 Yarong Zhao1,2,3
Yarong Zhao1,2,3 Chulan Huang1,2,3Rui Zeng1,2,3Peirong Chen1,2,3Kaihang Xu1,2,3Xiaomei Huang1,2,3Xu Wang1,2,3*
Chulan Huang1,2,3Rui Zeng1,2,3Peirong Chen1,2,3Kaihang Xu1,2,3Xiaomei Huang1,2,3Xu Wang1,2,3*- 1Institute of Quality Standard and Monitoring Technology for Agro-product of Guangdong Academy of Agricultural Sciences, Guangzhou, China
- 2Guangdong Provincial Key Laboratory of Quality & Safety Risk Assessment for Agro-Products, Guangzhou, China
- 3Key Laboratory of Testing and Evaluation for Agro-product Safety and Quality, Ministry of Agriculture and Rural Affairs, Guangzhou, China
Aflatoxins (AFs) are produced by fungi such as Aspergillus flavus and A. parasiticus and are one of the most toxic mycotoxins found in agricultural products and food. Aflatoxin contamination, which requires the control of A. flavus, remains problematic because of the lack of effective strategies and the exploration of new compounds that can inhibit A. flavus growth and mycotoxin production is urgently required to alleviate potential deleterious effects. Acetohydroxy acid synthase (AHAS) and dihydroxy acid dehydratase are important enzymes in the biosynthetic pathways of branched-chain amino acids (BCAAs), including isoleucine, leucine, and valine. Enzymes involved in BCAA biosynthesis are present in bacteria, plants, and fungi, but not in mammals, and are therefore, attractive targets for antimicrobial and herbicide development. In this study, we characterized AflaILVB/G/I and AflaILVD, which encode the catalytic and regulatory subunits of AHAS and dihydroxy acid dehydratase, from the pathogenic fungus Aspergillus flavus. The AflaILVB/G/I and AflaILVD deletion mutant grew slower and produced smaller colonies than the wild-type strain when grown on glucose minimal medium, potato dextrose agar, and yeast extract medium for three days at 28°C, and disruption of AflaILVB/G/I caused a significant reduction in conidia production when grown on all kinds of media. Cellular stress assays determined that all strains were sensitive to H2O2. Importantly, the pathogenicity and aflatoxin production were affected when AflaILVB/G/I and AflaILVD were knocked out, particularly AflaILVB/G/I. A series of genes that encoded enzymes involved in aflatoxin synthesis were downregulated, meaning that the knockout of AflaILVB/G/I influenced aflatoxin synthesis in A. flavus strain WT. Collectively, our results demonstrate the potential value of antifungals targeting AflaILVB/G/I in A. flavus.
1 Introduction
Mycotoxins are a wide variety of toxic secondary metabolites produced by fungi. Most are stable and heat-resistant, and are found in crops, feed, food, and other plant-derived products. Aflatoxins (AFs) are produced by fungi such as Aspergillus flavus and A. parasiticus. AFB1 is one of the most toxic mycotoxins and is classified as a 1A carcinogen by the International Center for Research on Cancer (Abdel-Wareth, 2023). It is the most common, serious, and widespread mycotoxin found in agricultural products and foods. Dietary exposure to AFs is estimated to affect more than five billion people globally (Strosnider et al., 2006). The most significant impact of AFs on human health is hepatocellular carcinoma (HCC). According to the World Health Organization (WHO), the number of deaths caused by HCC reached 830,000 globally in 2020, and AF is one of the most common risk factors for HCC in developing countries (WHO, 2022). AF pollution not only poses a serious threat to food safety but is also one of the factors affecting trade in various countries. In 2019, the EU Rapid Warning System for food and agricultural products from China to Europe recorded up to 30% of notifications associated with AFs. Therefore, research on control measures for AF pollution is of great significance for the food industry and consumer health protection.
Currently, the control of the AF pollution risk mainly focuses on detoxification technologies related to toxin pollution and the prevention and control of toxigenic fungi (Yu et al., 2020; Guan et al., 2021). Detoxification technology usually relies on physical, chemical, and biological methods to degrade toxins; however, such methods require the safety of the degradation products to be evaluated (Yang et al., 2022). Methods for controlling the growth of toxic fungi in edible agricultural products include the use of disease-resistant varieties, chemical agents, storage temperature and humidity controls, plant-derived bacteriostatic agents, and biological control (Xing et al., 2021). Although the development of antifungal agents has always been a focus of research, only a few agents are currently available. Therefore, the development of new agents is important for the continuous control of AF-producing fungi and AF pollution. AF contamination, which requires the control of A. flavus, remains problematic because of the lack of effective strategies. Thus, the exploration of new compounds that can inhibit A. flavus growth and mycotoxin production is urgently required to alleviate potential deleterious effects.
Amino acids are the basic building blocks of proteins that play an important role in the structure of body tissues and are essential for nutrition, metabolic regulation, immunity, and information transmission. Branched-chain amino acids (BCAAs) are synthesized in plants, fungi, bacteria, and algae but not animals. Therefore, a key enzyme in this synthetic pathway is a potential target for the development of non-toxic antimicrobial agents with limited toxicity in mammals.
Over the past 20 years, many drugs that have been developed and marketed worldwide, including sulfonylureas, imidazolinones, pyrimidine benzoates, triazolinones, sulfonamide-carbonyl triazolinones target acetohydroxy acid synthase (AHAS) in the BCAA pathway (Jeschke et al., 2019). Several studies have shown that some herbicide-targeting enzymes related to the BCAA pathway also exhibit antibacterial activity against human pathogenic bacteria. Lee et al. showed that chlorsulfuron (CE) had antibacterial effects on five different types of Candida and Cryptococcus neoformans. Garcia et al. further confirmed that the synergistic effect of CE and itraconazole effectively improves the survival rate of mice fed Candida albicans (Lee et al., 2013; Garcia et al., 2018). In addition, sulfonylureas exhibit antibacterial activity against Mycobacterium tuberculosis, which causes tuberculosis. For example, chloroxanthin has a stronger antibacterial effect against M. tuberculosis than streptomycin (Rehberg et al., 2018). In addition, studies have found that the BCAA synthesis pathway is an important part of mycotoxin biosynthesis, and the deletion of genes involved in the BCAA pathway causes mycotrophic deficiency, resulting in a significant reduction in pathogenicity and toxin biosynthesis (Du et al., 2013; Liu et al., 2019; Passalacqua et al., 2020).
In this study, we characterized the functions of AflaILVB/G/I and AflaILVD, which encode the catalytic and regulatory subunits of AHAS and dihydroxy acid dehydratase (DHD), respectively, using a strategy of targeted gene deletion and complementation in A. flavus to investigate their possible roles in various cellular processes, as well as their potential to serve as novel fungicide targets. Furtherly, transcriptome sequencing analysis of △AflaILVB/G/I was performed using the wild strain as a control group to investigate the regulatory mechanism.
2 Materials and methods
2.1 Strains, growth conditions, and phenotype assays
TJW 149.27 (pyrG1, Δku70::AfpyrG) was used as the parental wild-type (WT) strain. To assess mycelial growth and colony characteristics, the wild-type strain and mutants were cultured on different media, including glucose minimal medium (GMM), potato dextrose agar (PDA), yeast extract (YES), and yeast glucose trace (YGT), and incubated at 28°C. Colony diameter was measured at 28°C for three days. For the conidiation assay, three 5-mm mycelial plugs of the WT strain and mutants obtained from the edge of a three-day-old colony on PDA and YGT were placed in an Eppendorf tube containing 1 mL of normal saline, and the conidia were counted using a hemocytometer. The experiment was performed in triplicates.
2.2 Cellular sensitivity to different environmental stresses
To evaluate the sensitivity to various cellular stresses, 5-mm (diameter) mycelial plugs of the WT strain and mutants obtained from the edge of a three-day-old colony on PDA were transferred onto GMM with 8 mmol H2O2, GMM + 1 M NaCl, GMM +1 M KCl GMM + 0.6% Congo red, and GMM + 0.3% calcofluor white (CFW). Colony morphology was photographed and the diameter was measured after incubation at 28°C for three days. The relative inhibition rates were calculated.
