 Xia Xu1†
Xia Xu1† Liang Zhao1†
Liang Zhao1† Yanfei Chen1
Yanfei Chen1 Hancheng Wang2*Liuti Cai2
Hancheng Wang2*Liuti Cai2 Yanyan Wang1
Yanyan Wang1 Nalin N. Wijayawardene3
Nalin N. Wijayawardene3 Weihua Pan4Feng Wang2*
Weihua Pan4Feng Wang2* Yingqian Kang1*
Yingqian Kang1*- 1Key Laboratory of Environmental Pollution Monitoring and Disease Control, Ministry of Education of Guizhou & Institute of Health Research & Key Laboratory of Medical Microbiology and Parasitology, School of Basic Medical Sciences, Guizhou Medical University, Guiyang, China
- 2Guizhou Provincial Academician Workstation of Microbiology and Health, Guizhou Academy of Tobacco Science, Guiyang, China
- 3Center for Yunnan Plateau Biological Resources Protection and Utilization, College of Biological Resource and Food Engineering, Qujing Normal University, Qujing, Yunnan, China
- 4Department of Dermatology, Changzheng Hospital, Shanghai, China
The phyllospheric microbial composition of tobacco plants is influenced by multiple factors. Disease severity level is one of the main influencing factors. This study was designed to understand the microbial community in tobacco wildfire disease with different disease severity levels. Tobacco leaves at disease severity level of 1, 5, 7, and 9 (L1, L5, L7, and L9) were collected; both healthy and diseased leaf tissues for each level were collected. The community structure and diversity in tobacco leaves with different disease severity levels were compared using high-throughput technique and Biolog Eco. The results showed that in all healthy and diseased tobacco leaves, the most dominant bacterial phylum was Proteobacteria with a high prevalence of genus Pseudomonas; the relative abundance of Pseudomonas was most found at B9 diseased samples. Ascomycota represents the most prominent fungal phylum, with Blastobotrys as the predominant genus. In bacterial communities, the Alpha diversity of healthy samples was higher than that of diseased samples. In fungal community, the difference in Alpha diversity between healthy and diseased was not significant. LEfSe analysis showed that the most enriched bacterial biomarker was unclassified_Gammaproteobacteria in diseased samples; unclassified_Alcaligenaceae were the most enrich bacterial biomarker in healthy samples. FUNGuild analysis showed that saprotroph was the dominated mode in health and lower diseased samples, The abundance of pathotroph–saprotroph and pathotroph–saprotroph–symbiotroph increases at high disease levels. PICRUSt analysis showed that the predominant pathway was metabolism function, and most bacterial gene sequences seem to be independent of the disease severity level. The Biolog Eco results showed that the utilization rates of carbon sources decrease with increasing disease severity level. The current study revealed the microbial community’s characteristic of tobacco wildfire disease with different disease severity levels, providing scientific references for the control of tobacco wildfire disease.
1 Introduction
Tobacco (Nicotiana tabacum L.) is an extensively cultivated economic crop in China, whose growth and production are affected by a myriad of biotic and abiotic factors (Ma et al., 2023). Tobacco wildfire disease is caused by Pseudomonas syringae pv. tabaci, which results in heavy economical losses (Tumewu et al., 2022). The typical symptoms of tobacco wildfire disease include small brown or black lesions bordered by chlorotic halos on tobacco leaves (Sun et al., 2021). The characteristic chlorotic halos are produced by a toxin, tabtoxin (Ichinose et al., 2013).
The phyllosphere is an important habitat for many microorganisms, including commensal, beneficial, and pathogenic microbes affecting plant health and productivity (Lindow and Brandl, 2003; Xin et al., 2018; Zhu et al., 2022). It is notable to study the changes in microbial communities (natural inhabitants) when the host plant is stressed under an infection (Perreault and Laforest-Lapointe, 2022). The development of PCR and sequencing technologies (including high-throughput sequencing) has facilitated the exploration of microbial diversity and structure of microbial communities (Ahmed et al., 2022b; Fitzpatrick et al., 2020; Gong and Xin, 2021).
Tobacco bacterial wilt is caused by Ralstonia solanacearum and is reported to reduce the leaf fungal community abundance and diversity (Ahmed et al., 2022a), whereas a similar response was observed by Si et al. (2023) during the investigation of tobacco wildfire disease caused by P. syringae pv. tabaci. Chen et al. (2020a) noticed a much higher abundance of Rhizopus oryzae in cured tobacco leaves infected by tobacco pole rot than that of healthy leaves. The phyllospheric microbial community and composition are closely related to plant health (Zhang et al., 2019).
Disease severity level also affects the leaf microbiome, influencing the dynamics of phyllosphere microbial communities. Huang et al. (2021) studied the influence of the disease severity level on the community structure and diversity of the tobacco leaf spot disease caused by Didymella segeticola, identifying that the community diversity of phyllosphere fungi decreased with the increase in the disease severity level. Sun et al. (2022) studied the microbial community of tobacco leaves affected by target spot at different disease severity levels. The results indicate that fungal diversity and relative abundance increased with disease severity level, while bacterial diversity indices decreased. Luo et al. (2019) studied the microbial community structure of symptomatic cucumber leaves affected by angular leaf spot at different disease severity levels. The investigation showed that the pathogen caused an increase in the microbial community richness when disease pressure was higher. Biolog Eco can reflect the main types of carbon source utilization and the strength of metabolic functions of all microbial communities (Rutgers et al., 2016). This technique has been used to identify the carbon-source-associated metabolic pathways of phyllosphere microbial communities in both healthy and diseased tobacco plants (Feng et al., 2023; Shen et al., 2023). However, the effect of disease severity level on the structure and diversity of tobacco wildfire disease phyllosphere microbial community is still poor understood.
Tobacco wildfire disease can lead to serious damage. It is important to understand the microbial community changes in tobacco leaves. Therefore, the goals of this study were to identify the structure and diversity of phyllosphere bacterial and fungal communities and the metabolism of carbon sources of tobacco microorganisms in tobacco leaves with different disease severity levels. These results will provide theoretical reference for the prevention and control of wildfire disease in tobacco.
2 Materials and methods
2.1 Sampling sites and sampling strategy
In Jun 2022, one tobacco field (cultivar Yunyan 87) with symptoms of tobacco wildfire disease in Dafang County (27°12′15″ N, 105°57′18″ E), Bijie City, Guizhou Province of China, was selected as the sampling site. The tobacco leaves of different disease severity levels (L1, L5, L7, and L9) were randomly selected based on the Chinese National Standard (GB/T 23222-2008). For L1, L5, L7, and L9 groups, diseased lesion area ranged from 0% to 1%, 6% to 10%, 11% to 20%, and 11% to 100% of a leaf, respectively (Figures 1A–E). There were three biological replicates from each group. A total of 24 samples were collected. The four groups of leaves were separated into two parts, with and without visible leaf spots (Figure 1F), and labeled as diseased and healthy samples, respectively, as presented in Table 1. After sampling, the samples were immediately transported to the laboratory and stored at −80°C until further use.

Figure 1. Whole plant (A) and leaves displaying disease severity at (B) L1, (C) L5, (D) L7, and (E) L9, respectively. (F) sampling parts display of diseased and healthy samples on tobacco leaves. (G) map of sampling site.
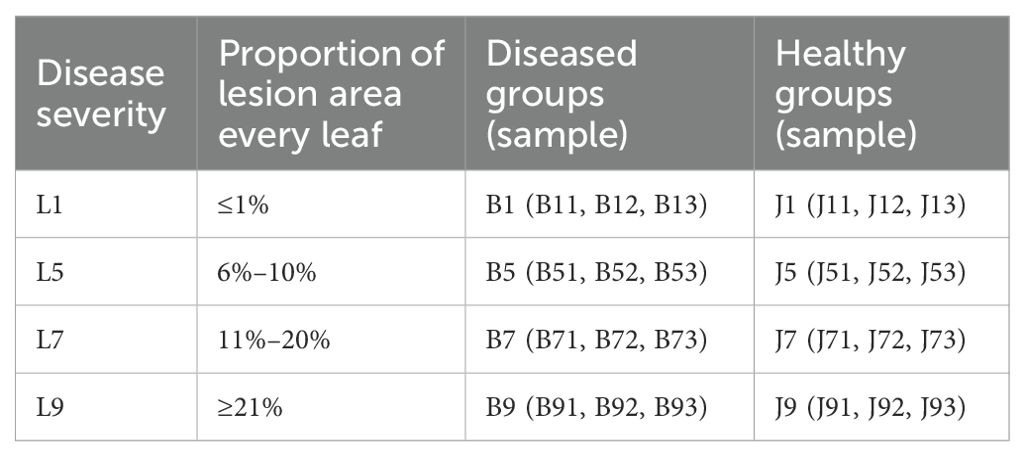
Table 1. Sample information for both diseased and healthy tobacco leaves with infected tobacco wildfire disease at different disease severity.
2.2 DNA extraction, PCR amplification, and high-throughput sequencing
Total DNA was extracted from the leaf samples using the PF Mag-Bind Stool DNA Kit (Omega Bio-Tek, GA, USA) according to the manufacturer’s instructions. The quality and concentration of DNA were determined by agarose gel electrophoresis. The hypervariable regions V5–V7 of the bacterial 16S rRNA gene were amplified with primer pairs 799F (5′-AACMGGATTAGATACCCKG-3′) and 1193R (5′-ACGTCATCCCCACCTTCC-3′) (Bulgarelli et al., 2015). Furthermore, the primer pair ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′) (Adams et al., 2013) were used to amplify the fungal ITS1 region. PCR products were checked by 2% agarose gel electrophoresis. The amplification products were amplified using an Illumina MiSeq platform and constructed the library.
2.3 Sequencing data processing
Qualitative filtering and merging of raw sequencing reads were performed using Flash (version 1.2.11) (Magoč and Salzberg, 2011). All sequences were clustered into operational taxonomic units (OTUs) by Uparse (version 11) (Edgar, 2013) with a 97% similarity cutoff. Subsequently, the OTUs were annotated for bacterial and fungal based on the Silva 16S rRNA (v138) and Unite (version 8.0) database, respectively, setting the confidence threshold to 70% (Huang et al., 2021; Sun et al., 2022). The Alpha and Beta diversities were performed by Mothur (version 1.30.2) and Qiime (2020.2.0), and the R statistics package (version 3.3.1) was utilized to draw the principal coordinate analysis (PCoA) diagram. The linear discriminant analysis (LDA) effect size (LEfSe) (Segata et al., 2011) was performed to detect the potential biomarker, using a linear discriminant analysis threshold of 4.0. The PERMANOVA test was used to calculate significant differences. PICRUSt and FUNGuild (Douglas et al., 2020) the databases were used to analyze enzyme functional and fungal trophic mode. Bacterial metabolic functions were analyzed with the bioinformatics software package PICRUSt (Barberán et al., 2012). The correlation of phyllosphere microorganisms was demonstrated using Spearman’ s rank analysis based on the absolute value of a correlation coefficient >0.6 and a p-value <0.05 (Nguyen et al., 2016).
