 Philip A. E. Pogge von Strandmann
Philip A. E. Pogge von Strandmann Chloe Tooley2
Chloe Tooley2 Phil Renforth
Phil Renforth- 1Mainz Isotope and Geochemistry Centre, Institute of Geosciences, Johannes Gutenberg University, Mainz, Germany
- 2London Geochemistry and Isotope Centre (LOGIC), Institute of Earth and Planetary Sciences, University College London and Birkbeck, University of London, London, United Kingdom
- 3Witteveen+Bos, Deventer, Netherlands
- 4School of Engineering & Physical Sciences, Heriot-Watt University, Edinburgh, United Kingdom
Crushed olivine was added to a soil core to mimic enhanced weathering, and water was continually dripped through for ~6 months. Our experiments were conducted at 4°C, and are compared to previously run identical experiments at 19°C. Olivine dissolution rates in both experiments start out similar, likely due to fines and sharp crystal corners. However, after >100 days of reaction, the dissolution rate at 4°C was two orders of magnitude lower than at 19°C. The accumulation of heavy metals, such as Ni and Cd, was low in both experiments, but soil retention of these elements was proportionally higher at higher temperatures, likely due to enhanced sorption and formation of clays. Overall, this study suggests that olivine dissolution rates in experiments that mimic natural settings are orders of magnitude slower than in normal laboratory experiments, and that enhanced weathering may be a considerably less efficient method of carbon dioxide removal at low climatic temperatures. Both of these conclusions have implications for the application of enhanced weathering as a CO2 removal method.
Introduction
Projections of future climate scenarios clearly show that to avoid climate warming above dangerous levels of 1.5°C, large quantities of CO2 will have to be removed from the atmosphere (IPCC, 2014, 2018), in addition to rapid and deep emissions reduction. Negative emissions technologies have been proposed for removing and storing atmospheric CO2 (The Royal Society, 2018), which have a range of carbon drawdown efficiencies, environmental requirements, energy inputs and costs (e.g., Shepherd et al., 2009; Matter et al., 2016; Taylor et al., 2016; Pogge von Strandmann et al., 2019a). Perhaps the processes that most lend themselves to be efficient and cost-effective are those that draw on the natural carbon cycles. Hence, techniques like ocean fertilization (e.g., Bowie et al., 2001; Williamson et al., 2012) and afforestation (e.g., Nilsson and Schopfhauser, 1995; Yosef et al., 2018) enhance the organic pathway of the long-term carbon cycle, while others like mineral carbonation (Gislason and Oelkers, 2014; Matter et al., 2016; Pogge von Strandmann et al., 2019a) and enhanced weathering increase the rates of the inorganic pathway of the long-term carbon cycle. This study examines the latter of these processes, enhanced weathering (Schuiling and Krijgsman, 2006; Hangx and Spiers, 2009; Koehler et al., 2010, 2013; Schuiling and de Boer, 2010; Renforth, 2012; Hartmann et al., 2013; Renforth et al., 2013, 2015; Renforth and Campbell, 2021).
Chemical weathering is the Earth's primary process of long-term atmospheric CO2 drawdown (Walker et al., 1981; Saenger and Wang, 2014). Dissolution of silicate rocks, in particular in rivers and soil pore waters, leads to carbon dissolution in water as alkalinity, and also carries critical cations such as Ca and Mg. When transported to the oceans, these initially contribute to elevated ocean alkalinity (Renforth and Henderson, 2017), then over ~10–100 ka precipitate as marine carbonate, sequestering CO2. At the same time, rock-derived cations such as P, Fe and Si are also dissolved and transported to the coastal oceans, where they fertilize organic carbon growth (Lalonde et al., 2012; Hawley et al., 2017). The burial of this organic carbon is also assisted by the supply of continent-derived particulate material (Kennedy and Wagner, 2011). Hence, silicate weathering influences both the inorganic and organic pathways of the long-term carbon cycle, on timescales from yearly (for organic carbon) to tens of kyr (for carbonate precipitation) (e.g., Colbourn et al., 2015; Pogge von Strandmann et al., 2017, 2021a).
The limiting steps on weathering reactions therefore significantly affects climate. In natural settings, these are both climate-based (temperature, runoff, etc.) and supply-based (availability of primary silicates for weathering) (Berner et al., 1983; Raymo and Ruddiman, 1992; West et al., 2005). Slow clay mineral precipitation rates have also been considered as limiting reactions (Maher, 2010; Zhang et al., 2010), although more recently both laboratory and field experiments suggest that the formation of clays (or at least their amorphous versions) can be fairly rapid, and on the order of days to months (e.g., Oelkers et al., 2019; Pogge von Strandmann et al., 2019b, 2021b). Enhanced weathering intends to circumvent especially the limitation of supply, by providing fine-grained silicates for weathering, for example by plowing mafic material into agricultural lands (Hartmann et al., 2013; Taylor et al., 2016; Beerling et al., 2020). The smaller the grain size and greater the surface area, the faster dissolution rates and CO2 drawdown will be, but the more CO2 will be emitted by grinding (Renforth, 2012). However, as yet little is known about precise dissolution rates of various silicate grain sizes in natural settings, in part due to surface passivation, where unreactive secondary minerals (e.g., carbonates and clays) precipitate on primary mineral surfaces, inhibiting dissolution (Taylor et al., 2016; Beerling et al., 2020). Laboratory-derived dissolution rates are generally several orders of magnitude higher than field-derived rates (White and Brantley, 2003), and dissolution rates of artificially-added silicates are even less well-known (Peters et al., 2004; ten Berge et al., 2012; Manning et al., 2013; Renforth et al., 2015). Effectively, at the fast end of this potential dissolution rate scale, enhanced weathering is a highly efficient carbon sequestration method, while at the slow end, less carbon would be sequestered than emitted during grinding, transport and installation.
In an attempt to reconcile laboratory- and field-derived weathering rates, Renforth et al. (2015) used natural soil columns and brought them into the laboratory to control and measure material inputs and outputs. Two identical soil cores were used, one as a control and the other with crushed olivine to mimic enhanced weathering. By monitoring dissolved output fluxes, a dissolution rate per grain size of olivine was determined for an air temperature of 19°C. Here, we determine the effect of temperature on olivine dissolution, by reproducing the Renforth et al. (2015) experiments at a temperature of 4°C. The goal is to assess to what degree temperature limits the geographical application of olivine as an enhanced weathering technique. While it is clear that temperature is a key control on the dissolution rate of silicate material, it is less clear whether temperature dominates in a system where there is no supply limitation (West et al., 2005). Also, it is not clear how the exchangeable fraction responds to differences in temperature.
Further, we have revisited the original experiments, and pre-concentrated solutions to better assess the behavior of toxic heavy metals (e.g., Ni, Cr) during the dissolution of olivine. This is in order to determine whether the use of olivine can lead to dangerous accumulations of such metals in either soils or surface waters (Haque et al., 2020).
