 Chunlei Wang1,2,3†Xiuli Zhai1,2,3†
Chunlei Wang1,2,3†Xiuli Zhai1,2,3† Shibo Wang4†Bohan Zhang2,3Caiqin Yang2,3Yanmei Song2,3
Shibo Wang4†Bohan Zhang2,3Caiqin Yang2,3Yanmei Song2,3 Hanping Li2,3Yongjian Liu2,3Jingwan Han2,3Xiaolin Wang2,3Jingyun Li2,3
Hanping Li2,3Yongjian Liu2,3Jingwan Han2,3Xiaolin Wang2,3Jingyun Li2,3 Mingyue Chen4*
Mingyue Chen4* Lei Jia2,3*
Lei Jia2,3* Lin Li1,2,3*
Lin Li1,2,3*- 1Department of Microbiology, School of Basic Medicine, Anhui Medical University, Hefei, Anhui, China
- 2Department of Virology, Beijing Institute of Microbiology and Epidemiology, Beijing, China
- 3State Key Laboratory of Pathogen and Biosecurity, Beijing, China
- 4National 111 Center for Cellular Regulation and Molecular Pharmaceutics, Key Laboratory of Fermentation Engineering, Hubei University of Technology, Wuhan, Hubei, China
Endogenous retroviruses (ERVs) originate from ancestral germline infections caused by exogenous retroviruses. Throughout evolution, they have become fixed within the genome of the animals into which they were integrated. As ERV elements coevolve with the host, they are normally epigenetically silenced and can become upregulated in a series of physiological and pathological processes. Generally, a detailed ERV profile in the host genome is critical for understanding the evolutionary history and functional performance of the host genome. We previously characterized and cataloged all the ERV-K subtype HML-8 loci in the human genome; however, this has not been done for the chimpanzee, the nearest living relative of humans. In this study, we aimed to catalog and characterize the integration of HML-8 in the chimpanzee genome and compare it with the integration of HML-8 in the human genome. We analyzed the integration of HML-8 and found that HML-8 pervasively invaded the chimpanzee genome. A total of 76 proviral elements were characterized on 23/24 chromosomes, including detailed elements distribution, structure, phylogeny, integration time, and their potential to regulate adjacent genes. The incomplete structure of HML-8 proviral LTRs will undoubtedly affect their activity. Moreover, the results indicated that HML-8 integration occurred before the divergence between humans and chimpanzees. Furthermore, chimpanzees include more HML-8 proviral elements (76 vs. 40) and fewer solo long terminal repeats (LTR) (0 vs. 5) than humans. These results suggested that chimpanzee genome activity is less than the human genome and that humans may have a better ability to shape and screen integrated proviral elements. Our work is informative in both an evolutionary and a functional context for ERVs.
1 Introduction
Endogenous retroviruses have played a role in primate evolution and result from exogenous retroviral infections, which integrate into the genome of the host germline and are subsequently inherited by the next generation (Stoye, 2012; Mager and Stoye, 2015; Jansz and Faulkner, 2021). ERVs can be found in all vertebrate genomes (Stoye, 2012; Jansz and Faulkner, 2021). For human endogenous retroviruses (HERVs), all residual components of HERVs have accounted for approximately 8% of the whole human genome (Venter et al., 2001; Jia et al., 2022; Liu et al., 2023). The proviral genome consists of a long terminal repeat (LTR) at both ends and four internal open reading frames. The LTRs at both ends contain functional regulatory elements, such as promoters, enhancers, and transcription factor-binding sites (Bannert and Kurth, 2004). The gag gene encodes structural proteins of the virus, including matrix (MA), capsid (CA), and nucleocapsid protein (NC). MA forms layer on the inside of the viral envelope and play important roles in virus assembly, as they form links or bridge between nucleocapsids/cores and the envelope. CA is the major structural component and plays a key role in the viral assembly and budding processes. NC is a small zinc finger protein that possesses nucleic acid chaperone activity that enables NC to rearrange DNA and RNA molecules into the most thermodynamically stable structures. The pro gene encodes a protease playing a central role in proteolytic processing. The pol gene encodes open reading frames for the proteins reverse transcriptase (RT) and integrase (IN). RT is responsible for converting RNA into complementary DNA, a key step in retrovirus replication. IN mediates the insertion of ERVs into the genome of the host cell. The env gene encodes surface and transmembrane proteins that participate in the assembly of retrovirus-like particles (Ono, 1986). Many of the coding regions of proviruses have lost the ability to encode functional proteins due to mutations, insertions, deletions, and rearrangements. In addition, the proviruses occasionally undergo homologous recombination between ancestral 5’ and 3’ proviral LTRs, where the intervening protein-coding sequence is deleted to form a separate solitary (or “solo”) LTR. It was reported that at least 85% of ERV cases are solitary (or “solo”) LTRs (Lander et al., 2001; Mager and Stoye, 2015). Surprisingly, there are few similarities between the LTRs of retroviruses from different genera (Benachenhou et al., 2013; Johnson, 2019).
There are many types of ERVs which can be classified according to their phylogenetic relationships. The three main categories are: Class I represents γ retrovirus-like elements, Class II represents β retrovirus-like elements, and Class III represents spuma virus-like elements (Vargiu et al., 2016). The ERV-K group, which belongs to Class II, contains 11 subtypes, which are called Human MMTV Like, so they are named HML with a number (HML1-11). The ERV-K is the most studied group (Barbulescu et al., 1999). In addition to HML-2, HML-6, HML-7, HML-8, and HML-9 have also attracted the attention of many researchers (Lavie et al., 2004; Flockerzi et al., 2005; Broecker et al., 2016; Scognamiglio et al., 2022).
Most sequences of ERVs have been mutated and inactivated, but some ERVs can still be expressed and play important roles in some physiological processes. Studies have shown that ERV transcription occurs in healthy cells and tissues, including embryos and placentas (Stoye, 2012). In addition, aberrant expression of ERVs occurs in several diseases, such as multiple sclerosis and breast cancer, and their proteins may contribute to disease etiology (Jansz and Faulkner, 2021). It has been reported that HERV-K (HML-2) is a risk factor for multiple sclerosis (Garcia-Montojo et al., 2018; Holloway et al., 2019). In addition, the transcription level of ERV is increased in breast cancer, teratoma, ovarian tumor, and melanoma (Garcia-Montojo et al., 2018; Johnson, 2019; Jansz and Faulkner, 2021; Chen et al., 2022; Jia et al., 2022; Liu et al., 2023). In summary, although many ERVs have acquired mutations and are not actively expressed, there are ERV loci that continue to have important biological functions.
Therefore, considering the substantial contribution of ERVs to the host genome and their emerging roles in shaping the host’s regulatory networks, exploring the dynamic expression and function of ERVs is important for understanding both human- and primate-specific aspects of gene regulation and development, including physiological and pathological processes (Kunarso et al., 2010; Grow et al., 2015). Before the dynamics of ERVs can be examined, it is essential to first determine the distribution and position of ERVs in the host genome. Many studies have focused on ERV elements in the human genome, but only a few have concentrated on these elements within the nonhuman primate genome. For chimpanzees, which are the closest living relative of humans, previous work revealed 45 HML-2 elements inserted specifically into the chimpanzee genome (Macfarlane and Badge, 2015). The results indicated that, compared with humans, the chimpanzee genome contains less chimpanzee-specific HML-2 integration. In addition, little work has been done to characterize ERVs in chimpanzees and compare these with those of other primates, such as gorillas and humans (Holloway et al., 2019). Previously, we performed comprehensive identification and characterization of the ERV-K (HML-8) group in the human genome (Liu et al., 2023). However, the distribution and function of HML-8 elements in other primates, such as chimpanzees remain unclear, and comparisons of the genomic distribution, integration time, and potential regulatory roles between the two hosts have not been performed. Chimpanzees are the closest living relative of human beings (Bannert and Kurth, 2006). Therefore, accurate and complete characterization of HML-8 elements in the chimpanzee genome is needed to compare the evolutionary forces underlying the 2 recent speciation patterns of mammalian groups. This work will facilitate the study of the existence, evolutionary relationship, and function of ERVs in primates, potentially helping to elucidate the pathogenesis of serious human diseases.
2 Materials and methods
21 HML-8 identification, localization, and genomic distribution
We used Jan. 2018 (Clint_PTRv2/panTro6) as the chimpanzee reference genome to determine the distribution of HML-8 remnants in the chimpanzee genome. The assembled MER11A-HERVK11-MER11A sequence from the Dfam database was used as a query for the HML-8 reference (Hubley et al., 2016) (https://dfam.org/home). There are typically two resources for reference: consensus representatives and single best representatives. Compared to the single best representative, which is a specific and high-quality ERV sequence for HML-8, the consensus sequence for HML-8 has a much broader representation. Therefore, consensus representatives are used as references or queries in most studies (Grandi et al., 2017; Pisano et al., 2019). The BLAT search tool in the UCSC genome browser database was used to detect the integrated HML-8 elements (Kent, 2002; Kent et al., 2002). BLAT on DNA is designed to quickly find sequences of 95% and greater similarity of length 25 bases or more. BLAT functions in DNA alignment by keeping an index of the entire genome in its memory. The index consists of all overlapping 11-mers stepping by 5 except for those heavily involved in repeats (http://genome.ucsc.edu/cgi-bin/hgBlat). Additionally, the expected distribution of HML-8 loci on each chromosome was calculated according to the Formula e=Cl × N/Tl (e is the expected integration number in the chromosome, Cl represents the nongap length of the chromosome, N represents the total number of actual HML-8 loci identified in the human genome, and Tl represents the total nongap length of all chromosomes) (Grandi et al., 2021; Jia et al., 2022; Liu et al., 2023). Chi-square (χ2) tests were performed to analyze the difference between the expected integration number and the actual number of HML-8 loci and to estimate the statistical significance based on the p value.
