 Marta Hernández-García1,2†
Marta Hernández-García1,2† Raquel Barbero-Herranz1†
Raquel Barbero-Herranz1† Natalia Bastón-Paz1
Natalia Bastón-Paz1 María Díez-Aguilar3
María Díez-Aguilar3 Eduardo López-Collazo4,5Francesc J. Márquez-Garrido6José María Hernández-Pérez7
Eduardo López-Collazo4,5Francesc J. Márquez-Garrido6José María Hernández-Pérez7 Fernando Baquero1,8
Fernando Baquero1,8 Miquel B. Ekkelenkamp9
Miquel B. Ekkelenkamp9 Ad C. Fluit9Víctor Fuentes-Valverde2,10
Ad C. Fluit9Víctor Fuentes-Valverde2,10 Miriam Moscoso2,10
Miriam Moscoso2,10 Germán Bou2,10
Germán Bou2,10 Rosa del Campo1,2
Rosa del Campo1,2 Rafael Cantón1,2*
Rafael Cantón1,2* José Avendaño-Ortiz1,2*
José Avendaño-Ortiz1,2*- 1Servicio de Microbiología, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Madrid, Spain
- 2CIBER de Enfermedades Infecciosas, Instituto de Salud Carlos III, Madrid, Spain
- 3Servicio de Microbiología y Parasitología, Hospital Universitario La Princesa, Madrid, Spain
- 4CIBER de Enfermedades Respiratorias, Instituto de Salud Carlos III, Madrid, Spain
- 5Innate Immune Response Group, IdiPAZ, Madrid, Spain
- 6Bruker Microbiology & Infection Diagnostics, Bruker Española S.A., Madrid, Spain
- 7Plataforma de Proteómica y Metabolómica, Instituto de Investigación Germans Trias i Pujol, Badalona, Spain
- 8CIBER de Epidemiología y Salud Pública, Instituto de Salud Carlos III, Madrid, Spain
- 9Department of Medical Microbiology, University Medical Center Utrecht, Utrecht, Netherlands
- 10Department of Microbiology, University Hospital A Coruña (CHUAC)-Biomedical Research Institute A Coruña (INIBIC), A Coruña, Spain
Introduction: Murepavadin is an antimicrobial peptide (AMP) in clinical development that selectively targets Pseudomonas aeruginosa LptD and whose resistance profile remains unknown. We aimed to explore genomic modifications and consequences underlying murepavadin and/or colistin susceptibility.
Methods: To define genomic mechanisms underlying resistance, we performed two approaches: 1) a genome-wide association study (GWAS) in a P. aeruginosa clinical collection (n=496), considering >0.25 mg/L as tentative cut-off of murepavadin acquired resistance; 2) a paired genomic comparison in a subset of 5 isolates and their isogenic murepavadin-resistant mutants obtained in vitro. Lipid-A composition, immunogenicity and cathelicidin and indolicidin effects on bacterial growth were also tested in this last subset of isolates. Murepavadin MICs were determined in ΔlpxL1 and ΔlpxL2 knock-out mutants obtained from a auxotroph PAO1 derivative.
Results: GWAS revealed a missense variant (A→G p.Thr260Ala in the hisJ gene) associated with murepavadin resistance although both resistant and susceptible strains harbored it (21% and 12% respectively, OR=1.92, p=0.012 in χ² test). Among the isolate subset, murepavadin-resistant mutants with deletions in lpxL1 and lpxL2 genes showed lower abundance of hexa-acylated lipid-A (m/z 1616, 1632). 4-aminoarabinose addition was found only in colistin-resistant isolates but not in the other ones, irrespective of murepavadin susceptibility. Accordingly, ΔlpxL1 and ΔlpxL2 mutants exhibited higher murepavadin MICs than parental PAO1 auxotroph strain (2 and 4 vs 0.5 mg/L respectively). Lipopolysaccharide from murepavadin-resistant mutants triggered lower inflammatory responses in human monocytes. Those with lpxL mutations and hexa-acylated lipid-A loss also exhibited greater growth reduction when exposed to host-derived AMPs cathelicidin and indolicidin.
Discussion: High murepavadin-resistance seems to be linked to lpxL1 and lpxL2 mutations and lower hexa-acylated lipid-A, corresponding to lower inflammatory induction and higher susceptibility to host-derived AMPs. Although GWAS identified one variant associated with the murepavadin-resistant phenotype, data revealed that there was no unique single genetic event underlying this phenotype. Our study provides insight into the mechanisms underlying murepavadin susceptibility.
1 Introduction
Antimicrobial peptides (AMPs) are small peptide-derived molecules (12-60 amino acids) with bactericidal activity (Huan et al., 2020; Talapko et al., 2022) that can be produced by host immune cells, such as cathelicidin or defensins, but also by microorganisms; colistin, vancomycin, and daptomycin are examples of the latter (Wang and Mechesso, 2022). AMPs represent a promising strategy for the discovery of new antimicrobial drugs (Mba and Nweze, 2022). Murepavadin (POL7080; Polyphor Ltd) is a new peptidomimetic, initially developed for systemic administration and currently being positioned for inhaled therapy (NCT03582007 and NCT03409679). Its target is the protein transporter, LptD, blocking lipopolysaccharide transport by the bacterium (Martin-Loeches et al., 2018). Murepavadin has notable and almost exclusive activity against Pseudomonas aeruginosa, including in biofilm (Díez-Aguilar et al., 2021), and it is considered an ecologic antibiotic for its lack of effect on the commensal microbiota. In addition, this antibiotic promotes the immune response, facilitating bacterial clearance (Amponnawarat et al., 2021). The mechanism underlying the decrease of susceptibility to murepavadin is not completely characterized, although previous studies have reported a tandem duplication of 6 residues in the periplasmic part of LptD (Andolina et al., 2018) and mutations in lipopolysaccharide/lipid A genes (Díez-Aguilar et al., 2021; Ghassani et al., 2024).
On the other hand, the polymyxin, colistin, is a cationic peptide that has been used in the last 2 decades as a last resort for severe infections caused by multidrug-resistant microorganisms, including P. aeruginosa. This positively charged antibiotic interacts electrostatically with the negatively charged lipid A phosphate group, causing destabilization of the outer membrane, pore formation, increased permeability, and cell lysis. In Enterobacterales, colistin resistance has been mainly associated with dissemination of mcr genes harbored in plasmids, but this is not a common mechanism in P. aeruginosa (Snesrud et al., 2018). In contrast, lipopolysaccharide modification and negative charge reduction by overproduction of 4-amino-L-arabinose (L-Ara4N) is one of the major causes of colistin resistance in P. aeruginosa (Lo Sciuto et al., 2020). The synthesis and the transport of L-Ara4N molecules are encoded by the large arnBCADTEF-UgD operon, simplified as arn, which has a complex regulatory network (Barrow and Kwon, 2009; Nowicki et al., 2015; Jochumsen et al., 2016; Bolard et al., 2019).
The ability to predict antimicrobial resistance using bioinformatics tools and bacterial genetic content has gained attention. In previous studies, we and others used whole genome sequencing (WGS) and single nucleotide polymorphisms (SNPs) analysis of specific relevant genes to explore their association with murepavadin resistance (Díez-Aguilar et al., 2021; Ghassani et al., 2024). Here, we propose an untargeted strategy by performing a genome-wide association study (GWAS), a recently successful approach in the study of genotype-phenotype associations in prokaryotes (Falush, 2016; Diaz Caballero et al., 2018; Bokma et al., 2021; Young et al., 2021). Furthermore, lipid A structural modifications, immunogenicity and the possible cross-resistance to host-derived AMPs were performed in a subset of murepavadin-resistant mutants previously obtained in vitro (Ekkelenkamp et al., 2020; Díez-Aguilar et al., 2021).
2 Material and methods
2.1 Bacterial strains, antibiotic susceptibility, and whole genome sequencing
A collection of 496 P. aeruginosa isolates previously obtained during the “Inhaled antibiotics in bronchiectasis and cystic fibrosis” (iABC) project from the Netherlands (n=240), Spain (n=139), Northern Ireland (n=94), and Australia (n=23) (https://www.imi.europa.eu/projects-results/project-factsheets/iabc) were used (Ekkelenkamp et al., 2020). Isolates were obtained from the airway of patients with cystic fibrosis (n=399), chronic obstructive pulmonary disease (n=29), bronchiectasis (n=30), or other respiratory pathologies (n=38). A single isolate per patient was considered. Susceptibility to murepavadin and colistin was determined by ISO-broth microdilution, following European Committee on Antimicrobial Susceptibility Testing (EUCAST) recommendations and interpretation (EUCAST v12.0, 2022) (The European Committee on Antimicrobial Susceptibility Testing, 2022). Colistin minimum inhibitory concentration (MIC) of >4 mg/L and murepavadin MIC of >0.25 mg/L were considered as a cut-off and tentative cut-off, respectively, to define resistant isolates (Ekkelenkamp et al., 2020, 2022; Díez-Aguilar et al., 2021; The European Committee on Antimicrobial Susceptibility Testing, 2022). WGS was obtained in an Illumina MiSeq or NextSeq platform (Illumina, San Diego, CA, USA), employing the Nextera XT library prep kit (Illumina) with 2x150 paired-end reads. All reads were trimmed with seqtk trimfq version 1.3 with an error rate threshold of 0.001. Sequence reads from each isolate were assembled with SPAdes v3.11.1. Sequences were deposited at the European Nucleotide Archive under BioProject ID: PRJNA527751 and PRJNA530912. Bacterial identification was confirmed by k-mer-based classification using Kraken (v1.1.1). All draft genomes were annotated by Prokka (v1.14.6). Resistance genes were screened with Abricate (v0.8) (ARG-ANNOT, CARD and ResFinder databases; threshold, 95% identity; 90% coverage). The Mash v. 2.3 and iTOL applications were employed to generate and trace a similarity tree based on a neighbor-joining algorithm.
