 Nina Zeyen1*
Nina Zeyen1* Baolin Wang1
Baolin Wang1 Sasha Wilson1
Sasha Wilson1 Carlos Paulo2Amanda R. Stubbs2Ian M. Power2
Carlos Paulo2Amanda R. Stubbs2Ian M. Power2 Matthew Steele-Maclnnis1Antonio Lanzirotti3Matthew Newville3
Matthew Steele-Maclnnis1Antonio Lanzirotti3Matthew Newville3 David J. Paterson4Jessica L. Hamilton4Thomas R. Jones5Connor C. Turvey6Gregory M. Dipple6
David J. Paterson4Jessica L. Hamilton4Thomas R. Jones5Connor C. Turvey6Gregory M. Dipple6 Gordon Southam5
Gordon Southam5- 1Department of Earth and Atmospheric Sciences, University of Alberta, Edmonton, AB, Canada
- 2Trent School of the Environment, Trent University, Peterborough, ON, Canada
- 3Centre for Advanced Radiation Sources, University of Chicago, Chicago, IL, United States
- 4The Australian Synchrotron, Australia's Nuclear Science and Technology Organisation (ANSTO), Clayton, VIC, Australia
- 5School of Earth and Environmental Sciences, The University of Queensland, St Lucia, QLD, Australia
- 6Department of Earth, Ocean and Atmospheric Sciences, The University of British Columbia, Vancouver, BC, Canada
Mineral carbonation of alkaline mine residues is a carbon dioxide removal (CDR) strategy that can be employed by the mining industry. Here, we describe the mineralogy and reactivity of processed kimberlites and kimberlite ore from Venetia (South Africa) and Gahcho Kué (Canada) diamond mines, which are smectite-rich (2.3–44.1 wt.%). Whereas, serpentines, olivines, hydrotalcites and brucite have been traditionally used for mineral carbonation, little is known about the reactivity of smectites to CO2. The smectite from both mines is distributed as a fine-matrix and is saponite, Mg3(AlxSi4−x)O10(OH)2·nH2O, where the layer charge deficiency is balanced by labile, hydrated interlayer cations (Mm+). A positive correlation between cation exchange capacity and saponite content indicates that smectite is the most reactive phase within these ultramafic rocks and that it can be used as a source of labile Mg2+ and Ca2+ for carbonation reactions. Our work shows that smectites provide the fast reactivity of kimberlite to CO2 in the absence of the highly reactive mineral brucite [Mg(OH)2]. It opens up the possibility of using other, previously inaccessible rock types for mineral carbonation including tailings from smectite-rich sediment-hosted metal deposits and oil sands tailings. We present a decision tree for accelerated mineral carbonation at mines based on this revised understanding of mineralogical controls on carbonation potential.
Introduction
Stabilization of atmospheric CO2 concentration is an ongoing challenge that is necessary to limit the adverse effects of climate change (IPCC, 2018). Mineral carbonation, also called carbon mineralization, is a natural weathering process that regulates atmospheric CO2 levels by forming carbonate rocks, which constitute the largest carbon reservoir on Earth (Berner, 1998). Accelerated mineral carbonation via enhanced weathering is a Carbon Dioxide Removal (CDR) strategy that can be employed by industries that generate alkaline wastes to offset their greenhouse gas emissions (Wilson et al., 2006, 2009a; Renforth et al., 2009). Mining companies, such as the De Beers Group of Companies (Mervine et al., 2018), are exploring integration of this strategy within their operations (e.g., Project CarbonVault™). This approach to Carbon Capture, Utilisation and Storage (CCUS) transforms atmospheric CO2 into environmentally benign carbonate minerals that are stable over geological time (Power et al., 2014). The mineral carbonation reaction has traditionally been viewed as silicate dissolution (1), releasing divalent cation(s) (M2+), followed by carbonate precipitation (2):
This mineral trapping of CO2 is generally described by the sum of reactions (1) and (2):
Key controls on the conversion of CO2 into solid carbonates relate to the nature of the target mineral used as a source of divalent cations, in particular the nature of its divalent cations (e.g., Ca2+, Mg2+, Fe2+) and its dissolution rate, which is a function of reactive surface area, the strength of metal–oxygen bonds (controlled by cation size and co-ordination number) and, in the case of silicates, the degree of silica polymerization (Daval et al., 2011; Power et al., 2013; Harrison et al., 2015). Parameters such as water content, solution chemistry [a(Ca2+), a(Mg2+), a(CO3)2−, pH], temperature and pressure also serve as important controls on carbonation rates and efficiency.
Mg2+ and Ca2+ are the most important cations for mineral carbonation because they compose the most common carbonate minerals at Earth's surface. Mg- and Ca-rich mafic and ultramafic rocks such as basalts, peridotites and serpentinites play an important role in chemical weathering and their carbonation acts as a major sink for atmospheric CO2 (Goff and Lackner, 1998; Dessert et al., 2003; Oelkers et al., 2008). Numerous experimental studies have described and/or measured the reactivity of the Mg-, Ca- and Fe-bearing phases found in (ultra)mafic rocks. Serpentine minerals, such as lizardite and chrysotile [Mg3Si2O5(OH)4], have received wide attention due to their high reactivity and their high Mg content (e.g., Hansen et al., 2005; Daval et al., 2013; Power et al., 2013). Olivine minerals [(Mg,Fe)2SiO4] have also been extensively studied because they are amongst (i) the most thermodynamically favorable substrates for carbonation and (ii) the fastest dissolving Mg-silicate minerals per unit of surface area (Giammar et al., 2005; Daval et al., 2011; Guyot et al., 2011; Bundeleva et al., 2014; Loring et al., 2015). Despite that, aqueous carbonation of olivine has shown negligible carbonate formation at temperatures below 100°C (e.g., Wang and Giammar, 2013). Wollastonite (CaSiO3) is one of the most reactive Ca-bearing silicate minerals and it has also been studied for mineral carbonation (Daval et al., 2009). Release of Mg or Ca from these silicate minerals is slowed due to the precipitation of passivating Si-rich layers at grain surfaces, which is why the use of organic compounds/chelators in soils, attrition grinding exfoliation or wave action on beaches are commonly studied and/or invoked in enhanced weathering of these silicate minerals (e.g., Sissmann et al., 2013; Julcour et al., 2015). The dissolution of silicate minerals also leads to the liberation of Si into solution and to a thermodynamic and kinetic competition between aqueous carbonate and silica species for aqueous Mg (Zeyen et al., 2021). Consequently, the effect of mineral carbonation can be limited by the precipitation of authigenic Mg-silicate phases (e.g., stevensite, kerolite) rather than Mg-carbonates (Alfredsson et al., 2013; Rigopoulos et al., 2018; Oelkers et al., 2019; Oskierski et al., 2019).
Alternatively, brucite [Mg(OH)2], which is a common component of some ultramafic rocks, is considered the easiest source of Mg for carbonation reactions because of its high reactivity and high solubility at Earth's surface temperatures (Wilson et al., 2010, 2014; Zhao et al., 2010; Pronost et al., 2011; Harrison et al., 2013; Power et al., 2020). Brucite content within ultramafic rocks is the single most important determinant of carbonation potential using atmospheric CO2, flue gas or CO2 streams from Direct Air Capture alone. Brucite carbonation in ultramafic rock is optimized under water undersaturated conditions (Assima et al., 2013b; Harrison et al., 2015; Hamilton et al., 2020), which affords an advantage over serpentine and olivine dissolution, since it does not require as large an investment of water. The full reactivity of brucite to CO2 can also be accessed at ambient temperature and pressure. Nesquehonite (MgCO3·3H2O), dypingite [Mg5(CO3)4(OH)2·~5H2O] and/or hydromagnesite [Mg5(CO3)4(OH)2·4H2O] are the dominant carbonation products (e.g., Zhao et al., 2010; Harrison et al., 2013) of brucite dissolution (4) and carbonate precipitation (5):
The sum of reactions (4) and (5) describes carbonation of brucite to nesquehonite:
Kimberlites are diamond-bearing ultramafic rocks containing a high quantity of reactive Mg- and Ca-silicates such as serpentine and olivine minerals (Wilson et al., 2009b; Mervine et al., 2018). They can also contain brucite so long as the kimberlites have not experienced substantial crustal contamination, which incorporates silica to form talc at the expense of brucite (Berg, 1989). An estimated range of 9–17 Gt/yr of mine tailings, including processed kimberlites, are produced worldwide (Bullock et al., 2021). Approximately 420 Mt/year of these tailings are ultramafic and suitable for mineral carbonation (Power et al., 2013). Although processed kimberlites have been recognized as suitable rocks for carbon sequestration (i.e., the multidisciplinary Carmex project, Bodénan et al., 2014), only a few studies on mineral carbonation in kimberlite exist, including those done at the Diavik mine, Northwest Territories, Canada (Wilson et al., 2009b, 2011; Power et al., 2011), the Ekati mine, Northwest Territories, Canada (Rollo and Jamieson, 2006) and the Kirkland Lake and Lake Timiskaming kimberlites, Ontario, Canada (Sader et al., 2007). The mineralogy of kimberlites from different diamond mines (i.e., Venetia, Voorspoed, Victor, Gahcho Kué and Snap Lake) varies both between mines and amongst facies within a single kimberlite pipe (Mervine et al., 2018). This mineralogical diversity—particularly the abundances and presence of serpentines, olivines, brucite, smectites, and pre-existing carbonates (calcite and dolomite)—must be carefully assessed and considered in choosing the most appropriate mineral carbonation strategy for a given mine.
In this study, we investigate the mineralogy and reactivity of kimberlite ore and processed kimberlites from the Venetia (South Africa) and Gahcho Kué (Canada) diamond mines, mimicking the first step of crushing during kimberlite ore processing. Highly reactive brucite is absent in samples from Venetia and either scarce or absent at Gahcho Kué, necessitating development of new strategies for mineral carbonation. Ore and processed kimberlite from these two mines consist of a clay-rich mineral assemblage that is dominated by serpentine minerals and Mg- and Ca-rich smectite(s). Carbonation of serpentine and brucite has long been a focus of CCUS in mine residues (e.g., Wilson et al., 2009a; Krevor and Lackner, 2011; Pronost et al., 2011; Harrison et al., 2013). Although serpentine minerals are abundant in ultramafic rocks, they react more slowly at Earth's surface temperatures, making the abundance of highly reactive brucite the major control on how much mineral carbonation can occur rapidly via enhanced weathering of mine tailings (Wilson et al., 2010; Pronost et al., 2011; Harrison et al., 2013).
Smectites are a group of minerals consisting of trioctahedral (e.g., stevensite and saponite) or dioctahedral (e.g., montmorillonite and non-tronite) swelling clays. Smectites can act as a source of magnesium and/or calcium for carbonation reactions via two possible mechanisms: (i) cation exchange, in which labile interlayer calcium and magnesium are released from the mineral in exchange for other less desirable cations (e.g., K+, ) from solution (7), avoiding silica dissolution and the precipitation of secondary silicate(s) and (ii) dissolution or direct replacement of smectites to form carbonate phases from recalcitrant magnesium located in octahedral sheets (Figure 1). The extracted cations Mm+ can then react with carbonate and/or bicarbonate ions to form carbonate minerals, MCO3 (8).
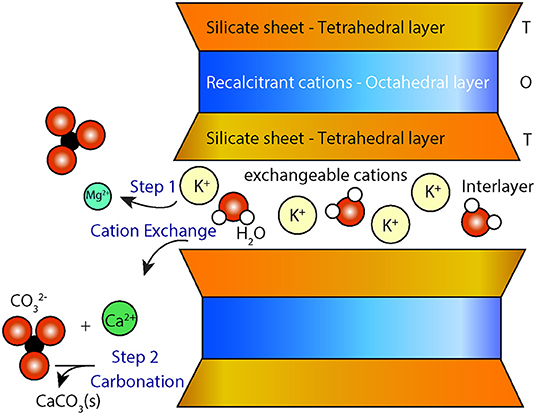
Figure 1. Conceptual diagram showing cation exchange between the interlayer of smectites and a solution that is saturated with a potassium salt (e.g., KCl). Divalent cations (Mg2+ and Ca2+) are exchanged with K+ in the interlayer space of the smectite (step 1) making them available for carbonation (step 2). T-O-T represents the layered configuration of smectite with an octahedral layer (O) between two tetrahedral silicate layers (T).
Equations (7) and (8) can be viewed as two distinct steps where the first step consists of the liberation of cations through the addition of a chemical amendment such as KCl (i.e., the cation exchange) and the second step consists of carbonation of the cation-rich leachate using a source of CO2 (Figure 1).
The use of smectites for mineral carbonation, sensu stricto (i.e., involving carbonate precipitation), has not previously been explored, but here we propose that they can be employed as fast-reacting alternatives to brucite. We then describe a framework for selecting technologies that are best suited to optimize carbonation of processed kimberlites and other mine residues based on their mineralogy.
Materials and Methods
Field Localities and Sampling
Fieldwork and sampling were conducted in 2017 at two kimberlite-hosted diamond mines, Venetia mine (Limpopo Province, South Africa) and Gahcho Kué mine (Northwest Territories, Canada), to assess the carbonation potential of processed kimberlite at these sites (Figure 2). Table 1 summarizes the samples that were collected, their corresponding lithofacies and the acronyms used to describe lithofacies.
Figure 2. Maps showing the location (marked by orange diamond symbols) of: (A) Venetia mine in South Africa; (B) Gahcho Kué mine in Canada. Modified after Mervine et al. (2018).
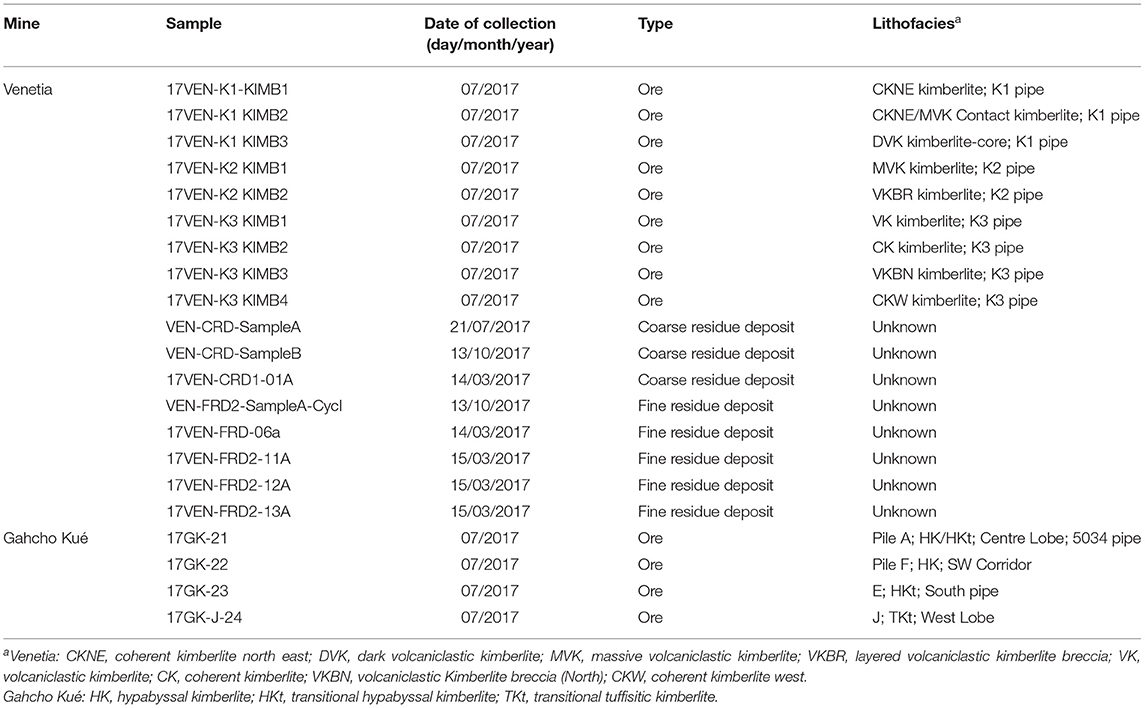
Table 1. Kimberlite facies studied.
