 Renhe Zhu1Rundong Cao1Lulu Wang1Yue Gong1Qian Cheng1Hu Long2Dong Xia1Qinqin Song1Zhiqiang Xia1Mi Liu1HaiJun Du1Juan Song1Jun Han1*Chen Gao1*
Renhe Zhu1Rundong Cao1Lulu Wang1Yue Gong1Qian Cheng1Hu Long2Dong Xia1Qinqin Song1Zhiqiang Xia1Mi Liu1HaiJun Du1Juan Song1Jun Han1*Chen Gao1*- 1National Key Laboratory of Intelligent Tracking and Forecasting for Infectious Diseases, National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 2Epidemic Prevention and Control Department, Guilin Center for Disease Control and Prevention, Guilin, China
Introduction: Seasonal human coronavirus NL63 (HCoV-NL63) is a frequently encountered virus linked to mild upper respiratory infections. However, its potential to cause more severe or widespread disease remains an area of concern. This study aimed to investigate a rare localized epidemic of HCoV-NL63-induced respiratory infections among pediatric patients in Guilin, China, and to understand the viral subtype distribution and genetic characteristics.
Methods: In this study, 83 pediatric patients hospitalized with acute respiratory infections and positive for HCoV-NL63 were enrolled. Molecular analysis was conducted to identify the viral subgenotypes and to assess genetic variations in the receptor-binding domain of the spiking protein.
Results: Among the 83 HCoV-NL63-positive children, three subgenotypes were identified: C4, C3, and B. Notably, 21 cases exhibited a previously unreported subtype, C4. Analysis of the C4 subtype revealed a unique amino acid mutation (I507L) in the receptor-binding domain of the spiking protein, which was also observed in the previously reported C3 genotype. This mutation may suggest potential increases in viral transmissibility and pathogenicity.
Discussion: The findings of this study highlight the rapid mutation dynamics of HCoV-NL63 and its potential for increased virulence and epidemic transmission. The presence of a unique mutation in the C4 subtype, shared with the C3 genotype, raises concerns about the virus’s evolving nature and its potential public health implications. This research contributes valuable insights into the understanding of HCoV-NL63’s epidemiology and pathogenesis, which is crucial for effective disease prevention and control strategies. Future studies are needed to further investigate the biological significance of the observed mutation and its potential impact on the virus’s transmissibility and pathogenicity.
1 Introduction
Acute Respiratory Infection (ARI) poses a significant public health challenge, leading to substantial morbidity and mortality on a global scale (Collaborators GDaI, 2020). Notably, acute lower respiratory infections (ALRI), encompassing pneumonia and bronchiolitis stemming from bacterial and viral sources, stand out as a primary cause of hospitalization and nosocomial deaths among young children, particularly in low- and middle-income countries (Collaborators GI, 2019). In the Chinese context, prevalent viruses in children hospitalized with ARI include respiratory syncytial virus, influenza virus, rhinovirus, human metapneumovirus, adenovirus, parainfluenza virus, bocavirus, and human seasonal coronavirus (Li et al., 2021).
Human seasonal coronavirus NL63 (HCoV-NL63) holds a prominent place among respiratory tract infections. Initially identified in Amsterdam, the Netherlands, in 2004, HCoV-NL63 belongs to the Coronaviridae family and is characterized by its enveloped, single-stranded, positively stranded RNA virus, encompassing 27,553 nucleotides (van der Hoek et al., 2004). Since the emergence of SARS-CoV-2 in late 2019, seven human coronaviruses (HCoVs) have been identified. Remarkably, HCoV-NL63 shares the same cellular receptor, ACE2, for cell entry with SARS-CoV and SARS-CoV-2, distinguishing it from the other three seasonal coronaviruses (HCoV-229E, HCoV-OC43, and HCoV-HKU1) (Lin et al., 2011; Lim et al., 2016). The spike glycoprotein (S) emerges as the primary antigenic protein, housing S1 and S2 domains responsible for viral binding to human angiotensin-converting enzyme 2 (ACE2) and membrane fusion, respectively. Notably, the S1 domain stands out as the most variable gene in the entire HCoV-NL63 genome, serving as a target in molecular epidemiological analyses (Lin et al., 2011).While HCoV-NL63 typically causes mild, self-limiting upper respiratory tract infections across all age groups, it occasionally leads to more severe lower respiratory tract infections, such as pneumonia and bronchiolitis, particularly in susceptible individuals like young children, the elderly, and immunosuppressed patients (Konca et al., 2017; Mayer et al., 2016; Oosterhof et al., 2010; van der Hoek, 2007).
Given the absence of a well-established molecular epidemiological surveillance system for HCoV-NL63 and limited research on its genotypic distribution and genetic characterization, our study aimed to contribute valuable sequence information and in-depth analysis of the molecular evolutionary features of HCoV-NL63, such as nucleotide homology, phylogeny, potential recombination events, N-glycosylation sites, major amino acid substitution sites, etc (Shao et al., 2022). In this study, Nasopharyngeal swabs from children hospitalized with acute respiratory infections were collected and subjected to viral profiling and whole-genome sequence amplification. Subsequent genomic phylogenetic and evolutionary analyses revealed the emergence of a new HCoV-NL63 genotype (type C4) in Guilin, China. This discovery sheds light on the evolutionary trajectory and potential epidemic risks associated with seasonal coronaviruses, offering novel insights for molecular epidemiological studies and control strategies for human coronavirus NL63.
2 Materials and methods
2.1 Clinical specimens
Nasopharyngeal swab samples from pediatric cases were collected between September 2021 and October 2022 at sentinel hospitals in Guilin, Guangxi Zhuang Autonomous Region. A total of 638 throat swab samples were obtained from children hospitalized for acute respiratory infections (ARI). Following collection, the throat swabs were immediately stored at -80°C and transported in dry ice throughout the cold chain to the laboratory of the Virus Resource Center at the Chinese Center for Disease Control and Prevention for further testing.
Clinical diagnostic criteria for pediatric ARI followed the “Sentinel Monitoring Plan for Severe Acute Respiratory Infection Cases in Hospitals (2011 edition)” issued by the Ministry of Health of the People’s Republic of China (Supplementary S1) (China MoHotPsRo, 2011).
Considering the subtropical monsoon climate of Guilin, clinical data for the children were grouped by season as follows: March to May (spring), June to August (summer), September to November (autumn), and December to February of the following year (winter).
2.2 Nucleic acids extraction and virus detection
Nucleic acids extraction and virus detection were performed using the GeneRotex96 fully automated nucleic acid extractor (Xi’an Tianlong Technology Co., Ltd.) and the corresponding nucleic acid extraction kit (ZTLJB-Y64T), following the manufacturer’s instructions. The extracted nucleic acids from nasal and pharyngeal swab specimens were subjected to detection for 15 common respiratory viruses using real-time quantitative polymerase chain reaction (qPCR) with the Applied Biosystems QuantStudio 5 instrument. The detected viruses included human rhinovirus (HRV), respiratory syncytial virus (RSV), human metapneumovirus (hMPV), influenza viruses A and B (FluA and FluB), parainfluenza virus types 1-3 (PIV1-3), and human coronaviruses (OC43, NL63, 229E, and HKU1), adenovirus (AdV), human bocavirus (HBoV), and human parvovirus B19 (HPV B19). Primers and probes for these 15 respiratory pathogens were designed in-house, synthesized, and validated by Shanghai Bioengineering Co., Ltd. The GoldStar Probe Mixture kit was used for DNA virus detection, and the GoldStar Probe One Step RT-qPCR Kit was used for RNA virus detection.
2.3 S1 domain gene amplification
Positive samples with a Ct value ≤30 from real-time RT-PCR nucleic acid detection of human coronavirus HCoV-NL63 were selected. The TAKARA PrimeScript™ One Step RT-PCR kit Ver. 2 was used for reverse transcription-polymerase chain reaction (RT-PCR) to amplify the nucleotide sequence of the S1 domain. The primer sequences are detailed in Table 1. The reaction program included incubation at 50°C for 30 min, initial denaturation at 94°C for 2 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 1 min, extension at 72°C for 2 min, and a final extension at 72°C for 10 min. The amplified fragments were sent to Qingke Biosciences Co., Ltd. for Sanger sequencing.