2.2 Variant calling
The allelic variants in the whole genome were determined in a first stage, with the second analysis restricted to a subset of genes previously associated with colistin and/or murepavadin resistance in P. aeruginosa (Díez-Aguilar et al., 2021; Hernández-García et al., 2021). SNPs and small insertions or deletions were extracted with Snippy (v4.6.0), using the P. aeruginosa PAO1 (NC_002516.2) genome as reference. Synonymous variants were excluded for further analysis.
2.3 Genome-wide association study analysis
An appropriate correction considering the clonal population structure was applied by pyseer (v1.3.10) according to the fixed effects model of software documentation, with the binary phenotype as susceptible or resistant. Genes, regions, or SNPs with a minor allele frequency of <0.01 were not tested. Performing sequence element enrichment analysis in pyseer can accurately detect associations with phenotype (in our case, antibiotic resistance) caused by SNPs in coding regions. The screening of allelic variants was performed independent of colistin or murepavadin MIC values. The clonal population structure was estimated/inferred employing a pairwise distance matrix generated with mash (v.3.2) that measures genetic similarity as the co-variance between the individual isolate genetic variant vectors. Bonferroni correction was applied in the statistical analysis, only considering results with p values <0.5.
2.4 Lipid A characterization
Lipid A characterization was performed on a subset of 5 P. aeruginosa isolate pairs, including 4 clinical isolates and the reference strain PAO1, and their corresponding murepavadin-resistant mutant derivatives previously reported (Díez-Aguilar et al., 2021). The selection of the isolates was based on their colistin MIC values, their ST lineage, and the growth mode (Supplementary Table S1). Lipid A was extracted with the MBT Lipid Xtract Kit (Bruker Daltonics GmbH) following the manufacturer’s instructions. Briefly, a 1-µL inoculation loop of bacteria from a fresh overnight culture of the bacterial strains grown on a Columbia blood agar plate was subjected to hydrolysis at 90°C for 10 min. The dry pellets were washed and left 2 min at 90°C for evaporation. Then, the pellet was resuspended in Lipid Xtract Matrix, and 2 µL were spiked into an MTP 384 target plate polished steel BC (Bruker Daltonics GmbH). Mass spectrometry runs were performed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) in an Autoflex maX spectrometer (Bruker Daltonics GmbH), in linear negative-ion mode. Spectra were analyzed with Clover MS Data Analysis Software v1.6.4 (CLOVER Bioanalytical Software S.L.).
2.5 Murepavadin MIC values determination of lpxL1 and lpxL2 auxotroph PAO1 mutants
The bacterial PAO1 mutant strains tested included PAO1 auxotroph mutants obtained in previous studies (Cabral et al., 2020; Fuentes-Valverde et al., 2022). The parental auxotroph PAO1 strain was a triple mutant including in-frame deletions of three genes (ΔmurI, Δalr and ΔdadX) making it auxotroph for D-glutamate and D-alanine (Cabral et al., 2020; Fuentes-Valverde et al., 2022). In this auxotrophic PAO1- strain, an additional deletion of lpxL1 and lpxL2 were used to form the ΔlpxL1 and ΔlpxL2 strains respectively. The MIC values of murepavadin in these strains was determined by the microdilution method in 96-well U-bottom plates (Corning). Briefly, a 0.5 McFarland scale inoculum was prepared in a sterile saline solution for each strain. This initial suspension was 1:100 diluted in LB broth supplemented with 8 mM of D-glutamate and 6 mM of D-alanine. Murepavadin concentrations tested ranged from 0.015 to 32 mg/L. After an overnight incubation at 37°C, the wells were examined for visible bacterial growth. The lowest concentration of murepavadin that prevented visible bacterial growth was defined as the MIC values. Experiment was repeated twice in independent experiments.
2.6 Lipopolysaccharide isolation and immunogenicity assay
Lipopolysaccharide was isolated using a lipopolysaccharide isolation kit (Sigma-Aldrich) according to the manufacturer’s protocol. Briefly, the 10 isolates (the selected 4 cystic fibrosis isolates plus PAO1 and their relative mutants) were grown overnight on Luria-Bertani (LB) medium agar plates (Difco). 200 mg of bacteria were taken with sterile 10 µL loops and resuspended in 10 mL of cold sterile PBS. The suspensions were centrifugated (2,500 x g for 10 min) to pellet the bacteria resuspended in 2 mL of lysis buffer and sonicated (3x30 s in continuous pulse). Then they were incubated on ice for 10 min washed (2,500 x g for 10 min, 4°C) and the lysate were transferred to a clean tube. Finally, proteinase K was added to a final concentration of 100 mg/L, heated 60 min at 60 °C then centrifuged (2,500 x g for 10 min, 4°C) and supernatant were transferred to a clean tube. Lipopolysaccharide in these supernatants was quantified with a total carbohydrate assay kit (Sigma-Aldrich). Blood from 8 healthy adult volunteers was obtained from the Transfusion Center of the Community of Madrid (n=4 males and n=4 females; mean age ± SD, 52.4 ± 8.2). Peripheral blood mononuclear cells were isolated by Ficoll-Paque Plus (Cytiva) gradient. Cells were counted, and monocyte percentages were checked by flow cytometry (BD FACSCalibur). The monocyte population was enriched by adherence protocol with 1 h of culture in serum-free Roswell Park Memorial Institute (RPMI) medium as previously described (del Fresno et al., 2009; Avendaño-Ortiz et al., 2019). After this period, the non-adherent cells were removed and the adherent cells were washed three times with PBS, and finally cultured with RPMI-10% fetal bovine serum medium. An average of monocyte purity percentage of 55.6 ± 10.4 defined by CD14+ monocytes in the adherent cells were obtained. Cells were stimulated with 10 mg/L lipopolysaccharide for 16 h in a humidified 5% CO2 atmosphere at 37°C. A commercial LPS from Escherichia coli O111:B4 (Sigma-Aldrich, cat. number L2630) were used as positive control. Cytokine production in cell culture supernatants was determined by flow cytometry (BD FACSCalibur), employing the LEGENDplex™ Human Essential Immune Response Panel. Cells were labeled with CD14-APC antibody (Immunostep) and apoptosis on gated CD14+ cells were determined by flow cytometry (BD FACSCalibur) using FITC Annexin V Apoptosis Detection Kit (Immunostep) following manufacturer’s instructions.
2.7 Cross-resistance with human antimicrobial peptides
Starting from an overnight culture on Columbia blood agar, a bacterial inoculum was adjusted to 0.5 McFarland diluted 1:100 (v/v) in Mueller Hinton II Broth (Liofilchem®). Based on previous reports in P. aeruginosa growth inhibition, a concentration of 128 mg/L for cathelicidin and indolicidin was chosen (Gooderham et al., 2008; Mohammed et al., 2019; Xiao et al., 2022). Thus, the effects on bacterial growth of 128 mg/L cathelicidin and 128 mg/L indolicidin (Anaspec, Fremont, CA, USA) were determined by incubating the bacterial suspensions with the two peptides separately, in 96-microwell plates at 30°C with orbital shaking. Cell density was monitored at OD600 every 15 min with a Synergy HTX (BioTek).
3 Results
3.1 Genomic clinical isolates characteristics
A collection of 496 P. aeruginosa clinical isolates from patients with cystic fibrosis or other respiratory pathologies were included in the analysis. Up to 348 (70.2%) were susceptible to both colistin and murepavadin, 15 (3.0%) colistin-resistant and murepavadin-susceptible, 116 (23.4%) murepavadin-resistant and colistin-susceptible, and 17 (3.4%) were resistant to both antibiotics. Five clusters were identified (Figure 1). A high genetic variability was observed in our collection, including 179 sequence types (STs). Of note: 42 (8.5%) isolates could not be assigned to any ST. Overall, 20.9% (94/450) of the isolates yielded unique STs, the most prevalent being ST406 (n=30, 6.7%), followed by ST146 (n=27, 6%). Only 3 STs (ST155, ST179 and ST253) were represented in the 4 countries.
