 Meng Xia1
Meng Xia1 Huajun Li
Huajun Li Chuanzhou Gao
Chuanzhou Gao Jingyi Han
Jingyi Han
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 15 May 2024
Sec. Intestinal Microbiome
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1333145
This article is part of the Research Topic The Role of Gut Microbes and Their Metabolites in Immune-related Diseases-Volume II View all 16 articles
Objective: To investigate the structure, composition, and functions of the gut microbiota in elderly patients with hyperlipidemia.
Methods: Sixteen older patients diagnosed with hyperlipidemia (M group) and 10 healthy, age-matched normal volunteers (N group) were included. These groups were further subdivided by sex into the male normal (NM, n = 5), female normal (NF, n = 5), male hyperlipidemia (MM, n = 8), and female hyperlipidemia (MF, n = 8) subgroups. Stool samples were collected for high-throughput sequencing of 16S rRNA genes. Blood samples were collected for clinical biochemical index testing.
Results: Alpha- and beta-diversity analyses revealed that the structure and composition of the gut microbiota were significantly different between the M and N groups. The relative abundances of Bacteroides, Parabacteroides, Blautia, Peptococcus, and Bifidobacterium were significantly decreased, while those of Lactobacillus, Helicobacter, and Desulfovibrio were significantly higher in the M group. There were also significant sex-related differences in microbial structure between the NM and NF groups, and between the MM and MF groups. Through functional prediction with PICRUSt 2, we observed distinct between-group variations in metabolic pathways associated with the gut microbiota and their impact on the functionality of the nervous system. Pearson’s correlation coefficient was used as a distance metric to build co-abundance networks. A hypergeometric test was used to detect taxonomies with significant enrichment in specific clusters. We speculated that modules with Muribaculaceae and Lachnospiraceae as the core microbes play an important ecological role in the intestinal microbiota of the M group. The relative intestinal abundances of Agathobacter and Faecalibacterium in the M group were positively correlated with serum triglyceride and low-density lipoprotein levels, while the relative abundance of Bifidobacterium was negatively correlated with the serum lipoprotein a level.
The prevalence of hyperlipidemia has increased in parallel with the development of the social economy and improvements in living standards (Teramoto et al., 2008). Hyperlipidemia is a medical condition characterized by the abnormal concentration beyond the normal range of one or more types of lipids in the bloodstream and is a result of metabolic disorder (He and Ye, 2020). Abnormal blood lipids affect normal physiological functions, and are closely related to a variety of chronic metabolic diseases (DeBose-Boyd, 2017). They are also considered a main predisposing factor of cardiovascular illnesses, including atherosclerotic disease, coronary heart disease, and myocardial infarction (Wang et al., 2013; Oliveira and Raposo, 2020).
There are about 100 trillion non-pathogenic microorganisms in the human intestine. The number of microorganisms per gram of colon contents can reach 1012 (Kim and Jazwinski, 2018). The gut microbial population collectively encodes millions of genes, which gives it the ability to modify and regulate the physiological functions of the host. Changes in the gut microbiota have typically been related to age, host genes, lifestyle, and epigenetic changes (Adak and Khan, 2019). There are differences in the gut microbiota of healthy people of different physiological ages. For example, the gut microbiota compositions of infants, adults, and the elderly are different (Milani et al., 2017; Salazar et al., 2017). The diversity of the gut microbiota in stool samples of children is significantly lower than that of adults (Iglesias-Vázquez et al., 2020). Studies have shown that the changes in the gut microbiota significantly with age (O’Toole and Jeffery, 2015).
Chronic metabolic diseases such as hyperlipidemia and hyperglycemia occur mostly in elderly people (Ducharme and Radhamma, 2008). Further research has revealed a close relationship between the gut microbiota and hyperlipidemia. Our previous animal experiments showed that the gut microbiota of mice with diet-induced hyperlipidemia is significantly different from that of normal mice, and that the gut microbiota also affects the metabolism of lipids and bile acids in mice (Chen et al., 2019). Another study found that material energy metabolism, the inflammatory response, and insulin resistance in the host all involve the participation of the gut microbiota (Tosti et al., 2018). Increasingly more scholars have come to believe that there is a correlation between hyperlipidemia and the gut microbiota, and the gut microbiota likely plays a role in the occurrence and development of metabolic diseases such as hyperlipidemia (Busnelli et al., 2018; Huang et al., 2019).
In this study, we used high-throughput sequencing of 16S rRNA genes in the fecal microbiota of elderly patients with hyperlipidemia and healthy elderly volunteers to examine differences in the gut microbiota between the two groups and to identify correlations between the gut microbiota and hyperlipidemia. This study may provide guidance and new ideas for the prevention and clinical treatment of hyperlipidemia.
The M group (n = 16) comprised patients who were previously diagnosed with hyperlipidemia (total cholesterol [TC] > 5.7mmo/L) and recruited from the First Affiliated Hospital of Dalian Medical University. The normal (N) group (n = 10) comprised age-matched healthy (TC < 5.2mmol/L) volunteers. The M and N groups were subdivided by sex into the male normal (NM, n = 5), female normal (NF, n = 5), male hyperlipidemia (MM, n = 8), and female hyperlipidemia (MF group, n = 8) groups.
The inclusion criteria were as follows: 1) male or female; 2) meeting diagnostic criteria for hyperlipidemia (M group only) without receiving any lipid-lowering treatment for hyperlipidemia; and 3) body mass index (BMI) within the range of 20–30 kg/m2 for all participants. The exclusion criteria for all participants were as follows: 1) a history of metabolic diseases, including diabetes and thyroid disease; 2) a history of peptic diseases, including intestinal inflammatory ulcers; and 3) use of antibiotics, probiotics, prebiotics, postbiotics, or immunosuppressive agents in the previous 2 months. The study was conducted in accordance with the principles of the Declaration of Helsinki, and the study protocol was approved by the First Affiliated Hospital Ethical Committee of Dalian Medical University (approval number: PJ-KS-KY-2021–90).
Blood samples were separated by centrifugation at 3000 rpm at 4°C for 20 min to obtain serum, which was used to measure levels of the following: uric acid (UA), homocysteine (HCY), fasting blood glucose (FBG), alanine aminotransferase(ALT), aspartate aminotransferase (AST), creatinine (Cre), prealbumin (PAB), albumin (ALB), globulin (GLB), total protein (TP), alkaline phosphatase (ALP), γ-glutamyl transpeptidase (γ-GT), total bilirubin (T-BIL), lipoprotein a (LPa), TC, triglycerides (TG), high -density lipoprotein (HDL), and low-density lipoprotein (LDL).
Fresh samples of feces (200 mg) were collected from each participant into a sterile container and immediately stored at −80°C until further processing. The Stool DNA Isolation Kit (Foregene, China) was used to extract genomic DNA from stool samples, in accordance with the manufacturer’s instructions.
The V3-V4 hypervariable variable regions of genomic DNA samples were amplified by polymerase chain reaction (PCR) using primers 338F (5’-ACTCCTACGGGAGGCAGCA-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’). PCR products were subjected to 16S rRNA gene high-throughput sequencing by Shanghai Maggi Biomedical Technology Co., Ltd., using an Illumina MiSeq PE300. The 16S rRNA gene sequences were defined as one operational taxonomic unit (OTU) based on 97% similarity. The abundance-based coverage estimator (ACE) index, observed species (Sobs) index, Shannon diversity index, and Faith’s phylogenetic diversity (PD) were used to reflect the alpha diversity of samples. Principal component analysis (PCA) and principal co-ordinates analysis (PCoA) were used to analyze beta diversity. Linear discriminant analysis (LDA) was used to screen for dominant microbial communities (Huang et al., 2021). Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was applied to predict functional profiles of the gut microbiota resulting from reference-based OTU picking against the Greengenes database. The predicted genes were then summarized by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway categorization. Pearson’s correlation coefficients were calculated using the difference-rich OTU, and network analysis was conducted. Gephi was used for topology analysis and visualization purposes.