Experimental Design
The original experiments of Renforth et al. (2015) took 1 m long soil cores from agricultural land in North Oxfordshire. This bedrock in this area is Jurassic limestone and mudstone, and the soils are calcareous. The cores span the transition from the plowed layer, though the B and C horizons, to the parent rock material. One core was used to examine the compositions of the bulk soil and leachable phases. The other two were used as column reactors, and a modified Hoagland nutrient solution (to mimic the addition of fertilizer) was dripped onto them at a rate of 15 ml/h (~200 μg/ml K, N, 30 μg/ml P, 37 ng/ml Ca, 30 ng/ml S; 69 ng/ml Mg). Crushed olivine (100 g), from Western Norway (Minelco Ltd.), was stirred into the top 10 cm of one core, while the other was stirred, but without olivine addition, and used as a control core. This addition amount of olivine equals ~12.7 kg/m2, which is around 2.5× higher than the highest rate of 5 kg/m2/yr proposed by the modeling study of Taylor et al. (2016). Effluent drip waters were collected periodically from the base of each core (Figure 1).
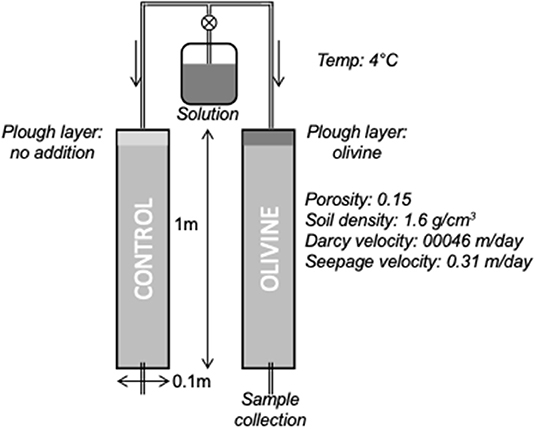
Figure 1. Diagram of the soil column experiments. Adapted from Pogge von Strandmann et al. (2021b) and Renforth et al. (2015).
In this study's experiments, additional cores drilled from the same site at the same time were used. The same olivine and identical Hoagland solution were used. The two primary differences compared to the original experiments were (1) our experiments were conducted in a cold room at 4° at the department of Earth Sciences, University College London rather than at 19°C; (2) the original experiments showed a general decrease in control core effluent Ca and Mg concentrations, due to the added Hoagland solution being out of equilibrium with the soil exchangeable fraction at the start of the experiment (Pogge von Strandmann et al., 2021b). To counteract this, we dripped Hoagland solution through our columns for 45 days before addition of the olivine. After this, solution was dripped through both columns for a further 6 months.
Methods
Major element concentrations in bulk soils and olivine powder were determined by XRF (Renforth et al., 2015). Elemental concentrations of the effluent solutions were determined by a Varian 820-MS inductively-coupled plasma mass spectrometer (ICP-MS) at the LOGIC laboratories at UCL. The analyses were calibrated using multi-element solutions mixed from single-element standards, and accuracy was assessed by analyzing the international reference standards SLRS-5 and TMDA. The analytical reproducibility was better than ±4%.
Calcium concentrations were analyzed by a Varian ICP-OES (Optical emission spectrometry), also at UCL's LOGIC laboratories. This was because Ca concentrations were very high due to the presence of Ca in the influent Hoagland solutions, and because of the carbonate rock in the columns dissolving.
In order to determine the concentrations of Cr, Ni, and Cu, 15 ml of solution from both these experiments, and those of Renforth et al. (2015), were dried down and also analyzed by ICP-MS.
Results
Data from the bulk soils and olivine composition, initially published by Renforth et al. (2015) are repeated in Supplementary Material.
Effluent Concentrations From the 4°C Experiment
As for the 19°C experiments (Renforth et al., 2015), the pH of the input solution was circumneutral, and the passage through the columns had little effect on the pH. Effluent pH was 7.1 ± 0.4 (1sd) for the olivine column and 7.3 ± 0.5 for the control column. All results are presented in Table 1. Overall, the effluent major element concentrations exhibit more scatter than observed in the 19°C experiment by Renforth et al. (2015). Magnesium and calcium concentrations in both of the columns' effluent show a general decrease with time (Figure 2). In most samples the concentrations of the olivine core's effluent are higher than in the control core's, and the difference between the cores decreases slightly with time. For example, initially, the Mg effluent concentrations from the olivine column were ~1.6 μg/ml higher than that of the control column. By day 134 this difference had decreased to ~0.1 μg/ml. Silicon concentrations, in contrast, are highly scattered in the effluent from both cores, with no straightforward trends.
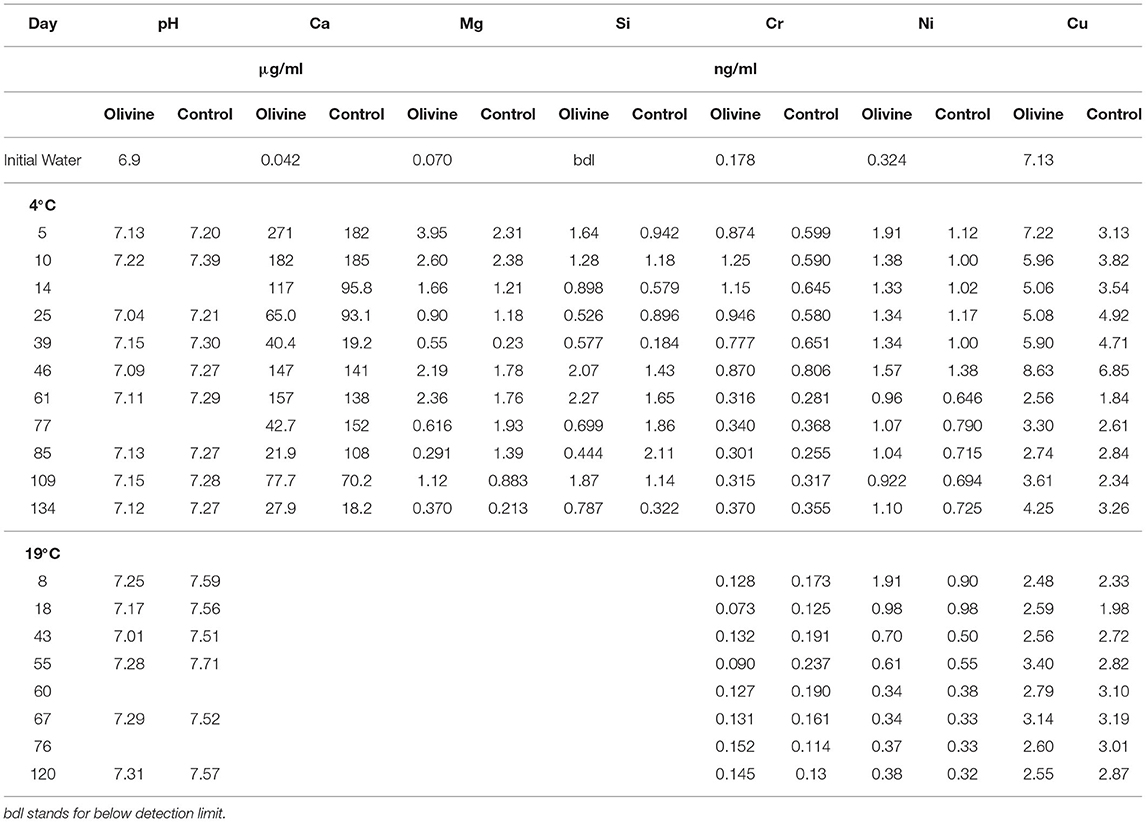
Table 1. Elemental concentrations from the 4°C experiments, and the heavy metal concentrations also from the 19°C experiment.
