
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 29 April 2024
Sec. Parasite and Host
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1385599
This article is part of the Research Topic Rising Stars in Parasite and Host: 2023 View all 7 articles
 Sandrine Musa1*
Sandrine Musa1* Theo Hemberle1
Theo Hemberle1 Staffan Bensch2
Staffan Bensch2 Vaidas Palinauskas3
Vaidas Palinauskas3 Laima Baltrūnaitė3
Laima Baltrūnaitė3 Friederike Woog4Ute Mackenstedt1
Friederike Woog4Ute Mackenstedt1Avian haemosporidian parasites are useful model organisms to study the ecology and evolution of parasite-host interactions due to their global distribution and extensive biodiversity. Detection of these parasites has evolved from microscopic examination to PCR-based methods, with the mitochondrial cytochrome b gene serving as barcoding region. However, standard PCR protocols used for screening and identification purposes have limitations in detecting mixed infections and generating phylogenetically informative data due to short amplicon lengths. To address these issues, we developed a novel genus-specific nested PCR protocol targeting avian haemosporidian parasites. The protocol underwent rigorous testing utilizing a large dataset comprising blood samples from Malagasy birds of three distinct Passeriformes families. Furthermore, validation was done by examining smaller datasets in two other laboratories employing divergent master mixes and different bird species. Comparative analyses were conducted between the outcomes of the novel PCR protocol and those obtained through the widely used standard nested PCR method. The novel protocol enables specific identification of Plasmodium, Haemoproteus (Parahaemoproteus), and Leucocytozoon parasites. The analyses demonstrated comparable sensitivity to the standard nested PCR with notable improvements in detecting mixed infections. In addition, phylogenetic resolution is improved by amplification of longer fragments, leading to a better understanding of the haemosporidian biodiversity and evolution. Overall, the novel protocol represents a valuable addition to avian haemosporidian detection methodologies, facilitating comprehensive studies on parasite ecology, epidemiology, and evolution.
The order Haemosporida, classified within the Apicomplexa, encompasses an important group of pathogens across all vertebrate groups. These unicellular blood parasites exhibit a heteroxenous life cycle, which requires alternation between vertebrate hosts and insect vectors (blood-sucking dipterans). In phylogenetic terms, avian Haemosporida of the genera Plasmodium, Haemoproteus, and Leucocytozoon are considered as the most ancient represantatives within their respective genera, signifying their ancestral status among vertebrate haemosporidian parasites (Pacheco et al., 2018a). Given their global distribution and substantial biodiversity (Clark et al., 2014), these genera are of scientific interest as model organisms to study the ecology and evolution of this parasite-host system (e.g. Bensch et al., 2000; Marzal et al., 2008; Pacheco et al., 2018a). Haemosporidian infections are often asymptomatic, can persist in the chronic phase within bird hosts over years (Valkiūnas, 2005), and the occurrence of mixed infections involving different genera or species of the same genus is common (Valkiūnas et al., 2006).
Before 2000, the detection of these parasites primarily relied on the microscopic examination of blood smears (Valkiūnas, 2005). However, with the emergence of molecular methods, PCR-based parasite detection gained popularity (Bensch et al., 2009). A fragment of the mitochondrial cytochrome b (cyt b) gene was identified as the target fragment that is now commonly used as a barcoding region of avian haemosporidian parasites. The identification is supported by the MalAvi database (Bensch et al., 2009), which contains data of over 5,000 unique avian haemosporidian lineages. The widely used detection method, a nested PCR targeting the barcoding region (Hellgren et al., 2004), is well-suited for simple screening due to its high sensitivity, although limitations have already been discovered in the detection of Haemosporida from Accipitridae (Harl et al., 2022, 2024). Additionally, there exist two further limitations: a short target fragment (479 bp), which limits its suitability for phylogenetic work, and insufficient detection of mixed infections with parasites of different genera (Bernotiene et al., 2016).
To address these limitations, alternative PCR protocols have been introduced. For instance, primer sets that enable amplifying longer gene fragments (Perkins and Schall, 2002; Schmid et al., 2017), or the entire mitochondrial genome (Pacheco et al., 2018a; Musa, 2023), are more suitable for phylogenetic studies. The inability of the standard detection method to adequately identify mixed infections is due to the primer pairs used. While there is a specific primer pair for Leucocytozoon, there is only one other pair that detects both Plasmodium and Haemoproteus parasites. In mixed infections, this often leads to one parasite genus being preferentially amplified due to the higher DNA concentration or more sensitive primer binding. Additional molecular methods, such as multiplex PCRs (Pacheco et al., 2018b; Ciloglu et al., 2019), have been introduced to uncover those mixed infections. Unfortunately, the target gene fragments of those methods cannot be used for lineage-level identification, as they only partially cover the barcoding region or not at all.
In response to these limitations, this study aims to devise a novel protocol using genus-specific primer sets for identifying avian haemosporidian parasites. This method is intended to ensure genus-specific amplification of the cyt b barcode, and still enable reliable identification of mixed infections between Plasmodium and Haemoproteus at the lineage level for the first time. The target fragment should also be larger than just the barcoding region to allow an improvement of phylogenetic analyses. Finally, the protocol should be designed in a way that is neither more complex nor cost-intensive compared to the standard method. The sensitivity and specificity of this new nested PCR assay, in comparison to the standard detection protocols, should be tested using different sample sets from different laboratories.
Using Geneious Prime 2023.2.1, a multiple alignment of 51 whole mitochondrial (mtDNA) genome sequences of avian Plasmodium, Haemoproteus, and Leucocytozoon isolates was prepared (list of accession numbers given in Supplementary Table 1). The sequences were derived from publications by Pacheco et al. (2018a) and Musa (2023). A manual search was performed for sequence motifs that occur in all genera and can be used as outer primers, as well as for motifs that are unique for each parasite genus to ensure genus-specific amplification in the nested PCR. The target fragment of all nested PCRs should be of comparable size and should contain the cyt b barcoding region. Since the subgenus Haemoproteus showed distinct sequence differences, different primer pairs were designed for the subgenera of Haemoproteus. Due to a lack of sample material for the evaluation of these primers, the primers of the subgenus Haemoproteus were not considered further in this study and only the primers for the subgenus Parahaemoproteus were evaluated. The binding sites of primers and sizes of the amplified fragments are shown in Figure 1.