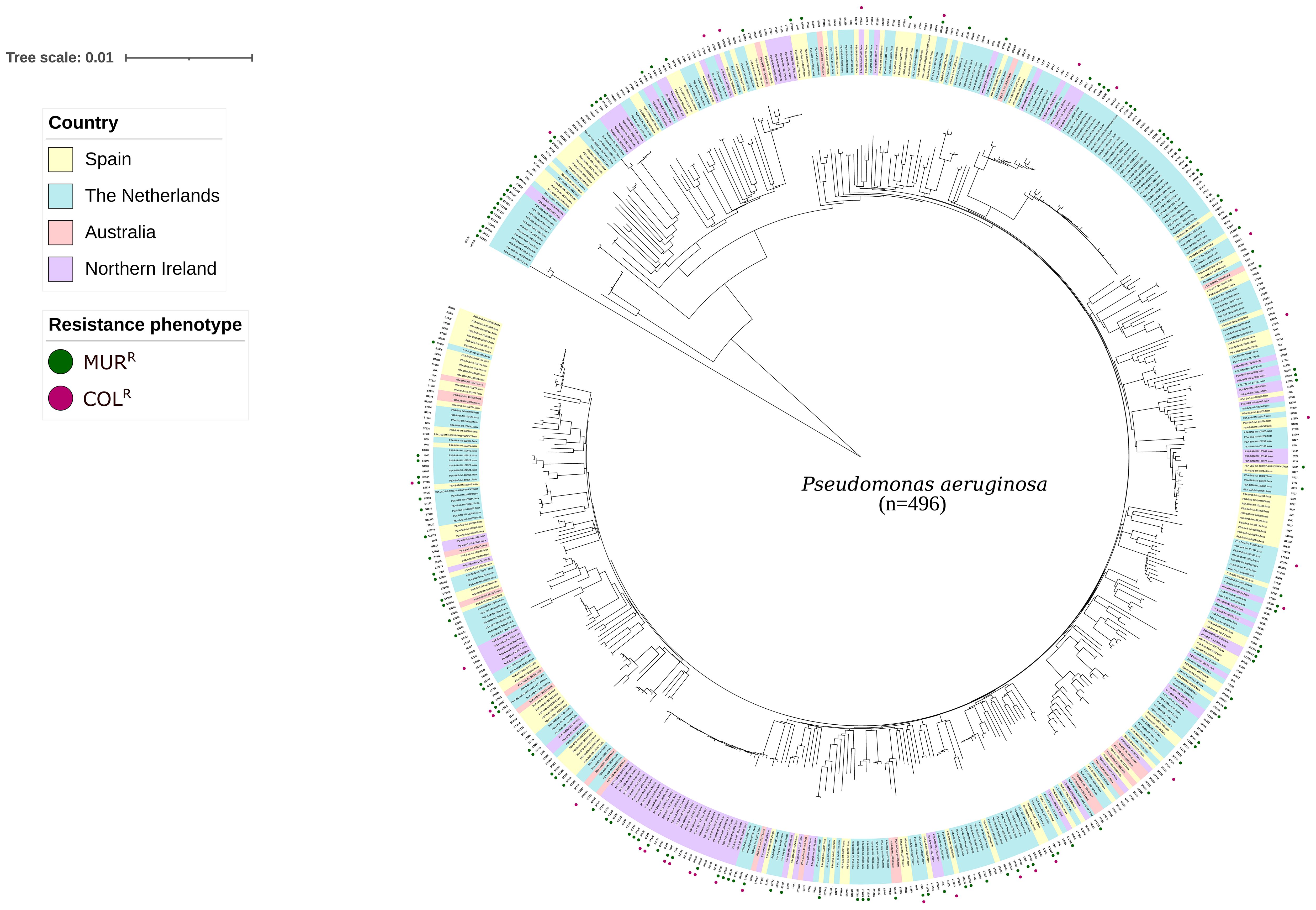
Figure 1. Similarity tree iTOL construction with the whole genome of the 496 P. aeruginosa isolates included in bioinformatic analysis. Legend differentiates the country of origin and their phenotypic susceptibility to murepavadin and colistin. MURR, murepavadin resistant (MIC >0.25 mg/L) and COLR, colistin resistant (MIC >4mg/L).
The average size of the de novo assembled genomes was 6.4 Mb, with a G + C content of 66.3%. All isolates resistant and susceptible to colistin carried the arnA gene, without other genes previously associated with colistin resistance. The pangenome of the 496 isolates included have 40,188 different orthologous genes, the core genome (genes in >95% of isolates) had 3,912 clusters (9.73% of all genes), and the accessory genome (genes in <15% of strains) had 2,629 clusters (6.54% of all genes).
3.2 Genome-wide association study analysis
An independent analysis was conducted for each antibiotic, excluding loci with a burden frequency below 0.01 and adjusting for population structure. Out of 777,515 allele variants, 102,008 non-synonymous were ultimately considered (Supplementary Figure S1). For colistin resistance, no allelic variants reached statistical significance although notable findings included a missense variant (C>T p.Asp921Asn) in the recC gene (p=2.25×10-5) and a variant in an intragenic region (p=1.97×10-5) (Supplementary Table S2). In the analysis of murepavadin resistance, a missense variant (A>G p.Thr260Ala) in the hisJ gene was significantly associated (p=7.77×10-8, OR=14.3, Supplementary Table S3) but it was not relevant for colistin resistance (p=0.28). This mutation was found in 21% of murepavadin-resistant strains, although it was also observed in 12% of the susceptible strains (OR=1.92, p=0.012 in χ² test) (Figure 2). A final GWAS was performed to identify variants associated with resistance to both colistin and murepavadin by comparing 17 isolates resistant to both antibiotics with the other 349 isolates. No significant variants were identified after Bonferroni correction (p=6.5×10-7). The highest associations were observed in intergenic regions, a protein of unknown function, and the YhjE and YofA3 genes (Supplementary Table S4).
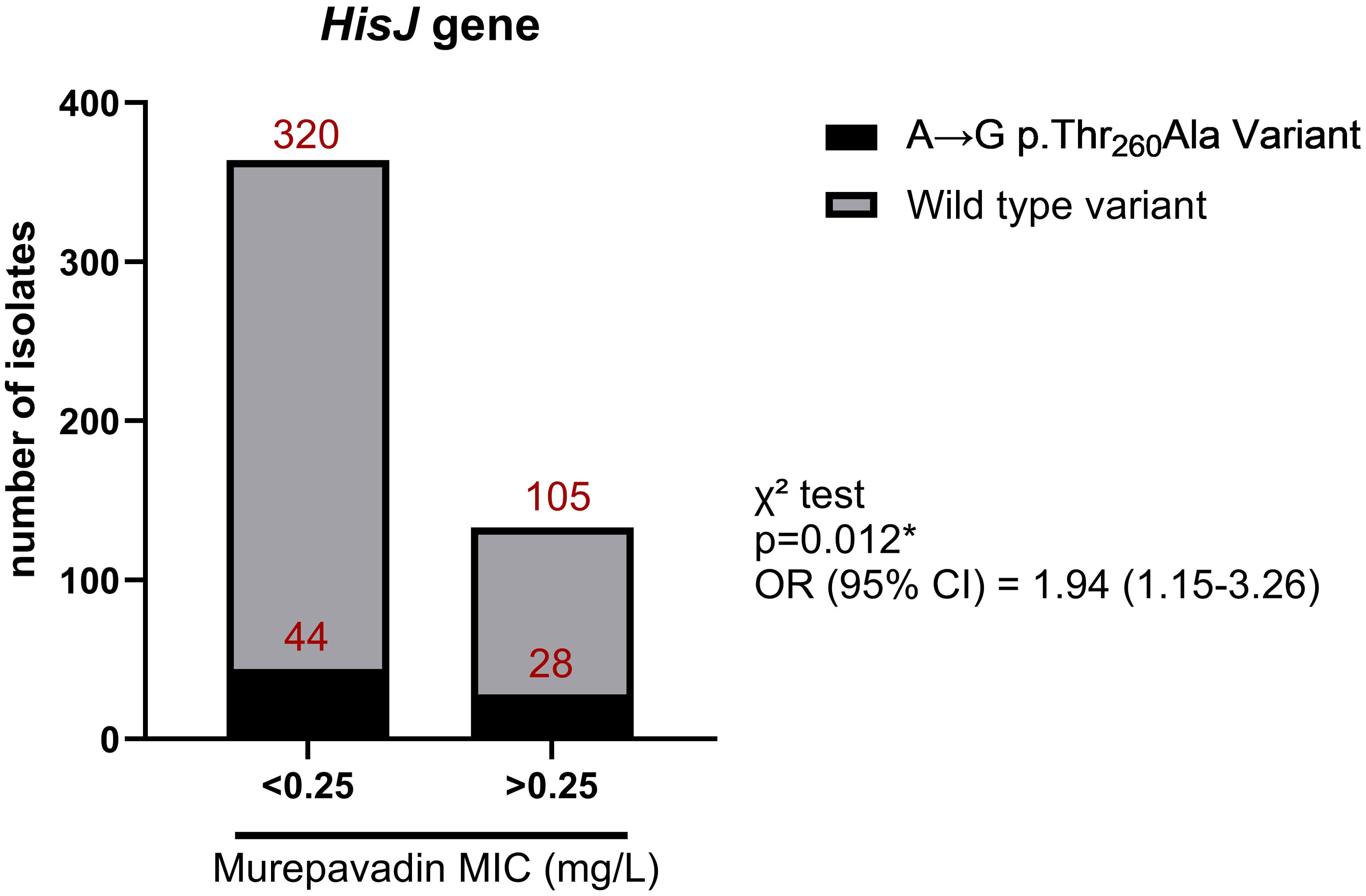
Figure 2. Number of P. aeruginosa isolates harboring wild type or A→G p.Thr260Ala variant according to their susceptibility to murepavadin. OR, odds ratio; *p-value < 0.05 in Chi-square (χ2) test.
3.3 Loss of lipid A acyl chains and lpxL1 and lpxL2 variation in murepavadin-resistant mutants
Since we did not identify any specific single genetic event that contributes to murepavadin resistance in the clinical isolate collection, we decided to study the mechanisms behind the resistance in in vitro obtained murepavadin-resistant mutants. In our previous study we obtained and sequenced murepavadin-resistant mutants from 4 P. aeruginosa clinical isolates susceptible to murepavadin (description in Supplementary Table S1) (Díez-Aguilar et al., 2021). The lipid A profiles of these P. aeruginosa isolates were characterized by hexa-acylated lipid A as the most abundant forms in 3 out of the 4 clinical isolates, while PAO1 exhibited a dominance of penta-acylated m/z 1447. Proposed lipid A structures are shown in Supplementary Figure S2. Apart from isolate P28, which exhibited the same hexa-acylated ions (m/z 1616 and 1632) in both susceptible and resistant strains, the murepavadin-resistant mutants were characterized by a decrease of the hexa-acylated forms, along with an increase of penta-acylated and/or tetra-acylated forms. This phenomenon was especially pronounced in P14 and P40 isolates, those that also carried allelic variants in the lpxL1 and lpxL2 genes (Figure 3). Note that these genes encode acyltransferases involved in lipid A biogenesis (Hittle et al., 2015). The addition of L-Ara4N or pEtN to lipid A was only detected in the P7 isolate, which was the unique isolate with colistin resistance. As three of the five murepavadin-resistant mutants exhibited lpxL1 and lpxL2 deletions compared to their parental, we move to test if these two genes could be involved in murepavadin resistance. We found that the ΔlpxL1 and ΔlpxL2 strains exhibited MICs to murepavadin that were 4 to 8 folds higher than the auxotrophic PAO1 parental strain (Table 1).
