
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 21 March 2024
Sec. Molecular Bacterial Pathogenesis
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1376289
This article is part of the Research Topic Research implications on microbial virulence factors, resistance, and new therapeutic strategies in the context of future infectious disease therapies. View all 9 articles
 Zhenzhou Huang1
Zhenzhou Huang1 Guozhong Zhang1Zhibei Zheng1Xiuqin Lou1Feifei Cao1Lingyi Zeng1
Guozhong Zhang1Zhibei Zheng1Xiuqin Lou1Feifei Cao1Lingyi Zeng1 Duochun Wang2Keyi Yu2*
Duochun Wang2Keyi Yu2* Jun Li1*
Jun Li1*Introduction: Kluyvera is a Gram-negative, flagellated, motile bacillus within the Enterobacteriaceae. The case reports of clinical infections shed light on the importance of this organism as an emerging opportunistic pathogen. The genus Phytobacter, which often be misidentified with Kluyvera, is also an important clinically relevant member of the Enterobacteriaceae. However, the identification of Kluyvera and Phytobacter is problematic, and their phylogenetic relationship remains unclear.
Methods: Here, 81 strains of Kluyvera and 16 strains of Phytobacter were collected. A series of comparative genomics approaches were applied to the phylogenetic relationship reconstruction, virulence related genes profiles description, and antibiotic resistance genes prediction.
Results: Using average nucleotide identity (ANI) and in silico DNA-DNA hybridization (isDDH), we offered reliable species designations of 97 strains, in which 40 (41.24%) strains were incorrectly labeled. A new Phytobacter genomospecies-1 were defined. Phytobacter and Kluyvera show great genome plasticity and inclusiveness, which may be related to their diverse ecological niches. An intergenomic distances threshold of 0.15875 was used for taxonomy reassignments at the phylogenomic-group level. Further principal coordinates analysis (PCoA) revealed 11 core genes of Kluyvera (pelX, mdtL, bglC, pcak-1, uhpB, ddpA-2, pdxY, oppD-1, cptA, yidZ, csbX) that could be served as potential identification targets. Meanwhile, the Phytobacter specific virulence genes clbS, csgA-C, fliS, hsiB1_vipA and hsiC1_vipB, were found to differentiate from Kluyvera. We concluded that the evolution rate of Kluyvera was 5.25E-6, approximately three times higher than that of Phytobacter. Additionally, the co-existence of ESBLs and carbapenem resistance genes were present in approximately 40% strains, suggesting the potential development of extensively drug-resistant or even fully drug-resistant strains.
Discussion: This work provided a better understanding of the differences between closely related species Kluyvera and Phytobacter. Their genomes exhibited great genome plasticity and inclusiveness. They not only possess a potential pathogenicity threat, but also a risk of multi-drug resistance. The emerging pathogens Kluyvera and Phytobacter warrant close attention.
Kluyvera is a Gram-negative, flagellated, motile bacillus within the Enterobacteriaceae. Members of this genus were phenotypically identical to Escherichia including the ability to use citrate, malonate, decarboxylate lysine, and ornithine and to produce large quantities of α-ketoglutaric acid during the glucose fermentation (Sarria et al., 2001). It can be isolated from various clinical and environmental sources such as freshwater, seawater, sewage, soil and rhizosphere (Sarria et al., 2001; Gessew et al., 2022; Paschoal et al., 2017; Li Y. et al., 2019; Yim et al., 2009). Since its redefinition in 1981 (Farmer et al., 1981), Kluyvera has been recognized as an important pathogen in human disease (Sarria et al., 2001), with reported infections in the gastrointestinal or urinary tract, the soft tissues, and cases of bacteremia and systemic infections involving multiple organs (Bolat et al., 2013; Erbin et al., 2020; Sierra-Madero et al., 1990; Sezer et al., 1996). Kluyvera also serves as a reservoir and communicator of the blaCTX-Ms genes, which are responsible for the production of Extended-Spectrum Beta-Lactamases (ESBLs). The chromosomal blaCTX-Ms genes from Kluyvera have been shown to be the progenitors of the plasmid-encoded blaCTX-Ms (Cantón et al., 2008).
In recent years, the taxonomy of Kluyvera has become increasingly complex with the identification of new genomospecies. Currently, there are five recognized species (K. ascorbata, K. cryocrescens, K. georgiana, K. intermedia, K. sichuanensis). Liu et al. identified four Kluyvera genomospecies according to the available sequences in Genbank (Liu et al., 2020). Rodríguez et al. proposed a new genomospecies-5 (Rodríguez et al., 2021). The classification of some genome sequences in NCBI has shown inconsistency with Phytobacter (Rodríguez et al., 2021; Smits et al., 2022), further emphasizing the need for clarification of genus taxonomy reassignments.
The taxonomic status of Phytobacter has also undergone a complex process. Originally described in 2008, Phytobacter and its type species Phytobacter diazotrophicus were isolated as endophytic nitrogen-fixing bacteria from wild rice in China (Zhang et al., 2008). However, subsequent research revealed that clinical isolates from a multi-state sepsis outbreak in Brazil also belonged to Phytobacter diazotrophicus (Pillonetto et al., 2018a). Moreover, a new Phytobacter species, Phytobacter ursingii, was identified among American clinical strains (Pillonetto et al., 2018a). Phytobacter has been found to occupy various ecological niches and can adapt to clinical environments, potentially becoming a multidrug-resistant microorganism (Smits et al., 2022; Sekizuka et al., 2018; Pillonetto et al., 2018b; Zhang et al., 2020).
Based on previous investigations, Phytobacter may have similar clinical relevance to Kluyvera (Smits et al., 2022). Despite their clinical significance, the genomic characteristics, virulence and resistance signature of Kluyvera and Phytobacter remain poorly understood. In this study, we conducted a comprehensive analysis of 97 high-quality draft genome sequences, including four newly sequenced strains, to provide new insights into the species status, genetic relatedness, potential pathogenicity and resistance genes diversity of these genera.
Ninety-three genomes belonging to the genus Kluyvera (originally labeled: n=77) and Phytobacter (originally labeled: n=16) were downloaded from the NCBI FTP server (ftp.ncbi.nih.gov) and SRA database (https://www.ncbi.nlm.nih.gov/sra). Only genomes with less than 200 contigs were selected for analysis. Additional four newly sequenced strains isolated from China (deposited in the Centre for Human Pathogenic Culture Collection, China CDC) were included in the study (GenBank accession: PRJNA899585). These strains were initially identified by 16S rRNA, API 20E and API 20NE characterization methods. Detailed information of selected strains was listed in Supplementary Table 1. Overall, a total of 93 publicly available genomes and four newly sequenced strains were used for the analysis.
The strains were cultured on LB-1% NaCl agar at a temperature of 37°C. The genomic DNA from the four isolated strains was performed using the Wizard@ Genomic DNA Extraction Kit (Madison, WI, Promega, USA) following the manufacturer’s instructions. Subsequently, the extracted DNA samples were subjected to 250-bp paired-end whole genome sequencing with 150× coverage using the HiSeq sequencer (Illumina HiSeq2000, San Diego, CA, USA). And the resulting reads were de novo assembled into contigs using SPAdes v3.13.0 (Bankevich et al., 2012).
Average nucleotide identity (ANI) calculations (Richter and Rosselló-Móra, 2009) and the genome-to-genome blast distance phylogeny (GBDP) algorithm were used for the phylogenomic analysis, which replicates the DNA-DNA hybridization digitally for species delineation (Meier-Kolthoff et al., 2013). Minimal cutoff points of 70% in silico DNA-DNA hybridization (isDDH) and 95% ANI values were considered to represent species delineation. Intergenomic distances among the 97 identified strains were calculated by using the genome-to-genome distance calculator web service (GGDC, http://ggdc.dsmz.de/) (Meier-Kolthoff et al., 2013). These distances were then transformed into a matrix and imported into FastMe v2.0 (Lefort et al., 2015) to build the neighbor-joining (NJ) phylogenomic tree. Aeromonas hydrophila ATCC 7966T was served as the outgroup. To assess strain identification at the genus or species level, the online TYGS platform (https://tygs.dsmz.de/user_requests/new) was utilized (Meier-Kolthoff and Göker, 2019). Clustering of intergenomic distances was examined using the OPTSIL v1.5 (Garrido-Sanz et al., 2020), which creates a non-hierarchical clustering using distance threshold T and a specific F value. F values ranging from 0 to 1 indicate the fraction of links required for cluster fusion. F value of 0.5 represents average-linkage clustering. In this study, the distance threshold (T) values ranging from 0 to 0.3 were used with a step size of 0.0005. The best T and F for both, phylogenomic-group level and species level were chosen based on the highest modified Rand Index (MRI) score.