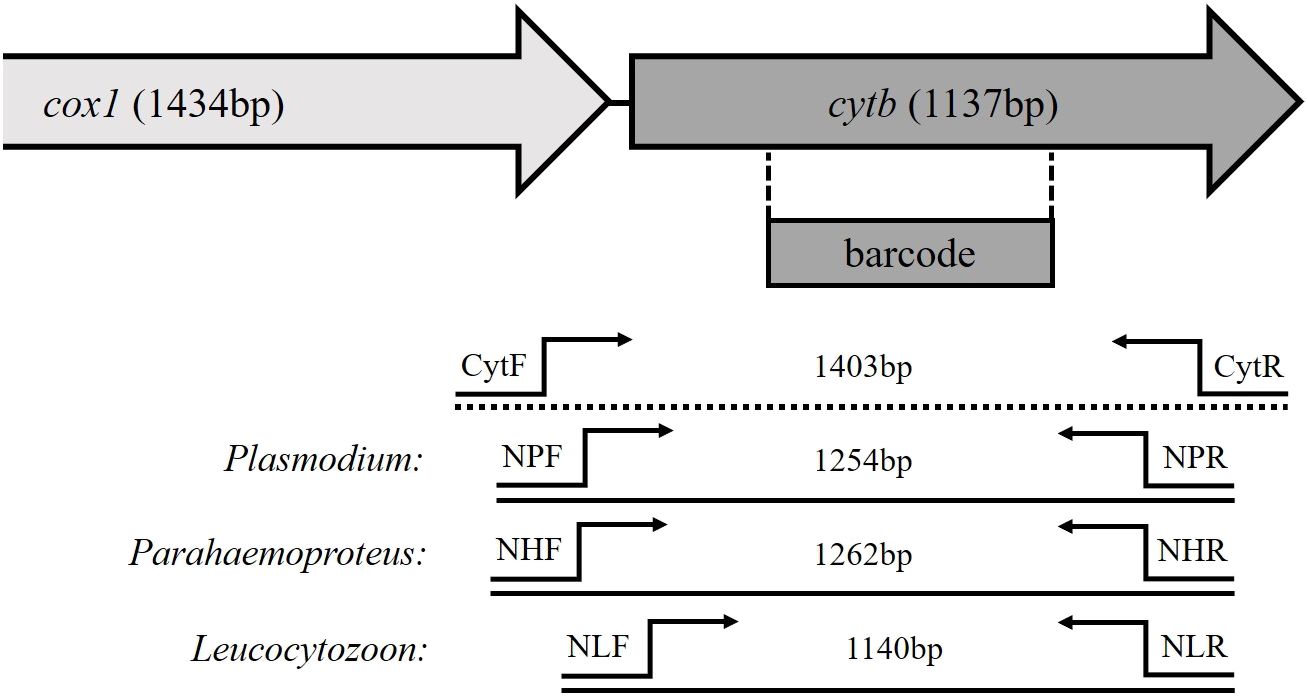
Figure 1 Schematic structure of partial mitochondrial genes of avian haemosporidian parasites. The location of the barcoding region is marked, as well as the primer binding sites of the newly developed genus-specific nested PCR and the length of their amplification products.
The primary dataset of this study consisted of 264 blood samples of Malagasy bird species (Table 1) collected between 2003 and 2022 as part of an ongoing ringing project in the Maromizaha rainforest in eastern Madagascar.
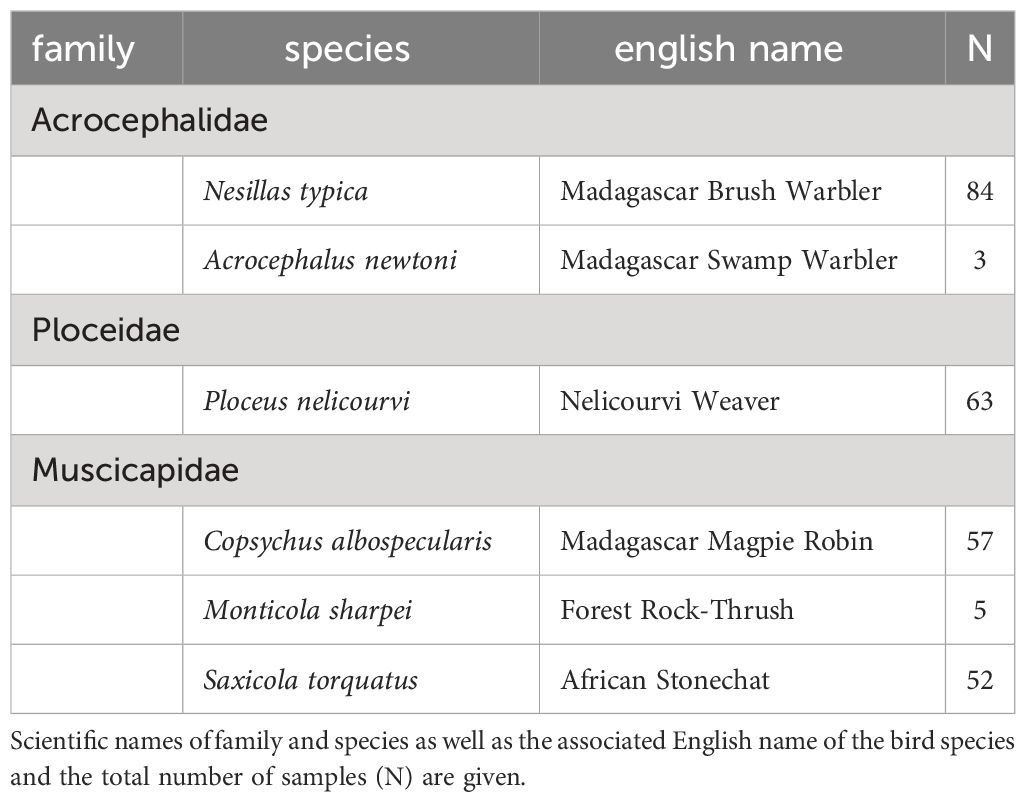
Table 1 Primary dataset comprising 264 Malagasy bird blood samples from different avian taxa.
The birds were caught in mist nets and a drop of blood was taken by puncturing the brachial vein. The protocol was approved by the Direction de la Préservation de la Biodiversité, Antananarivo, Madagascar. The blood was immediately stored in lysis buffer (Wink, 2006). Total DNA was extracted from the blood samples using the innuPREP Blood DNA Mini Kit (IST Innuscreen GmbH, Berlin, Germany) according to the manufacturer’s instructions. DNA concentration and purity were quantified using a NanoDrop N50 UV-Vis spectrophotometer (Implen GmbH, Munich, Germany) and samples were stored at –20°C until further use.
Two smaller datasets of naturally infected passerine birds were additionally tested, one at the Nature Research Centre, Vilnius, Lithuania and the other at the Molecular Ecology and Evolution Laboratory at Lund University in Lund, Sweden. The first dataset consisted of 54 blood samples collected at the Ventes Ragas ornithological station, Lithuania in 2019 – 2023 and the second of 52 samples collected at lake Krankesjön, Sweden in 2014-2017. For both these datasets, the blood was taken as described above from the brachial vein and stored in SET-buffer (0.05 M Tris, 0.15 M NaCl, 0.001 M EDTA, pH 8.0) (Hellgren et al., 2004). Total DNA extraction of blood samples was performed using an ammonium acetate protocol (Sambrook and Russel, 2001) or Quick-DNA Mini Prep (Zymo Research) DNA extraction kit and stored at –20°C until further use. The Swedish data set was previously analysed within a large host-parasite community study (Ellis et al., 2020) and because the extracted samples had been exposed to repeated thawing and freezing, the DNA might have become degraded. Therefore, DNA was re-extracted from ten samples that failed parasite detection in the first round of analyses.