The Venetia pipes were formed at 519 ± 6 Ma (Phillips et al., 1998) and emplaced into the Sand River Gneiss basement (3.1 Ga), which consists of granulites retrogressed to amphibolite facies, and the metamorphic Beit Bridge Complex (2.0–2.7 Ga), which consists of interlayered biotite gneiss, biotite schist, amphibolite, quartzite and dolomitic marble (Stripp et al., 2006). Samples of kimberlite ore were collected from stockpiles and the mine pit, whereas samples of processed kimberlite were obtained at the processing plants, Fine Residue Deposits 1 and 2 (FRD, particle size <0.8 mm) and Coarse Residue Deposit (CRD, particle size of 0.8–8 mm). Three crust samples, 17VEN-FRD2-11A,−12A and 13A were collected at the surface of FRD 2. Sampling at Venetia was done in March, July and October 2017.
The pipes at Gahcho Kué were emplaced into the Archaean basement granitoids of the Slave Craton (Hetman et al., 2004). Four samples of kimberlite ore, including hypabyssal kimberlite (17GK-22), transitional hypabyssal kimberlite (17GK-21, 17GK-23) and transitional tuffisitic kimberlite (17GK-J-24) from the 5034 kimberlite pipe, were collected from ore stockpiles in July 2017.
Bulk Analyses of Geochemical, Physical and Mineralogical Properties
Since most of the samples are hand-specimens of ore kimberlites, ~40 g of each sample were finely ground in a shatter box for 3 min, to mimic the first step in kimberlite ore processing. Aliquots of the same powdered sample were used for bulk analyses of geochemical composition and physical properties.
Major and Trace Element Geochemistry
Concentrations of major elements, total carbon and total inorganic carbon, total sulfur and trace elements were measured at SGS (Vancouver, Canada) on 21 samples (17 samples from Venetia and 4 samples from Gahcho Kué). Approximately 10 g of powder were used for each analysis. Major element analyses (14 elements) were performed using borate fusion for X-ray fluorescence (XRF) spectrometry. Trace elements (56 elements) were analyzed using sodium peroxide fusion combined with inductively coupled plasma—atomic emission spectroscopy (ICP-AES) and inductively coupled plasma—mass spectrometry (ICP-MS). Total carbon (TC) and total sulfur contents were determined using a carbon/sulfur analyzer by combustion. Total inorganic carbon (TIC) abundance was determined using coulometry. The amount of organic carbon was calculated as the difference between total carbon and total inorganic carbon.
Particle Size Distribution and Surface Area
The particle size distribution of 19 pulverized samples and one unpulverized sample, 17VEN-CRD-sampleB-13.10.2017, were obtained using a Vibratory Sieve Shaker AS 200 digit cA (Retsch) and a Horiba LA-950V2 laser scattering particle size distribution analyser (Teledyne, USA). Sodium hexametaphosphate (30 g/L), also known as Calgon, was used as a dispersant. Specific surface areas of the same samples were determined using the multipoint Brunauer–Emmett–Teller method (BET; Brunauer et al., 1938). Samples were degassed overnight at 200°C on a Smart VacPrepTM 067 instrument (Micromeritics, Norcross, GA, USA). BET analysis was performed at 77 K (−196°C) on a Micromeritics TriStar II Plus adsorption unit (Micromeritics, Norcross, GA, USA) and N2 isotherms were acquired using a p/p0 range of 0.01–0.90. All data processing was conducted with the MicroActive Interactive software (Micromeritics, Norcross, GA, USA). This provides an estimate of the reactive surface area of processed kimberlite and ore.
Cation Exchange Capacity
CEC is defined as a measure of the ability of a clay or a soil to adsorb cations in such a form that they can be readily desorbed by competing ions (Bache, 1976). CEC analyses were performed on 19 pulverized samples and one unpulverized sample, 17VEN-CRD-sample B-13.10.2017. Two of these 20 samples were analyzed in triplicate to check the precision of the analyses. In this study, we determined CEC in two ways: (1) using the sum of all exchangeable cations (denoted CEC-cations) in the NH4-acetate leachates and (2) using only the NH4-N concentration in the KCl leachates (denoted CEC-NH4-N), both of which are normalized to the BET surface area. More information about the CEC method is provided in the Supplementary Material S1.
Powder X-Ray Diffraction
Approximately 2 g of each pulverized sample was milled for 7 min under anhydrous ethanol using a McCrone micronizing mill. Following drying and disaggregation, micronized samples were Ca-exchanged by agitation on a shaker table for 24 h while suspended in a solution of 1 M CaCl2 following the method of Mervine et al. (2018). Ca-exchanged samples were rinsed three times with MilliQ water to prevent the formation of chloride salts as an artifact of this treatment during drying. Relative humidity (RH) strongly influences the structures of swelling clays (i.e., Bish et al., 2003). As such, samples were stored in a humidity vessel containing a slurry of NaCl (RH ~ 75%) for 24 h prior to X-ray diffraction (XRD) analysis, which served to stabilize the basal spacing of smectites to ~15 Å. A humidifier was used to maintain RH between 66.7 and 73.3% during acquisition of XRD patterns. Samples were back-loaded into cavity mounts and patterns were collected using a Bruker D8 Advance X-ray diffractometer equipped with a high-speed energy-dispersive LYNXEYE XE-T detector. Data acquisition was done using a Co X-ray tube operated at 35 kV and 40 mA. All patterns were collected over a 2θ range of 3–80° with a step size of 0.02°/step at a rate of 1 s/step. Mineral phase identification from all patterns was conducted using the DIFFRAC.EVA XRD phase analysis software (Bruker) with reference to the International Center for Diffraction Data Powder Diffraction File 4+ database (ICDD PDF4+). Rietveld refinements for samples from Venetia and Gahcho Kué, which contain high abundances of both serpentine and smectite, were done using the Poorly Ordered or No Known Crystal Structure (PONKCS) method (Scarlett and Madsen, 2006) using TOPAS 5 (Bruker). More information about this method is provided in Supplementary Material S2.
Thermogravimetric Analyses
Twenty-one samples were analyzed using TGA at the University of British Columbia. This technique was used as a complementary approach to XRD to confirm the absence of brucite in the samples. Approximately 50 mg aliquots of McCrone milled samples were analyzed using a Perkin Elmer TGA 4000 with Polyscience chiller and an AS 6000 autosampler. The samples were heated at a rate of 10°C/min from 40 to 900°C with and inert carrier gas (N2) flowing at a rate of 19.8 mL/min to prevent oxidation.
Fourier Transform Infrared Spectroscopy
FTIR analyses were performed on 21 samples to confirm the presence of the mineral phases detected by XRD and in particular to confirm the trioctahedral characteristic of the smectites. Approximately 10 mg aliquots of McCrone milled samples were analyzed using a Nicolet iS50 FTIR spectrometer (Thermo Fisher Scientific). The spectrometer was equipped with a diamond crystal attenuated total reflectance (ATR) cell outfitted with a potassium bromide (KBr) beam splitter and a deuterated triglycine sulfate (DTGS) detector. The sample was deposited directly on the ATR diamond crystal and was clamped to the crystal surface with a pressure arm to ensure a good optical contact between the sample and the crystal. Data were collected using the OMNIC software. ATR-FTIR spectra represent an average of 32 scans collected between 400 and 4,000 cm−1, with a data spacing of 0.482 cm−1 and a resolution of 4 cm−1. A background (ambient atmosphere) spectrum was collected between each sample and subtracted from sample spectra in all cases.
Microscopy
Scanning Electron Microscopy
SEM analyses were performed to describe the distributions, textures and elemental compositions of minerals. Analyses were performed on polished thin and thick sections made from kimberlite ore samples using a Zeiss Sigma 300 VP-field emission gun SEM. Backscattered electron (BSE) images were acquired under variable pressure (VP) mode at an accelerating voltage of 25 kV (for uncoated samples) and at 15 kV (for C- or Au-coated samples), a working distance of ~7.5 mm and with a 30 μm aperture. The elemental composition of mineral phases was determined using energy dispersive X-ray spectrometry (EDXS) with a Bruker detector. EDXS data were analyzed using the ESPRIT software package (Bruker).
Raman Microspectroscopy
Two polished petrographic thin sections, 17VEN-K2-KIMB1 and 17GK-J-24, were analyzed to confirm the nature of phases observed with optical microscopy and SEM. For the latter sample, luminescence induced by the incident laser precluded Raman analysis. Raman spectra were recorded using a HORIBA LabRam HR Evolution spectrometer. Analyses used a 532 nm laser focused on the sample using an Olympus 100X LWD objective (NA = 0.80) and a confocal pinhole aperture of 50 μm, to obtain a planar resolution of ~ 1 μm2. The signal was filtered by edge filters and dispersed by a diffraction grating with 1,800 grooves/mm and the signal was analyzed with a HORIBA Synapse Plus CCD (charge-coupled device; 1,024–256 pixels) detector. Before each session, the spectrometer was calibrated using a silicon standard. Spectra were collected using the LabSpec6 software provided by HORIBA.
X-Ray Fluorescence Microscopy
Two kimberlite samples from Venetia mine (17VEN-K1-KIMB-1 and 17VEN-K2-KIMB-1) were prepared as thin sections on quartz slides for trace element analysis using XFM at the Australian Synchrotron, Clayton, Australia. X-ray Fluorescence Microscopy (XFM) allows for rapid quantitative elemental analysis over large areas (such as a geological thin section) at high resolution (using pixel sizes as small as ~1 μm) with ppb-level sensitivity (Paterson et al., 2011; Ryan et al., 2014; Howard et al., 2020). Scans were conducted with an incident monochromatic X-ray beam of 12.9 keV focused to ~2 μm using Kirkpatrick-Baez mirrors, a step size of 5 μm, and a dwell time of 20 ms/pixel. Elemental abundance data were collected using a Maia detector (Ryan et al., 2010, 2014) and the data were processed using the GeoPIXE software program (Ryan, 2000).
In addition, XFM mapping data were acquired at the GSECARS X-ray microprobe beamline, 13-ID-E, at the Advanced Photon Source (APS), Argonne National Laboratory, Illinois, USA over the course of two beamtime allotments. Those analyses were performed on one kimberlite sample from Gahcho Kué (17GK-J-24) (regular borosilicate glass slide) and one sample from Venetia (17VEN-K2-KIMB1) (quartz slide). The 13-ID-E station uses an APS 3.6 cm period undulator source, a cryogenically-cooled double crystal monochromator and microfocusing mirrors in a Kirkpatrick–Baez (KB) geometry to produce focused X-ray beams of 1 (V) × 2 (H) μm (Sutton et al., 2017). For the experiments described here, a Si(111) monochromator crystal set was used to provide incident beam energies between 16.5 and 18 keV with an incident flux to the sample (I0) between ~5 × 1011 and 2 × 1012 photons per s. Energy dispersive XRF spectra were collected using a Canberra/Mirion SX7, 7-element silicon drift diode detector. Pulse processing was done with a high-speed Quantum Xpress3 digital spectrometer system. The resultant XRF maps were processed using the Larch software package (Newville, 2013). Note that the XRF maps are RGB maps corresponding to the distribution of three elements plotted with the brightest color indicating the highest measured count rate (and black corresponding to the lowest count rate), and the color ranges are not linked to the concentration of one element vs. another element within the same map.
Results
Bulk Chemistry
The bulk elemental chemistry of the samples from Gahcho Kué and Venetia mines is summarized in Supplementary Table S1. The kimberlite facies that comprise Venetia ore have average Mg contents of 15.30 ± 2.07 wt.% and Ca contents of 5.68 ± 0.53 wt.%. The Mg and Ca contents of kimberlite ore from Gahcho Kué differ in that the average Mg concentration is higher (18.60 ± 2.56 wt.%) whereas the Ca content is lower (2.18 ± 1.54 wt.%). The Mg and Ca contents of the processed kimberlites (FRD and CRD) from Venetia are lower compared to the unprocessed ore with average values of 12.10 ± 2.27 wt.% for Mg and 4.50 ± 0.21 wt.% for Ca (FRD) and 13.39 ± 2.97 wt.% for Mg and 5.00 ± 0.10 wt.% for Ca (CRD). These differences are likely due to the incorporation of more silica-rich country rocks (mainly biotite gneiss, biotite schist, and amphibolite) during the mining process.
It is also important to consider the first-row transition metal abundances of kimberlites and other ultramafic rocks because carbonation technologies can mobilize these valuable (and potentially toxic) elements (Hamilton et al., 2016, 2018, 2020). We compared the concentrations of these elements within kimberlites vs. their concentrations within CI chondrites (primitive meteorites) to describe weather kimberlites are enriched or depleted compared to the primitive mantle. Some of the first-row transition elements, such as Ti and V, are more concentrated in the studied kimberlites compared to CI chondrites (McDonough and Sun, 1995). Conversely, Fe, Mn, Ni, Cr are less concentrated in the kimberlites compared to CI chondrites. More generally, the concentrations of transition metals in kimberlites from Venetia and Gahcho Kué are comparable with average kimberlite compositions globally (Mitchell, 1986). The concentration of Cr is higher in Gahcho Kué ore (1,338 ± 160 ppm) compared to the Venetia ore (1,084 ± 270 ppm). Regarding the concentration of total carbon, the Venetia ores show a higher content compared to the Gahcho Kué ores (0.67 ± 0.36 wt.% vs. 0.12 ± 0.10 wt.%). The total carbon content of the Venetia CRD is greater than that of the Venetia FRD (0.73 ± 0.13 wt.% vs. 0.58 ± 0.09 wt.%).