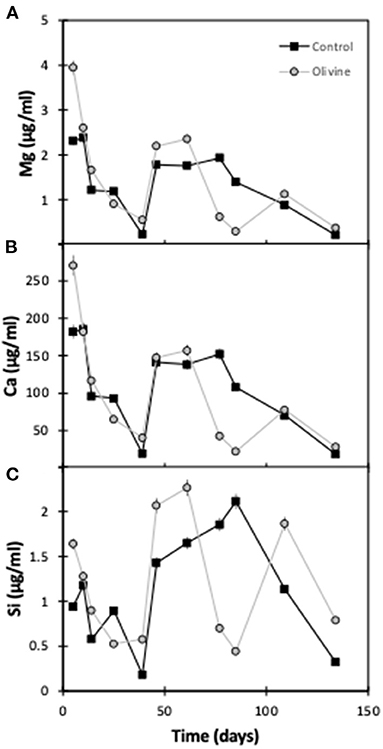
Figure 2. Effluent major element concentrations as a function of experimental time for both columns: (A) magnesium, (B) calcium, (C) silicon. The error bars represent the 2sd analytical uncertainties.
Heavy Metal Effluent Concentrations
In general, the concentrations of Cu, Ni, and Cr in the 4°C experiment start out with greater concentrations in the olivine core over the control core (Figure 3). After approximately 50 days the concentrations tend to decrease by ~50% (for Cr and Cu) to ~80% (for Ni). For Ni and Cu, the olivine core's effluent always is more concentrated, while for Cr, the difference between the cores is effectively zero after 60 days. The behavior of Ni between the 4° and 19°C experiment is fairly similar, in that both show a decline with time. In contrast, in the 19°C experiment there is little difference between the effluent from the olivine and control cores for Cr or Cu with time, meaning that the behavior of these elements is different at different temperatures.
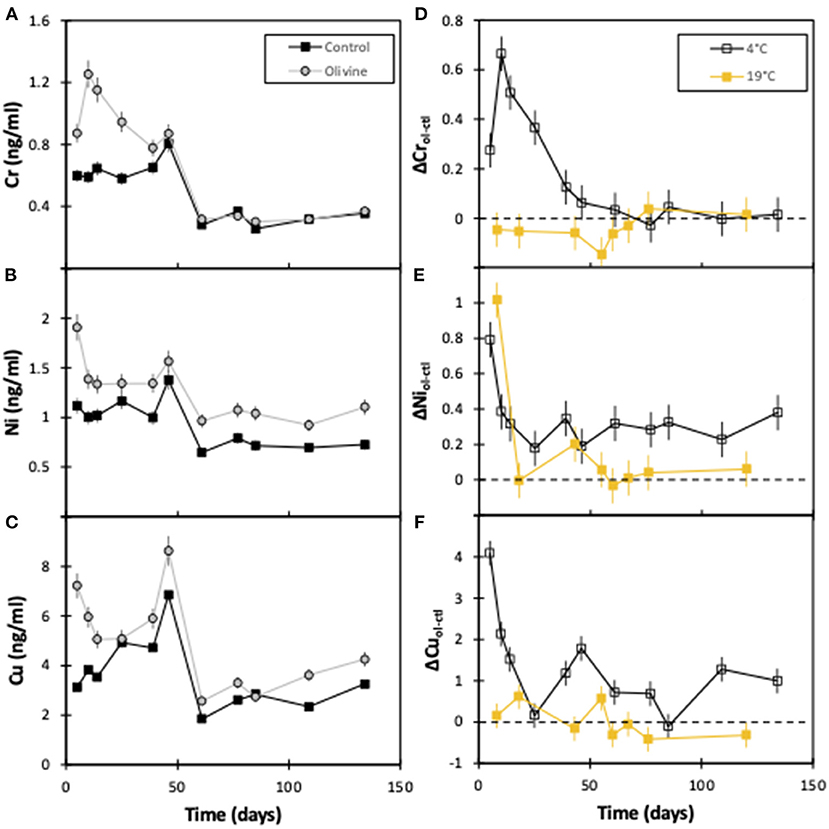
Figure 3. Effluent trace element concentrations as a function of experimental time for both columns: (A) chromium, (B) nickel, (C) copper. (D–F) show the difference in heavy metal concentration between the olivine and control core for this (4°C) and the 19°C experiments (Renforth et al., 2015). The error bars represent the 2sd analytical uncertainties. For the Δ panels (D–F), the analytical uncertainty has been compounded through the calculations.
Discussion
Source of Dissolved Cations
In order to determine the source of effluent cations such as Mg, a mass balance can be constructed. This is based on the assumptions that all (input-corrected) control core effluent Ca stems from the dissolution of carbonate rock, and all control core effluent Si is from the dissolution of silicates (clays, given the absence of primary silicates). We then use the Mg/Ca of carbonate leaches, and the Mg/Si of residual material (Renforth et al., 2015) to determine the amount of Mg coming from carbonates and secondary silicates. It is then assumed that all remaining Mg in the input-correct control core effluent stems from the exchangeable pool. Then we assume that the control and olivine cores are identical except for the added dissolution of olivine. According to these calculations, ~73% of Mg in the control core effluent stems from dissolution of carbonate by the end of the experiment, and ~26% from the exchangeable fraction, and only 1% from the dissolution of silicates. By comparison, in the 19°C experiment, these numbers are 65, 32, and 1%, respectively (Pogge von Strandmann et al., 2021b). In other words, increased temperature appears to decrease the proportion of carbonate contribution, and increase that of the exchangeable fraction. Error propagation of the analytical uncertainties yields ± ~ 6% relative error on the mass balance.
In the olivine core, by the end of the 4°C experiment, olivine dissolution only makes up ~3% of the total Mg, while in the 19°C experiment this number is ~45% (Pogge von Strandmann et al., 2021b). Overall, then, it is clear that lower temperatures do not favor the dissolution of olivine, but in relative terms the dissolution of carbonate is more favored. In general, a relative decrease in carbonate weathering and increase in silicate weathering as temperature increases has been observed before (West et al., 2005; Kasemann et al., 2014; Gaillardet et al., 2019; Romero-Mujalli et al., 2019). Further, the exchangeable pool is relatively less dominant in the colder experiments, likely due to kinetic effects on the exchange rate.