To evaluate the sensitivity and specificity of the new nested PCR assay in comparison to the broadly used standard diagnostic method, the blood samples of the Malagasy birds were at first screened for haemosporidian parasites using the following protocol. Each sample was separately tested for the presence of Plasmodium spp./Haemoproteus spp. using the primers HAEMNF/HAEMNR2 (Waldenström et al., 2004) and HAEMF/HAEMR2 (Bensch et al., 2000) and Leucocytozoon spp. using the specific primers HAEMNF/HAEMNR3 and HAEMFL/HAEMR2L (Hellgren et al., 2004). Reaction mixtures of the separate nested PCRs were identical. PCR reactions of the first run were carried out in a total volume of 25 μl containing 2.5 μl10X ReproFast Buffer with 20mM MgSO4 (Genaxxon bioscience GmbH, Ulm, Germany), 1 μl of each primer (HAEMNF/HAEMNR2 or HAEMNF/HAEMNR3; 10 mM), 0.5 μl of each dNTP (10 μmol), 0.125 μl ReproFast-DNA Polymerase (proofreading; 5 U/μl; Genaxxon bioscience GmbH, Ulm, Germany), 5 μl template DNA (10–100 ng/μl) and 14.875 μl nuclease-free water. The PCR amplification protocol started with an initial denaturation step of 94°C for 5 min, followed by 20 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s and extension at 72°C for 45 s. The final extension takes place at 72°C for 5 min. The reaction mixture of the nested PCRs consisted of 5 μl10X ReproFast Buffer with 20mM MgSO4 (Genaxxon bioscience GmbH, Ulm, Germany), 2 μl of each primer (HAEMF/HAEMR2 or HAEMFL/HAEMR2L; 10 mM), 1 μl of each dNTP (10 μmol), 0.25 µl ReproFast-DNA Polymerase (proofreading; 5 U/μl; Genaxxon bioscience GmbH, Ulm, Germany), 2 μl amplification product of the initial PCR and 37.75 μl nuclease-free water in a total volume of 50 μl. The cycling conditions included an initial denaturation step of 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 50°C (HAEMFL/HAEMR2L) or 55°C (HAEMF/HAEMR2) for 30 s and extension at 72°C for 45 s. The final extension was carried out at 72°C for 5 min.
Lithuanian samples were tested with the same primer sets as Malagasy birds using conditions described by Bensch et al. (2000) and Hellgren et al. (2004). PCR reactions were carried out in a total volume of 25 μl, in the first reaction containing 12.5 μl Dream Taq Green Master Mix (2X) (Thermo Fischer Scientific, Lithuania), 8.5 μl of ultra-pure water, 1 μl of each primer, and 2 μl of template DNA. 2 μl of the first reaction product was used as a DNA template in the second reaction.
The Swedish samples had previously been analysed (Ellis et al., 2020) following the same procedure as for the Lithuanian samples with the only difference in the use of DNA polymerase (AmpliTaq© DNA Polymerase, applied biosystems). Before testing the 52 samples with the GS-protocol, the samples were screened (in September 2023) using a multiplex PCR assay (Ciloglu et al., 2019) to verify the infections recorded by Ellis et al. (2020).
The newly established genus-specific nested PCR was performed running an unspecific PCR in the first place using the primers CytF (5´-GGARCAATAATTGSATTATTTAC-3´) and CytR (5´-TGCTTGDGAGCTGTAATCAT-3´) amplifying a fragment of 1,403bp, following by three separate, genus-specific nested PCRs using the amplification product of the first PCR as template. The primer pair NPF (5´-CGTGAAAAYTCAATAATAATAYTATGGTC-3´)/NPR (5´-AACGACCATATAAAATGTAAATATC-3´) was used for the detection of Plasmodium (1,254bp), NHF (5´-TAAACATTTACGTGATAATACAATAATTG-3´)/NHR (5´-CGACCATATAAAATGAAAATAGA-3´) for Haemoproteus (Parahaemoproteus; 1,262bp) and the primers NLF (5´-GACGTATACCTGATTATCCTGATAA-3´)/NLR (5´-TTTGTGGTAATTGATAACCTATAMMCAT-3´) for Leucocytozoon detection (1,1140bp). The reaction mixtures of the different PCRs were similar to those of the standard nested PCR protocol described above. Cycling conditions of the different PCRs are given in Table 2. Reaction mixtures of the PCRs for Lithuanian samples were using Dream Taq Green (2X) Master Mix (Thermo Fischer Scientific, Lithuania) as with standard protocol and following thermal conditions indicated in Table 2.
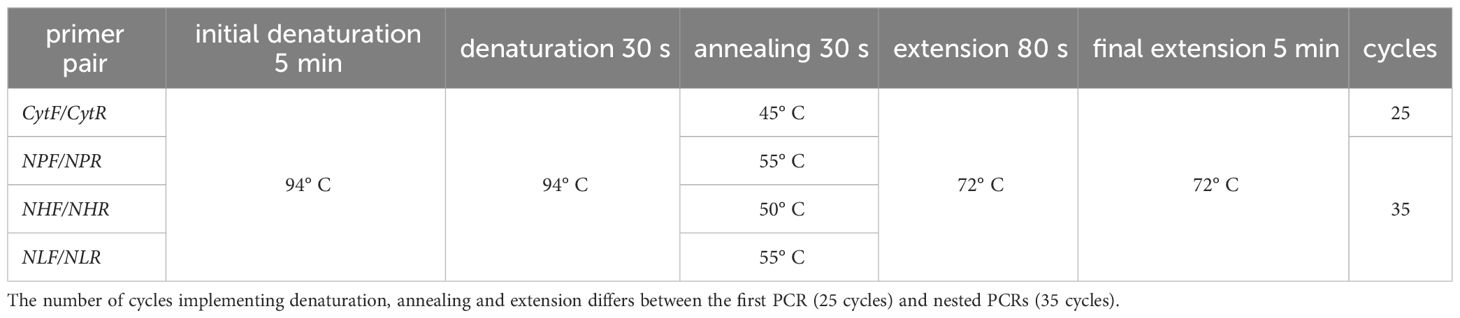
Table 2 Cycle conditions of newly established genus-specific nested PCR. First PCR was performed using primer pair CytF/CytR, whereas nested PCRs use genus-specific primer pairs (NPF/NPR for Plasmodium spp., NHF/NHR for Haemoproteus (Parahaemoproteus) spp. and NLF/NLR for Leucocytozoon spp.).
The amplification products (5 μl) of all nested PCR assays were mixed with GelRed™ stain and a loading buffer and then visualized on a 1.5% agarose gel after 20 min at 90 V. Amplification products (Figure 2) were then purified using the PCR Product Purification Kit (Roche, Mannheim, Germany) and after sequencing (Microsynth AG, Switzerland), the resulting sequence data were edited using Geneious v. 2021.1.1. The final sequences were then distinguished by identifying their closest match using a BLAST search of the MalAvi database (Bensch et al., 2009); as of November 2023. All newly detected lineages were deposited in GenBank (accession numbers OR901950-OR901956). To minimize false-negative results, the PCR was repeated for each sample that showed either no band in the agarose gel or insufficient sequencing results.