2.2 Structural characterization
The length and structure of all the HML-8 provirus remnants were characterized via multiple alignments with the Dfam reference MER11A-HERVK11-MER11A performed with MEGA 7 and the BioEdit software platform (Kumar et al., 2016; Tamura et al., 2021). All the structural details, including insertions and deletions, were annotated.
2.3 Phylogenetic analyses
To confirm the assignment of the identified HML-8 elements in the chimpanzee genome, maximum likelihood (ML) phylogenetic trees were constructed using MEGA 7 (Kumar et al., 2016). Elements containing many gaps were eliminated manually. Three proviral sequences (longer than 80% of the HML-8 reference length) were screened to determine their phylogenetic relationships. Using the model selection function in MEGA7, the best-fit model of nucleotide substitution for these near full-length proviruses was the general time reversible model with a gamma distribution and invariant sites (GTR+G+I). Additionally, elements longer than 90% of the corresponding 4 coding regions of HML-8 were screened to construct subregion phylogenetic trees, respectively. Based on the model selection model in MEGA7, the most suitable nucleotide substitution models for gag, pro, pol and env analysis are as follows: the Hasegawa-Kishino-Yano model with a gamma distribution and invariant sites (HKY+G+I); the general time reversible model with a gamma distribution and invariant sites (GTR+G+I); the general time reversible model with a gamma distribution (GTR+G); and the Hasegawa-Kishino-Yano model with a gamma distribution (HKY+G). The nearest neighbor interchange (NNI) procedure was used to search for the tree topology. The nearest neighbor interchange is a heuristic search to improve the likelihood of a tree by performing the following operation on it. If we have two unrooted trees, then we can specify a neighbor relation between the two of them and then swap their subtrees in an attempt to obtain a tree that has a higher likelihood (https://www.megasoftware.net/webhelp/centraldialogbox_hc/nearest_neighbor_interchange_nni_.htm). The confidence of each node in the phylogenetic trees was determined using the bootstrap test with 500 bootstrap replicates. The final trees were visualized by iTOL (Tamura et al., 2021).
2.4 Estimation of the integration time of HML-8 members in the chimpanzee genome
To estimate the integration time of each HML-8 element in the chimpanzee genome, we used a substitution rate of 0.2%/nucleotide/million years to evaluate the divergence effect on every HML-8 (Lebedev et al., 2000). For the 4 internal regions (gag, pro, pol, and env), the integration time was calculated based on the Formula T = D/0.2. For the flanking LTR regions, the integration time was calculated based on the Formula T = D/0.2/2. T represents the estimated time of integration (in million years). D represents the percentage of divergent nucleotides, and the D of each HML-8 element was estimated in two ways: (1) between the 5’ and 3’ LTRs of each provirus and (2) between each HML-8 internal element and its consensus generated. The divergence values were calculated with MEGA7.
2.5 Functional prediction of cis-regulatory regions and enrichment analysis
The noncoding LTR regions of HML-8 lack biological function annotations in the chimpanzee genome. To understand the biological significance of the HML-8 proviral LTRs, we performed functional prediction and enrichment analysis of the cis-regulatory regions of these HML-8 chimpanzees. Based on the Genomic Regions Enrichment of Annotations Tool (GREAT), gene annotations near HML-8 proviral LTRs were analyzed. The association rules were as follows: basal + extension, 5000 bp upstream, 1000 bp downstream, and 1000000 bp maximum extension; curated regulatory domains were included. When the potential regulatory genes were identified, the WEB-based Gene SeT Analysis Toolkit (WebGestalt) was subsequently used to analyze their functional enrichment (http://www.webge stage). org). This approach is crucial for interpreting the list of genes of interest. The enrichment method used here was overrepresentation analysis (ORA). The parameters for the enrichment analysis included the following: minimum number of IDs in the category: 5; maximum number of IDs in the category: 2000; FDR Method: Benjamini–Hochberg (BH); and significance level: top 10.
3 Results
3.1 Identification, localization, and distribution of HML-8 remnants in the chimpanzee genome [Jan.2018 (Clint_PTRv2/panTro6)]
The results showed that HML-8 elements pervasively invaded the chimpanzee genome. According to the BLAT results obtained for MER11A-HERVK11-MER11A in Jan. 2018 (Clint_PTRv2/panTro6), we identified a total of 76 HML-8 proviral elements (Table 1), as compared to the 40 proviral elements we identified in the human genome (Liu et al., 2023). Based on the integrated genomic loci, each HML-8 element was named according to the nomenclature previously proposed for HERV-K elements (Table 1) (Subramanian et al., 2011). First, we observed a notable feature: there was no complete full-length element of HML-8 in the chimpanzee genome. The longest proviral element was 9158 bp long, which accounted for 84.7% of the reference sequence. The length analysis revealed that the average length of these proviral elements was 4378 bp, with 9 elements being greater than 70% of the reference length, 21 elements being between 40 and 70% of the reference length, and the remaining 46 elements being between 8.14 and 37.49% (Table 1). Among them, the shortest proviral element was 875 bp long, which accounted for only 8.14% of the reference sequence. The longest and shortest HML-8 proviral elements in the human genome are 9162 and 874, respectively. This similarity suggested that the integration events of HML-8 simultaneously occurred before the divergence between humans and chimpanzees.
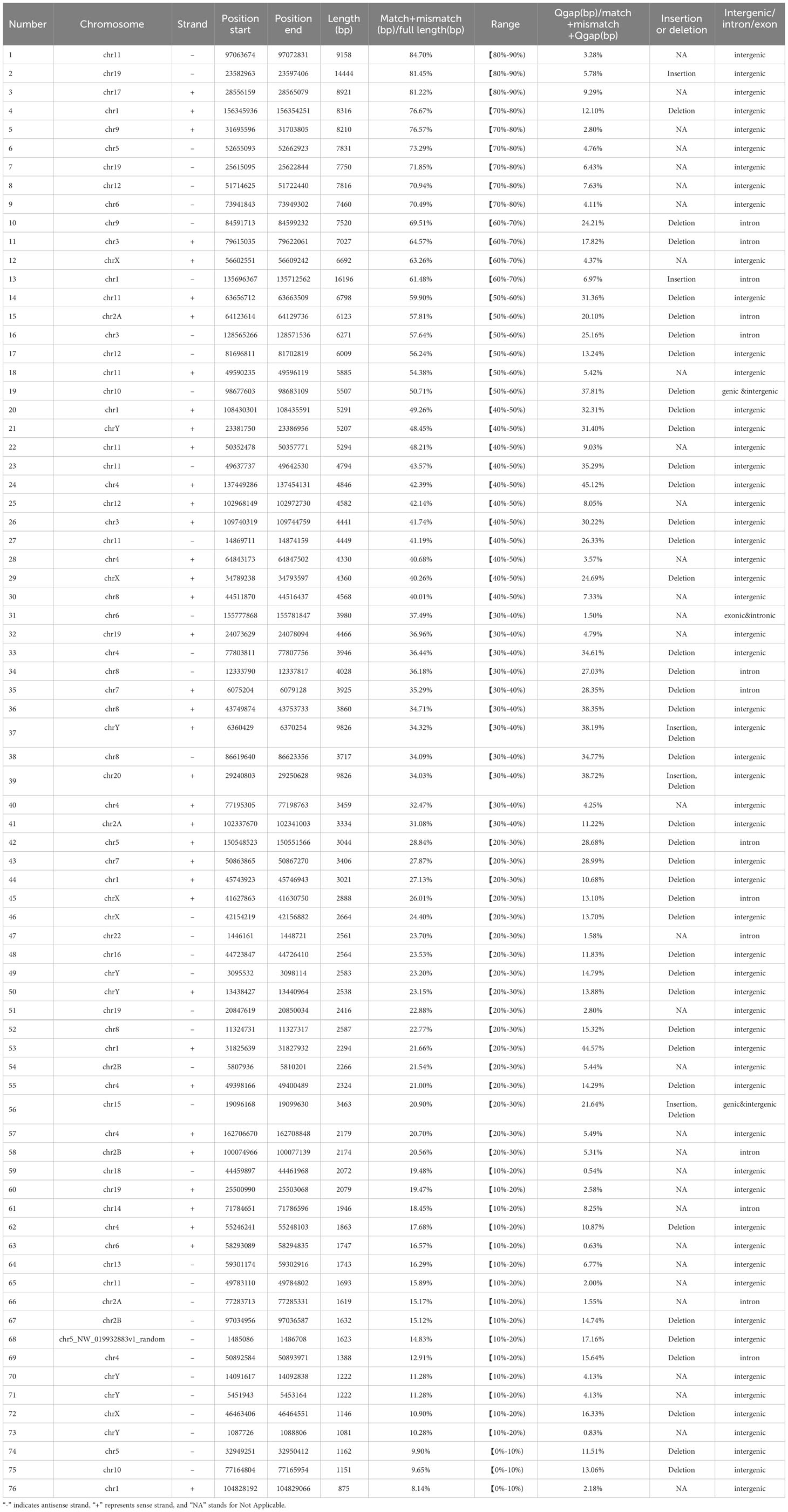
Table 1 HML-8 provirus distribution.
We did not find any solo HML-8 LTRs in the chimpanzee genome which is distinct from our findings in the human genome where there were 5 solo HML-8 LTRs. Although being short (approximately 75% of the representative reference MER11A), 5 solo LTRs exist in human genome. The nucleotide sequence of each proviral element is shown in Supplementary Dataset 1. The underlying distribution of HML-8 elements in the chimpanzee genome is shown in Figure 1A.