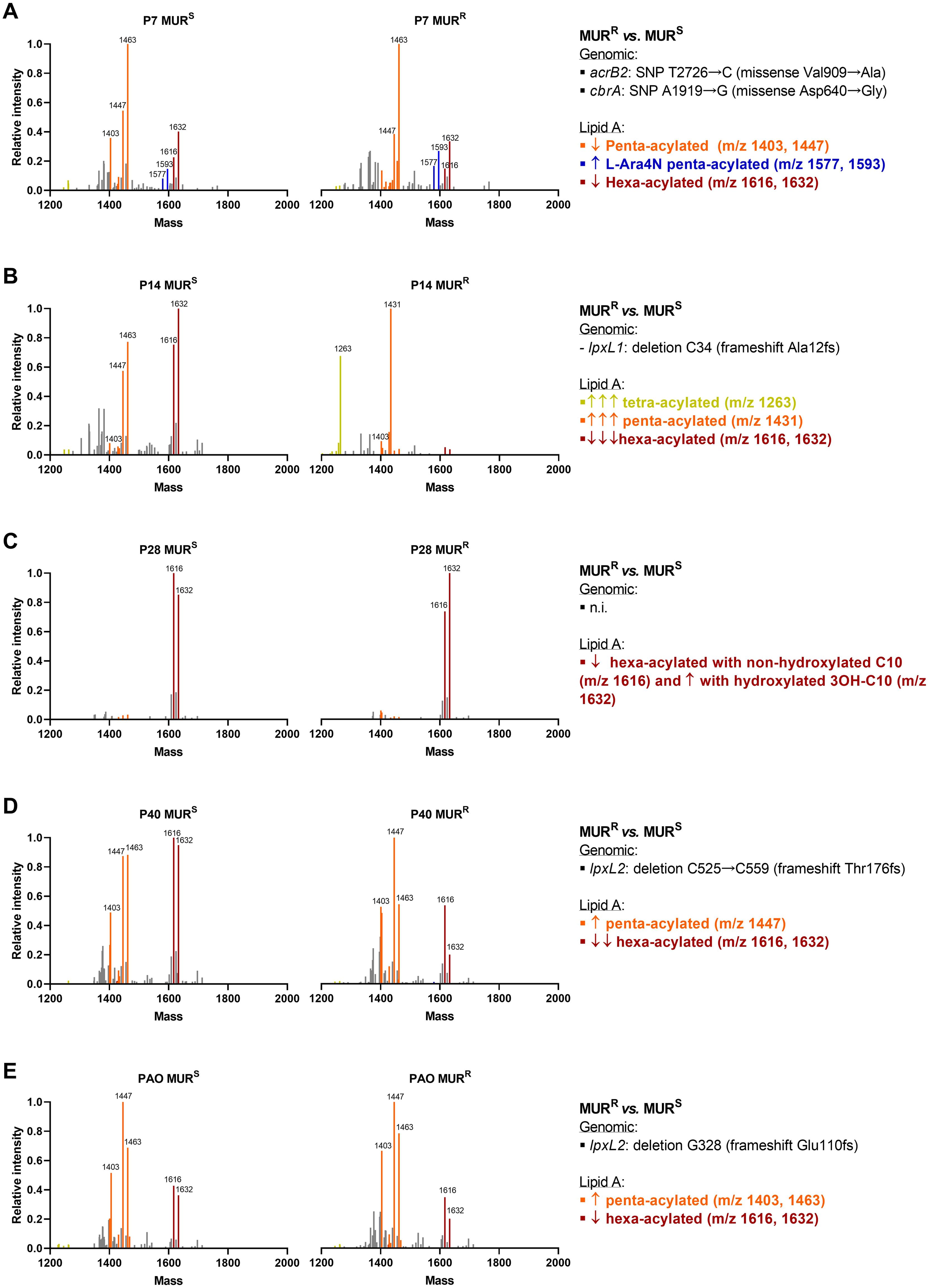
Figure 3. Lipid A profiles in the murepavadin resistant mutants (MURR) and murepavadin susceptible parental strains (MURS) of P7 (A), P14 (B), P28 (C), P40 (D) and PAO (E) strain pairs. Mass spectrum expressed in relative intensity against maximum intensity peaks of the 10 P. aeruginosa isolates. Peaks corresponding to tetra-acylated lipid A are in yellow, penta-acylated lipid A in orange, hexa-acylated lipid A in red and L-Ara4N modified lipid A in blue. n.i., not identified.
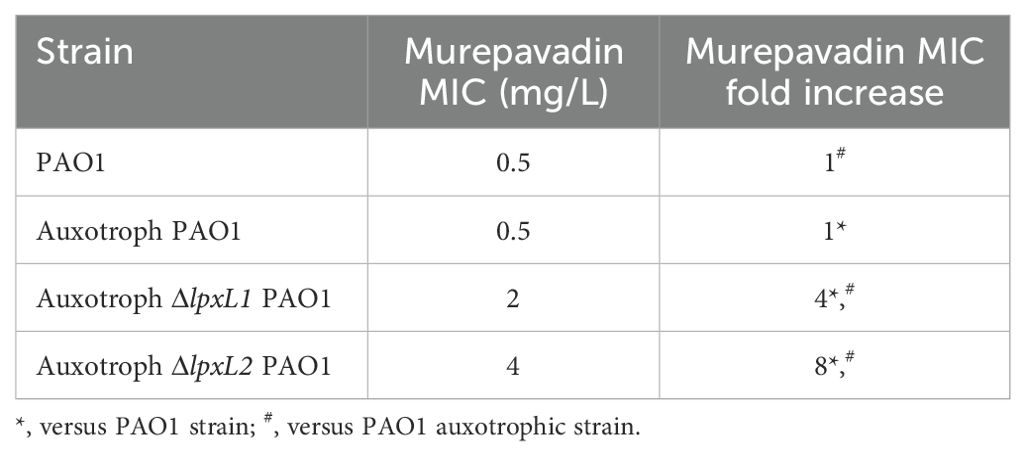
Table 1. Murepavadin MIC values of PAO1 and auxotroph derivative mutants.
3.4 Murepavadin-resistant mutants induce a lesser inflammatory response
Given that lipopolysaccharide is recognized by the immune system, the detected modifications of the murepavadin-resistant isolates could affect their inflammatory response. To measure the inflammatory response, human monocytes were stimulated with lipopolysaccharide from the 5 pairs of murepavadin-susceptible (parental) and murepavadin-resistant (mutant) isolates. The resulting murepavadin-resistant isolates appeared to induce lower expression of pro-inflammatory cytokines (tumor necrosis factor α, interleukin-1β and interleukin-6) although no clear pattern was found for interferon-γ (Figure 4). An exception was isolate P28, which had a small-colony-variant morphotype, and of which the resistant phenotype induced higher expression of cytokines. We confirmed that these differences were not due to apoptosis as we did not find differences when compared the apoptosis induction triggered by murepavadin-susceptible LPS to murepavadin-resistant LPS (Supplementary Figure S3).
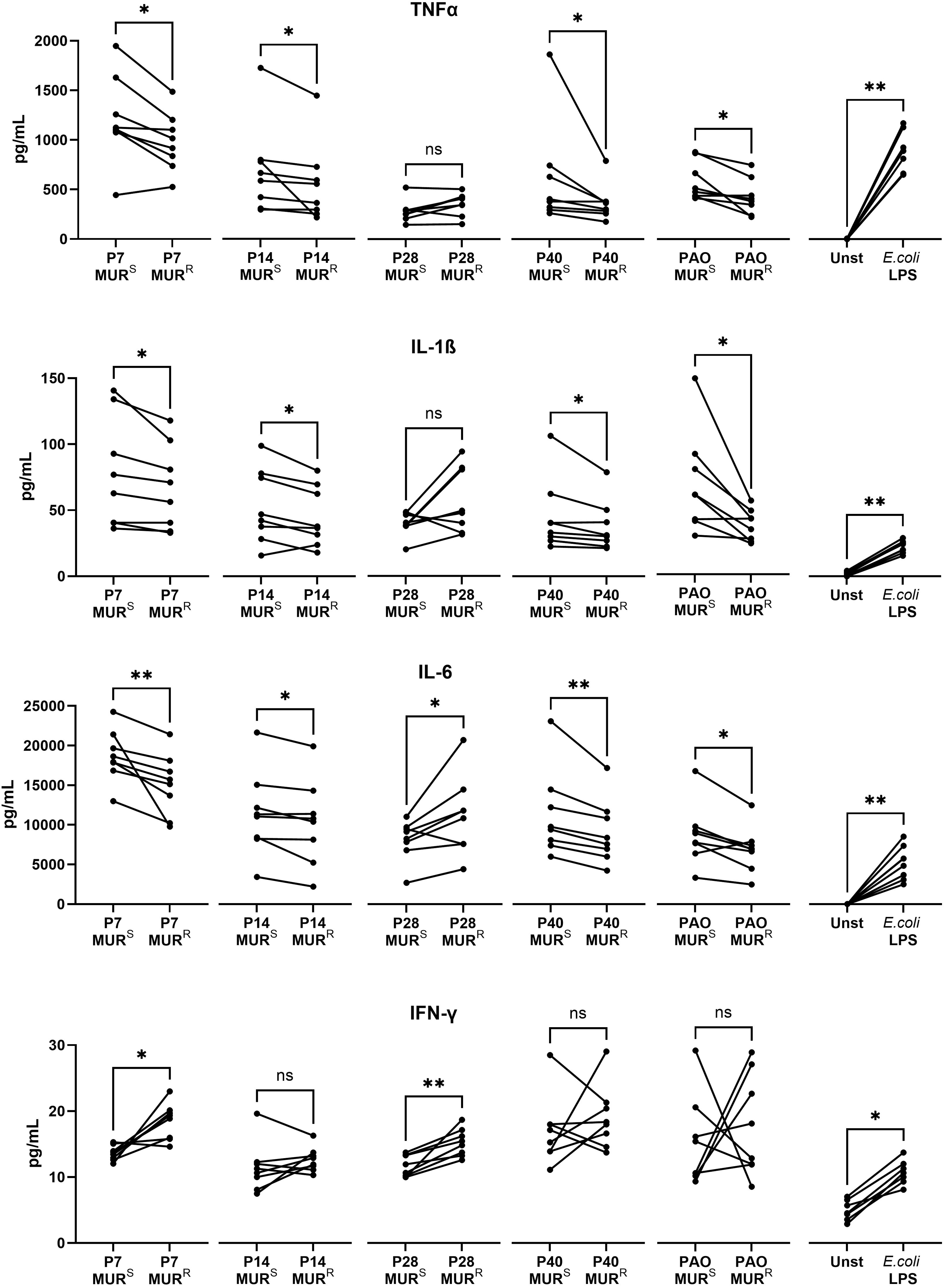
Figure 4. The lipopolysaccharide (LPS) from murepavadin-resistant strains (MURR) exhibit lower immunogenicity than their parental murepavadin susceptible strains (MURS). Human monocytes (from n=8 different healthy volunteers) were stimulated with 10 mg/L of purified LPS from MURR mutant and MURS parental strains strains for 16 h. Supernatant from unstimulated cells (Unst) and stimulated with 10 mg/L commercial E. coli O111:B4 LPS were used as negative and positive controls respectively. Levels of inflammatory cytokines TNFα, IL-1β, IL-6 and IFN-γ are shown. *p-value < 0.05; **p-value < 0.01; ns, no significant in Wilcoxon paired t-test.
