
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 31 January 2024
Sec. Virus and Host
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1338606
This article is part of the Research TopicUnsolved Challenges in Hepatitis B and Hepatitis C: Host-Virus Interaction, Molecular Pathogenesis and Treatment StrategiesView all 7 articles
 Maximilian Bunz1
Maximilian Bunz1 Mona Eisele1Dan Hu1Michael Ritter1Julia Kammerloher1,2Sandra Lampl2
Mona Eisele1Dan Hu1Michael Ritter1Julia Kammerloher1,2Sandra Lampl2 Michael Schindler1*
Michael Schindler1*The tetraspanin CD81 is one of the main entry receptors for Hepatitis C virus, which is a major causative agent to develop liver cirrhosis and hepatocellular carcinoma (HCC). Here, we identify CD81 as one of few surface proteins that are downregulated in HCV expressing hepatoma cells, discovering a functional role of CD81 beyond mediating HCV entry. CD81 was downregulated at the mRNA level in hepatoma cells that replicate HCV. Kinetics of HCV expression were increased in CD81-knockout cells and accompanied by enhanced cellular growth. Furthermore, loss of CD81 compensated for inhibition of pro-survival TBK1-signaling in HCV expressing cells. Analysis of functional phenotypes that could be associated with pro-survival signaling revealed that CD81 is a negative regulator of NF-κB. Interaction of the NF-κB subunits p50 and p65 was increased in cells lacking CD81. Similarly, we witnessed an overall increase in the total levels of phosphorylated and cellular p65 upon CD81-knockout in hepatoma cells. Finally, translocation of p65 in CD81-negative hepatoma cells was markedly induced upon stimulation with TNFα or PMA. Altogether, CD81 emerges as a regulator of pro-survival NF-κB signaling. Considering the important and established role of NF-κB for HCV replication and tumorigenesis, the downregulation of CD81 by HCV and the associated increase in NF-κB signaling might be relevant for viral persistence and chronic infection.
● CD81 is downregulated and transcriptionally silenced upon HCV genome replication.
● Loss of CD81 is associated with increased cell growth and HCV expression.
● CD81 suppresses NF-κB signaling.
● CD81 interferes with p65 activation and nuclear translocation.
Liver-related diseases are responsible for approximately 2 million deaths annually (Asrani et al., 2019; Moon et al., 2020; Global Burden of Disease Collaborative Network, 2021). Of those, an estimated 300,000 were caused by hepatitis C virus (HCV) in 2019 (World Health Organization, 2021). However, acute infection is not the major cause of HCV-related deaths, but liver cirrhosis and hepatocellular carcinoma (HCC) that can develop during chronic HCV infection (Manns et al., 2017). With no vaccine available and highly effective therapy options accessible to only a minority of the world’s population, greater efforts are required to decrease HCV-related disease burden (Bailey et al., 2019; World Health Organization, 2021; Manns and Maasoumy, 2022).
Many aspects of HCV molecular biology have been elucidated within the last decades, but the changes in cellular homeostasis during chronic HCV infection are less understood. It is well known that fibrogenesis and continuous inflammation of the liver are prerequisites for cancer development (Neumann-Haefelin and Thimme, 2013; Yamane et al., 2013; Manns et al., 2017). Several studies have found that chronic HCV infection leads to chronic liver inflammation (Neumann-Haefelin and Thimme, 2013; Yamane et al., 2013; Manns et al., 2017). However, the underlying mechanism that promotes the transition to cirrhosis and cancer has not been identified yet. One candidate described in the literature is cellular stress, which has been shown in some studies to be increased in patients with chronic HCV infection (Valgimigli et al., 2002; Shuda et al., 2003; Asselah et al., 2010). Other studies found a general dysregulation of pathways that are associated with cancer development such as the cell cycle, DNA repair, pro-survival signaling and apoptosis (Li et al., 2016; Gillman et al., 2021; El-Kafrawy et al., 2022).
The tetraspanin family of proteins is well known to serve as scaffolds for cell surface signaling complexes, for example, in the immunological synapse (Levy and Shoham, 2005; Charrin et al., 2014). Tetraspanins consist of four transmembrane helices connected by three domains: a large and a small extracellular loop (LEL/SEL), plus a short intracellular loop (Levy and Shoham, 2005; Charrin et al., 2014). The four transmembrane helices can form a cavity that binds cholesterol which can induce conformational changes (Zimmerman et al., 2016). Several tetraspanins have been connected to viral-related processes such as entry (CD151 for HPV) or budding (CD63 and CD81 for HIV) (Florin and Lang, 2018). CD81 is also a cellular receptor for HCV that is bound by the viral E1/E2 glycoprotein complex and mediates entry (Pileri et al., 1998; Cormier et al., 2004). Furthermore, CD81 is involved in several signaling events, such as B cell receptor signaling through interaction with CD19, NK cell activation via adhesion G-protein coupled receptor G1, and presumably EGFR signaling (Zona et al., 2013; Meyer et al., 2015; Chang et al., 2016; Susa et al., 2020; Susa et al., 2021).
We initiated this study based on an unbiased screening approach to characterize cell surface receptor modulation in HCV-expressing hepatoma cells. As a result of the screen, we identified members of the tetraspanin family as proteins that were downregulated by HCV. In particular, CD81 emerged as candidate tetraspanin. Inactivation of CD81 did not only impair HCV entry, but also affected kinetics of HCV expression. We then characterized the mechanism of HCV-mediated CD81 modulation and analyzed the functional role of CD81 in HCV-expressing cells.
HEK293T and HeLa cells were cultured in DMEM (Thermo Fisher) supplemented with 10% fetal calf serum (FCS; Thermo Fisher) and 1% Penicillin/Streptomycin (Life Technologies). Huh7.5 and Huh7-Lunet cells, originally obtained from Charles Rice (Rockefeller University, New York), were cultured in DMEM (Thermo Fisher) supplemented with 5% fetal calf serum (FCS; Thermo Fisher), 1% Penicillin/Streptomycin, 1% Non-essential amino acids and 1% Sodium pyruvate (all Life Technologies). Stably transduced cells were cultured with additional 1 µg/ml puromycin. Cells were serum starved by culturing them in medium without FCS.
To assess cell surface receptor modulation, the LEGENDScreen™ Human Cell PE Kit (Biolegend) was used. Huh7.5 cells were electroporated with Jc1_NS5A-mtagBFP. 48 h later, cells were detached and washed, before they were ntibody stained (5x104-2x105 cells per well). The staining and fixing procedure, as well as data analysis was performed as described previously (Businger et al., 2021), except that measurement of the samples was conducted with a BD FACS Canto II with high-throughput sampler. The complete measured mean fluorescence intensities (MFIs) and calculations of three biological replicate screens are summarized in Supplementary Table 1 (Table S1). In brief, for each receptor/antibody, the PE MFI of the non-fluorescent, i.e. HCV-negative cell population was divided by the MFI of the BFP, i.e. HCV-expressing cell population to calculate X-fold receptor downmodulation and vice versa to calculate receptor upregulation.
Plasmids were amplified in chemocompetent NEB10 or NEB Stbl3 (for CRISPR constructs) E. coli and isolated using the PureYield™Plasmid Midiprep System (Promega) according to the manual.
To generate pFK_Jc1_NS5A-mScarlet, the eGFP fluorescent protein of pFK_Jc1_NS5A-GFP (Schaller et al., 2007) was replaced by mScarlet. In brief, the mScarlet insert was amplified from pmScarlet-C1 (Addgene #85042) with primers adding XbaI (XbaI-mScarlet_fw; 5’- GTtctagaCCTCGAGCTATGGTGAGCAAGGGCGA-3’) and PmeI (meI-mScarlet_rev; 5’-CACgtttaaacCCCTTGTACAGCTCGTCCATGC-3’) restriction sites at the 5’- and 3’-ends, respectively. pFK_ Jc1_NS5A-GFP was digested with XbaI, PmeI and FastAP (Thermo Fisher) according to manufacturer’s instructions, separated by agarose gel electrophoresis, and the backbone band was cut out and isolated using NucleoSpin Gel and PCR cleanup Kit (Macherey-Nagel). Backbone and insert were ligated using T4 Ligase (Thermo Fisher) for 1 h at RT. Next, NEB10 chemocompetent bacteria were transformed with the ligation mix and plated on LB agar with 100 µg/ml ampicillin. Colonies were picked and a 5 ml culture was grown over night, followed by plasmid isolation (GeneJET Plasmid Miniprep System; Thermo Fisher). Isolated plasmids were test digested and sequenced.
pFK_Jc1_E2-mScarlet was generated according to Lee et al. (Lee et al., 2019), using a HCV genome with E2 N-terminally tagged with GFP. To generate a corresponding mScarlet expressing viral genome, a nucleotide sequence was synthesized (Genescript) starting at the Pfl23II restriction site in the E1 coding region, encoding mScarlet between E1 and E2 with the 3C peptide sequence connecting mScarlet and E2, flanked by XbaI. We additionally introduced an EcoRI restriction site between E1 and mScarlet and a BglII restriction site between mScarlet and the 3C peptide. The plasmid was created by cleaving the vector backbone [Jc1_E1(A4)_XbaI; similar to (Steinmann et al., 2007)] with Pfl23II and XbaI (Thermo Fisher), and ligated with the synthesized sequence as described above.
The fluorescent reporter construct where a fluorescent protein is N-terminally attached to the core coding region via a 2A self-cleaving peptide (pFK_Jc1_mScarlet-2A) analogous to the already described pFK_Jc1_R2A genome (Reiss et al., 2011) was generated via restriction-free cloning. The principle is described at https://www.rf-cloning.org/ and primers were designed according to this protocol (Bond and Naus, 2012). In brief, primers were designed that where half complementary to the plasmid insertion site, and half complementary to mScarlet (Table 1). A PCR was performed with the insert pFK_Jc1_E2-mScarlet as template to generate a megaprimer such as that the mScarlet, between EcoRI and BglII restriction sites, is flanked by sequences homologous to the targeted insertion site. After purification of the megaprimer, it was mixed with the target plasmid, and a rolling circle PCR was performed to generate a plasmid with the insertion. The template was digested with DpnI (NEB) and the new viral genome was transformed into bacteria. To increase the chance of successful rolling circle PCR, a truncated version of pFK_Jc1_R2A was generated that only encoded the HCV genome until the end of E2. For this, pFK_Jc1_R2A was digested with SdaI and Pfl23II, and the insert was purified for ligation into pFK_Jc1_p7-half (Steinmann et al., 2007) to generate pFK_Jc1_R2A_p7-half. Then, pFK_Jc1_R2A_p7-half was digested with BcuI, HindIII and XbaI (Thermo Fisher), and the longest fragment (containing the backbone plus the HCV genome until the end of E2) was purified. Then BcuI and XbaI matching overhangs were ligated, giving rise to Jc1_R2A_short, which was then used as template for the rolling circle PCR. The HCV genome with the new reporter gene (pFK_Jc1_mScarlet-2A_short) was then digested with SdaI and Pfl23II and ligated back into a full genome plasmid using pFK_Jc1_R2A as backbone. Correct sequence was confirmed by sequencing (Table 2).

Table 1 Cloning primers.

Table 2 Sequencing primers.
To generate 293T, HeLa or Huh7.5 knock-out cells for tetraspanins CD63 and CD81, the LentiCRISPRv2 plasmid was used as described (Sanjana et al., 2014; Shalem et al., 2014). In brief, oligonucleotides with the targeting sequence and specific overhangs for ligation were ordered (Metabion international); complementary oligonucleotides were annealed and phosphorylated, and then ligated into the LentiCRISPRv2 vector. The targeting sequences 5’-GAGGTGGCCGCAGCCATTGC-3’ (CD63) and 5’-CATCGGCATTGCTGCCATCG-3’ (CD81) were used. Subsequently, respective LentiCRISPRv2 constructs (3 µg/well) were transfected with lentiviral packaging (psPAX2; 2.25 µg/well) and envelope plasmids (pMD2G; 0.9 µg/well) in HEK293T cells using JetPRIME transfection reagent (Polyplus) according to the manufacturer’s instructions in a 6-well format. LentiCRISPRv2 without an integrated targeting sequence was used as control. 24-36 h after transfection, supernatant was harvested and spun at 3200 g for 10 min at RT to get rid of cellular debris. Cells were incubated with spun supernatant for 24 h and selected with puromycin (1 µg/ml) for 2 weeks.
HEK293T cells were transfected using polyethylenimine (PEI). Cells were seeded 24 h prior to transfection until they reached a confluency of 70-80%. In brief and exemplarily for a 12-well format, 1-2 µg plasmid DNA were added to 50 µl OptiMEM (Thermo Fisher) and another 50 µl PEI mix (OptiMEM with double the amount of PEI than DNA) was added. The transfection mixture was vortexed, spun down and incubated for 15 min at RT. 100 µl of the transfection mixture was dropped onto cells, and a medium change was performed 4-6 h later or the next day. DNA and PEI amounts were adjusted accordingly for transfection in other well formats.
HeLa and Huh7.5 were transfected using JetPRIME transfection reagent (Polyplus) according to manufacturer’s instructions.