2.3 Targeted gene deletion
AflaILVB/G/I and AflaILVD deletion mutants were obtained as follows: The upstream and downstream homologous arm fragments of AflaILVB/G/I and AflaILVD were connected to the arg B fragment of A. flavus to construct knockout fragments through overlapping PCR. The knockout fragments were imported into the protoplasts of the TJES 20.1(Δku70/ΔargB) strain (kindly provided by Professor Yang, Jiangsu Normal University) through homologous recombination. Transformants positive for the knockout were identified through PCR and Southern blotting using arginine as a screening marker.
2.4 Peanut infection
The ability of WT and mutant strains to infect peanuts was assessed. WT and mutant strain conidia were harvested from three-day-old PDA cultures, washed with sterilized water, and resuspended in 0.01% (v/v) Tween 20 solution. The concentration of the conidial suspension was adjusted to 1 × 107 conidia/mL. Peanut seeds weighing approximately 20 g were sterilized with 75% medical alcohol for 1 min and then rinsed five times with sterile water so that the water content of the seeds reached approximately 20%. Each peanut was soaked in spore suspension for 5 s, placed in a disposable petri dish, sealed, and placed in an incubator at 28°C for five days. The experiment was repeated three times.
2.5 AFB1 analysis
To determine AFB1 production, 10-mL aliquots of 1 × 106 spore/mL suspension of A. flavus conidia of the WT and mutant strains were incubated in YES and PDB media in the dark at 28°C for 3.5 d. Thin layer chromatography (TLC) was used to analyze AF production as previously described (Yang et al., 2016). AF was extracted from the 500-μL filtrate with an equal volume of chloroform. The chloroform layer was transferred to a new 1.5 mL tube, then evaporated to dryness at 70°C. Next, TLC was used to analyze AF biosynthesis. A solvent system consisting of acetone and chloroform (1:9, v/v) was used, and TLC results were obtained under ultraviolet light at 365 nm.
For the quantitative analysis of AF production, high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) was used to confirm the presence of AF in infected peanuts. The peanuts were sterilized at 121°C for 30 min before grinding, and the analysis performed as previously described (Zhao et al., 2017).
2.6 cDNA preparation and Illumina sequencing
Total RNA was extracted using a Total RNA Extractor (TRIzol) kit (B511311, Sangon Biotech Co. Ltd., Shanghai, China) according to the manufacturer´s protocol and treated with RNase-free DNase I to remove genomic DNA contamination. RNA integrity was evaluated using a 1.0% agarose gel electrophoresis. The quality and quantity of RNA were assessed using a NanoPhotometer ® spectrophotometer (Implen, Westlake Village, CA, USA) and a Qubit® 2.0 Fluorometer (Invitrogen). High-quality RNA samples were subsequently submitted to Sangon Biotech Co. Ltd., China) for library preparation and sequencing.
Briefly, mRNA was purified from the total RNA using poly-T oligo-attached magnetic beads. Sequencing libraries were generated using VAHTSTM mRNA-seq V2 Library Prep kit for Illumina® following manufacturer´s recommendations and index codes were added to attribute sequences to each sample. Fragmentation was conducted using divalent cations at elevated temperatures in VAHTSTM First Strand Synthesis Reaction Buffer (5X). First strand cDNA was synthesized using random hexamer primer and M-MuLV reverse transcriptase (RNase H-). Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activity. After adenylation of 3´ ends of DNA fragments, an adaptor was ligated to prepare the library. To select cDNA fragments preferentially 150–200 bp in length, the library fragments were purified using the AMPure XP system (Beckman Coulter, Brea, C A, USA). Then, 3 μL USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C before PCR. PCR was performed using the Phusion High-Fidelity DNA polymerase, universal PCR primers, and Index (X) Primer. Finally, the PCR products were purified (AMPure XP system), and library quality was assessed using an Agilent Bioanalyzer 2100 system. The libraries were quantified and pooled. Paired-end sequencing of the library was performed on a NovaSeq sequencer (Illumina, San Diego, CA, USA).
2.7 Expression analysis
The gene expression of the transcripts was calculated using StringTie (version 1.3.3b). A principal component analysis (PCA) was performed to determine the distance between samples. Transcripts per million (TPM) eliminated the influence of gene length and sequencing discrepancies to enable a direct comparison of gene expression between samples. DESeq2 (version 1.12.4) was used to determine differentially expressed genes (DEGs) between two samples. Genes were considered as significantly differentially expressed if the q-value was < 0.05 and |FoldChange| > 2.
2.8 Functional analysis of DEGs
Functional enrichment analyses, including Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG), were performed to identify which DEGs were significantly enriched in GO terms or metabolic pathways. GO is an international standard classification system for gene functions. DEGs were mapped to GO terms (biological functions) in the database, the number of genes in each term was calculated, and a hypergeometric test was performed to identify significantly enriched GO terms in the gene list from the background of the reference gene list. The KEGG database is a public database of pathway data, and KEGG pathway analysis identifies significantly enriched metabolic or signal transduction pathways enriched in DEGs compared to a reference gene background using the hypergeometric test. GO terms and KEGG pathways with a false discovery rate (q-value) < 0.05 were considered significantly altered.
2.9 Analysis of gene expression
Total RNA of the WT and △AflaILVB/G/I was extracted and cDNA was reverse transcribed using fungal RNA extraction kit (Coolaber, Beijing, China) and Hifair® III 1st Strand cDNA Synthesis kit (Yeasen Biotech Co. Ltd., Shanghai, China). Expression of the genes involved in aflatoxins synthesis was determined by quantitative real-time PCR (RT-PCR) with primes (Table 1). The RT-PCR amplifications were performed in a QuantStudio 3 (Thermo Fisher, Massachusetts, USA) using SYBR Green I fluorescent dye detection. Amplifications were conducted in a 20 ml volume containing 10 ml iQ SYBR Green Supermix (Bio-Rad Laboratories), 2 ml reverse transcription product and 0.4 ml of each primer. There were three replicates for each sample. RT-PCR amplifications were performed with the following parameters: an initial preheat at 95°C for 2 min, followed by 40 cycles at 95°C for 10 s, 60°C for 30 s. Once amplifications were completed, melting curves were obtained to identify PCR products. For each sample, PCR amplifications with primer pair actin of A.flavus for quantifying expression of the actin gene were performed as a reference. The experiment was repeated three times. Levels of expression of the genes of interest in the WT strain and deletion mutants were calculated using the 2-ΔΔCT method (Livak and Schmittgen, 2001).

Table 1 Prime sequence of gene for real-time PCR.
3 Results
3.1 Identification and analysis of AflaILVB/G/I and AflaILVD in A. flavus
Amino acids are important components in the metabolism of a variety of pathogens, plants, and animals. AFLA_000930 and AFLA_003530, orthologs of S. cerevisiae ILV2 and ILV3 and involved in valine, leucine, and isoleucine biosynthesis, were identified through BLAST analysis and named AflaILVB/G/I and AflaILVD. The regulatory subunit of AHAS AflaILVB/G/I is one of several enzymes that require thiamine pyrophosphate (TPP, vitamin B1) as a cofactor. Some of these enzymes were structurally related (PubMed: 8604141). All of them have an N-terminal central domain, and C-terminal TPP-binding domain for TPP enzymes. AflaILVD encodes a dihydroxy-acid dehydratase and catalyzes the fourth step in the biosynthesis of isoleucine and valine, that is, the dehydration of 2,3-dihydroxy-isovaleic acid into alpha-ketoisovaleric acid. The enzyme 6-phosphogluconate dehydratase catalyzes the first step in the Entner-Doudoroff pathway, that is, the dehydration of 6-phospho-D-gluconate into 6-phospho-2-dehydro-3-deoxy-D-gluconate. Another protein with this signature is Escherichia coli YjhG, which has been identified as a d-xylonate dehydratase. The N-terminal region of the protein contains a cysteine residue that may be involved in the binding of a 2Fe-2S iron-sulfur cluster.