Mineralogy of the Ore and Processed Kimberlites From Venetia and Gahcho Kué Mines
Quantitative mineralogical results determined by Rietveld refinement with XRD data are provided in Table 2. Serpentine minerals and smectites are the most abundant phases in most samples. Supplementary Figure S1 shows an example Rietveld refinement plot for the ore sample 17VEN-K2-KIMB1 (i.e., K2 pipe, MVK facies, Venetia mine). This sample contains 12 detectable mineral phases illustrating the mineralogical complexity of the samples. Lizardite is abundant in ore samples from Venetia in proportions varying from 10.8 wt.% (17VEN-K2-KIMB2, K2 pipe, VKBR facies) to 58.4 wt.% (17VEN-K3-KIMB4, K3 pipe, CKW facies). This mineral phase is also particularly abundant in the samples from Gahcho Kué with proportions varying broadly from 19.2 wt.% (17GK-J-24, TKt facies) to 90.0 wt.% (17GK-22, HK facies). Smectite is present in all samples from the two mines ranging from 6.2 wt. % (17VEN-K3-KIMB2, K3 pipe, CK facies) to 39.9 wt.% (17VEN-K2-KIMB2, K2 pipe, VKBR facies) at Venetia and from 2.3 wt.% (17GK-22, HK facies) to 44.1 wt.% (17GK-J-24, TKt facies) at Gahcho Kué.
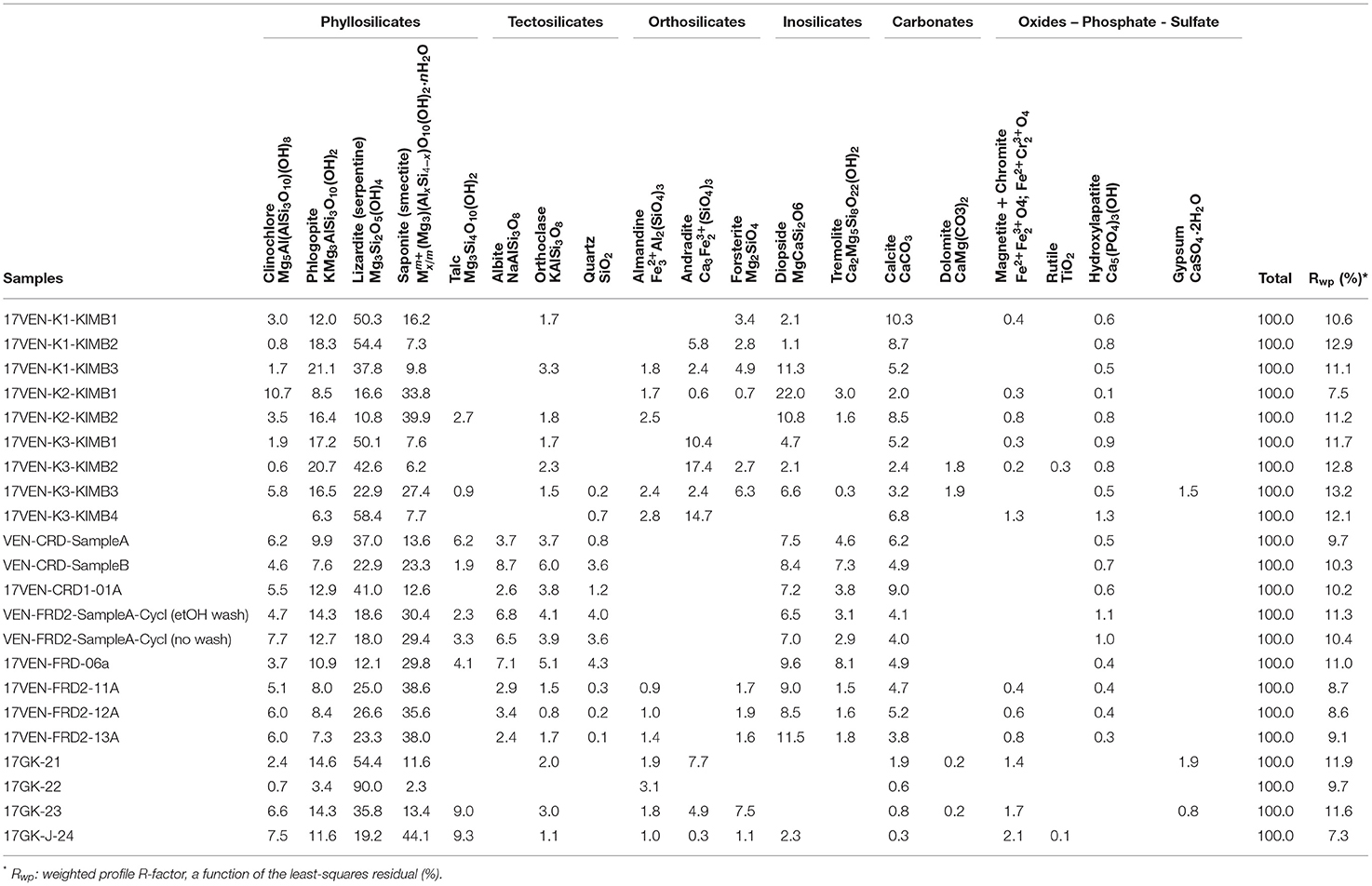
Table 2. Quantitative mineralogy results from Rietveld refinement.
Calcite (CaCO3) is present in all samples from Venetia with proportions varying from 2.0 wt.% (17VEN-K2-KIMB1, K2 pipe, MVK facies) to 10.3 wt.% (17VEN-K1-KIMB1, K1 pipe, CKNE facies). Calcite is likely to have been inherited from both the kimberlite ore and from the country rock in the FRD and CRD samples from Venetia. Notably, calcite is present at low abundance (<2 wt.%) in the four ore samples from Gahcho Kué. In addition to calcite, dolomite [CaMg(CO3)2] is either absent or present at low abundance (<2 wt.%) in the samples from the two mines. The calcite content determined via Rietveld refinement with XRD data and calculated from the bulk TIC analyses (measured by coulometry analyses) of all samples are in excellent agreement (Figure 3), supporting the accuracy of the Rietveld refinement results.
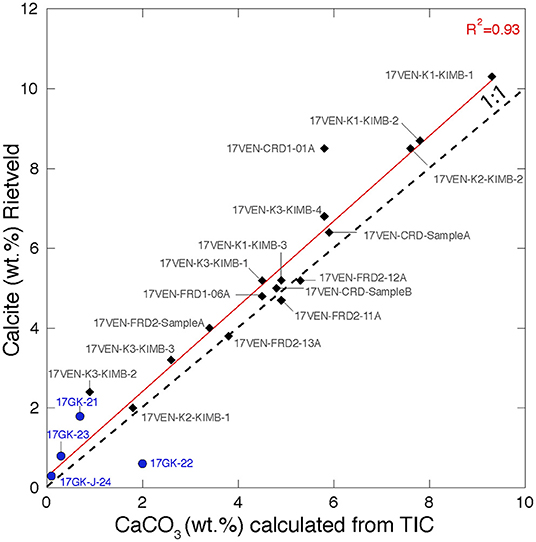
Figure 3. Biplot representing CaCO3 abundances calculated from bulk TIC analyses (measured by coulometry) vs. the calcite content (wt.%) determined by Rietveld refinement. The R2 of the regression line is 0.93 and close to the 1:1 line supporting the accuracy of the calcite estimation using Rietveld refinement. Samples from Venetia are represented by black diamonds and samples from Gahcho Kué are represented by blue circles. Samples from the Gahcho Kué mine are calcite and TIC-poor.
Brucite is difficult to detect with XRD in kimberlite samples since its most intense peak (i.e., 001 at 4.78 Å, 21.58 ° 2θ), and its second (i.e., 101 at 2.37 Å, 44.39 ° 2θ) and third (i.e., 102 at 1.80 Å, 59.71 ° 2θ) most intense peaks, all overlap with peaks of clinochlore [Mg5Al(AlSi3O10)(OH)8]. Clinochlore is absent in one sample (17VEN-K3-KIMB4) and is present in all the other samples from Venetia and Gahcho Kué with proportions varying from 0.6 wt.% (17VEN-K3-KIMB2, facies CK) to 10.7 wt. % (17VEN-K2-KIMB1, facies MVK). Brucite was not detected from XRD data in any of the samples from Venetia or Gahcho Kué. Nor could it be detected using any of the microscopy or spectroscopy techniques used in this study. Finally, TGA analyses were performed using the method of Assima et al. (2013a) to detect brucite at low abundances in mineralogically complex samples. None of the 21 samples showed TG weight loss between 250 and 400°C, which is the temperature range over which dehydroxylation of brucite occurs (Assima et al., 2013a) (Supplementary Figure S2).
The modal proportions of minerals differ substantially between samples of processed kimberlite and ore. For instance, quartz content is higher in processed kimberlites, reaching 4.3 wt.% for a FRD sample from Venetia (17VEN-FRD-06a), whereas quartz is absent or present at much lower abundance for ore from the two mines. Similarly, albite (NaAlSi3O8) is detectable in the FRD and CRD samples from Venetia while absent in Venetia and Gahcho Kué ore. Tremolite [□Ca2Mg5Si8O22(OH)2, where □ indicates a vacancy in the crystal structure], is also systematically present in the processed kimberlites while absent or present at trace or minor abundance in Venetia ore and absent in Gahcho Kué ore. Contrastingly, some minor phases that are detectable in ore (spinels, garnets, forsterite) are commonly undetectable in samples of processed kimberlites. The mineralogy of Venetia FRD (17VEN-FRD2-SampleA-Cyclone) and CRD (17VEN-CRD-SampleB) is similar. The most noticeable difference between these FRD and CRD samples is the smectite content, which is higher in the FRD sample (30.4 wt.% in FRD samples vs. 23.3 wt.% in CRD). This difference is consistent with fine-grained clay minerals concentrating preferentially into FRD.
The crust samples collected from FRD2 at Venetia (17VEN-FRD2-crust-11A,−12A, and−13A) contain particularly high proportions of smectite (35.6–38.6 wt.%). These three samples show a similar composition and can be distinguished from the three other FRD samples (VEN-FRD2-SampleA-Cyclone, VEN-FRD2-SampleB and 17VEN-FRD-06a) by a higher lizardite content, the trace presence of olivine, spinels (e.g., chromite) and the absence of talc. The six samples of processed kimberlite and crusts from Venetia have a similar mineralogical composition to that of the MVK ore, which is consistent with MVK being the dominant ore mined at the time of sampling. The mineralogy of the kimberlite ore from Gahcho Kué and Venetia differs. In particular, ore from Gahcho Kué is calcite- and diopside-poor which contrasts with most of the Venetia ore.
FTIR analyses are consistent with XRD results. Supplementary Figure S3 shows three FTIR spectra of samples with distinct mineralogical compositions and abundances: (1) a kimberlite ore (17VEN-K1-KIMB2, K1 pipe, facies CKNE/MVK) from Venetia containing major lizardite (54.4 wt.%), minor smectite (7.3 wt.%) and no talc, (2) a crust sample from FRD2 at Venetia (17VEN-FRD2-12A, MVK) containing smectite (35.6 wt.%), lizardite (26.6 wt.%) and no talc and (3) a kimberlite ore (17GK-J-24, J; TKt; West Lobe) from Gahcho Kué, containing smectite as the most abundant phase (44.1 wt.%), lizardite (19.2 wt.%) and talc (9.3 wt%). Lizardite is consistently detected in the samples with bands at ~939 and ~1,086 cm−1 (Supplementary Figure S3A), interpreted as two degenerate equatorial stretching modes of Si-O bonds and the symmetric stretch of apical Si-O, respectively (Balan et al., 2002). In the hydroxyl region (Supplementary Figure S3B), lizardite is characterized by a sharp peak at 3,683 cm−1, interpreted as in-phase stretching of the three inner surface OH groups (Balan et al., 2002). Two broad bands at ~3,580 and ~3,643 cm−1 are interpreted as the result of minor Fe substitution for Mg in octahedral sites and two degenerate out-of-phase stretching modes of the inner surface OH groups (Balan et al., 2002). The peak at 3,683 cm−1 is not observed for the kimberlite ore from Gahcho Kué (17GK-J-24). This might be due to the high amount of smectite and talc in this sample as quantified with Rietveld refinement (Table 2). A sharp band at 3,674 cm−1 is observed for this last sample and observed in sample 17VEN-FRD2-12A as well. This band is classically indicative of Mg3-OH stretching vibrations, characteristic of Mg in octahedral coordination such as in trioctahedral smectites (saponite and stevensite) and talc (Farmer and Russell, 1964; Madejová and Komadel, 2005). Additional broad bands at ~3,226, ~3,416–3,445, and ~3,620–3,630 cm−1 are observed in the hydroxyl region and are interpreted as the H2O bending vibration, the OH stretching associated with interlayer H2O and Al3-OH stretching, respectively, in trioctahedral smectites (Farmer, 1974; Tosca and Masterson, 2014; Kloprogge and Ponce, 2021). Finally, a band observed at ~1,640 cm−1 in the smectite-rich samples only is attributed to the H-O-H bending vibrations of H2O molecules located in the interlayer space of the smectite (Kloprogge et al., 2006). Consistent with XRD data, calcite is detected in some samples from Venetia (e.g., 17VEN-K1-KIMB2 and 17VEN-FRD2-12A, Supplementary Figure S3A) with bands at 713, 875, and 1,428 cm−1 (Huang and Kerr, 1960).
Cation Exchange Capacity of Venetia and GK Ores and Mine Residues
The two parameters describing the cation exchange capacity of each sample (i.e., CEC-cations and CEC-NH4-N), both normalized to specific surface area, lack a significant positive correlation (R2 = 0.3) (Figure 4A). The CEC-cation values lie above the 1:1 line indicating that additional processes are occurring in solution, potentially including dissolution of very soluble phases and/or desorption of cations. CEC determined using the NH4-N concentrations in leachates is positively correlated to smectite abundance as determined with Rietveld refinement (Figure 4B, R2 = 0.58), indicating that smectite content within kimberlite drives the reactivity of these rocks. A positive trend is observed between the calcite content of the samples and the Ca released following the NH4-acetate leach (Figure 4C), supporting the hypothesis that a fraction of bedrock carbonate may dissolve during the process.