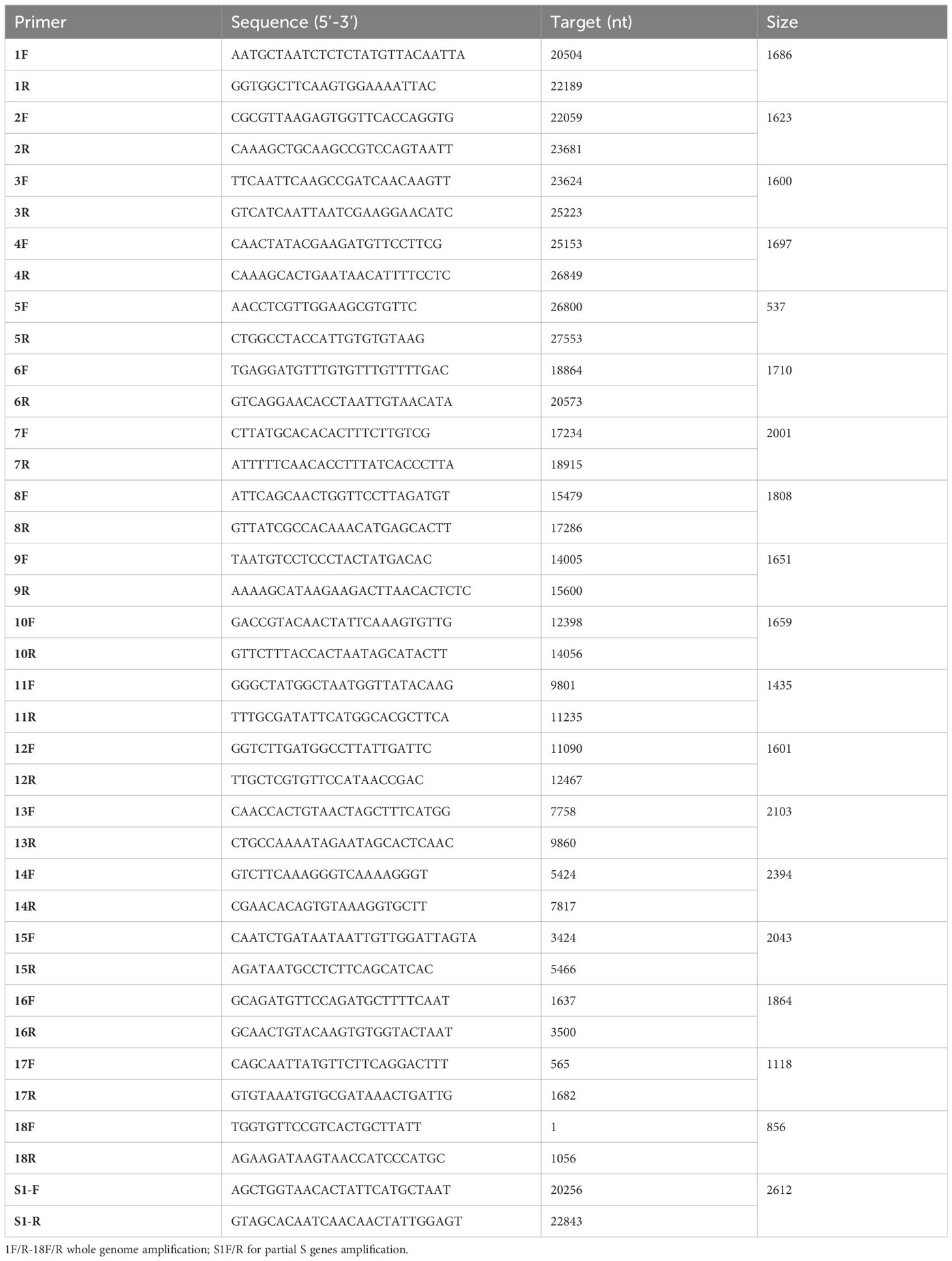
Table 1 RT-PCR primer sequences for amplifying the full-length genome and partial Spike gene of HCoV-NL63.
2.4 HCov-NL63 whole genome amplification
The HCoV-NL63 isolate from the Netherlands (HCoV-NL63_Amsterdam 1, GenBank accession number: NC_005831) served as the reference strain. Eighteen primer pairs covering overlapping regions were designed using the Blast-Primer web tool. RT-PCR was employed to amplify specific segments with overlapping regions, encompassing all protein-coding genes except the 5’ untranslated regions (UTR) and 3’ UTR. Primer pairs and amplification details are provided in Table 1. Samples with Ct values ≤25 from human coronavirus HCoV-NL63 real-time RT-PCR were selected, and the TAKARA PrimeScriptTM One Step RT-PCR kit Ver. 2 was used for RT-PCR amplification. The amplified segments were subjected to Sanger sequencing by Qingke Biosciences Co., Ltd.
2.5 Reorganization analysis
SimPlot 3.5.1.4 software was used for intraspecific whole-genome recombination analysis of the 13 HCoV-NL63 sequences obtained in this study against the reference sequence (Lole et al., 1999). A sliding window size of 400 bp with a step size of 40 bp was employed.
2.6 Phylogenetic analysis
As of October 11, 2023, all HCoV-NL63 S gene sequences were retrieved from NCBI to establish a dataset. Strain sequences with incomplete, excessively ambiguous bases, and unclear sampling years or countries were excluded. For identical sequences from the same region and year, only one was retained. The nucleic acid sequence alignment was performed using MAFFT software (Katoh et al., 2019). To confirm the completeness of the time signal of the HCoV-NL63 complete S gene database, IQ-TREE was used to build a maximum likelihood phylogenetic tree (ML-TREE). The phylogenetic tree utilized a non-molecular clock with 1000 repetitions of the best-fitting model. TempEst v1.5.3 was used to visualize a root-to-tip regression plot based on regression analysis.
Time-scaled phylogenetic tree inference for the complete HCoV-NL63 S gene sequence dataset with a time signal was performed using the Bayesian Markov Chain Monte Carlo (MCMC) method in BEAST v.1.10.4.3. The optimal nucleotide substitution model recommended by ModelFinder was selected based on Bayesian scores. The evolutionary model comprised TN93+Empirical+I+four gamma categories. The molecular clock model selected a log-normal distribution of uncorrelated relaxed clock (UCLN) based on previous references, and the tree prior model chose Bayesian SkyGrid. The time to the most recent common ancestor (tMRCA) was set at twice the height of the tree, with other parameters using default settings from BEAUti. The MCMC chain length was set to 200,000,000, with sampling every 20,000 steps. The XML file created in BEAUti v1.10.4 was run in BEAST, and the generated log file was loaded into Tracer v1.10.4 to confirm that all parameter estimates had effective sample sizes (ESS) exceeding 200, indicating sufficient convergence. The TreeAnnotator program in the BEAST package was used to construct the maximum clade credibility tree (MCC-tree) with a 10% burn-in, and FigTree v1.10.4 was employed to visualize the MCC-tree.
For the full-length coding sequence of HCoV-NL63, ORF1ab, and S1 domain, a neighbor-joining phylogenetic tree was constructed using MEGA v7.0 (Kumar et al., 2016) software with 1000 bootstrap repetitions and the Kimura2‐parameter nucleotide substitution model.
2.7 Amino acid variant sites analysis
Amino acid variation characteristics were analyzed based on the Spike protein amino acid sequence of the representative strain of HCoV-NL63. Amsterdam I strain (GenBank accession number: NC 005831) and HCoV-NL strain (GenBank accession number: AY518894) were chosen as reference strains for A and B genotypes, respectively.
2.8 Statistical analysis
Data organization and statistical analysis were conducted using Excel 2021 and SPSS 26.0 software. Differences in rates (or composition ratios) between groups were compared using the χ2 test or Fisher’s exact probability method. A two-sided test was applied with a significance level of α=0.05 (P<0.05).
3 Results
3.1 Clinical data of HCoV-NL63 infected patients
From 2021 to 2022, a total of 638 children hospitalized in Guilin, Guangxi Zhuang Autonomous Region, due to acute respiratory infections, were tested for HCoV-NL63. Among them, 83 cases tested positive for HCoV-NL63 (Table 2). Although the detection rate of seasonal coronaviruses in Guilin for the same period in previous years is lacking, the detection rate of 13.01% (83/638) is significantly higher than the national average detection level of four common seasonal HCoVs from 2009 to 2019 (Li et al., 2021). Considering the sharp increase in cases from July to September and the composition ratio of single infections, it suggests a localized outbreak of HCoV-NL63 in Guilin in 2022, which is relatively uncommon.