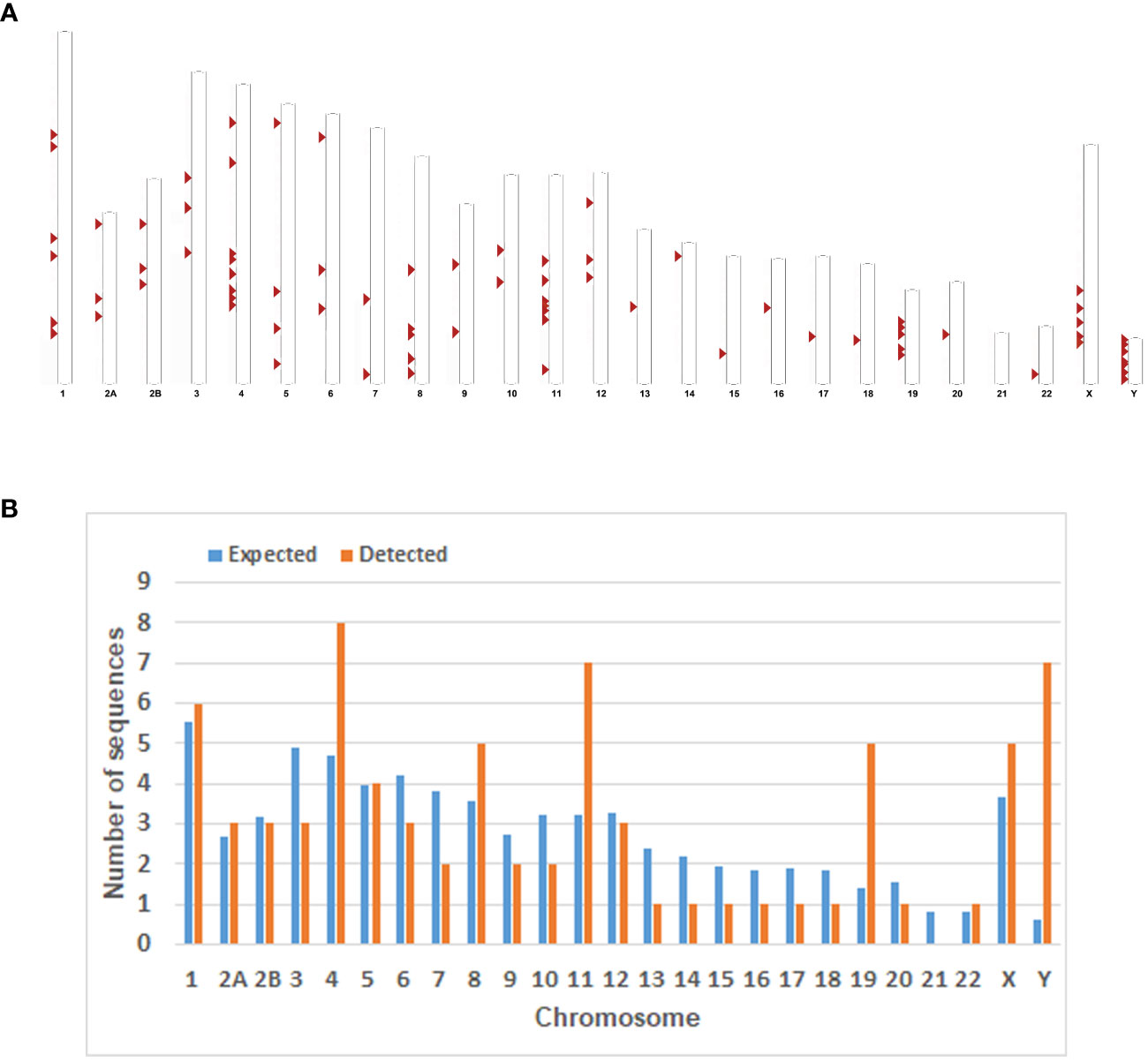
Figure 1 Chromosomal distribution of the HML-8 loci in the chimpanzee genome. (A) All the HML-8 elements (red arrows) are displayed on the chimpanzee karyotype (http://www.ensembl.org). (B) The number of HML-8 proviral elements integrated into each Chimpanzee chromosome was determined and compared to the expected number of insertion events. The expected number of sequences in each chromosome is marked in blue, and the actual number of detected sequences is marked in orange.
Furthermore, the expected number of HML-8 proviral elements in each chimpanzee chromosome was predicted. The expected number of HML-8 loci was subsequently compared with the actual number of HML-8 loci detected on each chimpanzee chromosome to evaluate whether HML-8 was randomly distributed in the chimpanzee genome. The results indicated that the number of observed HML-8 distribution events was significantly inconsistent with what was expected, thus supporting the nonrandom integration of HML-8 in the genome (Figure 1B). For proviral elements, the number of HML-8 insertions on chromosomes 4, 11, 19, and Y was greater than expected. In particular, the number of HML-8 proviral elements on the Y chromosome was 12 times greater than expected. In contrast, on chromosomes 3, 6, 7, 9, 10, 13, 14, 15, 16, 17, 18 and 20, the number of HML-8 locus integrations was lower than expected. Notably, we did not detect any HML-8 proviral integrations on chromosome 21 (Figure 1B). The analysis clearly showed that the integration of HML-8 into the chimpanzee genome was nonrandom. Furthermore, all 76 identified proviral elements were analyzed to determine their locations in intergenic regions, introns, or exons. The results showed that 59 proviral elements were located in intergenic regions, accounting for 77.63%; 14 proviral elements were located in introns, accounting for 18.42%; 2 proviral elements were located in both genic and intergenic regions, accounting for 2.63% (Table 1). Brady et al. previously validated that the accumulation of HML-2 proviruses in introns and intergenic regions is a selection against proviruses that integrate into exons and genic regions rather than a result of integration preference (Brady et al., 2009). Our study similarly revealed a nonrandom distribution and apparent bias for insertions into intergenic regions and introns.
3.2 Structural characterization
The analysis of the structural features of all 76 HML-8 proviruses, such as deletion and insertion events, can characterize the uniqueness of each proviral element and assess the potential for active expression. Thus, to define the structural characteristics of HML-8, the 76 proviral elements were first compared to the complete HML-8 reference (MER11A-HERVK11-MER11A). According to the annotation information in the Dfam database (https://www.dfam.org/family/DF0000193/features), the complete HML-8 reference exhibited a typical proviral structure containing 4 open reading frames (ORFs) and 2 flanking LTRs. Specifically, the 5’ LTR is located between nucleotides 1-1266, the coding sequence (CDS) range of the HERVK11 gag protein is from nucleotides 1422-3530, the CDS range of the HERVK11 pro protein is from nucleotides 3341-4345, the CDS range of the HERVK11 pol protein is from nucleotides 4303-7032, the CDS range of the HERVK11 env protein is from nucleotides 6890-9217, and the 3’ LTR is located between nucleotides 9220-10485.
All 76 HML-8 proviral sequences were aligned, and the positions of the insertions and deletions were annotated to describe the structure of each HML-8 provirus element (Figures 2, 3). We grouped HML-8 proviral loci based on their alignment to the consensus sequence. We found that all HML-8 loci in the chimpanzee genome were incomplete and lacked either some part of an LTR, internal coding sequences, or both. Among them, only 9 elements, including HML-8 chr11:97063674-97072831, chr19: 23582963-23597406, chr17:28556159-28565079, chr1:156345936-156354251, chr9:31695596-31703805, chr5:52655093-52662923, chr19:25615095-25622844, chr12:51714625-51722440, and chr6:73941843-73949302, were longer than 70% of the complete reference sequence in length and showed the typical proviral structure (Figure 2).
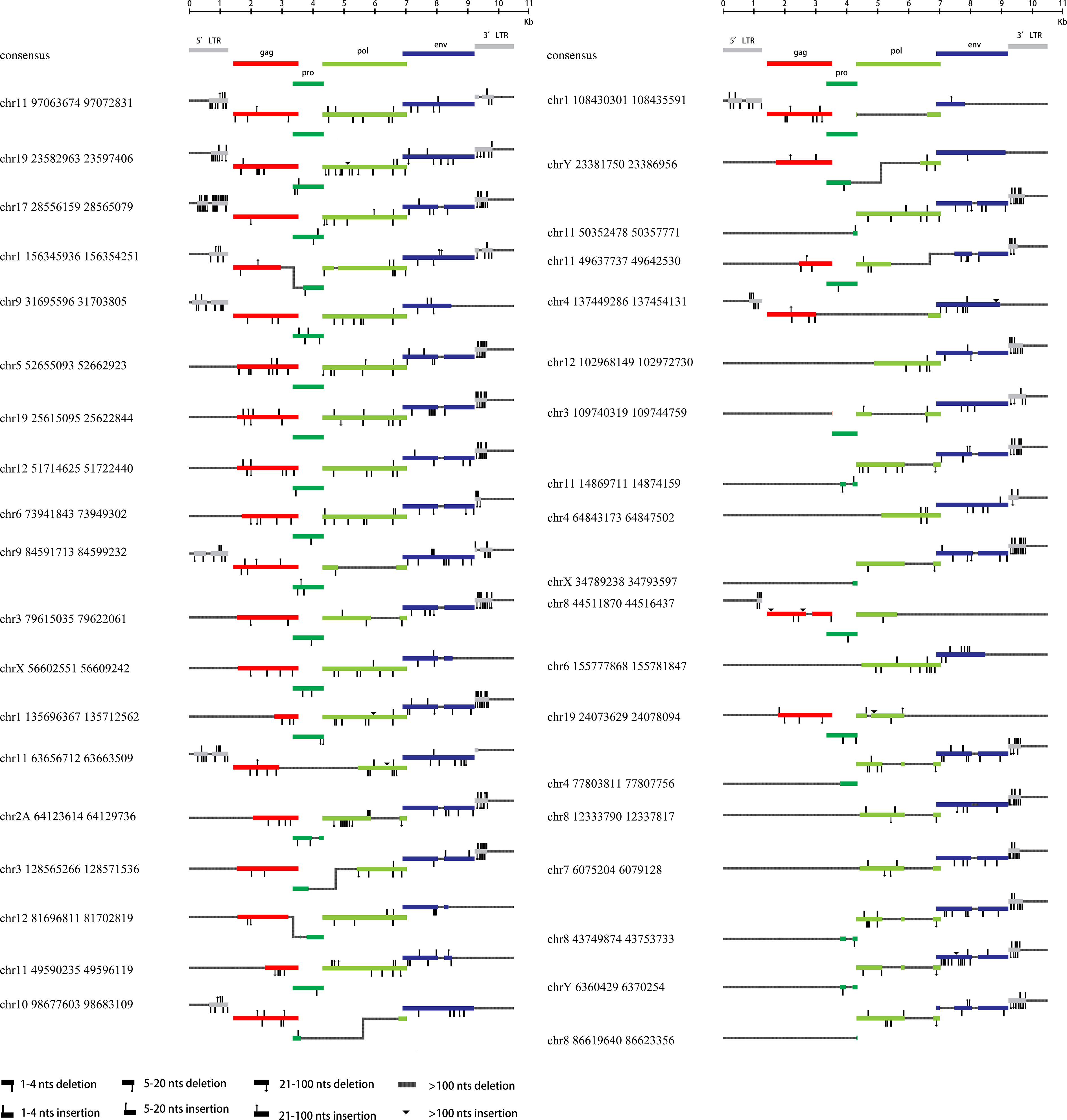
Figure 2 HML-8 proviruses structural characterization of elements 1-38. The front (1-38) HML-8 provirus elements were analyzed and compared with the Dfam reference sequence. All insertions and deletions have been annotated, as reported in the figure legend. The way the loci were grouped depended on the range of their sequence match to the consensus sequence.
