruppia L. (Ruppiaceae) in the Mediterranean region: an overview. Aquat. Bot. 124, 1–9. doi: 10.1016/j.aquabot.2015.02.005
Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220.
Marbà, N., and Duarte, C. M. (1998). Rhizome elongation and seagrass clonal growth. Mar. Ecol. Prog. Ser. 174, 269–280. doi: 10.3354/meps174269
Martínez-Garrido, J., Bermejo, R., Serrão, E. A., Sánchez-Lizaso, J., and González-Wangüemert, M. (2017). Regional genetic structure in the aquatic macrophyte Ruppia cirrhosa suggests dispersal by waterbirds. Estuar. Coasts 40, 1705–1716. doi: 10.1007/s12237-017-0247-9
McMahon, K. M., Evans, R. D., Van Dijk, K. J., Hernawan, U., Kendrick, G. A., Lavery, P. S., et al. (2017). Disturbance is an important driver of clonal richness in tropical seagrasses. Front. Plant Sci. 8:2026. doi: 10.3389/fpls.2017.02026
Michalakis, Y., and Excoffier, L. (1996). A generic estimation of population subdivision using distances between alleles with special interest to microsatellite loci. Genetics 142, 1061–1064.
Miller, M. P., Knaus, B. J., Mullins, T. D., and Haig, S. M. (2013). SSR-pipeline: a bioinformatic infrastructure for identifying microsatellites from paired-end illumina high-throughput DNA sequencing data. J. Hered. 104, 881–885. doi: 10.1093/jhered/est056
Nguyen Tien Lam (2002). A Preliminary Study on Hydrodynamics of the Tam Giang - Cau Hai Lagoon and Tidal Inlet System in the Thua Thien - Hue Province, Vietnam, 167.
Orth, R. J., Luckenbach, M., and Moore, K. A. (1994). Seed dispersal in a marine macrophyte: implications for colonization and restoration. Ecology 75,1927–1939. doi: 10.2307/1941597
Peakall, R., and Smouse, P. E. (2012). GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Phan, T. T. H., De Raeymaeker, M., Luong, Q. D., and Triest, L. (2017). Clonal and genetic diversity of the threatened seagrass Halophila beccarii in a tropical lagoon: resilience through short distance dispersal. Aquat. Bot. 142, 96–104. doi: 10.1016/j.aquabot.2017.07.006
Piry, S., Luikart, G., and Cornuet, J.-M. (1999). Bottleneck: a program for detecting recent effective population size reductions from allele data frequencies. J. Hered. 90, 502–503.
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
QGIS Development Team (2021). QGIS Geographic Information System. Open Source Geospatial Foundation Project. Available online at: http://qgis.osgeo.org. Qgisorg (accessed March 2, 2021).
Quiros, T. E. A. L., Croll, D., Tershy, B., Fortes, M. D., and Raimondi, P. (2017). Land use is a better predictor of tropical seagrass condition than marine protection. Biol. Conserv. 209, 454–463. doi: 10.1016/j.biocon.2017.03.011
R Core Team (2021). R: A Language and Environment for Statistical Computing. Vienna: R Foundation Statistical Computing.
Razanajatovo, M., Maurel, N., Dawson, W., Essl, F., Kreft, H., Pergl, J., et al. (2016). Plants capable of selfing are more likely to become naturalized. Nat. Commun. 7, 1–9. doi: 10.1038/ncomms13313
Reusch, T. (2001). New markers-old questions: population genetics of seagrasses. Mar. Ecol. Prog. Ser. 211, 261–274. doi: 10.3354/meps211261
Reusch, T. B. H. (2003). Floral neighbourhoods in the sea: how floral density, opportunity for outcrossing and population fragmentation affect seed set in Zostera marina. J. Ecol. 91, 610–615. doi: 10.1046/j.1365-2745.2003.00787.x
Short, F., Carruthers, T., Dennison, W., and Waycott, M. (2007). Global seagrass distribution and diversity: a bioregional model. J. Exp. Mar. Bio. Ecol. 350, 3–20. doi: 10.1016/j.jembe.2007.06.012
Strazisar, T., Koch, M. S., Dutra, E., and Madden, C. J. (2013). Ruppia maritima L. seed bank viability at the Everglades-Florida Bay ecotone. Aquat. Bot. 111, 26–34. doi: 10.1016/j.aquabot.2013.08.003
Sundqvist, L., Keenan, K., Zackrisson, M., Prodöhl, P., and Kleinhans, D. (2016). Directional genetic differentiation and relative migration. Ecol. Evol. 6, 3461–3475. doi: 10.1002/ece3.2096
Takebayashi, N., and Morrell, P. L. (2001). Is self-fertilization an evolutionary dead end? Revisiting an old hypothesis with genetic theories and a macroevolutionary approach. Am. J. Bot. 88, 1143–1150. doi: 10.2307/3558325
Taylor, M. L., Giffei, B. L., Dang, C. L., Wilden, A. E., Altrichter, K. M., Baker, E. C., et al. (2020). Reproductive ecology and postpollination development in the hydrophilous monocot Ruppia maritima. Am. J. Bot. 107, 689–699. doi: 10.1002/ajb2.1447
Tran, H. T. (2005). First simulation for two - dimensional currents in dry season of the Tam Giang - Cau Hai lagoon. Hue Univ. J. Res. Nat. Sci. 27, 57–64.