3.2 AflaILVB/G/I and AflaILVD are involved in colony growth and conidiation in A. flavus
To gain insight into the possible functions of AflaILVB/G/I and AflaILVD in the developmental stages of A. flavus, we generated △AflaILVB/G/I and △AflaILVD in the WT strain. As shown in Figures 1A, B, when grown on GMM, PDA, and YES medium for 3 days at 28°C, the △AflaILVB/G/I and △AflaILVD mutants grew slower and produced smaller colonies than the WT strain. When grown on YGT medium, however, the colony diameter of the △AflaILVD mutant was greater than that of the WT. In addition, △AflaILVB/G/I was significantly different compared with △AflaILVD.
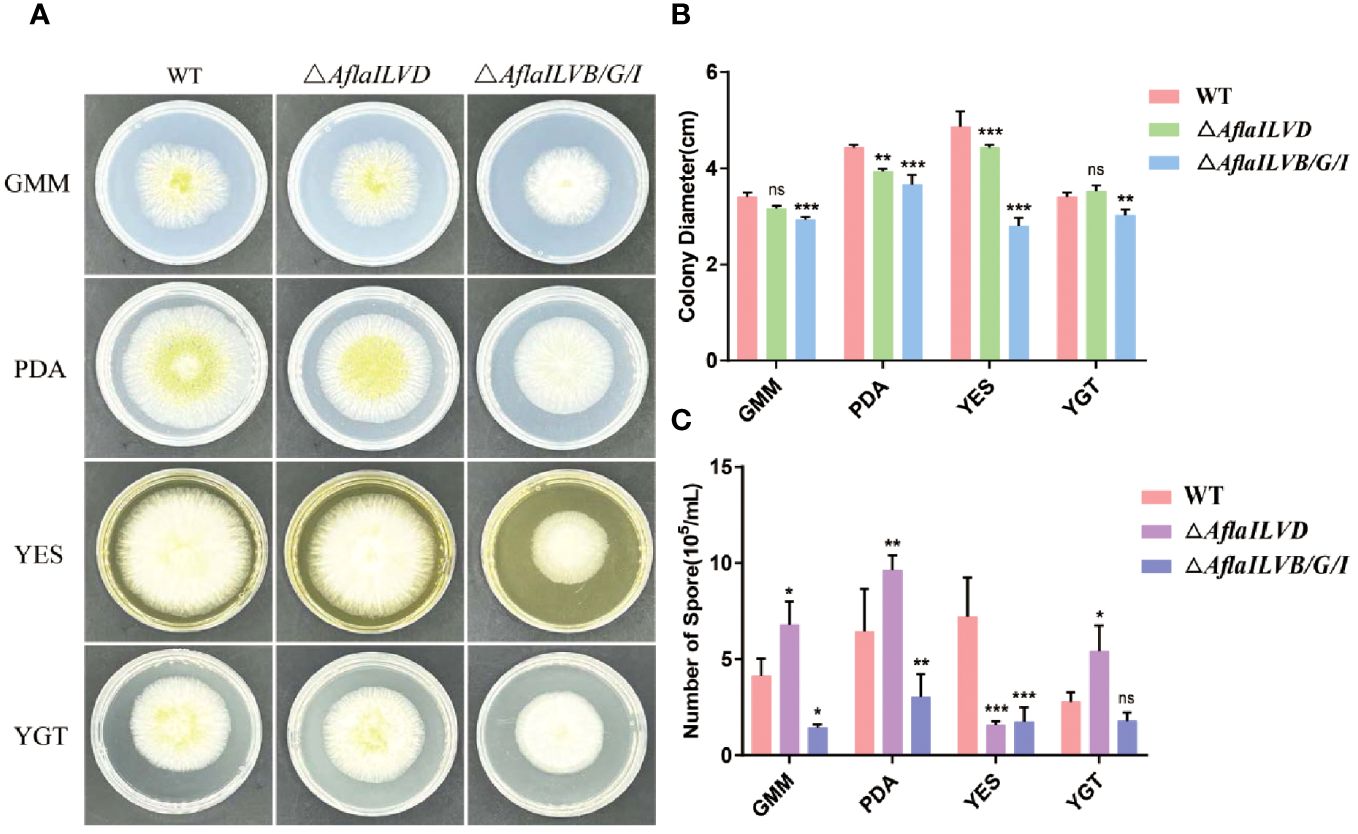
Figure 1 Influence of AflaILVB/G/I and AflaILVD on colony growth and conidiation. (A) Colonies formed by the WT, △AflaILVB/G/I, and △AflaILVD strains on GMM, PDA, YES, and YGT medium agar plates. (B) Colony diameters of WT, △AflaILVB/G/I, and △AflaILVD strains on GMM, PDA, YES, and YGT medium agar plates. (C) Number of spores of WT, △AflaILVB/G/I, and △AflaILVD strains on GMM, PDA, YES, and YGT media. *: p<0.05, **: p<0.01, ***: p<0.001.
Figure 1C shows that the disruption of AflaILVB/G/I caused a significant reduction in conidial production when grown on all media, but it is interesting that when grown on GMM, PDA, and YGT media, the △AflaILVD mutant produced more conidia than the WT strain, but it produced fewer on the YES medium.
3.3 AflaILVB/G/I and AflaILVD are required for adaptation to various cellular stresses in A. flavus
To test the responses of AflaILVB/G/I and AflaILVD to various environmental stresses, the sensitivities of the WT and mutant strains to osmotic and oxidative stresses were assessed. As shown in Figures 2A and 2B, when compared with WT, △AflaILVB/G/I and △AflaILVD strains did not exhibit recognizable changes to cell-wall-damaging agents Congo red (0.6%), 1 M NaCl and GMM + 1 M KCl. However, all strains were sensitive to 8 mmol H2O2 and 0.3% CFW. For H2O2, the relative inhibition rate of △AflaILVB/G/I was > 70%.
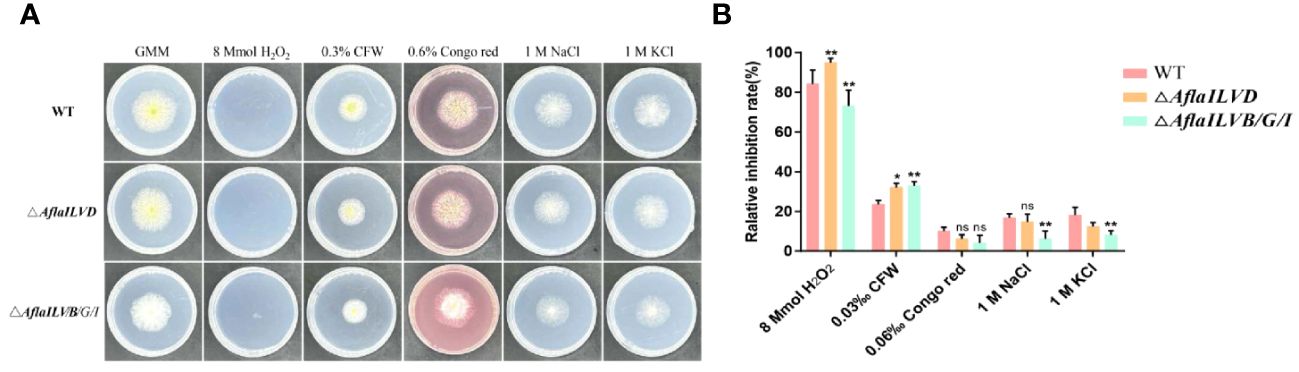
Figure 2 (A) Phenotype and (B) relative inhibition rate (%) of WT, △AflaILVB/G/I, and △AflaILVD A. flavus strains grown on GMM medium under various cellular stresses. *: p<0.05, **: p<0.01, ns: no significance.
3.4 AflaILVB/G/I and AflaILVD participate in full virulence and AFB1 biosynthesis in A. flavus
To explore the function of AflaILVB/G/I and AflaILVD genes in the pathogenicity and AF biosynthesis in A. flavus, we inoculated peanuts with WT and mutant strains. Peanut infection assays revealed the crucial role of AflaILVB/G/I and AflaILVD in full virulence and mycotoxin biosynthesis. As shown in Figure 3A, the pathogenicity of the mutants lacking AflaILVB/G/I and AflaILVD was reduced, and the former exhibited a greater loss of virulence. Figures 3B, C show that AFB1 production was detected by TLC and HPLC-MS/MS in the WT, △AflaILVB/G/I, and △AflaILVD strains. The synthesized AFB1 content was affected when AflaILVB/G/I and AflaILVD were knocked out, especially for AflaILVB/G/I, for which AFB1 production was significantly different from that of the WT.