For in vitro transcription of viral RNA, the respective DNA vector was linearized by digestion using MluI (Thermo Fisher) for 1 h at 37°C (Table 3). Linearized vector was then purified using the Wizard® DNA Clean-Up System (Promega) according to manufacturer’s instructions. Complete linearization was checked by agarose electrophoresis, and DNA concentration was measured. 1 µg linearized vector was used for in vitro transcription using the T7 RiboMAX™Express Large-Scale RNA Production System (Promega) according to the manual. After DNA vector digest, a phenol chloroform RNA extraction was performed. Samples were filled up to 200 µl with nuclease-free water and 200 µl phenol:chloroform:isoamylalcohol (25:24:1; Thermo Fisher) was added, then vortexed for 1 min and spun at max speed for 2 min. The upper phase was transferred to a new tube, and 200 µl chloroform:isoamylalcohol (24:1; Sigma Aldrich) was added. Again, the sample was vortexed for 1 min, spun at max speed for 2 min, and the upper phase was transferred to a new tube. Subsequently, 20 µl 3 M sodium acetate (pH 5.2) and 200 µl isopropanol were added and the sample placed in ice for 5 min. Then, the sample was spun for 10 min at max speed to pellet the RNA, supernatant was discarded and the pellet washed with 70% EtOH. Finally, the pellet was dried at 37°C for 5 min and resuspended in 40 µl nuclease-free water. RNA was stored at -80°C.
Table 3 Viral genome constructs.
Electroporation was performed using the Neon Electroporation System (Thermo Fisher). For each electroporation 1-4x105 (10 µl tip) or 1-4x106 (100 µl tip) cells were used. Cells were seeded at the respective density 24 h prior to electroporation. Then, cells were detached and washed three times with PBS. Subsequently, cells were resuspended in an appropriate volume of PBS (containing Ca2+ and Mg2+; Thermo Fisher), and viral RNA was added (0.25-1 µg/1x106 cells). The reaction chamber was filled with buffer E (buffer E2 for 100 µl tips). The cell/RNA mixture was put into an electroporation tip, and one pulse with 1300 V for 30 ms was applied. Electroporated cells were seeded accordingly in medium that did not contain any antibiotic.
Cellular RNA of 2-5x105 cells was extracted using the RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. For lysis, 1% 2-mercaptoethanol was added to the lysis buffer. 200 ng of extracted cellular RNA was used for cDNA transcription using the QuantiTect Reverse Transcription Kit (Qiagen) according to manufacturer’s instructions. cDNA samples were filled up to 60 µl with nuclease-free water. qRT-PCR measurements were carried out on a Lightcycler 480 (Roche) using Lightcycler 480 multiwell plates (Roche) and Luna Universial qPCR Master Mix (NEB) according to the manual. In brief, a master mix containing primers (final concentration 0.3 µM), Luna Universial qPCR Master Mix and nuclease free water was generated and added to the wells together with 2 µl of diluted cDNA in duplicates (Table 4). The ΔΔCp method was used for analysis.
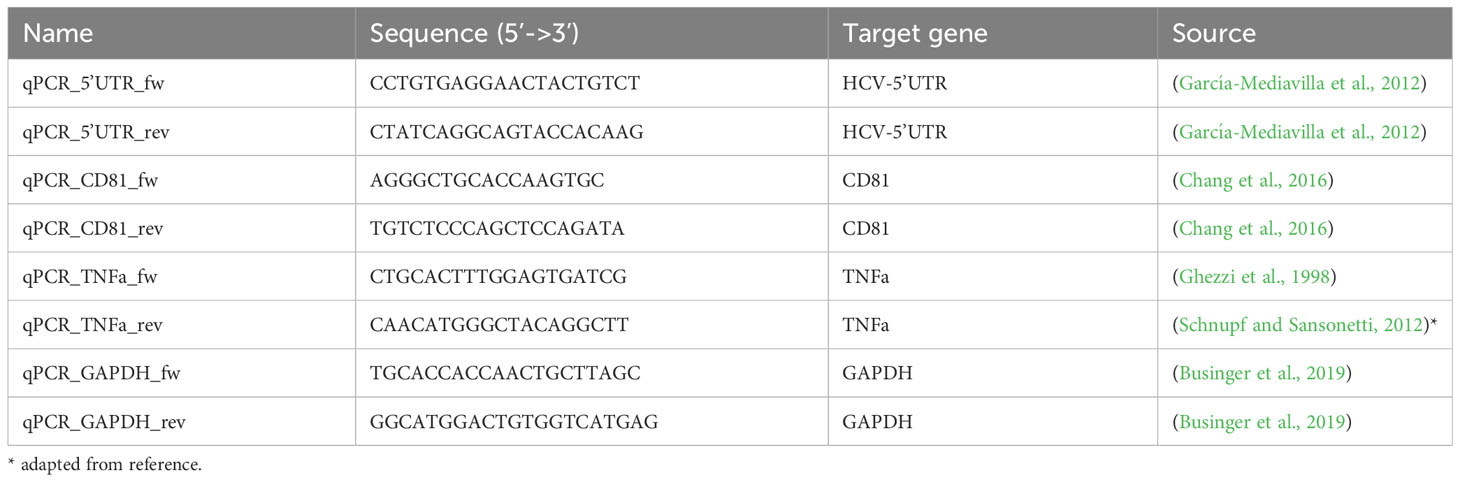
Table 4 qRT-PCR primers.
Cells were lysed with RIPA buffer (10 mM Tris-HCl (ph 7.4), 1 mM EDTA, 0.5 mM EGTA, 140 mM NaCl, 0.1% (v/v) Na-deoxychalate, 0.1% (v/v) SDS, 1% (v/v) Triton X-100, 1x protease and phosphatase inhibitor) for 20 min at 4°C. Subsequently, 6x sample buffer (0.5 M Tris (pH 6.8), 0.6 M DTT, 30% (v/v) glycerol, 10% (w/v) SDS, 2% (w/v) bromphenol blue) was added accordingly, and the lysate was heated to 95°C for 10 min. Then, samples were loaded on a 12% acrylamide gel and separated at 80-140 V for 90-150 min. Blotting was performed in a wet blotting chamber (BioRad) at 80 V for 90 min. Then, the membrane was blocked for 1 h at RT using 5% milk or 1% BSA in PBS or TBS. Primary antibodies were applied over night at 4°C. Secondary antibodies were applied 1h at RT. Between steps, the membrane was washed three times with PBS-T or TBS-T (PBS or TBS with 0.1% Tween 20; Table 5). An Odyssey Fc Imaging System (LI-COR Biosciences) was used for visualization.
Table 5 Anitbodies for western blot.
For flow cytometry, cells were detached, washed and fixed with 2% PFA for 10 min at RT. For intracellular staining, cells were permeabilized with 0.2% saponin in PBS or 80% acetone for 10 min at RT. Staining with primary antibodies was performed for 30min at 4°C, followed by two washing steps with FACS buffer (1% FCS in PBS). If applicable, staining with secondary antibody was performed for 30 min at 4°C in the dark, also followed by two washing steps with FACS buffer (Table 6). Measurements were performed using a MACSquant VYB flow cytometer (Miltenyi Biotech).
Table 6 Antibodies for flow cytometry and immunofluorescence.
HEK293T cells were seeded in a 12-well format one day prior to transfection. Two plasmids that encoded eCFP- or eYFP-tagged proteins of interest were used for transfection (Table 7). Cells were transfected with 1 µg of each plasmid using PEI as described above. Cells were harvested 24 h after transfection by detaching them with a pipet in 1 ml PBS. They were directly transferred to a 5 ml tube on ice. Cells were spun at 300 g for 5 min, supernatant was discarded, cell pellet was resuspended in 350 µl FACS buffer and cells were immediately measured for FRET signal using at a MACSquant VYB flow cytometer (Miltenyi Biotech).
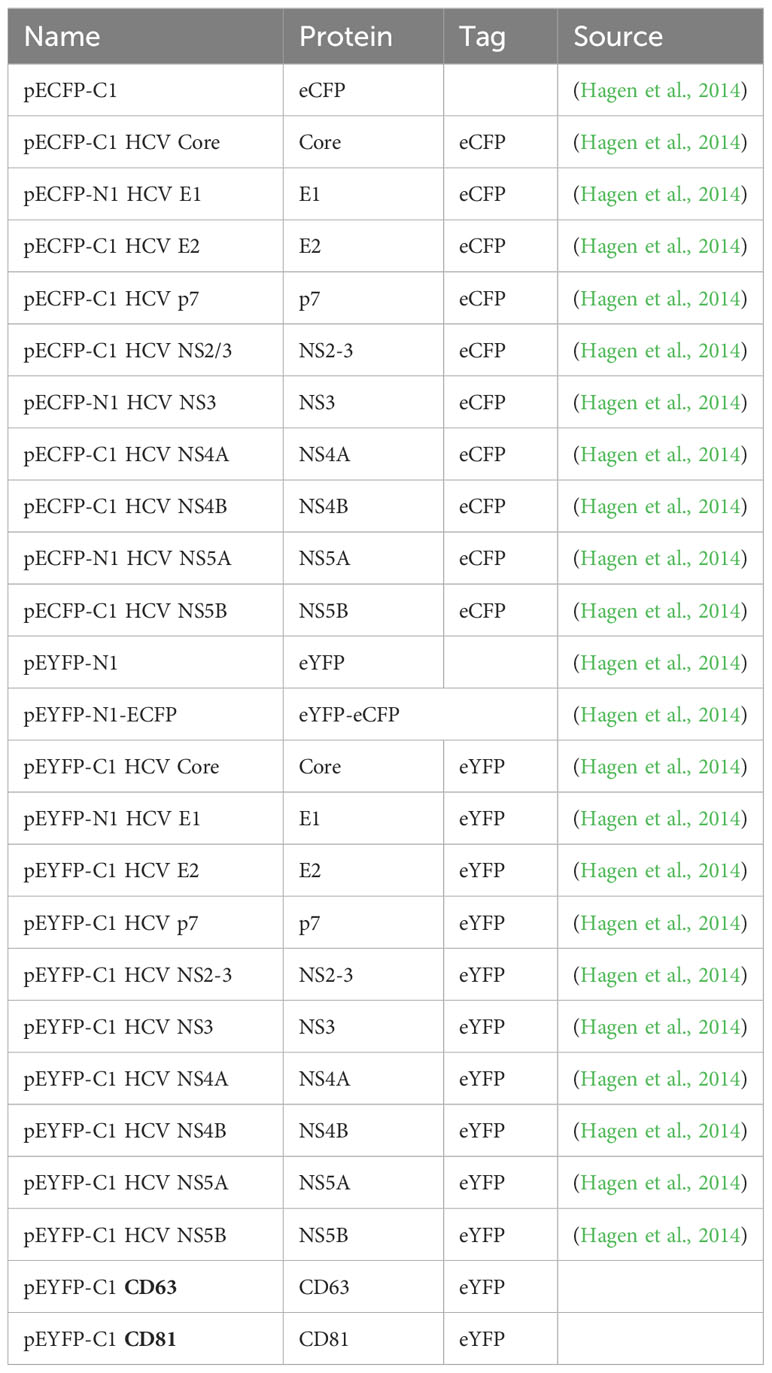
Table 7 Plasmids used for flow cytometry-based FRET.
HEK293T control and CD81KO (sorted for low CD81 expression) cells were transfected with plasmids encoding for fragments of p50 and p65 that were fused to parts of Kusabira green. 48h after transfection, cells were harvested as described above for flow cytometry-based FRET analysis and green fluorescence intensity was measured. Details are described in the CoralHue Fluo-chase Kit (MBL Int.corp.) manual.
Immunofluorescence experiments were performed in a 96-well format. Cells were fixed with 2% PFA for 10 min at RT before permeabilizing them with 80% acetone for 10 min at RT. Cells were then washed three times with PBS and blocked with 10% normal goat serum in PBS for 30 min at RT. After three washing steps, primary antibody (dilution 1:250) was applied for 1 h at RT, followed by three washing steps and secondary antibody incubation (dilution 1:250) for 1 h at RT. Then, nuclei were stained with DAPI (1:20,000, 10min, RT; Sigma Aldrich) or SiR-DNA dye (1 µM, 90 min, RT; Tebu-Bio). Three final washing steps were performed, and fluorescence was measured with a Cytation 3 plate imager (BioTek) or Incucyte plate imager (Sartorius).
Luciferase assay experiments were performed in a 96-well format and measured in a 96-well white opaque plate using a Cytation 3 plate reader (BioTek) or a TriStar2 S microplate reader (Bertold Technologies). In case of Jc1_R2A Renilla luciferase reporter constructs, cells were electroporated with Jc1_R2A viral RNA and seeded in a 96-well plate. After incubation, cells were washed and lysed as described elsewhere (Fischl and Bartenschlager, 2013). For NF-κB reporter activity, cells were transfected with several plasmids (Table 8). This included a NF-κB reporter plasmid (pNF-κB(3x)-FLuc), a Gaussia luciferase encoding plasmid as transfection control (pCMV-Gluc), and different plasmids encoding inducer proteins of NF-κB and IFN signaling cascades (p_human_IKKβ_ca, p(N)FLAG-CMV2 MAVS, pEF-Bos-RIG-I 1.211-flag). In some experiments, different chemical inducers of given pathways were used [phorbol 12-myristate 13-acetate (PMA; 10-100 ng/ml), TNFα (10 ng/ml), Lipopolysaccharide (LPS; 100 ng/ml), Ionomycin (0.25 µM), PolyIC (5 µg/ml, transfected)]. Cells were additionally transfected with a control plasmid (pWPI_BLR) or a plasmid encoding CD81 (pWPI_hCD81-HAHA_BLR), and plasmids encoding eYFP or eYFP-core (pEYFP, pEYFP-HCV-core). 4 h after transfection, cells were treated with chemical inducers and luciferase activity was determined 24 h after transfection. For this, cell culture supernatant was transferred to a 96-well white opaque plate and mixed with coelenterazine (final conc. 5 µM) to determine transfection efficiency. Then, cells were washed with PBS and lysed with 60 µl FLuc lysis buffer (0.1 M KH2PO4/K2HPO4 (pH 7.8), 1% (v/v) Triton X-100, 1 mM DTT before use) for 10 min at RT. 40 µl lysate was transferred to a 96-well white opaque plate and mixed with 40 µl FLuc assay buffer (0.1 M KH2PO4/K2HPO4 (ph 7.8), 15 mM MgSO4, 5 mM ATP) and 40 µl FLuc substrate buffer (0.28 mg/ml D-Luciferin in FLuc assay buffer). Firefly luciferase signal was measured immediately.
