 Runhua Han
Runhua Han Ye Qian
Ye Qian Chenguang Zheng3*
Chenguang Zheng3*- 1Department of Chemistry, University of Manitoba, Winnipeg, MB, Canada
- 2School of Basic Medical Sciences, North China University of Science and Technology, Tangshan, China
- 3School of Public Health, North China University of Science and Technology, Tangshan, China
Enterohemorrhagic Escherichia coli (EHEC) is a contagious foodborne pathogen that specifically colonizes the human large intestine, which is regulated by different environmental stimuli within the gut. Transcriptional regulation of EHEC virulence and infection has been extensively studied, while the posttranscriptional regulation of these processes by small RNAs (sRNAs) remains not fully understood. Here we present a virulence-regulating pathway in EHEC O157:H7, in which the sRNA EvrS binds to and destabilizes the mRNA of Z2269, a novel transcriptional regulator. In turn, Z2269 indirectly activates the expression of LEE (locus of enterocyte effacement) pathogenicity island through the master regulator Ler. Importantly, the expression of EvrS is modulated by environmental oxygen levels. EvrS also exhibits lower expression in the colon compared to the ileum, influencing the site-specific colonization of EHEC O157:H7 in mice. These results indicate that the oxygen status within the intestine may regulate the expression of EvrS, thereby modulating virulence factors of EHEC and contributing to successful infection in vivo. This study has broader implications for understanding sRNA functions in spatiotemporal virulence control of EHEC and may provide novel strategies to prevent EHEC infections.
Introduction
Escherichia coli (E. coli) is the predominant facultative commensal of the human intestine (Leimbach et al., 2013). However, some E. coli strains have evolved pathogenic mechanisms through the acquisition of large clusters of virulence factors encoded on prophages and plasmids. These genetic elements enable E. coli to cause both intraintestinal and extraintestinal diseases, including meningitis, sepsis, diarrhea, and urinary tract infections (Kaper et al., 2004; Leimbach et al., 2013). Enterohemorrhagic E. coli (EHEC) is one of the most notorious pathogenic E. coli, responsible for a series of diseases ranging from mild diarrhea to life-threatening hemolytic uremic syndrome (Mellies et al., 2007; Wong et al., 2011). EHEC is typically ingested through contaminated food or water, where it survives the harsh conditions of the stomach and travels to the intestine. There, EHEC precisely adheres to the epithelial cells of the large intestine. Once attached, EHEC secretes a range of virulence factors that manipulate various intracellular pathways, enabling the pathogen to colonize, proliferate, and cause disease (Mellies et al., 2007; Wong et al., 2011).
Adherence of EHEC to intestinal epithelial cells is the first crucial step of intestinal colonization. This is accompanied by the development of a type of ultrastructural lesion called attaching and effacing (A/E) lesion, which causes the destruction of microvilli and the re-arrangement of cytoskeletal (Connolly et al., 2015). The formation of A/E lesions is largely dependent on a chromosomal pathogenicity island (PAI) termed the locus for enterocyte effacement (LEE) (Wong et al., 2011). LEE comprises 41 open reading frames organized in five major polycistronic operons (LEE1-5). LEE1, LEE2, and LEE3 harbor the genes encoding the major components of the type III secretion system (T3SS) that exports effector molecules. LEE1 also encodes Ler, a main transcriptional regulator of the LEE region that modulates the expression of other LEE operons. LEE4 encodes additional effector proteins translocated via T3SS to the host cell. The LEE5 operon encodes the Tir and intimin proteins, which are necessary for intimate attachment to the host epithelium (Mcdaniel et al., 1995). In addition to LEE, the virulence armamentarium of EHEC pathogenesis also involves Shiga toxin (Stx) and flagella (Wong et al., 2011). Stx is produced in the lower intestine, where it is translocated across the intestinal epithelium. Once in the bloodstream, it targets vascular endothelial cells, inducing necrosis or apoptosis by inhibiting protein synthesis (Pacheco and Sperandio, 2012). Stx has also been shown to promote EHEC adhesion to host epithelial cells by upregulating surface expression of phosphatidylethanolamine and nucleolin (Robinson et al., 2006). Flagella act as adhesive appendages during the initial phase of EHEC colonization, facilitating attachment and enhancing the bacterium’s motility within the intestine. This motility provides a crucial advantage by helping EHEC reach favorable niches while avoiding less hospitable areas (Mahajan et al., 2009; Chaban et al., 2015).
The production of virulence factors in pathogens may reduce bacterial growth rates under certain conditions and simultaneously trigger the host’s immune response (Sturm et al., 2011). Therefore, the timing of expression of virulence genes in EHEC must be tightly controlled. Indeed, EHEC expertly navigates complex environmental cues, such as fluctuations in oxygen, pH, temperature, and nutrient availability, to effectively colonize and infect their hosts (Connolly et al., 2015; Cordonnier et al., 2016; Lackraj et al., 2016; Le Bihan et al., 2017; Gelalcha et al., 2022). Extensive research has revealed a plethora of proteinaceous regulators that sense these signals in the infected gut to control transcription of the virulence genes, especially LEE (Connolly et al., 2015). Notably, posttranscriptional regulatory mechanisms that converge on the EHEC pathogenicity by small regulatory RNAs (sRNAs) is becoming increasingly recognized (Sauder and Kendall, 2018; Sy and Tree, 2021). sRNAs (sRNA) are heterogeneous molecules ranging from ∼50 to 500 nucleotides in length and recognized as important players in many physiological and adaptive responses (Jørgensen et al., 2020). Their synthesis is tightly regulated and often induced by a specific stress or virulence condition. Most sRNAs function as regulators by directly base pairing with trans-encoded mRNAs over short regions with imperfect complementarity (Felden and Augagneur, 2021). The canonical mechanism of sRNA-mediated regulation involves interaction with the 5’ untranslated region (5’UTR) or the region near the ribosome binding site (RBS) of the target mRNAs. In this process, the sRNA sequesters the RBS, thereby affecting translation and/or mRNA stability (Gottesman and Storz, 2011; Papenfort et al., 2013). A few recent studies also revealed that sRNAs can directly interact within the coding region and affect the stability of a target mRNA (Pfeiffer et al., 2009; Melson and Kendall, 2019; Villa et al., 2021). Transcriptome sequencing (RNA-seq) and UV-induced RNA-protein cross-linking and analysis of cDNA by high-throughput sequencing (CRAC) experiments revealed hundreds of sRNAs specific to EHEC, many of which are encoded within prophages (Tree et al., 2014; Gruber and Sperandio, 2015; Han et al., 2017). One of these sRNAs, StxS, modulates the expression of Shiga toxin 1 and the general stress response sigma factor RpoS through direct interaction of their mRNAs (Sy et al., 2020). Several other sRNAs (e.g., Esr055, Esr41, MavR, DicF) also play important roles in EHEC pathogenesis by regulating different genes and pathways (Sudo et al., 2014; Han et al., 2017; Melson and Kendall, 2019; Sauder and Kendall, 2021). However, the mRNA targets and regulation mechanisms for the majority of sRNAs in EHEC are still unknown.
Oxygen is one of the most important signals that plays strong roles in flexibly modulating bacterial infection in the human intestine (Wallace et al., 2016). During its passage through the gastrointestinal tract, EHEC encounters relatively aerobic conditions that may suppress the expression of the LEE pathogenicity island and other virulence factors. This adaptation helps EHEC avoid constructing its T3SS in unfavorable environments, such as the small intestine, thereby conserving energy and preventing unnecessary activation of its virulence machinery. As EHEC moves into the large intestine, it encounters a drastically reduced oxygen environment (Espey, 2013). This change is largely due to the metabolic activities of the dense commensal microbiota that consume available oxygen, creating an anaerobic niche (Kendall and Sperandio, 2016). This reduction in oxygen levels can trigger the expression of LEE and T3SS, facilitating adherence to intestinal epithelial cells and enhancing its pathogenicity. Interestingly, a multi-copied sRNA, DicF, regulates the EHEC virulence through base pairing with the 5’UTR of the pchA mRNA under microaerobic condition (Melson and Kendall, 2019). This finding suggests that the ability to rapidly integrate oxygen sensing via sRNA-based regulation may be a key strategy for EHEC to establish infection. However, additional sRNA-mediated mechanisms of oxygen sensing and virulence in EHEC still remains uncovered.
In this study, we studied one of the previously discovered sRNAs and renamed it EvrS (EHEC virulence regulatory sRNA). We demonstrated that EvrS significantly impacts the bacterial adherence ability of EHEC O157:H7 through regulating the mRNA of Z2269, a novel transcriptional regulator that indirectly activates the expression of the master LEE regulator, Ler. We also found that the expression of EvrS is downregulated under oxygen-limited conditions, and its altered expression affects the site-specific colonization of EHEC O157:H7 in mice. Our study therefore enhances the understanding of the virulence mechanisms employed by EHEC and how this bacterium responds to diverse environmental stresses and regulates pathogenicity through sRNA-mediated control.
Materials and methods
Strains, growth conditions and genetic manipulations
The EHEC O157:H7 EDL933 strain and derivatives thereof were cultured at 37°C with shaking in Luria-Bertani (LB) broth or Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Scientific) unless otherwise specified. When necessary, antibiotics were used at the following concentrations: ampicillin (Sigma-Aldrich), 100 μg ml-1; chloramphenicol (Sigma-Aldrich), 15 μg ml-1; kanamycin (Sigma-Aldrich), 50 μg ml-1, nalidixic acid (Sigma-Aldrich), 50 μg ml-1.
Mutant strains were generated by substitution of a specific sRNA or gene with a kanamycin (kan) or a chloramphenicol acetyltransferase (cat) gene cassette using an optimized λ Red recombineering technology, as described previously (Datsenko and Wanner, 2000). Briefly, the chloramphenicol or kanamycin cassettes were amplified from pKD3 or pKD4, respectively, using primers carrying 39 bp of homology to the regions flanking the sRNA/gene to be deleted. The resulting PCR products were electroporated into bacteria carrying the plasmid pKD46 for homologous recombination. The mutants were selected on LB plates with chloramphenicol or kanamycin and verified by PCR amplification and Sanger sequencing. The pCP20 plasmids was then transformed into the mutant strains to remove the antibiotic cassettes.
The EvrS complementation strain (pEvrS) was constructed by cloning the EvrS sequence and its promoter region (100 bp upstream the transcriptional start site) into the SphI/HindIII digested pACYC184 plasmid. The resulting plasmid was then electroporated into the ΔevrS strain, and colonies were screened on LB plates with chloramphenicol. To construct the plasmid expressing the EvrS promoter-lux fusion, a PCR fragment corresponding to the EvrS promoter sequence (100 bp upstream the transcriptional start site and 10 bp downstream) was ligated into XhoI/BamHI digested pMS402 plasmid (Fan et al., 2020), which carries a promoter-less luxCDABE reporter gene. The recombinant plasmid was then transformed into the EDL933 WT strain, and colonies were screened on LB plates with kanamycin. The strain for Z2269 protein purification was constructed by cloning the z2269 gene into the NdeI/BamHI digested pET28a vector. The resulting plasmid was then electroporated into the E. coli BL21 strain. To construct the Z2269 overexpression plasmid, the coding sequence of Z2269 was amplified by PCR and cloned into the EcoRI/BamHI digested pTrc99a vector. The recombined plasmid was transformed into the WT and Δler strains, and colonies were screened on LB plates with amplicillin. To tag the Z2269 protein with 3×FLAG, the fragment corresponding to coding sequence of Z2269 (excluding the stop codon) and 3×FLAG was amplified and cloned into the pTrc99a vector between the EcoRI/BamHI cut site. The resulting plasmid was then transformed into the Δz2269 strain.