A local database containing 97 selected strains sequences was created for further analysis. The genomes were annotated using Prokka v1.12 (Seemann, 2014), which generated *.gff output files for each strain. The Roary pan-genome pipeline was then used with an identity cutoff of ≥95% (Page et al., 2015). Prodigal v2.6.3 was used for coding sequence prediction (Hyatt et al., 2010). To extract the core genes from all the selected strains, CD-HIT v4.6.6 was employed to create a non-redundant set of homologous genes (Fu et al., 2012). Next, BLAST+ was used to search for homologous genes in this non-redundant set with a minimum identity ≥90% and length coverage ≥60%. The identified core genes were aligned, and Gubbins (http://github.com/sanger-pathogens/gubbins) was used as a recombination-removal tool to reorganize the core genome. Phylogenetic trees were constructed using PhyML v3.1 by maximum-likelihood method with 1000 bootstrap replications (Guindon et al., 2010). Population structure was defined using FastBaps (https://github.com/gtonkinhill/fastbaps) by a fast hierarchical Bayesian analysis (Tonkin-Hill et al., 2019). Snippy v4.3.6 was used to extract core SNPs, which were then used to build a maximum-likelihood phylogenetic tree. Principal coordinate analysis (PCoA) based on Bray-Curtis distance and Jaccard distance was performed to assess the comprehensive composition of the accessory genes of these strains. Principal component analysis (PCA) was performed with prcomp function in R v3.6.1 based on the presence of common (5–95% prevalence) accessory genes.
We conducted evolutionary analysis on the core genes of Kluyvera and Phytobacter, and estimated the evolutionary origin time for each genus using BEAST v1.10.4 (Hill and Baele, 2019). The molecular clock was calibrated using the isolation date (by year of collection) for each strain. A GTR+Γ substitution model and uncorrelated log-normal relaxed clock were used in our analysis. The tree prior’s parameters were specified based on a normal distribution. For Kluyvera, we performed a Markov chain Monte Carlo (MCMC) of 1×10-8 iterations and sampled every 20,000 iterations to assure independent convergence of the chains. Similarly, for Phytobacter, we ran three independent Markov chain Monte Carlo of 1×10-8 iterations and sampled every 20,000 iterations. Convergence of the MCMC chains was assessed using Tracer v1.6.0, ensuring that all relevant parameters reached an effective sample size >200.
In this study, we analyzed the virulence-related gene profiles of 97 strains. The virulence-related genes were annotated according to the Virulence Factors Database (VFDB, http://www.mgc.ac.cn/VFs/). The protein sequences of selected strains were searched against the VFDB database using BLASTp with the parameters of E-value of 1e-5, identity ≥60% and coverage ≥70%. The pan virulence-related genes analyzed in this study included both core virulence-related genes, which were present in all strains, and accessory virulence-related genes (Li Z. et al., 2019), which were only present in some strains.
Antibiotic resistance genes were predicted using Comprehensive Antibiotic Research Database (CARD) (http://arpcard.mcmaster.ca), with the parameters of BLAST+ were E-value of 1e-5, identity ≥80% and coverage ≥80%.
Based on ANI and isDDH analysis, the multiple inconsistencies regarding the current assignment to species were observed (Figures 1A, B). A total of 40 (41.24%) strains were incorrectly labeled. Among them, 21 strains were misidentified within two genera and 19 strains were misidentified within the single genus. Finally, 60 strains belonging to the genus Kluyvera (K. ascorbata: n=16; K. cryocrescens: n=8; K. excreta: n=1; K. georgiana: n=4; K. intermedia: n=7; K. sichuanensis: n=3; K. chilikensis: n=1; Kluyvera genomospecies-1: n=1; Kluyvera genomospecies-2: n=10; Kluyvera genomospecies-3: n=4; Kluyvera genomospecies-4: n=2; Kluyvera genomospecies-5: n=2; Kluyvera sp.: n=1) and 37 strains belonging to the genus Phytobacter (P. diazotrophicus: n=14; P. ursingii: n=18; P. palmae: n=1; P. massiliensis: n=1; Phytobacter genomospecies-1: n=3) were involved in the taxonomy reassignments. Detailed information was listed in the Supplementary Table 1.
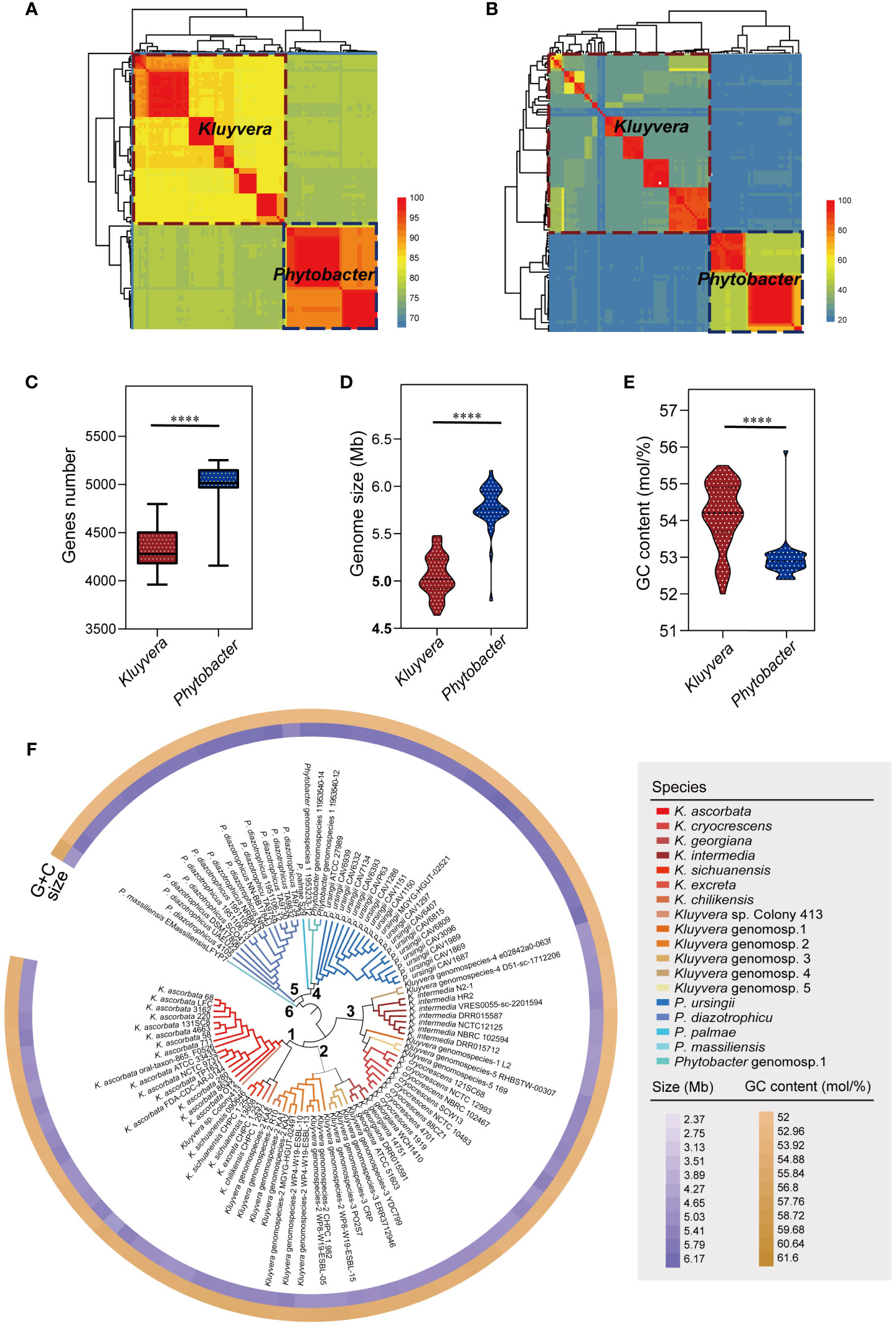
Figure 1 Strains re-identification and genomic characteristics analysis. (A) Species-level clusters with an ANI threshold at 95%. Aeromonas hydrophila ATCC 7966T served as an outgroup. (B) Species-level clusters with a isDDH threshold at 70%. A. hydrophila ATCC 7966T served as an outgroup. (C) Comparison of the genes number of Kluyvera and Phytobacter. (D) Comparison of the genome size of Kluyvera and Phytobacter. (E) Comparison of the GC content (mol/%) of Kluyvera and Phytobacter. Two groups with significant differences were marked with **** (P < 0.0001). (F) GBDP-based phylogeny of Kluyvera and Phytobacter genomes.