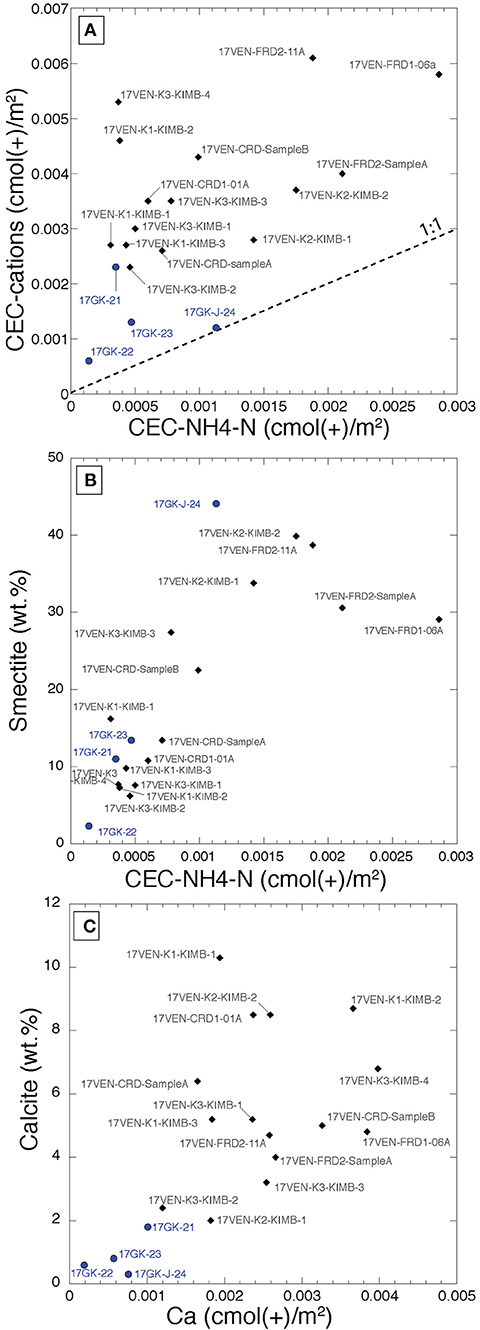
Figure 4. Biplots representing (A) the CEC determined by the NH4-N concentrations in the KCl leachates (CEC-NH4-N) vs. the CEC determined by the sum of the cations measured in the first NH4-acetate leachates (both BET corrected). No significant correlation exists (R2 = 0.3); (B) CEC-NH4-N (normalized to the BET) vs. smectite content (wt.%) determined by Rietveld refinement. The R2 of the linear fit is 0.58; (C) Ca contents of the NH4-acetate leachates normalized to the BET [cmol (+)/m2] vs. the calcite content (wt. %) determined by Rietveld refinement. The R2 of the fit is 0.39. Samples from Venetia are represented by black diamonds and samples from Gahcho Kué are represented by blue circles.
CEC values can also be expressed in g/kg and data are available for CEC for each of Mg, Ca, K and Na at Venetia and Gahcho Kué (Supplementary Table S2). Except for 17VEN-FRD2-11A, Ca is the most abundant element extracted from the calcite-rich Venetia samples, reaching a maximum of 10.43 g/kg for sample 17VEN-K2-KIMB2. The amount of Ca extracted is much lower in the non-pulverized sample, 17VEN-CRD-SampleB, compared to the pulverized samples (and the two other CRD samples). Ca is the main cation extracted in the calcite-poor samples from Gahcho Kué, reaching 6.74 g/kg for the smectite-rich sample, 17GK-J-24, suggesting that smectite is the most important source of Ca at Gahcho Kué. Mg is the second most important cation extracted from Gahcho Kué samples, with values ranging from 1.07 g/kg (17GK-23) to 2.38 g/kg (17GK-J-24). Sample 17VEN-FRD2-11A shows the highest amount of Na extracted (12.63 g/kg). The two kimberlite samples from the K2 pipe at Venetia also show high Na extraction of 2.82 and 3.93 g/kg. Processed kimberlite samples show Na extraction varying from 1.23 g/kg (17VEN-CRD1-01A) to 2.72 g/kg (17VEN-FRD2-cyclone-SampleA). No significant correlations were observed between smectite content and the Ca, Mg, Na and K concentrations measured in the leachates during CEC analyses (Supplementary Figure S4), suggesting that different cations, with different proportions are populating the interlayer spaces of the smectites.
Distribution of Mineral Phases and Transition Elements Within Kimberlites
Lizardite is an abundant mineral phase in samples from Venetia and Gahcho Kué. It is distributed as (1) pseudomorphosed forsterite macrocrysts with grain sizes between 200 μm and 1 cm and (2) microcrysts with grain sizes inferior to 200 μm in all the ore samples from Venetia and Gahcho Kué (Figures 5, 6). Relict forsterite was observed in particularly large macrocrysts where serpentinization was incomplete (Figure 5). This relict forsterite tends to be more Fe-rich (brighter in backscattered electron mode) compared to the lizardite (Figures 5C–E). Nickel is homogeneously distributed with lizardite as shown in the XFM map of sample 17GK-J-24 from Gahcho Kué (Figure 6B). Rims composed of talc [Mg3Si4O10(OH)2], Ti-rich particles (likely ilmenite, FeTiO3) and phlogopite [KMg3AlSi3O10(OH)2] are distributed around lizardite grains in sample 17GK-J-24 (Figure 6). Smectite is distributed as a fine-grained matrix around the lizardite pseudomorphs after forsterite micro- and macrocrysts (Figure 6). XFM maps show the distribution of (1) K, which is mainly borne by phlogopite, (2) Ca, which is associated with smectite and (3) Ni, which is associated with lizardite (Figure 6B). Smectite is also distributed around calcite macrocrysts (Figure 7).
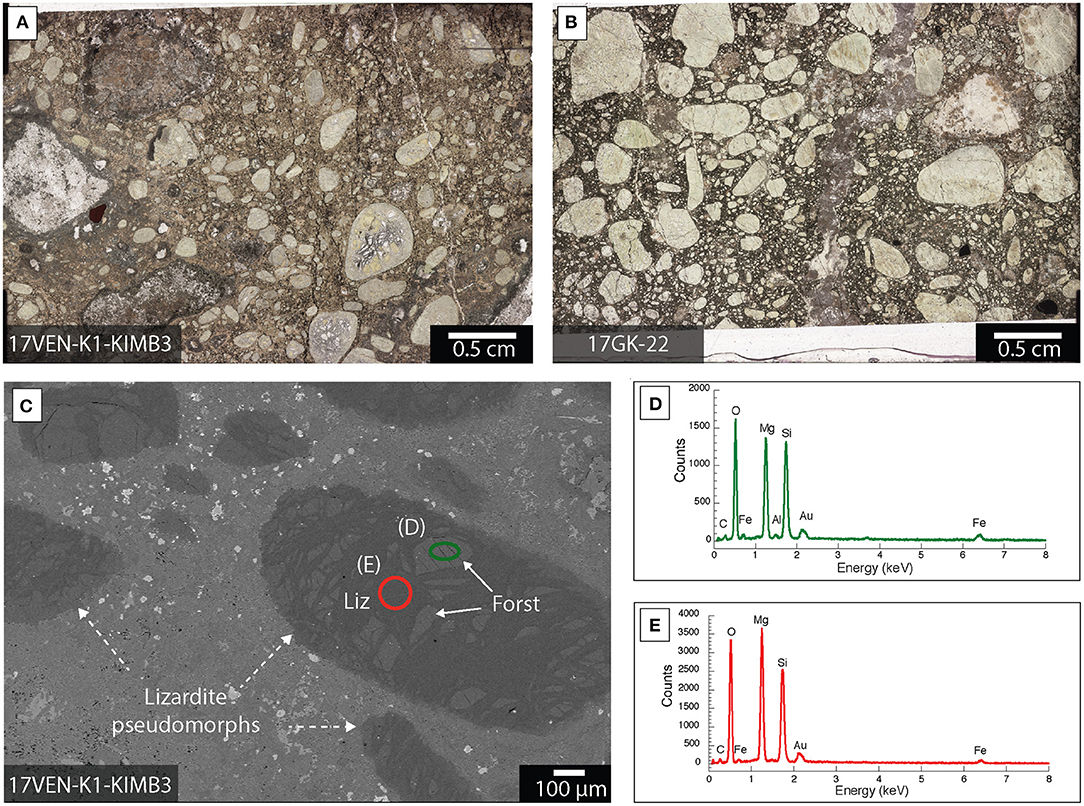
Figure 5. Lizardite distribution within kimberlite ores from Venetia and Gahcho Kué. (A,B) Optical microscope images of the entire petrographic thin section for each of two different kimberlite ores; (C) Backscattered electron image of a petrographic thick section showing large lizardite (Liz) pseudomorphs grains (dashed arrows) containing regions with relict forsterite (Forst); (D,E) EDXS spectra corresponding to the areas circled in (C). The sample was Au-coated.
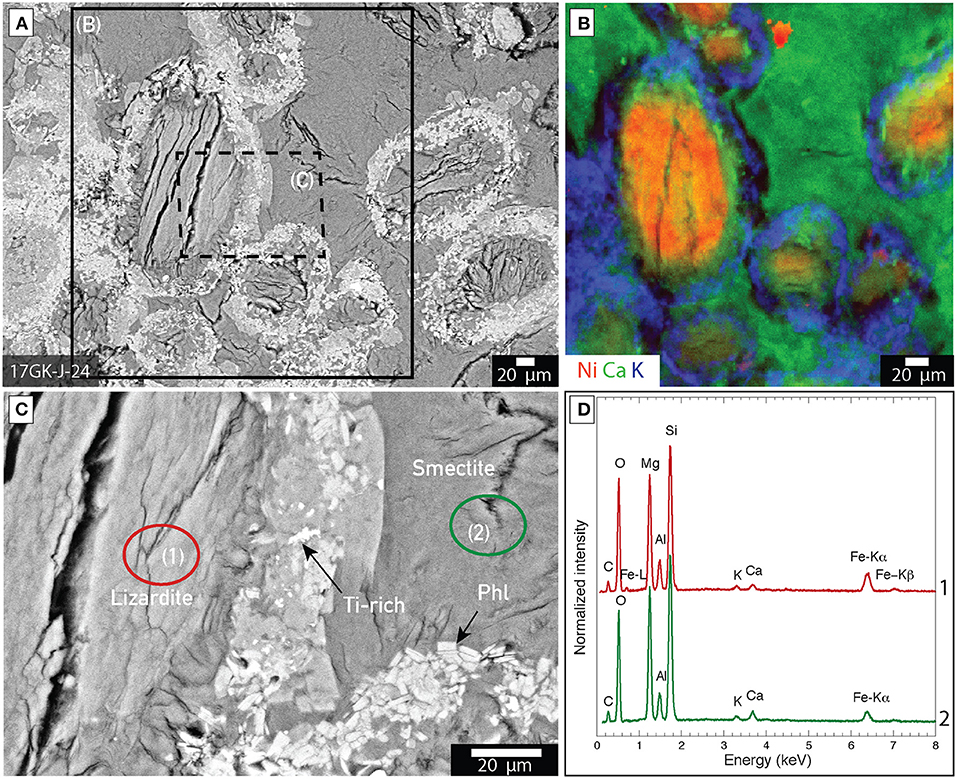
Figure 6. Distribution of mineral phases within kimberlite ores from Gahcho Kué (17GK-J-24). (A) Backscattered electron image of a petrographic thick section showing lizardite grains (after forsterite) surrounded by bright rims and a smectite matrix; (B) XFM map of Ni (red), Ca (green) and K (blue) of the area marked by a solid rectangle in (A). The red, green and blue colors correspond to the measured intensities of the Ni K-alpha, Ca K-alpha and K K-alpha emission lines, with the brightest color, the highest measured count rate and black corresponding to the lowest count rate; (C) Close up corresponding to the dashed rectangle in (A) showing that the rim is composed of talc, Ti-rich grains (Ti), likely ilmenite, and phlogopite (Phl); (D) SEM-EDXS spectrum of the circled area in (B).
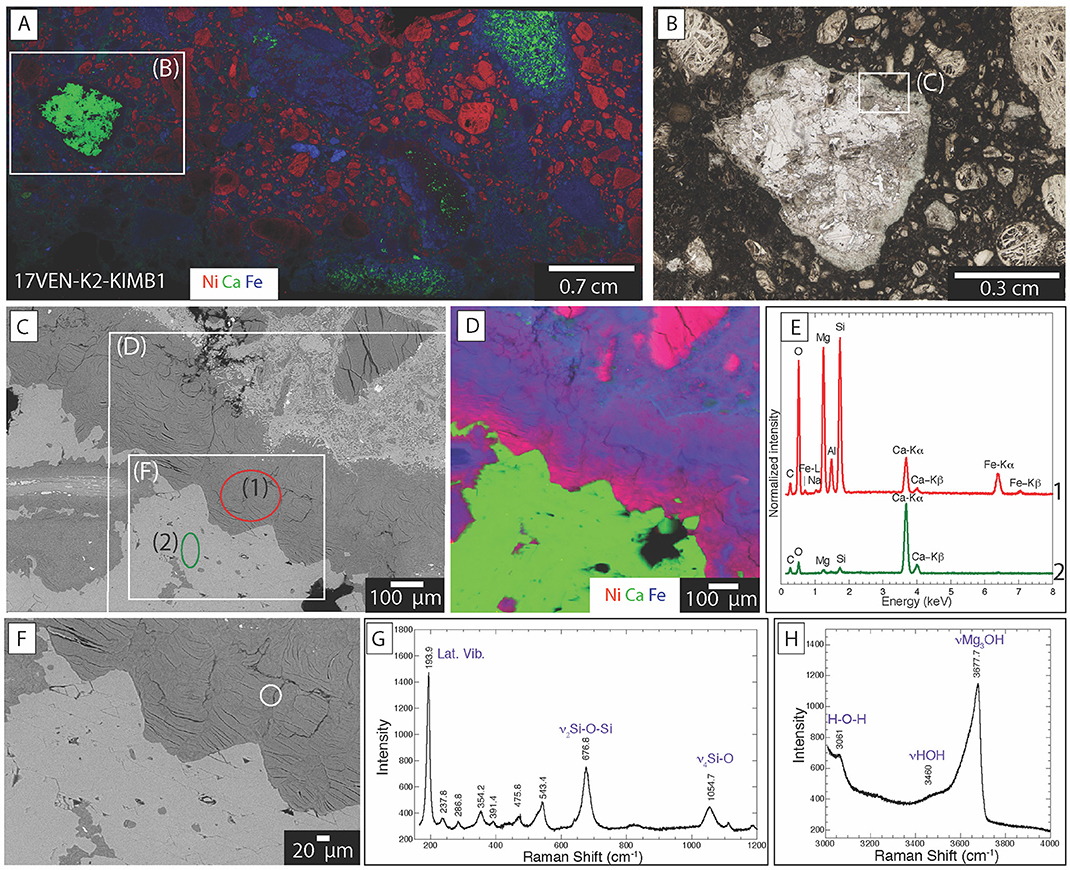
Figure 7. Distribution of mineral phases within kimberlite ores from Venetia (17VEN-K2-KIMB1) (A) XFM map showing the distribution of Ni (red), Ca (green) and Fe (blue) of the entire thin section. The red, green and blue colors correspond to the measured intensities of the Ni K-alpha, Ca K-alpha and Fe K-alpha emission lines, with the brightest color, the highest measured count rate and black corresponding to the lowest count rate; (B) Optical photograph of the area marked by a rectangle on (A) showing agglomeration of calcite crystals; (C) Backscattered electron image of the area marked by a rectangle in (B) at the interface between the matrix and the polycrystalline calcite grain; (D) XFM map showing the distribution of Ni, Ca and Fe. The red, green and blue colors correspond to the measured intensities of the Ni K-alpha, Ca K-alpha and Fe K-alpha emission lines, with the brightest color, the highest measured count rate and black corresponding to the lowest count rate. (E) Backscattered image of the area marked on (C); (G,H) Raman spectra of the areas circled in (F).