2.4 Biolog inoculation and analysis
Each of the 1-g samples from different disease severity levels was mixed with 50 ml of 0.85% NaCl and shaken (180 rpm) for 2 h at 28°C. The mixture was standing for 30 min, and 100 µL of dilution was added using an electronic pipette and incubated at 28°C in a Biolog OmniLog incubator for 7 days (Dai et al., 2022). The Biolog OmniLog system was equipped with a charge-coupled device camera system and an incubator (Feng et al., 2023). A Heatmap Illustrator (HemI 1.0.3.3) was used to analyze the microbe carbon metabolism of the samples.
2.5 Statistical analysis
The mean value and standard error were used to express the measured results. One-way ANOVA or nonparametric test Kruskal–Wallis was used to compare the differences between multiple groups. p ≤ 0.05 was considered to be statistically significant.
3 Results
3.1 Quality of total bacterial and fungal sequence data
A total of 2,277,441 bacterial sequences were obtained through sequencing of 24 samples. These sequences were classified into 845 OTUs at a 97% similarity level. For fungal sequences, a total of 1,681,293 sequences were classified into 389 OTUs across the 24 samples. Rarefaction curves for each sample reached a plateau, indicating sufficient sequencing depth to capture most microorganisms present (Figure 2). All the bacterial and fungal sequences were deposited in the SRA database under accession numbers PRJNA1096149 and PRJNA1096455, respectively.
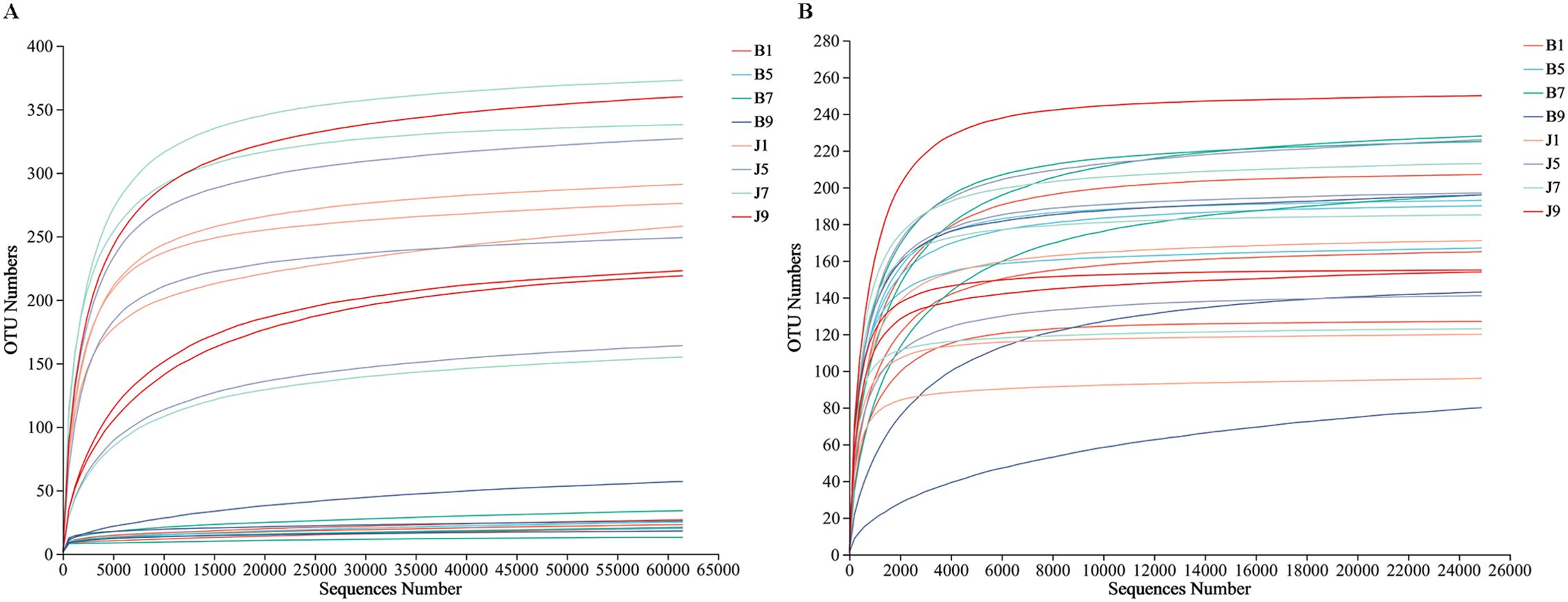
Figure 2. Rarefaction curves of bacterial (A) and fungal (B) OTUs across different tobacco leaf samples.
3.2 Distribution and diversity of bacterial and fungal operational taxonomic units
Venn diagrams (Figure 3) show the distribution of the OTUs in the four disease severity levels. For bacterial community, a total of 845 OTUs were obtained, including 111 belonging to the diseased group and 842 to the healthy group (Figures 3A, B). The number of shared OTUs among the four diseased groups was 20; there were 14, 15, 9, and 36 unique OTUs found only in B1, B5, B7, and B9, respectively. Meanwhile, the number of shared OTUs among the four health groups was 214, and the groups J1, J5, J7, and J9 only had 82, 46, 104, and 33 unique OTUs, respectively. Notably, the number of bacterial OTUs in the diseased samples was lower than that in the healthy samples. We compared the bacterial Alpha diversity of the tobacco leaves using the diversity index (Shannon index) and the richness indices (Ace and Chao1 index) in Table 2. The higher the Shannon value, the greater the level of diversity. The higher the Ace and Chao1 value, the greater the level of richness. The Shannon index of diseased and healthy samples ranged from 1.31 ± 0.33 to 1.44 ± 0.17 and 2.11 ± 0.87 to 2.66 ± 1.44, respectively. The Ace index ranged from 31.34 ± 14.73 to 55.72 ± 37.05 and 332.30 ± 103.30 to 404.90 ± 170.80 for diseased and healthy samples, respectively, whereas the Chao1 index ranged from 26.42 ± 12.45 to 49.11 ± 36.39 for diseased and 334.30 ± 106.50 to 404.20 ± 166.70 for healthy samples. Correspondingly, the diversity indices Ace, Chao1, and Shannon of healthy samples were greater than those of diseased samples, and the diversity index decreased significantly with increased disease severity level from L1 to L5 and then increased with the increase in the degree of disease from L5 to L9.
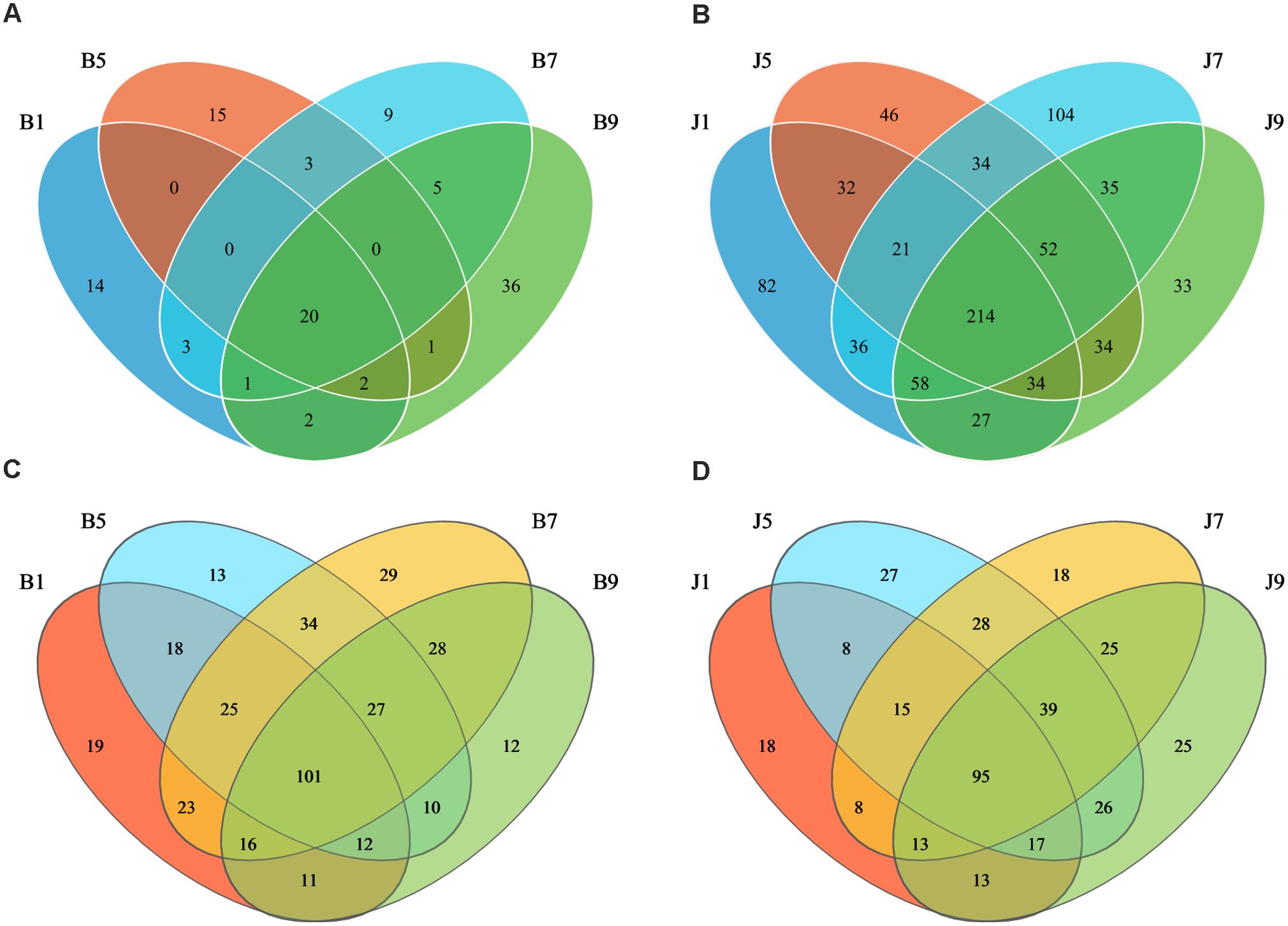
Figure 3. Venn diagram showing the number of bacterial and fungal OTUs observed in different groups at four different disease severity. Bacterial Venn diagram in diseased (A) and healthy (B) groups at four different disease severities. Fungal Venn diagram in diseased (C) and healthy (D) groups at four different disease severities. Numbers in the overlapping region indicated unique OTUs for two or three samples. Numbers in the non-overlapping regions indicated unique OTUs for the group.