3.5 Cross-resistance with human antimicrobial peptides
Growth differences between parental and mutant isolates were tested in presence of cathelicidin from human origin, and with cow neutrophil-derived indolicidin. A consistent delay in the growth of murepavadin-resistant P14 and P40 strains when exposed to human AMPs was observed when compared to their parental, murepavadin-susceptible counterparts (Figure 5). To note that these two strains exhibited lpxL mutations and a more pronounced loss of hexa-acylated lipid A forms (Figure 3).
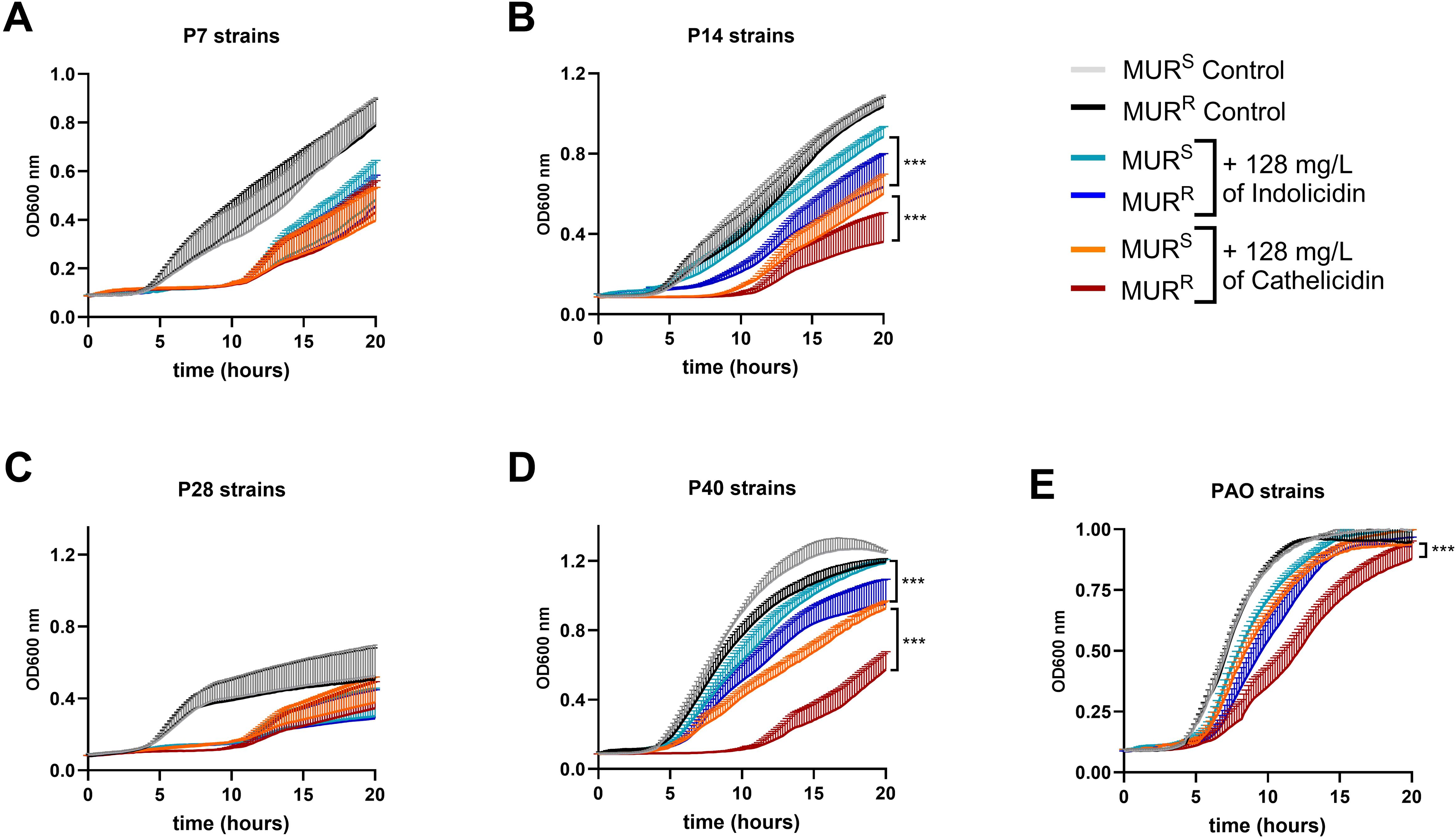
Figure 5. Murepavadin-resistant strains (MURR) strains are more susceptible to host-AMPs than the murepavadin susceptible strains (MURS). PA strains were grown in presence and absence of cathelicidin and indolicidin peptides (128 mg/L) and monitored by OD at 600 nm. (A-E) Growth curves of P7 (A), P14 (B), P28 (C), P40 (D), PAO1 (E) strains are shown. ***p-value < 0.001 in Two-way ANOVA and Tukey comparison test between murepavadin resistant mutants (MURR) and murepavadin susceptible parental strains (MURS) with the same treatment. Data expressed as median+SD from three independent replicates.
4 Discussion
Research on peptide-derived compounds has intensified in recent years as a consequence of increasing antimicrobial resistance. Murepavadin is a cyclic peptide active against P. aeruginosa, including strains that produce carbapenemases and/or those that are resistant to ceftolozane/tazobactam. This antibiotic has a narrow spectrum of action as specifically target the P. aeruginosa LptD thought its unique N-terminal domain, and thus it is considered an environment-friendly antimicrobial (Martin-Loeches et al., 2018; Robinson, 2019). Given that its resistance mechanisms have not yet been fully elucidated, we set out to analyze genetic and phenotypic variations in resistant P. aeruginosa isolates, including laboratory-induced resistant mutants. We complemented this investigation by comparing the immune responses and cross-resistance to human peptides of susceptible and resistant isolates.
The prediction of resistance using WGS could be crucial for the future development of murepavadin as treatment for multidrug-resistant P. aeruginosa, especially in colistin-resistant isolates. In the GWAS analysis, mutations significantly associated with colistin and/or murepavadin resistance were not observed within our collection of clinical isolates from several countries and with a differential genetic background. The main limitation of our study was the limited number of colistin-resistant isolates, which are rare in our countries. Nevertheless, our data confirm that this approach is feasible, although it requires a considerably larger number of bacterial genomes to reach good levels of statistical significance. Although GWAS has been widely used to investigate genotype-phenotype association in human (Uffelmann et al., 2021), its utility is less explored in microorganisms (Saber and Shapiro, 2020).
Our GWAS analysis revealed an SNP in hisJ associated with murepavadin resistance. Nevertheless, despite its association, this mutation was not a direct unique causative of murepavadin-resistance as some susceptible strains also harbored it. The hisJ gene codifies for a periplasmic binding protein involved in histidine transport and has never been associated with antibiotic resistance. A recent study showed that hisJ expression could be controlled by lasR in P. aeruginosa biofilm formation (Jeske et al., 2022). Additional studies are required to define the involvement of hisJ in antimicrobial resistance. Overall, our GWAS results manifested that there is not a unique solely genetic variation underlying murepavadin resistance and must be a combination of several mechanisms. This is another limitation of the GWAS study, since this analysis does not allow studying the effect of the accumulation of variants on resistance phenotypes.
Mutations identified by GWAS (hisJ) were different from those found in our in vitro-derived resistant mutant strains (lpxL1, lpxL2) or in previous reports (bamA, pmrB, msbA) (Romano et al., 2019; Díez-Aguilar et al., 2021; Ghassani et al., 2024). This difference could be explained by two factors. First, one of the limitations of our study is that there is no established MIC cut-off value for murepavadin. We defined a tentative cut-off of 0.25 mg/L finding 23.3% of resistant strains, while Ghassani et al. defined it as 4 mg/L with 9.1% of resistant strains. Our isolate collection came from patients with cystic fibrosis highly exposed to antibiotics, and the expression of resistance mechanisms such as efflux pumps could not be discarded. Consequently, the epidemiological cut-off value is likely higher than that in the wild-type and thus, we could overestimate the occurrence of the resistant phenotype. Additionally, the main difference between our study and previous studies is that GWAS is an untargeted analysis that includes mutations of diverse nature (SNP, insertions or deletions) in all genomes.