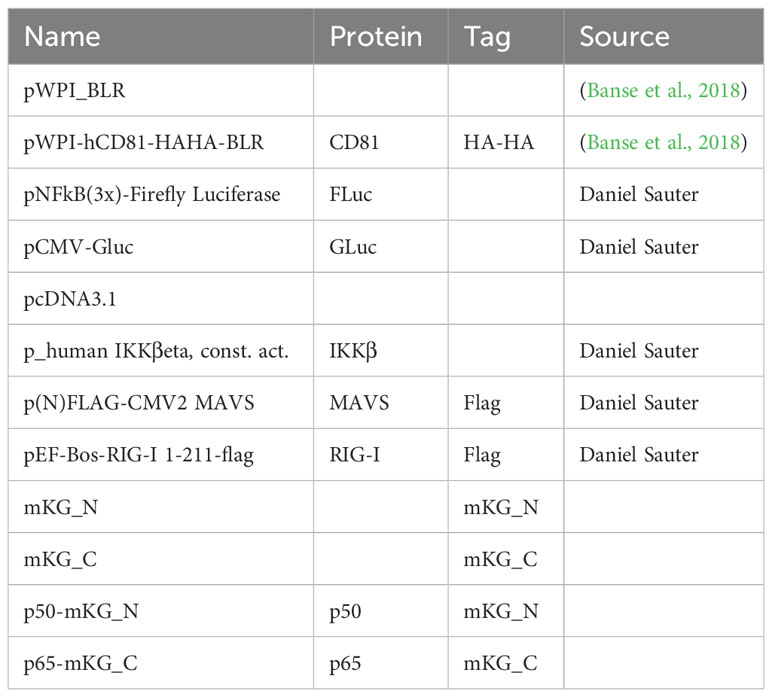
Table 8 Other plasmids.
Live cell imaging was performed in a 96-well format using an Incucyte plate imager (Sartorius). Images were taken every 2-4 h in the respective channels. Imaging of fixed plates was performed using a Cytation 3 plate imager (BioTek) for cells stained for HCV core and with an Incucyte plate imager for cells stained for p65. Translocation of p65 in images was analyzed using the Incucyte analysis software. In brief, nuclear area was defined by red fluorescence above an arbitrary threshold over background. Then, integrated green fluorescence intensity within the nuclear area was calculated and normalized to the nuclear area in total.
Design and alignment of DNA plasmids was done using SerialCloner v2.6.1 (SerialBasics) unless stated differently. Western blot membranes were analyzed using ImageStudio lite (LI-COR biosciences). Flow cytometry data was analyzed using Flowlogic v8.3 (Inivai Technologies). Microscopy images were analyzed using SoftWoRx 7.0 (Cytiva), Gen5 v3.10 (BioTek Instruments), IncuCyte GUI v2021A (Sartorius) according to instrument and subsequently handled with ImageJ. Statistical analysis and creation of graphs was done using GraphPad Prism 9 (GraphPad Spftware LLC) and Excel 2019 (Microsoft corp.). Arrangement of figures was done using CorelDraw X7 (Corel Corporation).
Viruses manipulate the plasma membrane of infected cells for antiviral immune evasion and manipulation of signaling cascades. In order to study alterations of the plasma membrane proteome upon HCV expression, a flow cytometry-based surface expression screen was performed. In brief, Huh7.5 hepatoma cells were electroporated with viral genomic RNA, which encodes for a fluorescently labeled NS5A fusion protein (Jc1_NS5A-mtagBFP) to distinguish cells with active HCV replication (BFP+) from non-HCV expressing cells (BFP-). Cells were then stained with an arrayed set of antibodies in a 96-well format, and the surface expression of 332 proteins was measured and compared between non-HCV and HCV expressing cells, to calculate fold receptor modulation. The raw data of the three independent biological replicate screens is summarized in Supplementary Table 1 (Table S1). Of surprise, receptor modulation was in general not very pronounced, with only seven proteins out of 332 whose levels were significantly lower upon HCV genome expression (Figure 1A). The two most down-regulated proteins, CD63 and CD81, which is one of the HCV entry receptors, were independently confirmed upon electroporation of Huh7.5 hepatoma cells with a viral genome RNA encoding for a fluorescently labeled NS5A fusion protein (Jc1_NS5A-eGFP) (Figure 1B). Notably, both are members of the family of tetraspanins. To get first insights on viral proteins involved in this phenotype, we transfected HEK293T cells to express single HCV proteins fused to eYFP and analyzed cell surface (Figure 1C) and total (Supplementary Figure 1A) CD63 and CD81 levels by flow cytometry. We employed E2, since it is known to directly interact with CD81, NS5A as multifunctional HCV accessory protein and the viral ion channel p7, that is incorporated into membranes. Both, cell surface and less pronounced total tetraspanin levels were differentially reduced by the viral proteins. However, efficient and dose dependent downregulation was observable only upon NS5A expression (Figure 1C). Therefore, HCV-mediated tetraspanin modulation seems to be exerted mainly by the action of NS5A.
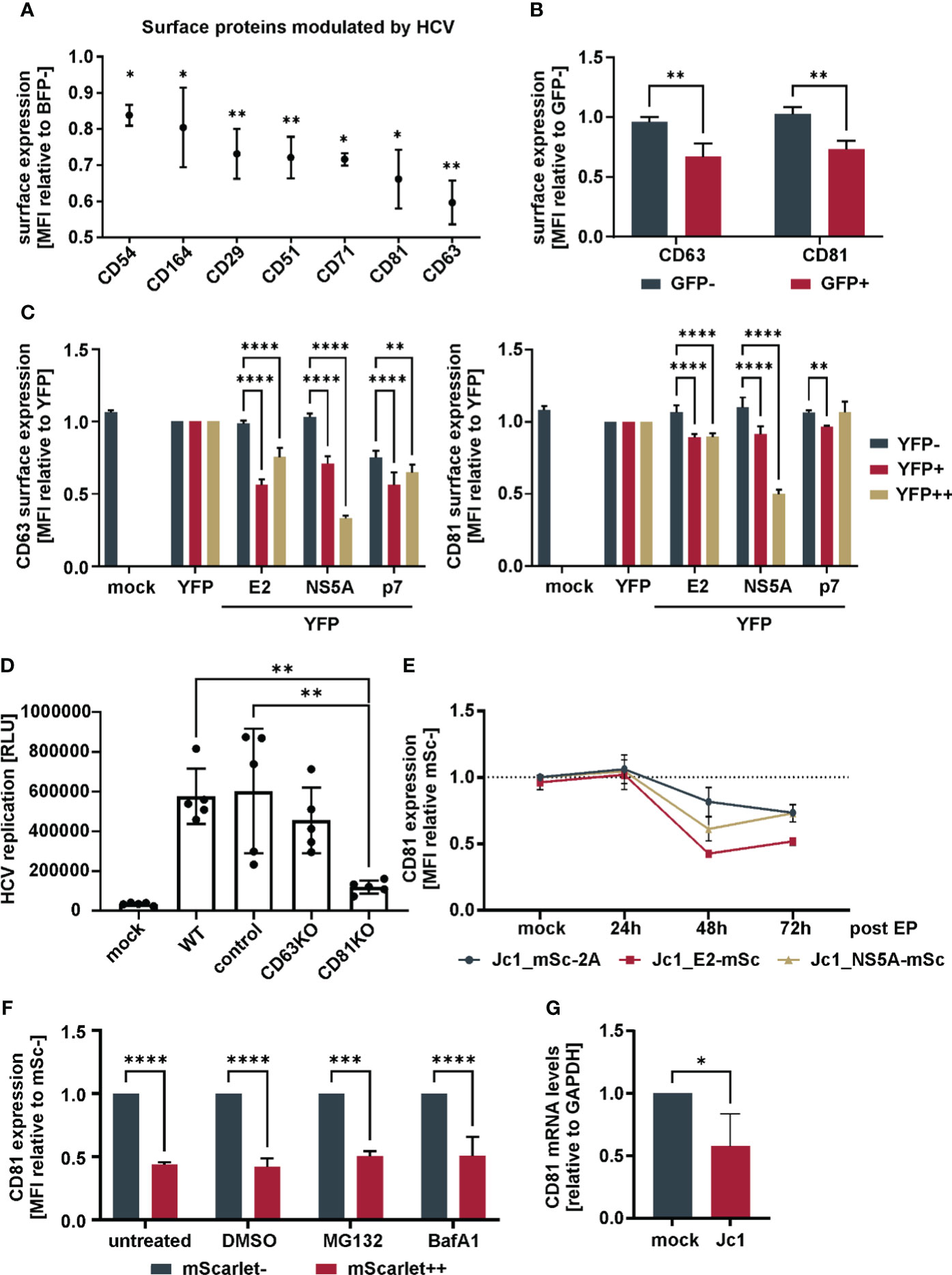
Figure 1 Downregulation of CD81 by HCV (A) Cell surface receptors downregulated in HCV-expressing versus non-infected bystander cells. Huh7.5 cells were electroporated with Jc1_NS5A-mtagBFP HCV RNA and surface receptor expression was screened with an arrayed panel of 332 PE-labeled antibodies 48h later by flow cytometry. Data from three independent screens (Supplementary Table 1). Shown are only receptors that were significantly modulated (p<0.05) in HCV expressing (BFP+) in comparison to bystander (BFP-) cells. (B) Cell surface receptor levels of tetraspanins CD63 and CD81 in HCV-expressing versus bystander cells. Huh7.5 cells were electroporated with Jc1_NS5A-GFP HCV RNA and cultivated for 72h. Cells were stained for tetraspanin surface expression and analyzed via flow cytometry. Depicted are data from 3 independent experiments. Significance was tested with a two-way ANOVA with Sidak’s multiple comparisons test. (C) Cell surface receptor levels of tetraspanins in cells transfected to express different viral proteins. HEK293T cells were transfected to express different YFP-tagged HCV proteins. 24h after transfection cells were harvested for flow cytometric analysis and stained for tetraspanins CD63 or CD81. Shown are cell surface tetraspanin levels of cells expressing no (YFP-) to medium (YFP+) and high (YFP++) levels of viral proteins. Depicted are data from 3 independent experiments. Significance was tested with a two-way ANOVA with Tukey’s multiple comparisons test. (D) Viral replication in Huh7.5 tetraspanin knock-out cells. Huh7.5 parental (WT), Crispr-control (control) and knock-out (KO) cells were electroporated with Jc1_R2A HCV RNA. 72h later cells were lysed and luciferase activity was measured as proxy for viral replication. Data from 5 independent experiments. Significance was tested with a one-way ANOVA with Tukey’s multiple comparisons test. (E) Relative total CD81 levels over time in cells expressing different mScarlet tagged viral genomes. Huh7.5 cells were electroporated with the indicated viral RNAs. After different time points, cells were fixed, permebilized, stained for CD81 and measured by flow cytometry. Data from 2-4 independent experiments. (F) Total CD81 levels in cells treated with proteasomal or lysosomal inhibitors. Huh7.5 cells were electroporated with Jc1_NS5A-mScarlet and 48h later treated with either MG132 (1µM), Bafilomycin A1 (100nM) or DMSO (1%) as control. 24h after treatment cells were harvested, fixed, permeabilized, stained for CD81 and measured via flow cytometry. Data from 3 independent experiments. (G) CD81 mRNA levels in Jc1 HCV and mock electroporated cells. Huh7.5 cells were electroporated with Jc1 HCV RNA. 48h later, cellular mRNA was extracted and qRT-PCR was performed. Data from 4 independent experiments. Significance was tested with an unpaired t-test. All data show mean values ± SD if not mentioned otherwise. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
To assess the functional role of tetraspanins for viral replication, an infectious HCV genomic RNA encoding a luciferase reporter was used [Jc1_R2A (Reiss et al., 2011)]. Huh7.5 hepatoma cells harboring gene knock-outs in CD63 and CD81 (Supplementary Figure 1B) were electroporated to express Jc1_R2A, and luciferase activity was measured as a proxy for viral genome replication and spread. Huh7.5 transduced with non-gRNA expressing lentivirus served as negative control (control cells, compare M&M). As expected, given the role of CD81 as HCV entry receptor, cells lacking CD81 showed a strongly decreased luciferase signal as compared to parental cells (WT), controls, or cells with CD63KO (Figure 1D).