To test the regulation of EvrS on the z2269 gene, the translational fusion containing the EvrS binding site on the z2269 mRNA and the lacZ reporter gene under the control of the arabinose promoter were generated in a similar way as previously described (Gruber and Sperandio, 2015). Briefly, the sequence of -100 bp from the translation start site to +48 bp of the z2269 gene and lacZ was synthesized by IDT and cloned into the pBAD24 vector between the NheI and HindIII restriction cut sites. The sRNA overexpression plasmids were constructed by cloning the EvrS sequence into the SacII/HindIII cut site on pBAD33. The plasmids with translational fusion, sRNA overexpression, or both were transformed into the WT EDL933 strain for further experiments. Site-directed mutagenesis was performed with the QuikChange site-directed mutagenesis kit (NEB) using these plasmids as the templates.
To test the expression of EvrS in vivo, the gfp coding sequence (synthesized by IDT) was used to replace the luxCDABE on the pMS402 plasmid by Gibson assembly. The recombinant plasmid containing PEvrS-gfp fusion was introduced into a EDL933 WT strain that constitutively expresses a mCherry gene under the promoter of the rpoD gene on the chromosome. The promoter of rpoD was selected because its activity remains unchanged throughout the murine gut (Armetta et al., 2021). The strain carrying the constitutively expressed mCherry gene was constructed using the λ Red recombineering technology by inserting the sequence of rpoD promoter and mCherry coding sequence as well as the sequence of a kanamycin cassette (synthesized by IDT) on chromosome between T5004191 and A5004192, where no sRNA or coding sequence was identified in previous studies (Tree et al., 2014; Gruber and Sperandio, 2015; Han et al., 2017; Waters et al., 2017). The pCP20 plasmid was finally transformed into the insertion strain to remove the antibiotic cassette.
All the bacterial strains and plasmids used are listed in Supplementary Table S1 and Supplementary Table S2. All the oligonucleotides used in this study are listed in Supplementary Table S3.
RNA isolation and handling
Bacterial samples were collected by centrifugation and mixed with 1/5 volume of the 95% ethanol, 5% phenol, snap-frozen in liquid nitrogen and thawed on ice. Total RNA was extracted using the TRIzol Reagent (Thermo scientific) according to the manufacturer’s protocol. RNA samples were further digested by DNase I (New England Biolabs) and purified using the RNA Clean & Concentrator™-5 (Zymo Research). RNA quantity was measured using the NanoDrop-2000 spectrophotometer, and the integrity of the RNA was checked by gel electrophoresis.
Rapid amplification of cDNA ends
RACE assays were carried out using the FirstChoice® RLM-RACE Kit (Thermo Scientific) according to the manufacturer’s protocol. For 5’-RACE, 5 μg of total RNA was treated by Calf Intestine Alkaline Phosphatase and Tobacco Acid Pyrophosphatase to remove free 5’-phosphates and cap structures. A 45 base RNA adapter oligonucleotide was then ligated to the treated RNA population using T4 RNA ligase. sRNA-specific antisense primer and M-MLV reverse transcriptase were used for the 5’ end amplification. For 3’-RACE, a poly(A) tail was added to 3’-end of the total RNA using the Poly(A) Polymerase Tailing Kit (Epicentre). First strand cDNA was generated from poly(A)-tailed RNA using the 3’ RACE Adapter and M-MLV Reverse Transcriptase. The cDNA was then served as the template for 3’-end amplification using other 3’ RACE Primers which are complimentary to the anchored adapter, and a sRNA-specific antisense primer. Nested PCRs were carried out with nested primers provided by the kit and sRNA-specific antisense primers if necessary. The PCR products were cloned into pGEM®-T Vector Systems (Promega), and the inserts were identified by Sanger sequencing. The sequences of the oligonucleotides used for RACE are listed in Supplementary Table S3.
Protein purification
The BL21 (DE3) strain carrying pET28a-Z2269 was grown in LB until the OD600nm reached 0.4~0.6. The protein was induced by 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for overnight at 18°C. The Z2269 protein was purified using the Ni-NTA Agarose (Qiagen) as described before (Han et al., 2022). The absorbance at 280nm for the purified protein was measured using Nanodrop, and the protein concentration was calculated using the extinction coefficient of the Z2269 approximated with Expasy.
Electrophoretic mobility shift assay
To explore if Z2269 can directly bind to the promoter of ler, electrophoretic mobility shift assays (EMSAs) were conducted using the purified Z2269 protein and PCR-amplified DNA fragments corresponding to the ler promoter region (150 bp upstream the transcriptional start site). 40 ng of the purified PCR fragment was incubated with different amounts of purified Z2269 in the EMSA binding buffer [20 mM Tris (pH 7.5), 50 mM KCl, 5 mM MgCl2, and 10% (w/v) glycerol] at 37°C for 30 min. The samples were then electrophoresed for approximately 1.5 h at 90 V and 4°C on a 10% native polyacrylamide gel. The gel was then stained with Ethidium Bromide (Sigma-Aldrich) for 30 min and visualized using a UV transilluminator.
Northern blotting
Northern blot assays were performed as previously described (Villa et al., 2021). 10 µg of total RNA was separated on 10%/7 M urea polyacrylamide gels at 120 V for 5~8 h. The gel was briefly rinsed in 0.5 × TBE buffer and electroblotted onto the Brightstar Plus nylon membranes (Applied Biosystems) at 15V for overnight and cross-linked with 254 nm UV light for 2 min. Crosslinked membranes were prehybridized for 1 h at 50°C in the PerfectHyb Plus hybridization buffer (Sigma-Aldrich). A ssDNA oligonucleotide specific to EvrS was labelled by γ-32P (PerkinElmer) and incubated with membranes in the PerfectHyb Plus hybridization buffer overnight at 50°C. After washes with 5×SSC once at 68°C for 15 min and with 1×SSC twice at 68°C for 20 min, signals were determined using a Typhoon FLA 7000 phosphorimager (GE Healthcare). The size of sRNA was determined by comparing its corresponding band to the ΦX174 DNA/HinfI Marker (Promega). The sequence of the specific probe is listed in Supplementary Table S3.
Mouse infection experiments
All animal experiments were performed according to the standards set forth in the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). The study was approved by the Institutional Animal Care and Use Committee of the North China University of Science and Technology Affiliated Hospital.
BALB/c mice (female, 6-8 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). Groups of 7 mice were maintained with sterilized food and water ad libitum for 7 days and deprived of food 14 h prior to infection. Mice were then administered orally by pipetting feeding of 109 CFU of WTNal, ΔevrSNal, and pEvrS strains growing in logarithmic phase (at an OD600nm of 0.6) suspended in 100 μl phosphate-buffered saline (pH 7.4). The sterilized diet and sterilized water were returned after the infection. Mice were euthanized by aether asphyxiation 6 h later. The ileum and colon were dissected and weighed after removing the luminal contents. The tissues were washed and homogenized in cold, sterile 1×PBS. The bacterial CFU was determined by plating of serially diluted homogenates on LB agar plates with appropriate antibiotics (50 μg ml-1 nalidixic acid for WTNal and ΔevrSNal; 15 μg ml-1 chloramphenicol for pEvrS).
Cell infection assays
Cell infection assays using human Caco-2 epithelial cells (ATCC HTB-37) were performed as described previously (Liu et al., 2020). Caco-2 cells were purchased from the Shanghai Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China) and maintained in Dulbecco’s Modified-Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin/gentamicin antibiotic mix and grown at 37°C and 5% CO2. WT, ΔevrS, and pEvrS strains were inoculated in DMEM and grown to an OD600nm of 0.6 at 37°C. Monolayers of Caco-2 cells were infected with bacteria cultured in DMEM at an m.o.i. (multiplicity of infection) of 100:1. After 3 h of incubation at 37°C in 5% CO2, cells were washed three times in pre-warmed PBS to remove unbound bacteria and lysed in 1 ml of 0.1% sodium dodecyl sulfate. Lysates were collected, diluted, and plated onto LB agar plates to determine bacterial colony-forming units (CFU). Each experiment was carried out at least three times.
Growth curve measurement
WT, ΔevrS and pEvrS strains were grown overnight in LB. The next morning, the strains were washed in DMEM media, and normalized to the OD600nm of 0.03 in fresh DMEM media. The strains were grown aerobically and shaking at 37°C. At the indicated time points, the OD600nm of each strain was monitored by the SCI-UV1100 Spectrophotometer (SCILOGEX, LLC). The average values and standard errors were calculated from the results of three experiments.
Real-time quantitative PCR
RT-qPCR was performed using the Luna® Universal One-Step RT-qPCR Kit (New England Biolabs) and the Applied Biosystems 7500 Real-Time PCR systems (Applied Biosystems). 50 ng RNA of each sample was first converted to cDNA at 55°C for 10 min. The cDNA was then denatured at 95°C for 1 min, followed by 45 cycles of 95°C (310 sec) and 60°C (30 sec). All data were normalized to levels of housekeeping gene 16S rRNA (rrsH), and the relative expression level was calculated as fold change using the 2-ΔΔCt method (Schmittgen and Livak, 2008). Each qRT-PCR analysis was performed in triplicates.
To evaluate the expression of EvrS in vivo, 6-week-old female BALB/c mice (n = 7) were infected intragastrically with 109 CFU of the EDL933Nal strain expressing PEvrS-fused GFP on the plasmid and mCherry on the chromosome (cells growing in DMEM with an OD600nm of 0.6 were collected). At 6 h following infection, the ileum and colon were removed aseptically. RNA was extracted, and the rRNAs were depleted using the Ribo-Zero Gold rRNA Removal Kit (Epicentre). qRT-PCR analysis was then carried out to compare the gfp expression in the two organs using the 2-ΔΔCt method. The expression of the mCherry gene was used for normalization.
All the primers used for qRT-PCR analysis are listed in Supplementary Table S3.