All experimental data are presented as the mean ± standard deviation (SD). SPSS version 22 (IBM, USA) was used to analyze and process data. Statistical analysis of KEGG pathway data was performed with STAMP v2.1.3 using Welsh’s t-test (P < 0.05). R software version 3.5.2 was used to analyze bioinformatics results. Graph Pad Prism Version 8 (Graph Pad Software Inc., USA) was used to draw statistical charts. One-way analysis of variance (ANOVA) and Duncan’s multiple range tests were used to analyze statistical data. A P value < 0.05 was considered to indicate statistical significance; *P < 0.05, **P < 0.01, ***P < 0.001.
Age and BMI were not significantly different between the M and N groups (P > 0.05; Table 1). Serum lipid levels (TC, TG, HDL, LDL, LPa) were significantly higher in the M group than in the N group (P < 0.05; Table 1). There were no significant between-group differences in other serum parameters (UA, HCY, FBG, ALT, AST, Cre, PAB, ALB, GLB, TP, ALP, γ-GT, T-BIL; P > 0.05; Table 1), indicating the comparability between the hyperlipidemia and healthy groups.
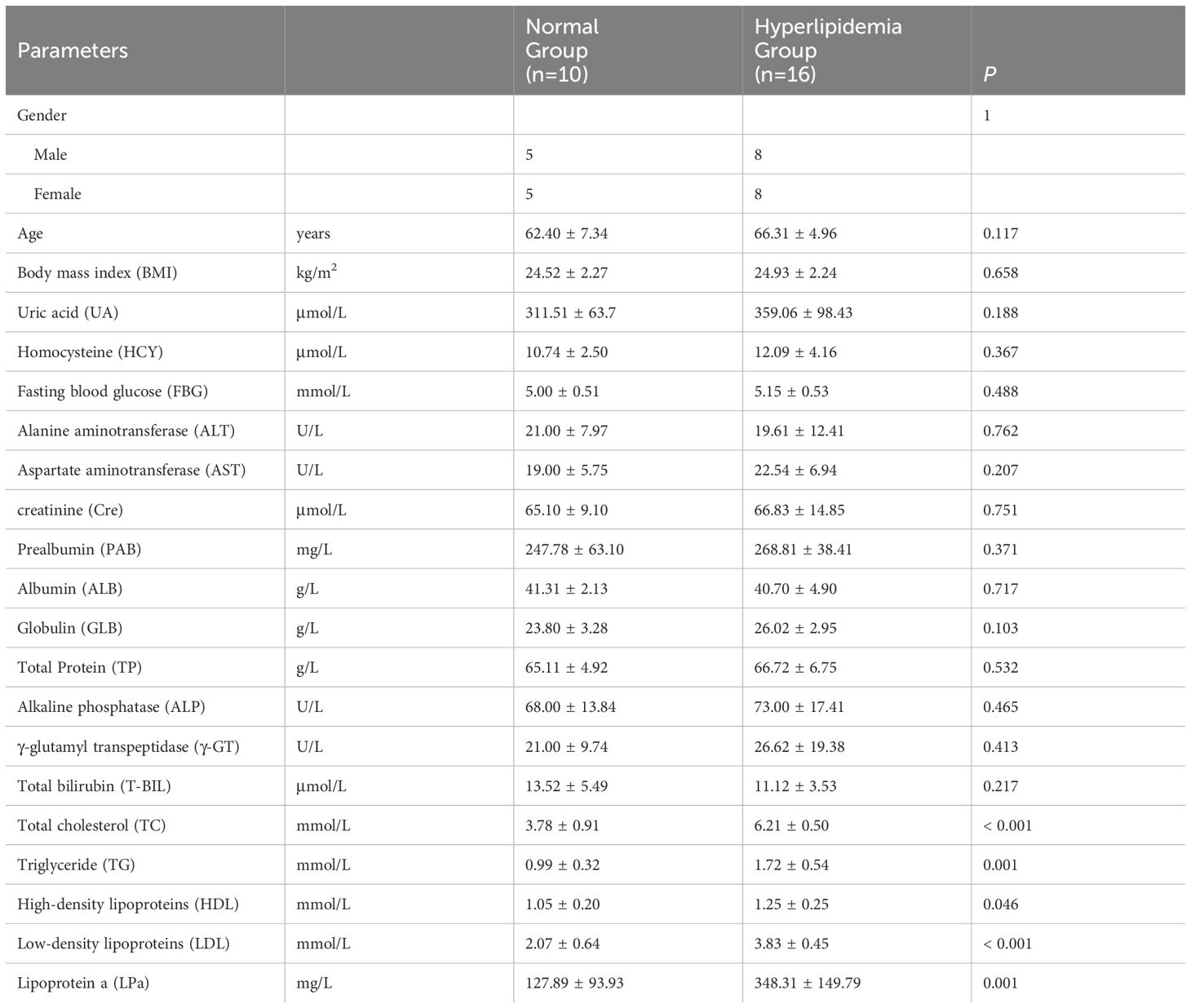
Table 1 Basic clinical information and biochemical indicators of the patient.
Fecal DNA samples with a concentration > 50 ng/µL and a purity ratio of A260/A280 > 1.8 were used to prepare libraries and conduct sequencing (Supplementary Table 1). We used the extracted data volume as the abscissa and the Sobs and Shannon index values as the ordinate to draw the rarefaction curve. Curve flatness was used to determine that the amount of sequence data was sufficient. As shown in Supplementary Figure 1, the sparse Sobs and Shannon curves of the M and N groups both tended to be stable, indicating that the depth of the sequencing data was sufficient to cover most of the microbial information.
The compositions of the gut microbiota at the phylum and genus levels are shown in Figure 1. There were obvious changes in the gut microbiota in the M group. At the phylum level, the top nine most prevalent phyla are shown in Figure 1A. Firmicutes and Bacteroidetes were the dominant phyla in all samples, followed by Desulfobacterota, Campilobacterota, Actinobacteriota and Deferribacterota. The abundances of Campilobacterota and Deferribacterota were significantly higher in the M group than in the N group (P < 0.0001). In the M group, the abundance of Proteobacteria and Cyanobacteria was significantly decreased (P < 0.0001). At the genus level the relative abundances of Bacteroides (P = 0.001), Parabacteroides (P < 0.0001), Blautia (P < 0.0001), Peptococcus (P < 0.0001), and Bifidobacterium (P < 0.0001) were significantly decreased, whereas those of Lactobacillus (P = 0.004), Helicobacter, and Desulfovibrio (P = 0.004) were significantly increased in the M group compared with those in the N group (Figure 1B).