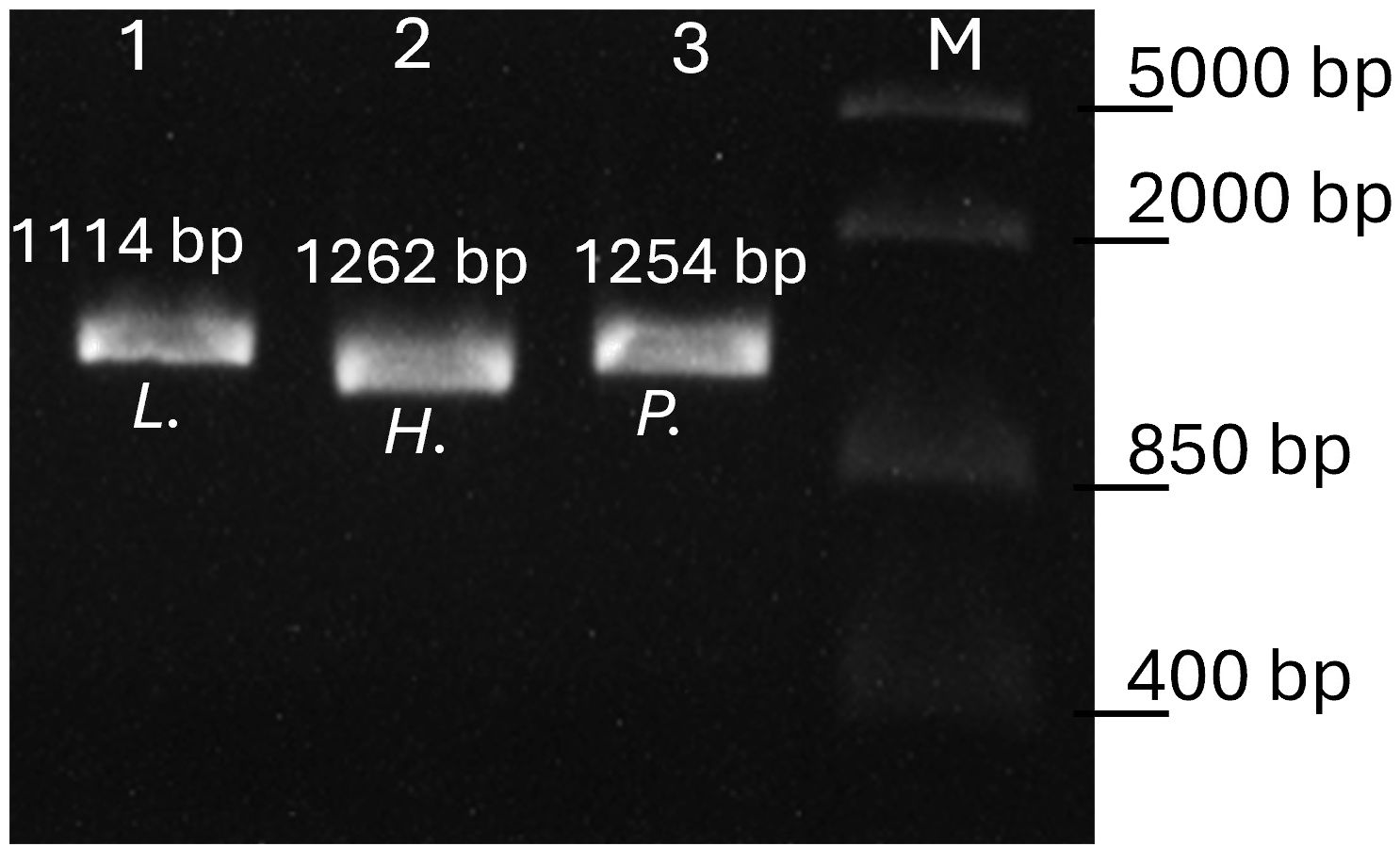
Figure 2 Agarose gel electrophoresis of the genus-specific PCR products. Lane 1 amplicon of Leucocytozoon (L.), lane 2 amplicon of subgenus Parahaemoproteus (H.) and lane 3 Plasmodium (P.) infection. Lane M, FastRuler Middle Range DNA Ladder (Thermo Scientific). Fragment sizes including primers are given.
To test for differences in phylogenetic analyses, due to the longer fragment size gained from the genus-specific nested PCR, two different phylogenetic analyses were performed. The first consisted of avian haemosporidian lineages from the standard nested PCR, each trimmed to the length of the barcoding region, 479 bp. The second dataset included sequences of the same lineages, amplified by the genus-specific nested PCR, each trimmed to a homologous fragment of 1,000 bp. Both datasets were phylogenetically analysed using MEGA v.10.2 (Kumar et al., 2018). Both analyses were performed running the general time reversible model with gamma distribution (GTR + G). All maximum likelihood methods were performed using 1000 replicates. The resulting phylograms were viewed and edited with MEGA v.10.2.
To compare the sensitivity of the genus-specific nested PCR method described here to that of the typical nested PCR protocol, both methods were used to assess the infection status of the Malagasy individual bird blood samples and the parasites lineage identity. The prevalence by each parasite genus and that of mixed infections was compared between the two methods using chi-squared tests. The number of additional positives in the second test run of each method was compared to elucidate the repeatability of results. Each detected haemosporidian parasite lineage was classified as either: a) detected in both methods, b) detected exclusively in this method, c) lineage of the same genus, but different lineage or d) lineage not identified due to sequencing errors. Those categories were compared between the two methods. Results of the sample sets from Lithuania and Sweden were separately analysed by comparing prevalences using chi-squared tests. Statistical analysis was performed using DATAtab.
The initial dataset, comprising 264 Malagasy bird blood samples, disclosed a haemosporidian parasite prevalence of 58.7% through the SN protocol, whereas the recently introduced GS protocol yielded a prevalence of 50.75%. The observed variance in detection probabilities did not attain statistical significance (χ2 = 3.371, df = 1, p = 0.066). The prevalence of Haemoproteus exhibited similar values between both methods (SN: 17.4%, GS: 18.2%), while Plasmodium was more frequently identified using the SN protocol (SN: 37.5%, GS: 31.1%), and Leucocytozoon was more prevalent with the GS protocol (SN: 25%, GS: 32.2%). Upon retesting negative samples, the genus-specific protocol demonstrated a lower incidence of false-negatives in the initial round, as illustrated in Table 3.
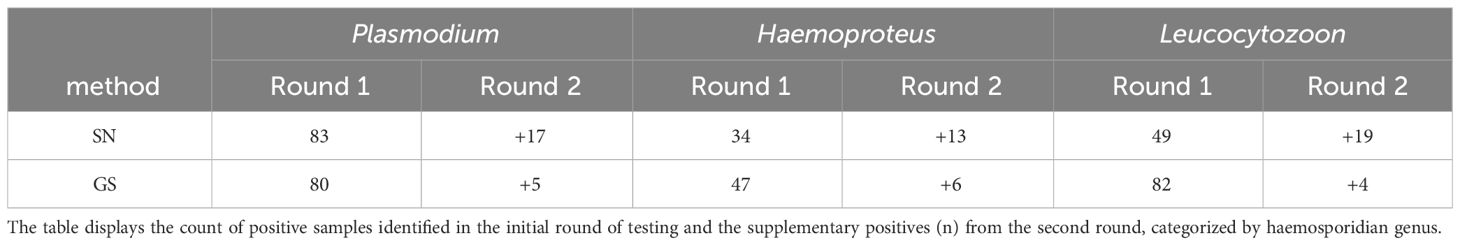
Table 3 Comparison of the number of positive tested samples (n) between the standard nested PCR approach (SN) and the genus-specific nested PCR (GS).
The genus-specific nested PCR identified a higher number of mixed infections compared to the standard nested PCR, although this difference is not statistically significant (χ2 = 1.255, df = 1, p = 0.263). Using the SN protocol, 58 double infections were identified, consisting of 43 Plasmodium/Leucocytozoon and 15 Haemoproteus/Leucocytozoon mixed infections. In addition, 31 samples contained sequences showing double peaks, suggesting mixed infections with species/haplotypes of the same genus. In contrast, the GS protocol successfully detected 55 double infections, including 39 Plasmodium/Leucocytozoon, 8 Haemoproteus/Leucocytozoon, and 8 Plasmodium/Haemoproteus mixed infections. Additionally, the GS protocol identified 14 samples containing DNA from all three haemosporidian genera. The sequences of 29 samples showed double peaks, indicating mixed infections of species/haplotypes of the same genus.
The combination of both methods revealed with 80.9% the highest prevalence for Ploceidae, namely Ploceus nelicourvi, and the lowest prevalence for Muscicapidae with 50%. Acrocephalidae showed a prevalence of 66.7% for haemosporidian parasite infections (Table 4). All avian species exhibited mixed infections, with a notable prevalence of co-infections involving both Plasmodium and Leucocytozoon spp. within samples of Ploceus nelicourvi.