Further analysis revealed significant differences in the genome size, GC content, and gene count between Kluyvera and Phytobacter strains (P < 0.05, Figures 1C–E). The genome sizes ranged from 2.37 to 6.17 Mb (Kluyvera: 2.37-5.48 Mb; Phytobacter: 4.79-6.17 Mb), while GC content ranged from 52 to 55.9% (Kluyvera: 52-55.5 mol%; Phytobacter: 52.4-55.9 mol%). Genes count ranged from 3961 to 5253 (Kluyvera: 3961-4797; Phytobacter: 4157-5253).
A phylogenomic tree was constructed based on the intergenomic distances (Figure 1F) using GBDP algorithm (Meier-Kolthoff et al., 2013). At the phylogenomic-group level, the distance threshold of 0.15875 was used for single-linkage clustering (F=0), resulting in complete consistency with the re-identification (i.e., MRI=0). The tree revealed six distinct clusters. Cluster 1 comprised K. ascorbata, K. sichuanensis, Kluyvera excreta, Kluyvera chilikensis. Cluster 2 included Kluyvera genomospecies-2, Kluyvera georgiana, Kluyvera genomospecies-3, with Kluyvera georgiana and Kluyvera genomospecies-3 showing a closer kinship. Cluster 3 encompassed several species with K. intermedia and Kluyvera genomospecies-4 clustered together, while Kluyvera genomospecies-1, Kluyvera genomospecies-5, and K. cryocrescens clustered together. Cluster 4 consisted of P. palma, Phytobacter genomospecies-1, and Phytobacter ursingii, while P. diazotrophicu formed its own cluster, Cluster 5. P. massiliensis showed a clear separation from other Phytobacter strains and formed Cluster 6. A distance threshold of T = 0.036, equivalent to 70% of isDDH, was used for taxonomy reassignments at the species level. This threshold resulted in 18 species clusters that showed complete consistency with the reference partition (i.e., MRI=0) when using single-lineage clustering (F=0). These findings validated the species-level re-identification in this study. Among the species clusters, 13 belong to the Kluyvera subgroup, while five belong to Phytobacter.
A total of 19,150 pan genes and 1201 core genes were identified among the 97 strains. For a single genus, the core genes (99% ≤ strains ≤ 100%) number of 60 Kluyvera strains was 1164, and that of 37 Phytobacter strains was 1886. The functions and gene numbers of core genes of Kluyvera and Phytobacter are depicted in Figure 2A. A maximum-likelihood phylogenetic tree based on core genes SNPs was constructed (Figure 2B), showing that the clustering pattern based on core genes SNPs of 97 strains closely resembled that based on whole-genome sequences. The two genera, Kluyvera and Phytobacter, were clearly separated, and the relative positions of the species remained unchanged. Notably, the clade of K. ascorbata showed deeper evolutionary roots, suggesting indicating a more ancient lineage. However, in terms of the phylogenetic tree topology, there were some differences in the accessory genome-based phylogenetic tree compared to the phylogenetic tree based on the core genome (Figure 2B). The branch length of the accessory genome-based phylogenetic tree was longer than that of the core genome, reflecting a longer genetic distance. In accessory genome-based phylogenetic tree, strains of the same species, those with similar niches were almost all clustered together, showing a more similar accessory genes composition. Besides, the relative position of some strains has changed. For example, the strain Colony413 was closed with K. ascorbata in the core genome-based phylogenetic tree, while it was closed with Kluyvera genomospecies-2 in the accessory genome-based phylogenetic tree (Figure 2B).
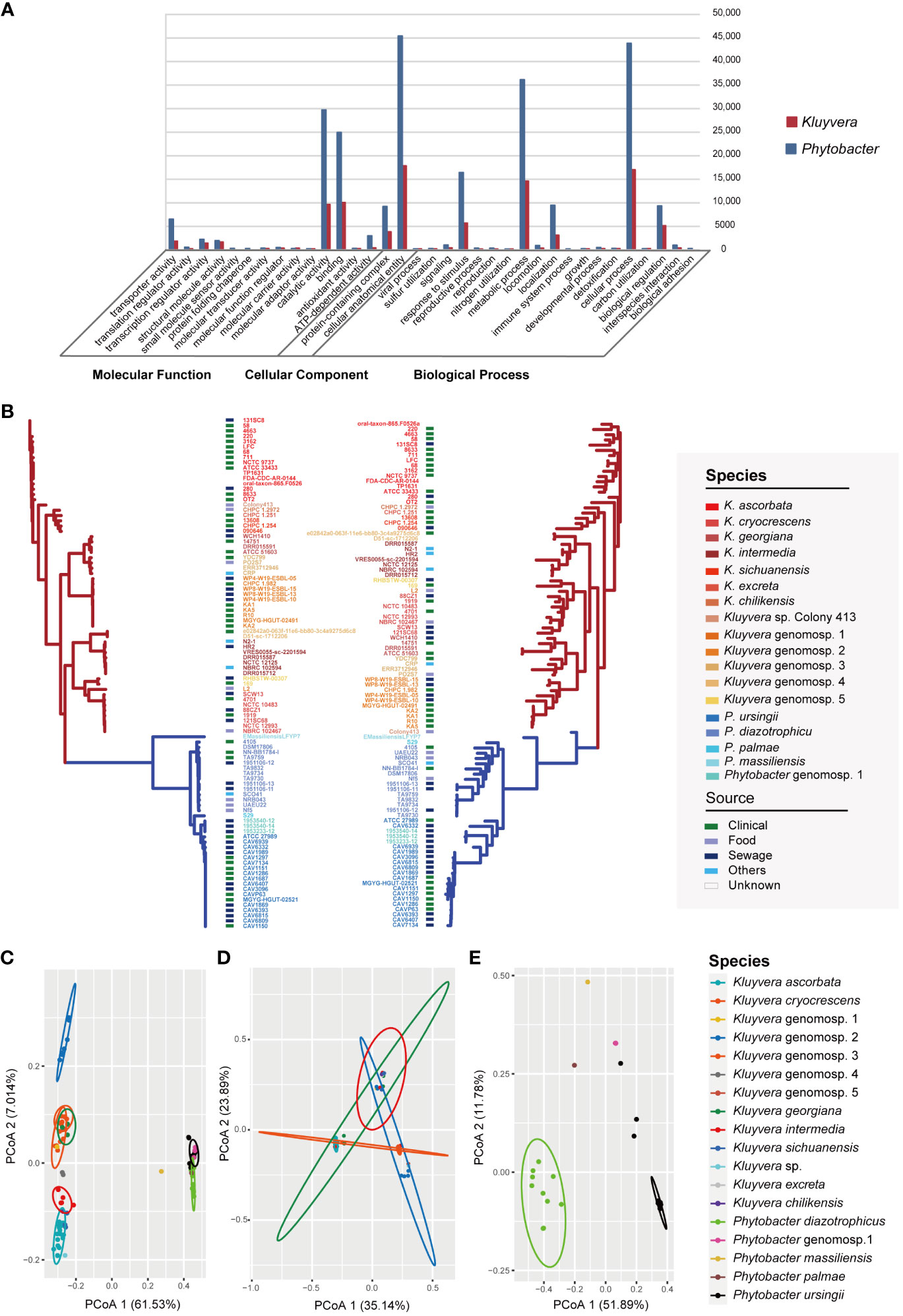
Figure 2 Pan-genome analysis of Kluyvera and Phytobacter strains. (A) Functions and gene numbers of core genes of Kluyvera and Phytobacter. (B) Phylogenetic tree of core genome sequences (left) and accessory genes (right) using the maximum likelihood method. (C) PCoA analysis of accessory genes of 97 strains. (D) PCoA analysis of 60 Kluyvera stains. (E) PCoA analysis of 37 Phytobacter stains.
The results of principal coordinates analysis (PCoA) exhibited clear separation among the strains based on the comprehensive composition of accessory genes (Figures 2C–E). Kluyvera genomospecies-2 was found to harbor sufficient species-specific genes to differentiate it from other species. Meanwhile, principal component analysis (PCA) was also performed based on the accessory genome, with the first and second principal components explaining 38.834% of the variation. Genus-specific genes of Kluyvera were identified, including pelX (Pectate disaccharide-lyase), mdtL (Multidrug resistance protein MdtL), bglC (Aryl-phospho-beta-D-glucosidase BglC), pcaK_1 (4-hydroxybenzoate transporter PcaK), uhpB (Signal transduction histidine-protein kinase/phosphatase UhpB), ddpa_2 (putative D,D-dipeptide-binding periplasmic protein DdpA), pdxY (Pyridoxal kinase PdxY), oppD_1 (Oligopeptide transport ATP-binding protein OppD), cptA (Phosphoethanolamine transferase CptA), yidZ (HTH-type transcriptional regulator YidZ), and csbX (Alpha-ketoglutarate permease).