Further evidence for very little dissolution of olivine at 4°C is shown when plotting [Mg] against [Ca] for all experiments. The 4°C control and olivine core effluents, as well as the 19°C control core effluent exhibit an identical gradient that is largely due to the dissolution of carbonate. The Mg/Ca ratio of the leached carbonate is 0.01 ± 0.005, while the Mg:Ca gradients of those three cores are 0.013–0.015 (Figure 4), demonstrating a likely carbonate source. In contrast, the 19°C olivine core's effluent has a similar gradient, but is considerably more Mg-enriched.
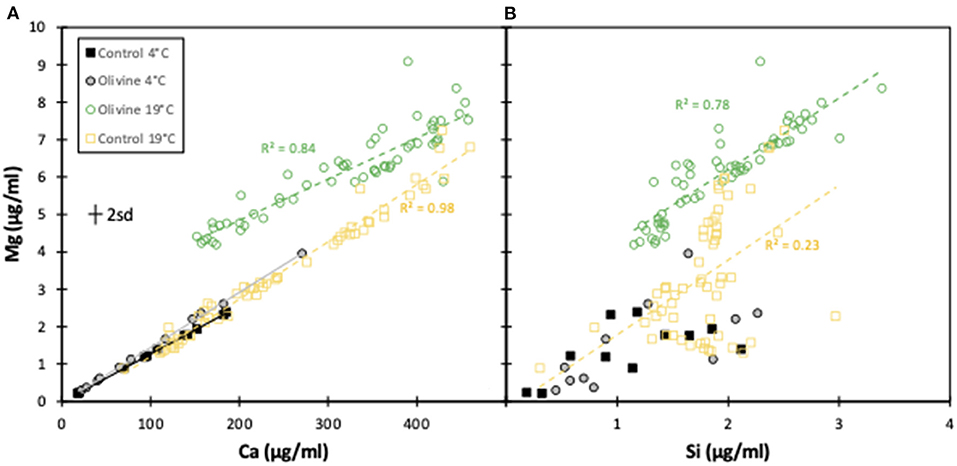
Figure 4. Mg concentrations plotted against Ca and Si concentrations for the effluents from this experiment and the 19°C experiment (Renforth et al., 2015). In all cases, only the effluent from the 19°C olivine column shows dissolution of olivine, and not the 4°C olivine column. The error bar represents the 2sd analytical uncertainty.
Magnesium vs. silicon trends follow a similar relationship, albeit more scattered (Figure 4). Combined, these trends clearly suggest that while a Mg- and Si-rich phase (olivine) is being dissolved in the 19°C experiment, this is occurring far less, if at all, in the 4°C experiment.
Olivine Dissolution Rates
Based on the assumption that the only difference between the effluent from the olivine and from the control core is due to olivine dissolution (Renforth et al., 2015; Pogge von Strandmann et al., 2021b), the rate of that dissolution can be calculated. Using the difference in effluent Mg concentrations, dissolution rates were calculated using a surface area normalized approach:
where Wr is the surface area normalized weathering rate, Q is the solution flux (in ml/s) and Mg is the concentration of Mg from the olivine and control cores, respectively. SSA is the surface area of the olivine, which is 3.04 × 104 cm2/g (Renforth et al., 2015).
Thus, the weathering rate of the 19°C experiment starts at 10−16.1 mol(Mg)/cm2/s and then increases slightly, before stabilizing at 10−15.8 mol(Mg)/cm2/s after ~50 days. However, while the 4°C experiment's weathering rate is initially almost identical to the 19°C experiment (10−16.0 compared to 10−16.1 mol(Mg)/cm2/s, respectively), it subsequently rapidly decreases with time. After 134 days the rate is 10−17.1 mol(Mg)/cm2/s, and there is over two orders of magnitude difference between the two experiments (Figure 5).
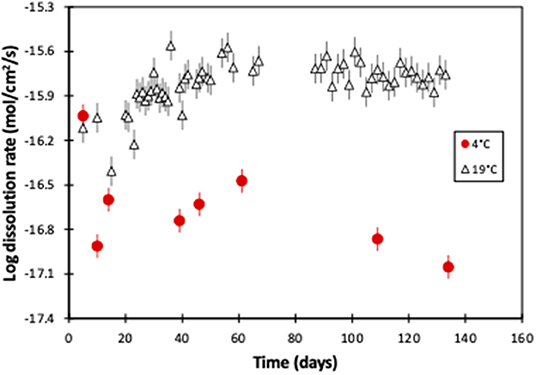
Figure 5. Magnesium-derived olivine dissolution rates of this 4°C experiment, and the 19°C experiment (Renforth et al., 2015), as a function of experimental time. The error bars represent the compounded 2sd uncertainty.
The similarity of the weathering rates between the two experiments at the start is likely due to initial dissolution of the fresh outer surface of the crushed olivine, including many relatively high surface area sharp corners (Anbeek, 1992). The subsequent reduction in weathering rate may be attributed to the reduced availability of reactive surface sites on the olivine surface (Orkoula and Koutsoukos, 2002; Appelo and Postma, 2004), suggesting dissolution rate is partially controlled by the mechanism of surface reaction (Renforth, 2012).
Using the dissolution rate law:
where R is the gas constant (8.3145 kJ mol−1 K−1), Ea is the activation energy (52.9 ± 6.9 kJ mol−1 K−1), A represents a pre-exponential factor (0.0854), n is the reaction order with respect to H+ = 0.46, T is temperature (K) and a is the activity (Pokrovsky and Schott, 2000; Hänchen et al., 2006), we can calculate the idealized temperature-dependent dissolution of forsterite (Figure 6). Interestingly, while these idealized dissolution rates show the same temperature-dependent slope as our two experiments, they are an order of magnitude faster. In conventional laboratory dissolution rate studies, the fluid to mineral ratio is much higher to ensure effective transport of the solutes from the mineral surface, and laboratory studies tend to be run at far-from-equilibrium conditions. In contrast, in our soil core experiments, there was likely only imperfect contact between minerals and fluid, as well as closer to equilibrium conditions, and the possibility that the surface of the olivine became less reactive with time as it was weathered. As such, this highlights the difference between pure dissolution experiments and soil core (and natural) experiments, and clearly shows that idealized laboratory experiments should not be used to estimate dissolution rates in enhanced weathering. Further, olivine dissolution rates (in laboratory experiments) are pH dependent, and decrease with increasing pH (Oelkers et al., 2018). In the experimentally analyzed pH range of 1–12, neutral pH causes an approximately median dissolution rate (Oelkers et al., 2018), but also highlights that olivine dissolution rates during enhanced weathering will also vary depending on the pH of the source water.
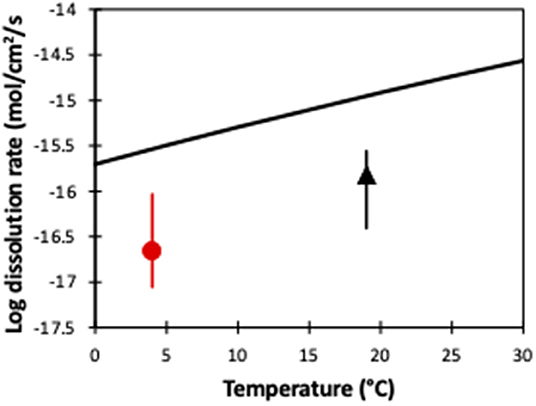
Figure 6. A comparison of olivine dissolution rates from pure laboratory experiments (black line) and our soil column experiments (this study and Renforth et al., 2015), as a function of temperature. The symbols represent the average value, while the error bars represent the entire observed range of dissolution rates (Figure 5).