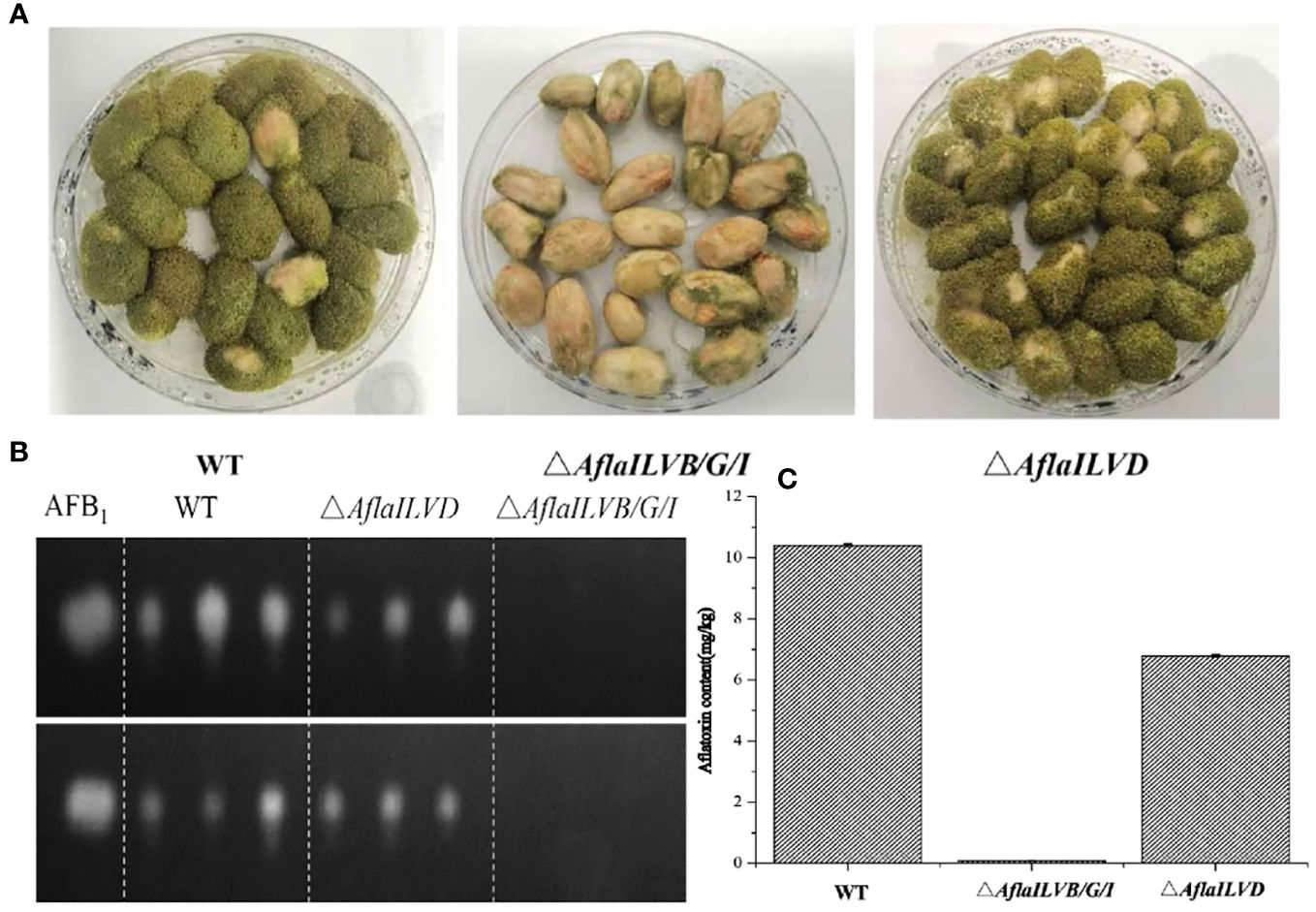
Figure 3 (A) Pathogenicity for peanut seed and (B) AFB1 production by WT, △AflaILVB/G/I, and △AflaILVD strains used TCL, (C) AF production by WT, △AflaILVB/G/I, and △AflaILVD strains used HPLC-MS/MS.
3.5 Quality control and gene expression analysis
The statistical findings of the sequencing data for the clean reads of the final obtained were Q20 > 95%. Q30 > 89%, indicating that the sequencing quality was good and that it could be used for subsequent comparative and data analyses. The results of the gene expression analysis and PCA are shown in Figure 4. The close distance between the samples within the groups indicated a high similarity between the samples, and the distance between the samples indicated that there were significant differences between the groups.
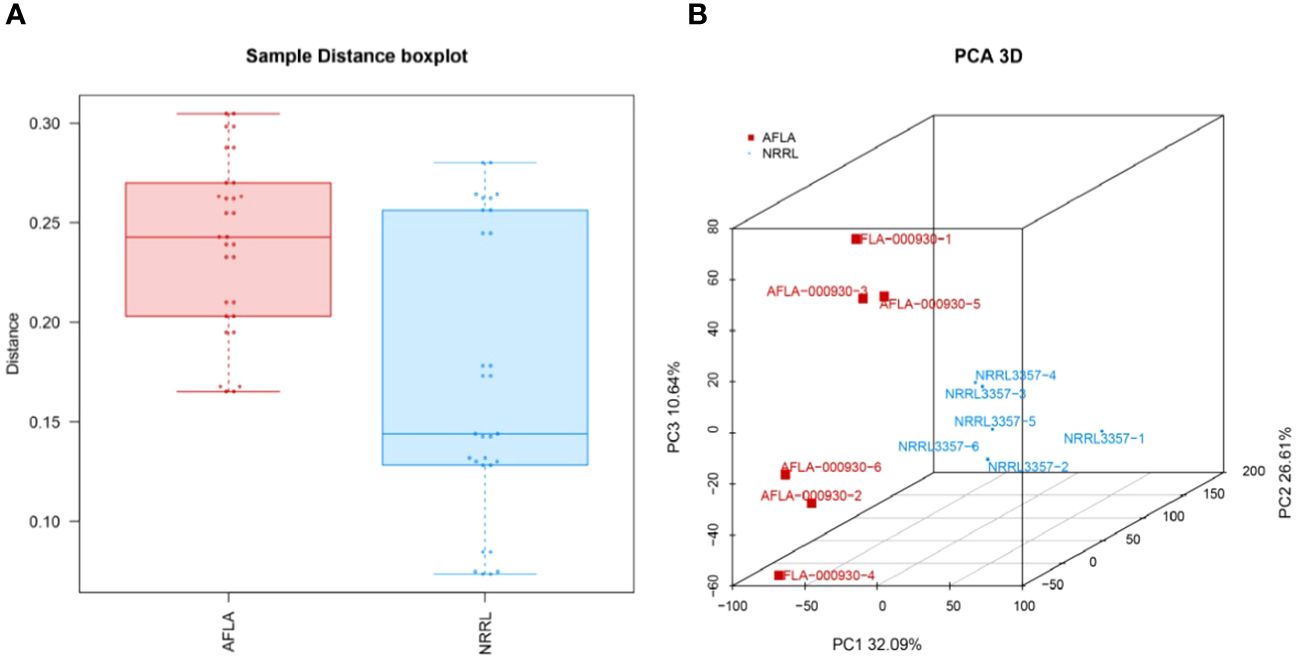
Figure 4 (A) TPM distribution boxplot and (B) PCA analysis. Points of different colors or shapes represent different groups, and the scale of the horizontal and vertical coordinate axes represents the relative distance, which has no practical significance.
3.6 DEGs identification and analysis
To identify the differences in molecular responses between the WT and mutant strains, TMM was used to standardize the read count and DEGseq was used to analyze the differences (q-value < 0.05, |FoldChange| > 2); 3929 DEGs were obtained. Among them, 2438 and 1491 genes were upregulated and downregulated, respectively (Figure 5A).