The isolation year of the strains was utilized to infer the location of ancestral nodes on the Bayesian family tree. Different colored branches on the tree represented different bacterial species, with the abscissa corresponding to the predicted divergence time (Figures 3B, C). The average evolution rate of Kluyvera was 5.25E-6, approximately three times higher than that of Phytobacter, which had an average evolution rate of 1.85E-6 (Figure 3A). The family tree of Kluyvera showed two main groups. Group A included K. ascorbata, K. sichuanensis, K. excreta, K. chilikensis, Kluyvera genomospecies-2, Kluyvera genomospecies-3, K. geogiana. And group B comprised K. intermedia, K. cryocrescens, Kluyvera genomospecies-1, and Kluyvera genomospecies-5. Regarding Phytobacter, group A solely consisted of P. diazotrophicus, while group B included P. diazotrophicus, P. ursingii, and Phytobacter genomospecies-1.
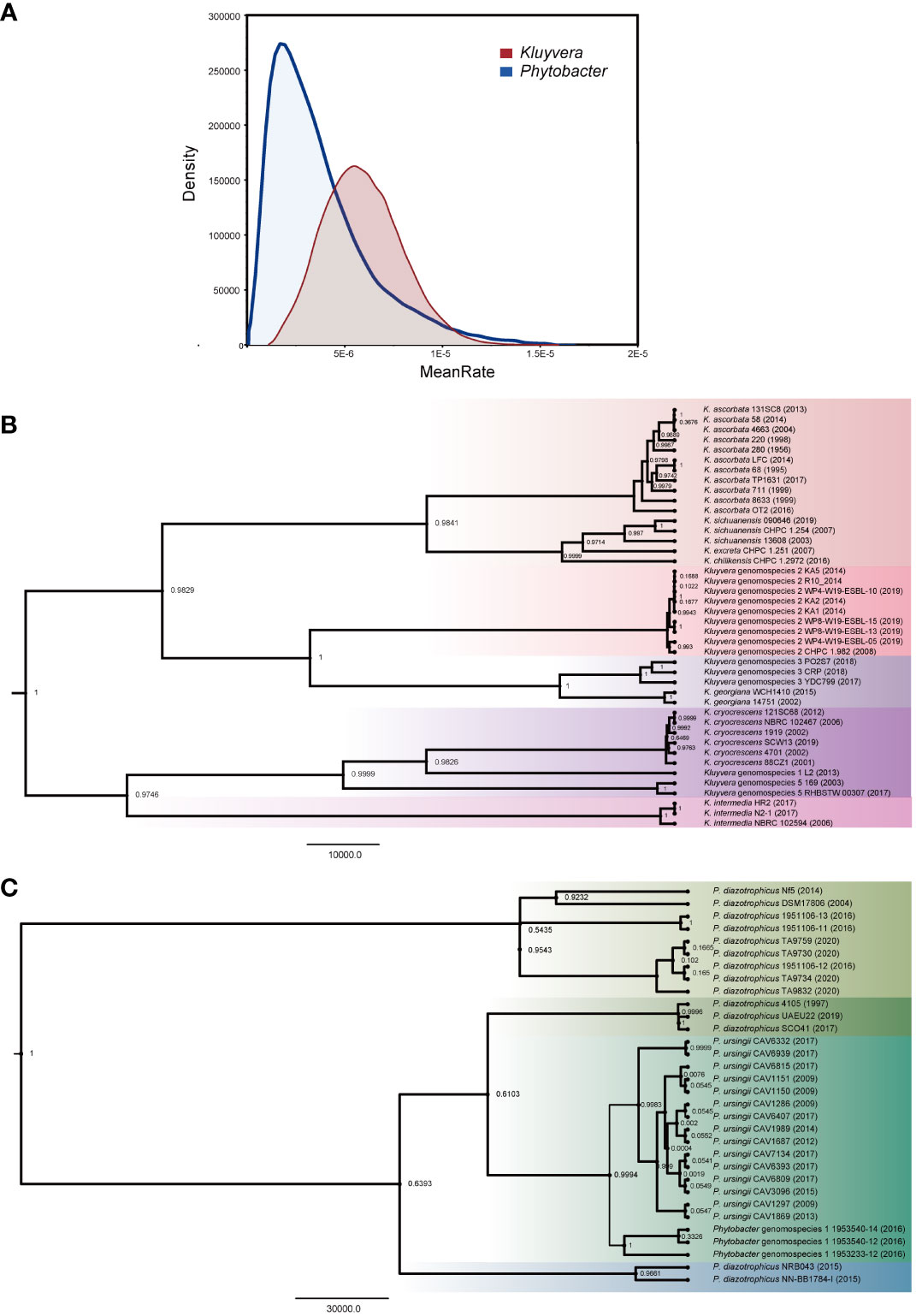
Figure 3 Phylogenetic analysis based on the core genes of Kluyvera and Phytobacter. The value on the node represents posterior. (A) The curve of evolutionary rate. (B) The Maximum Clade Credibility Tree (MCC Tree) of Kluyvera. (C) The Maximum Clade Credibility Tree (MCC Tree) of Phytobacter.
In total, 238 homologs of virulence-related genes were found to be present across the 97 strains, indicating a high level of divergence (Figure 4A). On average, each strain contained approximately 115 virulence-related genes. Among these strains, 23 virulence-related genes were shared, including acrA, acrB, algU, cheA, flhA, flhB, flhC, flhD, fliA, luxS, rcsB, tufa, galU, rfaD, rfaE, rfaF, rpe, gmd, wza, gnd, kdsA, motA, and motB. The genes acrA and acrB, categorized as Klebsiella antimicrobial activity/competitive advantage virulence factors, were involved in resistance against host-derived antimicrobial peptides (Padilla et al., 2010). The gene algU was a member of Pseudomonas aeruginosa alginate (mucoid exopolysaccharide) synthetic gene cluster, and was related with biofilm formation (Stapper et al., 2004; Nivens et al., 2001). The autoinducer-2 (AI-2) signaling molecule related gene luxS was presented in all 97 strains. Additionally, the gene tufA, which was homologous to Francisella tularensis elongation factor-Tu (EF-Tu), was identified as a core virulence gene in the 97 strains.