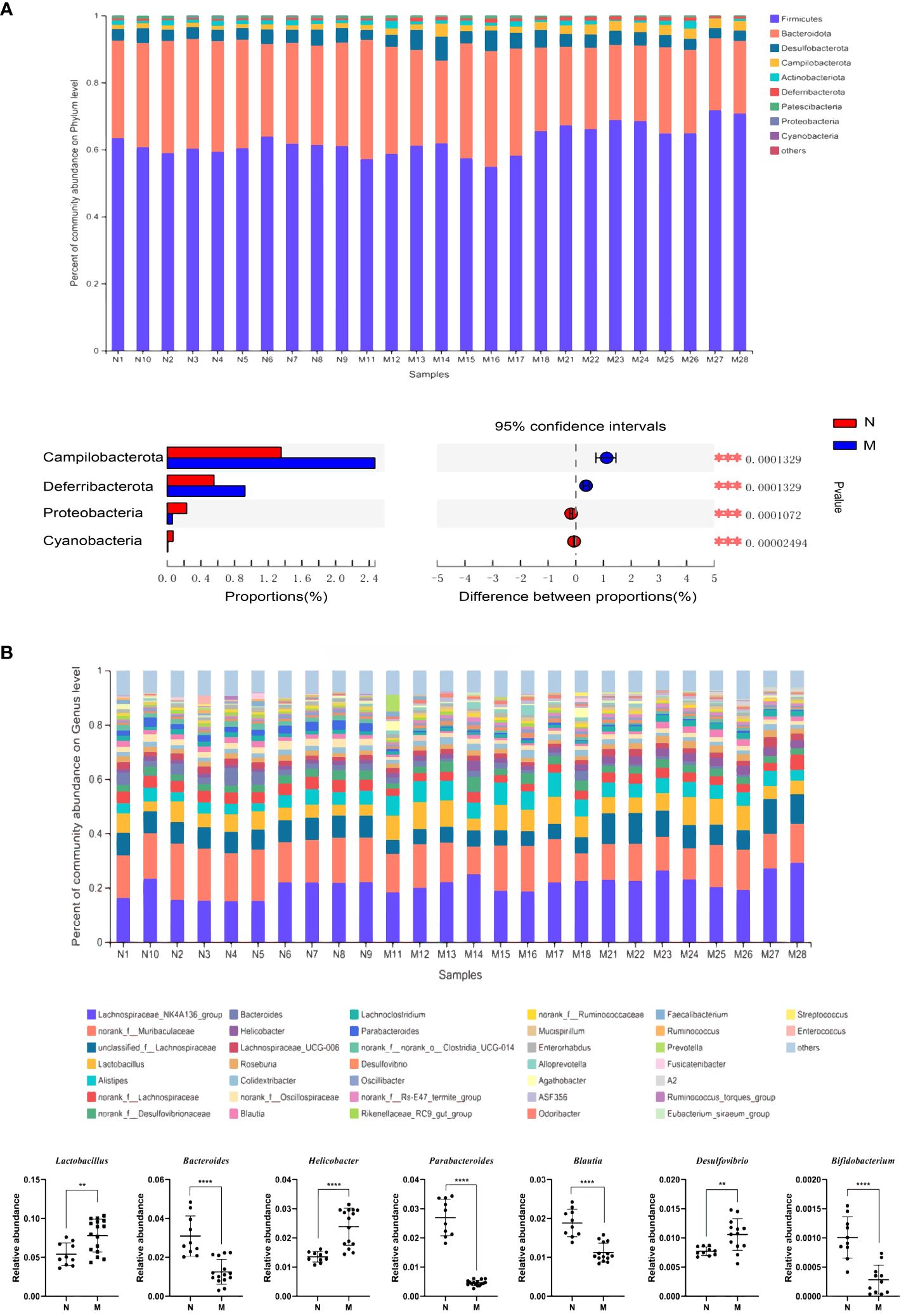
Figure 1 Analysis of the composition of gut microbiota in the N and M group. (A) Microbial distributions of different groups at the phylum level. (B) Microbial distributions of different groups at the genus level. N: normal group (n=10). M: hyperlipidemia group (n=16). **P<0.01, ***P<0.001, ****P<0.001 compared with the normal group.
Alpha-diversity analyses of the gut microbiota in the N and M groups are shown in Figures 2A–D. The Sobs (P = 0.008) and Shannon (P = 0.047) indices based on Kruskal–Wallis analysis showed that the richness and diversity of the gut microbiota were significantly lower in the M group compared with those in the N group. The ACE index (P = 0.023) and Faith’s PD (P = 0.002) metrics further confirmed this result.
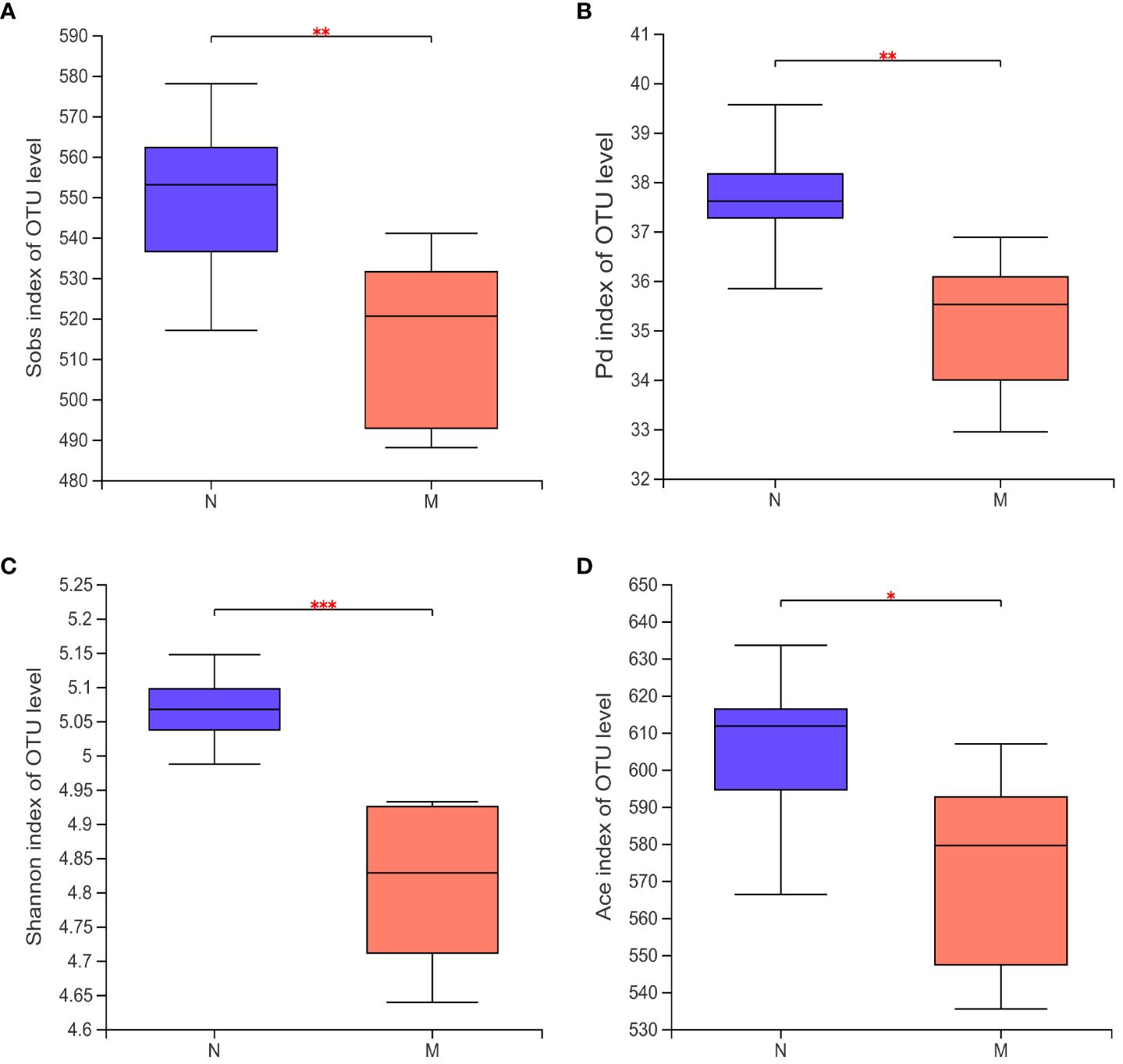
Figure 2 Alpha diversity of the gut microbiota in the N and M group. (A) Sobs index of OUT level. (B) Pd index of OUT level. (C) Shannon index of OUT level. (D) Ace index of OUT level. N: normal group (n=10). M: hyperlipidemia group (n=16). *P<0.05, **P<0.01, ***P<0.001, compared with the normal group.