Cationic AMP molecules may interact with the bacterial outer membrane; in this manner, lipopolysaccharide modifications normally reduce their antimicrobial activity (Barrow and Kwon, 2009; Jochumsen et al., 2016; Bolard et al., 2019). Isolates with the addition of the L-Ara4N motifs in lipid A are resistant to colistin (Lo Sciuto et al., 2020). Interestingly, in the murepavadin-resistant mutants, we identified a modification of lipid A compared with the parental strains. This modification was related to a lower number of acyl chains, particularly in derived isolates with mutations in lpxL genes. This observation was evident in strains P14 and P40. Whereas the parental strains exhibited the highest ionic peak at m/z 1632, corresponding to a hexa-acylated form of lipid A, their murepavadin-resistant mutants exhibited the penta-acylated chains m/z 1431 and 1441 as the most abundant form. Another relevant finding is that the parental isolates exhibited more abundance of the hexa-acylated lipid A form than the laboratory strain PAO1, whose main ion was the penta-acylated m/z 1447. These results agree with those of Ernst et al. who defined PAO1 lipid A as penta-acylated at m/z 1447 (Ernst et al., 2006). This and other studies revealed that the number of acyl chains are key mechanisms to P. aeruginosa adaptation in the cystic fibrosis airway (Ernst et al., 2006, 2007; Thaipisuttikul et al., 2014). These types of changes also affect immune recognition, eliciting various immune responses (Ernst et al., 2003; Pier, 2007; SenGupta et al., 2016; Avendaño-Ortiz et al., 2019). Our results indicated that the loss of hexa-acylated and subsequent increments of penta-acylated lipid A forms in murepavadin-resistant strains leads to a reduced inflammatory response. A reduction in the number of acyl chains in lipid A (e.g., from hexa-acylated to penta-acylated or tetra-acylated) has been linked to decreased stimulation of Toll-like receptor 4 (TLR4) and lower cytokine production for example in E. coli strains, where less acylated lipid A variants induced lower levels of inflammatory cytokines (Li et al., 2013).This outcome has also been used to explain the changes of inflammatory induction between species, e.g., the lower pro-inflammatory potency of penta-acylated Granulibacter bethesdensis lipid A compared with Escherichia coli lipid A (Lench et al., 2021). Another explanation for lower cytokine production could be the induction of cell death, such as apoptosis (Kell and Pretorius, 2015; Monguió-Tortajada et al., 2018). Since we did not find differences in LPS-induced apoptosis between resistant and susceptible strains, we can speculate that the change in the number of acyl chains might be the most plausible mechanism to explain our results in the inflammation experiments. One of the interesting aspects of our results in the inflammation experiments is that, while the main inflammatory cytokines (TNFα, IL-1β, and IL-6) decrease in the murepavadin-resistant strains compared to the susceptible ones, the production of IFN-γ were higher in two of the murepavadin-resistant strains. Consistent with our findings, it has been reported that monocytes are capable of producing low amounts of IFN-γ in response to LPS (Kraaij et al., 2014). The controversy over the source of IFN-γ production in cultures with various cell types has always existed (Darwich et al., 2009). Due to our isolation method (adherence), the basal production of IFN-γ by some Th1 T-cells that remained in the culture should not be ruled out. However, the LPS stimulation time in our experiments (16 hours) makes an antigen-specific response almost impossible, as it requires antigen presentation and several days to occur (Ulmer et al., 2000; Wölfl and Greenberg, 2014).
The number of acyl chains could also be relevant in murepavadin resistance. In the 3 strains with resistance to murepavadin that present alterations in the number of acyl chains, we observed deletions in the lpxL1 and lpxL2 genes. The lpxL1 gene is responsible for adding a laurate acyl chain to the lipid A, whereas the lpxL2 gene adds a second laurate chain at a different position. These modifications are critical for maintaining the structural integrity and functionality of the bacterial outer membrane (Hittle et al., 2015). Both lpxL1 and lpxL2 contribute to the biosynthesis of a hexa-acylated form of lipid A, which is essential for the bacterial membrane’s resistance to environmental stress and antimicrobial agents. Deletion or mutation of these genes can lead to altered acyl chain patterns in lipid A, affecting the bacterium’s ability to adapt to different environments (Lo Sciuto et al., 2019). Here, we found that the PAO1 strains with lpxL1 and lpxL2 gene deletions exhibited MICs to murepavadin that were four to eight folds higher than the parental strain. Thus, our findings supports previous studies describing the association between lpxL1 and lpxL2 genes mutations and murepavadin resistance (Ghassani et al., 2024).
Modifications in the lipopolysaccharide directly affect the interaction with the immune system; we previously demonstrated the tolerance of cystic fibrosis isolates after the modification of their lipopolysaccharide (Ernst et al., 2007; SenGupta et al., 2016). Herein, the murepavadin-resistant mutants reduced the number of the lipid A acyl chains, which is probably related to the downregulation of the immune response. In addition, lipid A plays a crucial role in the resistance of Gram-negative bacteria to AMPs from human origin. Understanding the mechanisms of AMP resistance has important implications for antimicrobial therapy and the development of new antimicrobial agents (Kidd et al., 2017; Assoni et al., 2020). Boll et al. demonstrated that the addition of an acyl palmitate chain to lipid A in E. coli and Salmonella spp. was crucial for resistance against cathelicidin (Boll et al., 2015). In addition, Nizet and Gallo discussed how modification of the bacterial wall can decrease the attractiveness of human peptides (Nizet and Gallo, 2003). The increase of acylation in lipopolysaccharide lipid A reduces the positive charge of the cell wall, and the number and length of acyl chains in lipid A also affect its physical properties (Gorman and Golovanov, 2022). Our data indicate the number of acyl chains in lipid A plays an important role in the resistance of bacteria to cathelicidin and indolicidin. Lipid A acyl chains increase bacterial resistance to peptides, while the physical properties of lipid A, influenced by the number and arrangement of acyl chains, might affect its stability and interaction with the immune system.
According to our results, murepavadin-resistant mutants with loss of hexa-acyl chains are more susceptible to cathelicidin and indolicidin, which could be crucial to achieving pathogen killing by the combined action of various AMPs. Cathelicidin is a family of diverse AMPs whose main mechanism of action is the binding to the target membrane, followed by permeabilization and/or disruption of the membrane, ultimately leading to oxidative stress and bacterial death (de Barros et al., 2019; Rowe-Magnus et al., 2019). Cathelicidin can adopt various membrane interaction models, including the carpet model, barrel model, and toroidal pore model (de Barros et al., 2019). In the case of human cathelicidin, the preform hCAP18 is processed to LL-37, which is the active antimicrobial peptide (Yamasaki et al., 2006). Indolicidin is a bovine AMP that exhibits activity against a variety of Gram-positive and Gram-negative bacteria, as well as fungi (Batista Araujo et al., 2022). Its mechanism of action involves membrane permeabilization; however, it is still not fully understood, and various models have been proposed. Indolicidin has also been shown to damage DNA, leading to an SOS response in bacteria (Rowe-Magnus et al., 2019; Vasilchenko and Rogozhin, 2019); and additionally, indolicidin inhibits DNA synthesis, leading to bacterial filamentation (Marchand et al., 2006).
In summary, using a challenging collection of clinical isolates of P. aeruginosa from patients with cystic fibrosis, we did not identify specific mutations associated with murepavadin resistance, alone or in combination with colistin resistance. However, in a subset of isogenic-derived resistant mutants we found significant changes in their Lipid A, mainly the loss of an acyl chain. These changes could lead to an increased immunological tolerance, but also with increased susceptibility to host AMPs.
Data availability statement
The genome datasets presented in this study can be found in online repositories. The names of the repository and accession number(s) can be found below: https://ncbi.nlm.nih.gov/bioproject/, PRJNA527751 and PRJNA530912. All the other data from the study will be provided by corresponding authors under reasonable request.
Ethics statement
The studies involving humans were approved by the Hospital Ramón y Cajal Ethics Committee (project code 366-22, approved on 2023 February 28th). Data were treated according to recommended criteria of confidentiality and the study was conducted in accordance with the ethical guidelines of the 1975 Declaration of Helsinki. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from healthy donation excedents (blood samples), obtained from the Centro de Transfusiones de la Comunidad de Madrid in accordance with the protocol approved by the Hospital Ramón y Cajal Ethics Committee (project code 366-22). Ethical review and approval was not required for the bacteria isolate collection in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
MH-G: Writing – original draft, Conceptualization, Investigation, Methodology. RB-H: Writing – original draft, Conceptualization, Investigation, Methodology. NB-P: Writing – review & editing, Investigation, Methodology. MD-A: Writing – review & editing, Methodology. EL-C: Writing – review & editing, Formal analysis. FM-G: Writing – review & editing, Investigation, Methodology. JH-P: Writing – review & editing, Investigation, Methodology. FB: Writing – review & editing, Formal analysis, Methodology. ME: Writing – review & editing, Funding acquisition. AF: Writing – review & editing, Formal analysis, Funding acquisition. VF-V: Writing – review & editing, Methodology, Resources. MM: Writing – review & editing, Methodology, Supervision, Resources. GB: Writing – review & editing, Methodology, Formal analysis, Resources. RdC: Writing – original draft, Conceptualization, Investigation, Methodology. RC: Writing – review & editing, Conceptualization, Formal analysis, Funding acquisition, Project administration, Supervision. JA-O: Writing – review & editing, Conceptualization, Formal analysis, Project administration, Supervision.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Innovative Medicines Initiative (IMI)-European Commission-funded project (iABC grant 115721-2), composed of financial contribution from the European Union Seventh Framework Program (FP7/2007-2013) and EFPIA companies in kind contribution, by CIBER de Enfermedades Infecciosas CIBERINFEC (CB21/13/00084), by IMP23_C10 (2024/0020) from FIBioHRC and also by Personalized and Precision Medicine grant (MePRAM Project, PMP22/00092), Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación. Funded by NextGenerationEU funds from the European Union that finance the actions of the Resilience and Recovery Facility. JA-O has a Sara Borrell contract and a M-AES mobility grant from Instituto de Salud Carlos III (references CD21/00059 and MV23/00036). RC is the recipient of a grant PI19/01043 co-financed by European Development Regional Fund ERDF “Away to achieve Europe”, Operative program Intelligent Growth 2014-2020. MH-G is supported by a postdoctoral contract funded by CIBERINFEC (CB21/13/00084).