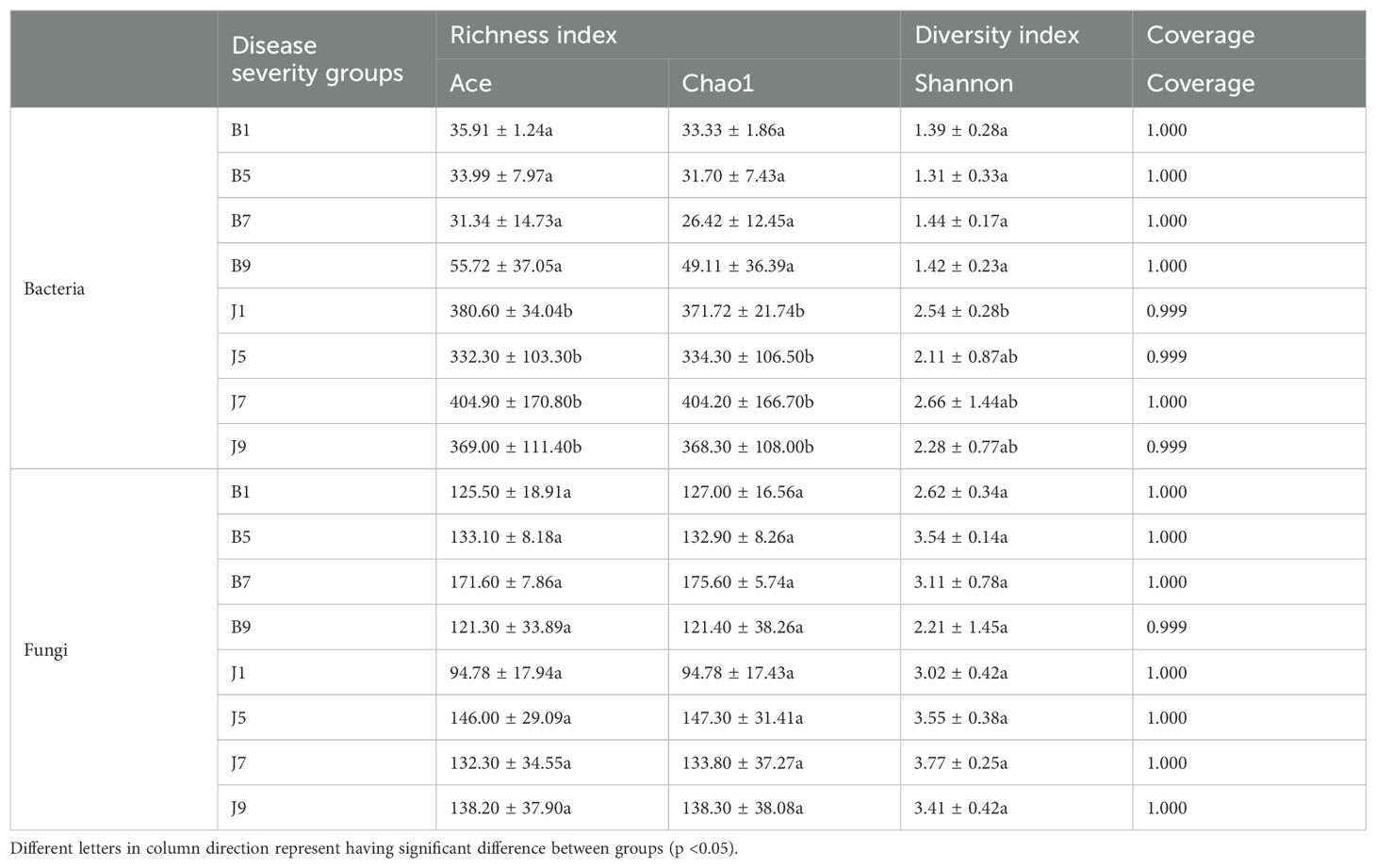
Table 2. Alpha-diversity indices of bacterial and fungal communities based on high-throughput sequencing in different samples.
In the fungal community, a total of 389 OTUs were obtained, with 378 OTUs in the diseased samples and 375 in the healthy group (Figures 3C, D). The four diseased and four healthy samples contained 101 and 95 shared OTUs, respectively. The unique OTU numbers in the four diseased groups were 19, 13, 29, and 12, respectively, while in the healthy group, the numbers were 18, 27, 18, and 25, respectively. The number of fungal OTUs in the diseased samples was higher than that in the healthy, but there was little difference. Similarly, the Shannon index of diseased and healthy samples ranged from 2.21 ± 1.45 to 3.54 ± 0.14 and 3.02 ± 0.42 to 3.77 ± 0.25, respectively (Table 2). The Ace index ranged from 121.30 ± 33.89 to 171.60 ± 7.86 and 94.78 ± 17.94 to 146.00 ± 29.09 for diseased and healthy samples, respectively, whereas the Chao1 index ranged from 121.40 ± 38.26 to 175.60 ± 5.74 for disease and 94.78 ± 17.43 to 147.30 ± 31.41 for healthy samples. The Ace and Chao1 indices were the highest at B7 and J5 in the disease and health groups, respectively. The Shannon index was the highest at B5 and J7 in the disease and health groups, respectively. Meanwhile, the Shannon index of healthy samples was greater than that of diseased samples, which indicated that the fungal diversity in the healthy group is higher than the diseased group.
3.3 Bacterial and fungal community composition
3.3.1 Bacterial community composition
The 16S dataset showed that the phyllosphere bacteria of diseased and healthy samples contain six phyla of bacteria (Table 3): Proteobacteria, Actinobacteria, Firmicutes, Bacteroidetes, Myxococcota, and Chloroflexi (Figure 4A). Proteobacteria was the predominant phylum, with relative abundances in the diseased groups (B1, B5, B7, and B9) being 99.95%, 99.97%, 99.25%, and 98.97%, respectively, while in healthy groups (J1, J5, J7, and J9), the relative abundance were 66.89%, 84.56%, 79.12%, and 86.06%, respectively. The relative abundances of Actinobacteria in the diseased groups (B1, B5, B7, and B9) were 0.04%, 0.02%, 0.02%, and 0.08%, respectively; the relative abundances in healthy groups (J1, J5, J7, and J9) were 14.17%, 7.49%, 10.72%, and 6.31%, respectively. Notably, the relative abundance of Proteobacteria in diseased samples were higher than those in healthy samples. Conversely, the relative abundance of Actinobacteria and Firmicutes in diseased samples were less than in healthy samples.
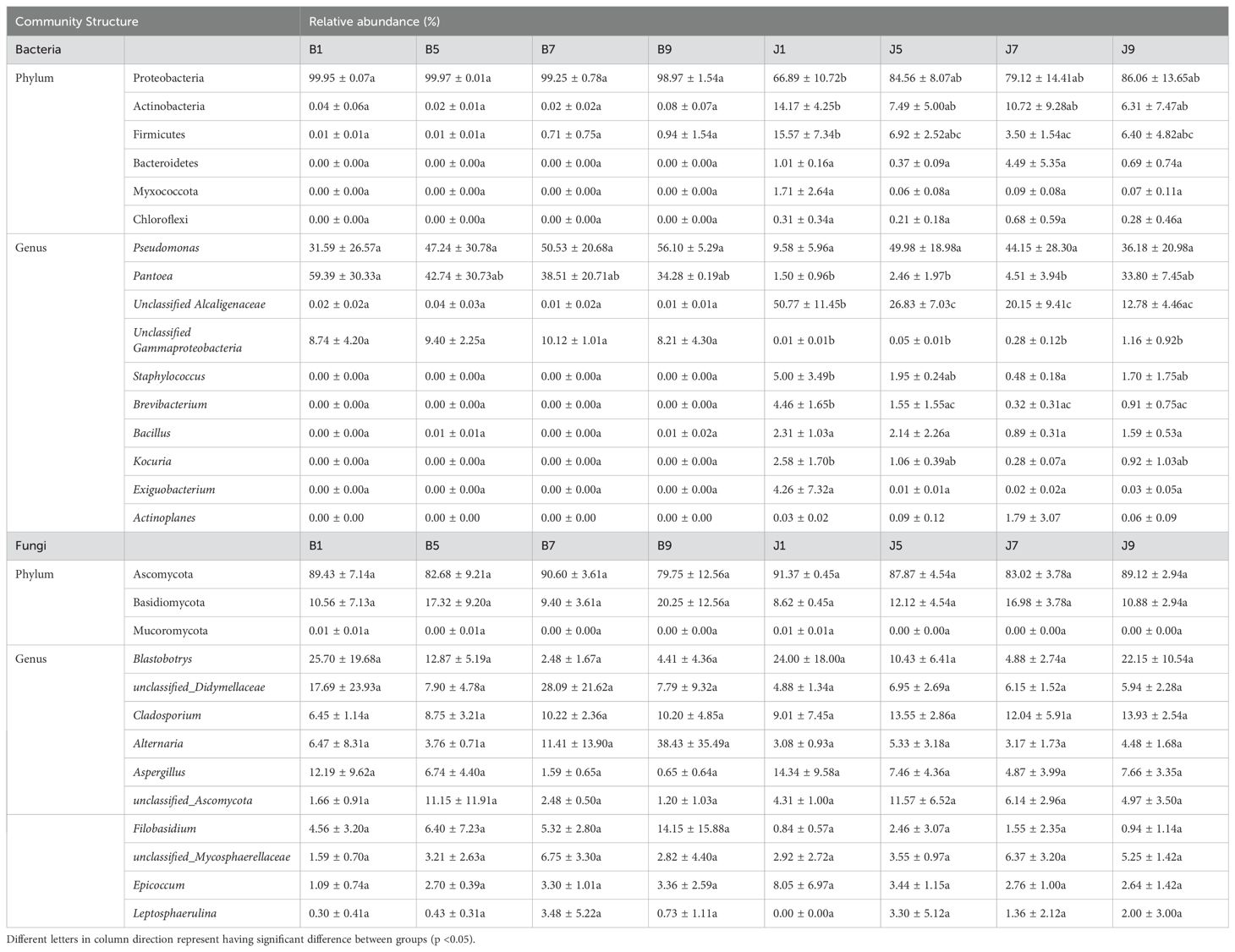
Table 3. List of top 10 dominant taxa and their relative abundance in the fungal and bacterial communities of the group.