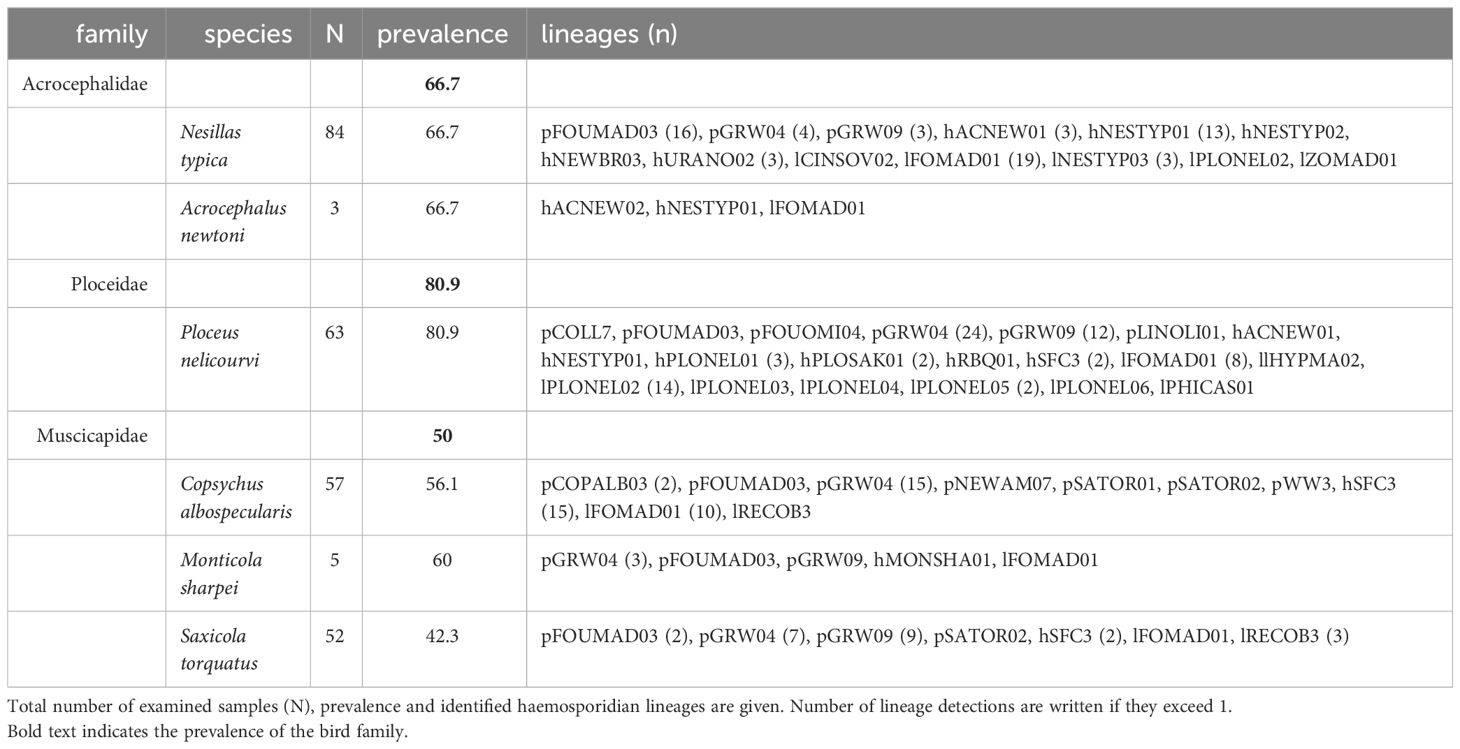
Table 4 Haemosporidian infections of Malagasy bird species.
Sequencing revealed 35 distinct haemosporidian lineages in the Malagasy sample set, encompassing 11 Plasmodium, 11 Haemoproteus, and 13 Leucocytozoon lineages. Notably, Ploceus nelicourvi exhibited the highest diversity, hosting a total of 20 different lineages, followed by Nesillas typica with 13 different lineages (Table 4). Four lineages (pFOUMAD03, pGRW04, pGRW09 and lFOMAD01) were detected in all three avian bird families, whilst two Haemoproteus lineages (hACNEW01 and hNESTYP01) were shared between Acrocephalidae and Ploceus nelicourvi and hSFC3 was found in Ploceus nelicourvi as well as in Muscicapidae. The detection rate of some haemosporidian lineages differed notably between the two nested PCR protocols. The standard nested PCR revealed more infections with pGRW09 and hNESTYP01, while the genus-specific PCR was able to detect more infections of hSFC3 and lFOMAD01 (Table 5).
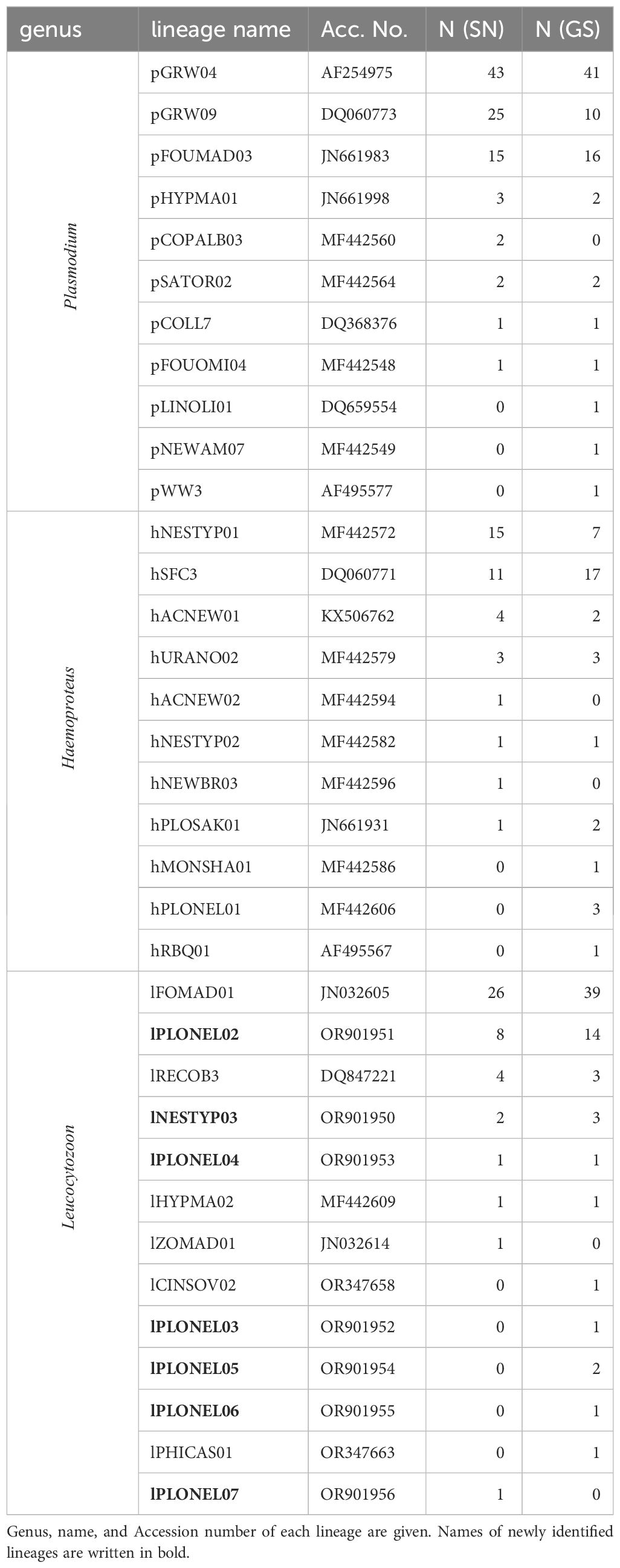
Table 5 Number of haemosporidian lineage detections within the Malagasy sample set using standard nested PCR protocol (SN) and genus-specific nested PCR protocol (GS).