Heavy Metals
One of the key areas of research surrounding the potential of enhanced weathering is the investigation into the build-up of toxic elements in soils and the water column (Renforth, 2012; Hartmann et al., 2013; Renforth et al., 2015; Haque et al., 2020). This potential environmental risk could limit the application of enhanced silicate weathering as a method of carbon sequestration. Both Cr and Ni have been used to assess this risk here. In the 4°C solutions, Cr concentrations in the olivine core effluent are up to 2× that of the control core for the first ~50 days. Following this, overall concentrations decrease, and are also similar in waters from both cores (Figure 3).
Using an Mg/Cr ratio of 3,000 for this olivine (Renforth et al., 2015), it is estimated that ~0.5 ± 0.07 μg Cr is released by the dissolution of ~5 mg of olivine at 4°C. This compares to a total difference between the Cr flux of the control and olivine cores for the whole experiment of 0.33 ± 0.02 μg, meaning that ~0.17 ± 0.01 μg was retained in the soil, which is a retention efficiency of 34 ± 4%. In the 19°C experiment, 37 ± 3 μg Cr was released from the olivine, and 9.6 ± 0.7 μg was retained in the soil column, giving a retention efficiency of 26 ± 4%.
Similarly, ~1 ± 0.07 μg Ni was in the effluent from the 4°C experiment, while the olivine dissolution led to release of ~14 ± 1 μg Ni. Hence Ni retention by the soil is ~90 ± 6%. Similarly, in the 19°C experiment, ~5 ± 0.4 μg Ni was in the effluent, compared to 1060 ± 70 μg released from olivine dissolution, leading to a retention efficiency of 99%. A high percentage build-up of Ni in soil has been observed in other experiments (Raveh-Rubin et al., 2015), where 90% of the initial Ni mass was extracted from the soil after experiment completion.
High retention of heavy metals in soils is observed throughout the world in relation to mature soils (Haque et al., 2020), both in secondary minerals (largely clays) and adsorbed. Some laterites have high Ni content (Lewis et al., 2006), with some serpentinite zones showing >2% Ni. Raveh-Rubin et al. (2015) show evidence of fast Ni retention at higher temperatures, with the expectation that Ni is not released into a mobile stage for a considerable time. In relation to temperature, the accumulation of Cr and Ni in soils will be slower in colder temperatures compared to warmer climates, due to slower dissolution rates. On the other hand, the build-up of Ni in the soil is considerably slower at 4°C compared to 19°C, which could be partially related to the increased availability of secondary clay minerals in the 19°C column providing easily sorbed surfaces for Ni (Raveh-Rubin et al., 2015).
The European Union's human drinking water standard (98/83/EC) is 50 ng/ml Cr. For the first 60 days in our experiments (before Cr concentrations in the control and olivine core became effectively identical), the 4°C experiment released 0.015 ng/ml Cr, which the 19°C experiment released 0.9 ng/ml. Hence, addition of this amount of olivine (for this Cr concentration in the olivine) is over an order of magnitude from breaching drinking water guidelines. Further, the Finland Ministry of the Environment has a soil Cr limit of 200 mg/kg. Given our experiments' retention of Cr, it would take over 2 million years at 4°C and ~44,000 years at 19°C of this level of enhanced weathering to breach these limits.
Equally, the Ni guidelines for drinking water is 20 ng/ml. The 4°C experiment released 0.05 ng/ml, while the 19°C experiment released 0.2 ng/ml. The higher Ni retention by soils, compared to Cr, means that this example of enhanced weathering would be even further from breaching drinking water guidelines. The soil limit for Ni is 100 mg/kg, and it would take ~15,800 years to reach this at 4°C, and ~200 years at 19°C.
It must be stressed, however, that using less pure olivine, or other silicates, for enhanced weathering (Taylor et al., 2016) could lead to a faster build-up of heavy metals. Equally, using materials whose weathering produces secondary minerals (clays, oxides, etc.), such as basalt, may also enhance the retention of heavy metals in soils. Further, the type of initial soil being seeded will have an effect, as of course the thicker application or faster dissolution of olivine. For example, Taylor et al. (2016) suggest the application of 1–5 kg/m2/yr of rock powder in their models of the global application of enhanced weathering. This amount is 2.5–13 times less than that applied during this experiment. This would suggest that olivine application on a global scale is unlikely to rapidly increase the heavy metal component of soils. However, it must be noted that fresh application on an annual basis would potentially cause a disproportionately greater heavy metal addition, because there would be a fairly constant supply of rapidly dissolving fresh mineral edges and fines. This is also further discussed below.
Possibility of Using Enhanced Weathering
The possible maximum carbon dioxide capture potential (enhanced weathering potential EWp, kgCO2 per ton) of the olivine used in this experiment has been calculated using the modified Steinour equation (see Renforth, 2019).
where CaO, MgO, SO3, P2O5, Na2O, and K2O, are the elemental concentrations of Ca, Mg, S, P, Na, and K, expressed as oxides, M is the molecular mass of those oxides; coefficients α, β,ϵ, θ (equal to +1), γ (equal to −1), and δ (equal to −2) consider the relative contribution of each oxide; and η is molar ratio of CO2 to divalent cation sequestered during enhanced weathering; we have used η = 1.5 in this study, which is a conservative global average.
EWp for the olivine used in this experiment is 0.79 tCO2/t. The land area normalized dissolution rate for the olivine has been calculated at 30 t/km2/yr for 4°C, and at 200 t/km2/yr at 19°C (Renforth et al., 2015).
However, these estimates are based on a constant olivine surface area, and do not consider the effects of comminution on dissolution rates. The feasibility of enhanced olivine weathering can be analyzed using a shrinking core model to estimate the reaction times for dissolution (Hangx and Spiers, 2009). Using the calculated weathering rates from this experiment and Renforth et al. (2015), a model for dissolution as a function of time for various grain sizes can be calculated (Figure 7). The model also assumes a distribution in the grain size, because a uniform grain size does not occur during grinding. Here we assume a relative standard deviation on the grain size distribution of 2.5, which occurs during ball milling of olivine.