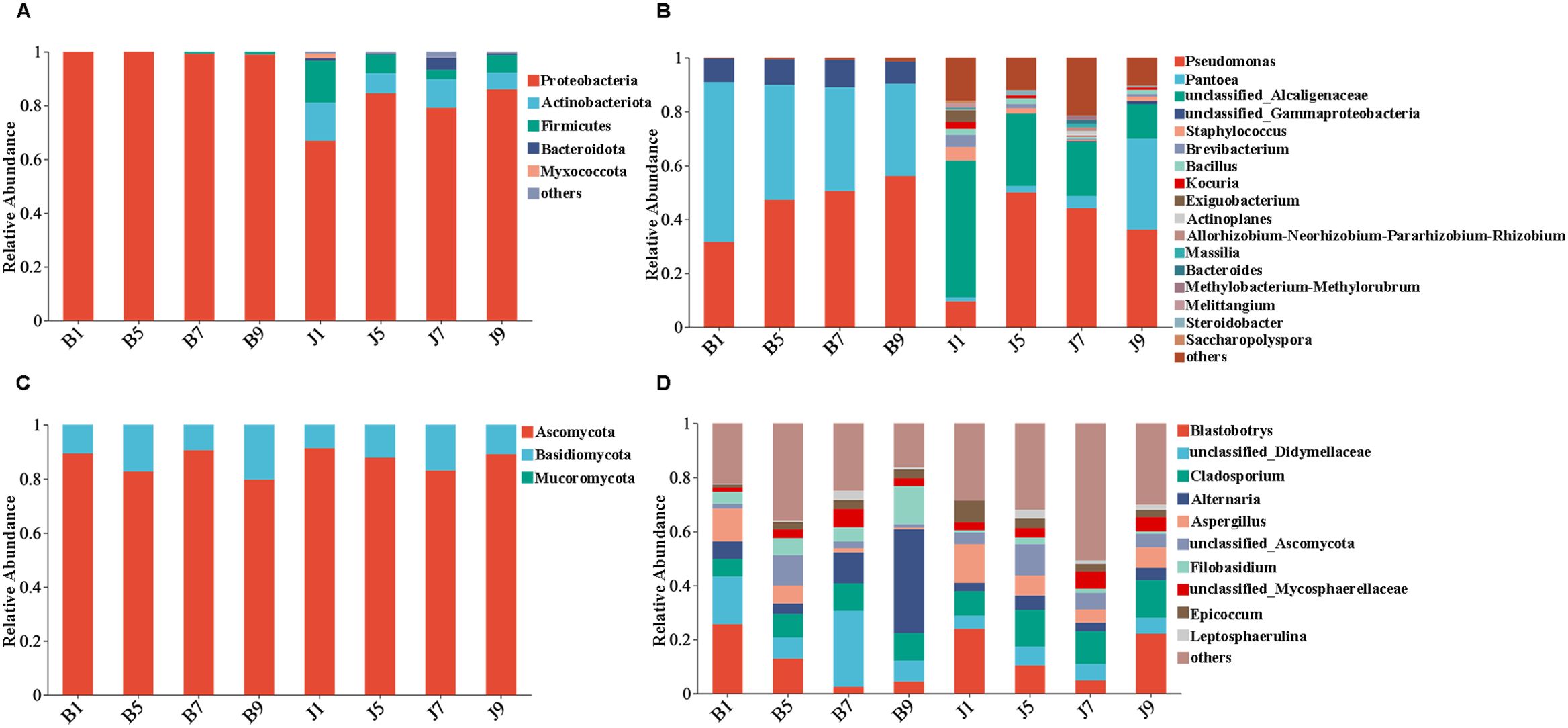
Figure 4. Microbial community composition of different groups at the phyla and genus levels. Bacterial community composition of different groups at the phyla (A) and genus (B) levels. Fungal community composition of different groups at the phyla (C) and genus (D) levels.
At the genus level, the top 10 genera included Pseudomonas, Pantoea, unclassified_Alcaligenaceae, unclassified_Gammaproteobaceria, Staphylococcus, Brevibacterium, Bacillus, Kocuria, Exiguobacteri, and Actinoplanes (Figure 4B). The abundance of Pseudomonas, Pantoea, and unclassified_Gammaproteobaceria in diseased samples was higher than that in healthy samples, whereas the abundance of unclassified_ Alcaligenaceae in diseased samples was less than that in healthy samples. The abundance of Pseudomonas increased with the increase in disease severity level and peaked at L9 sample. On the contrary, the abundance of Pantoea decreased with the increase in disease severity level (Table 3). In addition, with the increase in disease severity level, the relative abundance of unclassified_Alcaligenaceae decreased. The result revealed that the bacterial community composition was different for four disease severity levels.
3.3.2 Fungal community composition
The ITS dataset showed that the phyllosphere fungi of diseased and healthy samples contain three phyla of fungi: Ascomycota, Basidiomycota, and Mucoromycota (Figure 4C). The predominant fungal phylum was Ascomycota, with relative abundances in the diseased groups (B1, B5, B7, and B9) were 89.43%, 82.68%, 90.60%, and 79.75%, respectively, while in healthy groups (J1, J5, J7, and J9), the relative abundances were 91.37%, 87.87%, 83.02%, and 89.12%, respectively. The relative abundances of Basidiomycota in the diseased groups (B1, B5, B7, and B9) were 10.56%, 17.32%, 9.40%, and 20.25%, respectively; the relative abundances in healthy groups (J1, J5, J7, and J9) were 8.62%, 12.12%, 16.98%, and 10.88%, respectively (Table 3). The result indicates that the relative abundance of Ascomycota in diseased samples was lower than that in healthy samples, and the relative abundance of Basidiomycota in diseased samples was higher than that in healthy samples, except for L7 samples.
At the genus level, the top 10 genera included Blastobotrys, unclassified_Didymellaceae, Cladosporium, Alternaria, Aspergillus, unclassified_Ascomycota, Filobasidium, unclassified_Mycosphaerellaceae, Epicoccum, and Leptosphaerulina (Figure 4D). Similarly, the relative abundance was associated with the four disease severity levels samples, but the difference is not significant. The abundance of unclassified_Didymellaceae, Alternaria, and Filobasidium in diseased samples was higher than that in healthy samples, whereas the abundance of Cladosporium and Aspergillus in diseased samples was less than that in healthy samples (Table 3). The abundance of Aspergillus decreased with the increase in disease severity level in diseased samples.
3.4 Spatial distribution of bacterial and fungal communities
PCoA plots were used to reveal the spatial distribution of fungal and bacterial communities (Figure 5). In bacterial community, the result showed that diseased groups and healthy groups were obviously separate (Figure 5A). All diseased samples (B1, B5, B7, and B9) from four disease severity levels were irregularly separated from each other. On the contrary, healthy groups (J1, J5, J7, and J9) with four disease severity levels were clustered together. In the fungal community, healthy groups (J1, J5, J7, and J9) with four disease severity levels were clustered together, and diseased samples (B1, B5, B7, and B9) from four disease severity levels were irregularly separated from each other (Figure 5B). Notably, B1 and B5 of the diseased group were clustered together with healthy groups; B7 and B9 were separated from the healthy group. The result indicated that the spatial distributions of bacterial and fungal community composition were significantly different between disease and healthy samples. There were fewer differences in bacterial and fungal community composition of healthy samples compared to diseased samples.
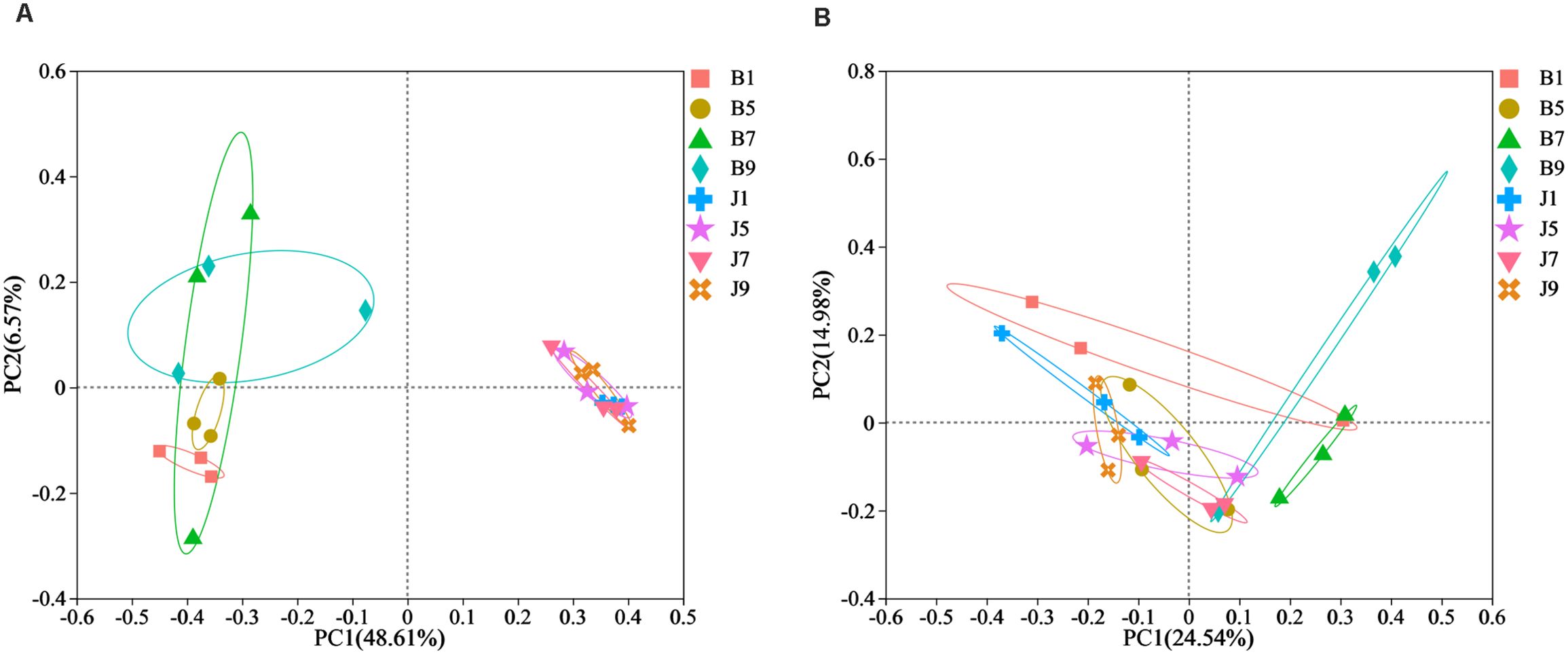
Figure 5. Principal Co-ordinate Analysis (PCoA) of the bacterial (A) and fungal (B) communities in the different group sample.
3.5 Significant differences in microbial communities
LEfSe analysis was used to detect the difference in fungal and bacterial communities between the diseased and healthy samples (Figure 6). For bacterial communities, 89 bacteria clades showed statistically significant differences when the LDA threshold was 4. In L1 leaf samples, 19 and 8 clades showed abundance advantage in healthy and diseased samples, respectively (Figures 6A, B). In L5 leaf samples, 13 and 8 clades present an abundance advantage in healthy and diseased samples, respectively (Figures 6C, D). In L7 leaf samples, 19 clades showed abundance advantage in healthy samples, while eight clades showed abundance advantage in diseased samples (Figures 6E, F). In L9 leaf samples, nine and five clades present abundance advantage in healthy and diseased samples, respectively (Figures 6G, H). At the genus level, Pantoea and unclassified_Gammaproteobacteria were enriched in B1, B5, and B7 samples, while the B9 sample only enriched unclassified_Gammaproteobacteria. Unclassified_Alcaligenaceae were enriched in J1, J5, J7, and J9 samples. Meanwhile, J1 healthy samples also were enriched with Staphylococcus, Brevibacterium, Bacillus, and Kocuria; J5 healthy samples also were enriched with Bacillus; and J7 healthy samples also were enriched with Methylobacterium–Methylorubrum. At the same time, significant differences were found between the different treatment groups using PERMANOVA test. The healthy groups had more characteristic species compared to the diseased groups. However, the B9 did not have characteristic species (Figure 7). Based on the results of LEfSe analysis, we found differences between healthy and diseased groups, and differences between samples of different disease severity levels. In addition, it was observed that the diversity of the enriched bacterial species was lower as the disease levels L1–L9 increased, indicating that the disease level has a strong influence on bacterial diversity. In contrast, there were no significant abundance differences for fungal communities between the diseased and healthy microbial communities of different disease severity levels.