The smectite at Venetia and Gahcho Kué systematically showed the same chemical composition when analyzed using SEM-EDXS: this mineral phase is Fe- Ca- Mg- and Al-rich (Figures 6, 7). However, SEM-EDXS (Figure 6D) cannot be used to discern which of these elements is found in the interlayer vs. the octahedral sheets of the smectite structure. Raman analyses reveal the presence of main bands at 193.9, 676.8, and 1,054.7 cm−1 (Figure 7G), corresponding to lattice mode vibrations, the ν2 internal symmetric stretching of Si-O-Si bonds and the ν4 internal symmetric stretching of the Si-O bonds of saponite, respectively (Wang et al., 2015; Kloprogge and Ponce, 2021). The exact position of the band at 676.8 cm−1 confirms the trioctahedral nature of the smectite which is different for dioctahedral smectite (e.g., 709 cm−1 for montmorillonite) (Wang et al., 2015). In the hydroxyl stretching region, an intense band at 3,675 cm−1 is observed and attributed to ν(Mg3OH) vibrations and the broad band near 3,460 cm−1 is attributed to ν(HOH) of interlayer H2O molecules (Kloprogge, 2017) (Figure 7H).
Calcite is distributed as polycrystalline aggregates, covering areas measuring up to 0.8 cm across (Figures 7A,B). Calcite is also observed as altered areas measuring from 10 μm up to several 100 μm (Supplementary Figure S5). Calcite was identified by Raman analyses based on a main band at 1,084.5 cm−1 corresponding to the ν1 internal symmetric stretching of the C-O bonds in the carbonate group (Perrin et al., 2016). Based on this exact position, the Mg content is almost zero. This is in accordance with SEM-EDXS analyses and the d(104) position of calcite at 3.04 Å in XRD patterns (Lumsden, 1979). Even though dolomite was detected in some samples with XRD, it was not possible to observe it in these samples due to its low abundance.
Discussion
Mineralogical Heterogeneity
The kimberlite ore and processed kimberlites from Gahcho Kué and Venetia mines display complex and diverse mineralogical compositions. This diversity was highlighted by Mervine et al. (2018) for kimberlites from different mines (Venetia, Gahcho Kué, Victor, Snap Lake) and from different lithofacies within a single mine or pipe. The mineralogical diversity of kimberlites from different locations in Russia (Archangelsk and Yakutia provinces) and China (Liaonin and Shandong provinces) has also been described by Posukhova et al. (2013). In particular, these last authors were able to subdivide kimberlites into four main groups based on their mineralogy: (i) saponite-rich, (ii) serpentine-rich, (iii) carbonate and phlogopite-rich and (iv) rocks with a complex mineralogical composition. The samples studied here belong to these four groups. This mineralogical diversity can be explained by differences in parent melt compositions, different degrees of hydrothermal metamorphism, the extent of meteoric alteration, the type of country rocks that the kimberlites intrude and variation in mineral processing techniques.
Whereas, kimberlites are commonly described as olivine-rich igneous rocks (e.g., Mitchell et al., 2019), olivine was either not detected or present at low abundance (<2 wt.%) in the majority of kimberlite samples. It reached maximum abundances of 7.5 wt.% (for 17GK-23) and 6.3 wt. % (17VEN-K3-KIMB3) for samples from Gahcho Kué and Venetia, respectively. Conversely, lizardite is the most abundant mineral in the majority of the samples. Kimberlite deposits are typically serpentinized as a result of the reaction of olivine and water during hydrothermal metamorphism, where olivine macrocrysts are pseudomorphed by lizardite (e.g., Stripp et al., 2006; Afanasyev et al., 2014). This hydrothermal metamorphism also results in the precipitation of carbonate minerals and other phyllosilicate phases such as smectites. Although the precise origin of smectites within kimberlites is still not clear, several hypotheses have been put forward to explain their formation. The source of Al3+ to form chlorite and smectite is attributed by Stripp et al. (2006) to alteration of plagioclase. Posukhova et al. (2013) suggested that saponite is a pseudomorph of olivine in autolith breccias of the Archangelsk, Pionerskaya, Lomonosov and Karpinsky pipes in Russia. White et al. (2012) also observed the partial or complete replacement of phlogopite by saponite. Since such replacements are not observed here, one hypothesis cannot be preferred.
An important difference between the kimberlite ore samples from Venetia and Gahcho Kué is their carbonate content. Mg-poor calcite and, to a lesser extent, dolomite are present in ore and processed kimberlites from Venetia. Calcite is present at 2.0 to 10.3 wt.% in ore from Venetia but is much less abundant (<2 wt.%) in ore from Gahcho Kué. The presence of carbonates is commonly invoked as support of either metasomatic CO2 enrichment of the source or derivation from low-degree partial melting of peridotite (e.g., Giuliani and Pearson, 2019). This mineralogical difference is of prime importance for choosing a mineral carbonation strategy (Paulo et al., 2021), which will be further discussed in Section A Decision Tree for Mineral Carbonation in Kimberlites.
Sources of Mg and Ca for Mineral Carbonation
Brucite
Carbonation of brucite-rich mine residues has been documented in tailings storage facilities at ambient temperature and atmospheric pCO2 (e.g., Oskierski et al., 2013, 2019; Wilson et al., 2014; Lechat et al., 2016; Gras et al., 2017; Hamilton et al., 2018; Turvey et al., 2018). This unassisted CO2 sequestration process is commonly described as passive carbonation. The lack of strong Si-O bonds within brucite makes it orders of magnitude more reactive than the more abundant Mg-silicates in ultramafic rocks (Harrison et al., 2013). Accelerated carbonation of high-purity brucite (Harrison et al., 2013, 2015) and brucite in serpentinite tailings (Hamilton et al., 2020) has been demonstrated in laboratory experiments using high-CO2 gases.
Despite the results of past studies, which reported the presence of brucite in kimberlites from South African mines, including the Venetia mine (Berg, 1989; Stripp et al., 2006), we detected no brucite using bulk methods (XRD, FTIR, TGA). Nor did we observe any brucite with the spatially resolved techniques used in this study (optical microscopy, SEM-EDSX, XFM). The absence of brucite in the samples from Venetia and Gahcho Kué mines necessitates the development of other strategies for mineral carbonation.
Serpentine Minerals
The serpentine polymorphs are of particular interest owing to their high Mg content, large surface areas, and relatively fast dissolution rates, which make them good feedstocks for carbonation reactions (e.g., Power et al., 2013). Acid leaching has long been used to accelerate silicate mineral dissolution (Park et al., 2003) and pH-swing methods (Park and Fan, 2004) allow optimization of conditions firstly (1) to enhance silicate dissolution at low pH (step 1), then (2) to promote carbonate mineral precipitation at high pH (step 2). Repeated heap leaching of ultramafic tailings from the Woodsreef chrysotile mine (Australia) with dilute sulfuric acid was tested by Hamilton et al. (2020) and shown to provide 14–25 × the CO2 sequestration potential of brucite carbonation alone using the same tailings. However, this method is not appropriate for carbonate-rich samples, which are already storing a substantial amount of CO2 in minerals that are highly soluble at low pH.
Other Silicate Minerals (Excluding Smectites)
Other phyllosilicate minerals present in kimberlites, such as talc, clinochlore and phlogopite are not sufficiently reactive (e.g., Brantley and Olsen, 2014) to contribute appreciably to the carbonation potential of processed kimberlites. Dissolution of these phyllosilicate minerals necessitates temperatures above 100°C (Saldi et al., 2007; Smith et al., 2013), injection of supercritical CO2 with SO2 and O2 (Pearce et al., 2016) or longer timeframes for reaction using approaches such as enhanced rock weathering in soils (Paulo et al., 2021; Stubbs et al., 2022). Tremolite has a slow dissolution rate under acidic pH (i.e., similar to talc) and due to its crystal structure, the removal of Ca tends to promote the breaking of tremolite crystals into small needle-shaped particles, potentially toxic to human lungs (Diedrich et al., 2014). Pyroxenes have received some attention as feedstocks for mineral carbonation (e.g., Stockmann et al., 2008; Daval et al., 2011), however their slow dissolution rates make them less suitable for mineral carbonation (Power et al., 2013). The reactivity of Mg-rich pyrope to CO2 is largely unknown, although, in theory, garnets should dissolve relatively quickly (e.g., Palandri and Kharaka, 2004) since they lack silica polymerization like the fast-dissolving olivine minerals.
Smectites as a New Resource for Mineral Carbonation in Kimberlites
Smectites are overlooked in petrographic descriptions of kimberlites, which tend to focus on primary minerals, such as forsterite, that have long been replaced rather than on the current mineralogical composition of kimberlites. As notable exceptions, smectite was detected with XRD by Posukhova et al. (2013) within kimberlites from the Chidviya pipe (Izhmozero, Archangelsk province), Snegopadnaya pipe (Daldyn) and Chernyshevsk pipe (Muna), Yakutia province, Russia. In addition, saponite was noted within the Wesselton kimberlite pipe (Kimberley, South Africa) (White et al., 2012). Smectite behavior in kimberlites was the focus of work by Morkel et al. (2006, 2007). The presence of saponite in kimberlite has mainly been highlighted in hydrometallurgy and water reclamation studies, where methods of cleaning saponite-containing water from diamond processing plants were tested (O'Gorman and Kitchener, 1974; Chanturiya et al., 2018). Saponite was observed as the dominant mineral of the slime fraction (i.e., very fine residues) of the kimberlites at the Cullinan mine (previously named Premier Mine), South Africa, where methods of coagulation and flocculation were investigated to simplify water recovery and recycling (O'Gorman and Kitchener, 1974). Saponite was also described as the main mineral of the clay fraction of kimberlites from the Lomonosov deposit, Archangelsk province, Russia (Chanturiya et al., 2018), where electrochemical separation of saponite from process water and the potential of saponite to host heavy metals were discussed. Our work calls for a better petrographic and crystallographic description of the smectite fraction of kimberlites owing to its potential utility for mineral carbonation. Smectites, not brucite, provide the labile Mg and Ca needed for mineral carbonation in kimberlites.
The smectite phase at Venetia and Gahcho Kué is the Al-, Fe-, Mg- and Ca-rich, trioctahedral species, saponite. Every kimberlite facies studied at both mines contains saponite. Specific facies of kimberlite ore from Gahcho Kué and Venetia are particularly saponite-rich: transitional tuffisitic kimberlite (Tkt) at Gahcho Kué and layered volcaniclastic kimberlite breccia (VKBR) and massive volcaniclastic kimberlite (MVK) at Venetia, with smectite abundances of 44.1, 39.9, and 33.8 wt.%, respectively. The reactivity of smectites has been well studied for industrial applications (e.g., Murray, 2000; Komadel, 2003; Krupskaya et al., 2017). For example, a traditional application of smectites is their use as a component of barrier systems for (radioactive) waste disposal (Krupskaya et al., 2017). In the context of CCUS, the smectite minerals, montmorillonite [(Al2−xMgx)Si4O10(OH)2·nH2O] and fluorohectorite [(Mg3−xLix)Si4O10F2·nH2O], are able to host CO2 within their interlayer spaces under conditions relevant to direct injection of supercritical CO2 into subsurface reservoirs (Loring et al., 2012; Michels et al., 2015). The dissolution rate of smectite is similar to that of chlorite (logk= −11 mol/m2/s) under acidic conditions at 25°C (Palandri and Kharaka, 2004). Dissolution of smectites poses the same kinetic restrictions as dissolution of serpentine and olivine minerals; it also produces highly reactive amorphous silica which initiates the thermodynamic battle between carbonate and silica for Mg. Alternatively, labile cations from the interlayer spaces of smectites are easily and rapidly exchanged using cation exchange reactions (Odom, 1984), such as the quick and routine CEC test used in this study.
The interlayer cations in smectites are easily exchanged without modifying the T-O-T structure, i.e., the octahedral layer (O) sandwiched between two tetrahedral silicate layers (T). The cation exchange capacity of smectites is high (Schiffman and Southard, 1996) and the exchange rate is rapid (Komadel, 2003). It is achievable in water undersaturated conditions and in humid air (Wilson et al., 2011; Wilson and Bish, 2012), meaning that low water–rock ratios can be used to drive cation exchange reactions. This process also leads to the liberation of Mg and Ca for mineral carbonation without releasing silica, offering the advantage of avoiding the precipitation of amorphous silica and/or Mg-silicate.
Kang and Roh (2013) experimentally explored the reactivity of vermiculite (another swelling clay with a greater layer charge than smectites) for mineral carbonation with a focus on cation exchange. To our knowledge, only Morkel et al. (2006, 2007) have previously published a detailed description of the crystal chemistry, CEC and abundances of smectites within kimberlites. The focus of their work was on the role of smectite swelling and cation exchange in kimberlite weathering, with the goal of making ore processing less energy intensive. We observed that saponite occurs as a fine matrix within the samples from Gahcho Kué and Venetia. The fine particle size of saponite is of prime importance for its increased surface reactivity and makes it a good target for producing high-Mg and -Ca leachates via cation exchange reactions.
Maximum Extractable Mg and Ca: Potential for CO2 Offsets
Previous studies have relied on the fast reactivity of brucite for mineral carbonation using serpentinites (e.g., Wilson et al., 2010; Pronost et al., 2011; Harrison et al., 2013; Wilson et al., 2014). The analogous Ca-hydroxide mineral, portlandite [Ca(OH)2], is also highly reactive to CO2; while it is vanishingly rare in nature, it is a key component of Portland cement and its carbonation in air is one of the reactions responsible for the setting of cement. We calculated the equivalent amount of brucite and portlandite reactivity that CEC from smectites provides (Table 3) to provide a comparator to previous estimates of serpentinite reactivity based on brucite content. Details of the calculations are provided in Supplementary Material S3. The CEC of smectites is equivalent to between 0.13 and 0.43 wt.% “brucite” at Venetia. At Gahcho Kué, the reactivity of smectites is equivalent to 0.26–0.51 wt.% “brucite”. In comparison, ore from the Baptiste nickel deposit (Decar district, British Columbia, Canada), which is dominated by serpentine minerals, contains 0.6–12.6 wt.% of brucite (Power et al., 2020) with an average value of 1.8 wt.% (Vanderzee and Dipple, 2019). The brucite content of the Woodsreef chrysotile mine (new South Wales, Australia) is very similar at 2.2 wt.% (Turvey et al., 2018). An average of 2.5 wt.% brucite is present in the tailings produced at Mount Keith nickel mine (Western Australia). This mine offsets ~11% of its annual greenhouse gas emissions via passive carbonation, although only half of the brucite is carbonated before being buried under freshly deposited tailings and cut off from atmospheric CO2.