Triest, L., Beirinckx, L., and Sierens, T. (2018a). Lagoons and saltwater wetlands getting more diversity: a molecular approach reveals cryptic lineages of a euryhaline submerged macrophyte (Ruppia). Aquat. Conserv. Mar. Freshw. Ecosyst. 28, 370–382. doi: 10.1002/aqc.2863
Triest, L., and Sierens, T. (2010). Chloroplast sequences reveal a diversity gradient in the Mediterranean Ruppia cirrhosa species complex. Aquat. Bot. 93, 68–74. doi: 10.1016/j.aquabot.2010.03.007
Triest, L., and Sierens, T. (2013). Is the genetic structure of Mediterranean Ruppia shaped by bird-mediated dispersal or sea currents? Aquat. Bot. 104, 176–184. doi: 10.1016/j.aquabot.2011.09.009
Triest, L., and Sierens, T. (2014). Seagrass radiation after Messinian salinity crisis reflected by strong genetic structuring and out-of-Africa scenario (Ruppiaceae). PLoS ONE 9:104264. doi: 10.1371/journal.pone.0104264
Triest, L., and Sierens, T. (2015). Strong bottlenecks, inbreeding and multiple hybridization of threatened European Ruppia maritima populations. Aquat. Bot. 125, 31–43. doi: 10.1016/j.aquabot.2015.05.001
Triest, L., Sierens, T., Menemenlis, D., and Van der Stocken, T. (2018b). Inferring connectivity range in submerged aquatic populations (Ruppia L.) along european coastal lagoons from genetic imprint and simulated dispersal trajectories. Front. Plant Sci. 9, 1–20. doi: 10.3389/fpls.2018.00806
Unsworth, R. K. F., McKenzie, L. J., Collier, C. J., Cullen-Unsworth, L. C., Duarte, C. M., Eklöf, J. S., et al. (2019). Global challenges for seagrass conservation. Ambio 48, 801–815. doi: 10.1007/s13280-018-1115-y
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M., and Shipley, P. (2004). MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538. doi: 10.1111/j.1471-8286.2004.00684.x
Van Vierssen, W., Van Kessel, C. M., and Van Der Zee, J. R. (1984). On the germination of Ruppia taxa in western Europe. Aquat. Bot. 19, 381–393. doi: 10.1016/0304-3770(84)90050-0
Verhoeven, J. T. A. (1979). The ecology of Ruppia-dominated communities in western Europe. I. Distribution of Ruppia representatives in relation to their autecology. Aquat. Bot. 6, 197–267. doi: 10.1016/0304-3770(79)90064-0
Waycott, M., Duarte, C. M., Carruthers, T. J. B., Orth, R. J., Dennison, W. C., Olyarnik, S., et al. (2009). Accelerating loss of seagrasses across the globe threatens coastal ecosystems. Proc. Natl. Acad. Sci. U. S. A. 106, 12377–12381. doi: 10.1073/pnas.0905620106
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-Statistics for the Analysis of Population Structure. Evolution (N. Y). 38:1358. doi: 10.2307/2408641
Wright, S. I., Kalisz, S., and Slotte, T. (2013). Evolutionary consequences of self-fertilization in plants. Proc. R. Soc. B Biol. Sci. 280:20130133. doi: 10.1098/rspb.2013.0133
You, F. M., Huo, N., Gu, Y. Q., Luo, M. C., Ma, Y., Hane, D., et al. (2008). BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinform. 9:253. doi: 10.1186/1471-2105-9-253
Yu, S., and den Hartog, C. (2014). Taxonomy of the genus Ruppia in China. Aquat. Bot. 119, 66–72. doi: 10.1016/j.aquabot.2014.08.003
Keywords: seagrass, inbreeding, selfing, genetic diversity, genetic structure, migration, microsatellites (SSR markers)
Citation: Triest L, Dierick J, Phan TTH, Luong QD, Huy NQ and Sierens T (2021) Low Genetic Connectivity of Strongly Inbred Ruppia brevipedunculata in Aquaculture Dominated Lagoons (Viet Nam). Front. Conserv. Sci. 2:723376. doi: 10.3389/fcosc.2021.723376
Received: 10 June 2021; Accepted: 23 August 2021;
Published: 17 September 2021.
Edited by:
Gustavo Maruyama Mori, São Paulo State University, BrazilReviewed by:
Yoshiaki Tsuda, University of Tsukuba, JapanCopyright © 2021 Triest, Dierick, Phan, Luong, Huy and Sierens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludwig Triest, bHRyaWVzdEB2dWIuYWMuYmU=; Jasper Dierick, amFzcGVyLmRpZXJpY2tAdnViLmJl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.