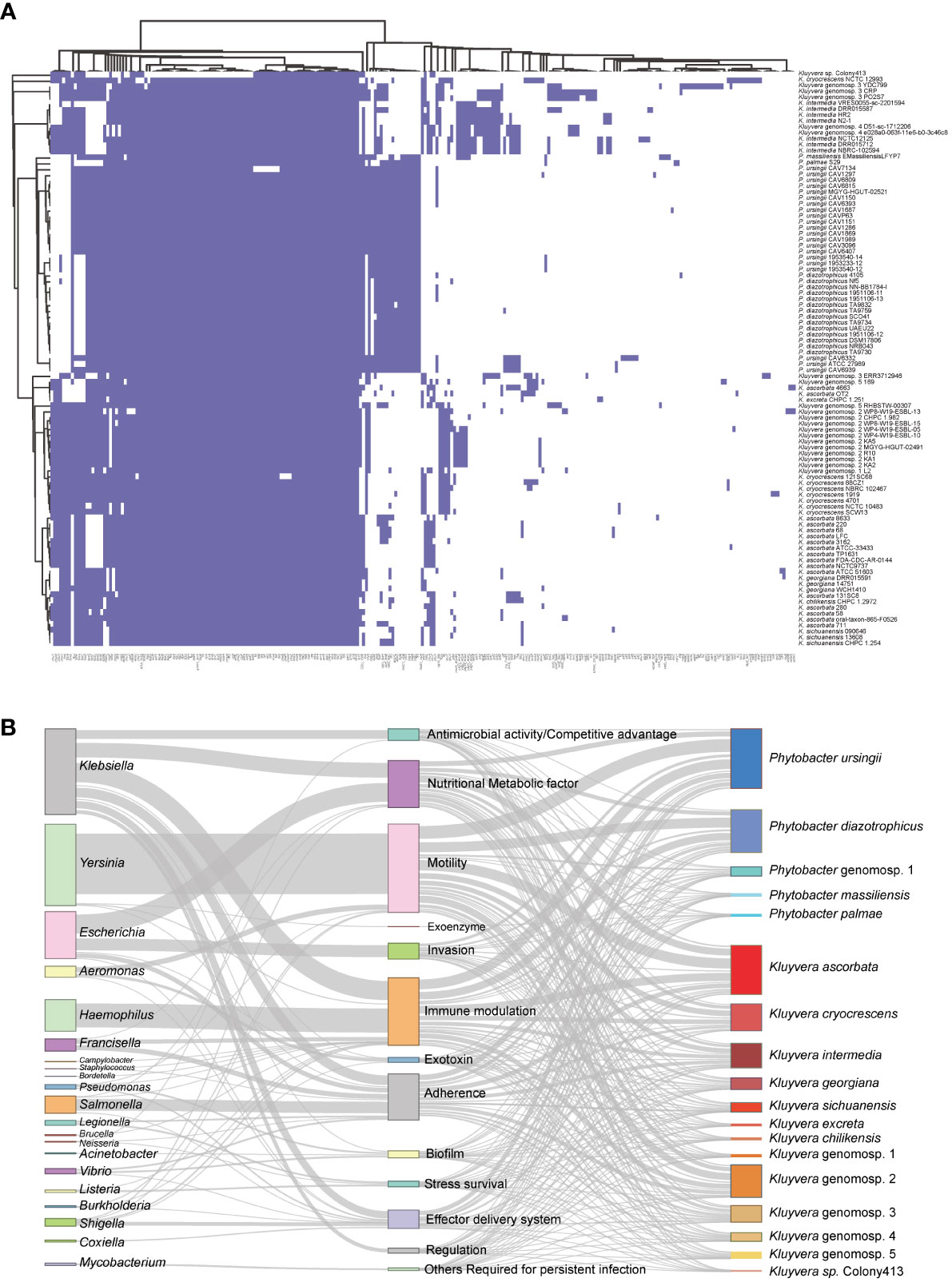
Figure 4 Pan virulence-related genes analysis of Kluyvera and Phytobacter genus. (A) The presence or absence of virulence related genes. (B) Virulence-related genes flow analysis. According to the BLASTp results derived from the VFDB database, virulence-related genes in Kluyvera and Phytobacter were linked to homologous genes in other species. The left column represented species share homologous virulence genes with Kluyvera and Phytobacter. The middle column represented different categories of virulence factors. The right column represented different species of Kluyvera and Phytobacter.
To explore the differences in pathogenicity, we further analyzed genus-specific and species-specific virulence factors. It was evident that the profiles of virulence genes did not conform to specific species, indicating their substantial diversity. Among the 60 Kluyvera strains, a total of 215 homologs of virulence-related genes were detected, with 37 genes belonging to the Kluyvera core genome. This suggests the conservative and necessary roles of these genes in Kluyvera pathogenesis. Among the 37 Phytobacter strains, 136 homologs of virulence-related genes were detected, with 72 of them belonging to the Phytobacter core genome. The Kluyvera-specific core virulence gene has not been identified thus far. However, the genes clbS, csgA-C, hsiB1_vipA, and hsiC1_vipB were found to be unique core virulence-related genes in Phytobacter.
We further analyzed the virulence flow to provide a comprehensive view of virulence profile. The 238 virulence-related genes mentioned above, which were of 21 different species, were divided into 13 virulence factors (Figure 4B). Certain genus shares fixed virulence genes with Kluyvera and Phytobacter. Escherichia coli-derived homologous virulence genes were the most numerous. The term of antimicrobial activity/competitive advantage was most similar to the virulence genes of the two genera (Klebsiella, Neisseria), of which Klebsiella accounted for 99.56% (448/450). The homologous genes of nutritional/metabolic factor were mainly derived from two genera (Klebsiella: 749/1847, 40.55%, Escherichia: 957/1847, 51.81%). The virulence factors of immune modulation had three larger sources, namely Haemophilus influenzae (46.80%), Klebsiella pneumoniae (37.5%), Francisella tularensis (8.1%). Remarkably, the virulence genes associated with invasion were derived only from Escherichia. H. influenzae shares only one virulence factor (immune modulation). The virulence genes homologous from Salmonella mainly belonged to the adherence, accounting for 32.94% (588/1785). The regulation related virulence genes were only homologous to Klebsiella. The flagella related genes mainly belonged to Yersinia enterocolitica, accounting for 90.0% (3062/3401). The exoenzyme related genes homologous to the Yersinia pestis were only found in the Phytobacter palmae strain.
In this study, we conducted the antibiotic resistance analysis of selected strains using both genomic and phenotypic approaches. A total of 120 ARGs were identified, associated with resistance to 27 different antimicrobial drugs (Figure 5A). Two antibiotic resistance ontology (ARO), H-NS and CRP, were identified in the core genome of the two genera, which represented common resistance mechanisms of the resistance-nodulation-cell division (RND) antibiotic efflux pump. Besides, the RND antibiotic efflux-related ARG baeR was identified as a Phytobacter-specific resistance gene, mediating resistance to multiple drugs. Many strains exhibited the potential to be multidrug-resistant, as they carried three or more classes of antimicrobial resistance genes. Extended-spectrum β-lactamases (ESBLs) genes were detected, including blaOXA-1, blaOXA-2, blaSHV-1, blaSHV-30, blaSHV-7, blaCTX-M-3, blaCTX-M-5, blaCTX-M-8, blaCTX-M-9, blaCTX-M-13, blaCTX-M-14, blaCTX-M-37, blaCTX-M-40, blaCTX-M-65, blaCTX-M-78, blaCTX-M-95, blaCTX-M-115, blaCTX-M-124-like, blaCTX-M-152, blaCTX-M-185, blaCTX-M-213, blaKLUC-1, blaKLUC-2, blaKLUC-5, blaSHV-134, blaCTX-M-15, and blaTEM-1. Co-existence of ESBLs were identified in 25 genomes, with 18 of them belonging to Phytobacter strains. Notably, the presence of ESBLs and carbapenem resistance genes (blaKPC, blaNDM, blaIMP, blaOXA-48) were simultaneously detected in the genomes of 38 bacterial strains (Figure 5B). Twenty-one strains contained blaKPC. The co-existence of antibiotic resistance genes was more frequently observed in strains isolated from sewage samples.
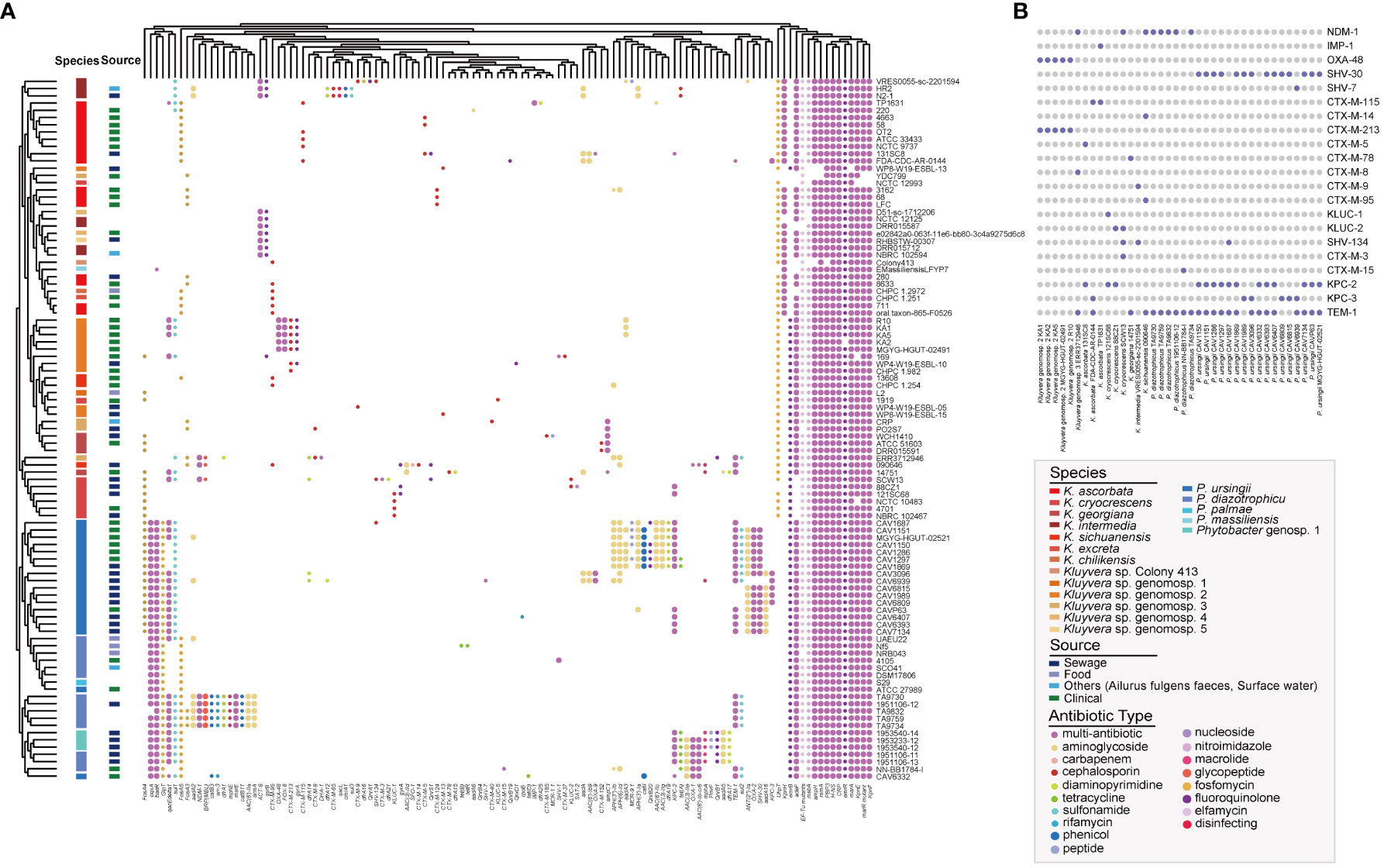
Figure 5 Antibiotic resistance genes (ARGs) landscape of Kluyvera and Phytobacter. (A) Absence and present of ARGs in Kluyvera and Phytobacter genus. (B) Coexistence patterns of ESBLs, blaNDM, blaKPC, and blaIMP in Kluyvera and Phytobacter genus.