As knock-out of CD63 did not have any impact on HCV replication (Figure 1D), we followed up on CD81 and analyzed in detail the kinetic of total cellular CD81 modulation in HCV-expressing cells. For this, we used an HCV reporter virus expressing the red fluorescent protein mScarlet, instead of the luciferase in front of the polyprotein (Jc1_mSc-2A), or Jc1-based constructs with internal E2 or NS5A mScarlet fusion proteins (Jc1_E2-mSc, and Jc1_NS5A-mSc, respectively). Monitoring mScarlet fluorescence allowed us to determine total CD81 levels in HCV replicating and bystander cells over time via intracellular CD81 staining and flow cytometry on a single cell level (Figure 1E; Supplementary Figure 2A). While total CD81 levels were not altered 24h post EP, a decrease was observed at 48h which stayed similar at 72h. In conclusion, HCV reduces not only cell surface CD81, but lowers total CD81 levels.
We next investigated if HCV degrades CD81 by proteasomal or lysosomal degradation. We electroporated Huh7.5 cells with Jc1_NS5A-mScarlet and treated the cells with a proteasomal (MG132) or a lysosomal inhibitor (Bafilomycin A1), stained for total CD81 and analyzed CD81 modulation by flow cytometry. Treatment with none of the inhibitors rescued CD81 levels, indicating that CD81 is not degraded by the proteasome or the lysosome in HCV-expressing cells (Figure 1F). Instead, RT-qPCR revealed that the levels of CD81 mRNA were reduced by approximately half 48 h after electroporation of Huh7.5 cells (Figure 1G). Of note, we here used non-modified infectious HCV Jc1, demonstrating that CD81 is also modulated by non-tagged viral genomes. Taken together, HCV reduces total CD81 expression at the mRNA level in Huh7.5 cells.
CD81 is transcriptionally silenced and downregulated upon the onset of viral genome replication (Figures 1E, G). This suggests, that downregulation of CD81 plays additional roles in HCV biology beyond serving as entry receptor. A common strategy of viruses is to prevent superinfection by downmodulating its entry receptors and this has been described for HCV and CD81 (Tscherne et al., 2007), even though another study reported superinfection exclusion in the absence of CD81 reduction (Schaller et al., 2007). In order to analyze if reduction of CD81 by HCV confers additional benefits in the viral life cycle, we took advantage of HCV reporter genomes producing virions that are either severely (Jc1_NS5A-mScarlet) or completely (Jc1_E2-mScarlet) compromised in their ability to de novo infect cells, due to the expression of fluorescent fusion proteins of mScarlet with either NS5A or E2 (Merz et al., 2011; Bayer et al., 2016). This allows to monitor intracellular viral RNA transcription and protein translation in the absence of viral spread. Infectious Jc1_mScarlet-2A was used as a positive control.
Crispr-control and CD81KO Huh7.5 cells were electroporated with the aforementioned genomes and then 96-well plate-based fluorescence microscopy live cell imaging was conducted over a period of six days to monitor the efficiency of HCV protein expression and viral spread using mScarlet as a surrogate marker for viral replication. This revealed, as expected, that infectious Jc1_mScarlet-2A was not able to spread in CD81-negative Huh7.5 cells (Figure 2A, left) whereas non-infectious Jc1_NS5A-mScarlet and Jc1_E2-mScarlet failed to induce spreading infection in both, control as well as CD81KO Huh7.5, confirming their loss of infectivity (Figure 2A, middle and right panel). However, of note, when using the two viral genomes devoid of inducing a spreading infection, we consistently observed higher numbers of HCV-expressing cells in the CD81KO cells in comparison to the CD81-positive Huh7.5 controls (Figure 2A, middle and right panel).
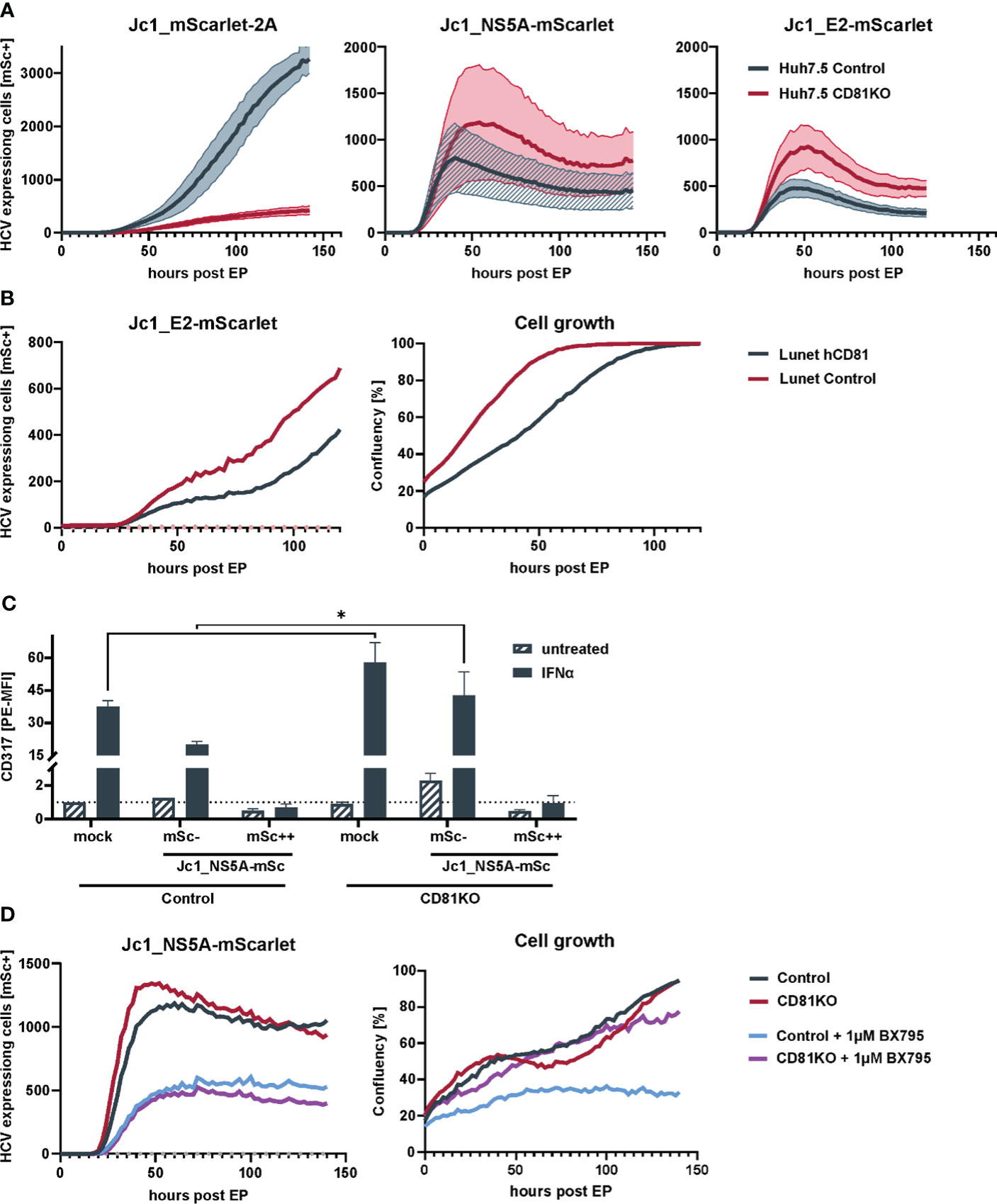
Figure 2 Loss of CD81 promotes growth of HCV-expressing cells. (A) Replication kinetics of different fluorescently-labeled HCV genomes in Huh7.5 Crispr control and CD81KO cells. Huh7.5 control and CD81KO cells were electroporated with the indicated viral genomes and the amount of HCV-expressing cells based on mScarlet fluorescence was quantified over time (every 2h) via live cell imaging. Representative data from three independent experiments. The lines represent mean values of images from the three technical replicates (4 images per well, three wells per condition) and their SD in semi-transparent area. (B) Cell growth and number of HCV expressing cells is reduced in CD81-expressing Huh7 cells. Huh7-Lunet and Huh7-Lunet-CD81 cells were electroporated with Jc1_E2-mScarlet. HCV expressing cells (red fluorescence count) and confluency was monitored over time via live cell imaging similar to the experiment shown in (A). One representative experiment from three biological replicates. (C) ISG counteraction by HCV is independent of CD81. Huh7.5 Crispr control and CD81KO cells were electroporated with Jc1_NS5A-mScarlet. 48h after EP cells were treated with IFNα (10ng/ml) for additional 24h, then harvested for flow cytometry and stained for surface expression of tetherin (CD317). Shown are tetherin surface expression levels relative to untreated mock electroporated Huh7.5 control cells. mSc- represent bystander cells while mSc++ represent cells expressing high levels of NS5A-mScarlet. See also Supplementary Figure 3A. Data from two biological replicates with duplicate electroporations. Significance was tested with a two-way ANOVA with Sidak’s multiple comparisons test), (D) Loss of CD81 compensates for pro-survival TBK1 signalling in HCV-expressing cells. Huh7.5 Crispr control and CD81KO cells were electroporated with Jc1_NS5A-mScarlet and treated with BX795 (1µM). The amount of HCV-expressing cells based on mScarlet fluorescence as well as confluency was quantified over time (every 2h) via live cell imaging. Shown is one representative of two independent biological replicates with triplicate electroporations. * p ≤ 0.05.
Moreover, this phenotype was recapitulated in CD81-negative Huh7 Lunet cells that were engineered to express hCD81 (Figure 2B). Similar to the conditions in which CD81 was knocked-out in Huh7.5, the number of HCV Jc1_E2-mScarlet replicating cells was high in CD81-negative parental Lunet cells and reduced upon reconstitution of hCD81. In addition, cellular proliferation and growth was reduced in Lunet cells upon expression of hCD81 (Figure 2B). In conclusion, CD81 negatively regulates intracellular HCV protein expression and cellular growth, explaining why HCV transcriptionally silences CD81 post entry. Altogether, as expected and considering its role as essential entry receptor, CD81 is clearly important for overall HCV infection and spread. However, at the cellular level and post entry, its absence seems beneficial for HCV genome replication and viral protein expression.
Among other functions, CD81 serves as a scaffold protein which is why we hypothesized that reduced CD81 levels might have an impact on intraviral protein interactions. We therefore used our established FRET assay (Banning et al., 2010; Hagen et al., 2014; Lim et al., 2022) to monitor HCV intraviral protein interactions and found that these were generally not altered in CD81KO cells (Supplementary Figure 2B). This indicates that higher intracellular HCV protein expression in CD81KO cells is due to other mechanisms.
Interferon (IFN) treatment triggers an antiviral state that renders cells largely resistant to HCV replication and viral protein expression. Nevertheless, HCV replicating cells can overcome early IFN-mediated antiviral immune response by expression of viral proteins that counteract induction of interferon stimulated genes (ISGs) (Lin et al., 2006). We hypothesized that CD81 might possibly alter IFN-signaling and thereby restrict intracellular HCV replication and protein expression. Therefore, Huh7.5 control and CD81KO cells were electroporated with Jc1_NS5A-mScarlet, stimulated with IFNα and stained for the cell surface expressed interferon-stimulated gene (ISG) tetherin (CD317), to analyze if Huh7.5 are responsive to IFNα despite the lack of PRRs and if HCV counteracts this response in our system (Figure 2C). Mock electroporated cells showed a clear induction of tetherin expression 24 h after IFNα stimulation. A similar effect was observed for Jc1_NS5A-mScarlet electroporated cells that were mScarlet-negative and did hence not express viral proteins (bystander cells, gated as mSc-, Figure 2C). In contrast, cells that actively replicate HCV, as evident by NS5A-mScarlet expression, had tetherin levels comparable to cells that were not treated with IFNα, demonstrating that the IFN-mediated antiviral immune response is indeed suppressed in HCV-expressing cells (Figure 2C). In addition, CD81 did not impair the ability of HCV to counteract the IFN-response, as tetherin induction was suppressed in control cells to a similar extent as in CD81KO Huh7.5. A remarkable difference was baseline induction of tetherin upon IFNα treatment. CD81KO cells that were mock electroporated or the mScarlet-negative bystander cells, both expressed higher tetherin levels as control cells, indicating that the absence of CD81 could sensitize cells for ISG induction (Figure 2C; Supplementary Figure 3).
The interferon signaling cascade involves activation of TBK1, which has a central role in innate immunity and ISG induction. In addition, TBK1 is involved in pro-survival and anti-apoptotic signaling and can activate NF-κB as downstream target (Shin and Choi, 2019). As HCV blunts ISG induction (Figure 2C), higher pro-survival signaling of TBK1 in CD81KO cells could be beneficial for HCV-gene expression and cellular growth. To test for this, we electroporated Huh7.5 control and CD81KO cells with Jc1_NS5A-mScarlet and treated with a TBK1 inhibitor (BX795). Then, live cell imaging was conducted and cellular growth as well as HCV-expression based on mScarlet-positive cells was monitored. Inhibition of TBK1 led to decreased viral gene expression in both, Huh7.5 control and CD81KO cells (Figure 2D, left). However, of note, HCV-expressing Huh7.5 CD81KO cells proliferated after being treated with BX795, whereas CD81-positive control cells showed impaired growth (Figure 2D, right, purple line as compared to blue line). This suggests that the absence of CD81 can compensate for TBK1-mediated inhibition of proliferation of Huh7.5 hepatoma cells.