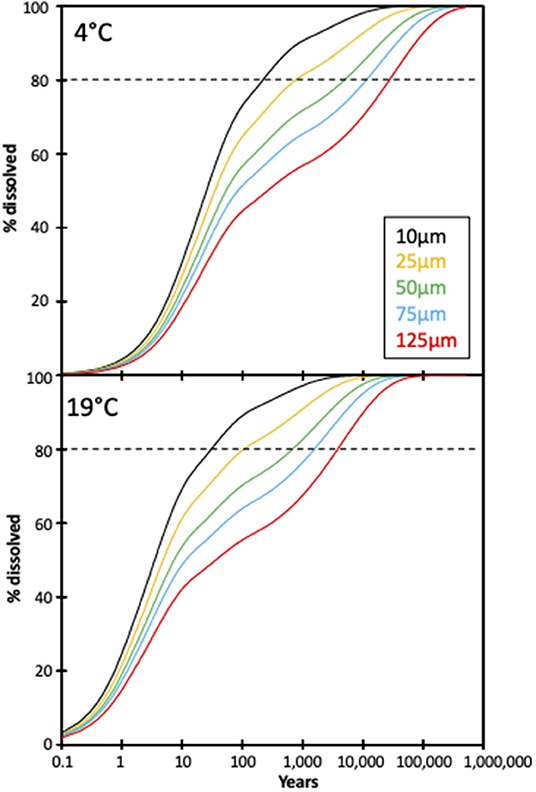
Figure 7. Modeled extents of dissolution for different olivine grain sizes based on a shrinking core model (see text for details). The horizontal dashed lines represent a target dissolution proportion of 80% (given that the final 20% take a significant amount of time to finally dissolve).
With an average grain size of 125 μm [80% of the grains in the olivine used in this experiment were >125 μm (Renforth et al., 2015)] dissolution of olivine at 4°C reaches ~20% in 10 years, sequestering ~0.2 kg CO2 /m2/yr for this experiment's application rate. For this grain size it takes >>10,000 years to reach 80% dissolution (Figure 7). In contrast, dissolution (of an average grain size of 125 μm) at 19°C reaches 20% after 1.5 years (sequestering ~1.3 kg CO2 /m2/yr), and 80% after ~4,000 years. The model of Taylor et al. (2016), which suggests global enhanced weathering in the tropics, assumes a uniform grain size of 10 μm. Our model suggests that 80% of olivine with an average grain size of 10 μm would take >200 years to dissolve at 4°C, and 20–30 years at 19°C, highlighting the importance of temperature and grain size on the dissolution rate and CO2 sequestration. This suggests that even at 19°C grain size must be reduced to ~1 μm to dissolve ~80% within <10 years. However, the extra processing (grinding) of olivine would then release additional CO2 (Renforth, 2012). A similar result was seen in Hangx and Spiers (2009), where olivine dissolution in a coastal setting was modeled for two temperatures using a shrinking core model, with the 15°C model showing rates three times longer than that of the 25°C simulation.
Clearly, if enhanced weathering is applied to natural settings, soil temperature will vary both diurnally and seasonally. Diurnal soil temperature changes at 1 m depth (the depth of our soil columns) are on the order of 1–2°C in Europe (German National Meteorological Service). Average monthly soil temperatures at 1 m depth vary between 1 and 22°C in, for example, central and western Europe (i.e., Germany, northern France, UK, Benelux; German National Meteorological Service), with mean annual soil temperatures of 8.8–12.7°C in the UK (Busby, 2015), which match fairly well to the range used in these experiments. At such temperatures (assuming a linear extrapolation between the dissolution rates at 4° and 19°C; Figure 6), it would take ~5,800 years for 80% of a mean grain size of 125 μm to dissolve (2.5 years for 20%). If the mean grain size were 10 μm, 20% would dissolve in ~1.2 years, and 80% in ~50 years. For a 1 μm mean grain size, these values are ~1 and ~14 years, respectively.
Overall, this study highlights that olivine dissolution rates are considerably lower in field-like experiments than in “normal” laboratory experiments. More experiments are needed to assess dissolution rates at higher, tropical, temperatures, but if our dissolution rates can be extrapolated to tropical temperatures, then a yearly application of material (Taylor et al., 2016) may not be feasible without accumulation of unweathered silicate material over time.
Conclusions
Soil core experiments with olivine addition, used to evaluate enhanced weathering at 4°C, show that olivine dissolution is almost two orders of magnitude lower than in identical experiments at 19°C. In general, the two experiments at different temperatures exhibit the same temperature-dependent gradient as idealized laboratory experiments, but offset to lower values. This is likely because soil columns do not represent ideal fluid-rock contact conditions, and suggests that, as observed previously, natural experiments show significantly lower dissolution rates.
Heavy metals, such as Cd and Ni, were examined to assess whether olivine addition can contaminate water or soils. While in this experiment concentrations in both phases were significantly lower than guidelines, retention of heavy metals in soils was higher at elevated temperatures. This represents an increased retention beyond that caused by higher dissolution of minerals and higher temperatures, and is likely due to enhanced secondary mineral (e.g., clay) formation at higher temperatures. In themselves, clays present a potential problem for enhanced weathering, as clay accumulation can inhibit plant growth.
Overall, as anticipated, enhanced weathering may be considerably less feasible in cold conditions. Kinetic-limitation of weathering reactions results in substantially longer dissolution times for olivine. For example, for an average grain size of 50μm, 50% of olivine would dissolve in <10 years at 19°C, but in ~50 years at 4°C. Thus, for example, enhanced weathering rates will be very low during northern and central European and American winters, and this must be factored into calculations.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
PP designed the project and wrote the manuscript. CT conducted the experiment and performed the analyses. PR also designed the project and interpreted the data. JM performed the dissolution calculations. All authors contributed to the article and approved the submitted version.
Funding
PP were funded by ERC Consolidator grant 682760 CONTROLPASTCO2. PR acknowledges UKRI funding under the UK Greenhouse Gas Removal Programme (NE/P019943/1, NE/P019730/1).