Figure 3A illustrates the beta-diversity of the gut microbiota in the two groups as hierarchical cluster trees at the OTU level based on Bray–Curtis dissimilarity (Figure 3A). PCA and PCoA revealed that the microbial structures of the M and N groups were significantly different (Figures 3B, C), supporting the difference in gut microbiota composition between healthy individuals and hyperlipidemia patients shown above. Next, we used LDA of effect size (LEfSe) to further screen for dominant microbial communities between the two groups at the genus level. The results indicated that Lactobacillus was enriched in the M group, whereas Parabacteroides and Lachnospiraceae-NK4A136 were enriched in the N group (Figures 3E, F). Bacterial typing analysis categorized the gut microbiota of the N and M groups into four types (Figure 3D). Coincidentally, when sex was considered as a grouping element, elderly healthy participants and hyperlipidemia patients were divided into four groups.
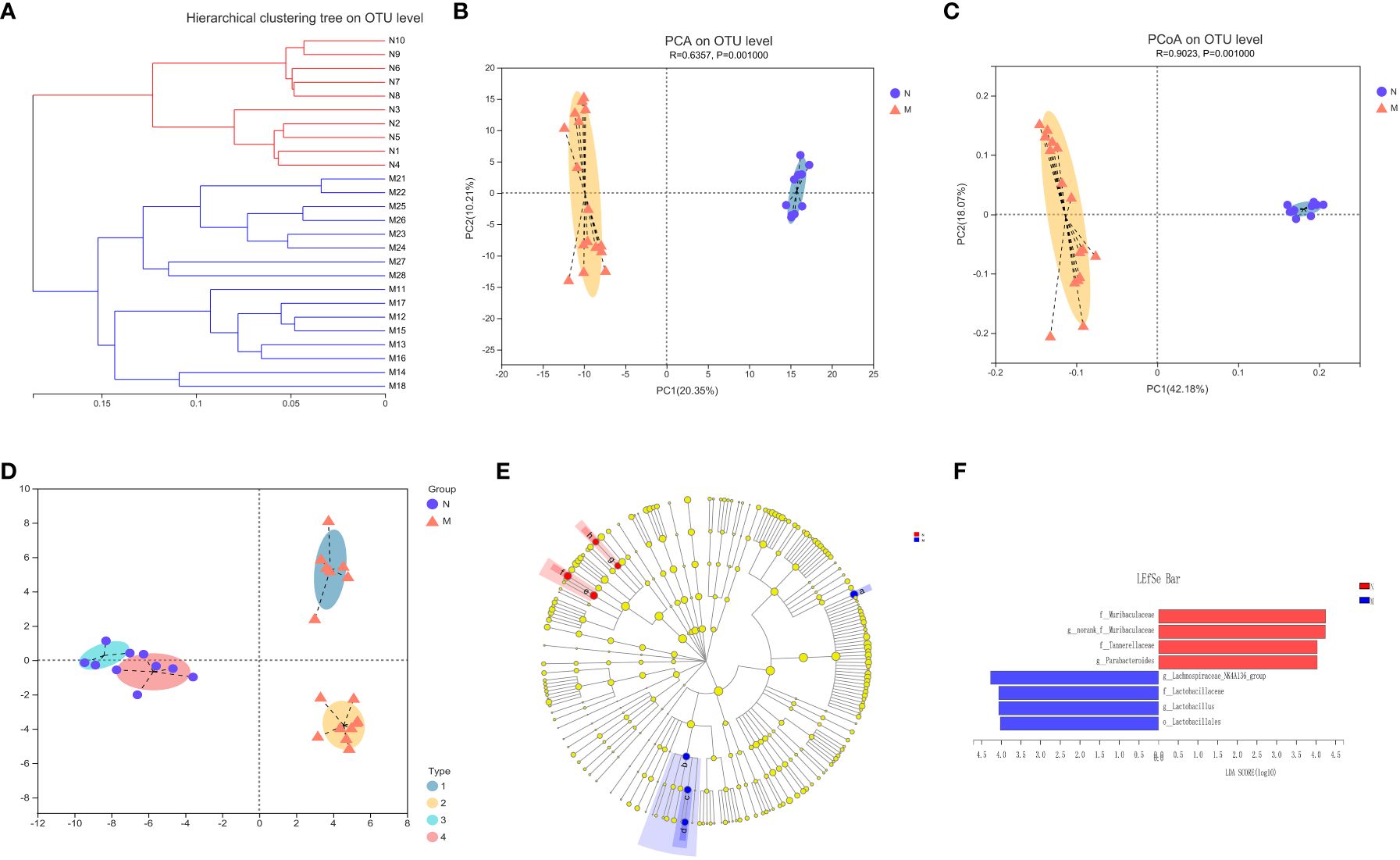
Figure 3 Analysis of the structure and communities of gut microbiota in the N and M group. (A) The microbial composition with the cluster at the OTU level. (B) Principal component analysis (PCA) with the cluster. (C) Principal co-ordinates analysis (PCoA) with cluster. (D) Bacteria typing analysis. (E) Cladogram. (F) LDA distribution. N: normal group (n=10). M: hyperlipidemia group (n=16).
To further investigate the potential impact of sex on the distribution of intestinal microbes, we analyzed the male and female subgroups of elderly healthy volunteers and hyperlipidemia patients (Figure 4). There were significant differences in gut microbial diversity between the male and female subgroups within both the N and M groups, with significant clustering observed on PCA and PCoA. Among the elderly healthy volunteers, the NF subgroup was rich in Parabacteroides, whereas the NM subgroup was rich in Bacteroides. Among the elderly patients with hyperlipidemia, Lachnospiraceae-NK4A136 was enriched in the MF subgroup, whereas Lactobacillus and Alistipes were enriched in the MM subgroup.