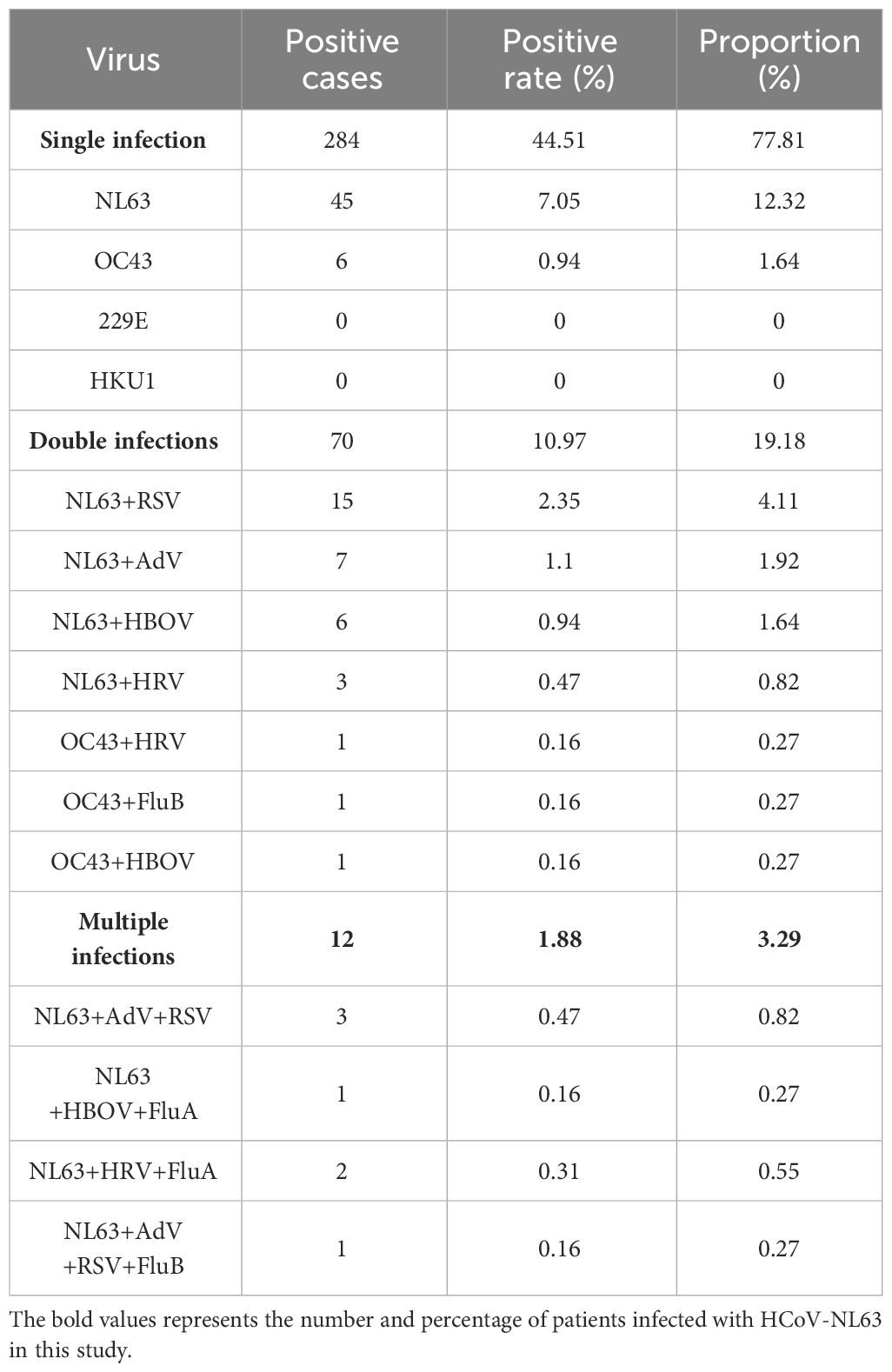
Table 2 Positive rate and proportion of four seasonal coronaviruses in ARI cases.
Most patients with HCoV-NL63 infection presented with upper respiratory tract infections, while 5 cases had pneumonia, 6 cases had bronchitis, and 2 cases had acute gastroenteritis. The age of the children ranged from 2 months to 12 years, and there was no statistically significant difference in detection rates between age groups and gender groups. Infections showed clear seasonality, with the peak of positive cases in August (Supplementary S2). The clinical information of HCoV-NL63-infected children is summarized in Table 2. The clinical manifestations included cough, fever, nasal congestion, shortness of breath, wheezing, abnormal lung breath sounds, and diarrhea. No significant statistical difference was observed in clinical presentation and diagnosis among different types of HCoV-NL63, including C3, C4, and B types (see Supplementary S3). Most children exhibited mild, self-limiting upper respiratory tract infections, with a small number experiencing gastrointestinal symptoms, consistent with previous reports (Kiyuka et al., 2018; Wen et al., 2022; Huang et al., 2017).
3.2 Co-infections
In addition to HCoV-NL63, samples from positive patients were tested for 14 other common respiratory viruses (Supplementary S4). Table 3 presents the detection results of the four seasonal coronaviruses. Among the 83 patients, 45 cases (54.22%) had HCoV-NL63 as a single infection, while 38 cases (45.78%) had co-infections with other viruses such as RSV, HBoV, and HRV (Table 3). Clinical characteristics, including age, gender, co-infection, and clinical symptoms, showed no significant differences among different genotypic infections. These data indicate that HCoV-NL63 primarily causes upper respiratory tract infections, and the prevalent strain in Guilin in this study is consistent with pathogenicity reported in previous studies (Zeng et al., 2018; Ye et al., 2023). However, the significantly increased infectivity suggests that the detected HCoV-NL63 epidemic strain may be a human respiratory pathogen with greater pathogenic potential than previously believed.
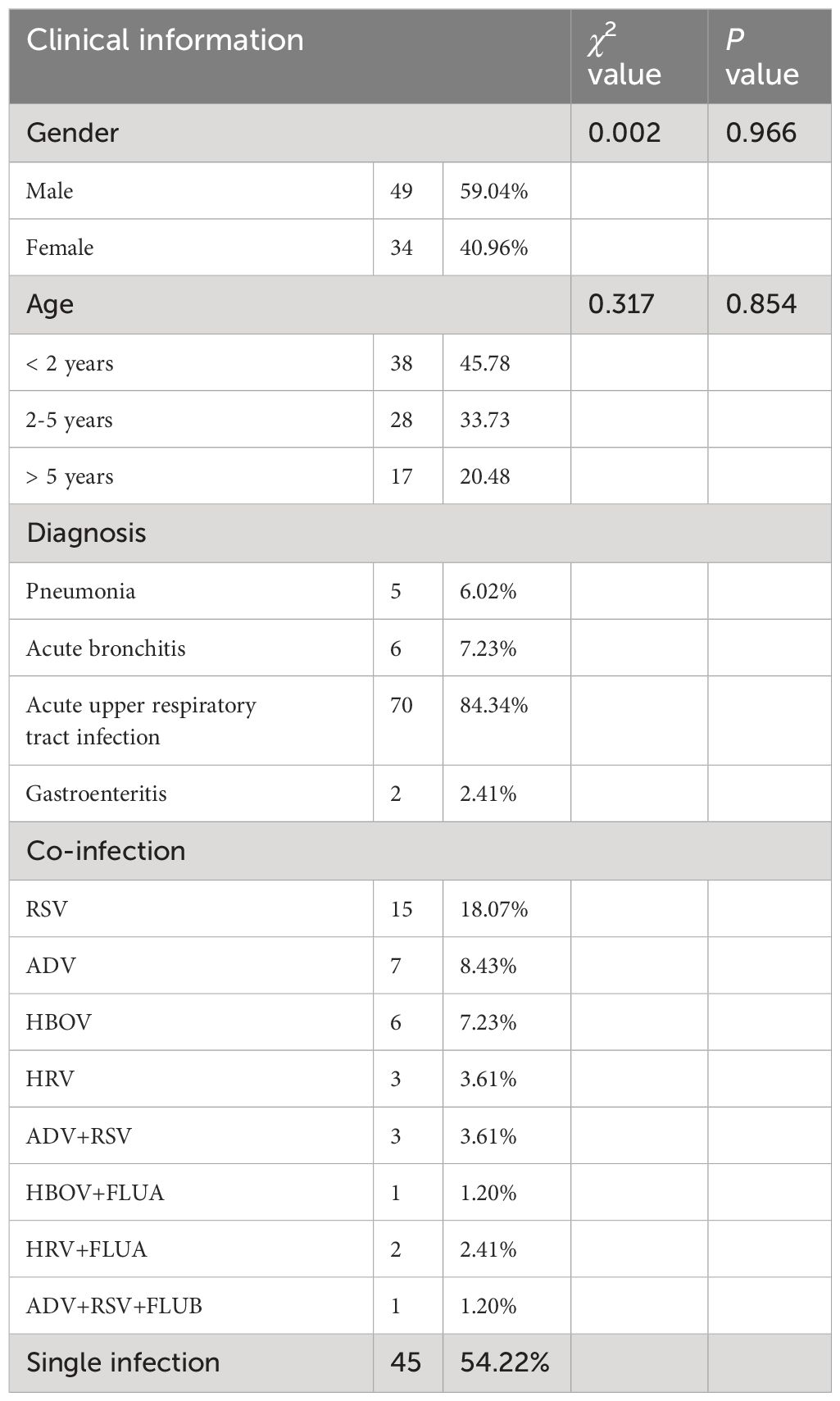
Table 3 Clinical data of children with acute respiratory infections caused by HCoV-NL63.