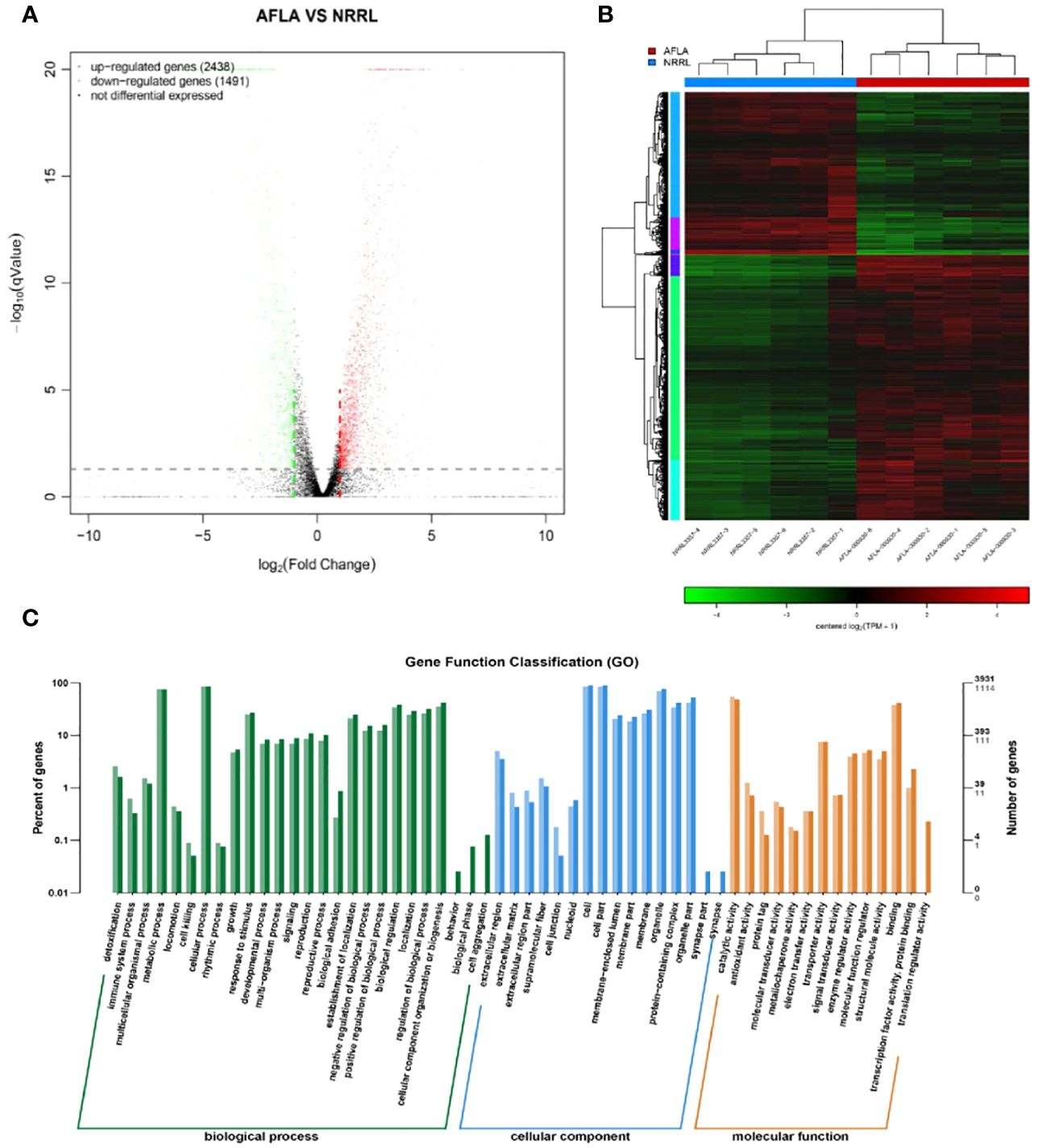
Figure 5 (A) Volcano map of the differentially expressed genes (B) Clustering results of AFLA and NRRL (C) GO functional enrichment analysis of differentially expressed genes.
The expression levels of differential genes under different experimental conditions were used for hierarchical clustering analysis. The biological function of a gene was predicted by grouping genes with high expression correlations in different groups. Genes with similar colors have similar expression patterns in the cluster area; the similarity of these patterns suggests that these genes may have similar functions or be involved in regulating the same metabolic pathway (Figure 5B).
Functional assignments were defined using GO terms, which provided a comprehensive functional classification of genes and their products based on various biological processes, cellular components, and molecular functions. GO functional enrichment analysis revealed several enriched terms for DEGs. Table 2 shows the top 10 most significantly different GO terms. Among them, small-molecule metabolic, carboxylic acid metabolic, oxoacid metabolic, organic acid metabolic, and small-molecule catabolic processes were the most significant. In addition, as shown in Figure 5C, according to the number of genes enriched in GO terms, most DEGs were enriched in cellular processes of biological processes, followed by cell and cell parts of cellular components.
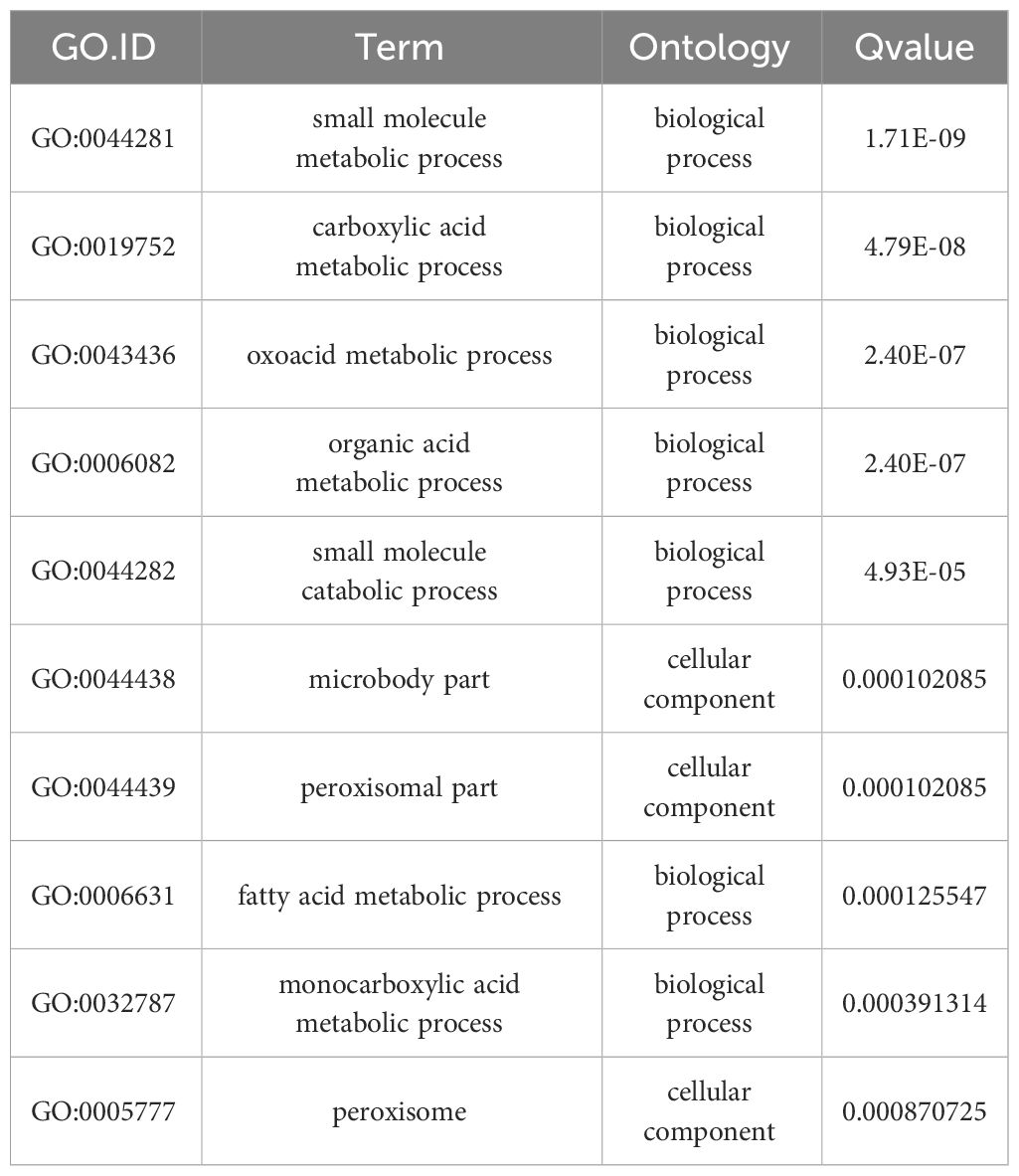
Table 2 GO functional enrichment of top 10 most significant differentially expressed genes.