Pro-survival signaling of TBK1 is mediated via NF-κB (Shin and Choi, 2019) and CD81KO cells showed enhanced survival and growth upon TBK1 inhibition (Figure 2D). We therefore hypothesized that CD81 suppresses NF-κB signaling which might be one of the reasons why CD81KO cells are resilient towards TBK1 inhibition. Furthermore, NF-κB is activated via HCV Core inducing proliferation of hepatoma cells and possibly tumor formation (You et al., 1999; Yoshida et al., 2001; Nguyen et al., 2006; Sato et al., 2006; Selimovic et al., 2012; Simonin et al., 2013). To study effects of Core and CD81 on NF-κB we first used HEK293T transfected to express a NF-κB luciferase reporter plasmid together with a control or CD81 plasmid. We then induced NF-κB signaling by co-transfection of HCV YFP-core plasmid with or without TNFα treatment. Indeed, HCV Core alone as well as TNFα induced NF-κB reporter activity ~20-fold, while both together led to a ~60-fold induction (Figure 3A). Of note, transfecting cells to express CD81 reduced NF-κB reporter activity induced by HCV Core and TNFα nearly to background levels. This confirms that the HCV Core protein induces NF-κB signaling and this activity can be counteracted by high levels of CD81.
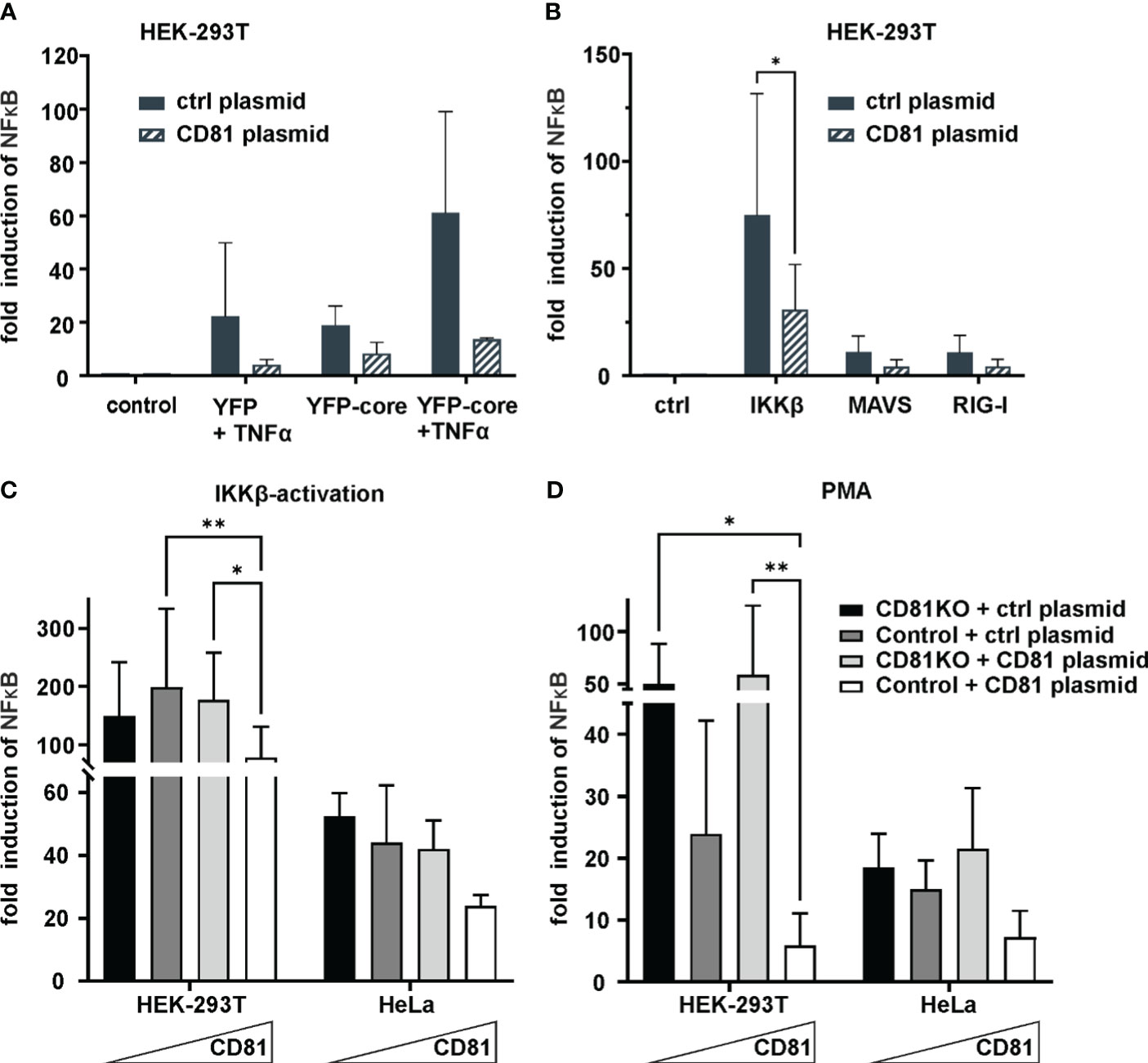
Figure 3 CD81 overexpression suppresses IKKβ and PMA-mediated NFκB activation. (A) CD81 suppresses NFκB activation by TNFa and HCV core. HEK293T cells were transfected to express a NFκB luciferase reporter together with a plasmid encoding for CD81 or a control plasmid (control vector). Additionally, cells were transfected to express HCV core (YFP-core) or a control plasmid (YFP alone). 4h after transfection, cells were treated with TNFa (10ng/ml). 24h after transfection cells were lysed and luciferase activity was measured. Data from 2 independent biological replicates with triplicate transfections. (B) CD81 reduces NFκB activation by IKKβ, MAVS and RIG-I. HEK293T cells were transfected to express a NFκB luciferase reporter and plasmids encoding either IKKβ, MAVS or RIG-I. Additionally, a plasmid encoding for CD81 (or empty control vector) was transfected. Data from 4 independent biological replicates. Significance was tested with a two-way ANOVA with Sidak’s multiple comparisons test. (C, D) CD81 reduces NFκB activation induced by IKKβ and PMA. HEK293T and HeLa Crispr control or CD81KO cells were transfected to express a NFκB luciferase reporter. Additionally, a plasmid encoding for CD81 (or empty control vector) was transfected. Either an IKKβ encoding plasmid (C) was transfected together with the other plasmids, or (D) cells were treated with PMA (10ng/ml) 4h after transfection. 24h after transfection cells were lysed and luciferase activity was measured. Data from 4 independent biological replicates with triplicate transfections. Significance was tested with a two-way ANOVA with Tukey’s multiple comparisons test. All data points show mean values ± SD. * p ≤ 0.05, ** p ≤ 0.01.
We next sought to more closely investigate effects of CD81 on NF-κB signaling induced by either IKKβ, MAVS or RIG-I. Activity of the NF-κB reporter was induced by all of them, with IKKβ being the most potent inducer and again upon expression of CD81 in this system, a clear reduction of NF-κB activity could be detected (Figure 3B). Subsequently, we decided to analyze cell type dependency by using HEK293T as well as HeLa cells, varied CD81 levels by employing cells with and without CD81KO that were additionally transfected to express CD81 or a control plasmid and used Phorbol 12-myristate 13-acetate (PMA) as an exogenous NF-κB trigger in addition to IKKβ (Figures 3C, D). In general, irrespective of the cell line or inducer used (IKKβ or PMA), increasing amounts of CD81 reduced NF-κB reporter activity, albeit the effect was only significant in HEK293T cells (Figures 3C, D).
To corroborate our results with more physiological triggers of NF-κB signaling, without transfecting IKKβ, we more closely investigated CD81KO HEK293T and HeLa cells upon induction of NF-κB via PMA (Figure 4). PMA mimics the second messenger lipid diacylglycerol (DAG) which activates protein kinase C (PKC). Some PKC isoforms require the second messenger Ca2+, to become fully activated (Reyland, 2009). Hence, cells transfected with the NF-κB reporter were additionally treated with Ionomycin to increase cytosolic Ca2+ levels. Ionomycin treatment did not increase NF-κB activity and did not induce NF-κB activity when administered alone (Figures 4A, B). However, in line with our previous results, NF-κB reporter activity in HEK293T (Figure 4A) and HeLa cells (Figure 4B) was strongly suppressed by CD81. Importantly, this phenotype was fully recapitulated in the absence of transfecting any overexpression plasmid in CD81KO cells, as we also observed suppression of PMA-induced NF-κB activity by endogenous CD81 (Figure 4C). Furthermore, when using a complete independent readout for NF-κB signaling and in the absence of any plasmid transfection upon KO of endogenous CD81 in HEK293T cells, we also found that levels of TNFα mRNA were increased at different time points post PMA stimulation, when cells were depleted for CD81 (Figure 4D). In conclusion, endogenous as well as overexpressed CD81 can suppress NF-κB activity induced by PMA of IKKβ in different cell lines.
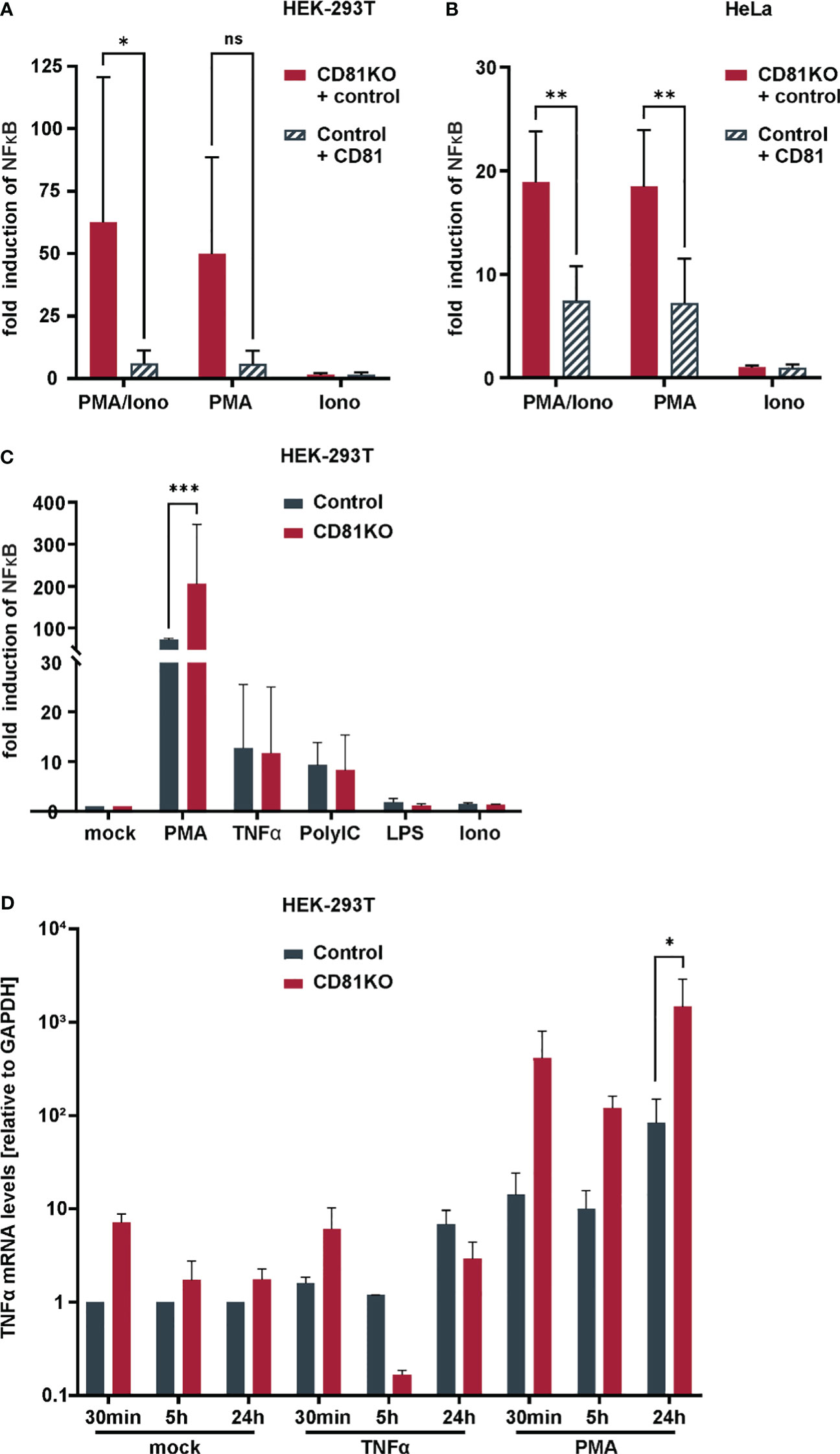
Figure 4 Suppression of NFκB and TNFα transcriptional activity by CD81 following PMA-stimulation. CD81 suppresses PMA-mediated NFκB activation in different cell lines. (A) HEK293T or (B) HeLa control and CD81KO cells were transfected to express a NFκB luciferase reporter. Additionally, cells were transfected with a plasmid encoding for CD81 or a control plasmid (empty control vector). 4h after transfection cells were treated with PMA (10ng/ml), Ionomycin (0.25µM), or both. 24h after transfection cells were lysed and luciferase activity was measured. Data from 4 independent biological replicates with triplicate transfections. Significance was tested with a two-way ANOVA with Sidak’s multiple comparisons test. (C) HEK293T control and CD81KO cells were transfected to express a NFκB luciferase reporter. 4h after transfection cells were treated with PMA (10ng/ml), TNFα (10ng/ml), LPS (100ng/ml), Ionomycin (0.25µM), or PolyIC (5µg/ml). 24h after transfection cells were lysed and luciferase activity was measured. Data from 3 independent biological replicates with triplicate transfections. Significance was tested with a two-way ANOVA with Sidak’s multiple comparisons test. (D) TNFα transcriptional activity is increased in CD81KO cells. HEK293T Crispr control and CD81KO cells were treated with PMA (10ng/ml), TNFα (10ng/ml) or left untreated (Mock) for the indicated time periods. Then, cellular RNA was extracted and TNFα mRNA was quantified via qRT-PCR. Data from 4 independent biological replicates. Shown are mean values ± SEM. All other data mean values ± SD. * p ≤ 0.05, ** p ≤ 0.01m *** p ≤ 0.001. ns, not significant.