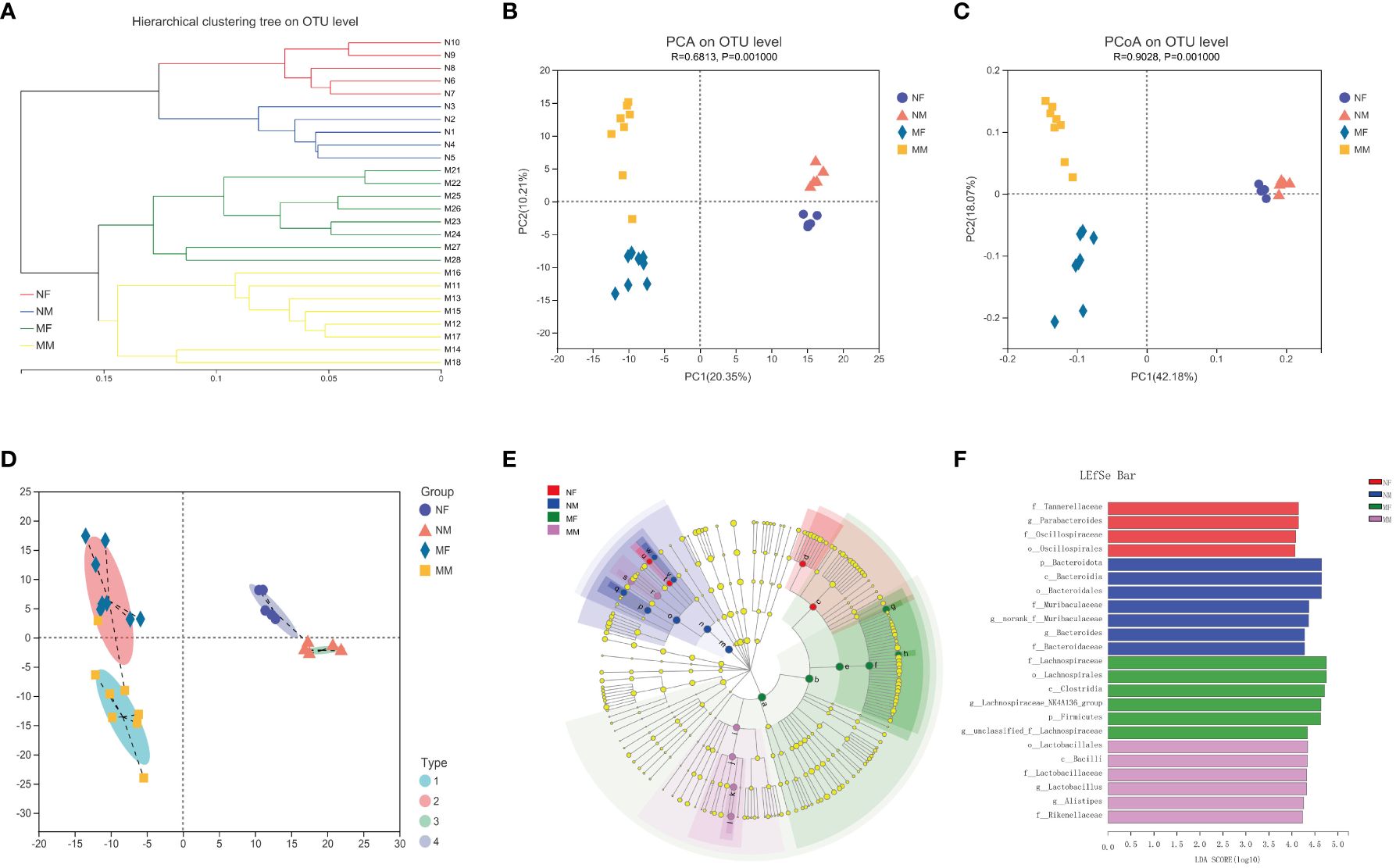
Figure 4 Analysis of the structure and communities of gut microbiota in the NF, NM, MF and MM groups. (A) microbial composition with the cluster at the OTU level. (B) principal component analysis (PCA) with cluster. (C) principal co-ordinates analysis (PCoA) with cluster. (D) bacteria typing analysis. (E) cladogram. (F) LDA distribution. NF group, the female normal group; NM group, the male normal group; MM group, the hyperlipidemia male patient group; MF group, the hyperlipidemia female patient group. NM, male normal group (n=5); NF, female normal group (n=5); MM, the hyperlipidemia male patient group (n=8); MF, hyperlipidemia female patient group (n=8).
KEGG level 2 functional pathway analysis indicated that the nervous system, amino acid metabolism, biosynthesis of other secondary metabolites, endocrine system, transport and catabolism, excretory system, signaling, and cellular processes were significantly reduced in the M group compared to those in the N group. Conversely, infectious disease (bacterial), genetic information processing, circulatory system, drug resistance (antineoplastic), and aging pathways were significantly increased in the M group compared to those in the N group (Figure 5A).
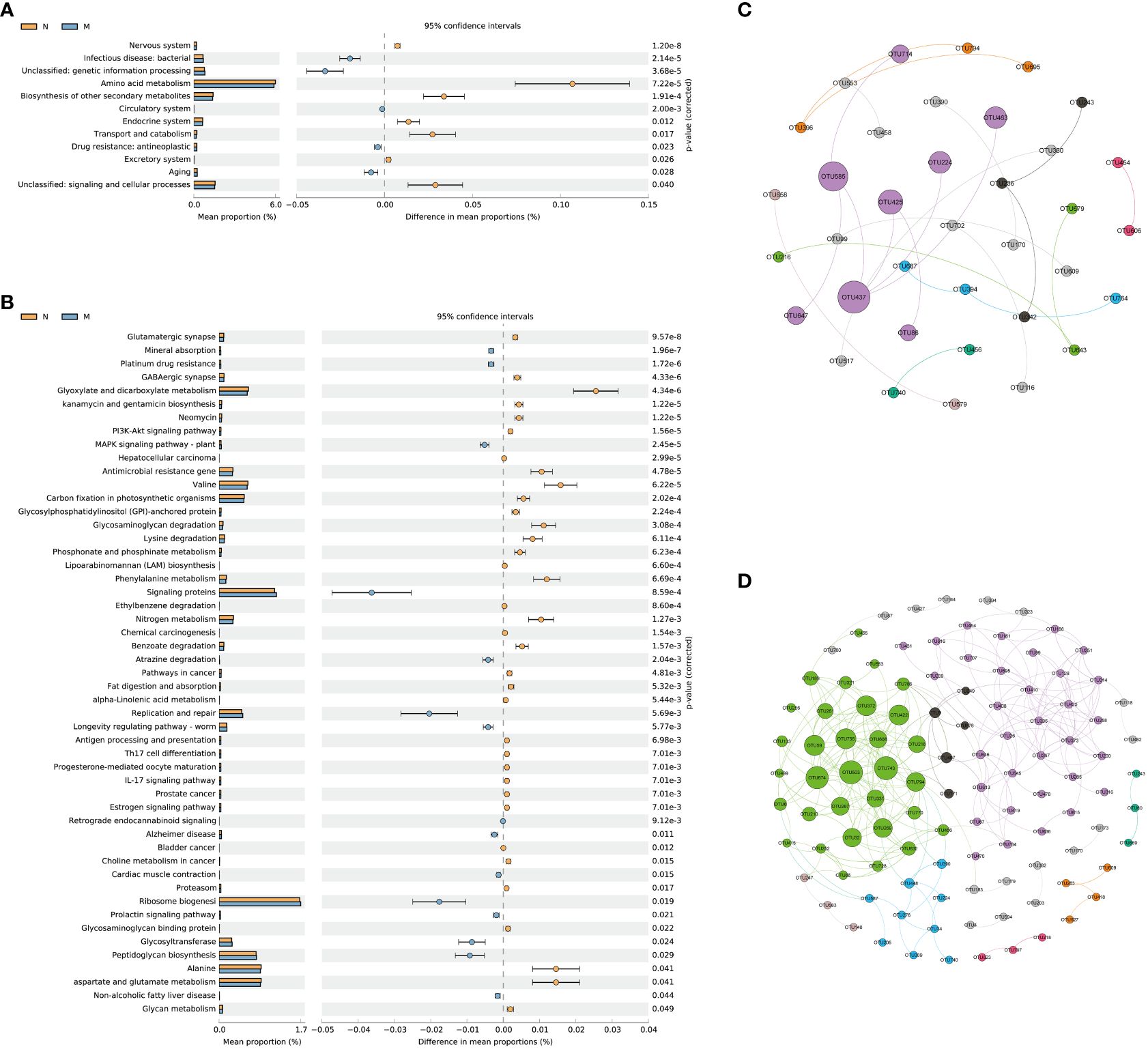
Figure 5 Functional prediction and co-abundance networks. (A) STAMP analysis for the inferred metabolic pathway in level 2. (B) STAMP analysis for the inferred metabolic pathway in level 3. (C) The co-abundance networks of the N group. (D) The co-abundance networks of the M group. N: normal group (n=10). M: hyperlipidemia group (n=16).