Kluyvera is part of the normal microbiota of the human digestive tract (Sarria et al., 2001). But in recent years, it has been increasingly implicated in infections, even fatal cases, highlighting its emerging opportunistic nature and underestimated pathogenicity (Erbin et al., 2020). At present, Kluyvera comprises five recognized species and five genomospecies. In our research, we further newly complemented the members of the five genomospecies based on whole genome sequences in public databases. Previous work found that the identification of Kluyvera and Phytobacter may be problematic (Rodríguez et al., 2021; Smits et al., 2022). Classification and understanding of the genus Phytobacter have undergone a complex and lengthy process (Smits et al., 2022). Until year 2018, the strains associated with a multistate outbreak in Brazil were officially included in the genus Phytobacter (Pillonetto et al., 2018b). The genus Phytobacter is now recognized as an important clinically relevant member of the Enterobacteriaceae. It can be found in various clinical samples, such as blood, sputum, the digestive tract, and bile (Smits et al., 2022). Currently, Phytobacter consists of four species, P. diazotrophicus, P. ursingii, P. palmae, and P. massiliensis. To address the issue of uncertain identification and potential clinical relevance of both genera, we performed a comparative genomic analysis, focusing on taxonomy, evolution, pathogenicity, and drug resistance.
In this work, we collected all Kluyvera and Phytobacter sequences available in public databases, along with four strains isolated in this study. Using ANI (Richter and Rosselló-Móra, 2009) and isDDH (Meier-Kolthoff et al., 2013), we provided reliable species designations of 97 strains. Surprisingly, 41.24% of these strains were incorrectly labeled. Also, we defined a new Phytobacter genomospecies, namely Phytobacter genomospecies-1, which includes three strains (1953233-12, 1953540-12, and 1953540-14) that require phenotypic confirmation.
We calculated intergenomic distances using the GBDP algorithm (Meier-Kolthoff et al., 2013) and found that under the distance threshold of 0.15875, the best clustering decision yielded results consistent with previous identification. The pan-genome analysis of the 97 strains revealed that both Phytobacter and Kluyvera exhibit great genome plasticity and inclusiveness, likely related to their diverse ecological niches. The clustering pattern based on the core genes SNPs aligned closely with those based on whole-genome sequences, showing two distinct clusters. The long branch length of the accessory genome-based phylogenetic tree indicated a greater genetic distance compared to that of the core genome-based tree, suggesting that strains of the same species may undergo more diversification in their accessory genes due to survival pressure. Further PCoA of two genera, we identified 11 differential core genes specific to Kluyvera (pelX, mdtL, bglC, pcak-1, uhpB, ddpA-2, pdxY, oppD-1, cptA, yidZ, csbX), which could be served as potential identification targets at the genus level. Evolutionary analyses revealed the origins of the two genera and the timing of species divergence, with results suggesting that Kluyvera evolved at a faster rate.
By analyzing the pan virulence-related genes, we observed significant variation and high diversity among the strains, implying different levels of pathogenicity. Our study characterized the variation of virulence-related genes in the genus of Kluyvera and Phytobacter. Pan virulence-related gene analysis could help to identify virulence gene pools and explore the fundamental virulence mechanism. We identified core homologous virulence-related genes unique to Phytobacter, such as clbS, csgA-C, fliS, hsiB1-vipA and hsiC1-vipB, suggesting their role in Phytobacter inherent pathogenicity. Furthermore, the Salmonella agf genes cluster was homology to csg of E. coli, which facilitated the bacterial attachment and the villi of enterocytes (Collinson et al., 1996; White et al., 2001; White et al., 2003). The absence of the Salmonella agf genes cluster in Kluyvera strains but the presence of the complete cluster in all Phytobacter strains indicated that Phytobacter may have a stronger ability to colonize the gut. The gene hsiB1-vipA and hsiC1-vipB were homology to the gene encoding the Pseudomonas HSI-I (Hcp1 secretion island I) tube-sheath complex. HSI-I was highly homologous to a group of genes found in many Gram-negative proteobacteria that encoded a secretory system to play a general role in chronic P. aeruginosa infections (Mougous et al., 2006). Among 15 virulence related genes present in all 97 strains, the genes acrA and acrB, homologous to the genes involved in the antimicrobial activity/competitive advantage in Klebsiella, were responsible for mediating resistance against host-derived antimicrobial peptides (Padilla et al., 2010). Bacteria growth with alginate production was frequently as mucoidy colony, which was be consistent with culture phenotypes of the collected strains. Alginate slime layer can make it more difficult for phagocytes to ingest and kill the bacteria, suggesting a potential pathogenesis. AI-2 was produced and detected by a wide variety of bacteria and was presumed to facilitate interspecies communications (Pereira et al., 2013). For Vibrio cholerae, information from AI-2 mediates LuxPQ receptor complex response to allow V. cholerae to leave the host, re-enter the environment in large numbers and initiate a new cycle of infection (Ng and Bassler, 2009). AI-2 related gene luxS was presented in all 97 strains, acting as an important virulence factor. Only the strain P. ursingii CAV 6332 contained allA-D, allR, allS, which were responsible for allantion utilization in Klebsiella. An allantoin utilization operon has been associated with hypervirulent K. pneumoniae strains that cause pyogenic liver abscesses, suggesting a high pathogenesis for the P. ursingii strain CAV 6332.
Virulence flow analysis revealed a comprehensive view of virulence genes shared among different bacterial genera. E. coli-derived homologous virulence genes accounted for the highest number. The invasion related genes were only from Escherichia. The virulence genes belonging to the term of ‘Antimicrobial activity/Competitive advantage and Regulation’ shared the highest number with Klebsiella. The accessory virulence-related genes may confer an evolutionary advantage under certain environmental conditions for the strains. Remarkably, the component chuA of Enterohemorrhagic E. coli (EHEC) gene cluster (related genes: chuA, chuS, chuT, chuU, chuW, chuX, chuY), responsible for iron uptake, were present in all strains of K. ascorbata, K. intermedia, Kluyvera genomospecies-4, and Kluyvera genomospecies-1. And K. ascorbata was the species most frequently isolated in clinical specimens (Sarria et al., 2001). The gene chuA showed high homology to shuA gene of S. dysenteriae type 1, appearing to be widely distributed among pathogenic E. coli strains (Torres and Payne, 1997). Other Kluyvera species except above contained the remaining components of Chu gene cluster but not chuA. Therefore, chuA was considered as an important potential virulence gene in the pathogenic process. This result also suggested that K. intermedia, Kluyvera genomospecies-1, and Kluyvera genomospecies-4 were potential pathogenic species. The chuA gene had not been found in Phytobacter genomes. However, limited by the number of strains, it remains challenge to link strains virulence to the molecular and cellular processes that explain the disease in the affected host.