Pathway-based analysis was performed using the KEGG pathway database to explore the biological functions and interactions in the genes. The results showed that 3929 DEGs were involved in 261 pathways. Table 3 shows the top ten upregulated and downregulated genes enriched in KEGG. Among the downregulated genes, the most enriched pathways were those involved in AF biosynthesis, followed by serotonergic synapses and pyruvate metabolism. This suggests that the expression of genes involved in AF synthesis was suppressed in the experimental group. In contrast, the most enriched pathway among the upregulated genes was peroxisomes, followed by purine and sulfur metabolisms. This indicates that genes encoding oxidation-reduction enzymes were highly expressed in the experimental groups. Furthermore, KEGG metabolic pathway analysis confirmed that the enrichment of peroxisomes, sulfur metabolism, and AF biosynthesis was significant (p < 0.05).
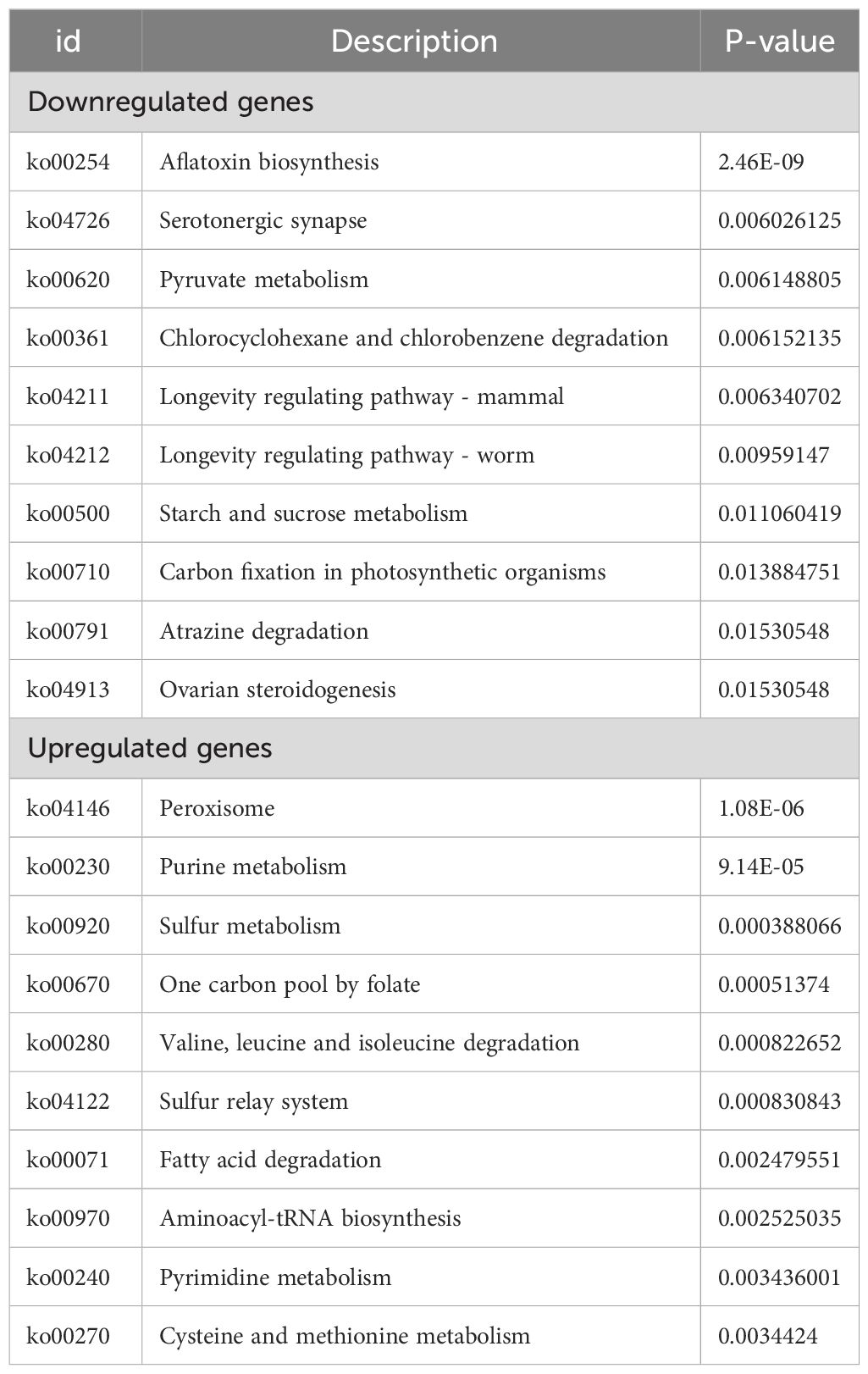
Table 3 Top 10 KEGG enrichment for down and upregulated genes.
3.7 Analysis of DEGs involved in AF biosynthesis
The DEGs involved in aflatoxin biosynthesis are shown in Figure 6, which shows that among the ten differentially expressed genes annotated in the two strains, eight genes were downregulated: hypC, aflK, aflP, aflO, aflH, aflD, aflC, aflQ and nor-1 was also upregulated, and one gene remained unchanged.
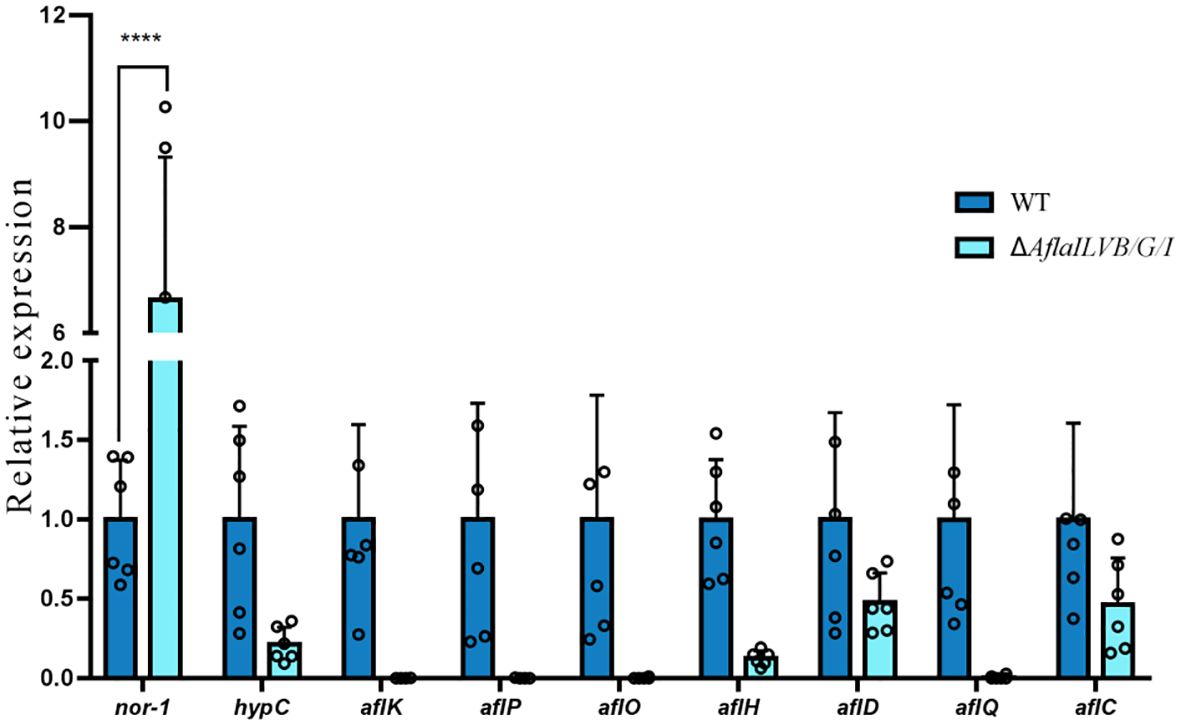
Figure 6 Differentially expressed genes involved in aflatoxin biosynthesis. ****: p<0.0001.