Chromatin immunoprecipitation
The pTrc99a vector carrying 3×FLAG-tagged z2269 were constructed and transformed into the Δz2269 mutation strain. The bacterial cultures were grown to an OD600nm of 0.6 at 37°C in DMEM and further incubated with 1 mM IPTG for 30 min at 37°C to induce the expression of Z2269-3×FLAG. For crosslinking, the cells were incubated with 1% formaldehyde for 25 min at room temperature. The cells were then washed with ice-cold 1×TBS and resuspended in 800 μL IP buffer [100 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA, 50 µg/mL RNase A, 1 mM PMSF]. The cells were then sonicated on ice with 15 cycles of 10 s on/off at 30% amplitude to generate DNA fragments of approximately 200-500 bp. After sonication, the samples were subjected to centrifugation at 13,000 g for 20 min at 4°C. The supernatant was split into two (400 μL each), with one aliquot incubated with 20 μL monoclonal ANTI-FLAG antibody (Sigma-Aldrich) at 4°C for 4 h as the ChIP sample, and the other without incubation with any antibody (as the mock sample). Both the ChIP and mock samples were then incubated with 50 µl protein A magnetic beads (Thermo Scientific) for 3 h at 4°C. The beads were washed with 1×PBS buffer and resuspended in 200 µL elution buffer [50 mM Tris-HCl (pH 8.0), 10 mM EDTA, 1% SDS]. The elution samples were incubated with 50 µg/mL RNaseA (Thermo Scientific) for 1 h at 37°C, and then with 100 µg/mL proteinase K (Thermo Scientific) for another 1 h at 55°C. DNA in the elution was further extracted using the QIAamp DNA Micro Kit (Qiagen). RT-qPCR was then carried out to detect if the ler promoter fragment is enriched in the ChIP sample relative to the mock sample. The experiments were performed in three biological replicates.
Enzyme-linked immunosorbent assay
Shiga toxin production was quantified using the RIDASCREEN Shiga toxin enzyme-linked immunosorbent assay (R-Biopharm) according to the manufacturer’s instructions. After growing the WT, ΔevrS, and pEvrS strains to an OD600nm of 0.6 in DMEM, supernatants were collected from 1 ml culture and sterilized by filtration (pore size, 0.22 µm). 100 µl of supernatants were incubated with 100 μl biotin-conjugated antibody for 60 minutes in a 96-well plate at room temperature. The wells were washed for five times using 300 µl wash buffer and incubated with 100 µl poly-streptavidin peroxidase conjugate for 30 minutes at room temperature. After washing five times, the wells were filled with 100 µl substrate, and the plate was incubated for 15 minutes in darkness at room temperature (20-25°C) before the addition of 50 µl stop reagent. Absorbances were measured at 450 nm using a Spark multilabel plate reader (Tecan). Three biological replicates were performed for each assay.
RNA decay assay
Overnight WT and ΔevrS strains were diluted 1:100 in fresh DMEM medium and cultured at 37°C. After OD600nm reached to 0.6, rifampin was added to the cultures at a final concentration of 250 µg/ml. At 2.5-, 5-, 10-, 20-, and 30-min posttreatment, 2 ml samples of each culture were collected and stored in liquid nitrogen immediately. Total RNA extraction and RT-qPCR were performed as described above for each sample. The half-life of z2269 was then calculated from three biological replicates.
Motility assays
1 μl of logarithmic-phase bacterial culture grown in DMEM media was spotted onto the center of 0.3% LB agar plates. Kanamycin and chloramphenicol were added to the plates when necessary. The plates were incubated at 30°C for 10 h before taking the images of the plates.
β-galactosidase activity assay
Translational β-galactosidase assays were performed as previously (Gruber and Sperandio, 2015). Overnight cultures of the strains containing the reporter plasmids were diluted in fresh DMEM medium and grown at 37°C until the OD600nm reached 0.6. The cell culture was then incubated with 0.2% arabinose for 30 min. 500 μl of culture was taken for β-galactosidase activity measurement using the β-Gal Assay Kit (Thermo Scientific) and a Spark multilabel plate reader (Tecan). β-Galactosidase activity is defined as (μmol of ortho-nitrophenol [ONP] formed min-1) × 106/[OD600nm] × ml of cell suspension. Each experiment was conducted in three replicates.
Luciferase activity assay
To determine the effects of environmental signals on EvrS expression, we performed a Luciferase activity assay. Bacteria harboring the promoter-luciferase fusion plasmid were grown in M9 minimal media until the OD600nm reached 0.6. Cells were further cultured with gentle shaking for another 30 min at 37°C with different “signals”, including pH 5.5, pH 8.5, low oxygen (microaerobic) condition, 200 nM biotin, 0.2% bile, 50 μM epinephrine, 50 μM norepinephrine, 500 μM indole, 200 mM NaCl, 200 µM α,α’-dipyridyl, 150 μM malate, 0.4% glucose, 20 µM riboflavin, 100 µM fucose, 10 mM N-acetylneuraminic acid, 10 mM N-acetylglucosamin, 1 mM pyruvate, 2.5 mM succinate, 2.5 mM fumarate, 100 mM acetate, 100 mM propionate, 100 mM butyrate, 5 mM choline, 150 nM vitamin B12, and 30 mM ethanolamine together with 150 nM vitamin B12. For the microaerobic condition, bacteria grown statically in M9 minimal media at 37°C in the presence of 5% CO2 in a 15 mL falcon tube with cap tightly closed until the OD600nm reached 0.6. After incubation with these signals/under these conditions, 100 μl of the bacterial culture was then taken to measure the luminescence intensity at a wavelength of 560 nm using a Spark multilabel plate reader (Tecan). The luciferase activity was normalized by dividing the luminescence intensities by OD600nm values. The average and standard error were calculated from the results of three experiments.
Statistical analysis
All the data were shown as mean ± standard deviations (SD). The statistical analysis was performed with the Graphpad Prism software using the Student’s t-test. For mouse infection experiments, the Mann-Whitney rank-sum test was performed. P ≤ 0.05, ≤ 0.005, or ≤ 0.0005 was considered to be statistically significant (*), highly significant (**), or extremely significant (***), respectively.
Results
The sRNA EvrS modulates EHEC adherence to epithelial cells
Through a previous transcriptomic analysis, a small RNA (Esr023) was found in the prophage CP-933N on the EDL933 genome (Han et al., 2017). We renamed it EvrS (EHEC virulence regulatory sRNA) and determined its 5’- and 3’-ends on the chromosome of the EHEC O157:H7 EDL933 strain by RACE. The transcription start site of EvrS mapped to C1641353, and a putative Rho independent terminator was found upstream of T1641507 (Figure 1A). The length of EvrS was determined to be 154 bp, which is consistent with the size of the RNA band shown by Northern blot analysis (Figure 1B). The gene flanking EvrS on one side encodes a transcriptional regulator (z1789) and the other encodes a novel esterase (z1793) (Figure 1A). To test the function of EvrS, a mutant strain (ΔevrS) was constructed by homologous recombination in the EDL933 wild-type strain (WT), and complementation of EvrS was achieved using the pACYC184 plasmid expressing the sRNA (pEvrS). Northern blotting and qRT-PCR confirmed the absence of EvrS expression in the ΔevrS strain and the restored EvrS level in the pEvrS strain (Figures 1C, D).
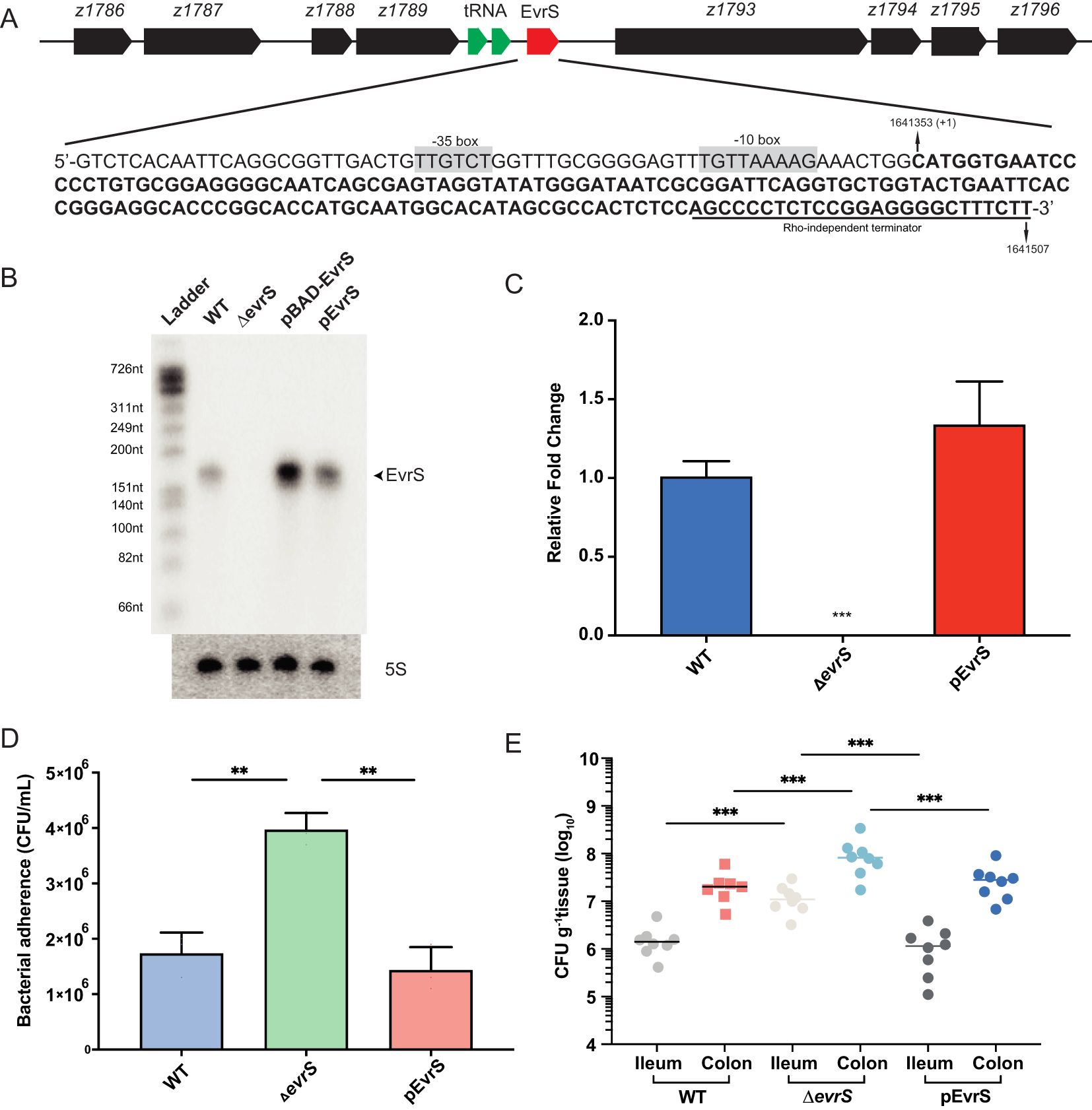
Figure 1. EvrS plays an essential role in pathogenesis of EHEC O157:H7. (A) Schematic diagram of the EvrS location on the chromosome of the EDL933 strain. The EvrS sequence is represented by bold. Putative -35 box and -10 box are highlighted in grey. The Rho-independent terminator is indicated by an underline. (B) Detection of EvrS by Northern blot analysis in the EDL933 wildtype (WT), EvrS deletion (ΔevrS), EvrS overexpression (pBAD-EvrS) (EvrS was induced with 0.2% arabinose for 10 min at OD600nm of 0.6), and EvrS complementation (pEvrS) strains at OD600nm of 0.6 grown in DMEM media. (C) RT-qPCR analysis of EvrS expression in EDL933 WT, EvrS deletion (ΔevrS), and EvrS complementation (pEvrS) strains. (D) Adherence capacity of the WT, ΔevrS and pEvrS strains to Caco-2 epithelial cells. CFU, colony-forming unit. (E) Adherence capacity of the WT, ΔevrS, and pEvrS strains in the ileum and colon of mice. CFU, colony-forming unit. For (C, D), **, P ≤ 0.01; ***, P ≤ 0.001 (Student t test). For (E), horizontal lines represent geometric means. ***, P ≤ 0.001 (Mann-Whitney rank-sum test).