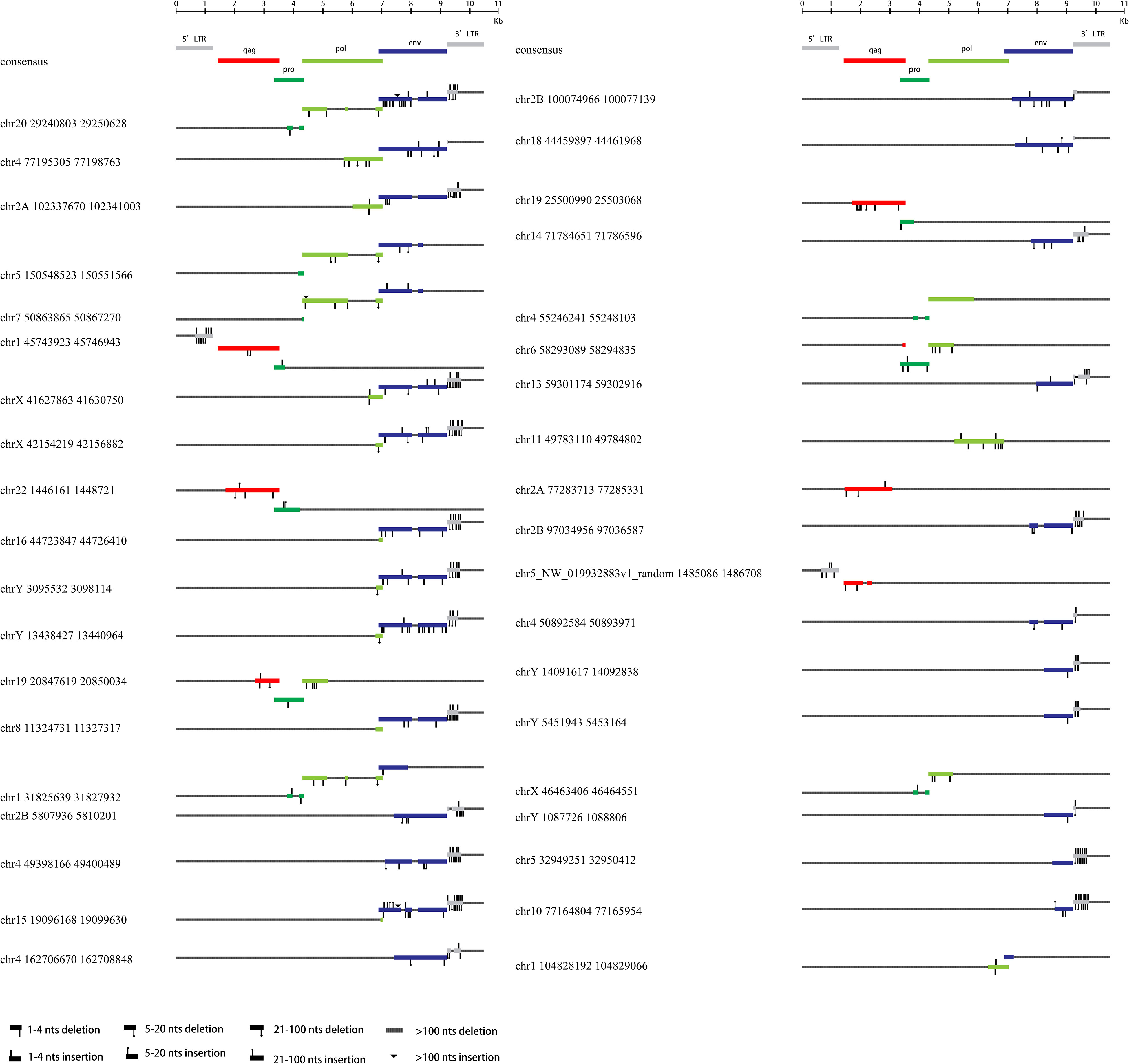
Figure 3 HML-8 proviruses structural characterization of elements 39-76. The following (39-76) HML-8 provirus elements were analyzed and compared with the Dfam reference sequence. All insertions and deletions have been annotated, as reported in the figure legend. The way the loci were grouped depended on the range of their sequence match to the consensus sequence.
Additionally, Table 2 summarizes the integrity of the 6 separate regions relative to the corresponding sections of the HML-8 reference sequence (5’ LTR, gag, pro, pol, env, and 3’ LTR), respectively. The results showed that among all 76 proviral elements, the 5’ LTR regions of 63 were missing. The longest 5’ LTR included 1023 base pairs out of 1266 base pairs (80.81%) relative to the corresponding reference region. The shortest 5’ LTR included 179 base pairs out of 1266 base pairs (14.14%). The remaining 11 5’ LTRs ranged from 33.49%-76.54% (Table 2). The 5’ LTR plays a crucial role in virus transcription and replication. Due to the truncation of the HML-8 proviral LTR sequences, it is unlikely these proviruses are actively expressed or able to retrotranspose into new locations in the chimpanzee genome. Among all 76 proviral elements, 43 gag regions have been deleted. The shortest gag gene accounts for 0.52%. The 15 gag loci range from 90.04%-99.72%. The remaining 17 gag loci ranged from 5.22%-86.58% (Table 2). Among all 76 proviral elements, 34 pro regions have been deleted. The shortest pro gene accounts for 3.28%. The 17 pro loci ranged from 91.84%-99.90%. The remaining 22 pro loci ranged from 7.06%-88.06% (Table 2). Among all 76 proviral elements, 19 pol regions have been deleted. The shortest pol gene accounted for 2.67%. The 15 pol loci range from 92.89%-99.82%. The remaining 41 pol loci ranged from 5.13%-78.46%. Among all 76 proviral elements, 11 env regions have been deleted. The shortest env gene accounted for 0.09%. The 33 env loci ranged from 90.21%-99.70%. The remaining 31 env loci ranged from 13.57%-89.99%. Among all 76 proviral elements, 25 3’ LTR regions have been deleted. The longest 3’ LTR element accounted for 41.47%. The shortest 3’ LTR element accounted for 3.40%. The remaining 49 3’ LTR loci ranged from 6.32%-40.84%. In summary, 63 5’ LTRs, 43 gag regions, 34 pro regions, 19 pol regions, 11 env regions, and 25 3’ LTR regions have been completely deleted. The loss of the 5’ LTR was the most severe and was much greater than that of the 3’ LTR. The 5’ LTR plays a crucial role in the transcription and replication of viruses. Therefore, the consistent truncation of the HML-8 5’ LTRs likely significantly impedes their expression and retrotransposition activity in the chimpanzee genome. In contrast, the env region has the smallest degree of absence. Only 11 have been deleted. Forty-four out of the 76 env regions accounted for ≥70.75%. Interestingly, a similar situation was also observed in human genome, suggesting that HML-8 was integrated before the divergence of human and chimpanzee ancestors. In the human genome, among all 40 proviral elements, 28 5’ LTR regions have been deleted. The longest 5’ LTR accounted for 73.93% of the total length relative to the corresponding reference region. The shortest 5’ LTR accounted for 28.2%. The remaining 10 5’ LTRs ranged from 32.94%-73.14%. Among all 40 proviral elements, the gag regions of 17 have been deleted. The shortest gag accounts for 39.02%. The 12 gag regions ranged from 92.89%-99.95%. The remaining 10 gag regions ranged from 49.64%-81.41%. Among all 40 proviral elements, the pro region of 12 was deleted. There were 3 complete pro regions. The shortest pro accounted for 8.06%. The 12 pro regions ranged from 94.93%-99.5%. The remaining 12 ranged from 13.23%-88.46%. Among all 40 proviral elements, the pol region of 6 was deleted. The shortest pol accounted for 6.7%. The 10 pol regions ranged from 93.33%-99.89%. The remaining 23 pol regions ranged from 10.29%- 78.35%. Among all 40 proviral elements, the env region of 6 has been deleted. The shortest env gene accounted for 13.57%. The 15 env loci ranged from 90.16%-99.05%. The remaining 18 env loci ranged from 30.07%- 89.73%. Among all 40 proviral elements, the 3’ LTR region of 16 was missing. The longest 3’ LTR accounted for 75.36%. The shortest 3’ LTR accounted for 8.93%. The remaining 22 3’ LTRs ranged from 9.64%-44.71%. In summary, 28 5’ LTR regions, 17 gag regions, 12 pro regions, 6 pol regions, 6 env regions, and 16 3’ LTR regions were completely missing.