To further confirm the results, we assayed the expression of aflQ, aflK, aflH, hypC, aflD by quantitative RT-PCR using RNA samples isolated from mycelia grown in PDB medium for 4 days at 28°C. The expression level of aflQ, aflK, aflH, hypC and aflD in the △AflaILVB/G/I was decreased by 92, 96, 86, 78 and 95%, respectively, as compared with levels in the WT (Figure 7).
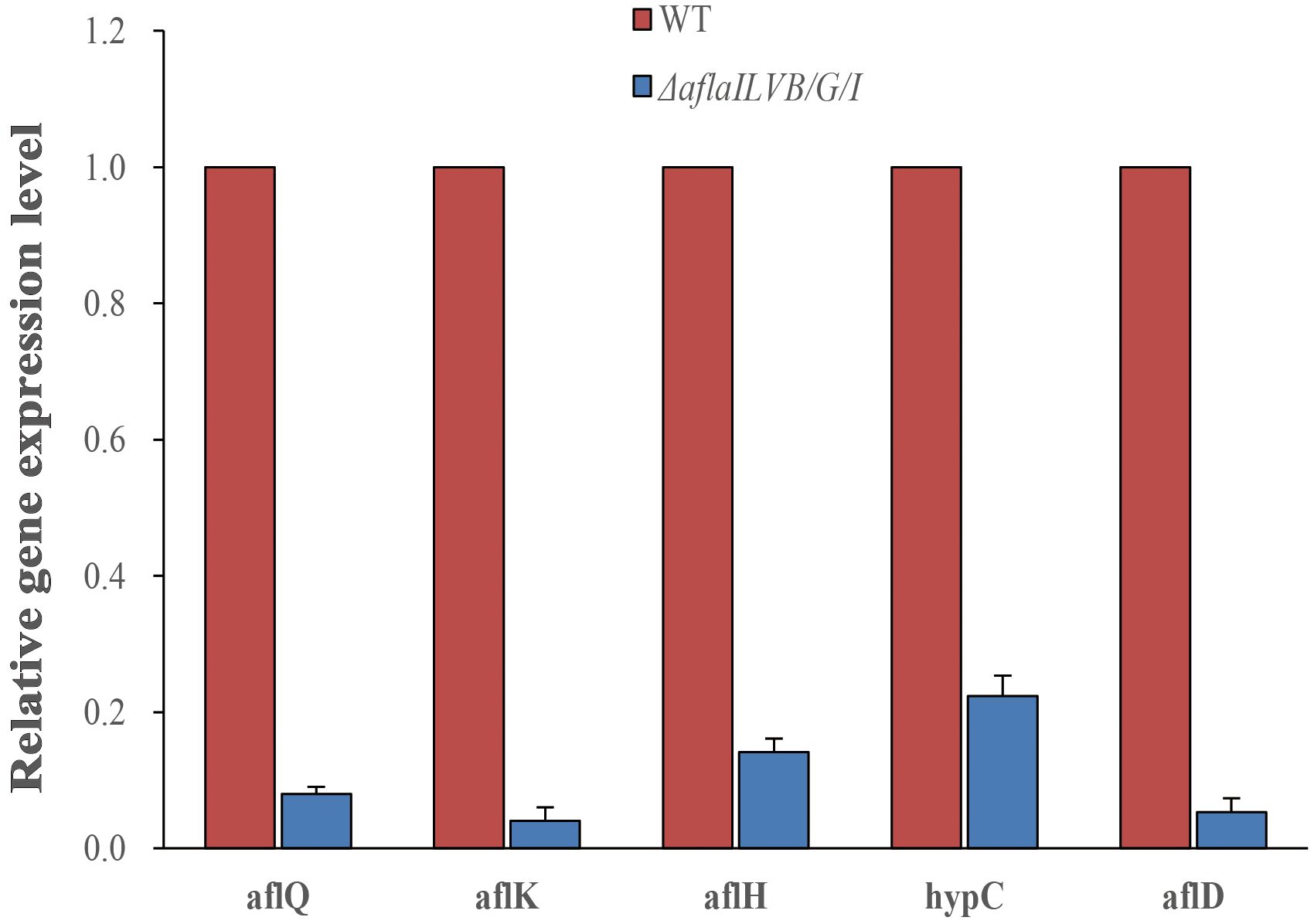
Figure 7 Relative expression of AFB1 synthetic genes in the △AflaILVB/G/I.
4 Discussion
AHAS is the first shared enzyme in the BCAA biosynthetic pathway and is an inhibitory target of the most widely used herbicides (AHAS inhibitors) (Lonhienne et al., 2018). Some AHAS-targeted herbicides exert inhibitory effects against several pathogenic bacteria and yeast, as reported in previous studies (Kreisberg et al., 2013), indicating a promising potential for AHAS as an anti-microorganism target. Functional analyses of S. cerevisiae, C. albicans, C. neoformans, M. oryzae, and F. graminearum have suggested that the AHAS catalytic subunit-encoding gene ILV2 has the potential to be an antifungal target, based on phenotypic defects caused by the disruption of ILV2 (Kingsbury et al., 2004, 2006; Kingsbury and McCusker, 2010; Liu et al., 2015). In this study, the blocking of BCAA biosynthesis caused by the deletion of AflaILVB/G/I and AflaILVD resulted in defects in vegetative growth and conidiation. Based on the degree of severity of phenotypic defects displayed by the mutants, AflaILVB/G/I may play a more important role in A. flavus than AflaILVD. In addition, pathogenicity and AFB1 accumulation were impaired in both deletion mutants. In the infection assay on peanut, the virulence of △AflaILVB/G/I and △AflaILVD strains was reduced and the former was almost completely lost.
In vitro, the deletion mutants were associated with severely infected phenotypes; therefore, it was not possible to assess their relative virulence. Compared with WT strains, △AflaILVB/G/I colony growth was reduced and fewer spores were observed when cultured on a medium. In addition, reactive oxygen species play essential roles in host-pathogen interactions. During a pathogen attack, organisms use an oxidative burst as an early defense mechanism (Liu et al., 2014). △AflaILVB/G/I showed significantly increased sensitivity to the oxidative agent H2O2, which may be related to the reduced virulence in peanut. The deletion mutants were less sensitive than the WT to osmotic stress mediated by Congo red, NaCl, and KCl.
Based on phenotypic analysis, △AflaILVB/G/I and WT strains were transcriptome sequenced, a total of 3929 differential genes were screened, of which 2438 upregulated and 1491 downregulated genes. GO and KEGG enrichment analyses were performed on DEGs. More importantly, in our study, a series of genes that encode enzymes involved in aflatoxin synthesis were downregulated, indicating that AflaILVB/G/I knockout influenced aflatoxin synthesis in A. flavus strain NRRL3557.
AF contamination remains a major global food safety challenge with serious implications for agriculture and public health (Nzuma and Mzera, 2023). A. flavus is a representative fungal species in Aspergillus section Flavi and has been used as a model system to gain insights into fungal development and toxin production. In this study, by identifying the key genes in the BCAA synthesis pathway of A. flavus and analyzing their biological functions, we clarified the roles of BCAAs in the important physiological processes of growth, sporulation, pathogenicity, and AF synthesis. This study provides a theoretical basis for developing new antimicrobials to reduce AF pollution.
In conclusion, ILV may be a key gene affecting the growth, development, pathogenicity, and toxin synthesis in A. flavus. The key genes in this pathway can be used to develop new, safe antimicrobial agents to prevent and control AF contamination.
Data availability statement
The data presented in the study are deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center repository, accession number CRA015215.
Author contributions
YZ: Conceptualization, Writing – original draft. CH: Methodology, Writing – review & editing. RZ: Formal analysis, Writing – review & editing. PC: Methodology, Writing – review & editing. KX: Validation, Writing – review & editing. XH: Validation, Writing – review & editing. XW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Guangdong Basic and Applied Basic Research Foundation (grant number 2022A1515110039).