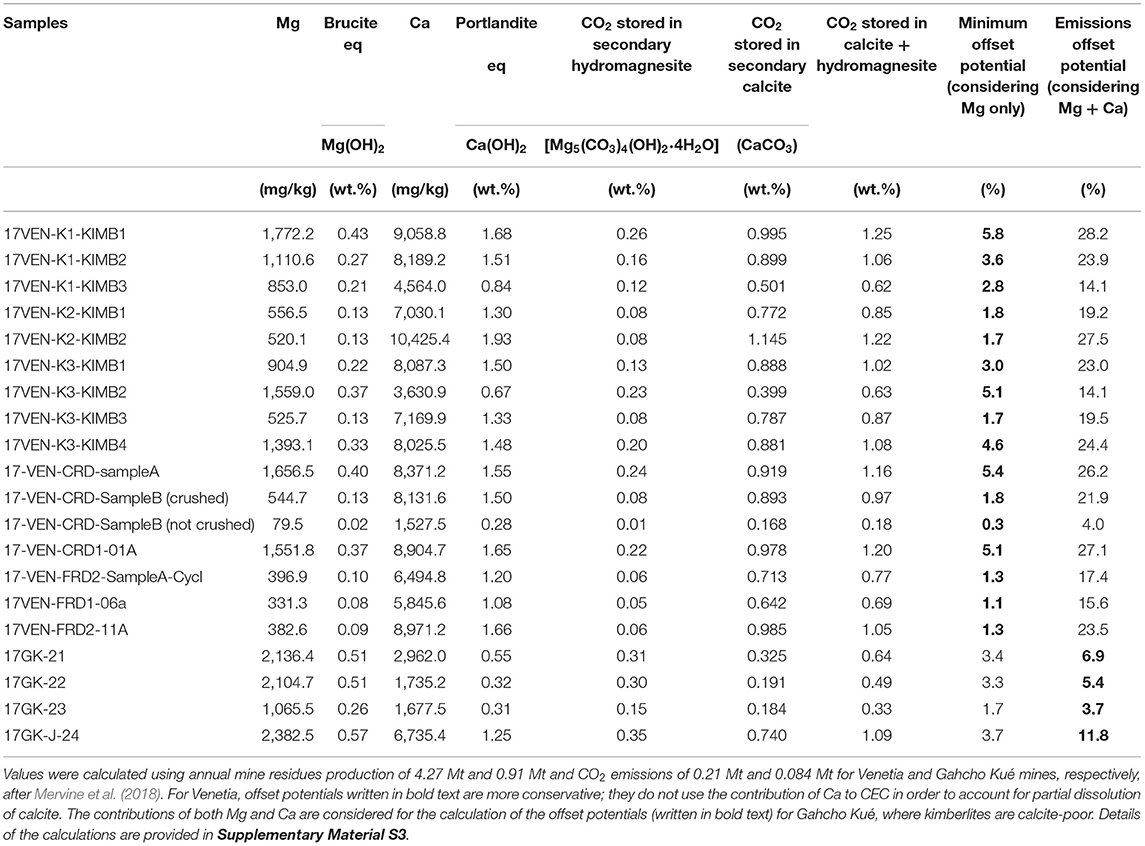
Table 3. Annual emissions offset potentials from cation exchange capacity at Venetia and Gahcho Kué mines.
Importantly, a positive trend between the Ca concentration of CEC leachates and the calcite content of the Venetia samples (Figure 4C) suggests liberation of Ca from calcite following the NH4-acetate leach. Partial dissolution of calcite must be accompanied by release of carbon as . As such, we have used the Ca component of CEC with caution when calculating the equivalent amount of “portlandite” reactivity, and the corresponding emissions offset potentials, at Venetia given its calcite-rich ore. Since the calcite from both mines is Mg-free, and given that dolomite [CaMg(CO3)2] is either absent or present in low abundance, almost all of the labile Mg comes from silicates rather than carbonates. Thus, we used the Mg component of CEC alone to determine the minimum offset potentials at Venetia. If all of the Mg could be fully extracted from the interlayers of smectites and carbonated on an annual basis, it would offset 1.1–5.8% of the current annual CO2 emissions at Venetia (Table 3). This calculation of the emissions offset potentials is based on current residue production and CO2 emissions as reported by Mervine et al. (2018) and the assumption that extracted Mg will precipitate as hydromagnesite [Mg5(CO3)4(OH)2·4H2O] and Ca as calcite, both of which are common carbonate minerals at Earth surface temperatures.
Since the ore samples from Gahcho Kué are calcite-poor, the emissions offset potentials were calculated considering both the Mg and Ca components of CEC. If all of the labile Mg and Ca could be fully extracted from the interlayers of smectites and carbonated on an annual basis, it would provide an offset potential of 3.7–11.8% of the annual CO2 emissions at Gahcho Kué (Table 3). Interestingly, sample 17VENK2-KIMB1 which is particularly carbonate-poor (2 wt.%) and particularly smectite-rich (33.8 wt.%), has an emissions offset potential (considering both Mg and Ca recovered via cation exchange reactions) of 19.2% of the annual CO2 emissions at Venetia (Table 3). This result suggests that smectite-rich and carbonate-poor kimberlites show the greatest emissions offset potentials using cation exchange while having a lower risk of calcite dissolution. It is noteworthy that crushed CRD shows a greater emissions offset potential using CEC compared to uncrushed material (21.9 vs. 4.0% for 17VEN-CRD-SampleB), suggesting that the use of FRD (or crushed CRD) is optimal for mineral carbonation using CEC in smectite-rich processed kimberlite.
A Decision Tree for Mineral Carbonation in Kimberlites
Here, we provide a decision tree based on the mineralogy (absence/presence of brucite, carbonate, serpentine, smectite) of processed kimberlite and the presence or absence of an on-site point source for CO2 (Figure 8). We propose four on-mine carbonation strategies: (1) CO2 injection and (2) passive carbonation for brucite-rich residues; (3) cation exchange for smectite-rich residues; and (4) acid leaching for serpentine-rich and carbonate-poor residues. The first two strategies combine cation extraction and carbonation in a single step, whereas the latter strategies necessitate two spatially separated steps: (i) a cation extraction step and (ii) a carbonation step. Power et al. (2014) proposed a decision tree that considers the presence or absence of highly reactive phases, such as brucite, and site-specific factors such as the availability of a CO2 point source or the availability of water to build carbonation bioreactors. Depending on these factors, they proposed different strategies for passive carbonation or enhanced carbonation (using a bioreactor and waste organics) when no CO2 point source is available. CO2 injection was the favored strategy where a point source for CO2 is available on site. This decision tree was built on knowledge and data from the Mount Keith nickel mine whose komatiitic ore is dominated by serpentine minerals with notably high abundances of brucite and hydrotalcite minerals. However, the reactivity of ultramafic rocks is variable and strongly dependent on mineralogy. The reactivity of serpentinites, kimberlites, peridotites, basalts, and gabbros is different.
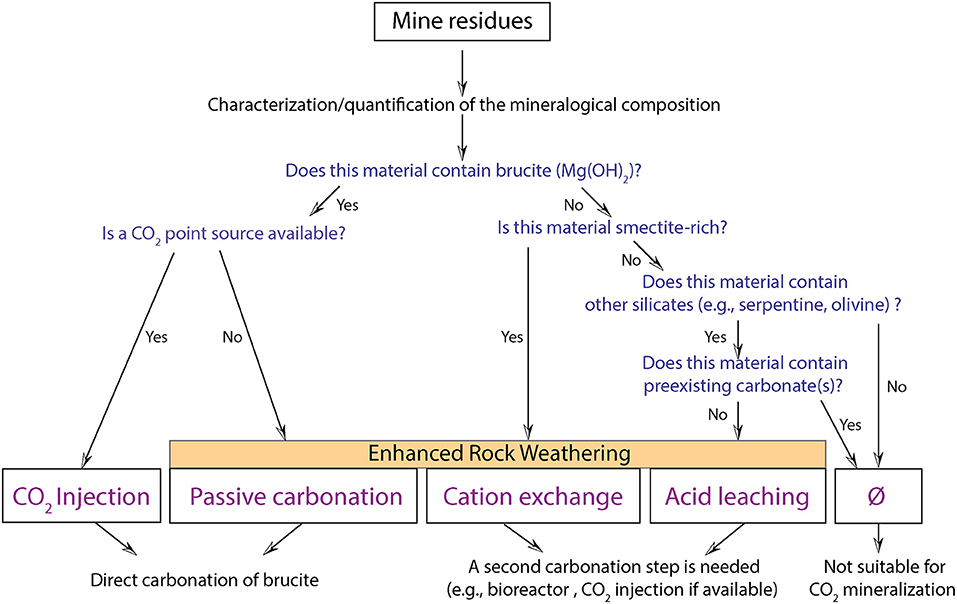
Figure 8. Decision tree for carbonation of mine residues. Whether CO2 injection, passive carbonation, cation exchange or acid leaching can be used depends on the mineralogy of the processed kimberlite and the availability of a CO2 point source.
Acid leaching could be a good strategy for accelerated mineral carbonation at Gahcho Kué where kimberlites are carbonate poor (<2 wt.% calcite). Although it would release some CO2 during calcite dissolution, it would make it possible to access the much greater carbonation potential of Mg in the octahedral sheets of lizardite and saponite. Venetia samples are rich in lizardite and saponite but also calcite (up to ~10 wt.%). This mineral assemblage necessitates another strategy to extract Mg and Ca for mineral carbonation—one that avoids dissolution of pre-existing (bedrock) calcite.
The more general model proposed here covers the most reactive and the most mined ultramafic rocks. The first and most critical decision point depends on careful characterization of ore and residue mineralogy using a combination of XRD, FTIR and TGA. The combination of these three techniques is of particular importance when the mineralogical composition is complex (many different phases ± the presence of amorphous phases). In addition, the quantification of mineral abundances using Rietveld refinements, or whole-rock chemical compositions (when the mineralogical composition is simple), will help to determine the most suitable carbonation strategy for a given ultramafic-hosted mine.
Since brucite is particularly reactive, its presence should be prioritized and systematically investigated. CO2 injection or passive carbonation (when no CO2 point source is available) should be the favored strategies for brucite-rich residues. If the mine residues do not contain brucite, but contain smectites, cation exchange should be considered given the lability of their interlayer Mg and Ca. This is the “lowest-hanging-fruit” strategy for on-mine carbonation of most of the residues and ore from Venetia and Gahcho Kué given that they are smectite-rich and do not contain brucite. Finally, in the case of serpentine-bearing residues characterized by the absence of both brucite and smectite, and great abundances of carbonate minerals, we recommend that the mineralogy is not suitable for carbon mineralization given the risk of calcite dissolution. This general framework for selecting technologies will need to be further adjusted considering the proportions of the minerals (brucite, carbonates, smectites) as well as considering life cycle assessments for implementation of such processes, including the estimation of the carbon footprints for the production and transportation of different chemical amendments such as KCl and NH4-Cl at a given mine.
Given the particular mineral assemblage of the kimberlites from Venetia and Gahcho Kué, cation exchange capacity in smectites opens up the fast reactivity of kimberlites to CO2. In addition to the existing mineral carbonation approaches, i.e., (1) dissolution of serpentines, olivines, wollastonite and other silicate minerals, (2) replacement of brucite and (3) anion exchange in hydrotalcites, we highlight here a new type of reactivity: cation exchange in smectites. The diversity of mechanisms and in particular the specific chemical conditions leading to cation exchange from smectites will need to be further assessed in laboratory-based experiments. Lastly, smectites are common and highly abundant in many types of rock and sediment on Earth (e.g., within marine and lacustrine sediments and in rocks found in sedimentary basins), which means that cation exchange in smectites could broaden the applicability of mineral carbonation to a variety of volcanic- and sediment-hosted ore deposits.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
NZ collected XRD, FTIR, Raman, SEM-EDXS and XFM data, interpreted the data, conducted Rietveld refinements, and wrote the manuscript. BW developed and optimized the Rietveld analysis and collected data on the 13IDE (APS) and XFM (ANSTO) beamlines. SW provided the idea for the project, participated in field sampling at Venetia, facilitated XRD data collection and Rietveld analysis and participated in data interpretation and writing. CP, AS, and IP participated in sampling at Gahcho Kué (IMP) and Venetia mines (ARS, IMP) and analyzed the particle size distributions and surface areas of the samples. MS-M collected the Raman data and contributed to interpretation. AL and MN provided technical support at the 13IDE beamline and contributed to the interpretation of data. CT collected the TGA data and CCT and GMD interpreted these results. DP and JH provided technical support and contributed to data acquisition and processing. GS and TJ contributed to the data acquisition on the XFM beamline at ANSTO. All authors contributed to the article and approved the submitted version.
Funding
Funding for this work was provided by De Beers Group Services, the Natural Resources Canada Clean Growth Program, the Natural Sciences and Engineering Research Council of Canada Discovery program, the Canada Research Chairs program and Mitacs Accelerate. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We acknowledge the support of GeoSoilEnviroCARS (Sector 13), which was supported by the National Science Foundation - Earth Sciences (EAR-1634415), and the U.S. Department of Energy, Geosciences (DE-FG02-94ER14466) (Proposal GUP 65798). Part of this research was undertaken on the X-ray fluorescence microscopy beamline at the Australian Synchrotron, part of ANSTO (AS193/XFM/15033 and AS1/XFM/15925).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Evelyn Mervine and Zandile Miya for logistical support and assistance with sampling at Venetia Diamond mine. Cation exchange capacity was measured at the Natural Resources Analytical Laboratory (University of Alberta) and we thank Allan Arms and Brett Feland for conducting the analysis and for their technical support. We thank Mark Labbe and Walter Harley for the preparation of petrographic thin sections at the Department of Earth and Atmospheric Sciences (University of Alberta), Sydney Evans for technical support in the Environmental Economic Geology Laboratory, and Liam Bullock and reviewer for their constructive and helpful reviews.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fclim.2022.913632/full#supplementary-material
References
Afanasyev, A. A., Melnik, O., Porritt, L., Schumacher, J. C., and Sparks, R. S. J. (2014). Hydrothermal alteration of kimberlite by convective flows of external water. Contribut. Mineral. Petrol. 168, 1038. doi: 10.1007/s00410-014-1038-y
Alfredsson, H. A., Oelkers, E. H., Hardarsson, B. S., Franzson, H., Gunnlaugsson, E., and Gislason, S. R. (2013). The geology and water chemistry of the Hellisheidi, SW-Iceland carbon storage site. Int. J. Greenhouse Gas Control 12, 399–418. doi: 10.1016/j.ijggc.2012.11.019
Assima, G. P., Larachi, F., Beaudoin, G., and Molson, J. (2013b). Dynamics of carbon dioxide uptake in chrysotile mining residues – effect of mineralogy and liquid saturation. Int. J. Greenhouse Gas Control 12, 124–135. doi: 10.1016/j.ijggc.2012.10.001
Assima, G. P., Larachi, F., Molson, J., and Beaudoin, G. (2013a). Accurate and direct quantification of native brucite in serpentine ores—New methodology and implications for CO2 sequestration by mining residues. Thermochimica Acta 566, 281–291. doi: 10.1016/j.tca.2013.06.006
Bache, B. W. (1976). The measurement of cation exchange capacity of soils. J. Sci. Food Agric. 27, 273–280. doi: 10.1002/jsfa.2740270313
Balan, E., Saitta, A. M., Mauri, F., Lemaire, C., and Guyot, F. (2002). First-principles calculation of the infrared spectrum of lizardite. Am. Mineral. 87, 1286–1290. doi: 10.2138/am-2002-1003
Berg, G. W. (1989). “The significance of brucite in South African kimberlites,” in Kimberlite and Related Rocks, v 2: Their Mantle/Crust Setting, Diamonds and Diamond Exploration (Cambridge. England: Geological Society of Australia Special Publication 14, Blackwell Scientific), 282–296.