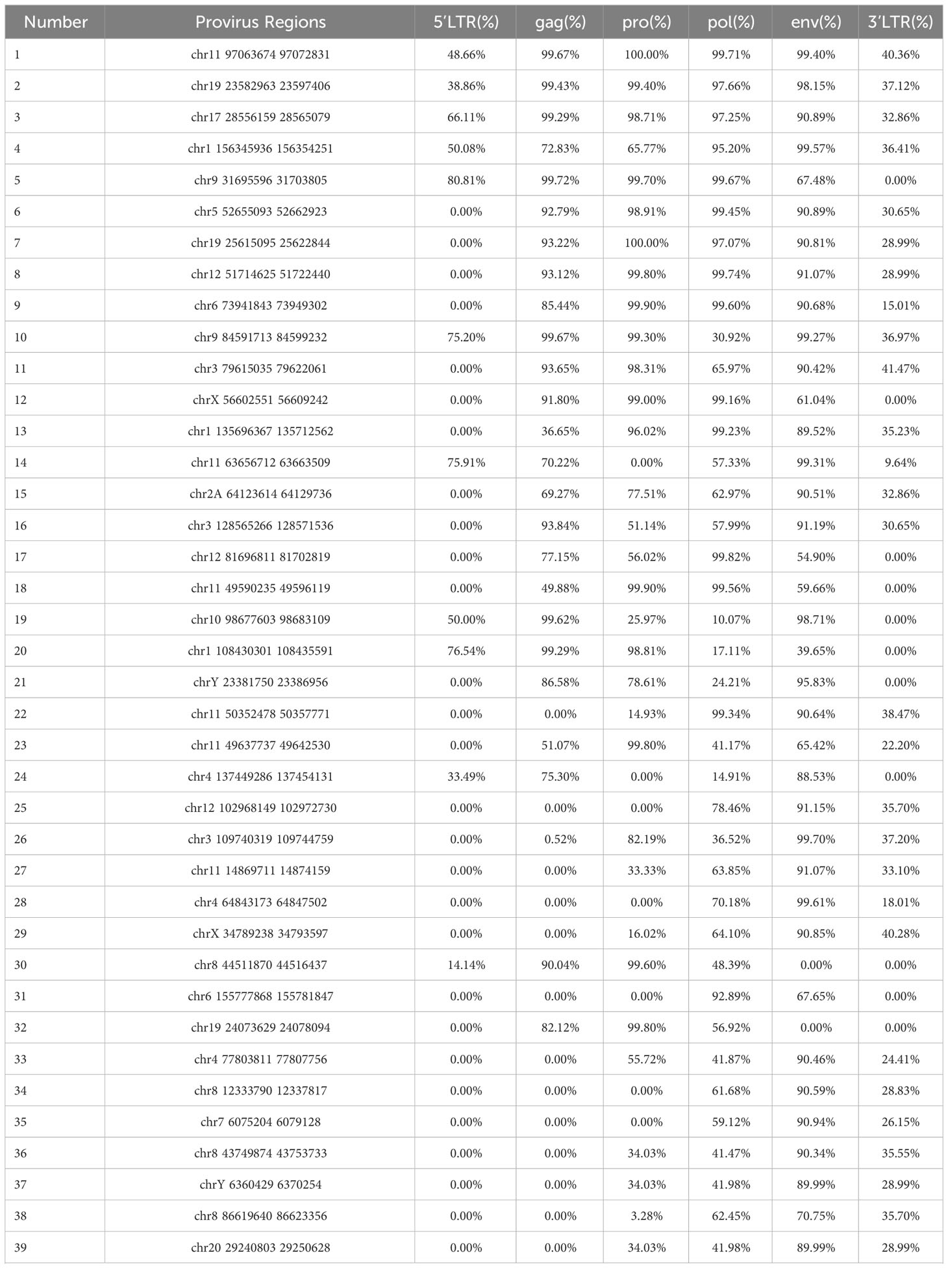
Table 2 The integrity of 6 separate regions relative to the corresponding sections of reference.
3.3 Phylogenetic analyses
To further confirm the assignment of identified HML-8 elements in the chimpanzee genome and characterize their phylogenetic relationships, an ML phylogenetic tree for near-full-length proviruses was first constructed. Three proviral sequences (longer than 80% of the HML-8 reference length) were screened to generate their phylogenetic relationships (Figure 4A). Next, 4 ML trees were constructed for subregions whose lengths were longer than 90% of the corresponding section of the reference sequence; these included 15 gag elements, 19 pro elements, 15 pol elements, and 33 env elements (Figures 4B–E). For comparison, the Dfam HERV-K group (HML-1–10) and 3 exogenous betaretroviruses were used as representatives and outgroups, respectively. These phylogenetic groups of different regions of HML-8 were all distinctly separated from the other HERV-K groups (HML1-7, 9-10) (Figures 4A-E). The 3 screened proviruses all clustered with the Dfam HML-8 reference supported by bootstrap support of 100%, indicating that they significantly more likely to be HML-8 than any other HML subtypes (Figure 4A). The phylogenetic groups for different regions of HML-8 all clustered together with their corresponding sections of the HML-8 reference, respectively (bootstrap support of 100% for gag, pol, and pro, 92.2% for env). Interestingly, two distinct clusters in the gag group were identified. The strains were statistically supported by ≥95% of bootstrap values and were named HML-8 type a and type b. The results showed that chr8 44511870 44516437, chr3 79615035 79622061, chr17 28556159 28565079, chrX 56602551 56609242, chr12 51714625 51722440, chr19 25615095 25622844, chr3 128565266 128571536, and chr5 52655093 52662923 were included in type a, whereas chr1 45743923 45746943, chr10 98677603 98683109, chr19 23582963 23597406, chr1 108430301 108435591, chr9 31695596 31703805, chr9 84591713 84599232, and chr11 97063674 97072831 were included in type b. HML-8 type b sequences included the Dfam HML-8 reference, whereas HML-8 type a elements showed more divergence relative to the HML-8 reference. There are no solo LTRs in the chimpanzee genome. Thus, no phylogenetic trees for solo LTRs have been constructed.

Figure 4 Phylogenetic analysis of the HML-8 near-full-length proviruses and 4 subregions by the maximum likelihood method. Phylogenetic analyses of 3 HML-8 proviral elements (A), 15 gag elements (B), 19 pro elements (C), 15 pol elements (D), and 25 env elements (E), along with reference sequences. The generated phylogenetic trees were all tested by the bootstrap method with 500 replicates. The branch length indicates the number of substitutions per site. The two intragroup clusters of the gag genes (types a and b) were annotated and depicted with different colors, respectively.
3.4 Estimated time of integration
Like the distribution dynamics and other characteristics of these remnants, the integration time of each chimpanzee HML-8 member is also a key clue to understanding the evolution of the group across primates. Given the serious lack of intact LTRs of the proviruses, i.e., no provirus has a 5’ LTR or 3’ LTR greater than 70%, the proviral LTRs were not used for the integration time calculation as previously described (Jia et al., 2022; Liu et al., 2023). Here, we estimated the age of the 46 HML-8 proviral elements in the chimpanzee genome based on the available gag, pro, pol, and env regions, respectively (Table 3). Each region whose length exceeds 90% of the corresponding reference sequence was used to calculate the integration time. Through the formula, an estimate of the integration time (T) can be obtained, namely, T = D/0.2, where D is the percentage of divergent nucleotides and 0.2 represents the host genome neutral mutation rate expressed in substitutions/nucleotide/million years. For each proviral region mentioned above, the ancestral sequences of each region were generated via MEGA7 based on multiple alignments of all the elements and the ML method. The details of the proviral formation periods are shown in Table 3. Overall, the HML-8 elements (gag, pro, pol, and env) found in the chimpanzee genome were integrated between 15 and 52.33 million years ago (mya). The average integration time was 35.86 mya, and the median was 37.25 mya. In our previous study, we performed a comprehensive identification and characterization of the HML-8 group in the human genome (Liu et al., 2023). Through comparison, it was found that the integration of human HML-8 elements mainly occurred between 23.5 and 52 mya. The average integration time was 37.11 mya, and the median was 37.42 mya. The divergence between human and chimpanzee ancestors is known to trace back to approximately 6.5–7.5 mya or earlier. The results indicated that the chimpanzee-specific insertion periods were indeed similar to the human-specific insertion periods and further confirmed that HML-8 was integrated into common ancestors before humans and chimpanzees diverged.
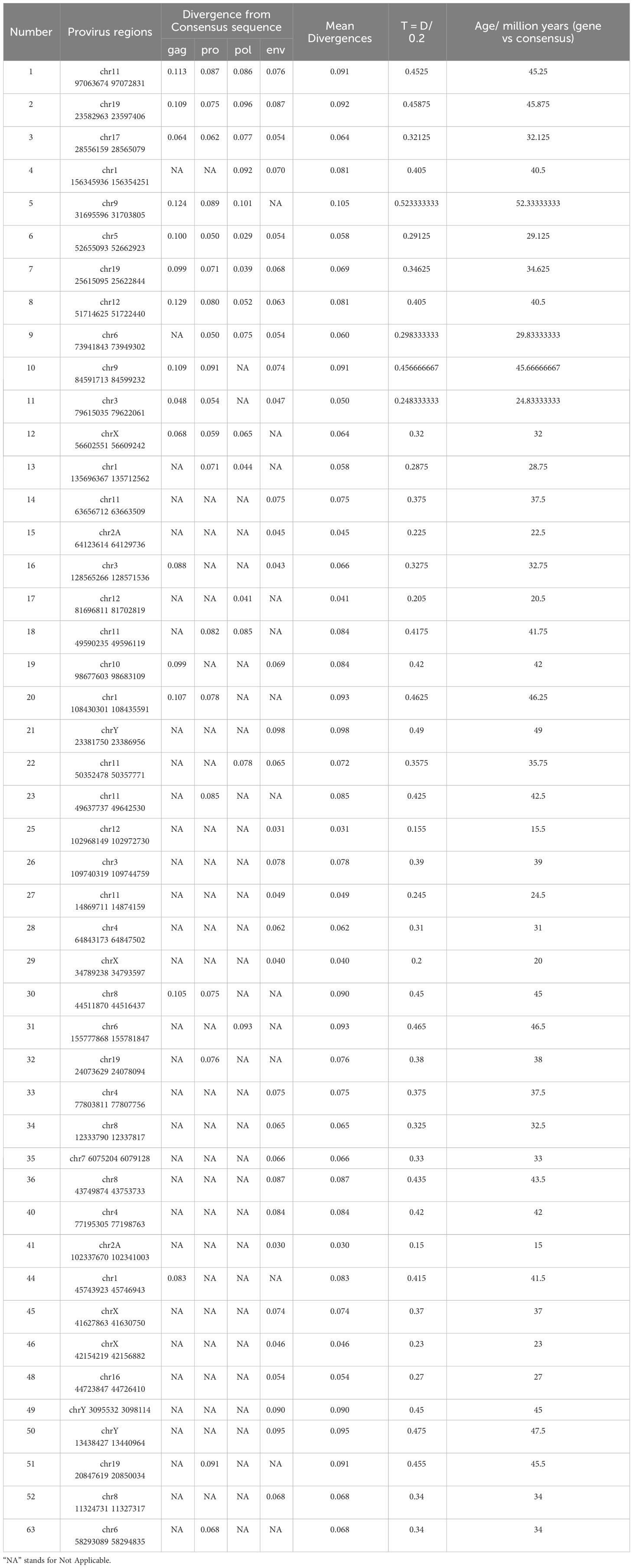
Table 3 Estimated time of HML-8 elements integration.