Phylogenetic examination of the larger fragment amplified by the genus-specific nested PCR resulted in an overall better-defined phylogenetic topology of lineages (Figure 3) and higher bootstrap values (Supplementary Material).
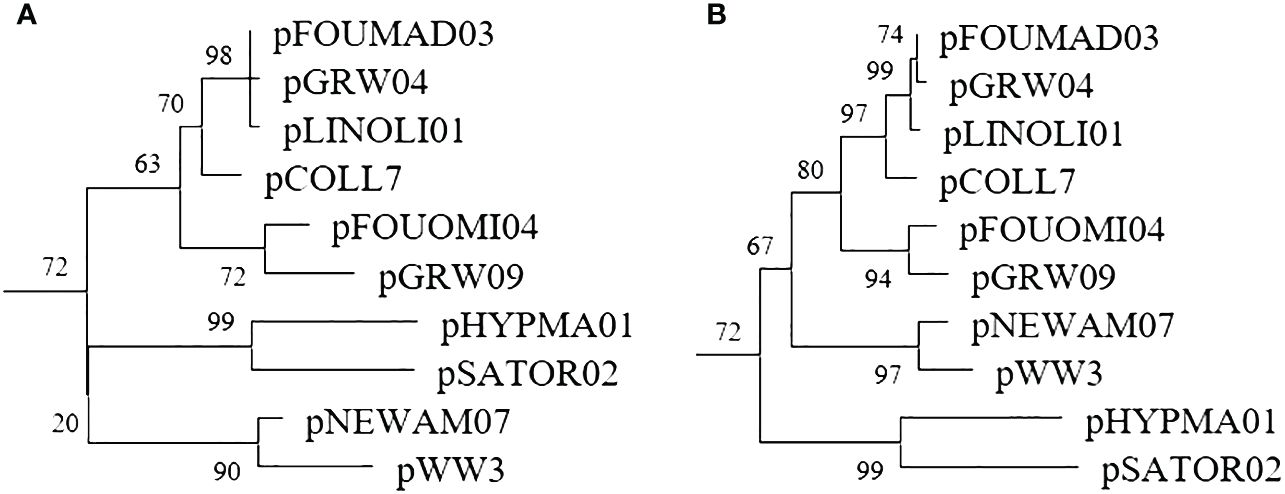
Figure 3 Plasmodium subtree of the phylogenetic trees constructed with a 479bp fragment amplified by the standard nested PCR (A) and a 1,000bp fragment amplified by the genus-specific nested PCR (B).
The analysis of the second dataset, encompassing 54 bird blood samples examined at the Nature Research Centre in Vilnius, unveiled marginally reduced detection rates for all haemosporidian genera when utilizing the genus-specific nested PCR method (Table 6). However, none of these differences reached statistical significance. Notably, with respect to Plasmodium detections, the genus-specific nested PCR exhibited a failure to identify three samples that had tested positive using the standard nested PCR. Conversely, this method successfully amplified two additional positive samples. Subsequent sequencing of these samples resulted in the identification of Plasmodium circumflexum (pTURDUS1) within this subset of samples.
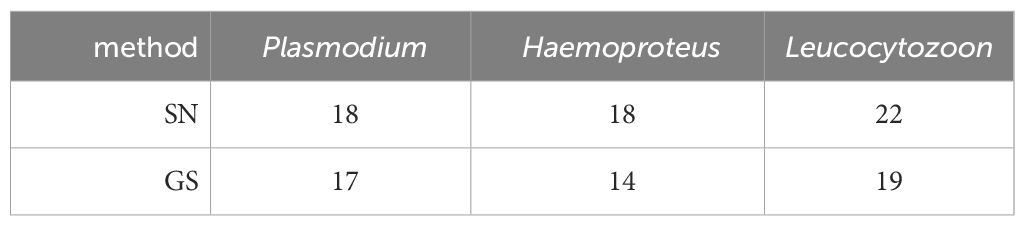
Table 6 Haemosporidian parasite infections detected in the Lithuanian sample set (N = 54) using standard nested PCR protocol (SN) and genus-specific nested PCR protocol (GS).
The third dataset, examined at the Molecular Ecology and Evolution Laboratory at Lund University, comprised 52 bird blood samples from passerines. The sample set was previously tested with the multiplex PCR assay and standard nested PCR. The success rate was ~50% lower for the GS protocol compared to the other two protocols (Table 7). The GS protocol detected one additional Haemoproteus infection, that was identified as H. fringillae hCCF3 (DQ060764).
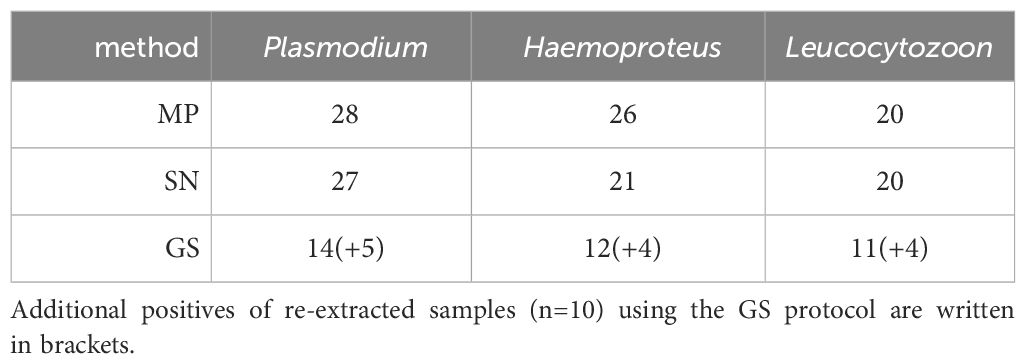
Table 7 Haemosporidian parasite infections detected in the Swedish sample set (N = 52) using multiplex PCR (MP), standard nested PCR protocol (SN) and genus-specific nested PCR protocol (GS).
To test whether this was due to degraded DNA, we tested the GS protocol again on re-extracted DNA from ten samples that failed parasite detection in the first test round. These ten samples had been scored to contain a total of 16 infections (6 single, 2 double and 2 triple infections) by the MP and SN protocols. In this second test with the GS protocol, we recovered 13 of these 16 infections.
In this study, novel genus-specific primers were designed to facilitate the specific identification of avian haemosporidian parasites belonging to the genera Plasmodium, Leucocytozoon, and the subgenus Parahaemoproteus. The newly developed protocol underwent rigorous testing utilizing a large dataset comprising blood samples from Malagasy birds of three distinct Passeriformes families. Furthermore, validation occurred through application to smaller datasets in two other laboratories employing divergent master mixes and different bird species. Comparative analyses were conducted between the outcomes of the novel PCR protocol and those obtained through the widely used standard nested PCR method. Notably, the latter was either run as an integral component of this investigation or had been previously carried out in other research projects.