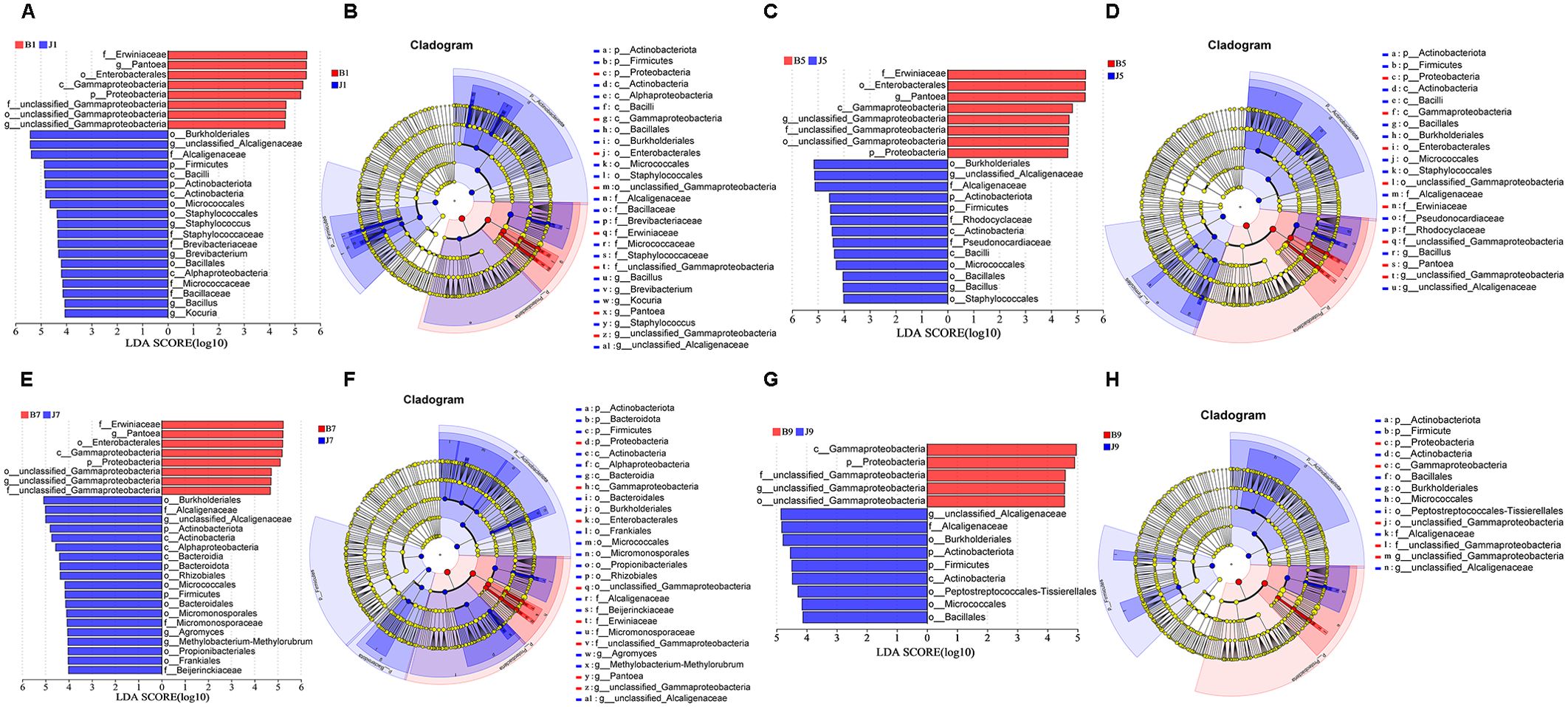
Figure 6. LEfSe analysis of bacteria abundance between diseased and healthy samples of different disease severities. (A), (C), (E), (G) are LDA score identified the size of differentiation between diseased and healthy groups of 1, 5, 7, 9 disease severity, respectively, with a threshold value of 4. (B), (D), (F), (H) are the cladogram of microbial communities of 1, 5, 7, 9 disease severity, respectively.
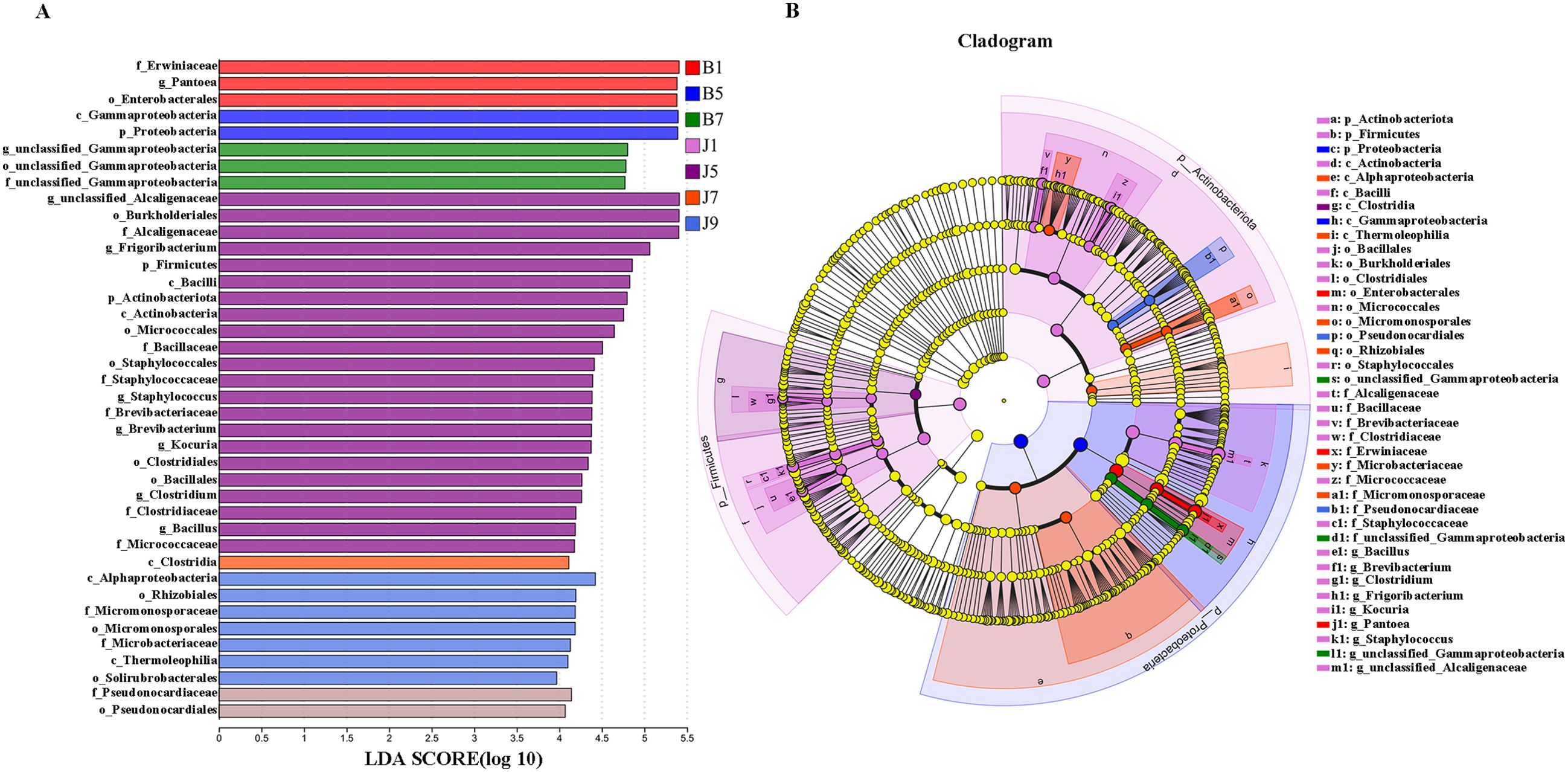
Figure 7. LEfSe analysis of bacteria abundance in all samples, LDA score (A) and cladogram of microbial communities (B).
3.6 Microbial community functional characteristics
PICRUSt and FUNGuild were used to predict the trophic modes of fungi in different disease severity levels, respectively (Figure 8). The PICRUSt analyses result showed that the top 6 enzymes were Adenosinetriphosphatase, L-arabinose isomerase, Glucan 1,4-alpha-glucosidase, DNA-directed RNA polymerase, Exo-alpha-sialidase, and unspecific monooxygenase. Meanwhile, most of the enzymes did not differ significantly between treatment groups. At the FUNGuild result, saprotroph, pathotroph–saprotroph, and pathotroph–saprotroph–symbiotroph were the dominant trophic modes in all samples. The abundance of saprotroph was higher in healthy groups than that in diseased groups and decreased with the increase in disease severity level. In addition, pathotroph–saprotroph–symbiotroph had a higher abundance in diseased groups compared to healthy groups, which increased with increasing disease severity level.
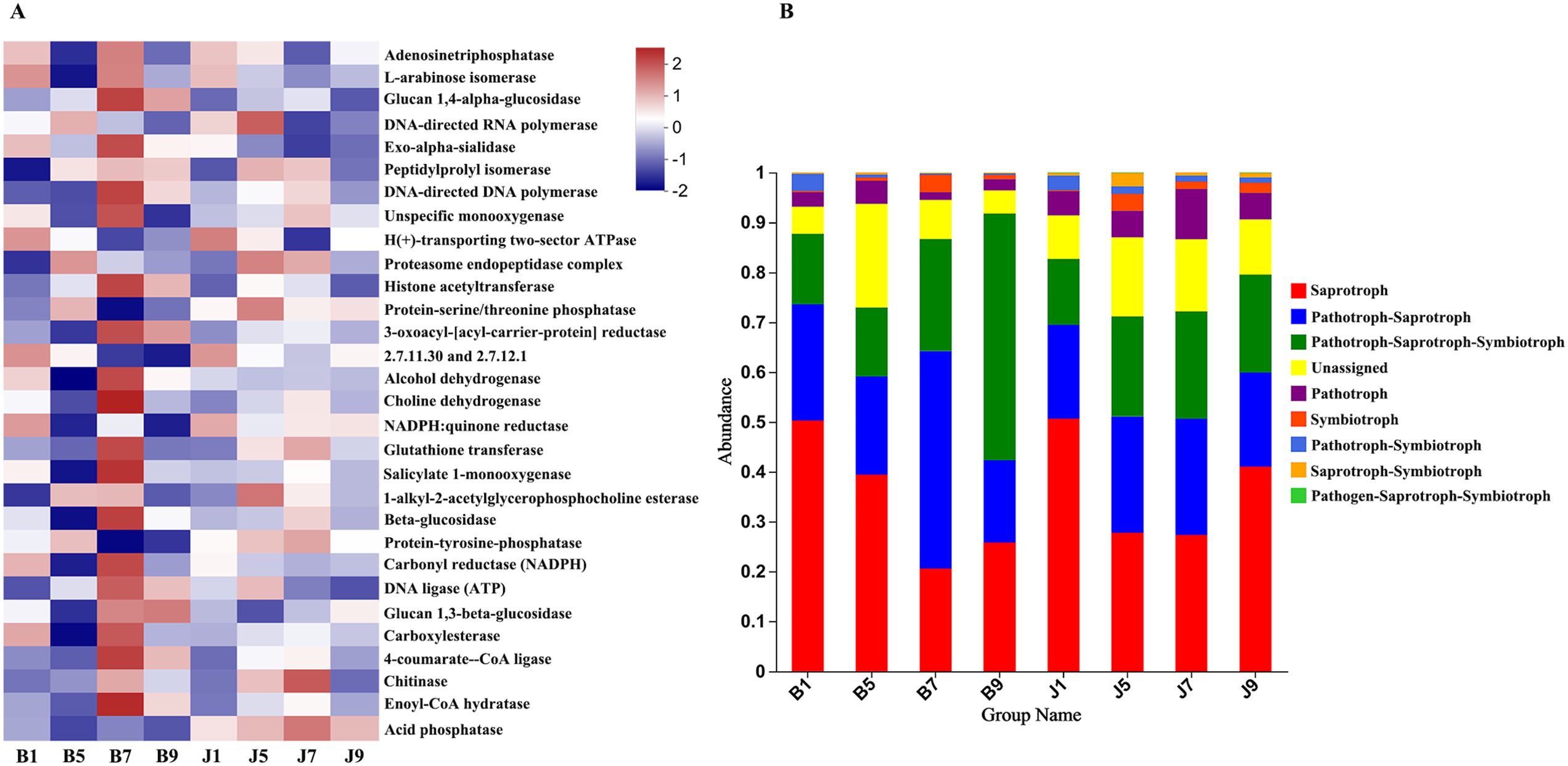
Figure 8. PICRUSt analyses of the enzyme functional of fungal in tobacco leaf samples (A). Relative abundance of fungal functional groups (modes) based on OTU annotation table with disturbance frequency level (B).