Despite all this, there are significant differences in distribution quantity and structural form. The chimpanzees included 76 HML-8 proviral elements and 0 solo LTRs. By comparison, there are only 40 proviruses in the human genome, almost half as many as in the chimpanzee genome. In addition, the human genome also contains 5 solo LTRs. Solo LTRs arise from recombination between LTRs and the removal of intermediate regions of a provirus, and these recombination events mainly occur during meiotic recombination (Jia and Li, 2018). This significant difference precisely indicated that even after integration, the interaction between the pathogen and its host did not stop. The host genome can retain helpful or select against harmful proviral integrations. The chimpanzee genome contains more HML-8 proviral elements (76 vs. 40) and fewer solo LTRs (0 vs. 5) than humans. Since HML-8 integration occurred more than 30 million years prior to the divergence of chimpanzees and humans, the different distribution and number of these elements is likely due to differences in selection on these proviruses in the different species. This suggests that HML-8 integrations were retained at a greater rate in the chimpanzee genome than in the human genome, perhaps due to selection pressure differences or different rates of recombination during meiosis. Our results may suggest the difference in genome response to proviral integration contributed to the speciation event, which created humans and chimpanzees as distinct species.
3.5 Functional prediction of cis-regulatory regions and enrichment analysis
The LTR plays a crucial role in virus transcription and replication. Although most HML-8 LTRs are severely truncated, any regulatory sites present in the remaining sequence can play a role in the host genome’s functional process as cis-regulatory regions. The tool of Genomic Regions Enrichment of Annotations Tool (GREAT) can predict the biological significance of these noncoding regions by analyzing annotations of nearby genes, i.e., based on spatial proximity. For the chimpanzee-specific HML-8 proviral LTRs, we selected LTR sequences larger than 70% of the reference sequence for further prediction. The results describing the associations between each proviral LTR and its putative-regulated gene(s) are shown in Supplementary Table S1. Seven genes were predicted in total. Among them, 1 LTR was associated with 1 gene, and 3 LTRs were associated with 2 genes (Figure 5A; Supplementary Table S1). No gene had an absolute distance from the transcription start site (TSS) of less than 5 kb. The absolute distance basically measures how far the gene is from the TSS. The absolute distances between the 4 genes and the TSS were 5 to 50 kb. The absolute distance between the 2 genes and the TSS was between 50 and 500 kb. The absolute distance between 1 gene and its TSS was greater than 500 kb (Figures 5B, C). To analyze the biological taxonomy of genes associated with LTRs, we produced GO Slim summaries to annotate these genes to functional categories. GO biological process (BP) analysis revealed that these genes were involved mainly in metabolic processes, responses to stimulus, localization, and biological regulation (Figure 5D). The GO Slim cellular component (CC) summary showed that these genes were significantly involved in the cytosol, mitochondrion, and endoplasmic reticulum, and the GO Slim molecular function (MF) summary revealed that these genes were significantly involved in protein binding, ion binding, and transferase activity (Figures 5E, F).
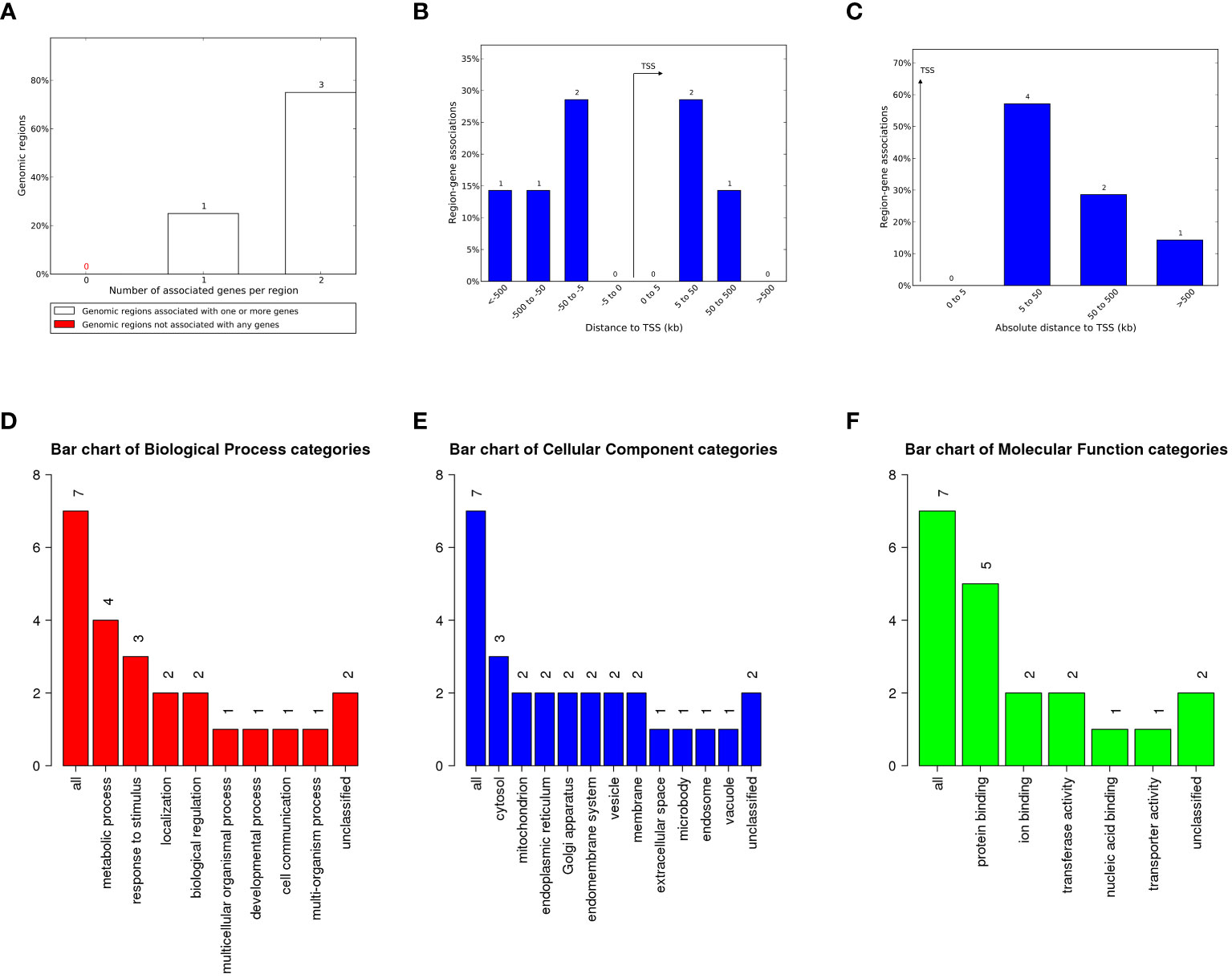
Figure 5 The genes associated with proviral LTRs and GO Slim summaries. (A) The number of associated genes per proviral LTR. (B) Binned by orientation and distance to the TSS. (C) Binned by the absolute distance to the TSS. The biological process (D), cellular component (E), and molecular function (F) categories are represented by red, blue, and green bars, respectively. The height of the bar represents the number of IDs in the gene list and in the category.
Moreover, these potential regulatory genes were subjected to enrichment analysis using WebGestalt. The top 10 most significant GO terms according to the FDR value for BPs included “response to iron(II)ion”, “detoxification of nitrogen compound”, “toll-like receptor 7 signaling pathway”, “glutathione derivative metabolic process”, “glutathione metabolic process”, “sulfur compound biosynthetic process”, “cellular modified amino acid metabolic process”, “peptide metabolic process”, and “cellular amide metabolic process” (Figure 6A).