Antibiotic resistance is a major challenge to global public health (Laxminarayan et al., 2013). Extended-spectrum β-lactamases (ESBLs) are enzymes that hydrolyze penicillin, cephalosporins and aztreonam, leading to treatment failure (Paterson and Bonomo, 2005). They can be inhibited by enzyme inhibitors, but were sensitive to cephamycin and carbapenem antibiotics. ESBLs include TEM, SHV, CTX-M, VEB, and GES enzymes, in which CTX-M family harbored the highest number of variants (Peirano and Pitout, 2019). At present, more than 200 genotypes have been discovered, distributing in at least five subgroups (CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, KLUC). Kluyvera chromosomally encoded blaCTX-Ms was found to be the progenitors of CTX-M resistance genes, for example, CTX-M-2 genotypes were from K. ascorbata, KLUC were from K. cryocrescens and CTX-M-8 were from K. georgiana (Cantón et al., 2012). In this study, we identified ESBL-producing strains carrying multiple resistance genes, including various variants of blaCTX-Ms, which were the most prevalent ESBLs. Along with the changes in Kluyvera taxonomy, the contribution of each species to the recruitment and dissemination of their β-lactamases should be re-evaluated.
The efflux pump was to pump out the antibacterial drugs that enter the bacterial cells, thereby reducing the effective drug concentration in the bacterial cells and causing drug resistance (Li et al., 2015; Lucaßen et al., 2021). In our study, efflux pump-related genes, such as H-NS and CRP, were observed in the core genome of both genera, indicating a common resistance mechanism of resistance-nodulation-cell division (RND) antibiotic efflux pump. The bacterial efflux pump and the low permeability of the outer membrane acted synergistically to protect the bacteria from drug damage and lead to an increase in the accumulation of antimicrobial drugs in the human body (Zwama and Nishino, 2021). This will undoubtedly put stronger antibiotic selection pressure on bacteria in the human body, prompting bacteria to become more resistant.
The co-existence of two or more β-lactamases in the single strain is possibility becoming a common bacterial strategy to enhance antibiotic resistance. In our study, each strain carried multiple resistance genes, suggesting that they had the higher opportunity to become more resistant. The co-existence of ESBLs happened in the 25 strains, of which 18 were Phytobacter strains. Since Phytobacter strains were usually detected in the environment, it explained the extraordinary dispersion trend of ESBLs genes. The five strains in Phytobacter still contained mcr-9.1 under the condition of containing several ESBLs resistance genes, all of which were derived from clinical samples. Carbapenems were generally considered the most effective option for infections caused by ESBLs-producing enterohepatic bacteria, but were hydrolyzed by carbapenemase (Li Y. et al., 2019). Importantly, we found co-existence of ESBLs and carbapenem resistance genes (blaKPC, blaNDM, blaIMP, blaOXA-48) in 38 strains, particularly in strains isolated from sewage. This suggests the potential development of extensively drug-resistant or even fully drug-resistant strains. Permanent selective force existing in complex environments like sewage drives diversification of the resistance mechanisms and reinforce horizontal transfer and acquisition of resistance genes. Due to the limited number of strains, the drug resistance phenotype of the strains cannot be verified. Although harboring ESBLs, two Kluyvera novel species identified in this study only resistant to azithromycin, a macrolide antibiotic, and sensitive to carbapenem antibiotic tested. The specific mechanism deserves further exploration. As important resistance genes reservoir, the two genera potentially become new, emerging threat as a multidrug-resistant microorganism to clinical treatment.
The current studies on the two genera, Kluyvera and Phytobacter, are limited. However, this study aims to provide a comprehensive understanding of the clinical relevance and validate the species status of these two genera through whole-genome analysis. Here, we provided a better understanding of the differences between closely related species and identified previously unrecognized species. This study explores the origin and divergence time of species through evolutionary analysis. By utilizing the population structure of Kluyvera and Phytobacter, we conducted a genome-wide screening of virulence gene and resistance gene profiles. These results shed light on the divergence of virulence gene profiles and resistance gene spectra within these genera. Additionally, based on the identification of differential virulence genes, potential pathogenic mechanisms and molecular biological targets for further exploration have been proposed. The importance of Kluyvera and Phytobacter as emerging causes of human disease has been emphasized in this study. Moreover, the study highlights the potential of these two genera as multidrug-resistant microorganisms, posing a threat to clinical treatment.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
ZH: Conceptualization, Data curation, Formal analysis, Software, Visualization, Writing – original draft. GZ: Data curation, Investigation, Methodology, Writing – original draft. ZZ: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft. XL: Formal analysis, Investigation, Resources, Writing – original draft. FC: Formal analysis, Investigation, Methodology, Writing – original draft. LZ: Formal analysis, Investigation, Resources, Writing – original draft. DW: Investigation, Resources, Supervision, Validation, Writing – review & editing. KY: Conceptualization, Formal analysis, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. JL: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Hangzhou Key Medicine Discipline Fund for Hygienic Microbiology Laboratory (2020-2024); Zhejiang Key Laboratory of Multi-Omics in Infection and Immunity; Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents (2019-2023).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1376289/full#supplementary-material
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bolat, F., Kilic, S. C., Abaci, C., Cevit, O., Gultekin, A. (2013). A neonate with kluyvera sepsis: a case report. Iran J. Pediatr. 23, 371–372.
Cantón, R., González-Alba, J. M., Galán, J. C. (2012). CTX-M enzymes: origin and diffusion. Front. Microbiol. 3, 110. doi: 10.3389/fmicb.2012.00110
Cantón, R., Novais, A., Valverde, A., MaChado, E., Peixe, L., Baquero, F., et al. (2008). Prevalence and spread of extended-spectrum beta-lactamase-producing Enterobacteriaceae in Europe. Clin. Microbiol. Infect. 14 Suppl 1, 144–153. doi: 10.1111/j.1469-0691.2007.01850.x
Collinson, S. K., Clouthier, S. C., Doran, J. L., Banser, P. A., Kay, W. W. (1996). Salmonella enteritidis agfBAC operon encoding thin, aggregative fimbriae. J. Bacteriol. 178, 662–667. doi: 10.1128/jb.178.3.662-667.1996
Erbin, A., Gozdas, H. T., Guler, Y., Canat, H. L. (2020). Urosepsis caused by Kluyvera Ascorbata in a pregnant woman. J. Coll. Physicians Surg. Pak. 30, 324–326. doi: 10.29271/jcpsp
Farmer, J. J., 3rd, Fanning, G. R., Huntley-Carter, G. P., Holmes, B., Hickman, F. W., Richard, C., et al. (1981). Kluyvera, a new (redefined) genus in the family Enterobacteriaceae: identification of Kluyvera ascorbata sp. nov. and Kluyvera cryocrescens sp. nov. in clinical specimens. J. Clin. Microbiol. 13, 919–933. doi: 10.1128/jcm.13.5.919-933.1981
Fu, L., Niu, B., Zhu, Z., Wu, S., Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Garrido-Sanz, D., Redondo-Nieto, M., Martín, M., Rivilla, R. (2020). Comparative genomics of the Rhodococcus genus shows wide distribution of biodegradation traits. Microorganisms 8. doi: 10.3390/microorganisms8050774
Gessew, G. T., Desta, A. F., Adamu, E. (2022). High burden of multidrug resistant bacteria detected in Little Akaki River. Comp. Immunol. Microbiol. Infect. Dis. 80, 101723. doi: 10.1016/j.cimid.2021.101723
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hill, V., Baele, G. (2019). Bayesian estimation of past population dynamics in BEAST 1.10 using the skygrid coalescent model. Mol. Biol. Evol. 36, 2620–2628. doi: 10.1093/molbev/msz172