The comprehensive evaluation of the Malagasy and Lithuanian datasets yielded a sensitivity comparable to, albeit slightly lower than, that of the standard nested PCR, although those differences were not statistically significant. In contrast, the Swedish dataset exhibited only a 50% success rate in detecting all known haemosporidian infections. It was hypothesized that this lower success rate might be attributable to the slight degradation of DNA within the samples, likely resulting from prolonged use. Degraded DNA is anticipated to have a more pronounced impact on the GS protocol compared to the SN and MP protocol due to the considerably longer amplicons (>1000 bp versus <500 bp). Subsequent analyses involving the extraction of freshly obtained DNA from samples that initially failed amplification yielded a recovery of the majority of the missed infections (81.25%). This highlights the importance of DNA quality in facilitating the amplification and sequencing especially of longer fragments (Freed and Cann, 2006), thus ensuring the reliability and comparability of results obtained through the GS protocol. To confirm the integrity of DNA samples, it is advisable to initially assess the total DNA quantity in the extracted samples, either through photometry or by examining the extractions on agarose gels (Reinoso-Pérez et al., 2020). In order to ensure a baseline level of DNA quality, it may be beneficial to conduct molecular amplification of host DNA simultaneously. However, it is important to note that this approach might not completely eliminate the possibility of moderate levels of DNA degradation which could affect the quality of haemosporidian DNA. Subtle variations in sensitivity are likely attributable to the primer specificity. Within the Malagasy dataset, the standard nested PCR demonstrated a notably higher detection rate for pGRW09, while the genus-specific nested PCR exhibited a greater frequency in detecting lFOMAD01. As the complete mitochondrial DNA (mtDNA) of lFOMAD01 (OR347660) is available, an analysis of primer binding sites was feasible. When considering the primers utilized in the GS protocol, only one mismatch is observed for the primer CytR. On the other hand, the primers employed in the NS protocol show three mismatches (2 for HaemNF and 1 with HaemNR2L). The primers used in these protocols demonstrate distinct affinities to the sequence of the respective lineage, leading to noticeable variations in prevalence. In the instance of Haemoproteus (Parahaemoproteus), where both protocols demonstrate a comparable prevalence, individual distinctions persist in the detection rates of specific lineages. For instance, hNESTYP01 was more frequently identified through the standard nested PCR, contrasting with the preference of the genus-specific nested PCR for detecting hSFC3. Given the great diversity among avian Haemosporida, the design of primers that are specific for all lineages within a genus presents an enormous challenge, if not an impossible one. The documented existence of diverse primer preferences in other protocols (Bernotiene et al., 2016) substantiates these findings. To comprehensively capture most lineages in mixed infections, the simultaneous application of at least three PCRs is recommended. Due to the significant effort needed in applying various PCR approaches, this recommendation has not been heeded. The standard nested PCR method remains the method of choice in the most recent studies conducted (e.g. Žiegytė et al., 2023; González-Olvera et al., 2023; Han et al., 2023). It was only complemented by the utilization of multiplex PCRs (Pacheco et al., 2018b; Ciloglu et al., 2019) in some studies (e.g. Musa et al., 2022; Barbon et al., 2023; Procházka et al., 2023) due to its limitation in detecting mixed infections of Plasmodium and Haemoproteus. However, those assays have failed to ensure a definitive identification at the parasite’s lineage level. The protocol presented in this study enables this identification for the first time by amplifying fragments that overlap with the haemosporidian barcoding region (Bensch et al., 2009). This allows for a thorough investigation into the exact composition of mixed infections. Furthermore, in the case of sequences exhibiting double peaks, usually only the genus can be determined, and it is assumed that a mixed infection of two lineages/species of the same genus is present. However, with the SN protocol, one cannot be entirely certain whether the double peaks detected in sequences resulting from the use of the primer pair HaemF/HaemR2 also represent a mixed infection of Plasmodium and Haemoproteus lineages. The use of the GS protocol ensures a more reliable identification of these double infections. Nevertheless, it is important to keep in mind that the observed frequency of mixed infections, prevalence, or biodiversity of haemosporidian parasites in a population is consistently underestimated (Valkiūnas et al., 2006).
Furthermore, it must be noted that the dataset of blood samples examined in this study, comes from Passeriformes. Haemosporidian parasites from other bird families show sometimes extreme differences in their mitochondrial genome. For example, the barcoding region of Haemoproteus elani hBUBT1, which infects Accipitriformes, exhibits only an 87% homology to other avian Haemosporida (Harl et al., 2022). Therefore, it cannot be ruled out that, in addition to the subgenus Haemoproteus, other haemosporidian lineages from birds of different bird families may not be detected by the primers developed here. Further studies should therefore investigate the detection rates more precisely with datasets from non-Passeriformes.
Methods for detecting avian haemosporidians can be categorized based on their primary purpose, such as screening, identification, or phylogeny (Bensch and Hellgren, 2020). Phylogenetic analyses are particularly crucial for investigating evolutionary factors, and the accuracy of such analyses relies heavily on the length of DNA fragments. Previous research has demonstrated that, for robust phylogenetic analyses, the barcoding region (479 bp) is insufficient for obtaining well-supported topologies, with only a few nodes showing bootstrap values >70% (Ellis and Bensch, 2018). When comparing phylogenetic trees constructed from the lineages detected in the Malagasy sample set, it was observed that 26.9% of nodes had bootstrap values <70% when using the barcoding region, whereas this decreased to 18.5% when using the 1,000 bp long fragment amplified by the genus-specific nested PCR. The utilization of these longer fragments enhances the resolution of the phylogenetic topology of lineages.
In conclusion, the newly described genus-specific nested PCR protocol addresses a crucial gap in the avian haemosporidian detection toolkit. It facilitates lineage-specific identification of Plasmodium, Haemoproteus (Parahaemoproteus), and Leucocytozoon parasites. The effectiveness of this innovative protocol has been demonstrated across various laboratories, yielding comparable results if DNA extract was of good quality. It utilizes the same equipment and requires nearly the same amount of time compared to the standard nested PCR, yet eliminates an additional step in identifying mixed infections of Plasmodium and Haemoproteus. Moreover, the length of the amplification product enhances the robustness of phylogenetic analyses. This holds significant importance for molecular epidemiological surveillance and biodiversity studies, enabling a thorough examination of mixed infections.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because Sample material was obtained in the course of other studies.
SM: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. TH: Conceptualization, Investigation, Writing – review & editing. SB: Investigation, Writing – review & editing. VP: Writing – review & editing. LB: Investigation, Writing – review & editing. FW: Writing – review & editing. UM: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. SM received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) -457213393.