3.3 Topology of the evolutionary tree reveals the emergence of new HCoV-NL63 genotypic subtypes
A total of 35 S1 domain genes and 13 HCoV-NL63 full-length genomes were obtained. To understand the genetic relationships between the prevalent HCoV-NL63 strain detected in this study and other HCoV-NL63 strains in GenBank, phylogenetic analyses were conducted based on the Spike gene, S1 domain gene, ORF1ab, and full genome sequences.
In the ARI hospitalized children’s cases in Guilin, both B and C genotypes of HCoV-NL63 were co-circulating, with C being the predominant genotype. Among the 35 HCoV-NL63 S1 domain sequences in this study, they clearly evolved into an independent cluster within the C subtype in the phylogenetic tree. This cluster occupied a relatively distinct position in the phylogenetic tree, different from known C1-C3 genotypic subtypes, and formed a unique lineage, tentatively named as C4 (Figure 1). The evolutionary tree showed that the HCoV-NL63 S1 domain gene sequences detected in Guilin exhibited both temporal and spatial clustering, confirming their common ancestry and continuous circulation. Additionally, 12 epidemic strains from Japan, the United States, and other parts of China belonged to the same lineage, indicating that this new lineage may have already spread widely worldwide. The discovery of the new lineage C4 suggests potential risks associated with the rapid mutation of HCoV-NL63, leading to an epidemic of acute respiratory infections. Notably, C4 shared a common origin with the C3 genotypic subtype.
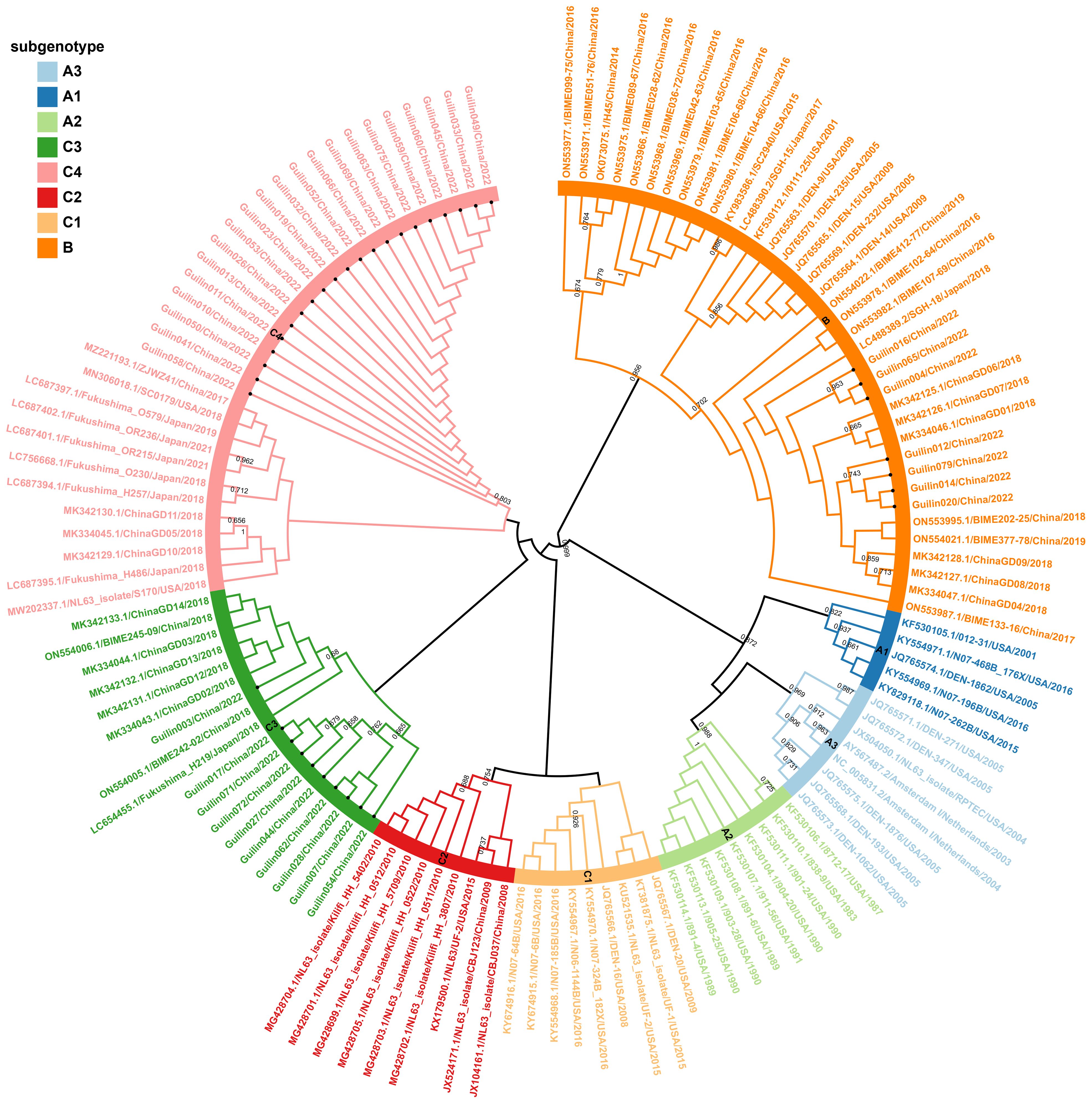
Figure 1 Phylogenetic analyses based on partial spike genes of HCoV-NL63 identified in this study. 38 HCoV-NL63 spike gene partial sequences detected in this study were used for phylogenetic analysis by MEGA 7.0 software using Neighbor-joining method and confirmed the presence of new subgenotype of HCoV-NL63. Bootstrap values greater than 65% were considered statistically significant for grouping. The sequence number in the tree with a circular symbol is the sequence obtained in this study.
For further evidence supporting this new lineage, phylogenetic analyses were conducted based on the full genome, ORF1ab, and Spike genes, showing similar topologies among the eight genotypic subtypes (Figures 2A–C). Systematic phylogenetic analysis further confirmed the emergence of the new lineage C4 of HCoV-NL63 in Guilin, China, suggesting its potential association with the increased cases of acute respiratory infections in 2022. Moreover, some epidemic strains obtained in this study belonged to the new genotypic subtype C3, discovered in 2018, as well as the B genotype. The shared ancestry between the new genotypic subtypes C4 and C3 in the evolutionary structure indicates a common origin. The discovery of the new genotypic subtype C4 suggests that HCoV-NL63 is undergoing continuous mutation, which could potentially contribute to the ongoing outbreak in Guilin.