PICRUSt2 was used to predict the bacterial community function. In this study, the most abundant pathway was metabolic (73.67%), followed by environmental information processing (9.14%), cellular processes (6.27%), genetic information processing (5.04%), and human disease (4.17%), with the lowest being the organic system (1.72%). B1 samples were the lowest for all functions, and J9 samples clustered with the diseased samples, which indicates that L9 samples were the most severely diseased, and the bacterial functions in their healthy samples were similar to those of the diseased samples (Figure 9A). There were significant differences between diseased and healthy samples at L1, except for cellular processes. However, most bacterial functions seem to be independent of the disease severity level (Figures 9B–G).
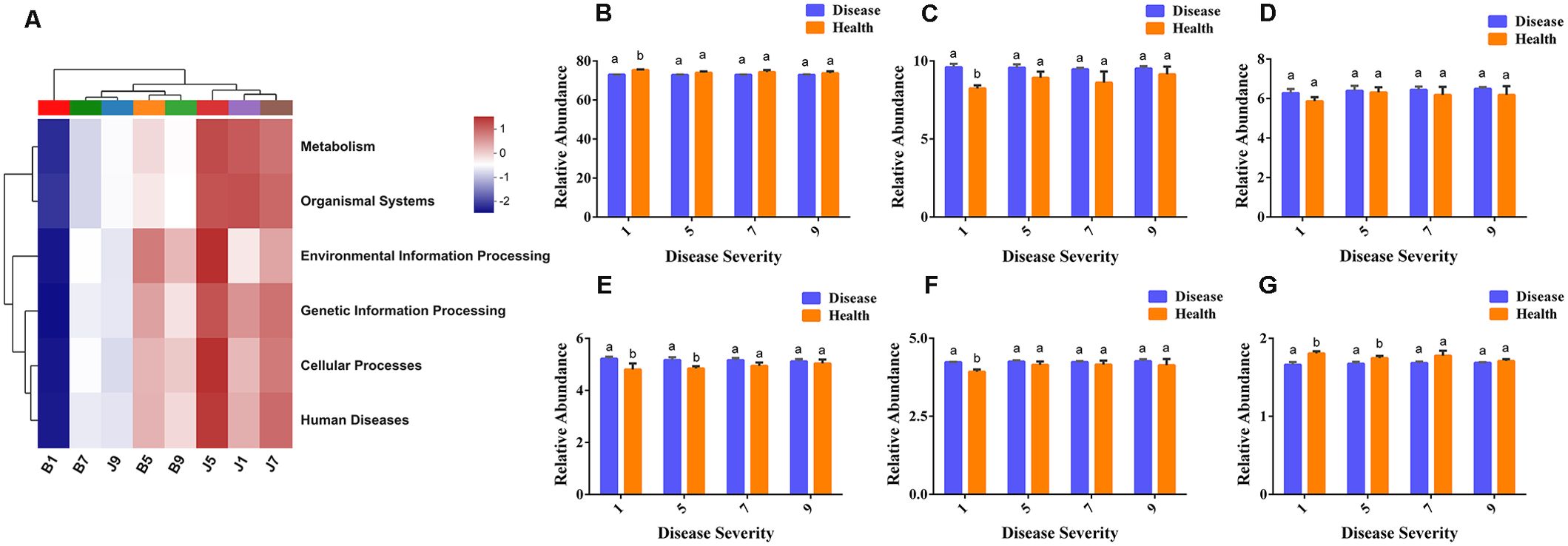
Figure 9. PICRUSt analyses of the KEGG level 1 functional categories of bacteria in tobacco leaf samples. (A) Dendrogram of bacterial functional categories, (B) Metabolism, (C) Environmental Information Processing, (D) Cellular Processes, (E) Genetic Information Processing, (F) Human Diseases, (G) Organismal Systems.
3.7 Microbial co-occurrence network analysis
Network analysis of co-occurrence patterns among the most abundant 50 bacterial and fungal genera presents that most of the bacterial genera showed positive correlation with each other, and unclassified_Gammaproteobacteria showed negative correlation with 19 bacterial genera such as unclassified_Alcaligenaceae, Kocuria, Bacillus, and Brevibacterium (Figure 10A). For fungal communities, most of the fungal genera were also positively correlated; Filobasidium was negatively correlated with six genera including Phaeosphaeria, Leptospora, and Exidia, and Alternaria was negatively correlated with Thermomyces and Sclerotiniaceae (Figure 10B).
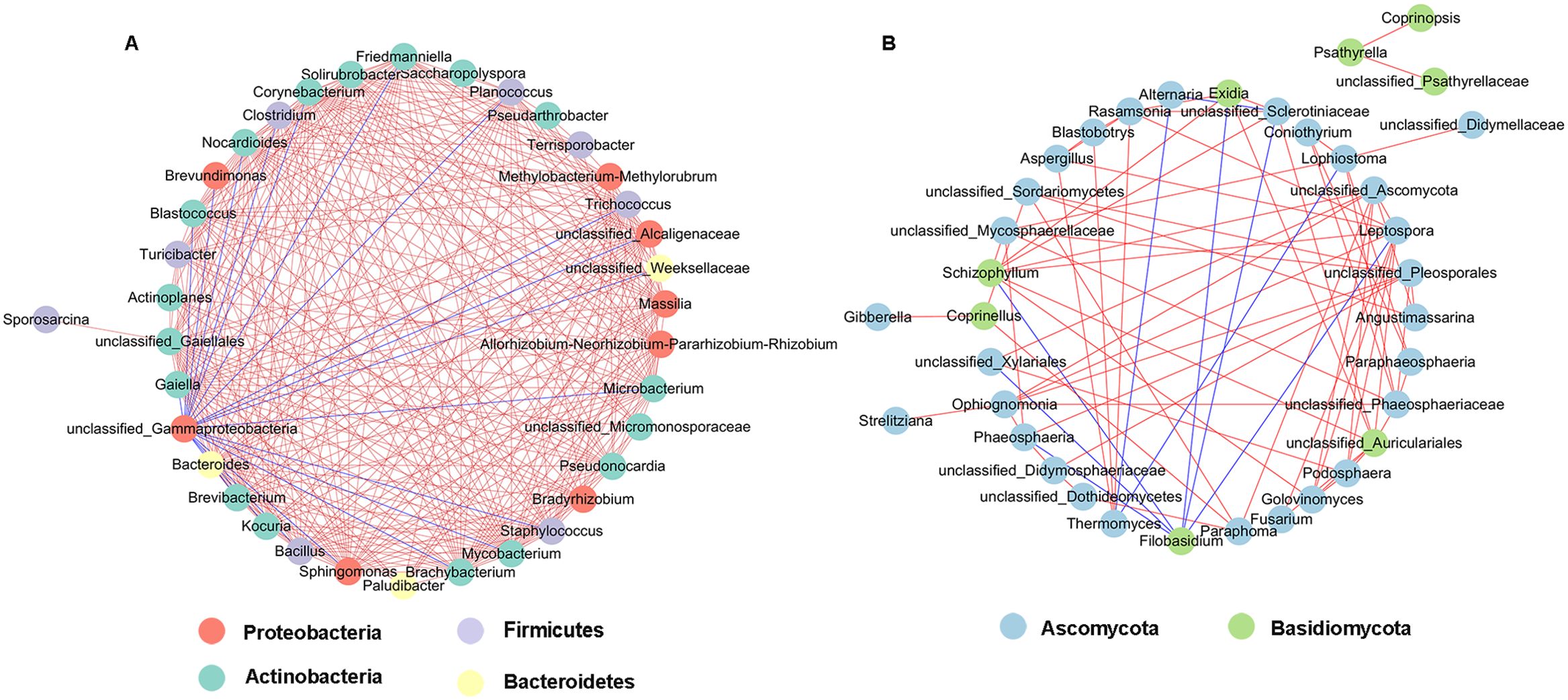
Figure 10. Network analysis of bacterial (A) and fungal (B) taxa showing significant positive or negative co-occurrences based on Spearman’s rank correlations. Red lines represent positive correlations and blue lines represent negative correlations.
3.8 Microbial community metabolic profiles
Phyllosphere microbial metabolic function analysis was performed using Biolog Eco metabolic 96-well plates (Figure 11). The metabolic capacity of carbon sources of different disease level samples decreased with increasing disease level, with B9 samples having the lowest metabolic capacity. The samples with different disease levels had lower capacity to utilize 2-hydroxybenzoic acid, α-cyclodextrin, α-butyronic acid, and phenylethyl-amine, but the B9 sample had a greater capacity to utilize these carbon sources than the other disease level samples.