Conflict of Interest
JM was employed by Witteveen+Bos.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Birkbeck, University of London is thanked for the MSc of CT. Gary Tarbuck is thanked for assistance during concentration analyses. We thank two reviewers for their useful comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fclim.2022.827698/full#supplementary-material
References
Anbeek, C. (1992). The dependence of dissolution rates on grain size for some fresh and weathered feldspars. Geochim. Cosmochim. Acta 56, 3957–3970. doi: 10.1016/0016-7037(92)90009-8
Appelo, C. A. J., and Postma, A. (2004). Geochemistry, Groundwater and Pollution. Rotterdam: A.A Balkema Publishers. doi: 10.1201/9781439833544
Beerling, D. J., Kantzas, E. P., Lomas, M. R., Wade, P., Eufrasio, R. M., Renforth, P., et al. (2020). Potential for large-scale CO2 removal via enhanced rock weathering with croplands. Nature 583, 242–248. doi: 10.1038/s41586-020-2448-9
Berner, R. A., Lasaga, A. C., and Garrels, R. M. (1983). The carbonate-silicate geochemical cycle and its effect on atmospheric carbon-dioxide over the past 100 million years. Am. J. Sci. 283, 641–683. doi: 10.2475/ajs.283.7.641
Bowie, A. R., Maldonado, M. T., Frew, R. D., Croot, P. L., Achterberg, E. P., Mantoura, R. F. C., et al. (2001). The fate of added iron during a mesoscale fertilisation experiment in the Southern Ocean. Deep Sea Res. II Top. Stud. Oceanogr. 48, 2703–2743. doi: 10.1016/S0967-0645(01)00015-7
Busby, J. (2015). UK shallow ground temperatures for ground coupled heat exchangers. Q. J. Eng. Geol. Hydrol. 48, 248–260. doi: 10.1144/qjegh2015-077
Colbourn, G., Ridgwell, A., and Lenton, T. M. (2015). The time scale of the silicate weathering negative feedback on atmospheric CO2. Glob. Biogeochem. Cycles 29, 583–596. doi: 10.1002/2014GB005054
Gaillardet, J., Calmels, D., Romero-Mujalli, G., Zakharova, E. A., and Hartmann, J. (2019). Global climate control on carbonate weathering intensity. Chem. Geol. 2019, 118762. doi: 10.1016/j.chemgeo.2018.05.009
Gislason, S. R., and Oelkers, E. H. (2014). Carbon storage in basalt. Science 344, 373–374. doi: 10.1126/science.1250828
Hänchen, M., Prigiobbe, V., and Storti, G. (2006). Dissolution kinetics of fosteritic olivine at 90-150°C including effects of the presence of CO2. Geochim. Cosmochim. Acta 70, 4403–4416. doi: 10.1016/j.gca.2006.06.1560
Hangx, S. J. T., and Spiers, C. J. (2009). Coastal spreading of olivine to control atmospheric CO2 concentrations: a critical analysis of viability. Int. J. Greenhouse Gas Control 3, 757–767. doi: 10.1016/j.ijggc.2009.07.001
Haque, F., Chiang, Y. W., and Santos, R. M. (2020). Risk assessment of Ni, Cr, and Si release from alkaline minerals during enhanced weathering. Open Agric. 5, 166–175. doi: 10.1515/opag-2020-0016
Hartmann, J., West, A. J., Renforth, P., Kohler, P., De la Rocha, C. L., Wolf-Gladrow, D. A., et al. (2013). Enhanced chemical weathering as a geoengineering strategy to reduce atmopsheric carbon dioxide, supply nutrients, and mitigate ocean acidification. Rev. Geophys. 51, 113–150. doi: 10.1002/rog.20004
Hawley, S. M., Pogge von Strandmann, P. A. E., Burton, K. W., Williams, H. M., and Gislason, S. R. (2017). Continental weathering and terrestrial (oxyhydr)oxide export: comparing glacial and non-glacial catchments in Iceland. Chem. Geol. 462, 55–66. doi: 10.1016/j.chemgeo.2017.04.026
Kasemann, S. A., Pogge von Strandmann, P. A. E., Prave, A. R., Fallick, A. E., Elliott, T., and Hoffmann, K. H. (2014). Continental weathering following a Cryogenian glaciation: evidence from calcium and magnesium isotopes. Earth Planet. Sci. Lett. 396, 66–77. doi: 10.1016/j.epsl.2014.03.048
Kennedy, M. J., and Wagner, T. (2011). Clay mineral continental amplifier for marine carbon sequestration in a greenhouse ocean. Proc. Natl. Acad. Sci. U.S.A. 108, 9776–9781. doi: 10.1073/pnas.1018670108
Koehler, P., Abrams, J. F., Voelker, C., Hauck, J., and Wolf-Gladrow, D. A. (2013). Geoengineering impact of open ocean dissolution of olivine on atmospheric CO2, surface ocean pH and marine biology. Environ. Res. Lett. 8:014009. doi: 10.1088/1748-9326/8/1/014009
Koehler, P., Hartmann, J., and Wolf-Gladrow, D. A. (2010). Geoengineering potential of artificially enhanced silicate weathering of olivine. Proc. Natl. Acad. Sci. U.S.A. 107, 20228–20233. doi: 10.1073/pnas.1000545107
Lalonde, K., Mucci, A., Ouellet, A., and Gelinas, Y. (2012). Preservation of organic matter in sediments promoted by iron. Nature 483, 198–200. doi: 10.1038/nature10855
Lewis, J. F., Draper, G., Proenza, J. A., Espaillar, J., and Jimenez, J. (2006). Ophiolite-related ultramafic rocks (serpentinites) in the Caribbean Region: a review of their occurrence, composition, origin emplacement and Ni-laterite soil formation. Geologica Acta. 4, 237–263.
Maher, K. (2010). The dependence of chemical weathering rates on fluid residence time. Earth Planet. Sci. Lett. 294, 101–110. doi: 10.1016/j.epsl.2010.03.010
Manning, D. A. C., Renforth, P., Lopez-Capel, E., Robertson, S., and Ghazireh, N. (2013). Carbonate precipitation in artificial soils produced from basaltic quarry fines and composts: an opportunity for passive carbon sequestration. Int. J. Greenhouse Gas Control 17, 309–317. doi: 10.1016/j.ijggc.2013.05.012
Matter, J. M., Stute, M., Snaebjornsdottir, S. O., Oelkers, E. H., Gislason, S. R., Aradottir, E. S., et al. (2016). Rapid carbon mineralization for permanent disposal of anthropogenic carbon dioxide emissions. Science 352, 1312–1314. doi: 10.1126/science.aad8132
Nilsson, S., and Schopfhauser, W. (1995). The carbon-sequestration potential of a global afforestation program. Clim. Change 30, 267–293. doi: 10.1007/BF01091928
Oelkers, E. H., Butcher, R., Pogge von Strandmann, P. A. E., Schuessler, J. A., von Blanckenburg, F., Snaebjornsdottir, S. O., et al. (2019). Using stable Mg isotope signatures to assess the fate of magnesium during the in situ mineralisation of CO2 and H2S at the CarbFix site in SW-Iceland. Geochim. Cosmochim. Acta 245, 542–555. doi: 10.1016/j.gca.2018.11.011
Oelkers, E. H., Declerqc, J., Saldi, G. D., Gislason, S. R., and Schott, J. (2018). Olivine dissolution rates: a critical review. Chem. Geol. 500, 1–19. doi: 10.1016/j.chemgeo.2018.10.008
Orkoula, M. G., and Koutsoukos, P. G. (2002). Variability of dissolution rates at constant undersaturation. J. Colloid Interface Science 253, 185–189. doi: 10.1006/jcis.2002.8536
Peters, S. C., Blum, J. D., Driscoll, C. T., and Likens, G. E. (2004). Dissolution of wollastonite during the experimental manipulation of Hubbard Brook Watershed 1. Biogeochemistry 67, 309–329. doi: 10.1023/B:BIOG.0000015787.44175.3f