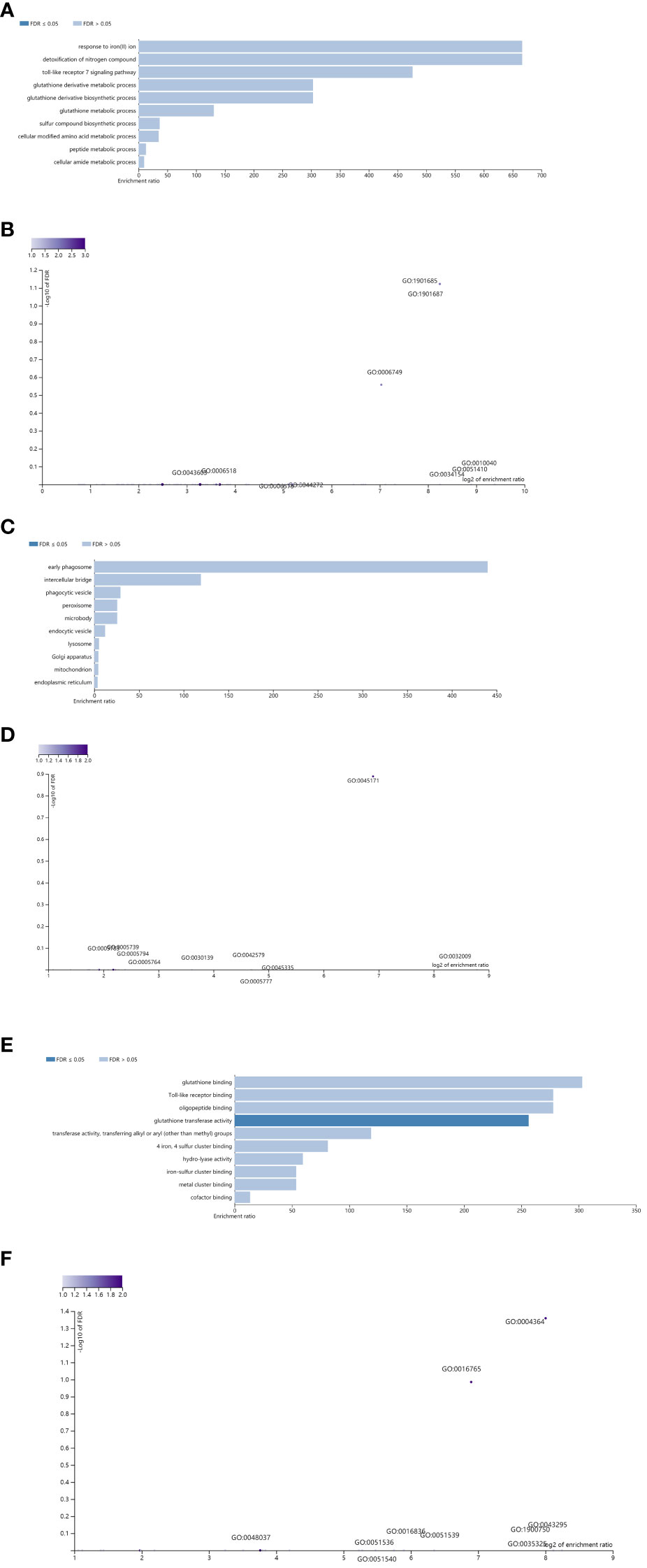
Figure 6 Enrichment result categories binned by biological process, cellular component, and molecular function. (A, B) Bar chart and customizable volcano plot of the biological process enrichment results. A bar graph showing the enrichment ratio of the results was constructed. Bars representing categories with an FDR ≤ 0.05 are shown in a darker shade (A). The volcano plot in (B) shows the log2 of the FDR versus the enrichment ratio for all the functional categories in the database, highlighting the degree to which the significant categories are separated from the background. The size and color of a dot are proportional to the number of overlaps (for ORA). The significantly enriched categories are labeled, and the labels are positioned automatically by a force field-based algorithm at startup. (C, D) Bar chart and customizable volcano plot of the cellular component enrichment results. (E, F) Bar chart and customizable volcano plot of the molecular function enrichment results.
The enrichment results for the CC and MF categories are shown in Figures 6C–F. As repeatedly emphasized in our previous papers, all these results are entirely prediction-based, and future biological research is needed to confirm any of the implied associations between proviral LTRs and nearby genes.
4 Discussion
ERV is an indispensable partner in the evolutionary process of primates. The integration and coevolution of ERVs can shape the host genome and participate in physiological and pathological processes (Johnson, 2019; Jansz and Faulkner, 2021; Chen et al., 2022). Therefore, it is critical to study the distribution of HML-8 loci in the chimpanzee genome to understand their evolutionary history and to inform future functional research. Previously, we conducted a comprehensive identification and characterization of the HML-8 group in the human genome (Liu et al., 2023). However, there is still a lack of comprehensive understanding of the evolutionary history of ERVs in other primates; for example, chimpanzees, which are the closest living genetic relatives to humans and share much of our genetic information, including ERV integrated in the genome. The distribution and function prediction of HML-8 in chimpanzees remain unclear and thus the comparisons of these elements between the two hosts cannot be carried out. We further characterized these remnants in chimpanzees and provided a detailed description of the HML-8 proviruses in the chimpanzee genome, including the HML-8 genome distribution, structural characteristics, phylogeny, integration time analysis, and regulatory function prediction.
We identified a total of 76 HML-8 proviral elements, and the results showed that the distribution of these proviral elements in the chimpanzee genome was nonrandom. Our previous studies have shown that the distribution of HML-8 loci in humans is not random (p<0.005). Our comparison between HML-8 elements in the human and chimpanzee genomes showed that there is great similarity in the distribution of proviral chromosomal positions between chimpanzees and humans. Both genomes showed significant enrichment of proviral integration in the 11, 19, and Y chromosomes of chimpanzees compared to the predicted number.
Like in humans, the number of proviral elements integrated into the Y chromosome of chimpanzees was significantly greater than that predicted (p<0.05). The Y chromosome is one of the two sex chromosomes that determines male sex. It not only is structurally complex but also the fastest-changing chromosome among human chromosomes. In addition to features related to sex determination, genes on the Y chromosome also have an impact on other traits and diseases in humans, such as the risk and severity of cancer (Rhie et al., 2023). There are several possible reasons for the insertions into the Y chromosome. The first possibility for additional provirus insertions may be due to the gene density on the Y chromosome, which became fixed in the population due to a decreased chance of gene disruption. An insertion on the Y chromosome may have a lower chance of being deleterious and, therefore, would be more likely to be retained and passed on to the next generation. In addition, the physical placement of the chromosome within the nucleus and the chromatin status also strongly influence whether a provirus can be inserted into that portion of the genome (Rhie et al., 2023). Anyhow, ERV enrichment on the Y chromosome could suggest that these elements may be deeply involved in reproduction, disease, and other unresolved processes.
Structural characterization revealed that no HML-8 members retained near full-length proviral structures. All the HML-8 elements have become fragmented due to insertion, deletion, or other mutations during the long history of evolution, including a total of 63 complete deletions of the 5’ LTR sequence and 25 complete deletions of the 3’ LTR of the proviruses. The middle four open reading frames (gag, pro, pol, and env) had 43, 34, 19, and 11 complete deletions, respectively. Such a large-scale deficiency reflects the host’s ability to reshape foreign elements, screening out harmful elements and leaving behind useful elements. Subregion phylogenetic analysis of 4 internal regions revealed that 15 gag elements, 19 pro elements, 15 pol elements, and 33 env sequences formed a unique cluster, each of which was supported by strong bootstrap values, confirming their assignment with great certainty.
The integration time of most HML-8 elements (gag, pro, pol, and env) found in the chimpanzee genome is mainly between 15 and 52.33 mya, with an average integration time of 35.86 mya and a median of 37.25 mya, which are very similar to those of humans. These results further confirmed that HML-8 was integrated before the divergence between human and chimpanzee ancestors, which occurred approximately 6.5–7.5 mya ago. The integration and coevolution of ERVs can reshape the host genome and participate in physiological and pathological processes (Johnson, 2019; Jansz and Faulkner, 2021; Chen et al., 2022). The significant differences in quantity and structure of HML-8 between humans and chimpanzees obtained from the present study indicated that, in turn, the host will also screen and reshape the external elements integrated from the outside. Even after proviral integration has completed, interactions between the host genome and the inserted provirus continue. Integrated exogenous retroviruses will undergo genetic recombination according to the evolutionary mechanisms of the host genome following meiotic recombination, site-specific recombination, and transpositional recombination (Jia et al., 2016; Jia and Li, 2018). A typical remnant of the original and complete provirus is solo LTR which arise from host homologous recombination between ancestral 5’ and 3’ proviral LTRs, where the intervening protein-coding sequence is deleted (Mager and Goodchild, 1989; Hughes and Coffin, 2004; Jia and Li, 2018; Thomas et al., 2018). It was reported that at least 85% of reference genome ERV instances are solo LTRs (Lander et al., 2001; Mager and Stoye, 2015; Thomas et al., 2018). Compared to humans, chimpanzees maintain many more proviral elements and fewer solo LTRs, indicating that the active interaction between the chimpanzee genome and the integrated proviruses is lower than that of the human genome which has a greater ability to shape integrated proviral elements.