To explore potential mechanisms of increased NF-κB signaling, we assessed the interaction of the NF-κB signaling components p50 and p65 via a split Kasubira-green assay and found that the interaction was increased in CD81KO vs control HEK293T cells (Figure 5A). Furthermore, moving to hepatoma cells, we found overall increased basal levels of p65 and phosphorylated p65, even though independently of TNFα (Figure 5B). To unambiguously directly analyze endogenous NF-κB signaling in Huh7.5 cells, nuclear translocation of p65, the subunit of the NF-κB transcription factor family, was followed upon treatment with PMA or TNFα (Figure 5C). PMA treatment induced p65 translocation after approximately 30 min which was strongly increased in CD81KO hepatoma cells (Figure 5C). Quantification of the nuclear intensity of the p65 staining revealed that while it reached a plateau in control cells at 30 min after treatment, it further increased in CD81KO Huh7.5 from 60 min post treatment on (Figure 5D). A similar trend was observed for TNFα-treated samples (Figures 5C, D). In conclusion, the cumulated results employing various NF-κB inducers, cell lines, overexpression and knockout conditions as well as different NF-κB readouts based on reporters and endogenous signals suggests a suppression of NF-κB activation by CD81.
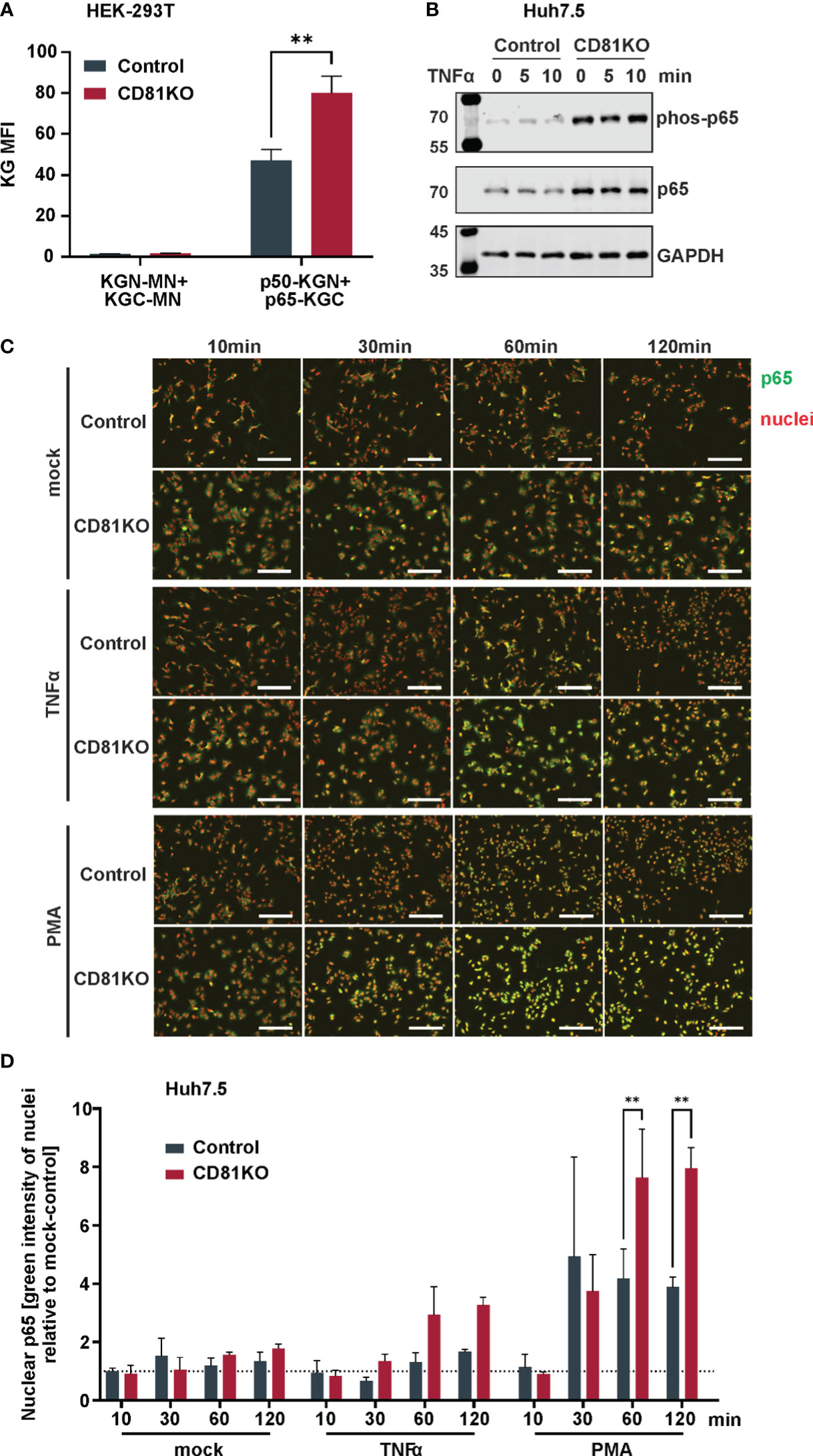
Figure 5 CD81 suppresses p65 nuclear translocation. (A) Interaction of NFκB subunits p50 and p65 assessed via a split Kusabira green assay. HEK293T CD81KO and Crispr control cells were transfected to express p50 or p65 fused to fragments of Kusabira green (KGN and KGC, respectively). 48 h after transfection cells were harvested for flow cytometry. Close proximity allows reconstitution of full Kusabira green and fluorescence, indicating interaction. Depicted are data from 3 independent biological replicates. Significance was tested with a two-way ANOVA with Sidak’s multiple comparisons test. **, p ≤ 0.01. (B) Protein amount and phosphorylation status of p65 in Huh7.5 cells. Huh7.5 Crispr control and CD81KO cells were treated with TNFa (10ng/ml) for the indicated time points. Then, cells were lysed and subjected to SDS-PAGE and western blot. Shown is one representative experiment out of two biological replicates. (C) Representative images of p65 nuclear translocation after stimulation with TNFα or PMA. Huh7.5 Crispr control and CD81KO cells were mock treated, stimulated with TNFα (10ng/ml) or PMA (10ng/ml) for the indicated time periods. Then, cells were fixed, permeabilized and IF stained for p65 (green) and nuclear DNA (red). Shown are representative images from 3 independent biological replicates. (D) Quantification of p65 nuclear translocation after stimulation with TNFα or PMA. Shown is the green integrated intensity (representing p65) in areas that overlap with red fluorescence (representing nuclear DNA) relative to unstimulated cells, to quantify p65 nuclear translocation. Data from 3 independent biological replicates. Shown are mean values ± SEM. Significance was tested with a two-way ANOVA with Sidak’s multiple comparisons test.
Here, we identify CD81 as repressor of NF-κB signaling in hepatoma and other cells. Apart from elucidating a novel thus far unknown cellular function of the tetraspanin CD81, this could be an intriguing mechanism of how HCV manipulates cells to promote persistent and chronic infection. By the downregulation of CD81 post cellular entry, HCV might relieve the CD81-mediated interference with NF-κB that subsequently promotes cell survival and growth and as well prevent superinfection of cells. Indeed, our data is in line with previous reports confirming transcriptional downregulation of CD81 in HCV replicon expressing cells or cells that stably express viral proteins and thereby prevent superinfection (Zheng et al., 2005; Tscherne et al., 2007; Zhang et al., 2010; Ke and Chen, 2013). Downregulation of entry receptors is a common strategy of viruses to prevent superinfection (Benson et al., 1993; Lu et al., 2022) even though superinfection exclusion in the context of HCV has also been observed in the absence of CD81 downregulation (Schaller et al., 2007).
However, superinfection exclusion is not a denominator of the effects observed here, as CD81 is downregulated post entry and the positive phenotype of CD81-depletion on proliferation of HCV-expressing cells was observed using tagged HCV-reporter genomes that produce non-infectious virions. Furthermore, the suppression of NF-κB via CD81 is completely independent of HCV-expression. It is thus remarkable, that by lowering levels of CD81, HCV independently mediates superinfection exclusion as well as enhancement of pro-survival intracellular signaling – two mechanisms that are highly likely to promote chronic and persistent infection. Indeed, and in line with this hypothesis, it was suggested previously that in the context of persistent HCV infection there is a selection towards maintenance of CD81-low cells, that are more resistant to cell death and apoptosis (Tscherne et al., 2007). We corroborate this hypothesis and extend it to mechanistic functions of CD81 in suppressing NF-κB signaling. Apart from that, it is noteworthy, that apparently from 332 surface receptors included in our screen, only seven were significantly lower in HCV-expressing cells (Figure 1A), which is in contrast to other viruses i.e. HCMV and HIV (Hsu et al., 2015; Sugden et al., 2016; Businger et al., 2021), that heavily dysregulate the plasma membrane of infected cells for evasion of antiviral immune responses. This indicates that HCV, for efficient persistent and chronic infection adopts a “stealth” mode in infected cells, instead of blunting adaptive and humoral cellular immune responses by cell surface receptor dysregulation.
While it is clear that HCV is generally sensitive to IFN and cannot overcome the antiviral state in the context of de novo infection, it highly efficiently blunts interferon signaling and thereby innate antiviral immune responses in actively replicating cells (Figure 2C) (Gale et al., 1997; Blindenbacher et al., 2003; Lin et al., 2006; Chandra et al., 2014). In such a scenario, NF-κB activation is pro-survival in the absence of innate immune activation (Zhang et al., 2021) (Figure 2D). This explains why higher NF-κB activity in HCV infected cells seems rather beneficial than detrimental and might ultimately support viral persistence and chronic infection.
A variety of interesting and important questions remain currently unanswered. Concerning the mechanism of CD81 downregulation by HCV, our data is in line with previous work reporting transcriptional silencing in cells stably expressing HCV NS4B or in HCV-replicon expressing cells via NS5A (Zheng et al., 2005; Ke and Chen, 2013). In our study, CD81 levels were mainly affected by NS5A (Figure 1C and Supplementary Figure 1A). Unfortunately, transient expression of NS4B was weak in our experiments, which is why we had not analyzed this viral protein. However, it is conceivable and highly likely that HCV evolved various mechanisms to downregulate CD81 as one of its main entry receptors. Similarly, for instance HIV, uses Nef, Vpu and Env to downregulate the primary receptor CD4 (Stevenson et al., 1988; Strebel et al., 1988; Garcia and Miller, 1991; Chen et al., 1996; Tanaka et al., 2003). Hence, it will be highly important to decipher how exactly CD81 suppresses NF-κB signaling. For instance it is known that there is a mechanistic interplay of the integrated stress response (ISR) and NF-κB (Schmitz et al., 2018). In this context, Fink et al. revealed that the activity of the ISR component IRE1α (Inositol-requiring transmembrane kinase/endoribonuclease 1α) is important for HCV replication as it regulates cell survival, presumably by degrading the pro-apoptotic miR-125a (Fink et al., 2017). They further showed that knock-out of another ISR factor, that is XBP1, with simultaneous activation of IRE1α by NS4B renders cells resistant to the intrinsic pathway of apoptosis. Together with the study of Tardif et al., this connects higher ISR activity to pro-survival NF-κB signaling (Tardif et al., 2004; Fink et al., 2017). As CD81 is a membrane-associated scaffold protein, it might well be involved in the regulation of the ISR as well.
CD81-mediated suppression of NF-κB was most prominent in PMA-stimulated cells indicating that CD81 represses mainly this signaling pathway. The downstream target of PMA are PKCs that are activated by diacylglycerol (DAG) which is mimicked by PMA. This stimulus is combined with ionomycin, as some PKC isoforms are only activated in the presence of Ca2+, released by Ionomycin (Reyland, 2009). However, Ionomycin was dispensable for NF-κB activation in our experimental settings indicating that CD81 suppresses the NF-κB signaling cascade by interfering with a subfamily of novel Ca2+ independent PKCs (Reyland, 2009). A hypothesis that requires further experimentation, but is supported by the fact that the serotonin receptor of the 5-HT2 family signals through DAG and PKC and interacts with CD81 (Mizuno and Itoh, 2009; Bruening et al., 2018).