The pathogenesis of EHEC O157:H7, to a considerable degree, depends on the adherence ability to intestinal epithelial cells (Wong et al., 2011). Therefore, we compared the adherence of the WT, ΔevrS, and pEvrS strains by incubating DMEM-grown bacteria with Caco-2 epithelial cells to analyze whether EvrS can affect bacterial adherence ability. We found that the adherence capacity of the ΔevrS strain to Caco-2 cells was significantly increased (by 5.25-fold) compared with the WT strain, which was restored in the pEvrS strain (Figure 1E). Importantly, no significant differences in growth rate were observed between the WT, ΔevrS, and pEvrS strains in DMEM (Supplementary Figure S1A). The deletion of EvrS did not alter significantly the expression of the surrounding ORFs as measured by qRT-PCR (Supplementary Figure S1B), indicating that the changed adherence ability of EvrS derivatives was not due to a growth difference or the altered expression of neighborhood genes.
We further tested the in vivo adherence capabilities of EvrS derivatives using a mouse model as described before (Liu et al., 2022). Groups of 7 BALB/c mice were infected with 109 CFU bacteria (the WT, ΔevrS, and pEvrS strains) by oral gavage, and the number of bacteria recovered from the ileum and colon after 6 h was determined to evaluate bacterial colonization capacity. We found that the number of adhered bacteria to ileum and colon from ΔevrS-infected mice (1.06 × 107 CFU g-1 tissue in ileum and 8.87 × 107 CFU g-1 tissue in colon) was significantly higher than the adhered bacteria from WT-infected mice (1.78 × 106 CFU g-1 tissue in ileum and 2.39 × 107 CFU g-1 tissue in colon) (Figure 1E). Importantly, the number of bacteria that adhered to the colon from WT-infected mice was 13.40-fold higher than the bacteria adhered to the ileum (P < 0.01) (Figure 1E), which is consistent with the fact that EHEC preferentially colonizes in the large intestine. Moreover, the numbers of adhered bacteria from pEvrS-infected mice (2.25 × 107 CFU g-1 tissue in the colon, and 1.36 × 106 CFU g-1 tissue in the ileum) restored to the WT level (Figure 1E). Taken together, we conclude that EvrS is a negative regulator in EHEC O157:H7 pathogenesis by affecting the bacterial adherence ability.
EvrS negatively regulates the expression of LEE via ler
The colonization ability of EHEC O157:H7 depends on a series of virulence factors (e.g., LEE), indicating that EvrS may affect the bacterial pathogenesis through regulating the expression of these factors (Foster, 2013; Carlson-Banning and Sperandio, 2018). To test this hypothesis, qRT-PCR was performed to measure the expression of seven representative LEE genes, including ler (the master regulator of LEE genes), escT (LEE1), escC (LEE2), escN (LEE3), espB (LEE 4), eae (intimin, LEE5), and tir (intimin receptor, LEE 5) in the WT, ΔevrS and pEvrS strains. As shown in Figure 2A, the expressions of all these LEE genes significantly increased in the absence of EvrS, whereas these changes were restored to wild-type levels in the pEvrS strain (Figure 2A). On the contrary, the bacterial motility and the production of Shiga toxin were not affected by the deletion and complementation of EvrS (Supplementary Figures S2A, S2B). These results suggested that the expression of EvrS only influences LEE expression but has no effect on Stx production and bacterial motility.
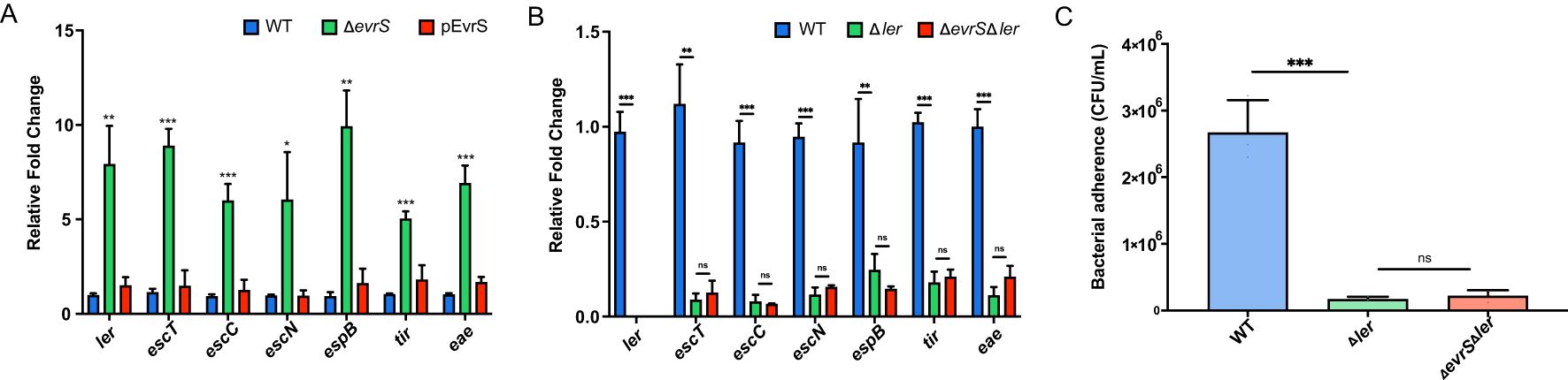
Figure 2. EvrS negatively affects locus of enterocyte effacement (LEE) gene expression through Ler. (A) RT-qPCR analysis of LEE expression in the WT, ΔevrS, and pEvrS strains. (B) RT-qPCR analysis of LEE expression in the WT, ler deletion (Δler) and EvrS+ler double deletion (ΔevrSΔler) strains. (C) Adherence capacity of WT, Δler, and ΔevrSΔler strains to Caco-2 epithelial cells. CFU, colony-forming unit. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns, non-significant (Student t test).
Considering that ler is one of the most important LEE regulators and that all LEE genes tested are affected by EvrS (Figure 2A), we assumed that EvrS may affect LEE expression via ler. To confirm this, we examined the EvrS regulation on LEE expression in the absence of ler. Deletion of ler in EHEC O157:H7 significantly reduced LEE gene expression and bacterial adherence to Caco-2 cells (Figures 2A, B), which is consistent with the role of Ler as a positive LEE regulator. Notably, the expression of LEE genes was comparable in the Δler strain and the ΔevrSΔler double deletion strain (Figure 2A). Furthermore, the cell adherence of the ΔevrSΔler strain was equivalent to the ΔevrS strain, which is much lower than the WT level (Figure 2B). These results suggested that the regulatory role of EvrS on bacterial adherence and LEE expression is mediated by Ler.
EvrS regulates the expression of z2269 mRNA by destabilizing the transcript
To find out the direct target(s) of EvrS, we used Target RNA2 (Kery et al., 2014) and predicted 25 potential mRNA targets of the sRNA (Supplementary Table S4). To identify these targets experimentally, we selected thirteen candidates and carried out qRT-PCR to test the expression of these genes under different EvrS backgrounds. These candidates were selected based on their potential relevance to EHEC pathogenesis as reported in previous studies (they showed differential expression between infection and in vitro conditions) (Connolly et al., 2015; Yang et al., 2015; Woodward et al., 2019) and/or their likely regulation by EvrS [the predicted binding sites located in the translation initiation region (-20 to +20 relative to the start codon)]. One gene, z2269, showed increased expression in the ΔevrS strain compared to the WT strain, and the complementation of EvrS in the ΔevrS strain restored z2269 expression to the wildtype level (Figure 3A). In contrast, the expressions of the other predicted EvrS target mRNAs did not change significantly upon EvrS deletion (Supplementary Figure S3). Interestingly, the predicted base pairing interactions between EvrS and z2269 were around the translation start site of the mRNAs (Figure 3B), suggesting that EvrS may bind directly to the mRNA of z2269 and affect its translation.
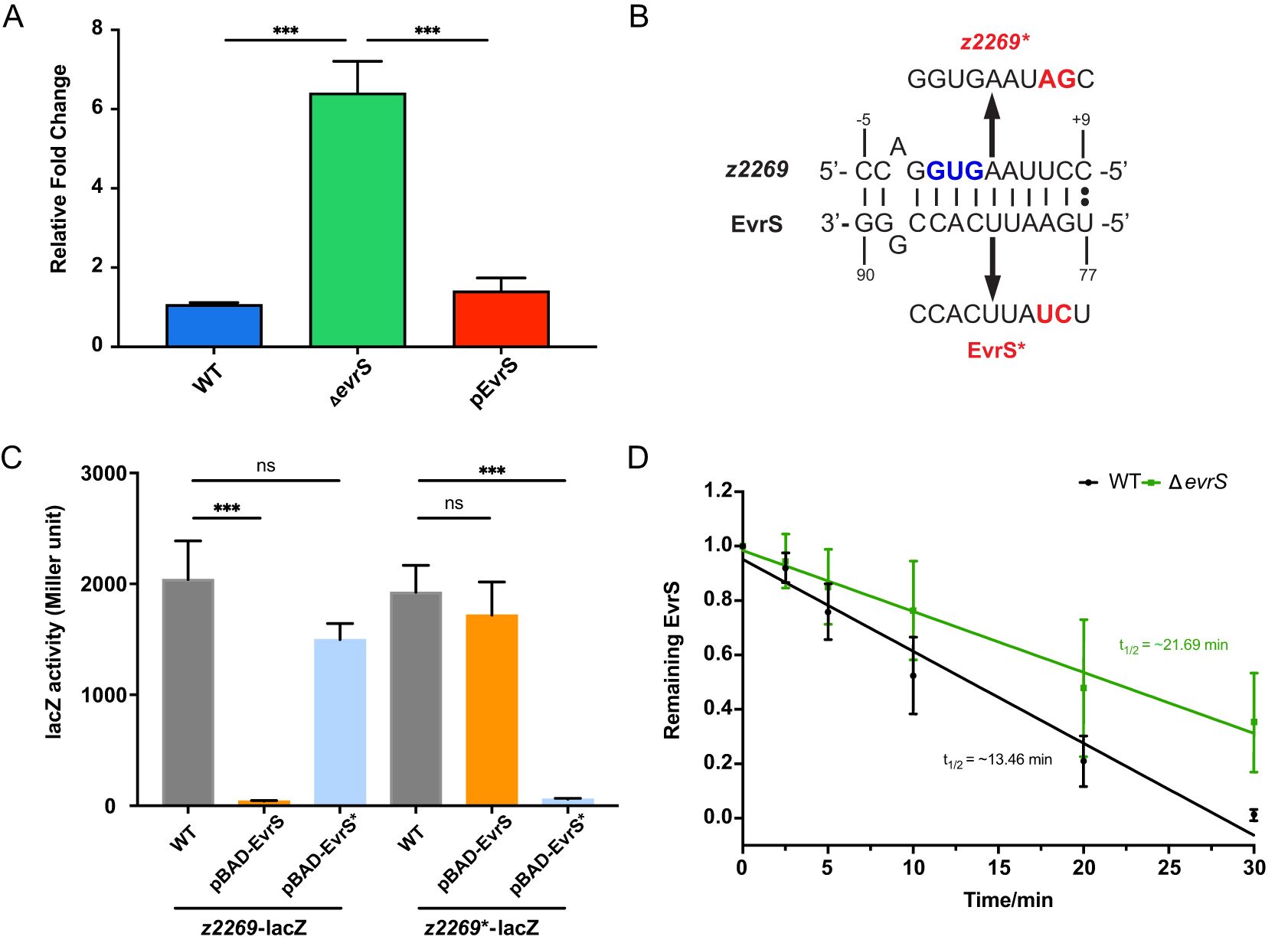
Figure 3. EvrS directly regulates and destabilizes the transcript of z2269. (A) RT-qPCR analysis of z2269 expression in the WT, ΔevrS, and pEvrS strains. (B) Predicted binding sites between EvrS and z2269 5’UTR. The start codon (GUG) is indicated in blue. The mutations introduced on z2269 or EvrS are indicated in red. (C) β-Galactosidase assays of the z2269 translational reporter in the WT, pBAD-EvrS, or pBAD-EvrS* strains. (D) Half-life measurement of z2269 in the WT and ΔevrS strains by RT-qPCR analysis. ***, P ≤ 0.001; ns, non-significant (Student t test).