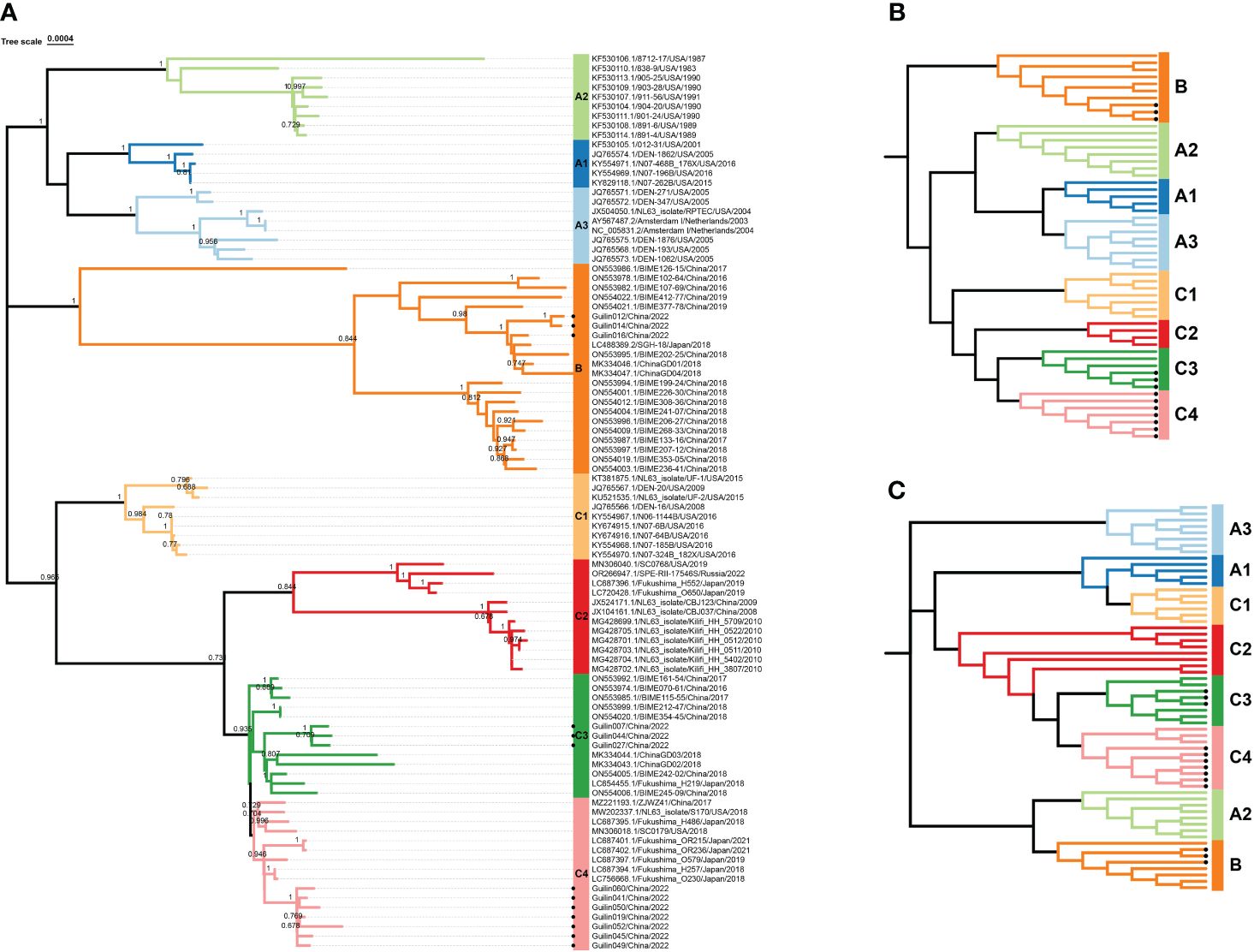
Figure 2 Phylogenetic analysis based on complete genome, S and ORF1ab genes of HCoV-NL63. All available HCoV-NL63 complete genomes from GenBank were collected and used for the evolutional analysis using MEGA 7.0 with 1000 bootstrap replications, Bootstrap values greater than 70% were considered statistically significant for grouping. (A). Five complete genomes derived from this study were in red. Nucleotide sequence alignments were created using MAFFT. Corresponding spike (B) and orf1ab (C) genes were used for the genotype identification. The sequence number in the tree with a circular symbol is the sequence obtained in this study.
3.4 Phylogenetic analysis of temporal signal analysis and time scale estimation
The Bayesian Evolutionary Analysis by Sampling Trees (BEAST) program is extensively utilized for exploring the spatiotemporal and evolutionary dynamics of viral pathogens through time-stamped nucleotide sequence datasets. Under the non-strict molecular clock model, the Bayesian MCC tree based on the HCoV-NL63 S gene sequence dataset is shown in Figure 3. The estimated evolution rate (4.37×10-4 substitutions per site per year) and TMRCA date (1978.674) results reveal ESS values exceeding 200, consistent with the results of the time signal analysis (Supplementary Figure 2). The MCC tree illustrates that the obtained 13 HCoV-NL63 strains belong to genotypic subtypes C3, C4, and B. They exhibit a certain temporal and spatial clustering, with subtype C4 being closely related to epidemic strains from Japan and the United States. This further confirms the results of the phylogenetic analysis based on the S1 domain, indicating the emergence of a new lineage of HCoV-NL63.
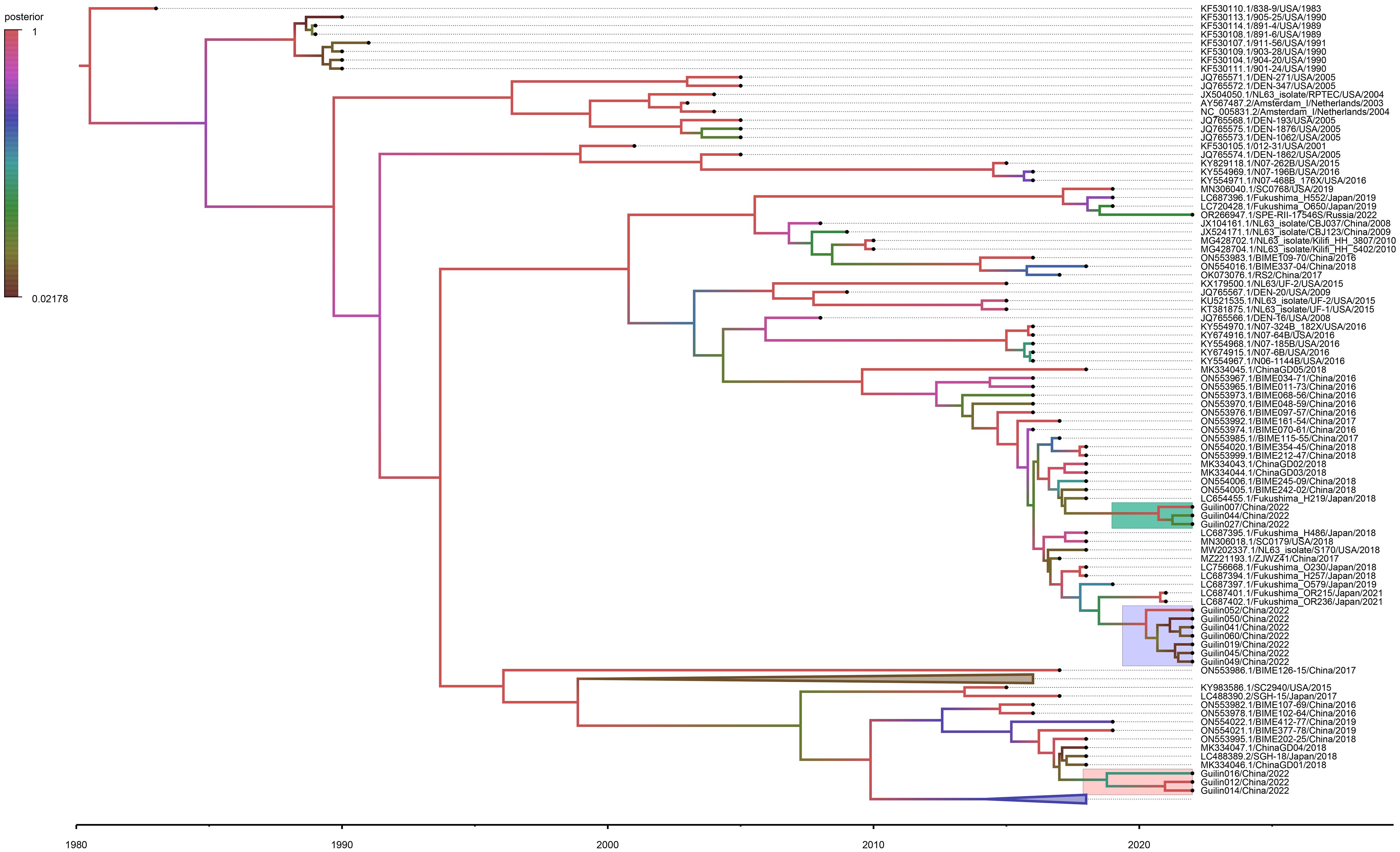
Figure 3 Time-resolved phylogenetic analysis of spike gene of HCoV-NL63 strains. The node label represents the posterior probability. The C4, C3, and B sub-genotype strains detected in the Guilin area are indicated in green, purple, and gray. BEAST software was used to estimate the nearest common ancestor (tMRCA) of the new subgenotype circulating in the Guilin area based on the nucleotide sequence of the Spike gene. Analyses were conducted under the best-fit nucleotide substitution model (TN93+F+I+G4) and using a relaxed (uncorrelated lognormal) molecular clock model.