Berner, R. A. (1998). The carbon cycle and CO2 over Phanerozoic time: the role of land plants. Phil. Trans. R. Soc. Lond. B. Biol Sci. 353, 75–82. doi: 10.1098/rstb.1998.0192
Bish, D. L., William Carey, J., Vaniman, D. T., and Chipera, S. J. (2003). Stability of hydrous minerals on the martian surface. Icarus 164, 96–103. doi: 10.1016/S0019-1035(03)00140-4
Bodénan, F., Bourgeois, F., Petiot, C., Augé, T., Bonfils, B., Julcour-Lebigue, C., et al. (2014). Ex situ mineral carbonation for CO2 mitigation: evaluation of mining waste resources, aqueous carbonation processability and life cycle assessment (Carmex project). Min. Eng. 59, 52–63. doi: 10.1016/j.mineng.2014.01.011
Brantley, S. L., and Olsen, A. A. (2014). “7.3 - Reaction kinetics of primary rock-forming minerals under ambient conditions,” in Treatise on Geochemistry. 2nd Edn, eds H. D. Holland and K. K. Turekian (Oxford: Elsevier), 69–113.
Brunauer, S., Emmett, P. H., and Teller, E. (1938). Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 60, 309–319. doi: 10.1021/ja01269a023
Bullock, L. A., James, R. H., Matter, J., Renforth, P., and Teagle, D. A. H. (2021). Global carbon dioxide removal potential of waste materials from metal and diamond mining. Front. Clim. 3, 694175. doi: 10.3389/fclim.2021.694175
Bundeleva, I. A., Ménez, B., Aug,é, T., Bodénan, F., Recham, N., and Guyot, F. (2014). Effect of cyanobacteria Synechococcus PCC 7942 on carbonation kinetics of olivine at 20°C. Min. Eng. 59, 2–11. doi: 10.1016/j.mineng.2014.01.019
Chanturiya, V., Minenko, V., Makarov, D., Suvorova, O., and Selivanova, E. (2018). Advanced techniques of saponite recovery from diamond processing plant water and areas of saponite application. Minerals 8, 549. doi: 10.3390/min8120549
Daval, D., Hellmann, R., Martinez, I., Gangloff, S., and Guyot, F. (2013). Lizardite serpentine dissolution kinetics as a function of pH and temperature, including effects of elevated pCO2. Chem. Geol. 351, 245–256. doi: 10.1016/j.chemgeo.2013.05.020
Daval, D., Martinez, I., Corvisier, J., Findling, N., Goffé, B., and Guyot, F. (2009). Carbonation of Ca-bearing silicates, the case of wollastonite: Experimental investigations and kinetic modeling. Chem. Geol. 265, 63–78. doi: 10.1016/j.chemgeo.2009.01.022
Daval, D., Sissmann, O., Menguy, N., Saldi, G. D., Guyot, F., Martinez, I., et al. (2011). Influence of amorphous silica layer formation on the dissolution rate of olivine at 90°C and elevated pCO2. Chem. Geol. 284, 193–209. doi: 10.1016/j.chemgeo.2011.02.021
Dessert, C., Dupré, B., Gaillardet, J., François, L. M., and Allègre, C. J. (2003). Basalt weathering laws and the impact of basalt weathering on the global carbon cycle. Chem. Geol. 202, 257–273. doi: 10.1016/j.chemgeo.2002.10.001
Diedrich, T., Schott, J., and Oelkers, E. H. (2014). An experimental study of tremolite dissolution rates as a function of pH and temperature: implications for tremolite toxicity and its use in carbon storage. Mineral. Mag. 78, 1449–1464. doi: 10.1180/minmag.2014.078.6.12
Farmer, V. C., and Russell, J. D. (1964). The infrared spectra of layer silicates. Spectrochimica Acta 20, 1149–1173. doi: 10.1016/0371-1951(64)80165-X
Giammar, D. E., Bruant, R. G., and Peters, C. A. (2005). Forsterite dissolution and magnesite precipitation at conditions relevant for deep saline aquifer storage and sequestration of carbon dioxide. Chem. Geol. 217, 257–276. doi: 10.1016/j.chemgeo.2004.12.013
Giuliani, A., and Pearson, D. G. (2019). Kimberlites: from deep earth to diamond mines. Elements 15, 377–380. doi: 10.2138/gselements.15.6.377
Goff, F., and Lackner, K. S. (1998). Carbon dioxide sequestering using ultramafic rocks. Environ. Geosci. 5, 89–102. doi: 10.1046/j.1526-0984.1998.08014.x
Gras, A., Beaudoin, G., Molson, J., Plante, B., Bussière, B., Lemieux, J. M., et al. (2017). Isotopic evidence of passive mineral carbonation in mine wastes from the Dumont Nickel Project (Abitibi, Quebec). Int. J. Greenhouse Gas Control 60, 10–23. doi: 10.1016/j.ijggc.2017.03.002
Guyot, F., Daval, D., Dupraz, S., Martinez, I., Ménez, B., and Sissmann, O. (2011). CO2 geological storage: the environmental mineralogy perspective. Comptes Rendus Geosci. 343, 246–259. doi: 10.1016/j.crte.2010.12.007
Hamilton, J. L., Wilson, S. A., Morgan, B., Harrison, A. L., Turvey, C. C., Paterson, D. J., et al. (2020). Accelerating mineral carbonation in ultramafic mine tailings via direct CO2 reaction and heap leaching with potential for base metal enrichment and recovery. Econ. Geol. 115, 303–323. doi: 10.5382/econgeo.4710
Hamilton, J. L., Wilson, S. A., Morgan, B., Turvey, C. C., Paterson, D. J., Jowitt, S. M., et al. (2018). Fate of transition metals during passive carbonation of ultramafic mine tailings via air capture with potential for metal resource recovery. Int. J. Greenhouse Gas Control 71, 155–167. doi: 10.1016/j.ijggc.2018.02.008
Hamilton, J. L., Wilson, S. A., Morgan, B., Turvey, C. C., Paterson, D. J., MacRae, C., et al. (2016). Nesquehonite sequesters transition metals and CO2 during accelerated carbon mineralization. Int. J. Greenhouse Gas Control 55, 73–81. doi: 10.1016/j.ijggc.2016.11.006
Hansen, L. D., Dipple, G. M., Gordon, T. M., and Kellett, D. A. (2005). Carbonated serpentinite (listwanite) at Atlin, British Columbia,: a geological analogue to carbon dioxide sequestration. Can. Mineral. 43, 225–239. doi: 10.2113/gscanmin.43.1.225
Harrison, A. L., Dipple, G. M., Power, I. M., and Mayer, K. U. (2015). Influence of surface passivation and water content on mineral reactions in unsaturated porous media: Implications for brucite carbonation and CO2 sequestration. Geochimica et Cosmochimica Acta 148, 477–495. doi: 10.1016/j.gca.2014.10.020
Harrison, A. L., Power, I. M., and Dipple, G. M. (2013). Accelerated carbonation of brucite in mine tailings for carbon sequestration. Environ. Sci. Technol. 47, 126–134. doi: 10.1021/es3012854
Hetman, C. M., Scott Smith, B. H., Paul, J. L., and Winter, F. (2004). Geology of the Gahcho Kué kimberlite pipes, NWT, Canada: root to diatreme magmatic transition zones. Lithos 76, 51–74. doi: 10.1016/j.lithos.2004.03.051
Howard, D. L., de Jonge, M. D., Afshar, N., Ryan, C. G., Kirkham, R., Reinhardt, J., et al. (2020). The XFM beamline at the australian synchrotron. J. Synchrotron Radiat. 27, 1447–1458. doi: 10.1107/S1600577520010152
Huang, C. K., and Kerr, P. F. (1960). Infrared study of the carbonate minerals. Am. Mineral. 45, 311–324.
IPCC (2018). “Global Warming of 1.5°C,” in An IPCC Special Report on the Impacts of Global Warming of 1.5°C Above Pre-industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty, eds V. Masson-Delmotte, P. Zhai, H.-O. Pörtner, D. Roberts, J. Skea, P. R. Shukla, et al.
Julcour, C., Bourgeois, F., Bonfils, B., Benhamed, I., Guyot, F., Bodénan, F., et al. (2015). Development of an attrition-leaching hybrid process for direct aqueous mineral carbonation. Chem. Eng. J. 262, 716–726. doi: 10.1016/j.cej.2014.10.031
Kang, I.-M., and Roh, K.-M. (2013). Mineral carbonation of gaseous carbon dioxide using a clay-hosted cation exchange reaction. Environ. Technol. 34, 3191–3195. doi: 10.1080/09593330.2013.821140
Kloprogge, J. T. (2017). “Raman spectroscopy of clay minerals”, in Surface and Interface Chemistry of Clay Minerals, eds R. Schoonheydt, C. T. Johnston, and F. Bergaya (Developments in Clay Science, book series, Elsevier), 150–199.
Kloprogge, J. T., Mahmutagic, E., and Frost, R. L. (2006). Mid-infrared and infrared emission spectroscopy of Cu-exchanged montmorillonite. J. Colloid Interface Sci. 296, 640–646. doi: 10.1016/j.jcis.2005.09.055
Kloprogge, J. T., and Ponce, C. P. (2021). Spectroscopic studies of synthetic and natural saponites: a review. Minerals 11, 112. doi: 10.3390/min11020112
Komadel, P. (2003). Chemically modified smectites. Clay Min. 38, 127–138. doi: 10.1180/0009855033810083
Krevor, S. C. M., and Lackner, K. S. (2011). Enhancing serpentine dissolution kinetics for mineral carbon dioxide sequestration. Int. J. Greenhouse Gas Control 5, 1073–1080. doi: 10.1016/j.ijggc.2011.01.006
Krupskaya, V. V., Zakusin, S. V., Tyupina, E. A., Dorzhieva, O. V., Zhukhlistov, A. P., Belousov, P. E., et al. (2017). Experimental study of montmorillonite structure and transformation of its properties under treatment with inorganic acid solutions. Minerals 7, 49. doi: 10.3390/min7040049
Lechat, K., Lemieux, J.-M., Molson, J., Beaudoin, G., and Hébert, R. (2016). Field evidence of CO2 sequestration by mineral carbonation in ultramafic milling wastes, Thetford Mines, Canada. Int. J. Greenhouse Gas Control 47, 110–121. doi: 10.1016/j.ijggc.2016.01.036
Loring, J. S., Chen, J., Bénézeth, P., Qafoku, O., Ilton, E. S., Washton, N. M., et al. (2015). Evidence for carbonate surface complexation during forsterite carbonation in wet supercritical carbon dioxide. Langmuir 31, 7533–7543. doi: 10.1021/acs.langmuir.5b01052
Loring, J. S., Schaef, H. T., Turcu, R. V. F., Thompson, C. J., Miller, Q. R. S., Martin, P. F., et al. (2012). In situ molecular spectroscopic evidence for CO2 intercalation into montmorillonite in supercritical carbon dioxide: Langmuir 28, 7125–7128. doi: 10.1021/la301136w
Lumsden, D. N. (1979). Discrepancy between thin-section and X-ray estimates of dolomite in limestone. J. Sedimentary Res. 49, 429–435. doi: 10.1306/212F7761-2B24-11D7-8648000102C1865D
Madejová, J., and Komadel, P. (2005). “Information available from infrared spectra of the fine fractions of bentonite” in The Application of Vibrational Spectroscopy to Clay Minerals and Layered Double Hydroxides, ed J. T. Kloprogge (Aurora, CO: CMS Workshop Lectures, v. 13, The Clay Mineral Society), 65–98.
McDonough, W. F., and Sun, S. S. (1995). The composition of the Earth. Chem. Geol. 120, 223–253. doi: 10.1016/0009-2541(94)00140-4
Mervine, E. M., Wilson, S. A., Power, I. M., Dipple, G. M., Turvey, C. C., Hamilton, J. L., et al. (2018). Potential for offsetting diamond mine carbon emissions through mineral carbonation of processed kimberlite: an assessment of De Beers mine sites in South Africa and Canada. Mineral. Petrol. 112, 755–765. doi: 10.1007/s00710-018-0589-4
Michels, L., Fossum, J. O., Rozynek, Z., Hemmen, H., Rustenberg, K., Sobas, P. A., et al. (2015). Intercalation and retention of carbon dioxide in a smectite clay promoted by interlayer cations. Sci. Rep. 5, 8775. doi: 10.1038/srep08775
Mitchell, R. H., Giuliani, A., and O'Brien, H. (2019). What is a kimberlite? Petrology and mineralogy of hypabyssal kimberlites. Elements 15, 381–386. doi: 10.2138/gselements.15.6.381
Morkel, J., Kruger, S. J., and Vermakk, M. K. G. (2006). Characterization of clay mineral fractions in tuffisitic kimberlite breccias by X-ray diffraction. J. Southern Afr. Inst. Mining Metall. 106, 397–406. doi: 10.10520/AJA0038223X_3102
Morkel, J., Pistorius, P. C., and Vermaak, M. K. G. (2007). Cation exchange behaviour of kimberlite in solutions containing Cu2+ and K+. Min. Eng. 20, 1145–1152. doi: 10.1016/j.mineng.2007.04.013
Murray, H. H. (2000). Traditional and new applications for kaolin, smectite, and palygorskite: a general overview. Appl. Clay Sci. 17, 207–221. doi: 10.1016/S0169-1317(00)00016-8
Newville, M. (2013). Larch: an analysis package for XAFS and related spectroscopies. J. Phys. Conf. Series 430, 012007. doi: 10.1088/1742-6596/430/1/012007
Odom, I. E. (1984). Smectite clay minerals: properties and uses: philosophical transactions of the royal society A. Math. Phys. Eng. Sci. 311, 391–409. doi: 10.1098/rsta.1984.0036
Oelkers, E. H., Butcher, R., Pogge von Strandmann, P. A. E., Schuessler, J. A., von Blanckenburg, F., Snæbjörnsdóttir, S. Ó., et al. (2019). Using stable Mg isotope signatures to assess the fate of magnesium during the in situ mineralisation of CO2 and H2S at the CarbFix site in SW-Iceland. Geochimica et Cosmochimica Acta 245, 542–555. doi: 10.1016/j.gca.2018.11.011
Oelkers, E. H., Gislason, S. R., and Matter, J. M. (2008). Mineral carbonation of CO2. Elements 4, 333–337. doi: 10.2113/gselements.4.5.333
O'Gorman, J. V., and Kitchener, J. A. (1974). The flocculation and de-watering of kimberlite clay slimes. Int. J. Min. Process. 1, 33–49. doi: 10.1016/0301-7516(74)90025-8
Oskierski, H. C., Beinlich, A., Mavromatis, V., Altarawneh, M., and Dlugogorski, B. Z. (2019). Mg isotope fractionation during continental weathering and low temperature carbonation of ultramafic rocks. Geochimica et Cosmochimica Acta 262, 60–77. doi: 10.1016/j.gca.2019.07.019
Oskierski, H. C., Dlugogorski, B. Z., and Jacobsen, G. (2013). Sequestration of atmospheric CO2 in a weathering-derived, serpentinite-hosted magnesite deposit: 14C tracing of carbon sources and age constraints for a refined genetic model. Geochimica et Cosmochimica Acta 122, 226–246. doi: 10.1016/j.gca.2013.08.029
Palandri, J. L., and Kharaka, Y. K. (2004). “A compilation of rate parameters of water-mineral interaction kinetics for application to geochemical modeling,” in U.S. Geological Survey Open-File Report, Menlo Park, Calif, U.S. Dept. of the Interior, U.S. Geological Survey.