We further used a translational reporter system (Gruber and Sperandio, 2014) to test the in vivo regulatory outcomes of the EvrS-z2269 interactions. In this assay, a DNA fragment spanning from -100 to +48 bp relative to the translation start site of the z2269 mRNA was amplified and cloned upstream of the lacZ gene, under the control of the PBAD promoter. This construct, along with another plasmid containing EvrS (also under the PBAD promoter), was transformed into the wild-type strain. This setup allows the regulatory effect of EvrS on z2269 translation to be assessed by measuring LacZ activity following induction of both the translational fusion and the sRNA (Supplementary Figure S4). Our results showed that the activity of z2269-lacZ fusion was repressed significantly upon EvrS overexpression (Figure 3C), indicating the translation of Z2269 is affected by EvrS. To examine whether the predicted binding sites are required for this regulation, we introduced mutations on the binding site without altering the codon preference (Figure 3B) and tested the effect of EvrS on the Z2269 fusion carrying the mutation (z2269*). We observed that the effect of EvrS on Z2269 was abolished when the mutation was introduced (Figure 3C). Moreover, the overexpression of EvrS* (carrying the corresponding mutations on the sRNA) significantly repressed the activity of z2269*-lacZ but has little effect on z2269-lacZ. These results indicate that the regulation of EvrS on z2269 is mediated by the direct interaction between the two RNAs.
To reveal mechanistic insights into the regulation of EvrS regulation on z2269, an RNA decay assay was performed to determine whether EvrS affects the stability of z2269 mRNA. The WT and ΔevrS cell cultures were treated with rifampicin to halt transcription. RNA samples were isolated at indicated time intervals posttreatment, and the abundance of z2269 transcript was then determined by RT-qPCR analysis. As shown in Figure 3D, the half-life of the z2269 mRNA was approximately 21.69 min in the ΔevrS strain, which is much longer than that in the WT strain (~13.46 min), indicating that the z2269 transcript is more rapidly degraded in the presence of EvrS. Altogether, these results suggest that EvrS directly binds to z2269 mRNA through the predicted binding sites and negatively regulates the stability of the transcript.
Z2269 activates LEE expression and bacterial adherence through ler
Since z2269 is the only identified target of EvrS, we assumed that the regulation of EvrS is mediated through this gene. We therefore investigated the contribution of z2269 to the LEE expression and bacterial adherence. We found that the expression of LEE genes and bacterial adherence decreased significantly in the Δz2269 strain compared to WT by qRT-PCR and cell infection assay (Figures 4A, B). However, comparable LEE gene expression and bacterial adherence ability were observed in the Δler strain and Δz2269Δler double deletion strain (Figures 4A, B). Moreover, overexpressing z2269 only resulted in elevated expression of LEE genes in WT but not in the Δler strain (Figure 4C). These results implied that Z2269 activates EHEC adherence and LEE gene expression via Ler.
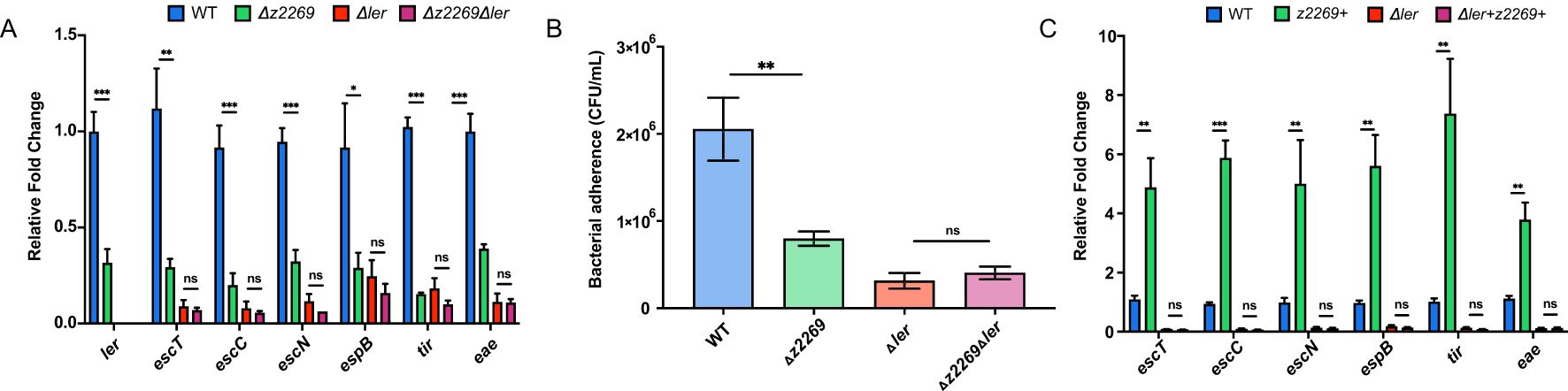
Figure 4. Z2269 activates locus of enterocyte effacement (LEE) gene expression and adherence to epithelial cells through Ler. (A) RT-qPCR analysis of LEE expression in WT, z2269 deletion (Δz2269), ler+z2269 double deletion (ΔlerΔz2269), and Δler strains. (B) Adherence capacity of WT, Δz2269, ΔlerΔz2269 and Δler strains to Caco-2 epithelial cells. (C) RT-qPCR analysis of the expression of LEE expression in WT, WT strain overexpressing z2269 (z2269+). ler deletion (Δler), and the Δler strain overexpressing z2269 (Δler+z2269+). CFU, colony-forming unit. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns, non-significant (Student t test).
We next tested how ler is regulated by z2269. Through bioinformatic prediction, we found that Z2269 contains an N-terminal DNA-binding helix-turn-helix (HTH) motif and a C-terminal ligand-binding domain (Supplementary Figure S5A), implying that Z2269 may serve as a transcriptional regulator of ler. To test if Z2269 could regulate ler by directly interacting with its promoter, we performed an Electrophoretic Mobility Shift Assay (EMSA) by incubating the purified Z2269 protein (Supplementary Figure S5B) with the DNA fragment corresponding to the ler promoter. As shown in Supplementary Figure S5C, we didn’t observe the complex formation between Z2269 and the ler promoter fragment in vitro, suggesting that Z2269 is unable to bind ler promoter. However, this lack of binding could also be due to the absence of a required ligand in the EMSA reaction. To further understand this, a chromatin immunoprecipitation (ChIP)-qPCR assay was carried out to examine if Z2269 can bind ler promoter in vivo. Consistent with the EMSA result, we did not detect any significant enrichment of the ler promoter in the ChIP sample compared to the mock sample (Supplementary Figure S5D). These results suggest that Z2269 does not bind to the promoter region of ler but regulates the ler expression indirectly, probably through an unknown intermediary factor.
The expression of EvrS is modulated by oxygen availability
The rapid response of sRNA to environmental signals is important for EHEC adaption and colonization of different intestinal sites (González Plaza, 2020; Felden and Augagneur, 2021; Sy and Tree, 2021). To test which signal is involved in the regulation of EvrS, we amplified a DNA fragment containing the EvrS promoter region and fused with a promoter-less luciferase operon on the low-copy-number plasmid pMS402 (Fan et al., 2020). The fusion construct was introduced into the EHEC O157:H7 EDL933 strain and luminescence was measured during growth in M9 minimal medium by changing 26 previously reported “signals” that play significant roles in regulating virulence genes (especially LEE) in EHEC (Bansal et al., 2007; Kus et al., 2011; Kendall et al., 2012; Pacheco et al., 2012; Gonyar and Kendall, 2014; Yang et al., 2015; Carlson-Banning and Sperandio, 2016, 2018; Cordonnier et al., 2016; Lackraj et al., 2016; Le Bihan et al., 2017; Connors et al., 2018; Melson and Kendall, 2019; Liu et al., 2020, 2022). Compared to the control condition (aerobic, pH 7.2), the activity of the EvrS promoter significantly decreased by ~3.74 fold under microaerobic condition (Figure 5A). The EvrS promoter activity also significantly changed after incubating with 50 μM epinephrine/norepinephrine, 2.5 mM fumarate, 100 mM propionate, or 30 mM ethanolamine (Figure 5A), though the change was not so remarkable as that observed under the microaerobic condition. In contrast, no significant change was observed with other treatments (Figure 5A). To reassure this finding, we performed qRT-PCR analysis to compare the expression of EvrS under the control and microaerobic conditions, or upon the addition of epinephrine, fumarate, propionate, or ethanolamine. In consistent with the Luciferase activity assay results, our data showed that the EvrS level was significantly reduced by 3.85-fold under the microaerobic condition but didn’t show significant change in the presence of epinephrine/norepinephrine, fumarate, and ethanolamine (Figure 5B). The expression of EvrS also significantly changed after incubating with propionate, but it only showed a slightly reduced level (~ 0.81-fold) compared to the control. These results suggest that the expression of EvrS is mainly modulated by oxygen availability in the environment.