We thank Jane Jönsson for assisting with PCR analyses in the laboratory and Dr. Carolina F. R. Chagas for providing part of the samples. This publication is based on work from COST Action WIMANET (Wildlife Malaria Network), CA22108, supported by COST (European Cooperation in Science and Technology).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1385599/full#supplementary-material
Barbon, H., Berthoud, J.-L., Woog, F., Musa, S. (2023). Haemosporidian parasite infections of Malagasy Philepittidae and Nectariniidae are driven by phylogeny rather than ecology. Parasitology 150(14), 1–46. doi: 10.1017/S0031182023001075
Bensch, S., Hellgren, O. (2020). “The Use of Molecular Methods in Studies of Avian Haemosporidians,” in Avian Malaria and Related Parasites in the Tropics (Springer International Publishing, Cham), 113–135. doi: 10.1007/978-3-030-51633-8_4
Bensch, S., Hellgren, O., Pérez-Tris, J. (2009). MalAvi: A public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol. Ecol. Resour. 9, 1353–1358. doi: 10.1111/j.1755-0998.2009.02692.x
Bensch, S., Stjernman, M., Hasselquist, D., Ostman, O., Hansson, B., Westerdahl, H., et al. (2000). Host specificity in avian blood parasites: a study of Plasmodium and Haemoproteus mitochondrial DNA amplified from birds. Proc. R. Soc. B: Biol. Sci. 267, 1583–1589. doi: 10.1098/rspb.2000.1181
Bernotiene, R., Palinauskas, V., Iezhova, T., Murauskaite, D., Valkiunas, G. (2016). Avian haemosporidian parasites (Haemosporida): A comparative analysis of different polymerase chain reaction assays in detection of mixed infections. Exp. Parasitol. 163, 31–37. doi: 10.1016/j.exppara.2016.01.009
Ciloglu, A., Ellis, V. A., Bernotienė, R., Valkiūnas, G., Bensch, S. (2019). A new one-step multiplex PCR assay for simultaneous detection and identification of avian haemosporidian parasites. Parasitol. Res. 118, 191–201. doi: 10.1007/s00436-018-6153-7
Clark, N. J., Clegg, S. M., Lima, M. R. (2014). A review of global diversity in avian haemosporidians (Plasmodium and Haemoproteus: Haemosporida): New insights from molecular data. Int. J. Parasitol. 44, 329–338. doi: 10.1016/j.ijpara.2014.01.004
Ellis, V. A., Bensch, S. (2018). Host specificity of avian haemosporidian parasites is unrelated among sister lineages but shows phylogenetic signal across larger clades. Int. J. Parasitol. 48, 897–902. doi: 10.1016/j.ijpara.2018.05.005
Ellis, V. A., Huang, X., Westerdahl, H., Jönsson, J., Hasselquist, D., Neto, J. M., et al. (2020). Explaining prevalence, diversity and host specificity in a community of avian haemosporidian parasites. Oikos 129, 1314–1329. doi: 10.1111/oik.07280
Freed, L. A., Cann, R. L. (2006). DNA quality and accuracy of avian malaria PCR diagnostics: A review. Condor 108, 459–473. doi: 10.1093/condor/108.2.459
González-Olvera, M., Hernandez-Colina, A., Chantrey, J., Allen, S., Lopez, J., Baylis, M. (2023). A non-invasive feather-based methodology for the detection of blood parasites (Haemosporida). Sci. Rep. 13, 16712. doi: 10.1038/s41598-023-43932-y
Han, Y., Hellgren, O., Wu, Q., Liu, J., Jin, T., Bensch, S., et al. (2023). Seasonal variations of intensity of avian malaria infection in the Thousand Island Lake System, China. Parasites Vectors 16, 1–12. doi: 10.1186/s13071-023-05848-4
Harl, J., Fauchois, A., Puech, M. P., Gey, D., Ariey, F., Izac, B., et al. (2024). Novel phylogenetic clade of avian Haemoproteus parasites (Haemosporida, Haemoproteidae) from Accipitridae raptors, with description of a new Haemoproteus species. Parasite 31, 1–18. doi: 10.1051/parasite/2023066
Harl, J., Himmel, T., Valkiūnas, G., Ilgūnas, M., Nedorost, N., Matt, J., et al. (2022). Avian haemosporidian parasites of accipitriform raptors. Malaria J. 21, 1–38. doi: 10.1186/s12936-021-04019-z
Hellgren, O., Waldenstrom, J., Bensch, S. (2004). A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J. Parasitol. 90, 797–802. doi: 10.1645/GE-184R1
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Marzal, A., Bensch, S., Reviriego, M., Balbontin, J., De Lope, F. (2008). Effects of malaria double infection in birds: One plus one is not two. J. Evolutionary Biol. 21, 979–987. doi: 10.1111/j.1420-9101.2008.01545.x
Musa, S. (2023). Mitochondrial genome amplification of avian haemosporidian parasites from single-infected wildlife samples using a novel nested PCR approach. Parasitol. Res. 122 (12), 2967–2975. doi: 10.1007/s00436-023-07986-1
Musa, S., Mackenstedt, U., Woog, F., Dinkel, A. (2022). Untangling the actual infection status: detection of avian haemosporidian parasites of three Malagasy bird species using microscopy, multiplex PCR, and nested PCR methods. Parasitol. Res. 121, 2817–2829. doi: 10.1007/s00436-022-07606-4
Pacheco, M. A., Cepeda, A. S., Bernotienė, R., Lotta, I. A., Matta, N. E., Valkiūnas, G., et al. (2018b). Primers targeting mitochondrial genes of avian haemosporidians: PCR detection and differential DNA amplification of parasites belonging to different genera. Int. J. Parasitol. 48, 657–670. doi: 10.1016/j.ijpara.2018.02.003
Pacheco, M. A., Matta, N. E., Valkiünas, G., Parker, P. G., Mello, B., Stanley, C. E., et al. (2018a). Mode and rate of evolution of haemosporidian mitochondrial genomes: Timing the radiation of avian parasites. Mol. Biol. Evol. 35, 383–403. doi: 10.1093/molbev/msx285
Perkins, S. L., Schall, J. J. (2002). A molecular phylogeny of malarial parasites recovered from cytochrome b gene sequences. J. Parasitol. 88, 972–978. doi: 10.1645/0022-3395(2002)088[0972:AMPOMP]2.0.CO;2
Procházka, P., Emmenegger, T., Bauer, S., Ciloglu, A., Dimitrov, D., Hansson, B., et al. (2023). The association between haemosporidian infection and non-breeding moult location in great reed warblers revisited by combining feather stable isotope profiles and geolocator data. Oecologia. 204, 107–118. doi: 10.1007/s00442-023-05491-x
Reinoso-Pérez, M. T., Dhondt, K. V., Sydenstricker, A. V., Heylen, D., Dhondt, A. A. (2020). Complex interactions between bacteria and haemosporidia in coinfected hosts: An experiment. Ecol. Evol. 10, 5801–5814. doi: 10.1002/ece3.6318
Sambrook, J., Russel, W. R. (2001). Molecular cloning: a laboratory manual. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Schmid, S., Dinkel, A., Mackenstedt, U., Tantely, M. L., Randrianambinintsoa, F. J., Boyer, S., et al. (2017). Avian malaria on Madagascar: bird hosts and putative vector mosquitoes of different Plasmodium lineages. Parasites Vectors. 10 (6), 1–7. doi: 10.1186/s13071-016-1939-x
Valkiūnas, G., Bensch, S., Iezhova, T. A., Križanauskiene, A., Hellgren, O., Bolshakov, C. V. (2006). Nested cytochrome b polymerase chain reaction diagnostics underestimate mixed infections of avian blood haemmosporidian parasites: Microscopy is still essential. J. Parasitol. 92, 418–422. doi: 10.1645/GE-3547RN.1
Waldenström, J., Bensch, S., Hasselquist, D., Östman, Ö. (2004). A new nested polymerase chain reaction method very efficient in detecting plasmodium and haemoproteus infections from avian blood. J. Parasitol. 90, 191–194. doi: 10.1645/GE-3221RN
Wink, M. (2006). Use of DNA markers to study bird migration. J. Ornithology 147, 234–244. doi: 10.1007/s10336-006-0065-5
Keywords: Plasmodium, Haemoproteus, Leucocytozoon, primers, mixed-infections
Citation: Musa S, Hemberle T, Bensch S, Palinauskas V, Baltrūnaitė L, Woog F and Mackenstedt U (2024) Raising the bar: genus-specific nested PCR improves detection and lineage identification of avian haemosporidian parasites. Front. Cell. Infect. Microbiol. 14:1385599. doi: 10.3389/fcimb.2024.1385599
Received: 13 February 2024; Accepted: 09 April 2024;
Published: 29 April 2024.
Edited by:
Olgica Djurkovic-Djakovic, University of Belgrade, SerbiaCopyright © 2024 Musa, Hemberle, Bensch, Palinauskas, Baltrūnaitė, Woog and Mackenstedt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandrine Musa, U2FuZHJpbmUubXVzYUB1bmktaG9oZW5oZWltLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.