At level 3, a total of 51 pathways exhibited significant differences between the two groups. Among the top 10 dominant KEGG pathways, the relative abundances of the glutamatergic synapse, GABAergic synapse, kanamycin and gentamicin biosynthesis, neomycin, the PI3K-Akt signaling pathway, and hepatocellular carcinoma were significantly lower in the M group than in the N group. Conversely, mineral absorption, platinum drug resistance, and the MAPK signaling pathway (plant) displayed significantly higher abundances in the M group than in the N group (Figure 5B).
Functional prediction with PICRUSt 2 revealed distinct variations in metabolic pathways associated with the gut microbiota and their impacts on the functionality of the nervous system between the two groups. Sequence-based characterization, which focuses on how individual taxa within the gut microbiome relate to the host, does not reveal the complex interactions that take place between taxa within microbial communities. Microbes cooperate in networks to provide critical nutrients for each other’s growth and survival. The identification of microbial communities is important for understanding their biological impact on the human body, but is hampered by our inability to culture most microbes. We hypothesized that generating OTU abundance modules from lists of differential OTUs between the two groups would allow us to identify associations with differential changes in function in elderly hyperlipidemia patients.
Figures 5C and D illustrate the more intricate network structure in group M compared to that in the N group, implying reduced stability of the intestinal microbiota in elderly patients with hyperlipidemia. Subsequent examination of the modules revealed that those containing Muribaculaceae and Lachnospiraceae as central microbial species hold significant ecological relevance in elderly patients with hyperlipidemia.
The Spearman’s correlation coefficient was used to assess the relationship between the gut microbiota and blood lipid levels. In the correlation heat map shown in Figure 6, blue represents positive correlations between bacteria and serum parameters, while red represents negative correlations (Figure 6). Specifically, Agathobacter and Faecalibacterium in the intestine of elderly patients with hyperlipidemia were positively correlated with serum TG and LDL levels, while Bifidobacterium was negatively correlated with the serum LPa level (Table 2).
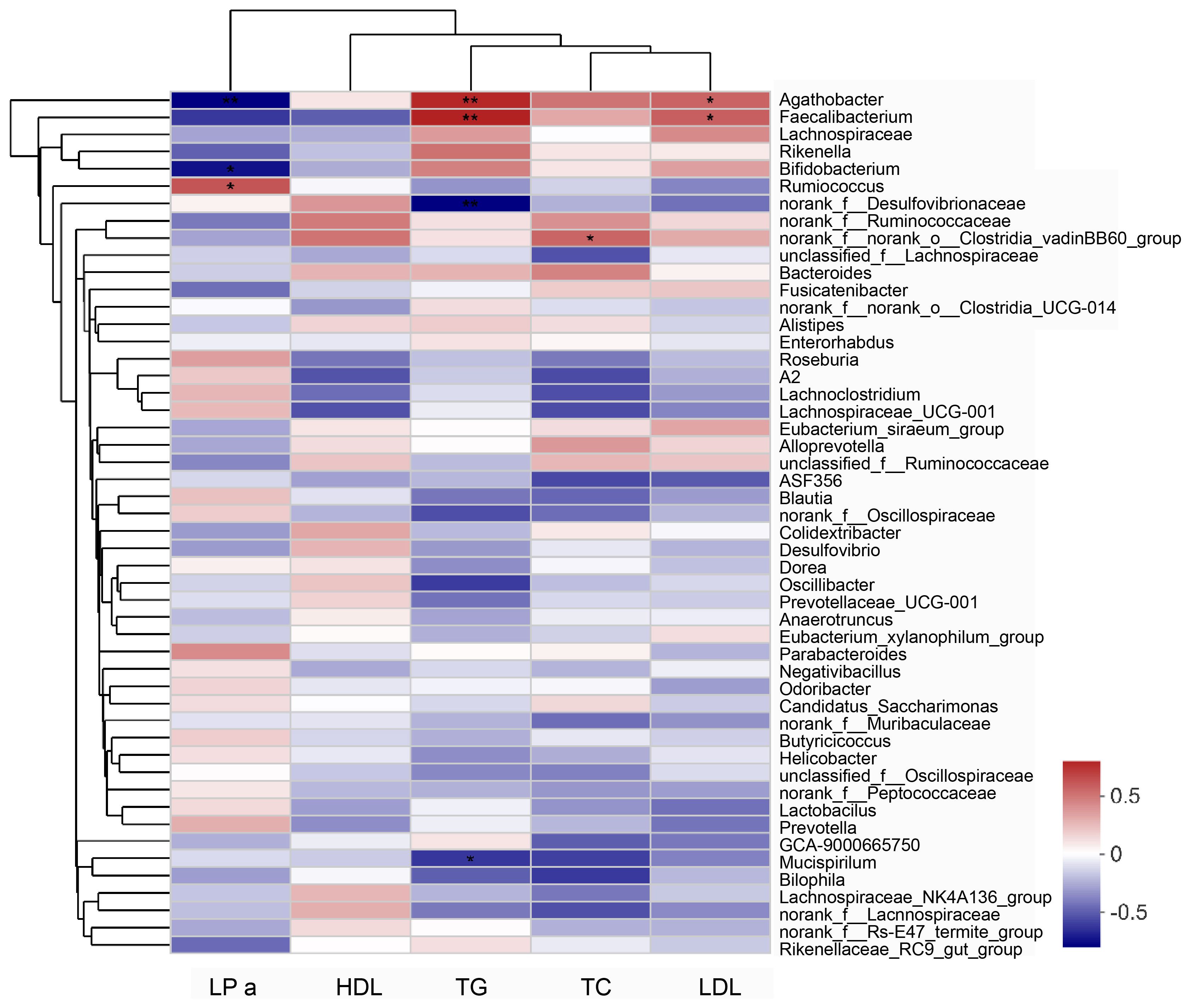
Figure 6 Analysis of the correlation between serum lipid parameters and gut microbiota in patients with hyperlipidemia. “0” means no correlation; “0-(0.5)” means positive correlation; “0-(-0.5)” means negative correlation. Data were analyzed by Spearman test. *P<0.05, **P<0.01. LPa, Lipoprotein a; TC, total cholesterol; TG, triglycerides; HDL, High-density lipoprotein; LDL, Low-density lipoprotein.
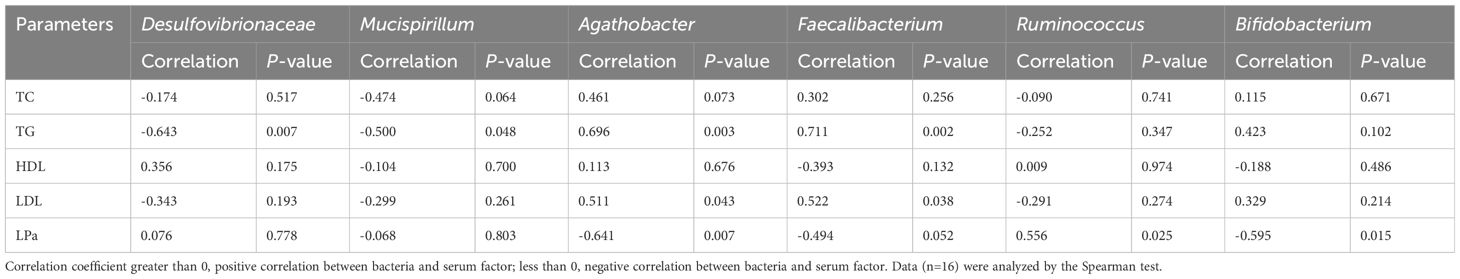
Table 2 Correlation between serum lipid parameters and gut microbiota in patients with hyperlipidemia.