According to the MCC time-calibrated tree estimation, although C3 and C4 share a common ancestor, they diverged around 2015, indicating different evolutionary paths. Overall, the results of phylogenetic analysis suggest that, in recent years (2021-2022), genotypic subtypes C3, C4, and B dominate among all prevalent HCoV-NL63 strains in the Guilin region. This pattern aligns with the predominant circulation of genotypic subtypes C and B of HCoV-NL63 in China.
3.5 Reorganization analysis
The 13 aligned sequences of HCoV-NL63 were imported into SimPlot software for subsequent recombination analysis. The results of the recombination analysis for HCoV-NL63, as shown in Figure 4, indicate no clear potential recombination regions in ORF1ab, S to N. Combining phylogenetic and recombination analyses, it can be suggested that the new genotypic subtype C4 of HCoV-NL63 may result from gene mutations rather than homologous gene recombination. No apparent intraspecific recombination signals were detected, suggesting that the new genotypic subtype C4 of HCoV-NL63 may have originated from gene mutations rather than intraspecific gene recombination.
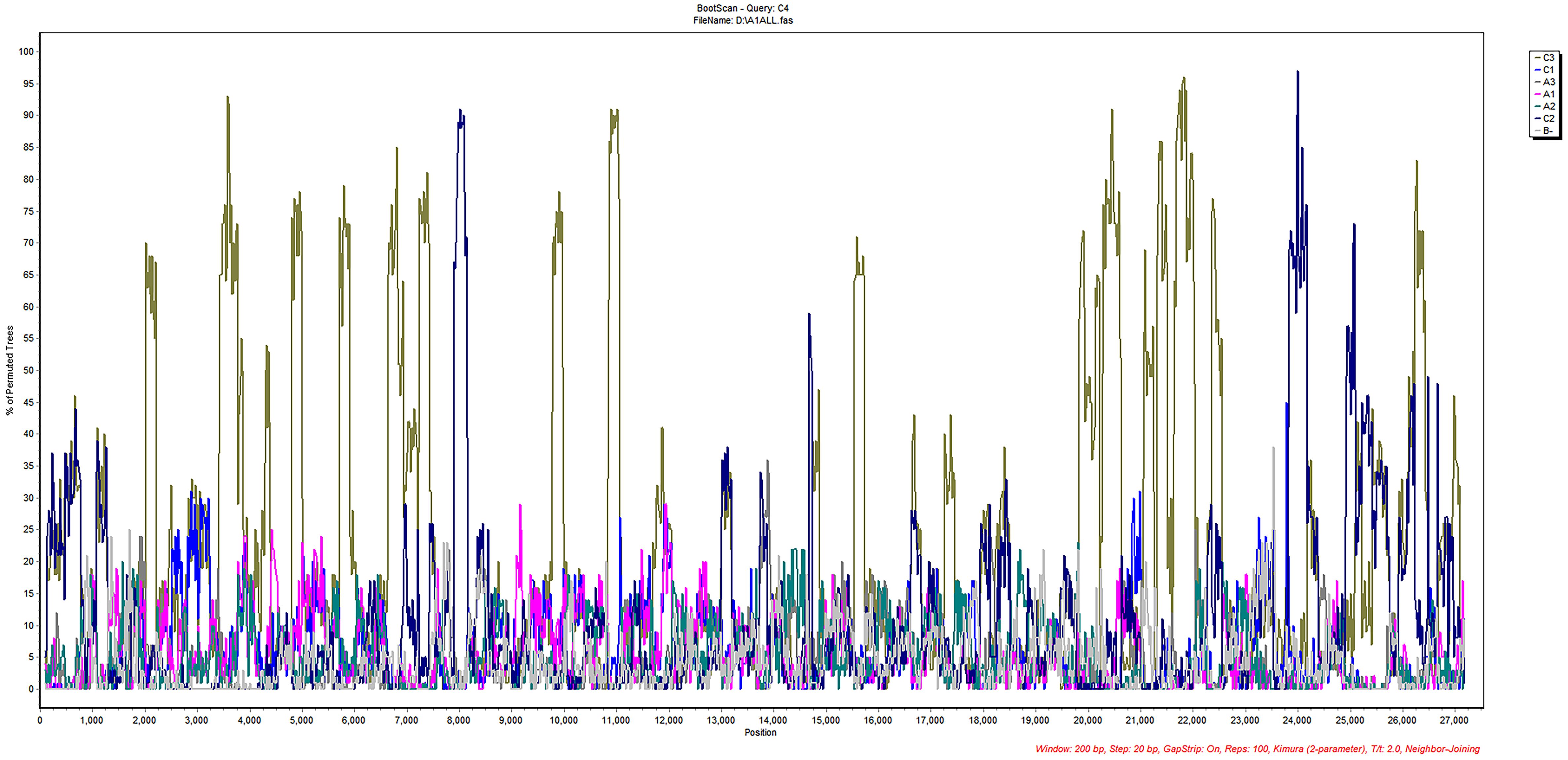
Figure 4 Recombination analyses of HCoV-NL63.Similarity plot and bootscan analysis were performed intraspecies. No obvious recombination events were detected in new subgenotype C4 using Simplot software analysis.
3.6 Amino acid mutations
The spike protein plays a crucial role in the entry and pathogenic mechanisms of coronaviruses. Specific mutations in the amino acid residues of the spike protein, particularly in the receptor-binding domain (RBD) responsible for specific binding to the ACE2 receptor protein, may alter the interaction levels between HCoV-NL63 and host cells, thereby affecting the virus’s virulence or transmission within the population. To further describe the antigenic features of the prevalent HCoV-NL63 strains detected in this study and understand the reasons for the localized outbreak in Guilin, a single amino acid polymorphism analysis (SAP) was conducted on the spike protein sequences, as shown in Figure 5.
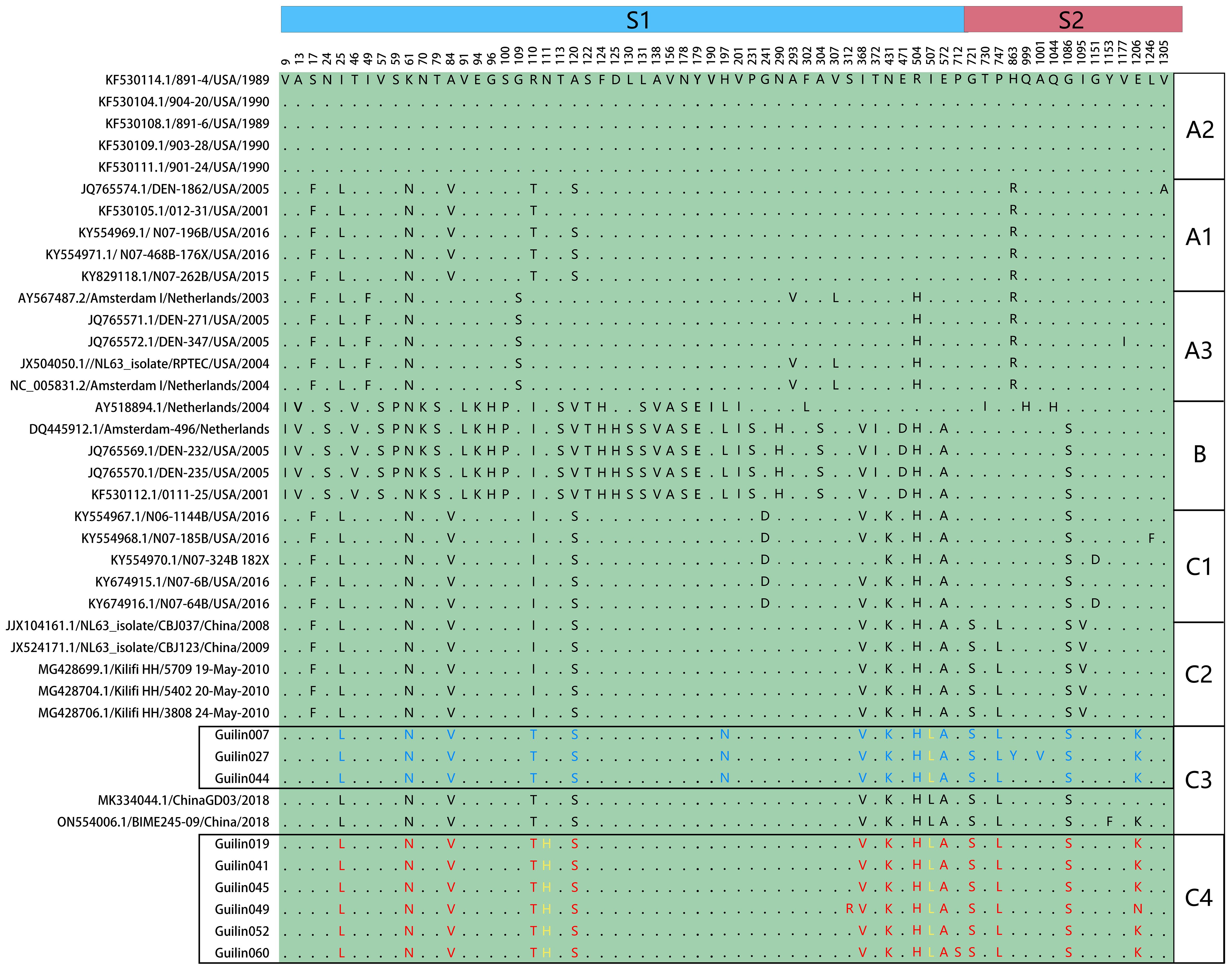
Figure 5 Single amino acid polymorphism analysis of HCoV-NL63 spike protein. All available HCoV-NL63 complete genomes were aligned, and corresponding spike proteins were retrieved and used for single amino acid polymorphism analysis. One unique mutation N111H was identified in yellow.