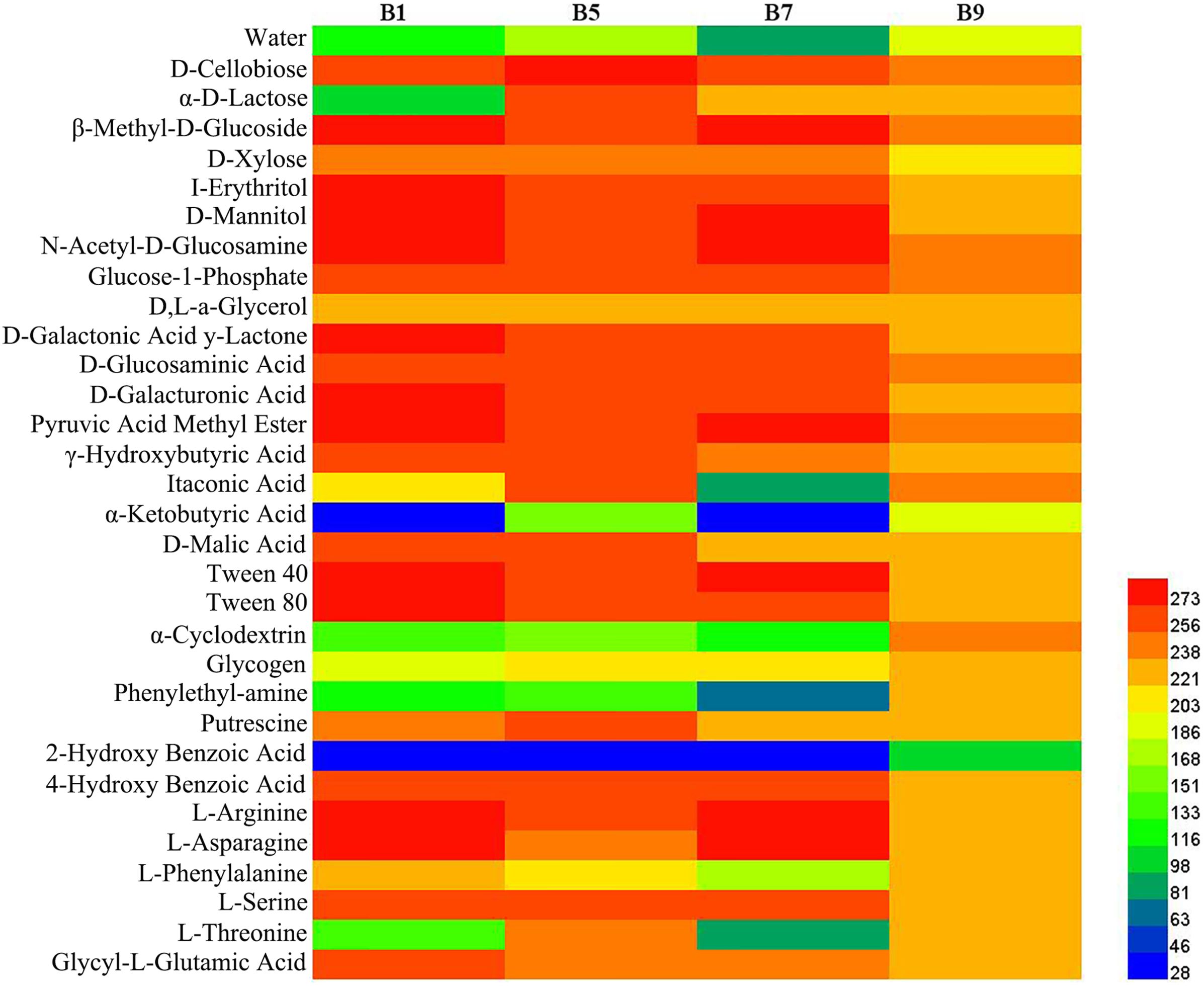
Figure 11. Heatmap of carbon sources in BIOLOG ECO-Plate of microbial communities from different samples.
4 Discussion
Disease severity level is an important factor to affect the structure of microbial communities in tobacco leaves. Understanding of the microbial characteristics of tobacco leaves with different disease severity levels is of great value to disease control. In this study, high-throughput sequencing and Biolog Eco were used to investigate the impact of disease severity level of tobacco wildfire disease on microbial communities and carbon source metabolizing capacity. It was shown that healthy plant leaves harbored greater microbial abundances and diversity compared to diseased leaves, with a highly diverse microbial diversity that tends to be conducive to resisting pathogen infection (De Mandal and Jeon, 2023). Similar results have been reported in this study, the Shannon, Ace, and Chao1 indices of bacterial community were higher in healthy groups than the diseased groups, and the Shannon index of the fungal community were higher in healthy groups than that diseased groups. The results suggested that the healthy phyllosphere has greater microbial diversity to resist disease expansion. Moreover, the Shannon index of bacteria decreased and then increased with increasing disease severity level, which was consistent with the conclusion of bacterial community diversity at different disease levels of powdery mildew in pumpkin reported by Luo et al. (2017). Conversely, Zhang et al. (2019) reported that the fungal diversity index showed increasing and then decreasing trend with the aggravation of the disease, which was consistent with the conclusion in this study about the diversity index of fungi. In general, the results suggested that there may be an interactive relationship between the bacteria and fungi in the leaf of tobacco wildfire disease.
The composition of the microbial community could be affected by the colonization of pathogens. In bacterial communities, Proteobacteria was the dominant phylum in both diseased and healthy samples at four disease severity levels. The abundance of Proteobacteria was higher in diseased samples than in the healthy samples (Guo et al., 2023), whereas the abundance of Actinobacteria and Firmicutes was much less in diseased than in healthy samples. The results of a previous study showed that Actinobacteria activates biosynthesis of salicylic acid to induce defense responses in plant leaves (Vergnes et al., 2020). Proteobacteria and Firmicutes are the common bacterial phyla in the phyllosphere; they always show opposite trends, which is thought to be due to the fact that Firmicutes were inhibited by Proteobacteria (Chen et al., 2020b). This may explain why the abundance of Firmicutes in diseased samples was lower than in healthy samples in tobacco wildfire disease. The dominant bacterial genus of both diseased and healthy leaves was Pseudomonas, which was the genus to which the pathogen of tobacco wildfire disease belongs. On the other hand, some strains of Pseudomonas are thought to produce a wide range of antibiotics that are effective in suppressing agricultural diseases.
The result of this study showed that a high abundance of Pseudomonas may be caused by pathogenic colonization or by antimicrobial active strains recruited in the phyllosphere. In addition, the relative abundance of Pseudomonas positively correlated with the disease severity level in diseased samples, which is similar to the results reported by Sun et al. (2022) who pointed out that the abundance of Thanatephorous (pathogen of tobacco target spot disease) increased with the increase in disease severity level. The abundance of unclassified_Alcaligenaceae was higher in healthy samples compared to diseased samples, which decreased with increasing disease severity level. Alcaligens has been heavily reported as a biocontrol agent against tobacco diseases. The result of this study suggests that there are several potentially beneficial genera in healthy samples, and whether plant health can be maintained by maintaining the abundance of Alcaligens is very much worthy of further investigation.
In fungal communities, the dominant phyla were Ascomycota and Basidiomycota in both diseased and healthy leaves, which is similar to Liu et al. (2022). The taxa unclassified_Didymellaceae, Alternaria, and Filobasidium were more enriched in diseased samples, whereas the abundance of Aspergillus was higher in healthy samples. Si et al. (2023) reported that Aspergillus was higher in healthy samples compared to diseased samples, which is consistent with our study. Among them, the relative abundance of Aspergillus negatively correlated with the disease severity level in diseased samples. Huang et al. (2021) found that the relative abundance of Alternaria increased with a disease severity level and deduced that Alternaria could benefit from the disease pressure caused by Didymella. The results of this study were consistent with them, and it is possible that the occurrence of wildfire disease is beneficial to Alternaria growth, but it remains to be investigated whether this genus accelerates or reduces wildfire disease occurrence. Based on the functional predictions of FUNGuild, most fungal communities were identified as saprotroph and pathotroph–saprotroph. Pathotroph–saprotroph and pathotroph–saprotroph–symbiotroph had a higher abundance in diseased samples. The data signify the presence of a large number of fungi in tobacco leaf, having pathogenic potential.
LEfSe analysis showed that the Pantoea and unclassified_Gammaproteobacteria were enriched in the L1, L5, and L7 diseased groups of different disease severity levels, but the L9 diseased groups were only enriched with unclassified_Gammaproteobacteria. The healthy groups were all enriched unclassified_Alcaligenaceae; moreover, the L1 healthy sample was enriched for the genera Brevibacterium, Bacillus, and Kocuria, the L5 healthy sample was also enriched for Bacillus, the L7 healthy sample Methylobacterium–Methylorubrum was enriched, while the L9 healthy sample did not have any other characterized genera. The results present that the diversity of enriched microbial communities were decreased with increasing disease severity level. In previous studies, Bacillus, Methylobacterium–Methylorubrum, Brevibacterium, and unclassified_Alcaligenaceae were considered to be biocontrol agents, beneficial to plant growth and to protect plants from pathogens (Madhaiyan et al., 2007; Son et al., 2014). Gammaproteobacteria are common bacteria in plants; in this study unclassified_Gammaproteobacteria were found to be in higher abundance in diseased samples compared to healthy samples, which is consistent with previous studies (Wang et al., 2022). Furthermore, the results of correlation network analysis also showed that unclassified_Gammaproteobacteria showed negative correlation with unclassified_Alcaligenaceae, Kocuria, Bacillus, and Brevibacterium in healthy groups. Gammaproteobacteria are eutrophic bacteria (Huang et al., 2017); we speculate that their lower abundance in healthy samples may be due to the higher microbial abundance and diversity in healthy samples, which compete with Gammaproteobacteria for nutrients.
The result of substrate utilization profiling displayed that the metabolizing ability of all samples to most carbon sources decreased with increasing disease severity level, but the metabolizing ability to α-butyronic acid, α-cyclodextrin, glycogen, phenylethylamine, and 2-hydroxybenzoic acid increased with increasing disease severity level, which was similar to the change in Pseudomonas abundance, but the relationship between these carbon sources and Pseudomonas needs to be further investigated. Meanwhile, we found that all samples had the lowest utilization of 2-2hydroxybenzoic acid, which has been reported to play an important role in the regulation of plant growth and development and defense response against pathogenic bacteria (Miura and Tada, 2014), which may explain its low utilization.
5 Conclusion
In this study, we found that the bacterial and fungal abundance and diversity from diseased samples was less than in healthy samples in the leaves affected by tobacco wildfire disease. Moreover, the abundance of bacterial and fungal community composition and diversity was affected by disease severity level. The relative abundance of Pseudomonas positively correlated with the disease severity level in diseased samples. The bacterial diversity and relative abundance decrease at the beginning and then increase with the tobacco wildfire disease severity level increases. Nevertheless, the fungal diversity and relative abundance increase and then decrease with increasing disease severity level. Finally, the metabolizing ability of all samples to most carbon sources decreased with increasing disease severity level. The most significant differences were found in β-methyl-D-glucoside, D-mannitol, N-acetyl-D-glucosamine, D-galacturonic acid, pyruvic acid methyl ester, and Tween 40. The obtained data from this study helps to infer that the composition and diversity of phyllosphere microbiome in tobacco wildfire disease. However, further study is needed to verify the interaction of phyllosphere microbiome. Hence, it is essential to direct attention toward these characteristic microorganisms in further studies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1096149 and PRJNA1096455.