In recent years, increasingly more studies have shown that the normal gut microbiota is inextricably linked with the metabolism of the body, and may regulate blood lipids (Patterson et al., 2016; Hills et al., 2019; Silva et al., 2020). In this research, we focused on exploring hyperlipidemia-related changes in the gut microbiota of elderly men and women. We found that the relative abundances of Bacteroides, Parabacteroides, Blautia, Peptococcus, and Bifidobacterium in the gut microbiota were significantly reduced, while those of Lactobacillus, Helicobacter, and Desulfovibrio were significantly increased with hyperlipidemia. Similar results have been observed in previous studies of patients with dyslipidemia, reporting varying degrees of changes in gut microbes, including Lactobacillus, Bifidobacterium, Bacteroidetes, Enterococcus, Enterobacteriaceae species, Clostridium, in which the proportion of Lactobacillus was upregulated (Song et al., 2017; Gargari et al., 2018; Jia et al., 2021; Guo et al., 2022).
Under normal circumstances, certain common intestinal bacteria produce cholesterol oxidase to accelerate the degradation of cholesterol, thereby participating in maintaining the normal level of cholesterol in the body. In addition, beneficial bacteria in the intestines, such as Clostridium, Bifidobacterium, Bacteroides, and Enterococcus produce bound bile acid hydrolase, which converts bound bile acid into free bile acid (Cai et al., 2022; Pushpass et al., 2022). However, under conditions of hyperlipidemia, the living environment of the gut microbiota undergoes significant changes, with the low relative abundances of Bifidobacterium and Bacteroides leading to an increase in accumulated cholesterol.
Some studies reporting the effects of lactic acid bacteria and fermented dairy products on blood lipids found that lactic acid bacteria, including Bifidobacteria lower serum cholesterol levels (Schoeler and Caesar, 2019). Most of these bacteria act on TG, TC, HDL, and LDL in the serum, thereby reducing blood lipids. It has also been reported that the content of TG in the serum of patients with hyperlipidemia is significantly negatively correlated with Bifidobacterium and Lactobacillus, and positively correlated with Enterobacteriaceae and Enterococcus (Osman et al., 2005). Our study found that the abundances of Agathobacter and Faecalibacterium in the intestines of elderly patients with hyperlipidemia were positively correlated with serum TG and LDL levels, while Bifidobacterium abundance was negatively correlated with the serum LPa level.
Gut microbial diversity changes throughout the human life span and is known to be associated with the sex of the host. The latest research has shown that many characteristics, including sex, age, TG and uric acid levels, obesity, and lifestyle, have significant impacts on the gut microbiota (Portune et al., 2017; Just et al., 2018; Ma et al., 2020). One study found that the gut microbiota is obviously dependent on sex, with higher alpha-diversity in women than in men (de la Cuesta-Zuluaga et al., 2019). Consistently, our research showed significant sex-related impacts on the microbiota structure and diversity of patients with hyperlipidemia. Beta-diversity analyses performed on the Bray-Curtis distance matrix showed different hierarchical cluster trees at the OTU level between healthy elderly males and females and between elderly males and females with hyperlipidemia. PCA and PCoA also showed significant differences in the microbial structure between elderly male and female patients with hyperlipidemia.
Numerous studies have established correlations between abundances of Muribaculaceae and Lachnospiraceae and metabolic diseases. Muribaculaceae bacteria are involved in the synthesis of short-chain fatty acids and influence the host’s metabolic function (Song et al., 2022; Bai et al., 2023). Lachnospiraceae bacteria possess the capacity to reduce inflammation and have been associated with cancer and neurological diseases (Shen et al., 2021; Du et al., 2023).
This study has one main limitation. While our aim was to explore sex- and hyperlipidemia-related changes in the gut microbiota in elderly patients, the number of participants was limited. We are currently conducting animal and cell experiments to further validate and explore the underlying mechanisms of our results.
In summary, the structure and composition of the gut microbiota in elderly patients with hyperlipidemia appear to undergo significant changes that are closely related to serum lipid levels and metabolic pathway activity. We also discovered sex-related differences in the distribution of the gut microbiota. Interestingly, modules with Muribaculaceae and Lachnospiraceae as the core microbes played an important ecological role in the gut microbiota of elderly patients with hyperlipidemia. Consideration of the relationship between the gut microbiota and hyperlipidemia should include the impact of sex differences.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject ID is PRJNA1063629.
The studies involving humans were conducted in accordance with the principles of the Declaration of Helsinki and approved by the ethics committee of the First Affiliated Hospital Ethical Committee of Dalian Medical University. Approval Number: PJ-KS-KY-2021-90. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
MX: Writing – original draft, Writing – review & editing. YX: Writing – review & editing. HL: Writing – review & editing, Methodology, Conceptualization. JuH: Writing – review & editing, Data curation, Writing – original draft. HZ: Data curation, Writing – review & editing, Writing – original draft. CG: Writing – review & editing. JiH: Writing – original draft, Writing – review & editing, Conceptualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Department of Education (Serving Local Project) of Liaoning Province (LF2017001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1333145/full#supplementary-material
Adak, A., Khan, M. R. (2019). An insight into gut microbiota and its functionalities. Cell Mol. Life Sci. 76, 473–493. doi: 10.1007/s00018-018-2943-4
Bai, Y. F., Yue, Z. L., Wang, Y. N., Li, Y. D., Li, C., Liu, X. T., et al. (2023). Synergistic effect of polysaccharides and flavonoids on lipid and gut microbiota in hyperlipidemic rats. Food Funct. 14, 921–933. doi: 10.1039/D2FO03031D
Busnelli, M., Manzini, S., Sirtori, C. R., Chiesa, G., Parolini, C. (2018). Effects of vegetable proteins on hypercholesterolemia and gut microbiota modulation. Nutrients 10, 1249. doi: 10.3390/nu10091249
Cai, J., Sun, L., Gonzalez, F. J. (2022). Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 30, 289–300. doi: 10.1016/j.chom.2022.02.004
Chen, Q., Liu, M., Zhang, P., Fan, S., Huang, J., Yu, S., et al. (2019). Fucoidan and galactooligosaccharides ameliorate high-fat diet-induced dyslipidemia in rats by modulating the gut microbiota and bile acid metabolism. Nutrition 65, 50–59. doi: 10.1016/j.nut.2019.03.001
DeBose-Boyd, R. A. (2017). Significance and regulation of lipid metabolism. Semin. Cell Dev. Biol. 81, 97. doi: 10.1016/j.semcdb.2017.12.003