The SAP analysis identified 65 polymorphic sites in the spike protein, highlighting genotypic-specific SAPs and confirming the rationale behind the new genotypic subtype classification (A1, A2, A3, B, C1, C2, C3, and C4). The amino acid mutation analysis revealed that the C4 subtype in the spike protein had a unique mutation, S574L, at the 574th amino acid residue in the receptor-binding domain (RBD), which is also detected in other C4 subtype epidemic strains in the N-terminal domain (NTD) of the spike protein. Furthermore, a specific mutation, I507L, associated with increased infection capability, was observed in the RBD of the central region of the spike protein in the C4 subtype, belonging to the C3 genotypic subtype. This mutation might be related to the increased number of acute respiratory infections caused by HCoV-NL63 in Guilin in 2022. These findings confirm the validity of the new genotypic subtype classification (A1, A2, A3, B, C1, C2, C3, and C4) and suggest an increased pathogenic risk of HCoV-NL63 with evolution.
4 Discussion
Until now, three highly pathogenic human coronaviruses have been identified: SARS-CoV, MERS-CoV, and SARS-CoV-2. Coronaviruses have caused two large-scale public health crises in the past 20 years. As of 2021, the global COVID-19 pandemic caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has resulted in approximately 6.9 million deaths, posing unprecedented challenges to global health (Drosten et al., 2003; Zaki et al., 2012; World Health Organization (2023)). As mentioned earlier, SARS-CoV, SARS-CoV-2, and HCoV-NL63 share the same receptor, angiotensin-converting enzyme (ACE)-2, when entering host cells. However, the consequences of virus invasion differ significantly. While SARS-CoV and SARS-CoV-2 can cause severe respiratory distress, HCoV-NL63 typically leads to mild respiratory tract infections limited to the upper respiratory tract in most individuals. This may be attributed to the lower affinity of NL63 spike protein for the receptor compared to SARS-CoV-2. Consequently, outbreaks of respiratory infections caused by HCoV-NL63 in children are rarely reported (Nyaguthii et al., 2021; Hand et al., 2018).
In this study, focusing on pediatric patients hospitalized in Guilin, Guangxi Zhuang Autonomous Region, due to acute respiratory infections in 2021-2022, we obtained a total of 13 nearly full-length HCoV-NL63 genome sequences and 25 S1 domain gene sequences. Molecular characteristics, including phylogenetic analysis, time signals, potential recombination events, and amino acid mutations, were described. Compared to national HCoV infection levels in the past 10 years, the number of HCoV-NL63 infection cases in Guilin significantly increased in 2021-2022, indicating a rare small-scale outbreak of acute respiratory infections caused by HCoV-NL63 in Guilin, China. Additionally, phylogenetic and recombination analyses suggested an association between this outbreak and three HCoV-NL63 subtypes (C3, C4, and B), with the identification of a new genetic subtype, C4. C4, homologous to C3, exhibited distinct evolutionary trends, and genetic variations may help the virus adapt to external environments to maintain prevalence in populations.
The spike protein plays a crucial role in coronavirus invasion of host cells and the resulting host disease process. Mutations in the amino acid residues of the spike protein, particularly in the receptor-binding domain (RBD) responsible for specific binding to the ACE2 receptor protein, may alter the interaction levels between HCoV-NL63 and host cells, affecting the virus’s virulence or transmission (Lin et al., 2011; Wu et al., 2009). The study identified specific mutations, such as I507L, in the RBD, which have been confirmed to enhance viral entry (Wang et al., 2020). This may be a contributing factor to the increased infectivity of the HCoV-NL63 C4 genetic subtype in the Guilin region. Furthermore, mutations in the N-terminal domain (NTD) of the spike protein, particularly at N111 and N119, were observed in C4, involving amino acid substitutions R110T and N111H. According to previous reports, the N-linked glycosylation sites at aa positions N111 and N119 made direct polar contacts, and they were linked to aa substitution sites R110I and A120S (Shao et al., 2022). While the importance of these mutations in the NTD region for SARS-CoV-2 has been reported (Cui et al., 2022), further evidence is needed to determine potential changes in spike protein antigenicity resulting from mutations in the N-glycosylation sites of the C4 genetic subtype. In summary, subtype-specific mutations in the spike protein further confirm the rationale behind the new subtype classification (A1, A2, A3, B, C1, C2, C3, and C4). It is important to emphasize that the emergence of the new subgenotype C4 is a natural consequence of virus evolution. Sustained genetic variation aids the virus in adapting to its external environment, thereby maintaining its prevalence in the population-a common and expected phenomenon among RNA viruses. The conclusions drawn in this study are based on limited datasets from specific geographic regions and time periods. Thus, it is premature to draw comprehensive conclusions regarding the global significance of C4 gene subtypes or their associated mutations.
HCoV-NL63 is generally highly susceptible to the human population, with most individuals experiencing their first infection in childhood and neutralizing antibodies against HCoV-NL63 are common in adult serum (Hofmann et al., 2005), but this protective immunity is not durable (Amoretti et al., 2002), leading to recurrent infections. Additionally, like other coronaviruses, HCoV-NL63 may be a zoonotic pathogen originating from bats (Huynh et al., 2012). Interspecies genetic recombination resulting from different HCoV strains carried by bats is one reason for the seasonal/recurrent infection pattern of HCoV-NL63 (Tao et al., 2017). Further research is necessary to elucidate the significance of the unique amino acid N111H mutation in the C4 subtype NTD as observed in this study.
While pathogenic changes are inevitable with the virus’s continuous spread and evolution, considering that HCoV-NL63 and SARS-CoV-2 share the same cell receptor, and given the severe consequences of the global spread of SARS-CoV-2, the potential risks of increased virulence or epidemic spread due to ongoing mutations of HCoV-NL63 are significantly elevated. Therefore, regular epidemiological and phylogenetic studies and the prompt establishment of a comprehensive HCoV-NL63 molecular epidemiological surveillance system are of particular practical importance.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Ethics Review Committee of National Institute for Viral Control and Prevention, Chinese Center for Disease Control and Prevention. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
RZ: Writing – original draft, Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Investigation, Software. RC: Investigation, Methodology, Software, Validation, Writing – original draft. LW: Conceptualization, Investigation, Software, Writing – original draft. YG: Formal analysis, Investigation, Methodology, Software, Writing – original draft. QC: Investigation, Methodology, Software, Visualization, Writing – original draft. HL: Investigation, Resources, Supervision, Writing – review & editing. DX: Investigation, Methodology, Supervision, Writing – original draft. QS: Investigation, Methodology, Resources, Supervision, Writing – original draft. ZX: Investigation, Methodology, Supervision, Writing – original draft. ML: Formal analysis, Investigation, Methodology, Supervision, Writing – original draft. HD: Investigation, Software, Supervision, Writing – review & editing. JS: Conceptualization, Investigation, Methodology, Supervision, Writing – review & editing. JH: Formal analysis, Funding acquisition, Methodology, Resources, Supervision, Writing – review & editing. CG: Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key R&D Program of China 2023YFC2605602, the Project of National Pathogen Resource Collection Center (Grant NO. NPRC-32), SKLID Development Grant (Grant NO. 2011SKLID104).