Acknowledgments
The author also want to thank Centro de transfusiones de la Comunidad de Madrid for the healthy blood supply for the in vitro experiments, and Carmen de la Vega for administrative and technical support.
Conflict of interest
Author FM-G is employee of Bruker Española S.A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1446626/full#supplementary-material
References
Amponnawarat, A., Chompunud Na Ayudhya, C., Ali, H. (2021).Murepavadin, a small molecule host defense peptide mimetic, activates mast cells via MRGPRX2 and mrgprB2 (Accessed July 20, 2023).
Andolina, G., Bencze, L.-C., Zerbe, K., Müller, M., Steinmann, J., Kocherla, H., et al. (2018). A peptidomimetic antibiotic interacts with the periplasmic domain of LptD from Pseudomonas aeruginosa. ACS Chem. Biol. 13, 666–675. doi: 10.1021/acschembio.7b00822
Assoni, L., Milani, B., Carvalho, M. R., Nepomuceno, L. N., Waz, N. T., Guerra, M. E. S., et al. (2020). Resistance mechanisms to antimicrobial peptides in Gram-positive bacteria. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.593215
Avendaño-Ortiz, J., Llanos-González, E., Toledano, V., Del Campo, R., Cubillos-Zapata, C., Lozano-Rodríguez, R., et al. (2019). Pseudomonas aeruginosa colonization causes PD-L1 overexpression on monocytes, impairing the adaptive immune response in patients with cystic fibrosis. J. Cyst. Fibros. 18, 630–635. doi: 10.1016/j.jcf.2018.11.002
Barrow, K., Kwon, D. H. (2009). Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 5150–5154. doi: 10.1128/AAC.00893-09
Batista Araujo, J., Sastre de Souza, G., Lorenzon, E. N. (2022). Indolicidin revisited: biological activity, potential applications and perspectives of an antimicrobial peptide not yet fully explored. World J. Microbiol. Biotechnol. 38, 39. doi: 10.1007/s11274-022-03227-2
Bokma, J., Vereecke, N., Nauwynck, H., Haesebrouck, F., Theuns, S., Pardon, B., et al. (2021). Genome-wide association study reveals genetic markers for antimicrobial resistance in Mycoplasma bovis. Microbiol. Spectr. 9, e0026221. doi: 10.1128/Spectrum.00262-21
Bolard, A., Schniederjans, M., Haüssler, S., Triponney, P., Valot, B., Plésiat, P., et al. (2019). Production of Norspermidine Contributes to Aminoglycoside Resistance in pmrAB Mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 63, e01044–e01019. doi: 10.1128/AAC.01044-19
Boll, J. M., Tucker, A. T., Klein, D. R., Beltran, A. M., Brodbelt, J. S., Davies, B. W., et al. (2015). Reinforcing lipid A acylation on the cell surface of Acinetobacter baumannii promotes cationic antimicrobial peptide resistance and desiccation survival. mBio 6. doi: 10.1128/mbio.00478-15
Cabral, M. P., Correia, A., Vilanova, M., Gärtner, F., Moscoso, M., García, P., et al. (2020). A live auxotrophic vaccine confers mucosal immunity and protection against lethal pneumonia caused by Pseudomonas aeruginosa. PloS Pathog. 16, e1008311. doi: 10.1371/journal.ppat.1008311
Darwich, L., Coma, G., Peña, R., Bellido, R., Blanco, E. J. J., Este, J. A., et al. (2009). Secretion of interferon-γ by human macrophages demonstrated at the single-cell level after costimulation with interleukin (IL)-12 plus IL-18. Immunology 126, 386–393. doi: 10.1111/j.1365-2567.2008.02905.x
de Barros, E., Gonçalves, R. M., Cardoso, M. H., Santos, N. C., Franco, O. L., Cândido, E. S. (2019). Snake venom cathelicidins as natural antimicrobial peptides (Accessed September 21, 2023).
del Fresno, C., García-Rio, F., Gómez-Piña, V., Soares-Schanoski, A., Fernández-Ruíz, I., Jurado, T., et al. (2009). Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J. Immunol. 182, 6494–6507. doi: 10.4049/jimmunol.0803350
Diaz Caballero, J., Clark, S. T., Wang, P. W., Donaldson, S. L., Coburn, B., Tullis, D. E., et al. (2018). A genome-wide association analysis reveals a potential role for recombination in the evolution of antimicrobial resistance in Burkholderia multivorans. PloS Pathog. 14, e1007453. doi: 10.1371/journal.ppat.1007453
Díez-Aguilar, M., Hernández-García, M., Morosini, M.-I., Fluit, A., Tunney, M. M., Huertas, N., et al. (2021). Murepavadin antimicrobial activity against and resistance development in cystic fibrosis Pseudomonas aeruginosa isolates. J. Antimicrob. Chemother. 76, 984–992. doi: 10.1093/jac/dkaa529
Ekkelenkamp, M. B., Cantón, R., Díez-Aguilar, M., Tunney, M. M., Gilpin, D. F., Bernardini, F., et al. (2020). Susceptibility of Pseudomonas aeruginosa recovered from cystic fibrosis patients to murepavadin and 13 comparator antibiotics. Antimicrob. Agents Chemother. 64, e01541–e01519. doi: 10.1128/AAC.01541-19
Ekkelenkamp, M. B., Díez-Aguilar, M., Tunney, M. M., Elborn, J. S., Fluit, A. C., Cantón, R. (2022). Establishing antimicrobial susceptibility testing methods and clinical breakpoints for inhaled antibiotic therapy. Open Forum Infect. Dis. 9, ofac082. doi: 10.1093/ofid/ofac082
Ernst, R. K., Adams, K. N., Moskowitz, S. M., Kraig, G. M., Kawasaki, K., Stead, C. M., et al. (2006). The Pseudomonas aeruginosa lipid A deacylase: selection for expression and loss within the cystic fibrosis airway. J. Bacteriol. 188, 191–201. doi: 10.1128/JB.188.1.191-201.2006
Ernst, R. K., Hajjar, A. M., Tsai, J. H., Moskowitz, S. M., Wilson, C. B., Miller, S. I. (2003). Pseudomonas aeruginosa lipid A diversity and its recognition by Toll-like receptor 4. J. Endotoxin Res. 9, 395–400. doi: 10.1179/096805103225002764
Ernst, R. K., Moskowitz, S. M., Emerson, J. C., Kraig, G. M., Adams, K. N., Harvey, M. D., et al. (2007). Unique lipid A modifications in Pseudomonas aeruginosa isolated from the airways of patients with cystic fibrosis. J. Infect. Dis. 196, 1088–1092. doi: 10.1086/521367
Falush, D. (2016). Bacterial genomics: Microbial GWAS coming of age. Nat. Microbiol. 1, 1–2. doi: 10.1038/nmicrobiol.2016.59
Fuentes-Valverde, V., García, P., Moscoso, M., Bou, G. (2022). Double auxotrophy to improve the safety of a live anti-Pseudomonas aeruginosa Vaccine. Vaccines (Basel) 10, 1622. doi: 10.3390/vaccines10101622
Ghassani, A., Triponney, P., Bour, M., Plésiat, P., Jeannot, K., MucoMicrobes study Group (2024). Mutations in genes lpxL1, bamA, and pmrB impair the susceptibility of cystic fibrosis strains of Pseudomonas aeruginosa to murepavadin. Antimicrob. Agents Chemother. 68, e0129823. doi: 10.1128/aac.01298-23
Gooderham, W. J., Bains, M., McPhee, J. B., Wiegand, I., Hancock, R. E. W. (2008). Induction by cationic antimicrobial peptides and involvement in intrinsic polymyxin and antimicrobial peptide resistance, biofilm formation, and swarming motility of PsrA in Pseudomonas aeruginosa. J. Bacteriol. 190, 5624–5634. doi: 10.1128/JB.00594-08
Gorman, A., Golovanov, A. P. (2022). Lipopolysaccharide structure and the phenomenon of low endotoxin recovery. Eur. J. Pharmaceut. Biopharmaceut. 180, 289–307. doi: 10.1016/j.ejpb.2022.10.006
Hernández-García, M., García-Castillo, M., García-Fernández, S., Melo-Cristino, J., Pinto, M. F., Gonçalves, E., et al. (2021). Distinct epidemiology and resistance mechanisms affecting ceftolozane/tazobactam in Pseudomonas aeruginosa isolates recovered from ICU patients in Spain and Portugal depicted by WGS. J. Antimicrobial Chemother. 76, 370–379. doi: 10.1093/jac/dkaa430
Hittle, L. E., Powell, D. A., Jones, J. W., Tofigh, M., Goodlett, D. R., Moskowitz, S. M., et al. (2015). Site-specific activity of the acyltransferases HtrB1 and HtrB2 in Pseudomonas aeruginosa lipid A biosynthesis. Pathog. Dis. 73, ftv053. doi: 10.1093/femspd/ftv053
Huan, Y., Kong, Q., Mou, H., Yi, H. (2020).Antimicrobial Peptides: Classification, design, application and research progress in multiple fields (Accessed July 24, 2023).