de la Cuesta-Zuluaga, J., Kelley, S. T., Chen, Y., Escobar, J. S., Mueller, N. T., Ley, R. E., et al. (2019). Age and sex dependent patterns of gut microbial diversity in human adults. mSystems 4, e00261–e00219. doi: 10.1128/mSystems.00261-19
Du, L., Wang, Q., Ji, S., Sun, Y., Huang, W., Zhang, Y., et al. (2023). Metabolomic and microbial remodeling by Shanmei capsule improves hyperlipidemia in high fat food-induced mice. Front. Cell Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.729940
Ducharme, N., Radhamma, R. (2008). Hyperlipidemia in the elderly. Clin. Geriatr. Med. 24, 471–487. doi: 10.1016/j.cger.2008.03.007
Gargari, G., Deon, V., Taverniti, V., Gardana, C., Denina, M., Riso, P., et al. (2018). Evidence of dysbiosis in the intestinal microbial ecosystem of children and adolescents with primary hyperlipidemia and the potential role of regular hazelnut intake. FEMS Microbiol. Ecol. 94 (5). doi: 10.1093/femsec/fiy045
Guo, L., Wang, Y. Y., Wang, J. H., Zhao, H. P., Yu, Y., Wang, G. D., et al. (2022). Associations of gut microbiota with dyslipidemia based on sex differences in subjects from Northwestern China. World J. Gastroenterol. 28, 3455–3475. doi: 10.3748/wjg.v28.i27.3455
He, N., Ye, H. (2020). Exercise and hyperlipidemia. Adv. Exp. Med. Biol. 1228, 79–90. doi: 10.1007/978–981-15–1792-1_5
Hills, R.D., Jr, Pontefract, B. A., Mishcon, H. R., Black, C. A., Sutton, S. C., Theberge, C. R. (2019). Gut microbiome: profound implications for diet and disease. Nutrients 11, 1613. doi: 10.3390/nu11071613
Huang, J., Huang, J., Li, Y., Wang, Y., Wang, F., Qiu, X., et al. (2021). Sodium alginate modulates immunity, intestinal mucosal barrier function, and gut microbiota in cyclophosphamide-induced immunosuppressed BALB/c mice. J. Agric. Food Chem. 69, 7064–7073. doi: 10.1021/acs.jafc.1c02294
Huang, F., Zheng, X., Ma, X., Jiang, R., Zhou, W., Zhou, S., et al. (2019). Theabrownin from Pu-erh tea attenuates hypercholesterolemia via modulation of gut microbiota and bile acid metabolism. Nat. Commun. 10, 4971. doi: 10.1038/s41467-019-12896-x
Iglesias-Vázquez, L., Van Ginkel Riba, G., Arija, V., Canals, J. (2020). Composition of gut microbiota in children with autism spectrum disorder: a systematic review and meta-analysis. Nutrients 12, 792. doi: 10.3390/nu12030792
Jia, X., Xu, W., Zhang, L., Li, X., Wang, R., Wu, S. (2021). Impact of gut microbiota and microbiota-related metabolites on hyperlipidemia. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.634780
Just, S., Mondot, S., Ecker, J., Wegner, K., Rath, E., Gau, L., et al. (2018). The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 6, 134. doi: 10.1186/s40168-018-0510-8
Kim, S., Jazwinski, S. M. (2018). The gut microbiota and healthy aging: a mini-review. Gerontology 64, 513–520. doi: 10.1159/000490615
Ma, J., Hong, Y., Zheng, N., Xie, G., Lyu, Y., Gu, Y., et al. (2020). Gut microbiota remodeling reverses aging-associated inflammation and dysregulation of systemic bile acid homeostasis in mice sex-specifically. Gut Microbes 11, 1450–1474. doi: 10.1080/19490976.2020.1763770
Milani, C., Duranti, S., Bottacini, F., Casey, E., Turroni, F., Mahony, J., et al. (2017). The first microbial colonizers of the human gut: composition, activities, and health Implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 81, e00036–e00017. doi: 10.1128/MMBR.00036-17
Oliveira, H. C. F., Raposo, H. F. (2020). Cholesteryl ester transfer protein and lipid metabolism and cardiovascular diseases. Adv. Exp. Med. Biol. 1276, 15–25. doi: 10.1007/978–981-15–6082-8_2
Osman, N., Adawi, D., Ahrné, S., Jeppsson, B., Molin, G. (2005). Probiotic strains of Lactobacillus and Bifidobacterium affect the translocation and intestinal load of Enterobacteriaceae differently after D-galactosamine-induced liver injury in rats. Microbial Ecol. Health Disease. 17, 40–46. doi: 10.1080/08910600510028433
O’Toole, P. W., Jeffery, I. B. (2015). Gut microbiota and aging. Science 350, 1214–1215. doi: 10.1126/science.aac8469
Patterson, E., Ryan, P. M., Cryan, J. F., Dinan, T. G., Ross, R. P., Fitzgerald, G. F., et al. (2016). Gut microbiota, obesity and diabetes. Postgrad Med. J. 92, 286–300. doi: 10.1136/postgradmedj-2015-133285
Portune, K. J., Benítez-Páez, A., Del Pulgar, E. M., Cerrudo, V., Sanz, Y. (2017). Gut microbiota, diet, and obesity-related disorders-the good, the bad, and the future challenges. Mol. Nutr. Food Res. 61, 1600252. doi: 10.1002/mnfr.201600252
Pushpass, R. G., Alzoufairi, S., Jackson, K. G., Lovegrove, J. A. (2022). Circulating bile acids as a link between the gut microbiota and cardiovascular health: impact of prebiotics, probiotics and polyphenol-rich foods. Nutr. Res. Rev. 35, 161–180. doi: 10.1017/S0954422421000081
Salazar, N., Valdés-Varela, L., González, S., Gueimonde, M., de Los Reyes-Gavilán, C. G. (2017). Nutrition and the gut microbiome in the elderly. Gut Microbes 8, 82–97. doi: 10.1080/19490976.2016.1256525
Schoeler, M., Caesar, R. (2019). Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 20, 461–472. doi: 10.1007/s11154-019-09512-0
Shen, T., Yue, Y., He, T., Huang, C., Qu, B., Lv, W., et al. (2021). The association between the gut microbiota and Parkinson’s disease, a meta-analysis. Front. Aging Neurosci. 13. doi: 10.3389/fnagi.2021.636545
Silva, Y. P., Bernardi, A., Frozza, R. L. (2020). The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. (Lausanne). 11. doi: 10.3389/fendo.2020.00025
Song, Y., Cai, Q., Wang, S., Li, L., Wang, Y., Zou, S., et al. (2022). The ameliorative effect and mechanisms of ruditapes philippinarum bioactive peptides on obesity and hyperlipidemia induced by a high-fat diet in mice. Nutrients 14, 5066. doi: 10.3390/nu14235066
Song, J. J., Tian, W. J., Kwok, L. Y., Wang, Y. L., Shang, Y. N., Menghe, B., et al. (2017). Effects of microencapsulated Lactobacillus plantarum LIP-1 on the gut microbiota of hyperlipidaemic rats. Br. J. Nutr. 118, 481–492. doi: 10.1017/S0007114517002380
Teramoto, T., Sasaki, J., Ueshima, H., Egusa, G., Kinoshita, M., Shimamoto, K., et al. (2008). Primary hyperlipidemia. J. Atheroscler Thromb. 15, 49–51. doi: 10.5551/jat.E590
Tosti, V., Bertozzi, B., Fontana, L. (2018). Health benefits of the mediterranean diet: metabolic and molecular mechanisms. J. Gerontol A Biol. Sci. Med. Sci. 73, 318–326. doi: 10.1093/gerona/glx227
Keywords: hyperlipidemia, gut microbiota, 16S rRNA gene high throughput sequencing, elderly people, lipid metabolism
Citation: Xia M, Xu Y, Li H, Huang J, Zhou H, Gao C and Han J (2024) Structural and functional alteration of the gut microbiota in elderly patients with hyperlipidemia. Front. Cell. Infect. Microbiol. 14:1333145. doi: 10.3389/fcimb.2024.1333145
Received: 04 November 2023; Accepted: 30 April 2024;
Published: 15 May 2024.
Edited by:
Alain Stintzi, University of Ottawa, CanadaReviewed by:
Maria Oana Sasaran, “George Emil Palade” University of Medicine, Pharmacy, Sciences and Technology of Târgu Mureş, RomaniaCopyright © 2024 Xia, Xu, Li, Huang, Zhou, Gao and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingyi Han, amluZ3lpNzIzNkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.