Acknowledgments
The authors would like to thank all physicians from Guilin Center for Disease Control and Prevention for collecting medical data and specimens.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1378804/full#supplementary-material
References
Amoretti, M., Amsler, C., Bonomi, G., Bouchta, A., Bowe, P., Carraro, C., et al. (2002). Production and detection of cold antihydrogen atoms. Nature 419, 456–459. doi: 10.1038/nature01096
China MoHotPsRo. (2011). Sentinel Monitoring Plan for Severe Acute Respiratory Infection Cases in Hospitals. Available online at: http://www.gov.cn/zwgk/2011-02/11/content_1801649.htm.
Collaborators GDaI (2020). Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222. doi: 10.1016/s0140-6736(20)30925-9
Collaborators GI (2019). Mortality, morbidity, and hospitalisations due to influenza lower respiratory tract infections, 2017: an analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 7, 69–89. doi: 10.1016/s2213-2600(18)30496-x
Cui, Z., Liu, P., Wang, N., Wang, L., Fan, K., Zhu, Q., et al. (2022). Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 185, 860–871. doi: 10.1016/j.cell.2022.01.019
Drosten, C., Günther, S., Preiser, W., van der Werf, S., Brodt, H. R., Becker, S., et al. (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976. doi: 10.1056/NEJMoa030747
Hand, J., Rose, E. B., Salinas, A., Lu, X., Sakthivel, S. K., Schneider, E., et al. (2018). Severe respiratory illness outbreak associated with human coronavirus NL63 in a long-term care facility. Emerg. Infect. Dis. 24, 1964–1966. doi: 10.3201/eid2410.180862
Hofmann, H., Pyrc, K., van der Hoek, L., Geier, M., Berkhout, B., Pöhlmann, S. (2005). Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U. S. A. 102, 7988–7993. doi: 10.1073/pnas.0409465102
Huang, S. H., Su, M. C., Tien, N., Huang, C. J., Lan, Y. C., Lin, C. S., et al. (2017). Epidemiology of human coronavirus NL63 infection among hospitalized patients with pneumonia in Taiwan. J. Microbiol. Immunol. Infect. 50, 763–770. doi: 10.1016/j.jmii.2015.10.008
Huynh, J., Li, S., Yount, B., Smith, A., Sturges, L., Olsen, J. C., et al. (2012). Evidence supporting a zoonotic origin of human coronavirus strain NL63. J. Virol. 86, 12816–12825. doi: 10.1128/jvi.00906-12
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kiyuka, P. K., Agoti, C. N., Munywoki, P. K., Njeru, R., Bett, A., Otieno, J. R., et al. (2018). Human coronavirus NL63 molecular epidemiology and evolutionary patterns in Rural Coastal Kenya. J. Infect. Dis. 217, 1728–1739. doi: 10.1093/infdis/jiy098
Konca, C., Korukluoglu, G., Tekin, M., Almis, H., Bucak, İ. H., Uygun, H., et al. (2017). The first infant death associated with human coronavirus NL63 infection. Pediatr. Infect. Dis. J. 36, 231–233. doi: 10.1097/inf.0000000000001390
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, Z. J., Zhang, H. Y., Ren, L. L., Lu, Q. B., Ren, X., Zhang, C. H., et al. (2021). Etiological and epidemiological features of acute respiratory infections in China. Nat. Commun. 12, 5026. doi: 10.1038/s41467-021-25120-6
Lim, Y. X., Ng, Y. L., Tam, J. P., Liu, D. X. (2016). Human coronaviruses: A review of virus-host interactions. Diseases 4(3):26. doi: 10.3390/diseases4030026
Lin, H. X., Feng, Y., Tu, X., Zhao, X., Hsieh, C. H., Griffin, L., et al. (2011). Characterization of the spike protein of human coronavirus NL63 in receptor binding and pseudotype virus entry. Virus Res. 160, 283–293. doi: 10.1016/j.virusres.2011.06.029
Lole, K. S., Bollinger, R. C., Paranjape, R. S., Gadkari, D., Kulkarni, S. S., Novak, N. G., et al. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160. doi: 10.1128/jvi.73.1.152-160.1999
Mayer, K., Nellessen, C., Hahn-Ast, C., Schumacher, M., Pietzonka, S., Eis-Hübinger, A. M., et al. (2016). Fatal outcome of human coronavirus NL63 infection despite successful viral elimination by IFN-alpha in a patient with newly diagnosed ALL. Eur. J. Haematol. 97, 208–210. doi: 10.1111/ejh.12744
Nyaguthii, D. M., Otieno, G. P., Kombe, I. K., Koech, D., Mutunga, M., Medley, G. F., et al. (2021). Infection patterns of endemic human coronaviruses in rural households in coastal Kenya. Wellcome Open Res. 6, 27. doi: 10.12688/wellcomeopenres.16508.1
Oosterhof, L., Christensen, C. B., Sengeløv, H. (2010). Fatal lower respiratory tract disease with human corona virus NL63 in an adult haematopoietic cell transplant recipient. Bone Marrow Transplant. 45, 1115–1116. doi: 10.1038/bmt.2009.292
Shao, N., Zhang, C., Dong, J., Sun, L., Chen, X., Xie, Z., et al. (2022). Molecular evolution of human coronavirus-NL63, -229E, -HKU1 and -OC43 in hospitalized children in China. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1023847
Tao, Y., Shi, M., Chommanard, C., Queen, K., Zhang, J., Markotter, W., et al. (2017). Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. J. Virol. 91:10.1128/jvi.01953-16. doi: 10.1128/jvi.01953-16
van der Hoek, L. (2007). Human coronaviruses: what do they cause? Antivir. Ther. 12, 651–658. doi: 10.1177/135965350701200S01
van der Hoek, L., Pyrc, K., Jebbink, M. F., Vermeulen-Oost, W., Berkhout, R. J., Wolthers, K. C., et al. (2004). Identification of a new human coronavirus. Nat. Med. 10, 368–373. doi: 10.1038/nm1024
Wang, Y., Li, X., Liu, W., Gan, M., Zhang, L., Wang, J., et al. (2020). Discovery of a subgenotype of human coronavirus NL63 associated with severe lower respiratory tract infection in China, 2018. Emerg. Microbes Infect. 9, 246–255. doi: 10.1080/22221751.2020.1717999
Wen, C., Sun, L., Zhao, M. C., Duan, S. X., Wang, L., Cui, X. W. (2022). Clinical study of human coronavirus NL63, OC43, 229E, HKU1 infentions in hospitalized children from 2015 to 2020. Infect. Drug Resist. 15, 1093–1101. doi: 10.2147/idr.S357193
World Health Organization. (2023). WHO Coronavirus Disease (COVID-19) Dashboard. Available at: https://data.who.int/dashboards/covid19/deaths.
Wu, K., Li, W., Peng, G., Li, F. (2009). Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U. S. A. 106, 19970–19974. doi: 10.1073/pnas.0908837106
Ye, R. Z., Gong, C., Cui, X. M., Liu, J. Y., Fan, H., Xie, H., et al. (2023). Continuous evolution and emerging lineage of seasonal human coronaviruses: A multicenter surveillance study. J. Med. Virol. 95, e28861. doi: 10.1002/jmv.28861
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D., Fouchier, R. A. (2012). Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820. doi: 10.1056/NEJMoa1211721
Zeng, Z. Q., Chen, D. H., Tan, W. P., Qiu, S. Y., Xu, D., Liang, H. X., et al. (2018). Epidemiology and clinical characteristics of human coronaviruses OC43, 229E, NL63, and HKU1: a study of hospitalized children with acute respiratory tract infection in Guangzhou, China. Eur. J. Clin. Microbiol. Infect. Dis. 37, 363–369. doi: 10.1007/s10096-017-3144-z
Keywords: human coronavirus NL63, new subgenotype, molecular epidemiology, clinical characteristics, whole-genome sequencing
Citation: Zhu R, Cao R, Wang L, Gong Y, Cheng Q, Long H, Xia D, Song Q, Xia Z, Liu M, Du H, Song J, Han J and Gao C (2024) Seasonal human coronavirus NL63 epidemics in children in Guilin, China, reveal the emergence of a new subgenotype of HCoV-NL63. Front. Cell. Infect. Microbiol. 14:1378804. doi: 10.3389/fcimb.2024.1378804
Received: 30 January 2024; Accepted: 11 March 2024;
Published: 26 April 2024.
Edited by:
Laura Petrarca, Sapienza University of Rome, ItalyReviewed by:
Mingbin Liu, Nanchang University, ChinaEllen Van Damme, Janssen Pharmaceutica NV, Belgium
Copyright © 2024 Zhu, Cao, Wang, Gong, Cheng, Long, Xia, Song, Xia, Liu, Du, Song, Han and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Gao, gaochen@ivdc.chinacdc.cn; Jun Han, hanjun_sci@163.com