In the context of HCV, Core induced NF-κB with TNFα (Chung et al., 2001). We confirm this mechanistic interplay and further demonstrate that CD81 efficiently suppresses this activation (Figure 3A). Given the importance of NF-κB for HCV replication and persistence, as well as its role in tumor development and hepatocellular carcinoma (You et al., 1999; Yoshida et al., 2001; Nguyen et al., 2006; Sato et al., 2006; Selimovic et al., 2012; Simonin et al., 2013), it is tempting to speculate that HCV-mediated downregulation of CD81 is involved in viral tumorigenesis (Tai et al., 2000; Bartosch et al., 2009; Jeong et al., 2021; El-Kafrawy et al., 2022; Kurden-Pekmezci et al., 2023). Indeed, low levels of CD81 correlate with HCC metastasis and tumor proliferation (Inoue et al., 2001; Mazzocca et al., 2008) and expression of CD81 suppresses hepatocellular carcinoma development (Li et al., 2020). Importantly, the role of CD81 in tumor development seems multifaceted with suppressive as well as protooncogenic functions (Vences-Catalán et al., 2017) and could be dependent on the tumor type as well as co-factors, for instance an ongoing HCV-infection that interferes with innate immune signaling in hepatocytes.
Our study has certain limitations that need to be addressed in future work. First of all, HCV exerts a large genotype and subtype dependent heterogeneity and we only addressed CD81-modulation by the Jc1 viral genome, which is derived from an acute fulminant hepatitis (Pietschmann et al., 2006). Hence, it will be exciting to address if CD81-modulation is a conserved feature of highly variable HCV genomes. Furthermore, even though we have witnessed CD81-dependent suppressive effects on NF-κB in different settings, including overexpression and KO as well as endogenous p65 translocation in different cell types (HeLa, 293T and Huh7.5) we have not assessed primary hepatocytes thus far, to verify this phenotype also in a non-tumorigenic setting. Finally, the mechanistic details, as discussed above, remain partly addressed and need to be carefully investigated. Nevertheless, altogether, we here identify an unprecedented role of CD81 in general and in the context of HCV infection biology, pathogenesis and beyond.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
MB: Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. ME: Investigation, Methodology, Writing – review & editing. DH: Investigation, Methodology, Writing – review & editing. MR: Investigation, Methodology, Writing – review & editing. JK: Investigation, Methodology, Writing – review & editing. SL: Investigation, Methodology, Writing – review & editing. MS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by a DFG German Research Foundation grant to MS (SCHI 1073/10-1, project number 399732171), as well as basic research support from the University Hospital Tübingen, Medical Faculty to MS.
We thank Gisa Gerold, Thomas Pietschmann and Daniel Sauter for the kind contribution of cell lines, reagents and plasmids and Daniel Sauter for critical reading of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1338606/full#supplementary-material
Supplementary Figure 1 | Downregulation of tetraspanins by HCV proteins and characterization of tetraspanin knock-out cells. (A) Total cellular tetraspanin levels in cells transfected to express different viral proteins. HEK293T cells were transfected to express YFP-tagged HCV proteins. 24h after transfection cells were harvested for flow cytometric analysis, permeabilized and stained for total expression of tetraspanins CD63 and CD81. Shown are total cellular tetraspanin levels of cells expressing no (YFP-) to medium (YFP+) and high (YFP++) levels of viral proteins. Data from 3 independent biological replicates. (B) Analysis of Crispr control and tetraspanin knock-out Huh7.5 cells for CD63 and CD81 cell surface expression via flow cytometry.
Supplementary Figure 2 | Reduction of total CD81 in HCV-expressing cells and intraviral HCV protein interaction in CD81KO cells. (A) Representative experiment showing reduction in total CD81 levels by different viral genomes. Huh7.5 cells were electroporated with indicated viral genome RNAs and harvested for flow cytometric analysis at the indicated time points. Cells were permeabilized and stained for CD81. Gates show bystander (mSc-; left gate), medium (mSc+; middle gate) and high (mSc++; right gate) HCV expressing cells. Numbers within gates give the mean fluorescent intensity (MFI) of PE-CD81. (B) Interaction network of HCV proteins in presence or absence of CD81. HEK293T Crispr control and CD81KO cells were transfected to express a pair of eCFP- and eYFP-tagged viral proteins. 24h after transfection, cells were harvested and FRET signals were measured via flow cytometry as described (Hagen et al., 2014). Data from 4-8 independent biological replicates. Shown are mean values ± SD.
Supplementary Figure 3 | CD81 does not impair the ability of HCV to counteract the interferon response. Representative experiment showing FACS plots of HCV expressing cells treated with IFNα, see also . Huh7.5 Crispr control and CD81KO cells were electroporated with Jc1_NS5A-mScarlet. 48h after EP cells were treated with IFNα (10ng/ml) for additional 24h, then harvested for flow cytometry and stained for surface expression of the ISG tetherin (CD317) as proxy for the interferon response. Gates show bystander cells (mSc-; left gate), and cells that express high (mSc++; right gate) levels of NS5A-mScarlet. Numbers within gates represent mean fluorescence intensity (MFI) of tetherin (PE-CD317) expression.
Asrani, S. K., Devarbhavi, H., Eaton, J., Kamath, P. S. (2019). Burden of liver diseases in the world. J. Hepatol. 70, 151–171. doi: 10.1016/j.jhep.2018.09.014
Asselah, T., Bièche, I., Mansouri, A., Laurendeau, I., Cazals-Hatem, D., Feldmann, G., et al. (2010). In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 221, 264–274. doi: 10.1002/path.2703
Bailey, J. R., Barnes, E., Cox, A. L. (2019). Approaches, progress, and challenges to hepatitis C vaccine development. Gastroenterology 156, 418–430. doi: 10.1053/j.gastro.2018.08.060
Banning, C., Votteler, J., Hoffmann, D., Koppensteiner, H., Warmer, M., Reimer, R., et al. (2010). A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PloS One 5, e9344. doi: 10.1371/journal.pone.0009344
Banse, P., Moeller, R., Bruening, J., Lasswitz, L., Kahl, S., Khan, A., et al. (2018). CD81 Receptor Regions outside the Large Extracellular Loop Determine Hepatitis C Virus Entry into Hepatoma Cells. Viruses 10, 207. doi: 10.3390/v10040207
Bartosch, B., Thimme, R., Blum, H. E., Zoulim, F. (2009). Hepatitis C virus-induced hepatocarcinogenesis. J. Hepatol. 51, 810–820. doi: 10.1016/j.jhep.2009.05.008
Bayer, K., Banning, C., Bruss, V., Wiltzer-Bach, L., Schindler, M. (2016). Hepatitis C virus is released via a noncanonical secretory route. J. Virol. 90, 10558–10573. doi: 10.1128/JVI.01615-16
Benson, R. E., Sanfridson, A., Ottinger, J. S., Doyle, C., Cullen, B. R. (1993). Downregulation of cell-surface CD4 expression by simian immunodeficiency virus Nef prevents viral super infection. J. Exp. Med. 177, 1561–1566. doi: 10.1084/jem.177.6.1561
Blindenbacher, A., Duong, F. H. T., Hunziker, L., Stutvoet, S. T. D., Wang, X., Terracciano, L., et al. (2003). Expression of hepatitis c virus proteins inhibits interferon α signaling in the liver of transgenic mice. Gastroenterology 124, 1465–1475. doi: 10.1016/S0016-5085(03)00290-7
Bond, S. R., Naus, C. C. (2012). RF-Cloning.org: an online tool for the design of restriction-free cloning projects. Nucleic Acids Res. 40, W209–W213. doi: 10.1093/nar/gks396
Bruening, J., Lasswitz, L., Banse, P., Kahl, S., Marinach, C., Vondran, F. W., et al. (2018). Hepatitis C virus enters liver cells using the CD81 receptor complex proteins calpain-5 and CBLB. PloS Pathog. 14, e1007111. doi: 10.1371/journal.ppat.1007111
Businger, R., Deutschmann, J., Gruska, I., Milbradt, J., Wiebusch, L., Gramberg, T., et al. (2019). Human cytomegalovirus overcomes SAMHD1 restriction in macrophages via pUL97. Nat. Microbiol. 4, 2260–2272. doi: 10.1038/s41564-019-0557-8
Businger, R., Kivimäki, S., Simeonov, S., Vavouras Syrigos, G., Pohlmann, J., Bolz, M., et al. (2021). Comprehensive analysis of human cytomegalovirus- and HIV-mediated plasma membrane remodeling in macrophages. mBio 12, e01770–e01721. doi: 10.1128/mBio.01770-21
Chandra, P. K., Bao, L., Song, K., Aboulnasr, F. M., Baker, D. P., Shores, N., et al. (2014). HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 184, 214–229. doi: 10.1016/j.ajpath.2013.10.005
Chang, G.-W., Hsiao, C.-C., Peng, Y.-M., Vieira Braga, F. A., Kragten, N. A. M., Remmerswaal, E. B. M., et al. (2016). The adhesion G protein-coupled receptor GPR56/ADGRG1 is an inhibitory receptor on human NK cells. Cell Rep. 15, 1757–1770. doi: 10.1016/j.celrep.2016.04.053
Charrin, S., Jouannet, S., Boucheix, C., Rubinstein, E. (2014). Tetraspanins at a glance. J. Cell Sci. 127(17), 3641–3648. doi: 10.1242/jcs.154906
Chen, B. K., Gandhi, R. T., Baltimore, D. (1996). CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J. Virol. 70, 6044–6053. doi: 10.1128/jvi.70.9.6044-6053.1996
Chung, Y. M., Park, K. J., Choi, S. Y., Hwang, S. B., Lee, S. Y. (2001). Hepatitis C virus core protein potentiates TNF-α-induced NF-κB activation through TRAF2-IKKβ-dependent pathway. Biochem. Biophys. Res. Commun. 284, 15–19. doi: 10.1006/bbrc.2001.4936
Cormier, E. G., Tsamis, F., Kajumo, F., Durso, R. J., Gardner, J. P., Dragic, T. (2004). CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl. Acad. Sci. 101, 7270–7274. doi: 10.1073/pnas.0402253101
El-Kafrawy, S. A., El-Daly, M. M., Bajrai, L. H., Alandijany, T. A., Faizo, A. A., Mobashir, M., et al. (2022). Genomic profiling and network-level understanding uncover the potential genes and the pathways in hepatocellular carcinoma. Front. Genet. 13, 880440. doi: 10.3389/fgene.2022.880440
Fink, S. L., Jayewickreme, T. R., Molony, R. D., Iwawaki, T., Landis, C. S., Lindenbach, B. D., et al. (2017). IRE1α promotes viral infection by conferring resistance to apoptosis. Sci. Signal 10, eaai7814. doi: 10.1126/scisignal.aai7814
Fischl, W., Bartenschlager, R. (2013). “High-throughput screening using dengue virus reporter genomes,” in Antiviral methods and protocols. Ed. Gong, E. Y. (Totowa, NJ: Humana Press), 205–219. doi: 10.1007/978-1-62703-484-5_17
Florin, L., Lang, T. (2018). Tetraspanin assemblies in virus infection. Front. Immunol. 9, 1140. doi: 10.3389/fimmu.2018.01140
Gale, M. J., Korth, M. J., Tang, N. M., Tan, S.-L., Hopkins, D. A., Dever, T. E., et al. (1997). Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230, 217–227. doi: 10.1006/viro.1997.8493
Garcia, J. V., Miller, A. D. (1991). Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350, 508–511. doi: 10.1038/350508a0
García-Mediavilla, M. V., Pisonero-Vaquero, S., Lima-Cabello, E., Benedicto, I., Majano, P. L., Jorquera, F., et al. (2012). Liver X receptor α-mediated regulation of lipogenesis by core and NS5A proteins contributes to HCV-induced liver steatosis and HCV replication. Lab. Invest. 92, 1191–1202. doi: 10.1038/labinvest.2012.88
Ghezzi, M. C., Raponi, G., Angeletti, S., Mancini, C. (1998). Serum‐Mediated enhancement of TNF‐α Release by human monocytes stimulated with the yeast form of candida albicans. J. Infect. Dis. 178, 1743–1749. doi: 10.1086/314484
Gillman, R., Lopes Floro, K., Wankell, M., Hebbard, L. (2021). The role of DNA damage and repair in liver cancer. Biochim. Biophys. Acta BBA. - Rev. Cancer 1875, 188493. doi: 10.1016/j.bbcan.2020.188493
Global Burden of Disease Collaborative Network (2021). Global burden of disease study 2019 (GBD 2019) reference life table. Available at: http://ghdx.healthdata.org/record/ihme-data/global-burden-disease-study-2019-gbd-2019-reference-life-table.