Author contributions
XX: Data curation, Investigation, Writing – original draft. LZ: Data curation, Writing – original draft. YC: Investigation, Writing – original draft. HW: Supervision, Writing – review & editing. LC: Supervision, Writing – review & editing. YW: Supervision, Writing – review & editing. NW: Data curation, Writing – review & editing. WP: Supervision, Writing – review & editing. FW: Supervision, Writing – review & editing. YK: Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by China National Tobacco Corporation (110202101048(LS-08),110202001035(LS-04)), the 111 Project (D20009), the National Natural Science Foundation of China (32060034, 32160522, 32460051, 32460698), China-Ukraine Intergovernmental Exchange Project (8); Talent Base Project of Guizhou Province, China [RCJD2018-22]; Hundred’ level Innovation Talent Project of Guizhou Province(GCC[2022]036-1, GCC[2022]028-1, GCC[2023]108); Foundation of Key Laboratory of Microbiology and Parasitology of Education Department, Guizhou (QJJ [2022] 019); Guizhou Province Applied Technology Research and Development Funding Post-subsidy Project, Guizhou Science Technology Foundation (Qiankehe Talent Platform – CXTD[2023]021), and Guizhou Tobacco Company Project (2024XM06). International Science and Technology Cooperation Base of Guizhou Province ([2020]4101); Scientists Workstation Guizhou Province (KXJZ[2024]009), Ministry of Education Project (07150120711).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JK declared a past collaboration with the authors NW, YK to the handling editor.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, R. I., Miletto, M., Taylor, J. W., Bruns, T. D. (2013). Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. Isme J. 7, 1262–1273. doi: 10.1038/ismej.2013.28
Ahmed, W., Dai, Z., Liu, Q., Munir, S., Yang, J., Karunarathna, S. C., et al. (2022a). Microbial cross-talk: dissecting the core microbiota associated with flue-cured tobacco (Nicotiana tabacum) plants under healthy and diseased state. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.845310
Ahmed, W., Dai, Z., Zhang, J., Li, S., Ahmed, A., Munir, S., et al. (2022b). Plant-microbe interaction: mining the impact of native Bacillus amyloliquefaciens WS-10 on tobacco bacterial wilt disease and rhizosphere microbial communities. Microbiol. Spectr. 10, e0147122. doi: 10.1128/spectrum.01471-22
Barberán, A., Bates, S. T., Casamayor, E. O., Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. Isme J. 6, 343–351. doi: 10.1038/ismej.2011.119
Bulgarelli, D., Garrido-Oter, R., Münch, P. C., Weiman, A., Dröge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Chen, Q. L., Cai, L., Wang, H. C., Cai, L. T., Goodwin, P., Ma, J., et al. (2020a). Fungal composition and diversity of the tobacco leaf phyllosphere during curing of leaves. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.554051
Chen, T., Nomura, K., Wang, X., Sohrabi, R., Xu, J., Yao, L., et al. (2020b). A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 580, 653–657. doi: 10.1038/s41586-020-2185-0
Dai, Y. F., Wu, X. M., Wang, H. C., Li, W. H., Cai, L. T., Li, J. X., et al. (2022). Spatio-temporal variation in the phyllospheric microbial biodiversity of alternaria alternata-infected tobacco foliage. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.920109
De Mandal, S., Jeon, J. (2023). Phyllosphere microbiome in plant health and disease. Plants (Basel) 12, 3481. doi: 10.3390/plants12193481
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Feng, R., Wang, H., Liu, T., Wang, F., Cai, L., Chen, X., et al. (2023). Response of microbial communities in the phyllosphere ecosystem of tobacco exposed to the broad-spectrum copper hydroxide. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1229294
Fitzpatrick, C. R., Salas-González, I., Conway, J. M., Finkel, O. M., Gilbert, S., Russ, D., et al. (2020). The plant microbiome: from ecology to reductionism and beyond. Annu. Rev. Microbiol. 74, 81–100. doi: 10.1146/annurev-micro-022620-014327
Gong, T., Xin, X. F. (2021). Phyllosphere microbiota: Community dynamics and its interaction with plant hosts. J. Integr. Plant Biol. 63, 297–304. doi: 10.1111/jipb.13060
Guo, M., Hu, J., Jiang, C., Zhang, Y., Wang, H., Zhang, X., et al. (2023). Response of microbial communities in the tobacco phyllosphere under the stress of validamycin. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1328179
Huang, W., Chen, X., Jiang, X., Zheng, B. (2017). Characterization of sediment bacterial communities in plain lakes with different trophic statuses. Microbiologyopen 6, e00503. doi: 10.1002/mbo3.503
Huang, Y., Wang, H. C., Cai, L. T., Li, W., Pan, D., Xiang, L., et al. (2021). Phyllospheric microbial composition and diversity of the tobacco leaves infected by Didymella segeticola. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.699699
Ichinose, Y., Taguchi, F., Mukaihara, T. (2013). Pathogenicity and virulence factors of Pseudomonas syringae. J. Gen. Plant Pathol. 79, 285–296. doi: 10.1007/s10327-013-0452-8
Lindow, S. E., Brandl, M. T. (2003). Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69, 1875–1883. doi: 10.1128/aem.69.4.1875-1883.2003
Liu, T., Guo, S., Wu, C., Zhang, R., Zhong, Q., Shi, H., et al. (2022). Phyllosphere microbial community of cigar tobacco and its corresponding metabolites. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1025881
Luo, L., Zhang, Z., Jin, D., Tan, X., Zhang, D., Tian, Y., et al. (2017). Phyllosphere bacterial diversities and community structures on pumpkin with different severities of powdery mildew. Acta Phytopathol 47, 688–695.
Luo, L., Zhang, Z., Wang, P., Han, Y., Jin, D., Su, P., et al. (2019). Variations in phyllosphere microbial community along with the development of angular leaf-spot of cucumber. AMB Express 9, 76. doi: 10.1186/s13568-019-0800-y
Ma, X., Zhu, X., Mu, Y., Gao, C., He, W., Ran, M., et al. (2023). Fabrication of polydopamine reduced CuO nanoparticle-alginate composite nanogels for management of Pseudomonas synringae pv. tabaci in tobacco. Pest Manag. Sci. 79, 1213–1224. doi: 10.1002/ps.7298
Madhaiyan, M., Poonguzhali, S., Sa, T. (2007). Metal tolerating methylotrophic bacteria reduces nickel and cadmium toxicity and promotes plant growth of tomato (Lycopersicon esculentum L). Chemosphere 69, 220–228. doi: 10.1016/j.chemosphere.2007.04.017
Magoč, T., Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Miura, K., Tada, Y. (2014). Regulation of water, salinity, and cold stress responses by salicylic acid. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00004
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal ecology. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Perreault, R., Laforest-Lapointe, I. (2022). Plant-microbe interactions in the phyllosphere: facing challenges of the anthropocene. Isme J. 16, 339–345. doi: 10.1038/s41396-021-01109-3
Rutgers, M., Wouterse, M., Drost, S. M., Breure, A. M., Mulder, C., Stone, D., et al. (2016). Monitoring soil bacteria with community-level physiological profiles using Biolog™ ECO-plates in the Netherlands and Europe. Appl. Soil Ecol. 97, 23–35. doi: 10.1016/j.apsoil.2015.06.007
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Shen, Y., Zhao, J., Zou, X., Shi, Z., Liao, Y., He, Y., et al. (2023). Differential responses of bacterial and fungal communities to siderophore supplementation in soil affected by tobacco bacterial wilt (Ralstonia solanacearum). Microorganisms 11, 1535. doi: 10.3390/microorganisms11061535
Si, H., Cui, B., Liu, F., Zhao, M. (2023). Microbial community and chemical composition of cigar tobacco (Nicotiana tabacum L.) leaves altered by tobacco wildfire disease. Plant Direct 7, e551. doi: 10.1002/pld3.551
Son, J.-S., Sumayo, M., Hwang, Y.-J., Kim, B.-S., Ghim, S.-Y. J. A. (2014). Screening of plant growth-promoting rhizobacteria as elicitor of systemic resistance against gray leaf spot disease in pepper. Appl. Soil Ecol. 73, 1–8. doi: 10.1016/j.apsoil.2013.07.016
Sun, S., Liu, C., Duan, C., Zhu, Z. (2021). Wildfire, a new bacterial disease of mung bean, caused by Pseudomonas syringae pv. tabaci. J. Plant Pathol. 103, 649–653. doi: 10.1007/s42161-021-00823-3
Sun, M., Shi, C., Huang, Y., Wang, H., Li, J., Cai, L., et al. (2022). Effect of disease severity on the structure and diversity of the phyllosphere microbial community in tobacco. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1081576
Tumewu, S. A., Watanabe, Y., Matsui, H., Yamamoto, M., Noutoshi, Y., Toyoda, K., et al. (2022). Identification of Aerotaxis Receptor Proteins Involved in Host Plant Infection by Pseudomonas syringae pv. tabaci 6605. Microbes Environ. 37, ME21076. doi: 10.1264/jsme2.ME21076
Vergnes, S., Gayrard, D., Veyssière, M., Toulotte, J., Martinez, Y., Dumont, V., et al. (2020). Phyllosphere colonization by a soil Streptomyces sp. Promotes plant defense responses against fungal infection. Mol. Plant Microbe Interact. 33, 223–234. doi: 10.1094/mpmi-05-19-0142-r
Wang, Z., Fu, C., Tian, J., Wang, W., Peng, D., Dai, X., et al. (2022). Responses of the bacterial community of tobacco phyllosphere to summer climate and wildfire disease. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1050967
Xin, X. F., Kvitko, B., He, S. Y. (2018). Pseudomonas syringae: what it takes to be a pathogen. Nat. Rev. Microbiol. 16, 316–328. doi: 10.1038/nrmicro.2018.17
Zhang, Z., Kong, X., Jin, D., Yu, H., Zhu, X., Su, X., et al. (2019). Euonymus japonicus phyllosphere microbiome is significantly changed by powdery mildew. Arch. Microbiol. 201, 1099–1109. doi: 10.1007/s00203-019-01683-3
Keywords: tobacco wildfire disease, microbial community, high-throughput sequencing, biolog-eco, disease severity
Citation: Xu X, Zhao L, Chen Y, Wang H, Cai L, Wang Y, Wijayawardene NN, Pan W, Wang F and Kang Y (2024) Phyllospheric microbial community structure and carbon source metabolism function in tobacco wildfire disease. Front. Cell. Infect. Microbiol. 14:1458253. doi: 10.3389/fcimb.2024.1458253
Received: 02 July 2024; Accepted: 19 September 2024;
Published: 01 November 2024.
Edited by:
Sinang Hongsanan, Shenzhen University, ChinaReviewed by:
Davide Palmieri, University of Molise, ItalyJaturong Kumla, Chiang Mai University, Thailand
Terd Disayathanoowat, Chiang Mai University, Thailand
Copyright © 2024 Xu, Zhao, Chen, Wang, Cai, Wang, Wijayawardene, Pan, Wang and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hancheng Wang, eGlhb2JhaXlhbmcxMjZAaG90bWFpbC5jb20=; Feng Wang, NTg0MjI4OTkyQHFxLmNvbQ==; Yingqian Kang, a2FuZ3lpbmdxaWFuQGdtYy5lZHUuY24=
†These authors share first authorship