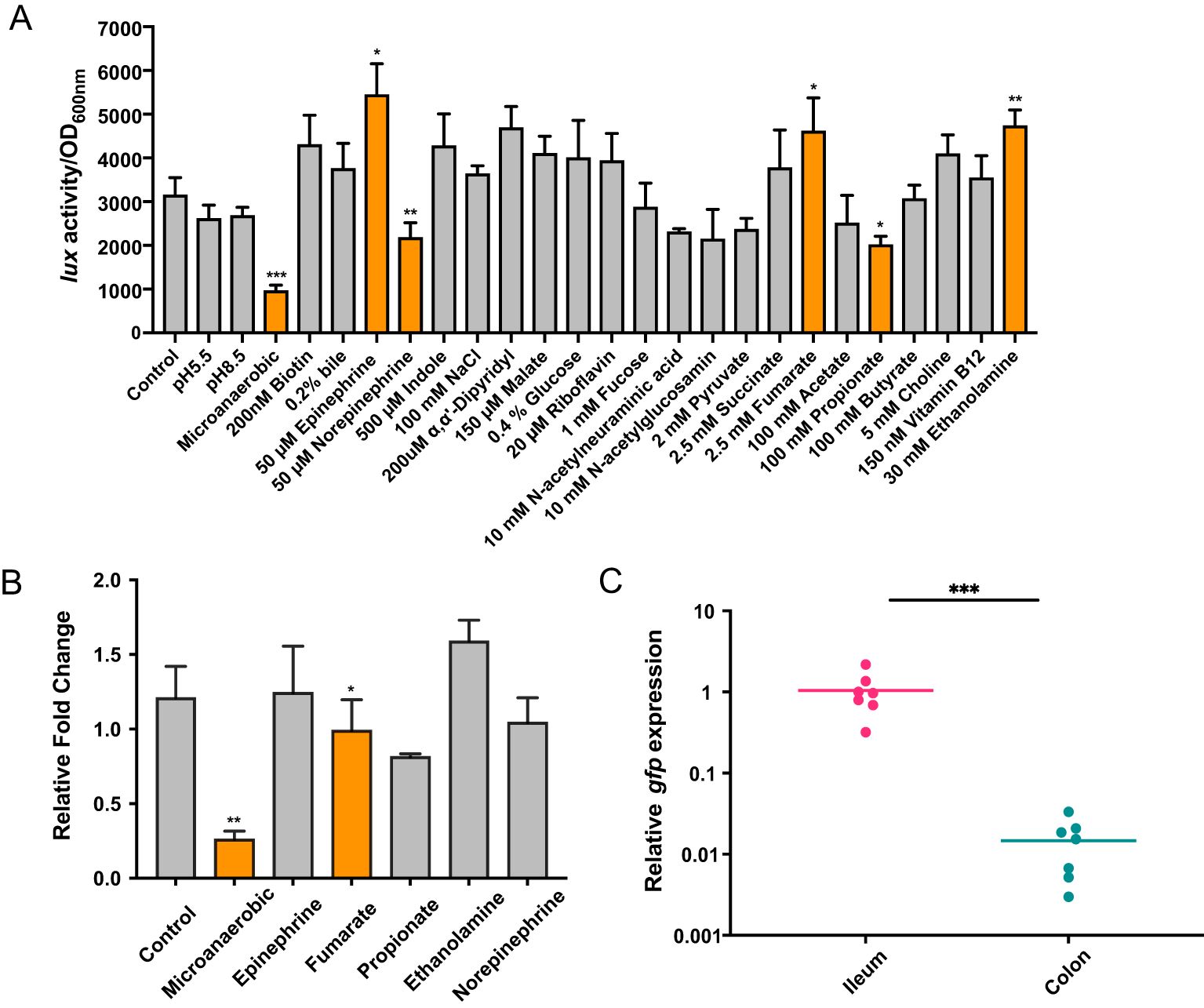
Figure 5. EvrS expression is affected by oxygen availability. (A) Activity of PEvrS-lux transcriptional fusions in EHEC EDL933 WT strain grown in M9 minimal media under the control condition (aerobic, pH 7.2) or under a series of other environmental conditions [pH 5.5, pH 8.5, low oxygen, or upon the incubation with 200 nM biotin, 0.2% bile, 50 μM epinephrine, 50 μM norepinephrine, 500 μM indole, 200 mM NaCl, 200 µM α,α’-dipyridyl, 150 μM malate, 0.4% glucose, 20 µM riboflavin, 100 µM fucose, 10 mM N-acetylneuraminic acid, 10 mM N-acetylglucosamin, 1 mM pyruvate, 2.5 mM succinate, 2.5 mM fumarate, 100 mM acetate, 100 mM propionate, 100 mM butyrate, 5 mM choline, 150 nM vitamin B12, or 30 mM ethanolamine (together with 150 nM vitamin B12)]. (B) RT-qPCR analysis of EvrS expression in WT under control or microaerobic conditions, or after adding 50 μM epinephrine/norepinephrine, 2.5 mM fumarate, 100 mM propionate, or 30 mM ethanolamine. (C) RT-qPCR analysis of gfp expression under EvrS promoter relative to mCherry under RpoD promoter in the ileum and colon of mice (n = 7). A higher gfp/mCherry value represents a higher EvrS expression. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (Student t test).
EvrS contributes to the site-specific colonization of EHEC O157:H7 in mice large intestine
Considering EHEC normally encounters an oxygen gradient in the human intestinal tract, we proposed that the expression of EvrS might be differentially regulated in different intestinal niches with varying oxygen availability. The expression of EvrS during EHEC O157:H7 colonization in the intestine of mice was subsequently analyzed. To achieve this, RT-qPCR analysis was performed to evaluate the expression of EvrS in ileum and colon by measuring the gfp expression fused to the EvrS promoter. We found that the activity of EvrS promoter is significantly reduced in the colon compared with that in the ileum, as indicated by the reduced relative expression of gfp in the colon (P < 0.001) (Figure 5C). The differential expression of EvrS in mice intestine indicates that this sRNA may influence the site-specific colonization of EHEC O157:H7 in the large intestine. Indeed, in contrast to the WT strain, the difference between the adhered bacteria in the ileum and colon reduced to 8.27-fold in the ΔevrS-infected mice compared to the WT strain (the colonization of which is 13.4-fold higher in the colon than in the ileum), although the difference was still significant (P < 0.001) (Figure 1E). The adherence of the pEvrS strain in the colon was 15.17-fold higher than that in the ileum, which resembled the WT level (Figure 1E). Collectively, these results indicated that EvrS is differentially expressed between the ileum and colon, and contributes to the site-specific colonization of EHEC in the large intestine in mice.
Discussion
The specific colonization of EHEC in the large intestine of mammals is highly regulated by different environmental stimuli in the intestine (Foster, 2013; Carlson-Banning and Sperandio, 2018). A deep understanding of this can provide insights into EHEC infection mechanisms and potential therapeutic interventions. Increasing evidence has indicated that these complex regulations possibly consist of multiple layers of regulation (transcriptional and posttranscriptional) coordinating responses to extracellular environments (Carlson-Banning and Sperandio, 2018; Sauder and Kendall, 2018; Melson and Kendall, 2019). These regulations are usually modulated by environmental changes in different niches. For instance, oxygen sensing is a key determinant for pathogens (including EHEC) in their ability to gauge their location within the host and effectively deploy their virulence arsenals (Wallace et al., 2016). During transit and colonization in the gastrointestinal (GI) tract, EHEC encounters an oxygen gradient with a decreasing trend from stomach to large intestine but faces an increasing oxygen trend from the anaerobic lumen to the epithelial border (Espey, 2013; Carlson-Banning and Sperandio, 2016). To date, several transcriptional regulators have been shown to play crucial roles in EHEC’s response to changes in oxygen levels in the environment. However, the mechanisms of oxygen sensing and virulence regulation by sRNAs in EHEC remain largely unknown.
Here, we demonstrated a signal transduction regulatory pathway in which EHEC utilizes a newly characterized sRNA to sense oxygen availability and regulate site-specific colonization in the host intestinal tract (Figure 6). Under oxygen-rich conditions, the expression of EvrS is upregulated, decreasing the abundance of z2269 by destabilizing its transcript, leading to the further repression of LEE and prevention of bacterial adherence. This allows the bacteria to conserve energy and avoid unnecessary host immune responses at non-specific colonization sites (e.g., small intestine). When the bacteria enter an environment with lower oxygen levels (e.g., the large intestine), the expression of EvrS is downregulated, resulting in the upregulation of z2269. This, in turn, activates LEE expression and promotes bacterial adherence at specific colonization sites. Interestingly, the significant difference of colonized bacteria between ileum and colon was still observed in the ΔevrS strain, indicating this sRNA is not the only effector contributing to the preferential colonization of EHEC in the large intestine. Indeed, other factors (e.g., Esr055, BirA, Fur) have been revealed to promote the preferential colonization of EHEC in the colon in response to different stimuli (Yang et al., 2015; Han et al., 2017).
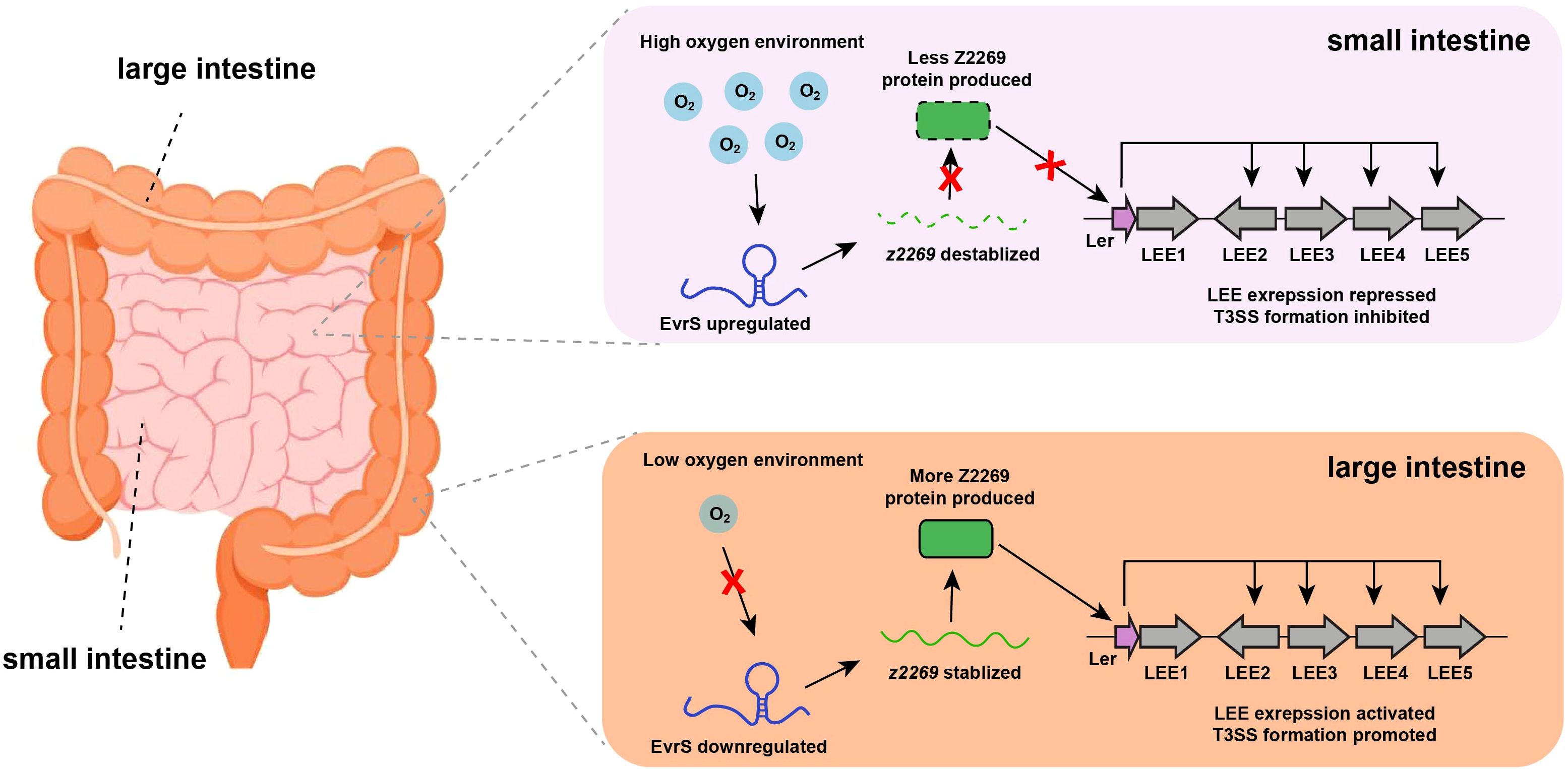
Figure 6. Proposed working model for EvrS regulation in different intestinal niches with varied oxygen availability.