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Laxminarayan, R., Duse, A., Wattal, C., Zaidi, A. K., Wertheim, H. F., Sumpradit, N., et al. (2013). Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 13, 1057–1098. doi: 10.1016/S1473-3099(13)70318-9
Lefort, V., Desper, R., Gascuel, O. (2015). FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Li, X. Z., Plésiat, P., Nikaido, H. (2015). The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418. doi: 10.1128/CMR.00117-14
Li, Y., Luo, L., Xiao, Z., Wang, G., Li, C., Zhang, Z., et al. (2019). Characterization of a carbapenem-resistant Kluyvera cryocrescens isolate carrying Bla(ndm-1) from hospital sewage. Antibiotics (Basel) 8. doi: 10.3390/antibiotics8030149
Li, Z., Pang, B., Wang, D., Li, J., Xu, J., Fang, Y., et al. (2019). Expanding dynamics of the virulence-related gene variations in the toxigenic Vibrio cholerae serogroup O1. BMC Genomics 20, 360. doi: 10.1186/s12864-019-5725-y
Liu, L., Feng, Y., Wei, L., Qiao, F., Zong, Z. (2020). Precise Species Identification and Taxonomy Update for the Genus Kluyvera With Reporting Kluyvera sichuanensis sp. nov. Front Microbiol. 11, 579306. doi: 10.3389/fmicb.2020.579306
Lucaßen, K., Müller, C., Wille, J., Xanthopoulou, K., Hackel, M., Seifert, H., et al. (2021). Prevalence of RND efflux pump regulator variants associated with tigecycline resistance in carbapenem-resistant Acinetobacter baumannii from a worldwide survey. J. Antimicrob. Chemother. 76, 1724–1730. doi: 10.1093/jac/dkab079
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10, 2182. doi: 10.1038/s41467-019-10210-3
Mougous, J. D., Cuff, M. E., Raunser, S., Shen, A., Zhou, M., Gifford, C. A., et al. (2006). A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312, 1526–1530. doi: 10.1126/science.1128393
Ng, W. L., Bassler, B. L. (2009). Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 43, 197–222. doi: 10.1146/annurev-genet-102108-134304
Nivens, D. E., Ohman, D. E., Williams, J., Franklin, M. J. (2001). Role of alginate and its O acetylation in formation of Pseudomonas aeruginosa microcolonies and biofilms. J. Bacteriol. 183, 1047–1057. doi: 10.1128/JB.183.3.1047-1057.2001
Padilla, E., Llobet, E., Doménech-Sánchez, A., Martínez-Martínez, L., Bengoechea, J. A., Albertí, S. (2010). Klebsiella pneumoniae AcrAB efflux pump contributes to antimicrobial resistance and virulence. Antimicrob. Agents Chemother. 54, 177–183. doi: 10.1128/AAC.00715-09
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paschoal, R. P., Campana, E. H., Corrêa, L. L., Montezzi, L. F., Barrueto, L. R. L., da Silva, I. R., et al. (2017). Concentration and variety of carbapenemase producers in recreational coastal waters showing distinct levels of pollution. Antimicrob. Agents Chemother. 61. doi: 10.1128/AAC.01963-17
Paterson, D. L., Bonomo, R. A. (2005). Extended-spectrum beta-lactamases: a clinical update. Clin. Microbiol. Rev. 18, 657–686. doi: 10.1128/CMR.18.4.657-686.2005
Peirano, G., Pitout, J. D. D. (2019). Extended-spectrum β-lactamase-producing enterobacteriaceae: update on molecular epidemiology and treatment options. Drugs 79, 1529–1541. doi: 10.1007/s40265-019-01180-3
Pereira, C. S., Thompson, J. A., Xavier, K. B. (2013). AI-2-mediated signalling in bacteria. FEMS Microbiol. Rev. 37, 156–181. doi: 10.1111/j.1574-6976.2012.00345.x
Pillonetto, M., Arend, L., Gomes, S. M. T., Oliveira, M. A. A., Timm, L. N., Martins, A. F., et al. (2018b). Molecular investigation of isolates from a multistate polymicrobial outbreak associated with contaminated total parenteral nutrition in Brazil. BMC Infect. Dis. 18, 397. doi: 10.1186/s12879-018-3287-2
Pillonetto, M., Arend, L. N., Faoro, H., D'Espindula, H. R. S., Blom, J., Smits, T. H. M., et al. (2018a). Emended description of the genus Phytobacter, its type species Phytobacter diazotrophicus (Zhang 2008) and description of Phytobacter ursingii sp. nov. Int. J. Syst. Evol. Microbiol. 68, 176–184. doi: 10.1099/ijsem.0.002477
Richter, M., Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodríguez, M. M., Power, P., Naas, T., Gutkind, G. (2021). Redefining the origin and evolution of chromosomally encoded bla(CTX-M/KLU) in the context of a revised taxonomy of genus Kluyvera. Antimicrob. Agents Chemother. 65, e0242420. doi: 10.1128/AAC.02424-20
Sarria, J. C., Vidal, A. M., Kimbrough, R. C., 3rd (2001). Infections caused by Kluyvera species in humans. Clin. Infect. Dis. 33, E69–E74. doi: 10.1086/322686
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sekizuka, T., Matsui, M., Takahashi, T., Hayashi, M., Suzuki, S., Tokaji, A., et al. (2018). Complete Genome Sequence of bla (IMP-6)-Positive Metakosakonia sp. MRY16-398 Isolate From the Ascites of a Diverticulitis Patient. Front. Microbiol. 9, 2853. doi: 10.3389/fmicb.2018.02853
Sezer, M. T., Gültekin, M., Günseren, F., Erkiliç, M. (1996). Ersoy F. A case of Kluyvera cryocrescens peritonitis in a CAPD patient. Perit. Dial. Int. 16, 326–327. doi: 10.1177/089686089601600317
Sierra-Madero, J., Pratt, K., Hall, G. S., Stewart, R. W., Scerbo, J. J., Longworth, D. L. (1990). Kluyvera mediastinitis following open-heart surgery: a case report. J. Clin. Microbiol. 28, 2848–2849. doi: 10.1128/jcm.28.12.2848-2849.1990
Smits, T. H. M., Arend, L., Cardew, S., Tång-Hallbäck, E., Mira, M. T., Moore, E. R. B., et al. (2022). Resolving taxonomic confusion: establishing the genus Phytobacter on the list of clinically relevant Enterobacteriaceae. Eur. J. Clin. Microbiol. Infect. Dis. 41, 547–558. doi: 10.1007/s10096-022-04413-8
Stapper, A. P., Narasimhan, G., Ohman, D. E., Barakat, J., Hentzer, M., Molin, S., et al. (2004). Alginate production affects Pseudomonas aeruginosa biofilm development and architecture, but is not essential for biofilm formation. J. Med. Microbiol. 53, 679–690. doi: 10.1099/jmm.0.45539-0
Tonkin-Hill, G., Lees, J. A., Bentley, S. D., Frost, S. D. W., Corander, J. (2019). Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 47, 5539–5549. doi: 10.1093/nar/gkz361
Torres, A. G., Payne, S. M. (1997). Haem iron-transport system in enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 23, 825–833. doi: 10.1046/j.1365-2958.1997.2641628.x
White, A. P., Collinson, S. K., Banser, P. A., Gibson, D. L., Paetzel, M., Strynadka, N. C., et al. (2001). Structure and characterization of AgfB from Salmonella enteritidis thin aggregative fimbriae. J. Mol. Biol. 311, 735–749. doi: 10.1006/jmbi.2001.4876
White, A. P., Gibson, D. L., Collinson, S. K., Banser, P. A., Kay, W. W. (2003). Extracellular polysaccharides associated with thin aggregative fimbriae of Salmonella enterica serovar enteritidis. J. Bacteriol. 185, 5398–5407. doi: 10.1128/JB.185.18.5398-5407.2003
Yim, W. J., Poonguzhali, S., Madhaiyan, M., Palaniappan, P., Siddikee, M. A., Sa, T. (2009). Characterization of plant-growth promoting diazotrophic bacteria isolated from field grown Chinese cabbage under different fertilization conditions. J. Microbiol. 47, 147–155. doi: 10.1007/s12275-008-0201-4
Zhang, Z., Li, D., Shi, X., Zhai, Y., Guo, Y., Zheng, Y., et al. (2020). Genomic characterization of an emerging Enterobacteriaceae species: the first case of co-infection with a typical pathogen in a human patient. BMC Genomics 21, 297. doi: 10.1186/s12864-020-6720-z
Zhang, G. X., Peng, G. X., Wang, E. T., Yan, H., Yuan, Q. H., Zhang, W., et al. (2008). Diverse endophytic nitrogen-fixing bacteria isolated from wild rice Oryza rufipogon and description of Phytobacter diazotrophicus gen. nov. sp. nov. Arch. Microbiol. 189, 431–439. doi: 10.1007/s00203-007-0333-7
Keywords: Kluyvera, Phytobacter, evolution, pathogenicity, drug-resistance
Citation: Huang Z, Zhang G, Zheng Z, Lou X, Cao F, Zeng L, Wang D, Yu K and Li J (2024) Genomic insights into the evolution, pathogenicity, and extensively drug-resistance of emerging pathogens Kluyvera and Phytobacter. Front. Cell. Infect. Microbiol. 14:1376289. doi: 10.3389/fcimb.2024.1376289
Received: 25 January 2024; Accepted: 08 March 2024;
Published: 21 March 2024.
Edited by:
Brankica Filipić, University of Belgrade, SerbiaReviewed by:
Alexey V. Rakov, Central Research Institute of Epidemiology (CRIE), RussiaCopyright © 2024 Huang, Zhang, Zheng, Lou, Cao, Zeng, Wang, Yu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Li, MTA0MDcwMzBAemp1LmVkdS5jbg==; Keyi Yu, eWt5MDQxNEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.