Jeske, A., Arce-Rodriguez, A., Thöming, J. G., Tomasch, J., Häussler, S. (2022). Evolution of biofilm-adapted gene expression profiles in lasR-deficient clinical Pseudomonas aeruginosa isolates. NPJ Biofilms Microbiomes 8, 6. doi: 10.1038/s41522-022-00268-1
Jochumsen, N., Marvig, R. L., Damkiær, S., Jensen, R. L., Paulander, W., Molin, S., et al. (2016). The evolution of antimicrobial peptide resistance in Pseudomonas aeruginosa is shaped by strong epistatic interactions. Nat. Commun. 7, 13002. doi: 10.1038/ncomms13002
Kell, D. B., Pretorius, E. (2015). On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr. Biol. 7, 1339–1377. doi: 10.1039/c5ib00158g
Kidd, T. J., Mills, G., Sá-Pessoa, J., Dumigan, A., Frank, C. G., Insua, J. L., et al. (2017). A Klebsiella pneumoniae antibiotic resistance mechanism that subdues host defences and promotes virulence. EMBO Mol. Med. 9, 430–447. doi: 10.15252/emmm.201607336
Kraaij, M. D., Vereyken, E. J. F., Leenen, P. J. M., van den Bosch, T. P. P., Rezaee, F., Betjes, M. G. H., et al. (2014). Human monocytes produce interferon-γ upon stimulation with LPS. Cytokine 67, 7–12. doi: 10.1016/j.cyto.2014.02.001
Lench, A., Zarember, K. A., Heiss, C., Shiloach, J., Berg, L. J., Audley, J., et al. (2021). Granulibacter bethesdensis, a pathogen from patients with chronic granulomatous disease, produces a penta-acylated hypostimulatory glycero-D-talo-oct-2-ulosonic acid–lipid A glycolipid (Ko-Lipid A). Int. J. Mol. Sci. 22, 3303. doi: 10.3390/ijms22073303
Li, Y., Wang, Z., Chen, J., Ernst, R. K., Wang, X. (2013). Influence of lipid A acylation pattern on membrane permeability and innate immune stimulation. Mar. Drugs 11, 3197–3208. doi: 10.3390/md11093197
Lo Sciuto, A., Cervoni, M., Stefanelli, R., Mancone, C., Imperi, F. (2020). Effect of lipid A aminoarabinosylation on Pseudomonas aeruginosa colistin resistance and fitness. Int. J. Antimicrob. Agents 55, 105957. doi: 10.1016/j.ijantimicag.2020.105957
Lo Sciuto, A., Cervoni, M., Stefanelli, R., Spinnato, M. C., Di Giamberardino, A., Mancone, C., et al. (2019). Genetic basis and physiological effects of lipid A hydroxylation in Pseudomonas aeruginosa PAO1. Pathogens 8, 291. doi: 10.3390/pathogens8040291
Marchand, C., Krajewski, K., Lee, H.-F., Antony, S., Johnson, A. A., Amin, R., et al. (2006). Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 34, 5157–5165. doi: 10.1093/nar/gkl667
Martin-Loeches, I., Dale, G. E., Torres, A. (2018). Murepavadin: a new antibiotic class in the pipeline. Expert Rev. Anti Infect. Ther. 16, 259–268. doi: 10.1080/14787210.2018.1441024
Mba, I. E., Nweze, E. I. (2022). Antimicrobial peptides therapy: an emerging alternative for treating drug-resistant Bacteria. Yale J. Biol. Med. 95, 445–463.
Mohammed, I., Said, D. G., Nubile, M., Mastropasqua, L., Dua, H. S. (2019). Cathelicidin-derived synthetic peptide improves therapeutic potential of vancomycin against Pseudomonas aeruginosa. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02190
Monguió-Tortajada, M., Franquesa, M., Sarrias, M.-R., Borràs, F. E. (2018). Low doses of LPS exacerbate the inflammatory response and trigger death on TLR3-primed human monocytes. Cell Death Dis. 9, 499. doi: 10.1038/s41419-018-0520-2
Nizet, V., Gallo, R. L. (2003). Cathelicidins and innate defense against invasive bacterial infection. Scandinavian J. Infect. Dis. 35, 670–676. doi: 10.1080/00365540310015629
Nowicki, E. M., O’Brien, J. P., Brodbelt, J. S., Trent, M. S. (2015). Extracellular zinc induces phosphoethanolamine addition to Pseudomonas aeruginosa lipid A via the ColRS two-component system. Mol. Microbiol. 97, 166. doi: 10.1111/mmi.13018
Pier, G. B. (2007). Pseudomonas aeruginosa lipopolysaccharide: A major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol. 297, 277–295. doi: 10.1016/j.ijmm.2007.03.012
Robinson, J. A. (2019). Folded synthetic peptides and other molecules targeting outer membrane protein complexes in Gram-negative bacteria. Front. Chem. 7. doi: 10.3389/fchem.2019.00045
Romano, K. P., Warrier, T., Poulsen, B. E., Nguyen, P. H., Loftis, A. R., Saebi, A., et al. (2019). Mutations in pmrB confer cross-resistance between the LptD Inhibitor POL7080 and colistin in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 63, e00511–e00519. doi: 10.1128/AAC.00511-19
Rowe-Magnus, D. A., Kao, A. Y., Prieto, A. C., Pu, M., Kao, C. (2019). Cathelicidin peptides restrict bacterial growth via membrane perturbation and induction of reactive oxygen species. mBio 10, e02021–e02019. doi: 10.1128/mBio.02021-19
Saber, M. M., Shapiro, B. J. (2020). Benchmarking bacterial genome-wide association study methods using simulated genomes and phenotypes. Microb. Genom 6, e000337. doi: 10.1099/mgen.0.000337
SenGupta, S., Hittle, L. E., Ernst, R. K., Uriarte, S. M., Mitchell, T. C. (2016). A Pseudomonas aeruginosa hepta-acylated lipid A variant associated with cystic fibrosis selectively activates human neutrophils. J. Leukoc. Biol. 100, 1047–1059. doi: 10.1189/jlb.4VMA0316-101R
Snesrud, E., McGann, P., Chandler, M. (2018). The Birth and Demise of the ISApl1-mcr-1-ISApl1 composite transposon: the vehicle for transferable colistin resistance. mBio 9. doi: 10.1128/mbio.02381-17
Talapko, J., Meštrović, T., Juzbašić, M., Tomas, M., Erić, S., Aleksijević, L. H., et al. (2022). Antimicrobial peptides—mechanisms of action, antimicrobial effects and clinical applications. Antibiotics 11. doi: 10.3390/antibiotics11101417
Thaipisuttikul, I., Hittle, L. E., Chandra, R., Zangari, D., Dixon, C. L., Garrett, T. A., et al. (2014). A divergent Pseudomonas aeruginosa palmitoyltransferase essential for cystic fibrosis-specific lipid A. Mol. Microbiol. 91, 158–174. doi: 10.1111/mmi.12451
The European Committee on Antimicrobial Susceptibility Testing (2022). Breakpoint tables for interpretation of MICs and zone diameters. Version 12.0, 2022 (http://www.eucast.org). (Sweden: EUCAST Development Laboratory, Central Hospital Växjö). Available at: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_12.0_Breakpoint_tables.pdf.
Uffelmann, E., Huang, Q. Q., Munung, N. S., de Vries, J., Okada, Y., Martin, A. R., et al. (2021). Genome-wide association studies. Nat. Rev. Methods Primers 1, 1–21. doi: 10.1038/s43586-021-00056-9
Ulmer, A. J., Flad, H., Rietschel, T., Mattern, T. (2000). Induction of proliferation and cytokine production in human T lymphocytes by lipopolysaccharide (LPS). Toxicology 152, 37–45. doi: 10.1016/s0300-483x(00)00290-0
Vasilchenko, A. S., Rogozhin, E. A. (2019). Sub-inhibitory effects of antimicrobial peptides. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01160
Wang, G., Mechesso, A. F. (2022). Realistic and critical review of the state of systemic antimicrobial peptides. ADMET DMPK 10, 91–105. doi: 10.5599/admet.1215
Wölfl, M., Greenberg, P. D. (2014). Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat. Protoc. 9, 950–966. doi: 10.1038/nprot.2014.064
Xiao, Q., Luo, Y., Shi, W., Lu, Y., Xiong, R., Wu, X., et al. (2022). The effects of LL-37 on virulence factors related to the quorum sensing system of Pseudomonas aeruginosa. Ann. Trans. Med. 10, 284–284. doi: 10.21037/atm-22-617
Yamasaki, K., Schauber, J., Coda, A., Lin, H., Dorschner, R. A., Schechter, N. M., et al. (2006). Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 20, 2068–2080. doi: 10.1096/fj.06-6075com
Young, B. C., Wu, C.-H., Charlesworth, J., Earle, S., Price, J. R., Gordon, N. C., et al. (2021). Antimicrobial resistance determinants are associated with Staphylococcus aureus bacteraemia and adaptation to the healthcare environment: a bacterial genome-wide association study. Microb. Genom 7, 700. doi: 10.1099/mgen.0.000700
Keywords: murepavadin, colistin, antibiotic resistance, host-derived antimicrobial peptides, monocytes, innate immune system
Citation: Hernández-García M, Barbero-Herranz R, Bastón-Paz N, Díez-Aguilar M, López-Collazo E, Márquez-Garrido FJ, Hernández-Pérez JM, Baquero F, Ekkelenkamp MB, Fluit AC, Fuentes-Valverde V, Moscoso M, Bou G, del Campo R, Cantón R and Avendaño-Ortiz J (2024) Unravelling the mechanisms causing murepavadin resistance in Pseudomonas aeruginosa: lipopolysaccharide alterations and its consequences. Front. Cell. Infect. Microbiol. 14:1446626. doi: 10.3389/fcimb.2024.1446626
Received: 10 June 2024; Accepted: 18 November 2024;
Published: 06 December 2024.
Edited by:
Bennett Tochukwu Amaechi, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Congran Li, Chinese Academy of Medical Sciences, ChinaRajesh Kumar Radhakrishnan, Saint Louis University, United States
Copyright © 2024 Hernández-García, Barbero-Herranz, Bastón-Paz, Díez-Aguilar, López-Collazo, Márquez-Garrido, Hernández-Pérez, Baquero, Ekkelenkamp, Fluit, Fuentes-Valverde, Moscoso, Bou, del Campo, Cantón and Avendaño-Ortiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Cantón, cmFmYWVsLmNhbnRvbkBzYWx1ZC5tYWRyaWQub3Jn; José Avendaño-Ortiz, am9zZWF2ZW5vcnRAZ21haWwuY29t
†These authors have share first authorship