Notably, a recent study showed that another sRNA in EHEC, DicF, is also affected by oxygen availability in vitro and promotes LEE expression through the direct interaction with the pchA, a LEE modulator (Melson and Kendall, 2019). Although the mechanism of DicF was thoroughly investigated, it is still not fully understood how this sRNA responds to fluctuating oxygen levels in the intestine to affect the in vivo colonization. By characterizing EvrS, our study expands the knowledge of how sRNAs regulate the specific colonization of EHEC in the large intestine. Intriguingly, the expression of DicF was only observed under microaerobic conditions, whereas EvrS can be detected under both aerobic and microaerobic conditions (Figures 1B, 5), suggesting other important roles of EvrS may exist under aerobic conditions. Unfortunately, we were unable to explain how oxygen modulates the expression of EvrS mechanistically. Upon examining the promoter sequence of the EvrS, we found a putative PdhR binding site and a putative CRP binding site (Supplementary Figure S6A). However, RT-qPCR analysis showed that both the mutant strains of crp and pdhR exhibited comparable EvrS levels to the WT strain (Supplementary Figure S6B). In addition, we examined the possible regulation of Fnr and ArcAB, two of the best-studied transcriptional regulators involved in adaptations to fluctuating oxygen levels in enteric pathogens (Salmon et al., 2003; Brown et al., 2022), on the expression of EvrS. As shown in Supplementary Figure S6B, no difference in EvrS level was observed in the Δfnr and ΔarcA strains compared to the WT strain, suggesting that the regulation of EvrS by oxygen is through some unknown factors.
Compared to DicF, which has four copies on the chromosome of EHEC O157:H7, EvrS only has a single copy located in the prophage CP-933N. Prophages are widely distributed in different bacterial genomes, and the genes or sRNAs in prophages enable host bacteria to adapt to various external environments (Fröhlich and Papenfort, 2016; Altuvia et al., 2018; Pleška et al., 2018). Given the importance of phage-mediated horizontal gene transfer on the evolution of bacteria-phage interplay, we wondered if EvrS would be conserved in other strains. Bioinformatics analysis revealed that putative EvrS homologs were found in the published genomes of many other EHEC strains, exhibiting a high level of conservation (Supplementary Figure S7). Interestingly, the homolog of EvrS in the EHEC Sakai strain was found in the Hfq CRAC data, suggesting that the function of EvrS may depend on Hfq. However, whether and how Hfq (and other sRNA binding proteins) modulate EvrS regulation in EHEC needs further investigation. It is worth noting that EvrS was also found in some gastrointestinal and extraintestinal pathogenic bacteria such as Enteropathogenic E. coli (EPEC), Uropathogenic E. coli (UPEC), Shigella flexneri, and Neonatal meningitis-causing E. coli (NMEC). These results indicate that EvrS is a widespread regulatory element to modulate the virulence in a range of human pathogens, even though the regulatory roles of EvrS in these pathogens have not been revealed.
Finally, we were only able to confirm one RNA target that is directly regulated by base pairing with EvrS due to the limitations of experimental and computational techniques used in this study. Considering that sRNAs typically interact with and regulate the expression of multiple target genes (Jørgensen et al., 2020), an expanded targetome and regulatory role of EvrS is possible, which however needs further study in the near future.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://doi.org/10.6084/m9.figshare.28067477.v1.
Ethics statement
The animal experiments were approved by the Laboratory Animal Ethics Committee of North China University of Science and Technology and complied with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals.
Author contributions
RH: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. YQ: Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. CZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Youth Scholars Promotion Plan of North China University of Science and Technology (QNTJ202408).
Acknowledgments
We gratefully appreciate Dr. Xin Deng from Hongkong City University for providing the pMS402 plasmid.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fcimb.2025.1642032.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1517328/full#supplementary-material
References
Altuvia, S., Storz, G., and Papenfort, K. (2018). “Cross-regulation between bacteria and phages at a posttranscriptional level,” in Regulating with RNA in Bacteria and Archaea (American Society of Microbiology), 501–514. doi: 10.1128/microbiolspec.rwr-0027-2018
Armetta, J., Schantz-Klausen, M., Shepelin, D., Vazquez-Uribe, R., Bahl, M. I., Laursen, M. F., et al. (2021). Escherichia coli Promoters with Consistent Expression throughout the Murine Gut. ACS Synth Biol. 10, 3359–3368. doi: 10.1021/ACSSYNBIO.1C00325
Bansal, T., Englert, D., Lee, J., Hegde, M., Wood, T. K., and Jayaraman, A. (2007). Differential effects of epinephrine, norepinephrine, and indole on Escherichia coli O157:H7 chemotaxis, colonization, and gene expression. Infect. Immun. 75, 4597–4607. doi: 10.1128/IAI.00630-07
Brown, A. N., Anderson, M. T., Bachman, M. A., and Mobley, H. L. T. (2022). The arcAB two-component system: function in metabolism, redox control, and infection. Microbiol. Mol. Biol. Rev. 86(2), e00110–21. doi: 10.1128/MMBR.00110-21
Carlson-Banning, K. M. and Sperandio, V. (2016). Catabolite and oxygen regulation of enterohemorrhagic Escherichia coli virulence. mBio 7.6 (2016): 10–1128. doi: 10.1128/mBio.01852-16
Carlson-Banning, K. M. and Sperandio, V. (2018). Enterohemorrhagic Escherichia coli outwits hosts through sensing small molecules. Curr. Opin. Microbiol. 41, 83–88. doi: 10.1016/j.mib.2017.12.002
Chaban, B., Hughes, H. V., and Beeby, M. (2015). The flagellum in bacterial pathogens: For motility and a whole lot more. Semin. Cell Dev. Biol. 46, 91–103. doi: 10.1016/j.semcdb.2015.10.032
Connolly, J. P. R., Brett Finlay, B., and Roe, A. J. (2015). From ingestion to colonization: The influence of the host environment on regulation of the LEE encoded type III secretion system in enterohaemorrhagic Escherichia coli. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00568
Connors, J., Dawe, N., and Van Limbergen, J. (2018). The role of succinate in the regulation of intestinal inflammation. Nutrients 11, 25. doi: 10.3390/nu11010025
Cordonnier, C., Le Bihan, G., Emond-Rheault, J. G., Garrivier, A., Harel, J., and Jubelin, G. (2016). Vitamin B12 uptake by the gut commensal bacteria bacteroides thetaiotaomicron limits the production of shiga toxin by enterohemorrhagic escherichia coli. Toxins (Basel) 8(1), 14. doi: 10.3390/toxins8010014
Datsenko, K. A. and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Espey, M. G. (2013). Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic. Biol. Med. 55, 130–140. doi: 10.1016/J.FREERADBIOMED.2012.10.554
Fan, L., Wang, T., Hua, C., Sun, W., Li, X., Grunwald, L., et al. (2020). A compendium of DNA-binding specificities of transcription factors in Pseudomonas syringae. Nat. Commun. 2020 11:1 11, 1–11. doi: 10.1038/s41467-020-18744-7
Felden, B. and Augagneur, Y. (2021). Diversity and versatility in small RNA-mediated regulation in bacterial pathogens. Front. Microbiol. 12. doi: 10.3389/FMICB.2021.719977
Foster, D. B. (2013). Modulation of the enterohemorrhagic E. coli virulence program through the human gastrointestinal tract. Virulence 4, 315–323. doi: 10.4161/viru.24318
Fröhlich, K. S. and Papenfort, K. (2016). Interplay of regulatory RNAs and mobile genetic elements in enteric pathogens. Mol. Microbiol. 101, 701–713. doi: 10.1111/mmi.13428
Gelalcha, B. D., Brown, S. M., Crocker, H. E., Agga, G. E., and Kerro Dego, O. (2022). Regulation mechanisms of virulence genes in enterohemorrhagic escherichia coli. Foodborne Pathog. Dis. 19, 598–612. doi: 10.1089/FPD.2021.0103
Gonyar, L. A. and Kendall, M. M. (2014). Ethanolamine and choline promote expression of putative and characterized fimbriae in enterohemorrhagic Escherichia coli O157: H7. Infect. Immun. 82, 193–201. doi: 10.1128/IAI.00980-13
González Plaza, J. J. (2020). Small RNAs as fundamental players in the transference of information during bacterial infectious diseases. Front. Mol. Biosci. 7. doi: 10.3389/fmolb.2020.00101
Gottesman, S. and Storz, G. (2011). Bacterial small RNA regulators: Versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 3.12 (2011): a00379. doi: 10.1101/CSHPERSPECT.A003798
Gruber, C. C. and Sperandio, V. (2014). Posttranscriptional control of microbe-induced rearrangement of host cell actin. mBio 5.1 (2014): 10–1128. doi: 10.1128/MBIO.01025-13
Gruber, C. C. and Sperandio, V. (2015). Global analysis of posttranscriptional regulation by GlmY and GlmZ in enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 83, 1286–1295. doi: 10.1128/IAI.02918-14
Han, R., Jiang, J., Fang, J., and Contreras, L. M. (2022). PNPase and rhlB interact and reduce the cellular availability of oxidized RNA in deinococcus radiodurans. Microbiol. Spectr. 10(4), e02140–22. doi: 10.1128/SPECTRUM.02140-22