Acknowledgments
We thank Kunlong Yang and Zhentong Sun for help with part of the experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdel-Wareth, M. T. A. (2023). Aflatoxins in food: a recent perspective. Int. J. Environ. Stud. 80, 243–245. doi: 10.1080/00207233.2022.2050586
Du, Y., Zhang, H., Hong, L., Wang, J., Zheng, X., Zhang, Z. (2013). Acetolactate synthases MoIlv2 and MoIlv6 are required for infection-related morphogenesis in Magnaporthe oryzae. Mol. Plant Pathol. 14, 870–884. doi: 10.1111/mpp.12053
Garcia, M. D., Chua, S. M. H., Low, Y. S., Lee, Y. T., Agnew-Francis, K., Wang, J. G., et al. (2018). Commercial AHAS-inhibiting herbicides are promising drug leads for the treatment of human fungal pathogenic infections. Proc. Natl. Acad. Sci. U.S.A. 115, E9649–E9658. doi: 10.1073/pnas.1809422115
Guan, Y., Chen, J., Nepovimova, E., Long, M., Wu, W., Kuca, K. (2021). Aflatoxin detoxification using microorganisms and enzymes. Toxins 13, 46. doi: 10.3390/toxins13010046
Jeschke, P., Witschel, M., Krmer, W., Schirmer, U. (2019). Acetohydroxyacid synthase inhibitors (AHAS/ALS). Mod. Crop Prot. Compd, 29–162. doi: 10.1002/9783527699261.ch2
Kingsbury, J. M., Goldstein, A. L., McCusker, J. H. (2006). Role of nitrogen and carbon transport, regulation, and metabolism genes for Saccharomyces cerevisiae survival in vivo. Eukaryot. Cell 5, 816–824. doi: 10.1128/EC.5.5.816-824.2006
Kingsbury, J. M., McCusker, J. H. (2010). Cytocidal amino acid starvation of Saccharomyces cerevisiae and Candida albicans acetolactate synthase (ilv2Δ) mutants is influenced by the carbon source and rapamycin. Microbiol. (Reading). 156, 929–939. doi: 10.1099/mic.0.034348-0
Kingsbury, J. M., Yang, Z., Ganous, T. M., Cox, G. M., McCusker, J. H. (2004). Cryptococcus neoformans Ilv2p confers resistance to sulfometuron methyl and is required for survival at 37 degrees C and in vivo. Microbiol. (Reading). 150, 1547–1558. doi: 10.1099/mic.0.26928-0
Kreisberg, J. F., Ong, N. T., Krishna, A., Joseph, T. L., Wang, J., Ong, C., et al. (2013). Growth inhibition of pathogenic bacteria by sulfonylurea herbicides. Antimicrob. Agents Chemother. 57, 1513–1517. doi: 10.1128/AAC.02327-12
Lee, Y. T., Cui, C. J., Chow, E. W., Pue, N., Lonhienne, T., Wang, J. G., et al. (2013). Sulfonylureas have antifungal activity and are potent inhibitors of Candida albicans acetohydroxyacid synthase. J. Med. Chem. 56, 210–219. doi: 10.1021/jm301501k
Liu, X., Han, Q., Xu, J., Wang, J., Shi, J. (2015). Acetohydroxyacid synthase FgIlv2 and FgIlv6 are involved in BCAA biosynthesis, mycelial and conidial morphogenesis, and full virulence in Fusarium graminearum. Sci. Rep. 5, 16315. doi: 10.1038/srep16315
Liu, X., Jiang, Y., Zhang, Y., Yu, M., Jiang, H., Xu, J., et al. (2019). FgIlv3a is crucial in branched-chain amino acid biosynthesis, vegetative differentiation, and virulence in Fusarium graminearum. J. Microbiol. 57, 694–703. doi: 10.1007/s12275-019-9123-6
Liu, X., Wang, J., Xu, J., Shi, J. (2014). FgIlv5 is required for branched-chain amino acid biosynthesis and full virulence in Fusarium graminearum. Microbiology 160, 692–702. doi: 10.1099/mic.0.075333-0
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2△△CT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lonhienne, T., Garcia, M. D., Pierens, G., Mobli, M., Nouwens, A., Guddat, L. W. (2018). Structural insights into the mechanism of inhibition of AHAS by herbicides. Proc. Natl. Acad. Sci. U.S.A. 115, E1945–E1954. doi: 10.1073/pnas.1714392115
Nzuma, J. M., Mzera, U. I. (2023). Evaluating aflatoxin contamination control practices among smallholder maize farmers in Kilifi County, Kenya: A Poisson regression analysis. Environ. Dev. Sustain., 1–13. doi: 10.1007/s10668-023-03133-z
Passalacqua, K. D., Zhou, T. H., Washington, T. A., Abuaita, B. H., Sonenshein, A. L., Riordan, M. X. D. O. (2020). The branched chain aminotransferase IlvE promotes growth, stress resistance and pathogenesis of Listeria monocytogenes. BioRxiv, 1–33. doi: 10.1101/2020.01.31.929828
Rehberg, N., Akone, H. S., Ioerger, T. R., Erlenkamp, G., Daletos, G., Gohlke, H., et al. (2018). Chlorflavonin targets acetohydroxyacid synthase catalytic subunit IlvB1 for synergistic killing of Mycobacterium tuberculosis. ACS Infect. Dis. 4, 123–134. doi: 10.1021/acsinfecdis.7b00055
Strosnider, H., Azziz-Baumgartner, E., Banziger, M., Bhat, R. V., Breiman, R., Brune, M. N., et al. (2006). Workgroup report: public health strategies for reducing aflatoxin exposure in developing countries. Environ. Health Perspect. 114, 1898–1903. doi: 10.1289/ehp.9302
WHO (2022) Cancer fact sheet. Available online at: https://www.who.int/en/news-room/fact-sheets/detail/cancer (Accessed February 2022).
Xing, F., Li, X., Zhang, C. (2021). Biosynthesis mechanisms and control strategies of aflatoxin. J. Food Sci. Technol. 39, 13–26. doi: 10.12301/j.issn.2095-6002.2021.01.002
Yang, B., Li, L., Geng, H., Wang, G., Zhang, C., Yang, S., et al. (2022). Detoxification of aflatoxin b1 by H2SO3 during maize wet processing, and toxicity assessment of the transformation product of aflatoxin b1. Food Control. 131, 108444. doi: 10.1016/j.foodcont.2021.108444
Yang, K., Liang, L., Ran, F., Liu, Y., Li, Z., Lan, H., et al. (2016). The DmtA methyltransferase contributes to Aspergillus flavus conidiation, sclerotial production, aflatoxin biosynthesis and virulence. Sci. Rep. 6, 23259. doi: 10.1038/srep23259
Yu, Y., Shi, J., Xie, B., He, Y., Qin, Y., Wang, D., et al. (2020). Detoxification of aflatoxin b1 in corn by chlorine dioxide gas. Food Chem. 328, 127121. doi: 10.1016/j.foodchem.2020.127121
Keywords: Aspergillus flavus, aflatoxin biosynthesis, branched-chain amino acids, AflaILVB/G/I, AflaILVD, fungal secondary metabolites
Citation: Zhao Y, Huang C, Zeng R, Chen P, Xu K, Huang X and Wang X (2024) AflaILVB/G/I and AflaILVD are involved in mycelial production, aflatoxin biosynthesis, and fungal virulence in Aspergillus flavus. Front. Cell. Infect. Microbiol. 14:1372779. doi: 10.3389/fcimb.2024.1372779
Received: 18 January 2024; Accepted: 11 March 2024;
Published: 26 March 2024.
Edited by:
Kunlong Yang, Jiangsu Normal University, ChinaReviewed by:
Yangyong Lv, Henan University of Technology, ChinaXin Liu, Jiangsu Academy of Agricultural Sciences (JAAS), China
Copyright © 2024 Zhao, Huang, Zeng, Chen, Xu, Huang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Wang, d2FuZ3h1Z3Vhbmd6aG91QDEyNi5jb20=