In summary, we have described in detail the existence and distribution of HML-8 elements in the chimpanzee genome, as well as the structural characterization and phylogenetic analysis of these remnants. In addition, we further predicted the potential biological function of the genes related to proviral LTRs via bioinformatics methods. Our work revealed that the chimpanzee genome contains fewer chimpanzee-specific HML-8 solo LTR integration but more chimpanzee-specific HML-8 provirus integration, suggesting that HML-8 elements evolved in different ways after the divergence of human and chimpanzee ancestors. The results of the present study could provide a comprehensive research background for the differences between human and chimpanzee genomes and the potential implications in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
CW: Writing – original draft, Data curation, Formal analysis. XZ: Writing – original draft, Formal analysis, Methodology. SW: Writing – original draft, Data curation, Methodology. BZ: Software, Writing – review & editing. CY: Software, Writing – review & editing. YS: Validation, Writing – review & editing. HL: Writing – review & editing, Validation. YL: Writing – review & editing, Validation. JH: Writing – review & editing, Visualization. XW: Writing – review & editing, Visualization. JL: Writing – review & editing, Visualization. MC: Writing – review & editing, Data curation, Methodology. LJ: Writing – review & editing, Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft. LL: Conceptualization, Writing – review & editing, Data curation, Formal analysis, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the State Key Laboratory of Pathogen and Biosecurity (SKLPBS2138).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1349046/full#supplementary-material
References
Bannert, N., Kurth, R. (2004). Retroelements and the human genome: new perspectives on an old relation. Proc. Natl. Acad. Sci. U. S. A. 101 Suppl 2, 14572–14579. doi: 10.1073/pnas.0404838101
Bannert, N., Kurth, R. (2006). The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genomics Hum. Genet. 7, 149–173. doi: 10.1146/annurev.genom.7.080505.115700
Barbulescu, M., Turner, G., Seaman, M. I., Deinard, A. S., Kidd, K. K., Lenz, J. (1999). Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans. Curr. Biol.: CB. 9, 861–868. doi: 10.1016/S0960-9822(99)80390-X
Benachenhou, F., Sperber, G. O., Bongcam-Rudloff, E., Andersson, G., Boeke, J. D., Blomberg, J. (2013). Conserved structure and inferred evolutionary history of long terminal repeats (LTRs). Mob. DNA 4, 5. doi: 10.1186/1759-8753-4-5
Brady, T., Lee, Y. N., Ronen, K., Malani, N., Berry, C. C., Bieniasz, P. D., et al. (2009). Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 23, 633–642. doi: 10.1101/gad.1762309
Broecker, F., Horton, R., Heinrich, J., Franz, A., Schweiger, M.-R., Lehrach, H., et al. (2016). The intron-enriched HERV-K(HML-10) family suppresses apoptosis, an indicator of Malignant transformation. Mobile. DNA 7, 25. doi: 10.1186/s13100-016-0081-9
Chen, M., Jia, L., Zheng, X., Han, M., Li, L., Zhang, L. (2022). Ancient human endogenous retroviruses contribute to genetic evolution and regulate cancer cell type–specific gene expression. Cancer Res. 82, 3457–3473. doi: 10.1158/0008-5472.CAN-22-0290
Flockerzi, A., Burkhardt, S., Schempp, W., Meese, E., Mayer, J. (2005). Human endogenous retrovirus HERV-K14 families: status, variants, evolution, and mobilization of other cellular sequences. J. Virol. 79, 2941–2949. doi: 10.1128/JVI.79.5.2941-2949.2005
Garcia-Montojo, M., Doucet-O’Hare, T., Henderson, L., Nath, A. (2018). Human endogenous retrovirus-K (HML-2): a comprehensive review. Crit. Rev. In. Microbiol. 44, 715–738. doi: 10.1080/1040841X.2018.1501345
Grandi, N., Cadeddu, M., Pisano, M. P., Esposito, F., Blomberg, J., Tramontano, E. (2017). Identification of a novel HERV-K(HML10): comprehensive characterization and comparative analysis in non-human primates provide insights about HML10 proviruses structure and diffusion. Mobile. DNA 8, 15. doi: 10.1186/s13100-017-0099-7
Grandi, N., Pisano, M. P., Pessiu, E., Scognamiglio, S., Tramontano, E. (2021). HERV-K(HML7) integrations in the human genome: comprehensive characterization and comparative analysis in non-human primates. Biol. (Basel). 10. doi: 10.3390/biology10050439
Grow, E. J., Flynn, R. A., Chavez, S. L., Bayless, N. L., Wossidlo, M., Wesche, D. J., et al. (2015). Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 522, 221–225. doi: 10.1038/nature14308
Holloway, J. R., Williams, Z. H., Freeman, M. M., Bulow, U., Coffin, J. M. (2019). Gorillas have been infected with the HERV-K (HML-2) endogenous retrovirus much more recently than humans and chimpanzees. Proc. Natl. Acad. Sci. U. S. A. 116, 1337–1346. doi: 10.1073/pnas.1814203116
Hubley, R., Finn, R. D., Clements, J., Eddy, S. R., Jones, T. A., Bao, W. (2016). The Dfam database of repetitive DNA families. Nucleic Acids Res. 44. doi: 10.1093/nar/gkv1272
Hughes, J. F., Coffin, J. M. (2004). Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: Implications for human and viral evolution. Proc. Natl. Acad. Sci. 101, 1668–1672. doi: 10.1073/pnas.0307885100
Jansz, N., Faulkner, G. J. (2021). Endogenous retroviruses in the origins and treatment of cancer. Genome Biol. 22, 147. doi: 10.1186/s13059-021-02357-4
Jia, L., Li, J. (2018). Transpositional recombination and site-specific Recombination May Be Initiated by Copy Choice during DNA Synthesis Rather Than Break/Join Mechanism. Preprints doi: 10.20944/preprints201808.0317.v1
Jia, L., Li, L., Gui, T., Liu, S., Li, H., Han, J., et al. (2016). Analysis of HIV-1 intersubtype recombination breakpoints suggests region with high pairing probability may be a more fundamental factor than sequence similarity affecting HIV-1 recombination. Virol. J. 13, 156. doi: 10.1186/s12985-016-0616-1
Jia, L., Liu, M., Yang, C., Li, H., Liu, Y., Han, J., et al. (2022). Comprehensive identification and characterization of the HERV-K (HML-9) group in the human genome. Retrovirology 19, 11. doi: 10.1186/s12977-022-00596-2
Johnson, W. E. (2019). Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 17, 355–370. doi: 10.1038/s41579-019-0189-2
Kent, W. J. (2002). BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., et al. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. doi: 10.1101/gr.229102
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kunarso, G., Chia, N. Y., Jeyakani, J., Hwang, C., Lu, X., Chan, Y. S., et al. (2010). Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 42, 631–634. doi: 10.1038/ng.600
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi: 10.1038/35057062
Lavie, L., Medstrand, P., Schempp, W., Meese, E., Mayer, J. (2004). Human endogenous retrovirus family HERV-K(HML-5): status, evolution, and reconstruction of an ancient betaretrovirus in the human genome. J. Virol. 78, 8788–8798. doi: 10.1128/JVl.78.16.8788-8798.200
Lebedev, Y. B., Belonovitch, O. S., Zybrova, N. V., Khil, P. P., Kurdyukov, S. G., Vinogradova, T. V. (2000). Differences in HERV-K LTR insertions in orthologous loci of humans and great apes. Gene 247. doi: 10.1016/s0378-1119(00)00062-7
Liu, M., Jia, L., Guo, X., Zhai, X., Li, H., Liu, Y., et al. (2023). Identification and characterization of the HERV-K (HML-8) group of human endogenous retroviruses in the genome. AIDS Res. Hum. Retroviruses 39, 176–194. doi: 10.1089/aid.2022.0084
Macfarlane, C. M., Badge, R. M. (2015). Genome-wide amplification of proviral sequences reveals new polymorphic HERV-K(HML-2) proviruses in humans and chimpanzees that are absent from genome assemblies. Retrovirology 12, 35. doi: 10.1186/s12977-015-0162-8
Mager, D. L., Goodchild, N. L. (1989). Homologous recombination between the LTRs of a human retrovirus-like element causes a 5-kb deletion in two siblings. Am. J. Hum. Genet. 45, 848–854.
Mager, D. L., Stoye, J. P. (2015). Mammalian endogenous retroviruses. Microbiology Spectrum 3 (1), MDNA3-0009-2014. doi: 10.1128/microbiolspec.mdna1123-0009-2014
Ono, M. (1986). Molecular cloning and long terminal repeat sequences of human endogenous retrovirus genes related to types A and B retrovirus genes. J. Virol. 58, 937–944. doi: 10.1128/jvi.58.3.937-944.1986
Pisano, M. P., Grandi, N., Cadeddu, M., Blomberg, J., Tramontano, E. (2019). Comprehensive characterization of the human endogenous retrovirus HERV-K(HML-6) group: overview of structure, phylogeny, and contribution to the human genome. J. Virol. 93. doi: 10.1128/jvi.00110-19
Rhie, A., Nurk, S., Cechova, M., Hoyt, S. J., Taylor, D. J., Altemose, N., et al. (2023). The complete sequence of a human Y chromosome. Nature 621, 344–354. doi: 10.1038/s41586-023-06457-y
Scognamiglio, S., Grandi, N., Pessiu, E., Tramontano, E. (2022). Identification, comprehensive characterization, and comparative genomics of the HERV-K(HML8) integrations in the human genome. Virus Res. 323, 198976. doi: 10.1016/j.virusres.2022.198976
Stoye, J. P. (2012). Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 10, 395–406. doi: 10.1038/nrmicro2783
Subramanian, R. P., Wildschutte, J. H., Russo, C., Coffin, J. M. (2011). Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8, 90. doi: 10.1186/1742-4690-8-90
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Thomas, J., Perron, H., Feschotte, C. (2018). Variation in proviral content among human genomes mediated by LTR recombination. Mobile. DNA 9, 36. doi: 10.1186/s13100-018-0142-3
Vargiu, L., Rodriguez-Tomé, P., Sperber, G. O., Cadeddu, M., Grandi, N., Blikstad, V., et al. (2016). Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 13, 7. doi: 10.1186/s12977-015-0232-y
Keywords: endogenous retroviruses, chimpanzee, human, characterization, evolution
Citation: Wang C, Zhai X, Wang S, Zhang B, Yang C, Song Y, Li H, Liu Y, Han J, Wang X, Li J, Chen M, Jia L and Li L (2024) Comprehensive characterization of ERV-K (HML-8) in the chimpanzee genome revealed less genomic activity than humans. Front. Cell. Infect. Microbiol. 14:1349046. doi: 10.3389/fcimb.2024.1349046
Received: 04 December 2023; Accepted: 06 February 2024;
Published: 22 February 2024.
Edited by:
Lei Huang, People's Liberation Army General Hospital, ChinaReviewed by:
Emanuela Balestrieri, University of Rome Tor Vergata, ItalyTara Theresa Doucet-O'Hare, National Institutes of Health (NIH), Bethesda, United States
Copyright © 2024 Wang, Zhai, Wang, Zhang, Yang, Song, Li, Liu, Han, Wang, Li, Chen, Jia and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingyue Chen, Y2hlbm15MjAwNzUyNUAxNjMuY29t; Lei Jia, MTUwMDExOTM0MDhAMTYzLmNvbQ==; Lin Li, ZGVhcndvb2RAc2luYS5jb20=
†These authors share first authorship