Han, R., Xu, L., Wang, T., Liu, B., and Wang, L. (2017). A small regulatory RNA contributes to the preferential colonization of escherichia coli O157:H7 in the large intestine in response to a low DNA concentration. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00274
Jørgensen, M. G., Pettersen, J. S., and Kallipolitis, B. H. (2020). sRNA-mediated control in bacteria: An increasing diversity of regulatory mechanisms. Biochim. Biophys. Acta (BBA) - Gene Regul. Mech. 1863, 194504. doi: 10.1016/J.BBAGRM.2020.194504
Kaper, J. B., Nataro, J. P., and Mobley, H. L. T. (2004). Pathogenic escherichia coli. Nat. Rev. Microbiol. 2, 123–140. doi: 10.1038/NRMICRO818
Kendall, M. M., Gruber, C. C., Parker, C. T., and Sperandio, V. (2012). Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic escherichia coli O157:H7. mBio 3(3), 10–1128. doi: 10.1128/MBIO.00050-12
Kendall, M. M. and Sperandio, V. (2016). What a dinner party! mechanisms and functions of interkingdom signaling in host-pathogen associations. mBio 7.2 (2016): 10–1128. doi: 10.1128/MBIO.01748-15
Kery, M. B., Feldman, M., Livny, J., and Tjaden, B. (2014). TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res. 42, W124–W129. doi: 10.1093/NAR/GKU317
Kus, J. V., Gebremedhin, A., Dang, V., Tran, S. L., Serbanescu, A., and Foster, D. B. (2011). Bile salts induce resistance to polymyxin in enterohemorrhagic escherichia coli O157:H7. J. Bacteriol 193, 4509–4515. doi: 10.1128/JB.00200-11
Lackraj, T., Kim, J. I., Tran, S. L., and barnett foster, D. E. (2016). Differential modulation of flagella expression in enterohaemorrhagic Escherichia coli O157: H7 by intestinal short-chain fatty acid mixes. Microbiol. (United Kingdom) 162, 1761–1772. doi: 10.1099/mic.0.000357
Le Bihan, G., Sicard, J.-F., Garneau, P., Bernalier-Donadille, A., Gobert, A. P., Garrivier, A., et al. (2017). The NAG sensor nagC regulates LEE gene expression and contributes to gut colonization by escherichia coli O157:H7. Front. Cell Infect. Microbiol. 7. doi: 10.3389/fcimb.2017.00134
Leimbach, A., Hacker, J., and Dobrindt, U. (2013). E. coli as an all-rounder: the thin line between commensalism and pathogenicity. Curr. Top. Microbiol. Immunol. 358, 3–32. doi: 10.1007/82_2012_303
Liu, B., Liu, Y., Yang, B., Wang, Q., Liu, X., Qin, J., et al. (2022). Escherichia coli O157:H7 senses microbiota-produced riboflavin to increase its virulence in the gut. Proc. Natl. Acad. Sci. U.S.A. 119, e2212436119. doi: 10.1073/PNAS.2212436119
Liu, Y., Han, R., Wang, J., Yang, P., Wang, F., and Yang, B. (2020). Magnesium sensing regulates intestinal colonization of enterohemorrhagic Escherichia coli O157:H7. mBio 11, 1–17. doi: 10.1128/MBIO.02470-20
Mahajan, A., Currie, C. G., Mackie, S., Tree, J., Mcateer, S., Mckendrick, I., et al. (2009). An investigation of the expression and adhesin function of H7 flagella in the interaction of Escherichia coli O157: H7 with bovine intestinal epithelium. Cell Microbiol. 11, 121–137. doi: 10.1111/j.1462-5822.2008.01244.x
Mcdaniel, T. K., Jarvis, K. G., Donnenberg, M. S., and Kaper, J. B. (1995). A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 92, 1664–1668. doi: 10.1073/pnas.92.5.1664
Mellies, J. L., Barron, A. M. S., and Carmona, A. M. (2007). Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect. Immun. 75, 4199–4210. doi: 10.1128/IAI.01927-06
Melson, E. M. and Kendall, M. M. (2019). The sRNA DicF integrates oxygen sensing to enhance enterohemorrhagic Escherichia coli virulence via distinctive RNA control mechanisms. Proc. Natl. Acad. Sci. U.S.A. 116, 14210–14215. doi: 10.1073/pnas.1902725116
National Research Council (2011). GUIDE LABORATORY ANIMALS FOR THE CARE AND USE OF Eighth Edition Committee for the Update of the Guide for the Care and Use of Laboratory Animals Institute for Laboratory Animal Research Division on Earth and Life Studies. Available online at: http://www.nap.edu (Accessed July 31, 2020).
Pacheco, A. R., Munera, D., Waldor, M. K., Sperandio, V., and Ritchie, J. M. (2012). Fucose sensing regulates bacterial intestinal colonization. Nature 492, 113–117. doi: 10.1038/NATURE11623
Pacheco, A. R. and Sperandio, V. (2012). Shiga toxin in enterohemorrhagic E.coli: regulation and novel anti-virulence strategies. Front. Cell Infect. Microbiol. 2. doi: 10.3389/FCIMB.2012.00081
Papenfort, K., Sun, Y., Miyakoshi, M., Vanderpool, C. K., and Vogel, J. (2013). Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153, 426–437. doi: 10.1016/j.cell.2013.03.003
Pfeiffer, V., Papenfort, K., Lucchini, S., Hinton, J. C. D., and Vogel, J. (2009). Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 16, 840–846. doi: 10.1038/nsmb.1631
Pleška, M., Lang, M., Refardt, D., Levin, B. R., and Guet, C. C. (2018). Phage-host population dynamics promotes prophage acquisition in bacteria with innate immunity. Nat. Ecol. Evol. 2, 359–366. doi: 10.1038/s41559-017-0424-z
Robinson, C. M., Sinclair, J. F., Smith, M. J., and O’Brien, A. D. (2006). Shiga toxin of enterohemorrhagic Escherichia coli type 0157:H7 promotes intestinal colonization. Proc. Natl. Acad. Sci. U.S.A. 103, 9667–9672. doi: 10.1073/pnas.0602359103
Salmon, K., Hung, S., Mekjian, K., Baldi, P., Hatfield, G. W., and Gunsalus, R. P. (2003). Global gene expression profiling in Escherichia coli K12: The effects of oxygen availability and FNR. J. Biol. Chem. 278, 29837–29855. doi: 10.1074/JBC.M213060200
Sauder, A. B. and Kendall, M. M. (2018). After the fact(or): Posttranscriptional gene regulation in enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol 200.19 (2018): 10–1128. doi: 10.1128/JB.00228-18
Sauder, A. B. and Kendall, M. M. (2021). A pathogen-specific sRNA influences enterohemorrhagic Escherichia coli fitness and virulence in part by direct interaction with the transcript encoding the ethanolamine utilization regulatory factor EutR. Nucleic Acids Res. 49, 10988–11004. doi: 10.1093/NAR/GKAB863
Schmittgen, T. D. and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Sturm, A., Heinemann, M., Arnoldini, M., Benecke, A., Ackermann, M., Benz, M., et al. (2011). The cost of virulence: retarded growth of salmonella typhimurium cells expressing type III secretion system 1. PLoS Pathog. 7, e1002143. doi: 10.1371/journal.ppat.1002143
Sudo, N., Soma, A., Muto, A., Iyoda, S., Suh, M., Kurihara, N., et al. (2014). A novel small regulatory RNA enhances cell motility in enterohemorrhagic Escherichia coli. J. Gen. Appl. Microbiol. 60, 44–50. doi: 10.2323/jgam.60.44
Sy, B. M., Lan, R., and Tree, J. J. (2020). Early termination of the Shiga toxin transcript generates a regulatory small RNA. Proc. Natl. Acad. Sci. U.S.A. 117, 25055–25065. doi: 10.1073/PNAS.2006730117
Sy, B. M. and Tree, J. J. (2021). Small RNA regulation of virulence in pathogenic escherichia coli. Front. Cell Infect. Microbiol. 10. doi: 10.3389/FCIMB.2020.622202
Tree, J. J., Granneman, S., McAteer, S. P., Tollervey, D., and Gally, D. L. (2014). Identification of bacteriophage-encoded anti-sRNAs in pathogenic escherichia coli. Mol. Cell 55, 199–213. doi: 10.1016/J.MOLCEL.2014.05.006
Villa, J. K., Han, R., Tsai, C. H., Chen, A., Sweet, P., Franco, G., et al. (2021). A small RNA regulates pprM, a modulator of pleiotropic proteins promoting DNA repair, in Deinococcus radiodurans under ionizing radiation. Sci. Rep. 11, 1–14. doi: 10.1038/s41598-021-91335-8
Wallace, N., Zani, A., Abrams, E., and Sun, Y. (2016). The impact of oxygen on bacterial enteric pathogens. Adv. Appl. Microbiol. 95, 179–204. doi: 10.1016/BS.AAMBS.2016.04.002
Waters, S. A., McAteer, S. P., Kudla, G., Pang, I., Deshpande, N. P., Amos, T. G., et al. (2017). Small RNA interactome of pathogenic E. coli revealed through crosslinking of RNase E. EMBO J. 36, 374–387. doi: 10.15252/embj.201694639
Wong, A. R. C., Pearson, J. S., Bright, M. D., Munera, D., Robinson, K. S., Lee, S. F., et al. (2011). Enteropathogenic and enterohaemorrhagic Escherichia coli: Even more subversive elements. Mol. Microbiol. 80, 1420–1438. doi: 10.1111/j.1365-2958.2011.07661.x
Woodward, S. E., Krekhno, Z., and Finlay, B. B. (2019). Here, there, and everywhere: How pathogenic Escherichia coli sense and respond to gastrointestinal biogeography. Cell Microbiol. 21.11 (2019): e13107. doi: 10.1111/cmi.13107
Keywords: enterohemorrhagic Escherichia coli, O157:H7, oxygen, sRNA (small RNA), gene regulation, LEE, site-specific colonization
Citation: Han R, Qian Y and Zheng C (2025) A novel small RNA regulates Locus of Enterocyte Effacement and site-specific colonization of enterohemorrhagic Escherichia coli O157:H7 in gut. Front. Cell. Infect. Microbiol. 14:1517328. doi: 10.3389/fcimb.2024.1517328
Received: 25 October 2024; Accepted: 11 December 2024;
Published: 15 January 2025; Corrected: 10 July 2025.
Edited by:
Fernando Navarro-Garcia, National Polytechnic Institute of Mexico (CINVESTAV), MexicoReviewed by:
Luis Caetano Martha Antunes, University of Kansas, United StatesPaul Briaud, Georgia Institute of Technology, United States
Copyright © 2025 Han, Qian and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chenguang Zheng, Y2d6aGVuZ0BuY3N0LmVkdS5jbg==
†These authors have contributed equally to this work