Park, A.-H. A., and Fan, L.-S. (2004). CO2 mineral sequestration: physically activated dissolution of serpentine and pH swing process. Chem. Eng. Sci. 59, 5241–5247. doi: 10.1016/j.ces.2004.09.008
Park, A.-H. A., Jadhav, R., and Fan, L.-S. (2003). CO2 mineral sequestration: chemically enhanced aqueous carbonation of serpentine. Can. J. Chem. Eng. 81, 885–890. doi: 10.1002/cjce.5450810373
Paterson, D., de Jonge, M. D., Howard, D. L., Lewis, W., McKinlay, J., Starritt, A., et al. (2011). “The X-ray fluorescence microscopy beamline at the Australian Synchrotron,” in AIP Conference Proceedings, Vol. 1365, 219–222.
Paulo, C., Power, I. M., Stubbs, A. R., Wang, B., Zeyen, N., and Wilson, S. A. (2021). Evaluating feedstocks for carbon dioxide removal by enhanced rock weathering and CO2 mineralization. Appl. Geochem. 129, 104955. doi: 10.1016/j.apgeochem.2021.104955
Pearce, J. K., Dawson, G. K. W., Law, A. C. K., Biddle, D., and Golding, S. D. (2016). Reactivity of micas and cap-rock in wet supercritical CO2 with SO2 and O2 at CO2 storage conditions. Appl. Geochem. 72, 59–76. doi: 10.1016/j.apgeochem.2016.06.010
Perrin, J., Vielzeuf, D., Laporte, D., Ricolleau, A., Rossman, G. R., and Floquet, N. (2016). Raman characterization of synthetic magnesian calcites. Am. Mineral. 101, 2525–2538. doi: 10.2138/am-2016-5714
Phillips, D., Kiviets, G. B., Barton, E. B., Smith, C. B., and Fourie, L. F. (1998). “40Ar/39Ar dating of kimberlites and related rocks: problems and solutions,” in Proceedings of the 7th International Kimberlite Conference, Cape Town, South Africa, Vol. 7, 690–692.
Posukhova, T. V., Dorofeev, S. A., Garanin, K. V., and Siaoin, G. (2013). Diamond industry wastes: mineral composition and recycling. Moscow Univ. Geol. Bull. 68, 96–107. doi: 10.3103/S0145875213020087
Power, I. M., Dipple, G. M., Bradshaw, P. M. D., and Harrison, A. L. (2020). Prospects for CO2 mineralization and enhanced weathering of ultramafic mine tailings from the Baptiste nickel deposit in British Columbia, Canada. Int. J. Greenhouse Gas Control 94, 102895. doi: 10.1016/j.ijggc.2019.102895
Power, I. M., Harrison, A. L., Dipple, G. M., Wilson, S. A., Kelemen, P. B., Hitch, M., et al. (2013). Carbon mineralization: from natural analogues to engineered systems. Rev. Mineral. Geochem. 77, 305–360. doi: 10.2138/rmg.2013.77.9
Power, I. M., McCutcheon, J., Harrison, A. L., Wilson, S. A., Dipple, G. M., Kelly, S., et al. (2014). Strategizing carbon-neutral mines: a case for pilot projects. Minerals 4, 399–436. doi: 10.3390/min4020399
Power, I. M., Wilson, S. A., Small, D. P., Dipple, G. M., Wan, W., and Southam, G. (2011). Microbially mediated mineral carbonation: roles of phototrophy and heterotrophy. Environ. Sci. Technol. 45, 9061–9068. doi: 10.1021/es201648g
Pronost, J., Beaudoin, G., Tremblay, J., Larachi, F., Duchesne, J., Hébert, R., et al. (2011). Carbon sequestration kinetic and storage capacity of ultramafic mining waste. Environ. Sci. Technol. 45, 9413–9420. doi: 10.1021/es203063a
Renforth, P., Manning, D. A. C., and Lopez-Capel, E. (2009). Carbonate precipitation in artificial soils as a sink for atmospheric carbon dioxide. Appl. Geochem. 24, 1757–1764. doi: 10.1016/j.apgeochem.2009.05.005
Rigopoulos, I., Harrison, A. L., Delimitis, A., Ioannou, I., Efstathiou, A. M., Kyratsi, T., et al. (2018). Carbon sequestration via enhanced weathering of peridotites and basalts in seawater. Appl. Geochem. 91, 197–207. doi: 10.1016/j.apgeochem.2017.11.001
Rollo, H. A., and Jamieson, H. E. (2006). Interaction of diamond mine waste and surface water in the Canadian Arctic. Appl. Geochem. 21, 1522–1538. doi: 10.1016/j.apgeochem.2006.05.008
Ryan, C. G. (2000). Quantitative trace element imaging using PIXE and the nuclear microprobe. Int. J. Imaging Syst. Technol. 11, 219–230. doi: 10.1002/ima.1007
Ryan, C. G., Siddons, D. P., Kirkham, R., Dunn, P. A., Kuczewski, A., Moorhead, G., et al. (2010). The new Maia detector system: methods for high definition trace element imaging of natural material. AIP Conf. Proc. 1221, 9–17. doi: 10.1063/1.3399266
Ryan, C. G., Siddons, D. P., Kirkham, R., Li, Z. Y., Jonge, M. D. D., Paterson, D. J., et al. (2014). Maia X-ray fluorescence imaging: capturing detail in complex natural samples. J. Phys. Conf. Series 499, 012002. doi: 10.1088/1742-6596/499/1/012002
Sader, J. A., Leybourne, M. I., McClenaghan, M. B., and Hamilton, S. M. (2007). Low-temperature serpentinization processes and kimberlite groundwater signatures in the Kirkland Lake and Lake Timiskiming kimberlite fields, Ontario, Canada: implications for diamond exploration. Geochem. Explor. Environ. Anal. 7, 3–21. doi: 10.1144/1467-7873/06-900
Saldi, G. D., Köhler, S. J., Marty, N., and Oelkers, E. H. (2007). Dissolution rates of talc as a function of solution composition, pH and temperature. Geochimica et Cosmochimica Acta 71, 3446–3457. doi: 10.1016/j.gca.2007.04.015
Scarlett, N. V. Y., and Madsen, I. C. (2006). Quantification of phases with partial or no known crystal structures. Powder Diff. 21, 278–284. doi: 10.1154/1.2362855
Schiffman, P., and Southard, R. J. (1996). Cation exchange capacity of layer silicates and palagonitized glass in mafic volcanic rocks: a comparative study of bulk extraction and in situ techniques. Clays Clay Min. 44, 624–634. doi: 10.1346/CCMN.1996.0440505
Sissmann, O., Daval, D., Brunet, F., Guyot, F., Verlaguet, A., Pinquier, Y., et al. (2013). The deleterious effect of secondary phases on olivine carbonation yield: insight from time-resolved aqueous-fluid sampling and FIB-TEM characterization. Chem. Geol. 357, 186–202. doi: 10.1016/j.chemgeo.2013.08.031
Smith, M. M., Wolery, T. J., and Carroll, S. A. (2013). Kinetics of chlorite dissolution at elevated temperatures and CO2 conditions. Chem. Geol. 347, 1–8. doi: 10.1016/j.chemgeo.2013.02.017
Stockmann, G., Wolff-Boenisch, D., Gíslason, S. R., and Oelkers, E. H. (2008). Dissolution of diopside and basaltic glass: the effect of carbonate coating. Mineral. Mag. 72, 135–139. doi: 10.1180/minmag.2008.072.1.135
Stripp, G. R., Field, M., Schumacher, J. C., Sparks, R. S. J., and Cressey, G. (2006). Post-emplacement serpentinization and related hydrothermal metamorphism in a kimberlite from Venetia, South Africa. J. Metamorphic Geol. 24, 515–534. doi: 10.1111/j.1525-1314.2006.00652.x
Stubbs, A. R., Paulo, C., Power, I. M., Wang, B., Zeyen, N., and Wilson, S. A. (2022). Direct measurement of CO2 drawdown in mine wastes and rock powders: implications for enhanced rock weathering. Int. J. Greenhouse Gas Control 113:103554. doi: 10.1016/j.ijggc.2021.103554
Sutton, S. R., Lanzirotti, A., Newville, M., Rivers, M. L., Eng, P., and Lefticariu, L. (2017). Spatially resolved elemental analysis, spectroscopy and diffraction at the GSECARS sector at the advanced photon source. J. Environ. Qual. 46, 1158–1165. doi: 10.2134/jeq2016.10.0401
Tosca, N. J., and Masterson, A. L. (2014). Chemical controls on incipient Mg-silicate crystallization at 25°C: Implications for early and late diagenesis. Clay Min. 49, 165–194. doi: 10.1180/claymin.2014.049.2.03
Turvey, C. C., Wilson, S. A., Hamilton, J. L., Tait, A. W., McCutcheon, J., Beinlich, A., et al. (2018). Hydrotalcites and hydrated Mg-carbonates as carbon sinks in serpentinite mineral wastes from the Woodsreef chrysotile mine, New South Wales, Australia: Controls on carbonate mineralogy and efficiency of CO2 air capture in mine tailings. Int. J. Greenhouse Gas Control 79, 38–60. doi: 10.1016/j.ijggc.2018.09.015
Vanderzee, S. S. S., and Dipple, G. M. (2019). Targeting Highly Reactive Labile Magnesium in Ultramafic Tailings for Greenhouse-Gas Offsets and Potential Tailings Stabilization at the Baptiste Deposit, Central British Columbia (NTS 093K/13, 14), Geoscience BC Summary of Activities 2018: minerals and mining, Report 2019-1, 109–118.
Wang, A., Freeman, J. J., and Jolliff, B. L. (2015). Understanding the Raman spectral features of phyllosilicates: Raman spectral features of phyllosilicates. J. Raman Spectrosc. 46, 829–845. doi: 10.1002/jrs.4680
Wang, F., and Giammar, D. E. (2013). Forsterite dissolution in saline water at elevated temperature and high CO2 pressure. Environ. Sci. Technol. 47, 168–173. doi: 10.1021/es301231n
White, J. L., Sparks, R. S. J., Bailey, K., Barnett, W. P., Field, M., and Windsor, L. (2012). Kimberlite sills and dykes associated with the Wesselton kimberlite pipe, Kimberley, South Africa. South Afr. J. Geol. 115, 1–32. doi: 10.2113/gssajg.115.1.1
Wilson, S. A., Barker, S. L. L., Dipple, G. M., and Atudorei, V. (2010). Isotopic disequilibrium during uptake of atmospheric CO2 into mine process waters: implications for CO2 sequestration. Environ. Sci. Technol. 44, 9522–9529. doi: 10.1021/es1021125
Wilson, S. A., and Bish, D. L. (2012). Stability of Mg-sulfate minerals in the presence of smectites: Possible mineralogical controls on H2O cycling and biomarker preservation on Mars. Geochimica et Cosmochimica Acta 96, 120–133. doi: 10.1016/j.gca.2012.08.008
Wilson, S. A., Dipple, G. M., Power, I. M., Barker, S. L. L., Fallon, S. J., and Southam, G. (2011). Subarctic weathering of mineral wastes provides a sink for atmospheric CO2. Environ. Sci. Technol. 45, 7727–7736. doi: 10.1021/es202112y
Wilson, S. A., Dipple, G. M., Power, I. M., Thom, J. M., Anderson, R. G., Raudsepp, M., et al. (2009a). Carbon dioxide fixation within mine wastes of ultramafic-hosted ore deposits: examples from the clinton creek and cassiar chrysotile deposits, Canada. Econ. Geol. 104, 95–112. doi: 10.2113/gsecongeo.104.1.95
Wilson, S. A., Harrison, A. L., Dipple, G. M., Power, I. M., Barker, S. L. L., Ulrich Mayer, K., et al. (2014). Offsetting of CO2 emissions by air capture in mine tailings at the Mount Keith Nickel Mine, Western Australia: Rates, controls and prospects for carbon neutral mining. Int. J. Greenhouse Gas Control 25, 121–140. doi: 10.1016/j.ijggc.2014.04.002
Wilson, S. A., Raudsepp, M., and Dipple, G. M. (2006). Verifying and quantifying carbon fixation in minerals from serpentine-rich mine tailings using the Rietveld method with X-ray powder diffraction data. Am. Mineral. 91, 1331–1341. doi: 10.2138/am.2006.2058
Wilson, S. A., Raudsepp, M., and Dipple, G. M. (2009b). Quantifying carbon fixation in trace minerals from processed kimberlite: a comparative study of quantitative methods using X-ray powder diffraction data with applications to the Diavik Diamond Mine, Northwest Territories, Canada. Appl. Geochem. 24, 2312–2331. doi: 10.1016/j.apgeochem.2009.09.018
Zeyen, N., Benzerara, K., Beyssac, O., Daval, D., Muller, E., Thomazo, C., et al. (2021). Integrative analysis of the mineralogical and chemical composition of modern microbialites from ten Mexican lakes: what do we learn about their formation? Geochimica et Cosmochimica Acta 305, 148–184. doi: 10.1016/j.gca.2021.04.030
Keywords: carbon sequestration, mineral carbonation, saponite, cation exchange, kimberlite, smectite
Citation: Zeyen N, Wang B, Wilson S, Paulo C, Stubbs AR, Power IM, Steele-Maclnnis M, Lanzirotti A, Newville M, Paterson DJ, Hamilton JL, Jones TR, Turvey CC, Dipple GM and Southam G (2022) Cation Exchange in Smectites as a New Approach to Mineral Carbonation. Front. Clim. 4:913632. doi: 10.3389/fclim.2022.913632
Received: 06 April 2022; Accepted: 18 May 2022;
Published: 23 June 2022.
Edited by:
Valentina Prigiobbe, Stevens Institute of Technology, United StatesReviewed by:
Rafael Mattos Dos Santos, University of Guelph, CanadaLiam Bullock, University of Oxford, United Kingdom
Copyright © 2022 Zeyen, Wang, Wilson, Paulo, Stubbs, Power, Steele-Maclnnis, Lanzirotti, Newville, Paterson, Hamilton, Jones, Turvey, Dipple and Southam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Zeyen, zeyen@ualberta.ca