Pogge von Strandmann, P. A. E., Burton, K. W., Snaebjornsdottir, S. O., Sigfusson, B., Aradottir, E. S. P., Gunnarsson, I., et al. (2019a). Rapid CO2 mineralisation into calcite at the CarbFix storage site quantified using calcium isotopes. Nat. Commun. 10:1983. doi: 10.1038/s41467-019-10003-8
Pogge von Strandmann, P. A. E., Fraser, W. T., Hammond, S. J., Tarbuck, G., Wood, I. G., Oelkers, E. H., et al. (2019b). Experimental determination of Li isotope behaviour during basalt weathering. Chem. Geol. 517, 34–43. doi: 10.1016/j.chemgeo.2019.04.020
Pogge von Strandmann, P. A. E., Jones, M. T., West, A. J., Murphy, M. J., Stokke, E. W., Tarbuck, G., et al. (2021a). Lithium isotope evidence for enhanced weathering and erosion during the Paleocene-Eocene Thermal Maximum. Sci. Adv. 7, eabh4224. doi: 10.1126/sciadv.abh4224
Pogge von Strandmann, P. A. E., Renforth, P., West, A. J., Murphy, M. J., Luu, T.-H., and Henderson, G. M. (2021b). The lithium and magnesium isotope signature of olivine dissolution in soil experiments. Chem. Geol. 560, 120008. doi: 10.1016/j.chemgeo.2020.120008
Pogge von Strandmann, P. A. E., Vaks, A., Bar-Matthews, M., Ayalon, A., Jacob, E., and Henderson, G. M. (2017). Lithium isotopes in speleothems: temperature-controlled variation in silicate weathering during glacial cycles. Earth Planet. Sci. Lett. 469, 64–74. doi: 10.1016/j.epsl.2017.04.014
Pokrovsky, O. S., and Schott, J. (2000). Kinetics and mechanism of forsterite dissolution at 25 degrees C and pH from 1 to 12. Geochim. Cosmochim. Acta 64, 3313–3325. doi: 10.1016/S0016-7037(00)00434-8
Raveh-Rubin, S., Edery, Y., Dror, I., and Berkowitz, B. (2015). Nickel migration and retention dynamics in natural soil columns. Water Resour. Res. 51, 7702–7722. doi: 10.1002/2015WR016913
Raymo, M. E., and Ruddiman, W. F. (1992). Tectonic forcing of late cenozoic climate. Nature 359, 117–122. doi: 10.1038/359117a0
Renforth, P. (2012). The potential of enhanced weathering in the UK. Int. J. Greenhouse Gas Control 10, 229–243. doi: 10.1016/j.ijggc.2012.06.011
Renforth, P. (2019). The negative emission potential of alkaline materials. Nat. Commun. 10, 1401. doi: 10.1038/s41467-019-09475-5
Renforth, P., and Campbell, J. S. (2021). The role of soils in the regulation of ocean acidification. Philos. Trans. R. Soc. B Biol. Sci. 376, 1834. doi: 10.1098/rstb.2020.0174
Renforth, P., and Henderson, G. (2017). Assessing ocean alkalinity for carbon sequestration. Rev. Geophys. 55, 636–674. doi: 10.1002/2016RG000533
Renforth, P., Jenkins, B. G., and Kruger, T. (2013). Engineering challenges of ocean liming. Energy 60, 442–452. doi: 10.1016/j.energy.2013.08.006
Renforth, P., Pogge von Strandmann, P. A. E., and Henderson, G. M. (2015). The dissolution of olivine added to soil: implications for enhanced weathering. Appl. Geochem. 61, 109–118. doi: 10.1016/j.apgeochem.2015.05.016
Romero-Mujalli, G., Hartmann, J., and Börker, J. (2019). Temperature and CO2 dependency of global carbonate weathering fluxes - implications for future carbonate weathering research. Chem. Geol. 527, 118874. doi: 10.1016/j.chemgeo.2018.08.010
Saenger, C., and Wang, Z. (2014). Magnesium isotope fractionation in biogenic and abiogenic carbonates: implications for paleoenvironmental proxies. Q. Sci. Rev. 90, 1–21. doi: 10.1016/j.quascirev.2014.01.014
Schuiling, R. D., and de Boer, P. L. (2010). Coastal spreading of olivine to control atmospheric CO2 concentrations: a critical analysis of viability. Comment: Nature and laboratory models are different. Int. J. Greenhouse Gas Control 4, 855–856. doi: 10.1016/j.ijggc.2010.04.012
Schuiling, R. D., and Krijgsman, P. (2006). Enhanced weathering: an effective and cheap tool to sequester CO2. Clim. Change 74, 349–354. doi: 10.1007/s10584-005-3485-y
Shepherd, J., Caldeira, K., Cox, P., Haigh, J. E., Keith, D., Launder, B., et al. (2009). Geoengineering the Climate: Science, Governance and Uncertainty. The Royal Society.
Taylor, L. L., Quirk, J., Thorley, R. M. S., Kharecha, P. A., Hansen, J., Ridgwell, A., et al. (2016). Enhanced weathering strategies for stabilizing climate and averting ocean acidification. Nat. Clim. Change 6, 402–406. doi: 10.1038/nclimate2882
ten Berge, H. F. M., van der Meer, H. G., Steenhuizen, J. W., Goedhart, P. W., Knops, P., and Verhagen, J. (2012). Olivine weathering in soil, and its effects on growth and nutrient uptake in ryegrass (Lolium perenne L.): a pot experiment. PLoS ONE 7, e42098. doi: 10.1371/journal.pone.0042098
Walker, J. C. G., Hays, P. B., and Kasting, J. F. (1981). A negative feedback mechanism for the long-term stabilization of earths surface-temperature. J. Geophys. Res. Oceans Atmos. 86, 9776–9782. doi: 10.1029/JC086iC10p09776
West, A. J., Galy, A., and Bickle, M. (2005). Tectonic and climatic controls on silicate weathering. Earth Planet. Sci. Lett. 235, 211–228. doi: 10.1016/j.epsl.2005.03.020
White, A. F., and Brantley, S. L. (2003). The effect of time on the weathering of silicate minerals: why do weathering rates differ in the laboratory and field? Chem. Geol. 202, 479–506. doi: 10.1016/j.chemgeo.2003.03.001
Williamson, P., Wallace, D. W. R., Law, C. S., Boyd, P. W., Collos, Y., Croot, P., et al. (2012). Ocean fertilization for geoengineering: a review of effectiveness, environmental impacts and emerging governance. Process Saf. Environ. Protect. 90, 475–488. doi: 10.1016/j.psep.2012.10.007
Yosef, G., Walko, R., Avisar, R., Tatarinov, F., Rotenberg, E., and Yakir, D. (2018). Large-scale semi-arid afforestation can enhance precipitation and carbon sequestration potential. Sci. Rep. 8,996. doi: 10.1038/s41598-018-19265-6
Keywords: weathering, carbon sequestration, weathering experiment, enhanced weathering of minerals, temperature effect on weathering
Citation: Pogge von Strandmann PAE, Tooley C, Mulders JJPA and Renforth P (2022) The Dissolution of Olivine Added to Soil at 4°C: Implications for Enhanced Weathering in Cold Regions. Front. Clim. 4:827698. doi: 10.3389/fclim.2022.827698
Received: 02 December 2021; Accepted: 18 January 2022;
Published: 10 February 2022.
Edited by:
Mathilde Hagens, Wageningen University and Research, NetherlandsReviewed by:
Fatima Haque, National Taiwan University, TaiwanAndrew Lenton, Commonwealth Scientific and Industrial Research Organization, Australia
Copyright © 2022 Pogge von Strandmann, Tooley, Mulders and Renforth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip A. E. Pogge von Strandmann, cHBvZ2dldm9AdW5pLW1haW56LmRl