Hagen, N., Bayer, K., Rösch, K., Schindler, M. (2014). The intraviral protein interaction network of hepatitis C virus. Mol. Cell. Proteomics 13, 1676–1689. doi: 10.1074/mcp.M113.036301
Hsu, J.-L., Van Den Boomen, D. J. H., Tomasec, P., Weekes, M. P., Antrobus, R., Stanton, R. J., et al. (2015). Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by HCMV US2 in cooperation with UL141. PloS Pathog. 11, e1004811. doi: 10.1371/journal.ppat.1004811
Inoue, G., Horiike, N., Onji, M. (2001). The CD81 expression in liver in hepatocellular carcinoma. Int. J. Mol. Med. 7, 67–71. doi: 10.3892/ijmm.7.1.67
Jeong, S., Lee, Y., Kim, K., Yoon, J., Kim, S., Ha, J., et al. (2021). 2-O-methylhonokiol suppresses HCV replication via TRAF6-mediated NF-kB activation. Int. J. Mol. Sci. 22, 6499. doi: 10.3390/ijms22126499
Ke, P.-Y., Chen, S. S.-L. (2013). Active RNA replication of hepatitis C virus downregulates CD81 expression. PloS One 8, e54866. doi: 10.1371/journal.pone.0054866
Kurden-Pekmezci, A., Cakiroglu, E., Eris, S., Mazi, F. A., Coskun-Deniz, O. S., Dalgic, E., et al. (2023). MALT1 paracaspase is overexpressed in hepatocellular carcinoma and promotes cancer cell survival and growth. Life Sci. 323, 121690. doi: 10.1016/j.lfs.2023.121690
Lee, J.-Y., Cortese, M., Haselmann, U., Tabata, K., Romero-Brey, I., Funaya, C., et al. (2019). Spatiotemporal coupling of the hepatitis C virus replication cycle by creating a lipid droplet- proximal membranous replication compartment. Cell Rep. 27, 3602–3617.e5. doi: 10.1016/j.celrep.2019.05.063
Levy, S., Shoham, T. (2005). The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 5, 136–148. doi: 10.1038/nri1548
Li, Y., Yu, S., Li, L., Chen, J., Quan, M., Li, Q., et al. (2020). KLF4-mediated upregulation of CD9 and CD81 suppresses hepatocellular carcinoma development via JNK signaling. Cell Death Dis. 11, 299. doi: 10.1038/s41419-020-2479-z
Li, Y.-L., Zheng, M.-X., Wang, G. (2016). A personalized approach identifies disturbed pathways and key genes in hepatitis C virus-cirrhosis with hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 20, 4266–4273.
Lim, J., Petersen, M., Bunz, M., Simon, C., Schindler, M. (2022). Flow cytometry based-FRET: basics, novel developments and future perspectives. Cell. Mol. Life Sci. CMLS. 79, 217. doi: 10.1007/s00018-022-04232-2
Lin, W., Kim, S. S., Yeung, E., Kamegaya, Y., Blackard, J. T., Kim, K. A., et al. (2006). Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J. Virol. 80, 9226–9235. doi: 10.1128/JVI.00459-06
Lu, Y., Zhu, Q., Fox, D. M., Gao, C., Stanley, S. A., Luo, K. (2022). SARS-CoV-2 down-regulates ACE2 through lysosomal degradation. Mol. Biol. Cell 33, ar147. doi: 10.1091/mbc.E22-02-0045
Manns, M. P., Buti, M., Gane, E., Pawlotsky, J.-M., Razavi, H., Terrault, N., et al. (2017). Hepatitis C virus infection. Nat. Rev. Dis. Primer. 3, 17006. doi: 10.1038/nrdp.2017.6
Manns, M. P., Maasoumy, B. (2022). Breakthroughs in hepatitis C research: from discovery to cure. Nat. Rev. Gastroenterol. Hepatol. 19, 533–550. doi: 10.1038/s41575-022-00608-8
Mazzocca, A., Liotta, F., Carloni, V. (2008). Tetraspanin CD81-regulated cell motility plays a critical role in intrahepatic metastasis of hepatocellular carcinoma. Gastroenterology 135, 244–256.e1. doi: 10.1053/j.gastro.2008.03.024
Merz, A., Long, G., Hiet, M.-S., Brügger, B., Chlanda, P., Andre, P., et al. (2011). Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286, 3018–3032. doi: 10.1074/jbc.M110.175018
Meyer, K., Kwon, Y.-C., Liu, S., Hagedorn, C. H., Ray, R. B., Ray, R. (2015). Interferon-α inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis C virus infection. Sci. Rep. 5, 9012. doi: 10.1038/srep09012
Mizuno, N., Itoh, H. (2009). Functions and regulatory mechanisms of gq-signaling pathways. Neurosignals 17, 42–54. doi: 10.1159/000186689
Moon, A. M., Singal, A. G., Tapper, E. B. (2020). Contemporary epidemiology of chronic liver disease and cirrhosis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 18, 2650–2666. doi: 10.1016/j.cgh.2019.07.060
Neumann-Haefelin, C., Thimme, R. (2013). “Adaptive immune responses in hepatitis C virus infection,” in Hepatitis C virus: from molecular virology to antiviral therapy. Ed. Bartenschlager, R. (Berlin, Heidelberg: Springer Berlin Heidelberg), 243–262. doi: 10.1007/978-3-642-27340-7_10
Nguyen, H., Sankaran, S., Dandekar, S. (2006). Hepatitis C virus core protein induces expression of genes regulating immune evasion and anti-apoptosis in hepatocytes. Virology 354, 58–68. doi: 10.1016/j.virol.2006.04.028
Pietschmann, T., Kaul, A., Koutsoudakis, G., Shavinskaya, A., Kallis, S., Steinmann, E., et al. (2006). Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A 103, 7408–7413. doi: 10.1073/pnas.0504877103
Pileri, P., Uematsu, Y., Campagnoli, S., Galli, G., Falugi, F., Petracca, R., et al. (1998). Binding of hepatitis C virus to CD81. Science 282, 938–941. doi: 10.1126/science.282.5390.938
Reiss, S., Rebhan, I., Backes, P., Romero-Brey, I., Erfle, H., Matula, P., et al. (2011). Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9, 32–45. doi: 10.1016/j.chom.2010.12.002
Reyland, M. E. (2009). Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front. Biosci. 2386. doi: 10.2741/3385
Sanjana, N. E., Shalem, O., Zhang, F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. doi: 10.1038/nmeth.3047
Sato, Y., Kato, J., Takimoto, R., Takada, K., Kawano, Y., Miyanishi, K., et al. (2006). Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut 55, 1801–1808. doi: 10.1136/gut.2005.070417
Schaller, T., Appel, N., Koutsoudakis, G., Kallis, S., Lohmann, V., Pietschmann, T., et al. (2007). Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 81, 4591–4603. doi: 10.1128/JVI.02144-06
Schmitz, M., Shaban, M., Albert, B., Gökçen, A., Kracht, M. (2018). The crosstalk of endoplasmic reticulum (ER) stress pathways with NF-κB: complex mechanisms relevant for cancer, inflammation and infection. Biomedicines 6, 58. doi: 10.3390/biomedicines6020058
Schnupf, P., Sansonetti, P. J. (2012). Quantitative RT-PCR profiling of the Rabbit Immune Response: Assessment of Acute Shigella flexneri Infection. PloS One 7, e36446. doi: 10.1371/journal.pone.0036446
Selimovic, D., El-Khattouti, A., Ghozlan, H., Haikel, Y., Abdelkader, O., Hassan, M. (2012). Hepatitis C virus-related hepatocellular carcinoma: An insight into molecular mechanisms and therapeutic strategies. World J. Hepatol. 4, 342–355. doi: 10.4254/wjh.v4.i12.342
Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelson, T., et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. doi: 10.1126/science.1247005
Shin, C., Choi, D.-S. (2019). Essential roles for the non-canonical IκB kinases in linking inflammation to cancer, obesity, and diabetes. Cells 8, 178. doi: 10.3390/cells8020178
Shuda, M., Kondoh, N., Imazeki, N., Tanaka, K., Okada, T., Mori, K., et al. (2003). Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 38, 605–614. doi: 10.1016/S0168-8278(03)00029-1
Simonin, Y., Vegna, S., Akkari, L., Grégoire, D., Antoine, E., Piette, J., et al. (2013). Lymphotoxin signaling is initiated by the viral polymerase in HCV-linked tumorigenesis. PloS Pathog. 9, e1003234. doi: 10.1371/journal.ppat.1003234
Steinmann, E., Penin, F., Kallis, S., Patel, A. H., Bartenschlager, R., Pietschmann, T. (2007). Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PloS Pathog. 3, e103. doi: 10.1371/journal.ppat.0030103
Stevenson, M., Meier, C., Mann, A. M., Chapman, N., Wasiak, A. (1988). Envelope glycoprotein of HIV induces interference and cytolysis resistance in CD4+ cells: Mechanism for persistence in AIDS. Cell 53, 483–496. doi: 10.1016/0092-8674(88)90168-7
Strebel, K., Klimkait, T., Martin, M. A. (1988). A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science 241, 1221–1223. doi: 10.1126/science.3261888
Sugden, S., Bego, M., Pham, T., Cohen, É. (2016). Remodeling of the host cell plasma membrane by HIV-1 nef and vpu: A strategy to ensure viral fitness and persistence. Viruses 8, 67. doi: 10.3390/v8030067
Susa, K. J., Rawson, S., Kruse, A. C., Blacklow, S. C. (2021). Cryo-EM structure of the B cell co-receptor CD19 bound to the tetraspanin CD81. Science 371, 300–305. doi: 10.1126/science.abd9836
Susa, K. J., Seegar, T. C., Blacklow, S. C., Kruse, A. C. (2020). A dynamic interaction between CD19 and the tetraspanin CD81 controls B cell co-receptor trafficking. eLife 9, e52337. doi: 10.7554/eLife.52337
Tai, D.-I., Tsai, S.-L., Chang, Y.-H., Huang, S.-N., Chen, T.-C., Chang, K. S. S., et al. (2000). Constitutive activation of nuclear factor kB in hepatocellular carcinoma. Cancer 89, 2274–2281. doi: 10.1002/1097-0142(20001201)89:11<2274::AID-CNCR16>3.0.CO;2-2
Tanaka, M., Ueno, T., Nakahara, T., Sasaki, K., Ishimoto, A., Sakai, H. (2003). Downregulation of CD4 is required for maintenance of viral infectivity of HIV-1. Virology 311, 316–325. doi: 10.1016/s0042-6822(03)00126-0
Tardif, K. D., Mori, K., Kaufman, R. J., Siddiqui, A. (2004). Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279, 17158–17164. doi: 10.1074/jbc.M312144200
Tscherne, D. M., Evans, M. J., von Hahn, T., Jones, C. T., Stamataki, Z., McKeating, J. A., et al. (2007). Superinfection exclusion in cells infected with hepatitis C virus. J. Virol. 81, 3693–3703. doi: 10.1128/JVI.01748-06
Valgimigli, M., Valgimigli, L., Trerè, D., Gaiani, S., Pedulli, G. F., Gramantieri, L., et al. (2002). Oxidative stress EPR measurement in human liver by radical-probe technique. Correlation with etiology, histology and cell proliferation. Free Radic. Res. 36, 939–948. doi: 10.1080/107156021000006653
Vences-Catalán, F., Duault, C., Kuo, C.-C., Rajapaksa, R., Levy, R., Levy, S. (2017). CD81 as a tumor target. Biochem. Soc Trans. 45, 531–535. doi: 10.1042/BST20160478
World Health Organization (2021). Global progress report on HIV, viral hepatitis and sexually transmitted infections, 2021: accountability for the global health sector strategies 2016–2021: actions for impact: web annex 1: key data at a glance (Geneva: World Health Organization). Available at: https://apps.who.int/iris/handle/10665/342808.
Yamane, D., McGivern, D. R., Masaki, T., Lemon, S. M. (2013). “Liver injury and disease pathogenesis in chronic hepatitis C,” in Hepatitis C virus: from molecular virology to antiviral therapy. Ed. Bartenschlager, R. (Berlin, Heidelberg: Springer Berlin Heidelberg), 263–288. doi: 10.1007/978-3-642-27340-7_11
Yoshida, H., Kato, N., Shiratori, Y., Otsuka, M., Maeda, S., Kato, J., et al. (2001). Hepatitis C virus core protein activates nuclear factor kappa B-dependent signaling through tumor necrosis factor receptor-associated factor. J. Biol. Chem. 276, 16399–16405. doi: 10.1074/jbc.M006671200
You, L. R., Chen, C. M., Lee, Y. H. (1999). Hepatitis C virus core protein enhances NF-kappaB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J. Virol. 73, 1672–1681. doi: 10.1128/JVI.73.2.1672-1681.1999
Zhang, T., Ma, C., Zhang, Z., Zhang, H., Hu, H. (2021). NF‐κB signaling in inflammation and cancer. MedComm 2, 618–653. doi: 10.1002/mco2.104
Zhang, Y.-Y., Zhang, B.-H., Ishii, K., Liang, T. J. (2010). Novel function of CD81 in controlling hepatitis C virus replication. J. Virol. 84, 3396–3407. doi: 10.1128/JVI.02391-09
Zheng, Y., Ye, L.-B., Liu, J., Jing, W., Timani, K. A., Yang, X.-J., et al. (2005). Gene expression profiles of heLa cells impacted by hepatitis C virus non-structural protein NS4B. BMB. Rep. 38, 151–160. doi: 10.5483/bmbrep.2005.38.2.151
Zimmerman, B., Kelly, B., McMillan, B. J., Seegar, T. C. M., Dror, R. O., Kruse, A. C., et al. (2016). Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell 167, 1041–1051.e11. doi: 10.1016/j.cell.2016.09.056
Keywords: hepatitis C virus, hepatocellular carcinoma, HCC, CD81, tetraspanin, NF-κB
Citation: Bunz M, Eisele M, Hu D, Ritter M, Kammerloher J, Lampl S and Schindler M (2024) CD81 suppresses NF-κB signaling and is downregulated in hepatitis C virus expressing cells. Front. Cell. Infect. Microbiol. 14:1338606. doi: 10.3389/fcimb.2024.1338606
Received: 14 November 2023; Accepted: 15 January 2024;
Published: 31 January 2024.
Edited by:
Ming Yue, Nanjing Medical University, ChinaReviewed by:
Nadjet Lebsir, Catholic University of Lyon, FranceCopyright © 2024 Bunz, Eisele, Hu, Ritter, Kammerloher, Lampl and Schindler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Schindler, bWljaGFlbC5zY2hpbmRsZXJAbWVkLnVuaS10dWViaW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.